

Abstract
Z-DNA Binding protein 1 (ZBP1) activates Receptor Interacting Protein Kinase 3 (RIPK3) - dependent cell death during lytic infection by members of the orthomyxovirus, herpesvirus and poxvirus families. ZBP1 possesses two Zα domains capable of selective binding to Z-DNA, as well as to Z-RNA. We have now unveiled Z-RNA as the ligand that activates ZBP1 in cells infected with orthomyxoviruses (influenza A and B viruses) and the poxvirus vaccinia virus (VACV). Orthomyxovirus Z-RNA is sensed by ZBP1 in the nucleus of infected cells, resulting in nuclear activation of RIPK3, consequent rupture of the nucleus, and hyper-inflammatory ‘nuclear necroptosis’. VACV-generated Z-RNA accumulates in the cytoplasm, where it is sequestered from ZBP1 by E3, the viral E3L gene product. In viruses where the E3 Zα domain has been mutated, ZBP1 senses Z-RNA and triggers RIPK3-dependent necroptosis in the cytoplasm. Z-RNA is thus a new viral pathogen-associated molecular pattern (PAMP).
Keywords: Z-NA, DAI, DLM-1, RIPK3, MLKL, cell death, necroptosis, influenza A virus, vaccinia virus, poxvirus, herpesvirus
“No one here gets out alive”
– Jim Morrison (of The Doors)
Introduction.
Virus replication dictates the fate of the infected cell. Some lytic viruses destroy the cell within hours, releasing enormous amounts of progeny. Other viruses establish persistent or latent infections that may not impact - or may even improve - the viability of the host cell. Most poxviruses and all orthomyxoviruses initiate lytic infections, while herpesviruses initiate either lytic or latent infections depending on the cell type that becomes infected. For example, herpes simplex virus-1 and -2 (HSV-1 and HSV-2), while lytic in epithelial cells, initiate a non-lytic infection of neurons, switching to a latent life cycle lasting for the remaining lifespan of the host and often accompanied by recurrence at the epithelial surface.
Cell death triggered by lytic infection can promote both virus clearance and pathology. The ‘poxes’ caused by variola virus, a poxvirus and the etiological agent of smallpox, and the vesicular rashes of HSV or varicella zoster viruses largely result from lysis of infected cells, sometimes in combination with the host response to infection. Similarly, bronchioalveolar injury characterized by epithelial cell death are hallmarks of severe influenza A virus (IAV), allowing secondary bacterial pneumonia to take hold and predisposing to IAV-associated acute respiratory distress syndrome (ARDS). In many of these settings, cell death may be delayed until after viral progeny have been produced, favoring the virus, or death may cut short infection by eliminating cells before progeny are generated, acting in defense of the host.
Virus-activated pathways of apoptosis and necroptosis.
For decades, apoptosis was considered the dominant, if not sole, mechanism of programmed cell death activated by virus infections in multicellular organisms [1]. This caspase-mediated pathway systematically dismantles infected cells and produces discrete membrane-bound apoptotic bodies that are then rapidly engulfed, phagocytosed, and destroyed, thus preventing or slowing virus spread [2]. The importance of apoptosis to host defense and virus control is underscored by the myriad viral mechanisms capable of blocking this mode of death. For example, virus-encoded caspase inhibitors (e.g., baculovirus p35, cowpox virus CrmA, VACV B13R, cytomegalovirus UL36, HSV ICP6) and anti-apoptotic proteins which function like host Bcl-2 family cell death suppressors (e.g., adenovirus E1B-19K, VACV F1L, cytomegalovirus UL37x1) prevent apoptosis of infected cells [3–6]. Indeed, blocking apoptosis by simply overexpressing Bcl-2 can change a lytic alphavirus infection into a persistent one [7], and eliminating viral suppression of apoptosis during latency results in reactivation of HSV-1 [8]. Thus, simply interfering with the apoptosis machinery can covert the infected host cell into a factory capable of sustained or recurrent progeny virion production! Such elegant findings demonstrate the importance of apoptosis in limiting virus production and spread to uninfected tissues [9–12].
Host adaptation to viral anti-apoptotic strategies likely resulted in the evolution, mostly within mammalian orders, of caspase-independent forms of death [6,13]. Indeed, apoptotic caspase inhibition in some settings does not prevent cell death, but instead switches the mode of death from apoptosis to (caspase-independent) programmed necrosis. Such necrotic death is observed when death receptors of the TNF receptor superfamily are activated in the presence of caspase inhibitors, or when cells are exposed to innate-immune stimuli such as double-stranded (ds)RNA (a virus mimetic) or to antiviral cytokines, such as interferons (IFNs) [14–21]. No matter the trigger, this programmed cell death pathway, called necroptosis, is mediated by the protein kinase RIPK3 and its substrate, the Mixed Lineage Kinase-Like protein MLKL [22–26].
