

Abstract
Background
Ferroptosis, a programmed cell death characterized by lipid peroxidation, is implicated in various diseases including cancer. Although cell density-dependent E-cadherin and Merlin/Neurofibromin (NF2) loss can modulate ferroptosis, the role of ferroptosis and its potential link to NF2 status and E-cadherin expression in meningioma remain unknown.
Methods
Relationship between ferroptosis modulators expression and NF2 mutational status was examined in 35 meningiomas (10 NF2 loss and 25 NF2 wild type). The impact of NF2 and E-cadherin on ferroptosis were examined by lactate dehydrogenase (LDH) release, lipid peroxidation, and western blot assays in IOMM-Lee, CH157, and patient-derived meningioma cell models. Luciferase reporter and chromatin immunoprecipitation assays were used to assess the ability of MEF2C (myocyte enhancer factor 2C) to drive expression of NF2 and CDH1 (E-cadherin). Therapeutic efficacy of Erastin-induced ferroptosis was tested in xenograft mouse models.
Results
Meningioma cells with NF2 inactivation were susceptible to Erastin-induced ferroptosis. Meningioma cells grown at higher density increased expression of E-cadherin, which suppressed Erastin-induced ferroptosis. Maintaining NF2 and E-cadherin inhibited ferroptosis-related lipid peroxidation and meningioma cell death. MEF2C was found to drive the expression of both NF2 and E-cadherin. MEF2C silencing enhanced Erastin-induced ferroptotic meningioma cell death and lipid peroxidation levels in vitro, which was limited by forced expression of MEF2C targets, NF2 and E-cadherin. In vivo, anti-meningioma effect of Erastin was augmented by MEF2C knockdown and was counteracted by NF2 or E-cadherin.
Conclusions
NF2 loss and low E-cadherin create susceptibility to ferroptosis in meningioma. MEF2C could be a new molecular target in ferroptosis-inducing therapies for meningioma.
Keywords: E-cadherin, ferroptosis, MEF2C, meningioma, NF2
Key Points.
1. Deficient NF2 and E-cadherin sensitize meningioma to ferroptosis.
2. MEF2C promotes transcription of both NF2 and E-cadherin.
3. MEF2C silencing enhances ferroptosis-mediated inhibition of meningioma growth in vivo.
Importance of the Study.
Identification of aberrant genetic alterations driving tumor progression, and discovery of novel molecular therapeutic targets are essential to improve the management of aggressive meningiomas. Ferroptosis is a cell death process that can be affected by NF2 and cell density. However, the role of ferroptosis in meningioma biology and therapy remains largely unknown. Here, we show for the first time that meningiomas with NF2 inactivation and diminished E-cadherin expression are more prone to ferroptosis. We identify the transcription factor MEF2C as a driver of transcription of both NF2 and CDH1 (E-cadherin) in meningioma and demonstrate that MEF2C silencing augments ferroptosis-mediated inhibition of meningioma growth in vitro and in vivo. Our study highlights ferroptosis as a novel therapeutic vulnerability in the major subset of meningioma carrying NF2 inactivation. Our work also supports the development of MEF2C inhibitors that could maximize ferroptosis-inducing therapies in aggressive meningioma.
Meningioma is the most common primary neoplasms in the central nervous system (CNS) (CBTRUS 2020),1 which is classified into 3 histological grades and 15 subtypes according to the 2016 WHO CNS tumor grading criterion.2 About 80% of the meningiomas are benign accompanied by a lower histological grade and usually have satisfying outcomes. However, a subset of higher-grade tumors manifests a more aggressive biological behavior and is more prone to relapse.3–5 Due to the lack of effective chemotherapeutic or targeted therapies, patients with aggressive meningiomas, especially with WHO grade III tumors, currently carry an extremely poor prognosis. Therefore, identification of aberrant genetic alterations driving meningioma progression, and discovery of novel molecular therapeutic targets are significant unmet clinical needs for aggressive meningioma patients.
