

Abstract
Epithelial ovarian cancer (EOC) is a leading cause of death from gynecologic malignancies and requires new therapeutic strategies to improve clinical outcomes. EOCs metastasize in the abdominal cavity through dissemination in the peritoneal fluid and ascites, efficiently adapt to the nutrient-deprived microenvironment, and resist current chemotherapeutic agents. Accumulating evidence suggests that mitochondrial oxidative phosphorylation is critical for the adaptation of EOC cells to this otherwise hostile microenvironment. Although chemical mitochondrial uncouplers can impair mitochondrial functions and thereby target multiple, essential pathways for cancer cell proliferation, traditional mitochondria uncouplers often cause toxicity that precludes their clinical application. In this study, we demonstrated that a mitochondrial uncoupler, specifically 2,5-dichloro-N-(4-nitronaphthalen-1-yl)benzenesulfonamide, hereinafter named Y3, was an antineoplastic agent in ovarian cancer models. Y3 treatment activated AMP-activated protein kinase and resulted in the activation of endoplasmic reticulum stress sensors as well as growth inhibition and apoptosis in ovarian cancer cells in vitro. Y3 was well tolerated in vivo and effectively suppressed tumor progression in three mouse models of EOC, and Y3 also induced immunogenic cell death of cancer cells that involved the release of damage-associated molecular patterns and the activation of antitumor adaptive immune responses. These findings suggest that mitochondrial uncouplers hold promise in developing new anticancer therapies that delay tumor progression and protect ovarian cancer patients against relapse.
Keywords: epithelial ovarian cancer, mitochondrial uncoupler, endoplasmic reticulum stress, unfolded protein response, immunogenic cell death
Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynecological cancer. Patients often develop resistance to chemotherapy, which prompts new therapeutic strategies for improving clinical outcomes (1). While immunotherapy offers promising treatment for many cancers, clinical trials in EOC have not achieved satisfactory results (2,3). This underscores the continuing need of new treatments for EOC.
We previously reported that N-aryl benzenesulfonamides functioned as mitochondrial uncouplers (4). Similar to a classic mitochondrial uncoupler, FCCP (5), they possessed a hydrophobic substructure and an ionizable, nitrogen-hydrogen bond that participates in proton translocations. They facilitated proton influx across the mitochondrial membrane without generating ATP, consequently reducing ATP production, activating AMP-activated protein kinase (AMPK), and inhibiting the proliferation of colon cancer cells (4).
In this study, we investigated the bioactivity of a leading compound from this family of N-aryl benzenesulfonamides. This compound named Y3 can induce endoplasmic reticulum (ER) stress-associated apoptosis of EOC cells. Cellular response to ER stress, also called unfolded protein response (UPR), is triggered by accumulation of unfolded proteins in the ER lumen. UPR initiates the dissociation of ER chaperon protein BIP1 (binding immunoglobin protein), also known as GRP78 (78kDa glucose-regulated protein), from three ER stress sensors, activating transcription factor 6 (ATF6), inositol-requiring enzyme 1a (IRE1α), and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) (6). ATF6 translocates to the Golgi and undergoes cleavage to gain transcriptional activity; IRE1α induces X-box binding protein 1 (XBP1) mRNA splicing and regulated-IRE1-dependent decay of mRNA; and PERK phosphorylates eukaryotic translation initiation factor 2A (eIF2α) leading to the translation of ATF4. This signaling cascade restores homeostasis by accelerating protein folding, transport, and degradation. As stress signals intensify, UPR switches towards activating pro-apoptotic pathways, including PERK-eIF2α-ATF4-CHOP and IRE1α-TRAF2-ASK1-JNK pathways (7,8).
UPR is essential for the adaptation of cancer cells to rapid growth, hypoxia, nutrition deprivation, and chemotherapies. In EOC, UPR promotes chemoresistance and inhibits anti-tumor immune response (9,10). However, irreversible ER stress can cause apoptotic UPR of cancer cells. ER stress-inducing compounds have been developed as anti-cancer agents (11). Bortezomib (a proteasome inhibitor), IPI-504 (a heat shock protein 90 inhibitor), and tunicamycin (a glycosylation inhibitor) increase misfolded proteins and create severe ER stress (12-14). Thapsigargin (an ER calcium pump inhibitor), GSK2606414 (a PERK inhibitor), and IT-139 (a BIP1 inhibitor) directly inhibit components of the UPR pathway (15-17). The challenges of these approaches lie in the almost obvious finding that excessive ER stress also causes toxicity to normal cells. New approaches must selectively target UPR in cancer cells without damaging normal cells.
Clinical agents that induce ER stress can induce immunogenic cell death (ICD) of cancer cells, including doxorubicin (18), bortezomib (19), mitoxantrone (20), and oxaliplatin (21). During ICD, the dying cancer cells release a spatiotemporally defined set of damage-associated molecular patterns (DAMPs) that are recognized by the innate immune cells, such as dendritic cells (DC). ICD can promote DC maturation and antigen processing or presentation (22,23) and thereby elicit tumor antigen-specific adaptive immune response (24). DAMPs include cell-surface exposure of endoplasmic reticulum chaperone calreticulin (CALR), secretion of extracellular ATP, and the release of high mobility group box 1 (HMGB1) protein (23,25,26). We demonstrated that Y3 functioned as an antineoplastic ER stress inducer with ICD-inducing activity in EOC in vitro and in vivo models.
Methods
Reagents
Y3 and Y3-M were synthesized as previously reported (4). Details of compounds (Y3, Y3-M, A-769662, AICAR, and 4μ8c), primers and antibodies used in this study are all listed in the supplemental information.
Cell lines
EOC cell lines were provided by Dr. Anirban Mitra (Indiana University School of Medicine) or obtained from ATCC (ATCC, VA, USA). Patient-derived EOC cell lines were isolated from tumor samples of patients diagnosed with serous epithelial ovarian cancer. They were characterized previously (27,28) and provided by Dr. Gil Mor and Dr. Alessandro Santin (Yale University School of Medicine). This study was conducted in accordance with the standards of Institute Research Ethics Committee of Yale University. FT240 and FT246 (29) were provided by Dr. Ronny Drapkin (University of Pennsylvania). Cell lines were propagated in RPMI 1640 or DMEM/F12 medium (Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (Seradigm, Radnor, PA) and penicillin/streptomycin (Life Technologies). Cells were maintained at 37°C and 5% CO2, authenticated by short tandem repeat DNA profiling, and tested for mycoplasma contamination.
Seahorse assay
Cells were treated in Seahorse XF modified media with 25 mM glucose, 1 mM pyruvate, 1 μM oligomycin A, 1 μM FCCP, and a mixture of 1 μM rotenone and 1 μM antimycin A in standard mitochondrial stress test conditions. ATP production rate was analyzed using Seahorse XF Real-Time ATP Rate Assay (Agilent, Santa Clara, CA).
Cell viability assay
Cells were plated at the concentration of 3,000 cells/well in 96-well plates 24 h before the treatments. After 48-h treatment cell viability was determined using CellTiter-Glo reagent and a GloMax Navigator Microplate Luminometer (Promega, Madison, WI).
Flow cytometry analysis of p-S6, AnnexinV/ Propidium Iodide (PI), and CALR
Cells were fixed in 4% formaldehyde for 15 min at room temperature (RT) and permeabilized in ice-cold 100% methanol for 30 min. Cells were resuspended and incubated in 100 μL incubation buffer (0.5% bovine serum albumin in PBS) containing Alexa Fluor 488-conjugated anti-p-S6 ribosomal protein antibody (Cell Signaling Technology, Danvers, MA) for 1 h at RT. The apoptotic cell population was assessed using Annexin V-fluorescein isothiocyanate (FITC)/PI propidium iodide (PI) Apoptosis Detection kit (Cell Signaling Technology). Annexin V-binding buffer containing Annexin V-FITC Conjugate and PI (1 μl: 12.5 μl per 200 μl) were used to resuspend 105 cells. Samples were incubated on ice for 10 min followed by flow cytometry analysis. To detect cell-surface CALR, cells were incubated on ice for 45 min with anti-CALR antibody or isotype control antibody diluted in cold incubation buffer followed by washing and flow cytometry analysis.