Necroptosis is a potent host defense mechanism capable of bypassing caspase-8 inhibition by viruses and promoting the elimination of infected cells [6]. Indeed, work over the past decade has demonstrated that the ‘lytic’ death triggered by diverse virus families in many cell types is dependent on RIPK3 and can manifest as either apoptosis, necroptosis, or both, dictated by the presence or absence of virus-encoded inhibitors of each pathway. For example, the herpesvirus MCMV encodes an inhibitor of caspase-8 (M36-encoded Inhibitor of Caspase-8 Activation, or vICA) as well as an inhibitor of necroptosis (M45-encoded Inhibitor of RIP kinase Activation, or vIRA). A mutant of MCMV lacking vICA selectively induces apoptosis, the vIRA mutant primarily triggers necroptosis, and a mutant virus deficient in vICA and vIRA activates both death pathways [27]. Notably, orthomyxoviruses such as IAV lack obvious inhibitors of either pathway and therefore activate both death modalities in infected cells [28,29]. IAV-infected epithelial cells and fibroblasts may undergo either apoptosis or necroptosis; apoptosis is dictated by RIPK3-dependent recruitment and activation of caspase-8, and necroptosis by RIPK3-dependent phosphorylation of MLKL [29]. Of note, pyroptosis has also been reported in IAV-infected myeloid cells [30]. Apoptosis and necroptosis are mutually exclusive cell fates, indicating that a molecular switch may dictate downstream events leading to one mode of death or another [31].
ZBP1 as cell-death activating sensor of viruses.
A breakthrough in our understanding of RIPK3-dependent activation of apoptotic or necroptotic death came with the discovery that the host sensor protein ZBP1 (also called DAI or DLM-1) controls activation of RIPK3 during infection with herpesviruses such as MCMV and HSV-1, the poxvirus VACV, and the orthomyxoviruses IAV and IBV [30,32–35]. ZBP1 was initially described as a sensor of cytosolic dsDNA in the pathway leading to activation of a type I IFN response [36]. But soon afterwards, ZBP1 was shown to be dispensable for induction of type I IFNs by dsDNA [37,38]. Instead, we and others discovered that ZBP1 contains a functional RIP homotypic interaction motif (RHIM), and associates with RIPK3 and RIPK1 [39,40]. We subsequently showed that MCMV encodes a specific inhibitor of RHIM-mediated signal transduction, the aforementioned necroptosis suppressor vIRA [41]. vIRA prevents ZBP1-mediated recruitment of RIPK3 and consequent necroptosis during infection. When vIRA is mutated or deleted, ZBP1 associates with RIPK3 and triggers necroptosis [33]. Homotypic RHIM:RHIM interactions between ZBP1 and RIPK3 initiates RIPK3 activation and cell death, but how does ZBP1 sense virus infections to activate RIPK3 in the first place?
ZBP1 possesses two N-terminal Zα domains (Fig. 1A) and is a member of the Zα family of proteins. The founding member of this family, adenosine deaminase RNA-dependent (ADAR) p150, binds Z-form (i.e., left-handed) dsDNA or dsRNA (Z-DNA or Z-RNA) in vitro [42,43]. (‘Z’ is short for ‘zig-zag’, reflective of the appearance of the left-handed double helical conformations of dsRNA or dsDNA, compared to either A-RNA or B-DNA, the right-handed duplexes of these nucleic acids; Fig. 1B). Z-DNA and Z-RNA were not thought to readily occur in natural settings because they are energetically less-favorable conformations of dsDNA or dsRNA. We have recently shown that Z-RNA produced during orthomyxovirus and VACV infection is sensed via ZBP1, demonstrating for the first time that Z-RNA is a naturally occurring ligand for ZBP1[44,45]. In this article, we summarize our discovery of Z-RNA as a novel PAMP, and briefly outline our current mechanistic understanding of how Z-RNA is sensed by ZBP1 during both RNA (IAV and IBV) and DNA (VACV) virus infection. We also provide a concluding perspective on current unknowns and future directions.
Fig. 1. Z-RNA is a ZBP1-activating PAMP.
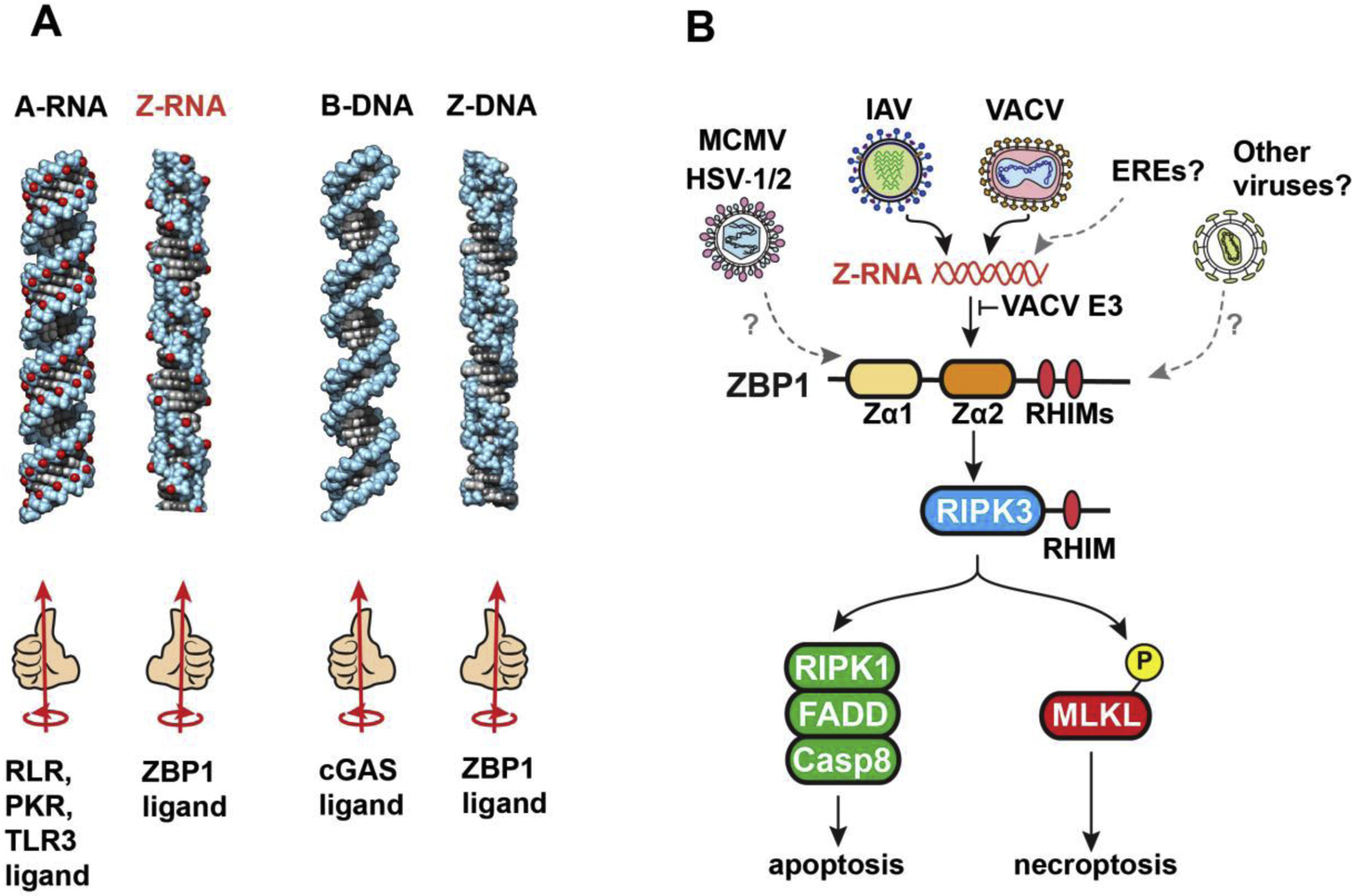
A. Structures of the left- and right-handed conformations of dsRNA and dsDNA. The ‘handedness’ of each duplex, and their cellular receptors, are shown below each structure. B. Orthomyxoviruses (IAV/IBV) and poxviruses (VACV) produce Z-RNAs, which are sensed by ZBP1 in a manner requiring the second of its two Zα domains. VACV E3 contains a Zα domain and prevents ZBP1 activation by competing with ZBP1 for Z-RNA. ZBP1 activates the kinase RIPK3 via RHIM:RHIM interactions between these proteins. RIPK3 then activates parallel pathways of apoptosis and necroptosis in infected cells. Herpesviruses (MCMV, HSV-1/2) also activate ZBP1, but the ZBP1-triggering ligand(s) produced by these viruses is currently unknown. It remains to be seen if members of other virus families activate ZBP1, and if cellular sources of Z-RNA, for example those originating from transcription of endogenous retroviral elements (EREs), contribute to ZBP1 activation during acute infections.