Mitochondrial membrane potential assays
Mitochondrial membrane potential was evaluated using Mitochondrial Membrane Potential Assay kits (I) and (II) (Cell Signaling Technology). Cells were resuspended in PBS at 106 cells/mL. Tetraethylbenzimidazolylcarbocyanine iodide (JC-1) was added to a final concentration of 2 μM. Tetramethylrhodamine, ethyl ester (TMRE) was added to a final concentration of 200 nM. For both assays, cells were incubated at 37°C for 20 min before flow cytometry analysis.
Caspase-3 and caspase-9 activity assays
Caspase-3 or caspase-9 activity was evaluated using Caspase-Glo 3/7 Assay kit or Caspase-Glo 9 Assay kit (Promega). Protein lysate (10 μg) was diluted to a final volume of 50 μL. An equal volume of Caspase-Glo 3/7 or Caspase-Glo 9 Reagent was added to the lysate and incubated at room temperature for 1 h before they were recorded using a Navigator Microplate Luminometer.
Quantitative real-time PCR (QPCR) and XBP1 mRNA splicing PCR
Total RNA was extracted using Total RNA Purification Kit (Norgen Biotek, Thorold, ON, Canada). cDNA was synthesized with qScript cDNA SuperMix Kit (Quantabio, Beverly, MA). Quantitative PCR was performed using SYBR Green Supermix (Bio-Rad, Hercules, CA) and CFX Connect QPCR detection system (Bio-Rad). GAPDH was used as a reference gene. Relative expression was calculated using the comparative ΔΔCT method. All reactions were performed with three biological replicates. Each replicate included three technical replicates. PCR reactions for the detection of XBP1 mRNA splicing were conducted under the following conditions with Taq DNA polymerase (Qiagen, Hilden, Germany): 94°C for 2 min; 35 cycles of 98°C for 30 sec, 52°C for 30 sec, and 72°C for 60 sec; and 72°C for 10 min. The amplified DNA fragments representing spliced and unspliced XBP1 (289 bp and 263 bp, respectively) were visualized on 4% agarose gels with ethidium bromide staining.
Western blotting
Cell lysates were prepared with cell lysis buffer (1% Triton X-100, 0.05% SDS, 100 mM Na2HPO4, and 150 mM NaCl). Protein lysate was electrophoresed on a 12% SDS-polyacrylamide gel and transferred onto Amersham Hybond 0.45 PVDF membranes (GE Healthcare, Chicago, IL). After blocking with 5% non-fat milk in PBS-0.05% Tween 20, the membranes were incubated with primary antibodies at 4°C overnight, and then secondary antibodies for 1 h at room temperature. The blots were developed using Clarity or Clarity Max Western ECL Blotting Substrates (Bio-Rad).
Gene knockdown by siRNA, esiRNA, or GapmeR
siRNAs and esiRNAs were transfected using Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA). siRNAs and esiRNAs (Millipore-Sigma, Woodlands, TX) include the validated negative control siRNA (#SIC001), siRNAs targeting PERK (#SASI_Hs01_00096846 and #SASI_Hs01_00096846), siRNAs targeting AMPKa1 (#SASI_Hs01_00092447), control esiRNA targeting EGFP (ESIRNA1-EHUEGFP) and esiRNA targeting BIP1 (ESIRNA1-EHU003061). GapmeRs targeting AMPKa1 and the negative control GapmeR (#LG00227216-DDA and LG00000002-DDA, Qiagen) were delivered to cells via gymnosis following the manufacturer’s instruction.
Co-immunoprecipitation (co-IP) assay
Co-IP was conducted using SureBeads Protein G Magnetic Beads (Bio-Rad). One μg of antibody was first linked to 100 μL beads for 10 min at room temperature. Cell lysate was added to the beads and incubated for 1 h at room temperature on a rotator. After the beads were washed three times with PBST, the IP product was eluted using 40 μL Laemmli buffer by incubating at 70°C for 10 min.
In vivo tumorigenic assays
Animal experiments were approved by the Yale University Institutional Animal Care and Use Committee. A patient-derived EOC cell line OVC1 labeled with red fluorescence protein (RFP) was intraperitoneally injected (3 × 106 cells/mouse) into Hsd:Athymic Nude-Foxn1nu mice as previously described (28). Y3 was dissolved in polyethylene glycol (PEG)/DMSO/saline (7:2:1) vehicle and intraperitoneally injected as 20 mg/kg, the highest safe dose of Y3 in nude mice, twice per week starting from three days after the injection of cancer cells. Mice in the control group were injected with vehicle solution. Tumor growth was monitored using Spectrum In Vivo Imaging System (PerkinElimer, Waltham, MA). ID8 mouse ovarian cancer cell line (30) was IP injected to C57BL/6 mice (6 × 106 cells/mouse). Twenty-one days after injecting cells, Y3 was IP injected to mice twice per week. The Maximum Tolerated Dose (MTD) of IP-injected Y3 in the C57BL6 mice is 10 mg/kg. We used half of this MTD and IP injected 5 mg/kg Y3 in C57BL6 mice. The control group mice were injected with vehicle solution. Abdominal circumference was measured using a measuring tape. Tumors in the peritoneal cavity were collected during necropsy to calculate the numbers of intraperitoneal tumors. The organs with tumors and the total weight of tumors from each mouse were documented. Mouse ovarian cancer cell line was derived from tumors formed in Dicerflox/flox-Ptenflox/flox-Tpr53LSL-R172H/+ Amhr2cre/+ triple mutant mice (31,32) and labeled with lentiviral RFP vector. The triple mutant ovarian cancer cells (TKO cells) were injected subcutaneously (SC) into the flank of C57BL/6 mice (6 × 106 cells/mouse). Y3 was IP injected (5 mg/kg) to mice once per week after the tumors reached detectable size. Mice in the control group were injected with vehicle solution. Tumors were measured with a vernier caliper every three days. Mice were euthanized to collect the xenografts. In the control group of the vaccination assay, each mouse was SC injected with 105 necrotic TKO cells that were frozen and thawed in liquid nitrogen and at 37°C. The same numbers of TKO cells were treated with 10μM Y3 in vitro for 24 h before they were SC injected to the left flank of C57BL/6 mice. One week later, normal live TKO cells were injected to the contralateral flank of all the mice (6 × 106 cells/mouse). Tumors were measured with a vernier caliper every three days.
Immunofluorescence staining
The formalin-fixed paraffin-embedded tumor tissue sections were deparaffinized and hydrated by three washes of xylene for 5 min each, two washes of 100% ethanol for ten min each, two washes of 95% ethanol for 10 min each, and two washes with water for 5 min each. Slides were heated in 1X citrate unmasking solution (10mM Citric Acid, 0.05% Tween 20, pH 6.0) in a steam cooker for ten min at 95-98°C. After cooling, the slides were incubated in blocking buffer (5% normal serum and 0.3% Triton X-100 in 1X PBS) for 60 min and then in primary antibody in antibody dilution buffer (1% BSA and 0.3% Triton X-100 in 1X PBS) overnight at 4°C. After rinsing three times in 1X PBS for 5 min each, the slides were incubated with fluorochrome-conjugated secondary antibody diluted for 1 h at RT in the dark. Prolong Gold Antifade Reagent with DAPI was used to mount the slides (Cell Signaling Technology).