Z-RNA as ZBP1 ligand in orthomyxovirus infections.
In 2016, we showed that IAV and IBV strains activated RIPK3-dependent cell death in fibroblasts and type I alveolar epithelial cells [29]. The same year, we and others reported that ZBP1 was the host sensor which detected replicating IAV and activated RIPK3-mediated necroptosis or apoptosis in non-immune cell types, as well as pyroptosis in myeloid cells [30,32]. Cells lacking ZBP1 or RIPK3 were mostly resistant to IAV-induced cell death, implicating ZBP1-dependent, RIPK3-regulated death modalities as the primary means by which IAV-infected primary cells succumb to infection.
We found that the Zα2 domain of ZBP1 was essential for IAV-induced cell death signaling [32]. Mutating just two RNA contact residues (N122 and Y126) [46] in the Zα2 domain of murine ZBP1 abolished death signaling, strongly suggesting that direct sensing of IAV RNA by ZBP1 instigated cell death. We then discovered that ZBP1 associated with several IAV genomic RNAs, and that these RNAs fell into two classes: Class I, which are smaller (<500 bp) RNA species, mapping to the very 3’ and 5’ ends of the polymerase gene segments; and Class II, which are somewhat longer RNAs (1000–1500 bp) representing full-length viral gene segments [32,45]. Interestingly, Class I RNAs were derived from defective viral genomes (DVGs) that form when the IAV polymerase falls off its template RNAs but re-engages to copy shorter subgenomic RNA segments. These truncated subgenomic RNAs nonetheless retain their 3’ and 5’ packaging signals, and are packaged into so-called defective interfering (DI) particles [47]. IAV stocks low in DVG content did not trigger ZBP1 as much as those with high DVG content, and a DVG derived from the PA gene segment robustly bound ZBP1 in infected cells, indicating that DVGs are virus-produced ligands for this sensor [45]. Notably, the 5’ and 3’ ends of IAV gene segments are semi-complementary to each other, so DVGs may adopt panhandle, corkscrew, and other dsRNA structures [47]. When these dsRNAs are in their A-conformation, they are established ligands for RLRs [48,49]. Whether they can also adopt the Z-conformation to serve as ZBP1 ligands was not known.
Z-RNAs are notoriously unstable in vitro, making their isolation from infected cells technically challenging. Previous studies have, however, used in situ antibody-based staining approaches to show that Z-RNAs do indeed form under natural conditions (for example, in the cytoplasm of the protozoan Tetrahymena), and that an immunofluorescence approach to detecting Z-RNA in fixed cells is feasible [50]. No antibodies to Z-RNA are currently commercially available, but Z-RNA and Z-DNA share very similar structures, and several antibodies (both monoclonal and polyclonal) raised to Z-DNA cross-react with Z-RNA [51–53]. To determine whether these antibodies also detect Z-RNA in cells, we first synthesized a Z-RNA hairpin by introducing 2′-O-methyl-8-methyl modified guanosine nucleosides (m8Gm) to stabilize CG-repeat dsRNA in the Z-conformation under physiological salt concentrations in vitro [54]. We then used these modified synthetic Z-RNAs to screen anti-Z-DNA antibodies for their capacity to selectively detect Z-RNA. From this screen, we identified a polyclonal antiserum and a monoclonal antibody (clone Z22) capable of specifically recognizing the Z-RNA hairpin in vitro as well as within transfected cells [45].