Blood cell counts
Blood was collected from the venous sinus of mice. White blood cell count and platelet count were assessed as previously described (33,34)
Detection of urea nitrogen, creatinine, and HMGB1 in fluid samples
Blood urea kit (MyBioSource, San Diego, CA, USA) was used to evaluate the levels of urea in plasma samples. Urinary creatinine colorimetric assay kit (Cayman, Ann Arbor, MI, USA) was used to detected creatinine in mouse urine samples that were collected as previously described (35). HMGB1 in cell culture medium was determined using a chemiluminescence ELISA kit (Novus, Centennial, CO, USA).
Multiplex cytokine assay
Plasma samples were shipped to Eve Technologies on dry ice (Calgary, AB, Canada) for analyses using a Multiplexing LASER Bead Assay (Mouse Cytokine Array/Chemokine Array 31-Plex, Eve Technologies).
Extracellular ATP detection assay
The supernatant of cell culture medium was collected and mixed with RealTime-Glo Extracellular ATP Assay Reagent (Promega) to incubate for ten min. The luminescent signal was detected using the GloMax Navigator Microplate Luminometer (Promega).
Statistical analysis
Prism 7 (GraphPad Software, La Jolla, CA) was used to perform statistical analyses. An unbiased elimination of outliers was performed using ROUT’s method. Statistical differences between two groups were analyzed using Student’s t-test. One-way or two-way ANOVA tests were used compare multiple treatment groups depending on the experimental design. *P < 0.05 was considered significantly different.
Results
Mitochondrial uncoupler Y3 inhibited EOC cell proliferation and induced apoptosis.
We previously reported a family of N-aryl benzenesulfonamides with mitochondrial uncoupling activity (4). We confirmed this function of a leading compound, namely Y3 in an EOC cell line, OVCAR8, in a Seahorse assay. Y3 increased the oxygen consumption rate that was otherwise inhibited by oligomycin, a finding consistent with reduced electron transport and phosphorylation uncoupling (Figure 1A). The N-methylated analog of Y3, namely Y3-M (Figure 1A) was inactive as an uncoupler. This finding established that the nitrogen-hydrogen bond of the sulfonamide subunit of Y3 was essential for its activity and the inactivity of the nitrogen-methyl subunit in Y3-M was consistent with our previous observations in colon cancer cells (4).
Figure. 1. Y3 induces apoptosis and inhibits proliferation of EOC cells.
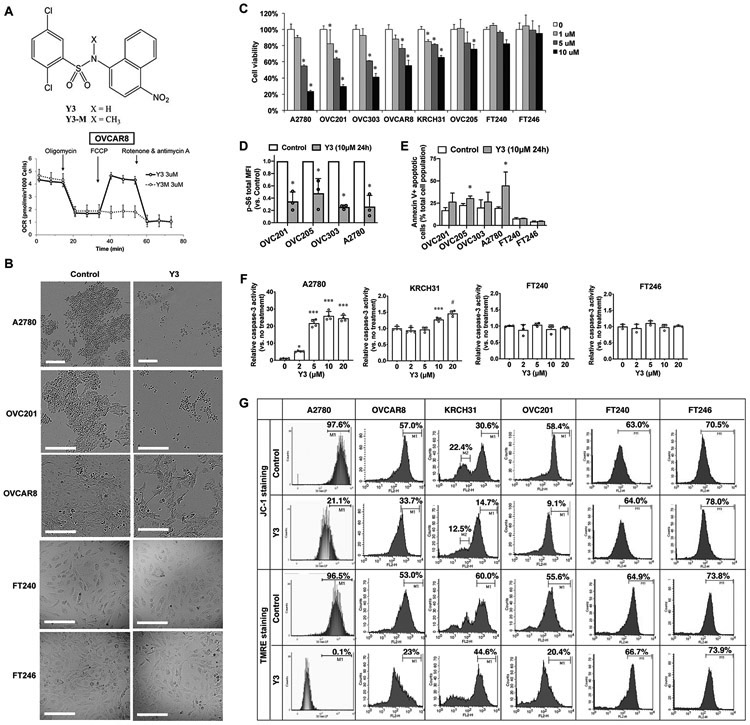
A, Chemical structure of Y3/Y3-M and the oxygen consumption rate (OCR) of OVCAR8 cells. The final concentration of Y3 or Y3-M was 3μM. B, Morphology of 10 μM Y3-treated and untreated EOC cell lines. Scale bar = 200 μm. C, Cell viability of Y3 treated cells. Two-way ANOVA followed by Sidak HSD test, n=5, *P<0.005. D, p-S6 flow cytometry analysis. Cancer cells were treated with 10 μM Y3 for 24 hs. MFI, median fluorescence intensity. Two-way ANOVA followed by Sidak HSD test, n=3, *P<0.05. E, AnnexinV-FITC/PI flow cytometry analysis. Cancer cells were treated with 10 μM Y3 for 24 h. Two-way ANOVA followed by Sidak HSD test, n=3, * P<0.05. The connecting lines indicate the paired data points that were generated from one experiment. F, Caspase-3 activity of EOC cell lines treated with 10 μM Y3 for 48 h. One-way ANOVA followed by Tukey’s HSD test, n=4, *P<0.05, ***p<0.0005, and #p<0.0001. G, Histograms of flow cytometry analysis in EOC cell lines stained with JC-1 or TMRE. The staining-positive cell populations were labeled with marker 1 (M1).
Y3-treatment (10 μM for 48 h) induced unhealthy morphologies in EOC cells (Figure 1B) and reduced the viability of two common EOC cell lines, A2780 and OVCAR8, and the patient-derived EOC cell lines, OVC201, OVC303, OVC205, and KRCH31 (Figure 1C). We used the immortalized noncancerous fallopian tube cell lines, FT240 and FT246 (29), as normal counterpart controls since fallopian tube epithelial cells were previously identified as a main origin of high-grade serous ovarian carcinoma, the most common type of EOC (36,37). The Y3-treated FT240 and FT246 cells did not change their morphologies (Figure 1B) and had IC50 values of 52.93 μM and 67.33 μM, respectively (Figure 1C and Supplemental Table 1), suggesting that they were more resistant to Y3 treatment than the EOC cell lines, A2780, OVCAR8, OVC201, OVC303, OVC205, and KRCH31, with IC50 values of 4.379 μM, 9.643 μM, 4.632 μM, 7.014 μM, 15.44 μM, and 14.84 μM, respectively (Supplemental Table 1).
To characterize the effects of Y3 on EOC cells, we quantified the level of phosphorylated ribosomal protein S6 (p-S6) that is required for protein synthesis and cell growth as a cell proliferation marker (38). Y3 treatment significantly lowered the levels of p-S6 in EOC cells in comparison with the untreated controls (Figure 1D). Secondly, we demonstrated that 24h Y3-treatment increased the AnnexinV+ apoptotic cell populations in EOC cell lines, but not in FT240 and FT246 cells (Figure 1E). Thirdly, we detected the activation of caspase-3 in A2780 and KRCH31 cells after 48h Y3-treatment, and the caspase-3 activity of FT240 and FT246 cells was not affected by Y3-treatment (Figure 1F). Finally, using JC-1 and TMRE staining we assessed mitochondrial membrane potential (Δψm) and showed that 24h Y3-treatment (10 μM) caused a substantial loss of Δψm in EOC cell lines, but not in FT240 and FT246 cells (Figure 1G). Taken together, Y3 treatment induced apoptosis and inhibited proliferation in EOC cells but not in FT240 and FT246 noncancerous cell lines.
Y3 activated three main ER stress sensors in EOC cells.
We identified the UPR pathway as one of the major altered pathways in Y3-treated EOC cells using RNA microarrays (Supplemental Tables 2 and 3). Using real-time QPCR in EOC cells lines, we confirmed the induction of UPR genes by Y3-treatment (10 μM for 24 h, Figure 2A). Among these genes, CHOP (C/EBP-Homologous Protein or DNA Damage Inducible Transcript 3), GADD34 (Growth Arrest And DNA-Damage-Inducible protein 34) and PUMA (p53 Upregulated Modulator of Apoptosis) are genes that mediate the ER stress-induced apoptosis. We also confirmed the downregulation of PCNA (Proliferating Cell Nuclear Antigen) mRNA by Y3-treatment (Figure 2A and Supplemental Table 3). The Y3-induced changes of BAK, BIP1, and PERK mRNA levels were not statistically significant. In a time-course experiment, we detected the induction of UPR genes as early as four hours after initiating Y3 treatment in A2780 cells followed by the decrease of PCNA mRNA at a later time point (Figure 2B), suggesting that ER activation was triggered as an early responding event.
Figure 2. Y3 treatment induces UPR in EOC cells.
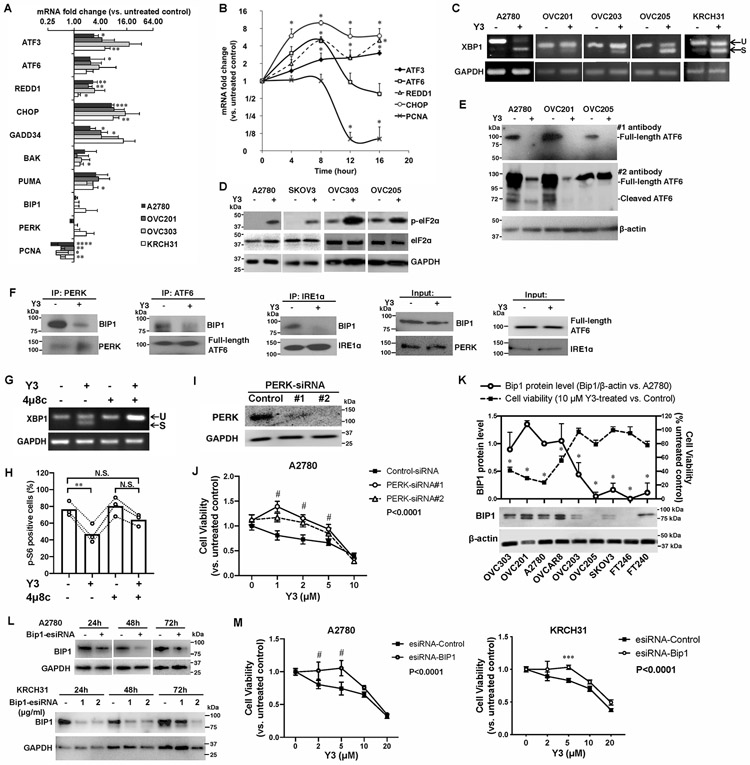
A, RT-QPCR of UPR genes in EOC cell lines. Cells were treated with 10 μM Y3 for 24 h. GAPDH was used as a house-keeping gene. Two-way ANOVA followed by Sidak HSD test, n=3, *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001. B, RT-QPCR of UPR target genes in A2780 cells at different time points of Y3 treatment. A2780 cells were treated with 10 μM Y3. Two-way ANOVA followed by Sidak HSD test, n=3, *P<0.05. C, XBP1 mRNA splicing analyzed by RT-PCR and agarose gel. EOC cells were treated by 10 μM Y3 for 24 h. U, unspliced. S, spliced. D, Y3 treatment (10 μM, 24h) induces eIF2a phosphorylation. GAPDH was used as a loading control. E, Y3 treatment (10 μM, 24h) induces ATF6 cleavage. β-actin was used as a loading control. F, Co-IP of BIP1, PERK, ATF6, and IRE1α. A2780 cells were treated with 10 μM Y3 for 6 h. G, XBP1 mRNA splicing analyzed by RT-PCR and agarose gel. 4μ8c inhibits Y3-induced XBP1 mRNA splicing. A2780 cells are treated by 10 μM Y3 and/or 10 μM 4μ8c for 24 h. H, Flow cytometry analysis of p-S6. A2780 cells are treated by 10 μM Y3 and/or 10 μM 4μ8c for 24 h. Two-way ANOVA followed by Sidak HSD test, n=3, **P<0.005. I, Western blot images of PERK protein knocked down by two siRNAs (#1 and #2). GAPDH was used as a loading control. J, PERK siRNAs inhibit the response of A2780 cells to Y3. Two-way ANOVA followed by Sidak HSD test, n=6, #P<0.0001. K, BIP1 protein level is associated with sensitivity to Y3. Student’s t-test, *P<0.05. β-actin was used as a loading control. L, BIP1-targeting esiRNAs inhibit the expression of BIP1 in A2780 and KRCH31 cells. The negative control esiRNAs target GFP. GAPDH was used as a loading control. M, BIP1-targeting esiRNA inhibits the response of A2780 and KRCH31 cells to Y3. Two-way ANOVA followed by Sidak HSD test, n=6, ***P<0.0005 and #P<0.0001 for comparisons between Y3-treated and untreated groups at the indicated concentrations. P values for the comparisons between the entire control and knockdown groups were indicated in the legend.
Y3 induced XBP1 mRNA splicing in EOC cells (Figure 2C), indicating the activation of IRE1α. The same treatment also increased the levels of phosphorylated eIF2α (Figure 2D), a downstream target of PERK. To examine the effect of Y3 on ATF6 protein expression using western blot, we detected full-length ATF6 protein in the untreated EOC cells but did not detect cleaved ATF6 in any sample (Figure 2E). We used another anti-ATF6 antibody to confirm the result. The second anti-ATF6 antibody detected both full-length and cleaved ATF6 in the untreated cells. Both forms of ATF6 were significantly downregulated by 24 h Y3-treatment (Figure 2E). These results suggest that Y3 treatment affected the protein stability and function of ATF6. Finally, we performed co-immunoprecipitation (co-IP) using cell lysate collected after six-hour Y3 treatment. At this early time point, the full-length ATF6 was not yet downregulated. Our result demonstrated the dissociation of BIP1 from IRE1α, PERK, and ATF6 (Figure 2F). In conclusion, Y3 induced UPR in EOC cells.
UPR pathway was involved in the anti-cancer activity of Y3.
To confirm the involvement of UPR pathway, we determined how the inhibition of components in UPR pathway affected the activity of Y3. We inhibited the IRE1α arm of UPR pathway using an IRE1 inhibitor, 4μ8c. Treatment of 4μ8c completely blocked Y3 from inducing XBP1 mRNA splicing (Figure 2G). In the absence of 4μ8c, Y3 treatment caused p-S6 positive OVC201 cells to decrease from 76.6±8.7% to 46.9±11.2% (Figure 2H). In the presence of 4μ8c, 64.1±7.1% of Y3-treated cells were p-S6 positive, which was not statistically different from the untreated group or 4μ8c alone treated group (Figure 2H). The inhibition of IRE1α attenuated the activity of Y3 to downregulate p-S6. Secondly, we knocked down PERK expression with two siRNAs (Figure 2I). These siRNAs attenuated the cytotoxic effect of Y3 and increased the viability of Y3-treated A2780 cells (Figure 2J). This result suggested that PERK also played an important role in Y3-mediated cellular changes. Finally, we discovered that EOC cell lines expressing high levels of BIP1 (OVC201, OVC303, A2780 and OVCAR8) were more sensitive to Y3 treatment than those expressing lower levels of BIP1 (OVC203, OVC205, and SKOV3) and noncancerous cell lines (FT240 and FT246, Figure 2K). We knocked down BIP1 expression with esiRNAs (Figure 2L) comprised of a heterogeneous pool of siRNAs targeting the same gene. They led to highly specific and effective gene knockdowns with lower off-target effects than single chemically synthesized siRNA (39). BIP1 knockdown reduced the sensitivity of A2780 and KRCH31 to Y3 treatment (Figure 2M). These results support our observation (Figure 2K) that BIP1 levels can correlate with sensitivity or resistance to Y3. Our results support that components of the UPR pathway played critical roles in the activity of Y3.