Using these antibodies, we observed a modest RNase-sensitive nuclear signal in IAV-infected cells, but not in uninfected cells [45]. In agreement with previous work showing that viral RNAs are masked by cellular or viral proteins [55], protease treatment of IAV-infected cells post-fixation was required to fully unmask viral Z-RNAs. A nuclear Z-RNA signal that was first observed 2 to 4 hr p.i. and increased in intensity over the course of infection. In support of the idea that IAV DVGs are a dominant source of Z-RNA, the Z-NA-specific antibodies detected more Z-RNAs in cells infected with DVG-enriched IAV stocks than in cells infected with equivalent amounts of IAV low in DVG content. Other orthomyxoviruses, including seasonal strains of IAV and IBV, also produced nuclear Z-RNA [45]. ZBP1 is primarily a cytoplasmic protein in most uninfected cell types, but rapidly accumulates in the nucleus following IAV infection, where it senses viral Z-RNAs via its Zα2 domain. Binding viral Z-RNAs triggers ZBP1 activation, likely by inducing its oligomerization. Once activated in the nucleus, ZBP1 initiates ‘inside-out’ (i.e., nucleus-to-cytoplasm) cell death signaling by stimulating RIPK3 and activating MLKL. Active (phosphorylated) MLKL triggers disruption of the nuclear envelope, releasing DNA and other nuclear material, and culminating in hyper-inflammatory ‘nuclear necroptosis’. Such nuclear necroptosis is a very effective antiviral mechanism in mild cases of ‘the flu’, where it not only prevents virus spread by eliminating infected cells, but also promotes adaptive immunity by releasing viral antigen (for potential cross-presentation to T cells by APCs) and DAMPs (for generating effective adaptive immune responses) [29,31]. Either apoptosis or necroptosis signaling is equally effective at protecting mice from IAV, such that only combined elimination of both pathways in mice (e.g., ZBP1-, RIPK3- or combined FADD/MLKL-deficiency) compromises host defense [29,32]. In mice lacking both apoptosis and necroptosis, infected cells are not promptly eliminated and become virus factories. Consequently, virus clearance is compromised and the animal succumbs to infection at doses that are sub-lethal in wild-type mice with intact ZBP1-RIPK3 signaling [29,32,45].
Z-RNA as ZBP1 ligand in VACV infections.
Another remarkable instance of Z-RNA acting as a PAMP emerged from studies of the VACV strain Western Reserve (WR). This strain is known to be susceptible to the antiviral impact of TNFα-mediated necroptosis [22]. VACV WR encodes a potent suppressor protein kinase R (PKR) activation, E3, that also subverts death signaling initiated by ZBP1. E3 is composed of two functional domains: an N-terminal Zα domain [46], and a C-terminal dsRNA binding domain (dsRBD), which selectively binds A-RNA [56]. The dsRBD has long been known to compete with PKR for A-RNA, and to prevent PKR activation late in infection [57]. The dramatic attenuation in virulence manifested by the E3L mutant virus in vivo [58], however, is not driven by PKR, but rather by ZBP1-RIPK3-mediated necroptosis [59]. Our laboratories have now shown that E3 Zα-deficient viruses produce Z-RNA, which accumulates in the cytoplasm within a few hours after VACV infection [44]. When the E3 Zα domain is intact, it outcompetes ZBP1 for Z-RNA and prevents the antiviral consequences of necroptosis. However, when the N-terminal Zα-containing region of E3 is deleted, or when Z-RNA binding is disrupted by the introduction of specific point mutations (Y48A or P63A) into Zα, E3 fails to prevent ZBP1 activation and necroptosis. One surprise from these studies was the discovery that the E3 A-RNA binding dsRBD was necessary for Z-RNA accrual in the cytoplasm, and for activation of ZBP1. VACV mutants with a complete deletion of E3 did not induce necroptosis; only Zα mutants with an intact dsRBD did. These findings identify an unanticipated role for the C-terminal dsRBD of E3 in the formation or stabilization of Z-RNA [44]. Although it remains to be seen how the dsRBD promotes Z-RNA binding by E3, these new insights bring to closure expectations of biological activity for Zα domains first suspected when VACV E3 Zα mutants were shown almost two decades ago to be nonpathogenic [58].
Concluding perspectives.
We end by highlighting a few areas meriting further exploration. First, the mechanisms by which viral Z-RNA forms in infected cells is currently unclear. Drawing on studies examining Z-DNA formation in cells, we suggest that Z-RNAs may arise as a consequence of torsional strain generated in viral dsRNAs by polymerase activity during virus replication [60,61]. The Z-conformation may also be stabilized as a consequence of binding to the Zα domains of ZBP1, given their known ability to induce an A→Z transition in Z-prone dsRNA [46]. Unclear is whether covalent modifications to RNA (such as m6A) impact Z-RNA formation. It is notable that not all RNA viruses produce detectable Z-RNA or appear to activate ZBP1 during infection, although the same viruses (e.g., VSV or EMCV) may generate substantial amounts of A-RNA [45]. These observations indicate that additional virus-specific co-factors, or aspects of virus biology unique to orthomyxoviruses and other ZBP1-activating virus families, are required for Z-RNA formation in cellulo; these await discovery. Second, although our work implicates Z-RNA as ZBP1-activating ligand during orthomyxovirus and poxvirus infections, the Zα domain also binds Z-DNA [43] and has been shown to associate with G-quadruplexes [62]. Whether other ZBP1-activating viruses (such as the herpesviruses HSV-1/2 and MCMV) produce Z-DNA ligands, and whether RNA-DNA hybrids or other non-B conformations of dsDNA (such as G-quadruplexes) are physiologically relevant ligands for ZBP1 during virus infections remains to be seen. Relatedly, whether other viruses besides those mentioned in this review produce Z-NAs and activate ZBP1 is not known. Third, emerging evidence