Y3 induced UPR through AMPK activation.
AMPK pathway responses to ATP reduction (4) and Y3 induced AMPK phosphorylation (p-AMPK) in EOC cells (Figure 3A). We found that small-molecule activators of AMPK, A-769662 and AICAR, induced eIF2α phosphorylation and XBP1 mRNA splicing (Figure 3B). When we inhibited the upregulation of p-AMPK using an siRNA targeting AMPKa1, the Y3-induced eIF2α phosphorylation and XBP1 mRNA splicing were suppressed (Figure 3C). We validated this result using a GapmeR (40) that is a locked nucleic acid-conjugated chimeric single-strand antisense oligonucleotide specifically silencing AMPKa1 (Figure 3D). When AMPK was inhibited by siRNA or GapmeR, the viability of A2780 cells was increased under Y3 treatment (0.5-4 μM) when compared to the cells expressing negative control siRNA or GapmeR (Figure 3E and 3F). These results demonstrate that the activation of AMPK was upstream of Y3-induced UPR, and AMPK activation contributed to inducing apoptosis and inhibiting proliferation of Y3-treated cells.
Figure 3. Y3 induces UPR through activating AMPK in EOC cells.
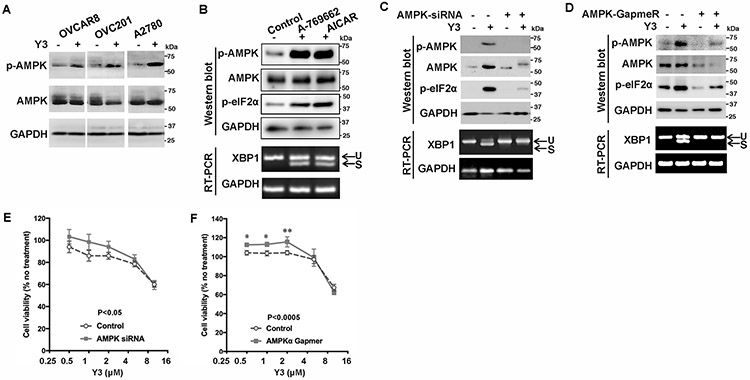
A, Representative images of western blot. Y3 induced AMPK phosphorylation in ovarian cancer cells. p-AMPK, phosphorylated AMPK. AMPK, total AMPK. GAPDH was used as a loading control. B, Representative images of western blot and agarose gel electrophoresis. A2780 cells were treated with AMPK activators A-769662 (10 μM) or AICAR (1 mM) for 24 h. Whole cell lysate was analyzed with western blot. The splicing of XBP1 mRNA was analyzed by RT-PCR and agarose gel electrophoresis. U, unspliced. S, spliced. GAPDH was used as a loading control. C, AMPK siRNA knocks down AMPK expression and inhibits Y3-induced eIF2α phosphorylation and XBP1 mRNA splicing. U, unspliced. S, spliced. GAPDH was used as a loading control. D, AMPK GapmeR knocks down AMPK expression and inhibits Y3-induced eIF2α phosphorylation and XBP1 mRNA splicing. U, unspliced. S, spliced. GAPDH was used as a loading control. E and F, AMPK siRNA (E) and GapmeR (F) partially inhibit the response of A2780 cells to Y3. Two-way ANOVA followed by Sidak HSD test, n=5, p values for the comparisons between the entire control and knockdown groups as indicated in the graph. *P<0.05 and **P<0.005 for comparisons between control and knockdown groups at the indicated concentrations.
Y3 suppressed ovarian cancer progression and was well tolerated in vivo
We first evaluated the in vivo efficacy of Y3 in a patient-derived xenograft (PDX) EOC model. Intraperitoneally (IP) injected-Y3 (20 mg/kg) suppressed tumor growth (Figure 4A and 4B). When we compared tumors from Y3-treated and control mice, we found that the Y3-treated group had lower overall tumor weight (Figure 4C) and fewer tumors (Figure 4D). Y3-treated tumors showed significantly higher levels of caspase-9 and 3 activities (Figure 4E), indicating that Y3 induced apoptosis in these tumors. Y3 treatment also increased the expression of UPR genes in the tumors, including the spliced XBP1, BIP1, PERK, and PUMA (Figure 4F). Immunostaining of tumor tissue sections showed that Y3 treatment significantly decreased the levels of Ki67-positive proliferating cells (Figure 4G), increased the levels of CHOP staining (Figure 4H), and caused tumors to develop large necrotic areas (Figure 4I), again demonstrating the activation of ER stress-induced apoptosis.
Figure 4. Y3 suppresses EOC progression in nude mice.

A, Live imaging of red fluorescence protein (RFP) of tumors in nude mice. B, Tumor growth curves based on RFP signals. Normalized tumor fluorescence signal was calculated as the fluorescence signal relative to signal of the tumor on the first day of imaging. Two-way ANOVA followed by Sidak HSD test, *P<0.05, ***P<0.0005, #P<0.0001. C, Tumor weight. Student’s t-test, **P<0.005. D, Numbers of tumors in each mouse. Student’s t-test, **P<0.005. E, Caspase-9 and caspase-3 activity in tumor protein lysate. Student’s t-test, **P<0.005, ***P<0.0005. F, Expression of spliced XBP1 mRNA, PERK, BIP1 and PUMA in mouse ovarian cancer xenografts. The levels of mRNA are assessed with RT-QPCR. GAPDH is used as a housekeeping gene. Student’s t-test, *P<0.05, **P<0.005, ***P<0.0005, #P<0.0001. G, Ki67 staining and quantification of fluorescence positive cells in tumors from nude mice. DAPI was used to stain nuclear DNA. Scale bar = 40 μm. Student’s t-test, #P<0.0001. H, CHOP staining and quantification of fluorescence positive cells in tumors from nude mice. Scale bar = 35 μm. Student’s t-test, #P<0.0001. I, H&E staining of tumor, kidney, and liver tissues from the untreated control and Y3-treated nude mice. J, Y3 does not affect body weight of nude mice. K, White blood cell (WBC) count of control and Y3-treated nude mice. L, Platelet count of control and Y3-treated mice. M, Blood urea nitrogen of control and Y3-treated mice assessed by ELISA. N, Urine creatinine of control and Y3-treated mice. N.S., not significant for student’s t-test.
Histopathology analysis of the livers and kidneys of Y3-treated mice did not reveal morphological damages that can cause hepatotoxicity or nephrotoxicity (Figure 4I). Y3 treatment did not cause differences in the body weight (Figure 4J), white blood cell count (Figure 4K), platelet count (Figure 4L), blood urea nitrogen level (Figure 4M), or urine creatinine level (Figure 4N) in comparison with untreated mice. Taken together, our data suggests that Y3 suppressed tumor growth in this EOC mouse model without significant side-effects.
Y3 induced immunogenic cell death of ovarian cancer cells
We evaluated Y3 in two immunocompetent syngeneic mouse EOC models. The first model was generated by IP injection of ID8 mouse ovarian cancer cells into C57BL/6 mice. IP injection of Y3 at a dose of 5 mg/kg did not affect body weight (Figure 5A) or cause damage to the livers or kidneys of Y3-treated mice (Figure 5B). Mice in this model usually develop widespread peritoneal tumors and ascites causing distended abdomens; therefore, we monitored their abdominal circumferences and demonstrated the effect of Y3 treatment in delaying the accumulation of ascites (Figure 5C). This treatment improved the survival of Y3-treated mice (Figure 5D). A closer examination of their abdominal organs showed that Y3 treatment significantly decreased the numbers of peritoneal tumor implants and organs with tumors (Figure 5E, 5F, 5G, and Supplemental Table 4). The overall tumor weight was significantly lower in the Y3-treated mice (Figure 5H). H&E staining of tumor sections revealed that in contrast to the typical intact cancer cell structure with distinct nuclei and a thin uniform cytoplasmic rim around each individual cell in control tumors, Y3-treated tumors showed a nonuniform thickened rim of cytoplasm around damaged nuclei and significant structural collapse and deformity in tissues (Figure 5I). Y3-treated tumors also had significantly decreased percentages of Ki67-positive cells and increased percentages of tumor-infiltrating CD4+ and CD8+ T cells in comparison with the control tumors (Figure 5J and 5K).
Figure 5. Y3 inhibits tumor development in ID8 syngeneic mouse EOC model.

A, Y3 does not affect body weight of wild type C57/BL6 mice. B, H&E staining of kidneys and livers from C57/BL6 mice. C, Y3 delayed abdominal distension in mice, a sign of accumulation of ascites. D, Y3 improved the survival of mice injected with ID8 mouse ovarian cancer cells. E, Representative images of peritoneal tumors in control and Y3-treated mice. F, Total tumor numbers. Student’s t-test, ****P<0.0001. G, Numbers of organs with tumor implants. Student’s t-test, **P<0.005. H, Total tumor weight. Student’s t-test, **P<0.005. I, H&E staining of intraperitoneal tumor tissues from untreated control and Y3-treated C57/BL6 mice. J, Immunofluorescence staining of Ki67, Cd4, and Cd8 in tumor tissues. DAPI was used to stain nuclear DNA. K, Quantification of fluorescence positive cells. Student’s t-test, ***P<0.005, ****P<0.0005.
In the second syngeneic mouse model, we subcutaneously (SC) injected p53-Dicer-Pten triple-mutant (TKO) mouse EOC cells (31) into C57BL/6 mice. Weekly Y3 IP injection (5 mg/kg) effectively inhibited tumor growth (Figure 6A and 6B). Using multiplex array, we assessed the plasma levels of 31 cytokines of these mice and identified that Y3 upregulated the levels of Cxcl1, Il-6, Il-5, Il-4 and G-CSF in tumor-bearing mice 12 days after initiating Y3 treatment comparing to the untreated mice (Figure 6C). Y3 treatment did not affect these cytokines the same way in healthy mice without tumor cell inoculation. This result suggested that the cytokine production was a response specific to the Y3-treated tumors. The release of these cytokines was associated with immunogenic cell death (ICD) and the activation of antitumor immune response (Supplemental references). Furthermore, the level of IL-17 in blood was lower in the Y3-treated tumor-bearing mice than the untreated group. The inhibition of IL-17 also was known to enhance adaptive immune response against tumors (41). Together with data from the ID8 mouse model, this result led us to hypothesize that Y3 treatment induced ICD of EOC cells.
Figure 6. Y3 induces immunogenic cell death in TKO syngeneic mouse EOC model.
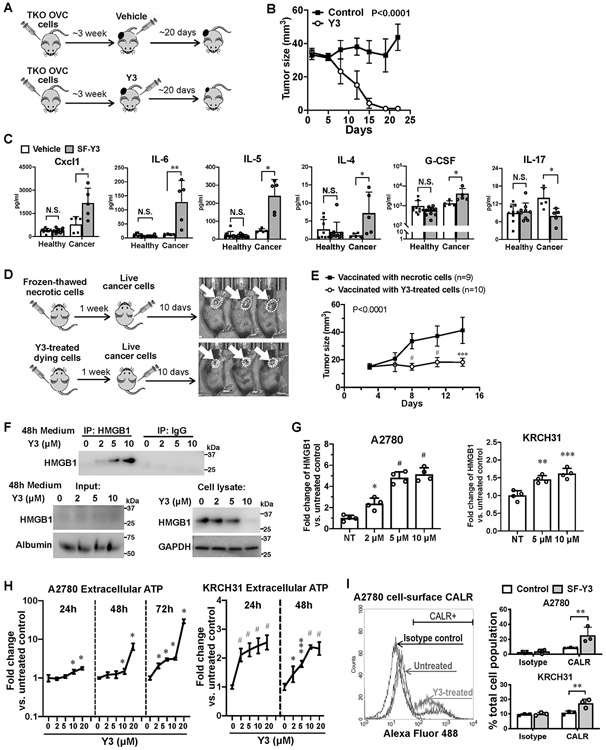
A, Flowchart of TKO tumor formation and Y3 treatment. B, Growth curves of tumors formed by TKO mouse ovarian cancer cells in C57BL/6 mice. Two-way ANOVA analysis, n=5. C, Levels of cytokine in mouse plasma samples. Healthy and tumor-bearing mice are both injected with Y3 (5mg/kg, IP once per week) or vehicle. Two-way ANOVA followed by Sidak HSD test, *P<0.05, **P<0.005. D, Flowchart of vaccination assay. Mice in the control group were injected with necrotic TKO cells that were frozen and thawed in liquid nitrogen and at 37°C. The same numbers of TKO cells were treated with 10μM Y3 in vitro for 24 h before they were injected to the left flank of C57BL/6 mice in the test group. One week later, normal live TKO cells were injected to the contralateral flank of all the mice. E, Growth curves of tumors in the right flank of mice. Two-way ANOVA followed by Sidak HSD test, ***P<0.0005, #P<0.0001. F, Immunoprecipitation of HMGB and western blot. A2780 cells are treated with 10 μM Y3 for 48 h. HMGB is precipitated from the culture medium of A2780 cells and detected using anti-HMGB antibody. Albumin was used as a loading control for medium samples. GAPDH was used as a loading control for cell lysate samples. G, HMGB in the culture medium of A2780 cells assessed using ELISA. One-way ANOVA followed by Tukey’s HSD test, *p<0.05, **p<0.005, and #p<0.0001. H, Levels of ATP in cell culture medium of A2780 and KRCH31 cells. Two-way ANOVA followed by Sidak HSD test, n=5, *P<0.05, ***p<0.0005, #p<0.0001. I, Flow cytometry analysis of cell-surface CALR. Cells are treated with 10 μM Y3 for 24 h. Two-way ANOVA followed by Sidak HSD test, n=3, **P<0.005.
We utilized a vaccination assay to determine whether Y3 could induce ICD in vivo. In the control group, we vaccinated C57BL/6 mice by SC injecting necrotic TKO cells that were killed by repeatedly freezing and thawing into their left flank. In the test group, we vaccinated the mice by SC injecting Y3-treated TKO cells (10 μM for 24 h). One week after vaccination, we injected untreated live TKO cells into their right flank (Figure 6D). Tumor growth in the right flank was suppressed in the mice vaccinated with Y3-treated cells (Figure 6D and 6E).
To test our hypothesis in human cells, we examined whether Y3 induced human EOC cells to release DAMPs that were critical markers for ICD. First, we detected a dose-dependent increase of HMGB1 protein in cell culture medium of Y3-treated A2780 cells via immunoprecipitation and western blot of HMGB1 (Figure 6F), which was confirmed by HMGB1 ELISA (Figure 6G). We also observed a dose-dependent decrease of intracellular HMGB1 (Figure 6F). Second, Y3 treatment induced the secretion of extracellular ATP (Figure 6H). Finally, we detected increased levels of CALR on the surface of Y3-treated EOC cells using flow cytometry (Figure 6I). Our findings demonstrated the ability of Y3 to induce the emission of DAMPs in human EOC cells.