suggests that endogenous (i.e., host cell-derived) Z-RNAs can activate ZBP1 in certain settings, such as when the threshold for triggering necroptosis is lowered by deletion or mutation of RIPK1 [63,64]. These endogenous Z-RNAs are repressed by the p150 subunit of ADAR1 [65–68], which is the only other known mammalian protein (besides ZBP1) with a Zα domain[43]. Whether endogenous Z-RNAs contribute to ZBP1 activation during acute virus infections, and whether ADAR1 p150 regulates ZBP1 function in these settings is unknown. Finally, the necroptosis machinery is absent in invertebrates, conserved in most mammals (but not across all vertebrate species), and curiously missing in mammalian carnivores [69]. Neither ZBP1 nor RIPK3 are found in birds [69], which are the major orthomyxovirus reservoirs in the wild. These observations indicate that the necroptosis machinery, while capable of robust antiviral activity as a standalone cell death mechanism [31], can also pose a selective disadvantage during host-virus coevolution, when the benefits of its antiviral activity are outweighed by the downsides to its hyperinflammatory consequences [70]. How viruses [and other potential sources of ZBP1-activating ligands, such as endogenous retroelements [64,71,72]] impact both positive and negative selection of necroptosis genes, and, by corollary, how an intact necroptosis machinery dictates cross-species spread of necroptogenic viruses [73–75], are each largely unexplored frontiers.
Acknowledgements.
We thank Chaoran Yin and Mark Andrake for figure preparation. We are grateful to Heather Koehler and Carly DeAntoneo for comments on the manuscript. Research relevant to this article was supported by NIH grants AI135025 and AI144400 (to SB) and AI020211 (to ESM). SB is also supported by Cancer Center Support Grant P30CA006927.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: none
References.
- 1.Yatim N, Albert ML: Dying to replicate: the orchestration of the viral life cycle, cell death pathways, and immunity. Immunity 2011, 35:478–490. [DOI] [PubMed] [Google Scholar]
- 2.Green DR: Apoptotic pathways: the roads to ruin. Cell 1998, 94:695–698. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G: Viral control of mitochondrial apoptosis. PLoS Pathog 2008, 4:e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Upton JW, Chan FK: Staying alive: cell death in antiviral immunity. Mol Cell 2014, 54:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardwick JM: Viral interference with apoptosis. Semin Cell Dev Biol 1998, 9:339–349. [DOI] [PubMed] [Google Scholar]
- 6.Mocarski ES, Upton JW, Kaiser WJ: Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol 2012, 12:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM: Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature 1993, 361:739–742. [DOI] [PubMed] [Google Scholar]
- 8.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, et al. : Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287:1500–1503. [DOI] [PubMed] [Google Scholar]
- 9.Best SM: Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol 2008, 62:171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kepp O, Senovilla L, Galluzzi L, Panaretakis T, Tesniere A, Schlemmer F, Madeo F, Zitvogel L, Kroemer G: Viral subversion of immunogenic cell death. Cell Cycle 2009, 8:860–869. [DOI] [PubMed] [Google Scholar]
- 11.Neumann S, El Maadidi S, Faletti L, Haun F, Labib S, Schejtman A, Maurer U, Borner C: How do viruses control mitochondria-mediated apoptosis? Virus Res 2015, 209:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danilova N: The evolution of immune mechanisms. J Exp Zool B Mol Dev Evol 2006, 306:496–520. [DOI] [PubMed] [Google Scholar]
- 13.Upton JW, Shubina M, Balachandran S: RIPK3-driven cell death during virus infections. Immunol Rev 2017, 277:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P: Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998, 187:1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P: Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 1998, 188:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Beg AA: Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol 2000, 74:7470–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khwaja A, Tatton L: Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J Biol Chem 1999, 274:36817–36823. [DOI] [PubMed] [Google Scholar]
- 18.Jaattela M, Tschopp J: Caspase-independent cell death in T lymphocytes. Nat Immunol 2003, 4:416–423. [DOI] [PubMed] [Google Scholar]
- 19.Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X, Vandenabeele P: Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ 2002, 9:981–994. [DOI] [PubMed] [Google Scholar]
- 20.Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G: The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 1997, 15:1573–1581. [DOI] [PubMed] [Google Scholar]
- 21.Laster SM, Wood JG, Gooding LR: Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 1988, 141:2629–2634. [PubMed] [Google Scholar]
- 22.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK: Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X: Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137:1100–1111. [DOI] [PubMed] [Google Scholar]
- 24.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J: RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325:332–336. [DOI] [PubMed] [Google Scholar]
- 25.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG: Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 2012, 109:5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. : Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148:213–227. [DOI] [PubMed] [Google Scholar]
- 27.*.Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, Mocarski ES: Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci U S A 2017, 114:E2786–E2795. [DOI] [PMC free article] [PubMed] [Google Scholar]; A herpesvirus (MCMV) activates both apoptosis and necroptosis, when virus-encoded inhibitors of these pathways are disabled.