Discussion
This study demonstrated the anti-cancer activity of Y3 in ovarian cancer models. We provided evidence that Y3 activated AMPK and UPR pathways in EOC cells in addition to inducing ICD and the release of DAMPs. The tumor-suppressing activity and minimal to nonexistent toxicity of Y3 in vivo demonstrated its therapeutic potential. Chemical uncouplers modulate multiple pathways to induce apoptosis of cancer cells and suppress tumor progression. Among them, AMPK is activated by the reduced ATP production (Supplemental references). Our data provided evidence that mitochondrial uncoupling triggered UPR through the activation of AMPK. Crosstalk between the AMPK and UPR pathway was implicated to play a critical role in cancer and metabolic diseases, however, the mechanism is unclear. Honokiol, a natural polyphenol isolated from the genus Magnolia, is an ER stress inducer that also activated AMPK to inhibit the growth of melanoma cells (42,43). Metformin and phenformin were identified as AMPK activators that also mediated UPR (44,45). Using Y3 and ovarian cancer cells, we have identified that the effects on AMPK were upstream of UPR.
The UPR is a crucial adaptive mechanism of cancer cells. Therapeutic agents that inhibit pro-survival UPR and/or induce apoptotic UPR provide promising treatments to suppress tumor progression. Besides common ER stress inducers, a growing number of anticancer compounds were linked to ER-mediated apoptosis through molecular mechanisms that lack complete knowledge, including compounds such as dihyfroartemisinin, diindolylmethane, versipelostatin, and vorinostat (Supplemental references). The molecular mechanisms of how ER stress elicits apoptosis are not fully understood. Our study on Y3 identified a new approach to induce ER stress-associated cell death in cancer cells through mitochondrial uncoupling. The low toxicity of Y3 in noncancerous cells and in vivo suggests its therapeutic potential.
Y3 is more effective in cancer cells that express higher levels of BIP1 than cells with low levels of BIP1. BIP1 is an anti-apoptotic central regulator of ER membrane signaling molecules. It enhances cell survival and drug-resistance (46,47). BIP1 is overexpressed by cancer stem cells in ovarian, breast, and head and neck cancers (Supplemental references). Our findings indicate the ability of Y3 to target drug-resistant cells and cancer stem cells in which the level of BIP1 expression can be used to predict the response to Y3 treatment.
The tumor microenvironment blocks the maturation and function of antigen presenting cells and suppresses the activation of tumor-specific cytotoxic T cells (48). Y3 has the potential to enhance adaptive antitumor immune response by inducing ICD. The activation of UPR is a known key denominator for ICD induction (49). UPR, in particular the PERK arm of the pathway, is critical for the ICD-derived emission of DAMPs (Supplemental references). Among the Y3-induced DAMPs, ATP binds to P2Y2/P2X7 receptors on innate immune cells, CALR binds to CD91 receptor on phagocytes, and HMGB1 binds to TLR2 and TLR4 on DCs (Supplemental references). These DAMPs stimulate pro-inflammatory cytokine production, the engulfing of dying cancer cells, and antigen presentation (Supplemental references). Y3 treatment stimulates the release of DAMPs and potentially helps to present tumor antigens to CD4+ and CD8+ T lymphocytes and to enable the adaptive immune response. This was supported by our observation that Y3 treatment eliminated tumors in immunocompetent mice and established immunological memory in the in vivo vaccination assay. Since mitochondria and UPR pathway are essential for the cellular functions of immune cells, future studies on how Y3 treatment directly affects immune cells will advance the development of mitochondria-targeting compounds as therapeutic agents.
Mitochondrial uncouplers have therapeutic potential; however, this potential requires that we address the challenge of developing selective uncouplers for cancer cells versus normal cells. Our findings suggest that these challenges are within our grasp in part because Y3 dispayed little to no toxicity in mouse models. The results of Y3 toxicity evaluation in the mouse models were promising. The next step towards the clinical translation of Y3 and its analogs requires the characterization of their pharmacokinetics and bioavailability as well as a complete toxicity assessment. Chemical modification and preclinical in vivo evaluation are also necessary to achieve optimal bioactivity with minimal off-target toxicity. Our research into these compounds will contribute to the development of anticancer therapies that eliminate cancer cells and provide long-term protection against relapse.
Supplementary Material
Acknowledgements
We thank Drs. Ronny Drapkin (University of Pennsylvania), Anirban Mitra (Indiana Univeristy School of Medicine), Alessandro Santin (Yale University School of Medicine), and Gil Mor (Yale University School of Medicine) for providing cell lines used in this study.
Funding
The research is supported by Rivkin Center Cookie Laughlin Bridge Award, Discovery To Cure Ovarian Cancer Research Grant, Colleen’s Dream Foundation, and the Office of the Assistant Secretary of Defense for Health Affairs through the Ovarian Cancer Research Program under Award No. W81XWH-15-1-0221. Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under Award Number K12CA215110. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AMPK
AMP-activated protein kinase
- ASK1
apoptosis signal-regulating kinase 1
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- eIF2α
eukaryotic translation initiation factor 2A
- CHOP
C/EBP-Homologous Protein, or DNA Damage inducible transcript 3 (DDIT3)
- co-IP
co-immunoprecipitation
- DAPI
4′,6-diamidino-2-phenylindole
- ER
Endoplasmic reticulum
- ERAD
ER-associated Protein Degradation
- FITC
fluorescein isothiocyanate
- GADD34
Growth Arrest And DNA-Damage-Inducible 34
- BIP1
binding immunoglobin protein, heat shock protein 70 family protein 5, glucose-regulated protein 78kDa, or GRP78
- IRE1α
Endoplasmic Reticulum To Nucleus Signaling 1
- JNK
c-Jun N-terminal kinase
- PARP
polyADP-ribose polymerase
- PCNA
Proliferating Cell Nuclear Antigen
- PERK
protein kinase R (PKR)-like endoplasmic reticulum kinase
- p-S6
phosphorylated ribosomal protein S6
- PUMA
P53 Up-Regulated Modulator Of Apoptosis
- RIDD
Regulated-IRE1-Dependent Decay of mRNA
- siRNAs
small interfering RNAs
- TRAF2
TNF Receptor Associated Factor 2
- XBP1
X-Box Binding Protein 1
- Δψm
mitochondrial membrane potential
Footnotes
Competing interests
The authors declare no potential conflicts of interest.
Availability of data and materials
All materials, data and protocols used in this manuscript are provided in the manuscript text or as supplementary material.