- 28.Thomas PG, Shubina M, Balachandran S: ZBP1/DAI-Dependent Cell Death Pathways in Influenza A Virus Immunity and Pathogenesis. Curr Top Microbiol Immunol 2020: 10.1007/1082_2019_1190. [DOI] [PubMed] [Google Scholar]
- 29.*.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. : RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]; An orthomyxovirus (influenza A virus) triggers both apoptosis and necroptosis in infected cells.
- 30.**.Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardana S, Place DE, Neale G, Vogel P, Kanneganti T-D: ZBP1/DAI is an Innate Sensor of Influenza Virus Triggering the NLRP3 Inflammasome and Programmed Cell Death Pathways. Science Immunology 2016, 1:aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first indication, together with Thapa et al. below, that ZBP1 functions as cell death-activating sensor of influenza A virus
- 31.*.Shubina M, Tummers B, Boyd DF, Zhang T, Yin C, Gautam A, Guo XJ, Rodriguez DA, Kaiser WJ, Vogel P, et al. : Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J Exp Med 2020, 217:e20191259. [DOI] [PMC free article] [PubMed] [Google Scholar]; Necroptosis by itself is a very efficient antiviral host defense mechanism.
- 32.**.Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al. : DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20:674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first indication, together with Kuriakose et al et al. above, that ZBP1 functions as cell death-activating sensor of influenza A virus
- 33.**.Upton JW, Kaiser WJ, Mocarski ES: DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]; The landmark study was the first to report that ZBP1 activates RIPK3-dependent cell death in virus-infected cells.
- 34.Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, Dovey CM, Carette JE, Upton JW, Mocarski ES, et al. : Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis 2018, 9:816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pham TH, Kwon KM, Kim YE, Kim KK, Ahn JH: DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J Virol 2013, 87:3076–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. : DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448:501–505. [DOI] [PubMed] [Google Scholar]
- 37.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, et al. : TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451:725–729. [DOI] [PubMed] [Google Scholar]
- 38.Dempsey A, Bowie AG: Innate immune recognition of DNA: A recent history. Virology 2015, 479–480:146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaiser WJ, Upton JW, Mocarski ES: Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 2008, 181:6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J: DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 2009, 10:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Upton JW, Kaiser WJ, Mocarski ES: Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem 2008, 283:16966–16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Athanasiadis A: Zalpha-domains: at the intersection between RNA editing and innate immunity. Semin Cell Dev Biol 2012, 23:275–280. [DOI] [PubMed] [Google Scholar]
- 43.Herbert A, Alfken J, Kim YG, Mian IS, Nishikura K, Rich A: A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci U S A 1997, 94:8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.**.Koehler H, Cotsmire S, Zhang T, Balachandran S, Upton JW, Langland J, Kalman D, Jacobs BL, Mocarski ES: Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 2021, 29:1266–1276 e1265. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first evidence that Z-RNAs are ligands for ZBP1 during VACV infections, and that a virus-encoded product, E3, prevents ZBP1 activation by sequestering Z-RNA.
- 45.**.Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, Ragan KB, Ishizuka T, Crawford JC, Tummers B, et al. : Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180:1115–1129 e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first evidence that Z-RNAs are ligands for ZBP1 during orthomyxovirus infections.
- 46.Ha SC, Kim D, Hwang HY, Rich A, Kim YG, Kim KK: The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc Natl Acad Sci U S A 2008, 105:20671–20676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nayak DP, Chambers TM, Akkina RK: Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr Top Microbiol Immunol 1985, 114:103–151. [DOI] [PubMed] [Google Scholar]
- 48.Liu G, Park HS, Pyo HM, Liu Q, Zhou Y: Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J Virol 2015, 89:6067–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baum A, Sachidanandam R, Garcia-Sastre A: Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 2010, 107:16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zarling DA, Calhoun CJ, Hardin CC, Zarling AH: Cytoplasmic Z-RNA. Proc Natl Acad Sci U S A 1987, 84:6117–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardin CC, Zarling DA, Puglisi JD, Trulson MO, Davis PW, Tinoco I Jr.: Stabilization of Z-RNA by chemical bromination and its recognition by anti-Z-DNA antibodies. Biochemistry 1987, 26:5191–5199. [DOI] [PubMed] [Google Scholar]
- 52.Hardin CC, Zarling DA, Wolk SK, Ross WS, Tinoco I Jr.: Characterization of anti-Z-RNA polyclonal antibodies: epitope properties and recognition of Z-DNA. Biochemistry 1988, 27:4169–4177. [DOI] [PubMed] [Google Scholar]
- 53.Zarling DA, Calhoun CJ, Feuerstein BG, Sena EP: Cytoplasmic microinjection of immunoglobulin Gs recognizing RNA helices inhibits human cell growth. J Mol Biol 1990, 211:147–160. [DOI] [PubMed] [Google Scholar]
- 54.Balasubramaniyam T, Ishizuka T, Xiao CD, Bao HL, Xu Y: 2’-O-Methyl-8-methylguanosine as a Z-Form RNA Stabilizer for Structural and Functional Study of Z-RNA. Molecules 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Son KN, Liang Z, Lipton HL: Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J Virol 2015, 89:9383–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langland JO, Kash JC, Carter V, Thomas MJ, Katze MG, Jacobs BL: Suppression of proinflammatory signal transduction and gene expression by the dual nucleic acid binding domains of the vaccinia virus E3L proteins. J Virol 2006, 80:10083–10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang HW, Watson JC, Jacobs BL: The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A 1992, 89:4825–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, Rich A: A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci U S A 2003, 100:6974–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.*.Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, Mocarski ES, Jacobs BL: Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci U S A 2017, 114:11506–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first indication that VACV E3 blocks ZBP1-initiated necroptosis signaling.
- 60.Wittig B, Dorbic T, Rich A: Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc Natl Acad Sci U S A 1991, 88:2259–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rich A, Zhang S: Timeline: Z-DNA: the long road to biological function. Nat Rev Genet 2003, 4:566–572. [DOI] [PubMed] [Google Scholar]
- 62.Kang HJ, Le TV, Kim K, Hur J, Kim KK, Park HJ: Novel interaction of the Z-DNA binding domain of human ADAR1 with the oncogenic c-Myc promoter G-quadruplex. J Mol Biol 2014, 426:2594–2604. [DOI] [PubMed] [Google Scholar]
- 63.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose-Girma M, Warming S, Solon M, et al. : RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540:129–133. [DOI] [PubMed] [Google Scholar]
- 64.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M: RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Reuver R, Dierick E, Wiernicki B, Staes K, Seys L, De Meester E, Muyldermans T, Botzki A, Lambrecht BN, Van Nieuwerburgh F, et al. : ADAR1 interaction with Z-RNA promotes editing of endogenous double-stranded RNA and prevents MDA5-dependent immune activation. Cell Rep 2021, 36:109500. [DOI] [PubMed] [Google Scholar]
- 66.Nakahama T, Kato Y, Shibuya T, Inoue M, Kim JI, Vongpipatana T, Todo H, Xing Y, Kawahara Y: Mutations in the adenosine deaminase ADAR1 that prevent endogenous Z-RNA binding induce Aicardi-Goutieres-syndrome-like encephalopathy. Immunity 2021, 54:1976–1988 e1977. [DOI] [PubMed] [Google Scholar]
- 67.Tang Q, Rigby RE, Young GR, Hvidt AK, Davis T, Tan TK, Bridgeman A, Townsend AR, Kassiotis G, Rehwinkel J: Adenosine-to-inosine editing of endogenous Z-form RNA by the deaminase ADAR1 prevents spontaneous MAVS-dependent type I interferon responses. Immunity 2021, 54:1961–1975 e1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maurano M, Snyder JM, Connelly C, Henao-Mejia J, Sidrauski C, Stetson DB: Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity 2021, 54:1948–1960 e1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJ, Vandenabeele P: An evolutionary perspective on the necroptotic pathway. Trends Cell Biol 2016, 26:721–732. [DOI] [PubMed] [Google Scholar]
- 70.Balachandran S, Rall GF: Benefits and Perils of Necroptosis in Influenza Virus Infection. J Virol 2020, 94:e01101–01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J: Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J 2017, 36:2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Herbert A: Mendelian disease caused by variants affecting recognition of Z-DNA and Z-RNA by the Zalpha domain of the double-stranded RNA editing enzyme ADAR. Eur J Hum Genet 2020, 28:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES: Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, et al. : RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 2015, 17:229–242. [DOI] [PubMed] [Google Scholar]
- 75.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, et al. : Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A 2014, 111:15438–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]