References
- 1.McGee J, Bookman M, Harter P, Marth C, McNeish I, Moore KN, et al. Fifth Ovarian Cancer Consensus Conference: individualized therapy and patient factors. Ann Oncol 2017;28(4):702–10 doi 10.1093/annonc/mdx010. [DOI] [PubMed] [Google Scholar]
- 2.Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol 2015;33(34):4015–22 doi 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 3.Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol 2019;30(7):1080–7 doi 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Sviripa VM, Kril LM, Yu T, Xie Y, Hubbard WB, et al. An Underlying Mechanism of Dual Wnt Inhibition and AMPK Activation: Mitochondrial Uncouplers Masquerading as Wnt Inhibitors. J Med Chem 2019;62(24):11348–58 doi 10.1021/acs.jmedchem.9b01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.To MS, Aromataris EC, Castro J, Roberts ML, Barritt GJ, Rychkov GY. Mitochondrial uncoupler FCCP activates proton conductance but does not block store-operated Ca(2+) current in liver cells. Arch Biochem Biophys 2010;495(2):152–8 doi 10.1016/j.abb.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 2013;5(3):a013169 doi 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007;129(7):1337–49 doi 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 2004;279(44):45495–502 doi 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, Wang C, Su J, Xie Q, Ma L, Zeng L, et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol Rep 2015;34(6):3051–60 doi 10.3892/or.2015.4283. [DOI] [PubMed] [Google Scholar]
- 10.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015;161(7):1527–38 doi 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limonta P, Moretti RM, Marzagalli M, Fontana F, Raimondi M, Montagnani Marelli M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int J Mol Sci 2019;20(4) doi 10.3390/ijms20040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006;107(12):4907–16 doi 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banerjee A, Lang JY, Hung MC, Sengupta K, Banerjee SK, Baksi K, et al. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J Biol Chem 2011;286(33):29127–38 doi 10.1074/jbc.M110.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson J, Palombella VJ, Fritz C, Normant E. IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer Chemother Pharmacol 2008;61(6):923–32 doi 10.1007/s00280-007-0546-0. [DOI] [PubMed] [Google Scholar]
- 15.Janssen K, Horn S, Niemann MT, Daniel PT, Schulze-Osthoff K, Fischer U. Inhibition of the ER Ca2+ pump forces multidrug-resistant cells deficient in Bak and Bax into necrosis. J Cell Sci 2009;122(Pt 24):4481–91 doi 10.1242/jcs.055772. [DOI] [PubMed] [Google Scholar]
- 16.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}−2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 2012;55(16):7193–207 doi 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 17.Schoenhacker-Alte B, Mohr T, Pirker C, Kryeziu K, Kuhn PS, Buck A, et al. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett 2017;404:79–88 doi 10.1016/j.canlet.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Huang FY, Lei J, Sun Y, Yan F, Chen B, Zhang L, et al. Induction of enhanced immunogenic cell death through ultrasound-controlled release of doxorubicin by liposome-microbubble complexes. Oncoimmunology 2018;7(7):e1446720 doi 10.1080/2162402X.2018.1446720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC, et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol 2012;189(6):3209–20 doi 10.4049/jimmunol.1103826. [DOI] [PubMed] [Google Scholar]
- 20.Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 2015;6(29):26841–60 doi 10.18632/oncotarget.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bains SJ, Abrahamsson H, Flatmark K, Dueland S, Hole KH, Seierstad T, et al. Immunogenic cell death by neoadjuvant oxaliplatin and radiation protects against metastatic failure in high-risk rectal cancer. Cancer Immunol Immunother 2019. doi 10.1007/s00262-019-02458-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007;13(9):1050–9 doi 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 23.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007;13(1):54–61 doi 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 24.Feng H, Zeng Y, Graner MW, Likhacheva A, Katsanis E. Exogenous stress proteins enhance the immunogenicity of apoptotic tumor cells and stimulate antitumor immunity. Blood 2003;101(1):245–52 doi 10.1182/blood-2002-05-1580. [DOI] [PubMed] [Google Scholar]
- 25.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009;15(10):1170–8 doi 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 26.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418(6894):191–5 doi 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 27.Perrone E, Lopez S, Zeybek B, Bellone S, Bonazzoli E, Pelligra S, et al. Preclinical Activity of Sacituzumab Govitecan, an Antibody-Drug Conjugate Targeting Trophoblast Cell-Surface Antigen 2 (Trop-2) Linked to the Active Metabolite of Irinotecan (SN-38), in Ovarian Cancer. Front Oncol 2020;10:118 doi 10.3389/fonc.2020.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craveiro V, Yang-Hartwich Y, Holmberg JC, Joo WD, Sumi NJ, Pizzonia J, et al. Phenotypic modifications in ovarian cancer stem cells following Paclitaxel treatment. Cancer Med 2013;2(6):751–62 doi 10.1002/cam4.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karst AM, Drapkin R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat Protoc 2012;7(9):1755–64 doi 10.1038/nprot.2012.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Yang N, Garcia JR, Mohamed A, Benencia F, Rubin SC, et al. Generation of a syngeneic mouse model to study the effects of vascular endothelial growth factor in ovarian carcinoma. Am J Pathol 2002;161(6):2295–309 doi 10.1016/s0002-9440(10)64505-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Coffey DM, Ma L, Matzuk MM. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 2015;156(6):1975–81 doi 10.1210/en.2014-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim O, Park EY, Klinkebiel DL, Pack SD, Shin YH, Abdullaev Z, et al. In vivo modeling of metastatic human high-grade serous ovarian cancer in mice. PLoS Genet 2020;16(6):e1008808 doi 10.1371/journal.pgen.1008808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aurbach K, Spindler M, Haining EJ, Bender M, Pleines I. Blood collection, platelet isolation and measurement of platelet count and size in mice-a practical guide. Platelets 2019;30(6):698–707 doi 10.1080/09537104.2018.1528345. [DOI] [PubMed] [Google Scholar]
- 34.O'Connell KE, Mikkola AM, Stepanek AM, Vernet A, Hall CD, Sun CC, et al. Practical murine hematopathology: a comparative review and implications for research. Comp Med 2015;65(2):96–113. [PMC free article] [PubMed] [Google Scholar]
- 35.Chew JL, Chua KY. Collection of mouse urine for bioassays. Lab Anim (NY) 2003;32(7):48–50 doi 10.1038/laban0803-48. [DOI] [PubMed] [Google Scholar]
- 36.Folkins AK, Jarboe EA, Saleemuddin A, Lee Y, Callahan MJ, Drapkin R, et al. A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecologic oncology 2008;109(2):168–73 doi 10.1016/j.ygyno.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhn E, Kurman RJ, Vang R, Sehdev AS, Han G, Soslow R, et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J Pathol 2012;226(3):421–6 doi 10.1002/path.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cappella P, Gasparri F. Highly multiplexed phenotypic imaging for cell proliferation studies. J Biomol Screen 2014;19(1):145–57 doi 10.1177/1087057113495712. [DOI] [PubMed] [Google Scholar]
- 39.Ali MM, Akhade VS, Kosalai ST, Subhash S, Statello L, Meryet-Figuiere M, et al. PAN-cancer analysis of S-phase enriched lncRNAs identifies oncogenic drivers and biomarkers. Nat Commun 2018;9(1):883 doi 10.1038/s41467-018-03265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fazil MH, Ong ST, Chalasani ML, Low JH, Kizhakeyil A, Mamidi A, et al. GapmeR cellular internalization by macropinocytosis induces sequence-specific gene silencing in human primary T-cells. Sci Rep 2016;6:37721 doi 10.1038/srep37721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma YF, Chen C, Li D, Liu M, Lv ZW, Ji Y, et al. Targeting of interleukin (IL)-17A inhibits PDL1 expression in tumor cells and induces anticancer immunity in an estrogen receptor-negative murine model of breast cancer. Oncotarget 2017;8(5):7614–24 doi 10.18632/oncotarget.13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin S, Lamb HK, Brady C, Lefkove B, Bonner MY, Thompson P, et al. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br J Cancer 2013;109(2):433–43 doi 10.1038/bjc.2013.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaushik G, Kwatra D, Subramaniam D, Jensen RA, Anant S, Mammen JM. Honokiol affects melanoma cell growth by targeting the AMP-activated protein kinase signaling pathway. Am J Surg 2014;208(6):995–1002; discussion 1-2 doi 10.1016/j.amjsurg.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin YC, Wu MH, Wei TT, Lin YC, Huang WC, Huang LY, et al. Metformin sensitizes anticancer effect of dasatinib in head and neck squamous cell carcinoma cells through AMPK-dependent ER stress. Oncotarget 2014;5(1):298–308 doi 10.18632/oncotarget.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L, Sha H, Davisson RL, Qi L. Phenformin activates the unfolded protein response in an AMP-activated protein kinase (AMPK)-dependent manner. J Biol Chem 2013;288(19):13631–8 doi 10.1074/jbc.M113.462762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett 2007;581(19):3641–51 doi 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal 2009;11(9):2307–16 doi 10.1089/ARS.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011;208(10):1989–2003 doi 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012;31(5):1062–79 doi 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.