Ferroptosis is a programmed cell death process characterized by aberrant, iron-dependent accumulation of lipid peroxidation. Ferroptosis was first described in 2012 as an unusual form of cell death induced by Erastin, a small molecule that inhibits the cellular import of cysteine, leading to glutathione depletion and inactivation of the phospholipid peroxidase glutathione peroxidase 4 (GPX4).6,7 On the other hand, acyl-CoA synthetase long-chain family member 4 (ACSL4) is essential in ferroptosis execution and mediates ferroptosis sensitivity by shaping the cellular lipid composition.8 Thus, GPX4 and ACSL4 are the 2 well-known key proteins involved in the induction of ferroptosis. Exploiting ferroptosis is a new promising approach to inhibit tumor growth or kill therapy-resistant cancers.9
Heterozygous deletion and loss-of-function mutations of the Merlin-encoding gene NF2, which is located at the 22q12.2 locus, are detected with high frequency in sporadic high-grade meningiomas. Inactivating mutation in NF2 is associated with a worse prognosis in meningioma patients.10 Altered NF2, however, is not directly actionable, and extensive efforts have been made to identify vulnerabilities linked to NF2-mutant meningiomas.11 Previously, studies identified NF2 as a regulator of ferroptosis.12 Moreover, cell density-dependent ferroptosis has been observed in several different cancer cell lines.12 Being secreted on the surface of cell membranes, E-cadherin is upregulated as the cell density increases, participating in the lipid peroxidase pathway in a cell density-dependent manner.13
The aim of this study was to evaluate the potential role of ferroptosis in meningioma. We found that NF2 loss sensitized meningioma cells to Erastin-induced ferroptosis, and that E-cadherin alleviated ferroptosis-induced cell death. We further discovered that the transcription factor, myocyte enhancer factor 2C (MEF2C) regulated both NF2 and E-cadherin, and limited ferroptosis. Given our findings and the importance of ferroptosis-related lipid metabolism in CNS tumors,14,15 investigating the impact of ferroptosis on meningioma growth should increase understanding of the biology of meningioma progression and might provide new therapeutic opportunities.
Materials and Methods
Tumor Samples
A number of 35 meningiomas surgically treated in 2018 were enrolled in the study (Supplementary Table 1). See the Supplementary Methods for ethics statement and details.
RT-PCR and Sanger Sequencing
Quantitative RT-PCR was performed following the manufacturer’s instructions. See the Supplementary Methods for details.
Cell Culture and Treatment
The source and culture of CH157-MN, IOMM-Lee, and patient-derived PM3 meningioma cells were described in the Supplementary Methods for details.
Western Blotting
Proteins were isolated from cells using a KGP2100 kit (KeyGene, Biotech), and levels of relative proteins were detected according to the manufacturer’s instructions. See the Supplementary Methods for details.
Immunohistochemistry (IHC) Assay
The detailed protocol was described in our previous studies.16 See the Supplementary Methods for details.
Immunofluorescence Staining
E-cadherin and tumor-associated epithelial membrane antigen (EMA) expressions were detected by immunofluorescence staining. See the Supplementary Methods for details.
Lentivirus Transduction of Cells
Vectors expressing NF2 (pLV-Ef1a-NF2-Puro), MEF2C (pLV-Ef1a-MEF2C-Puro), E-cadherin (pLV-Ef1a-E-cadherin-Puro), and control plasmid (pLV-Ef1a-Puro) were obtained from Genepharma (Shanghai, China). See the Supplementary Methods for details.
Dual-Luciferase Reporter Assay
Luciferase vectors driven by promoter sequences containing mutated putative MEF2C binding sites were generated. NF2 and E-cadherin transcriptional activity was measured by dual-luciferase reporter assay following the manufacturer’s instructions. See the Supplementary Methods for details.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were performed using the ChIP assay kit (Millipore), following the manufacturer’s instructions and as described previously.17 See the Supplementary Methods for details.
Lipid Peroxidation Assay
Lipid peroxidation levels were tested by the reagent BODIPY 581/591 C11 (D3861, Invitrogen), MDA Assay Kit (ab118970, Abcam), and 12/15-HETE ELISA kits (ab133034/ab133035, Abcam). See the Supplementary Methods for details.
Cytotoxicity Assay
Cytotoxicity was quantified by measuring released LDH activity using the Cytotoxicity Detection Kit (LDH) (Beyotime Biotech, China). See the Supplementary Methods for details.
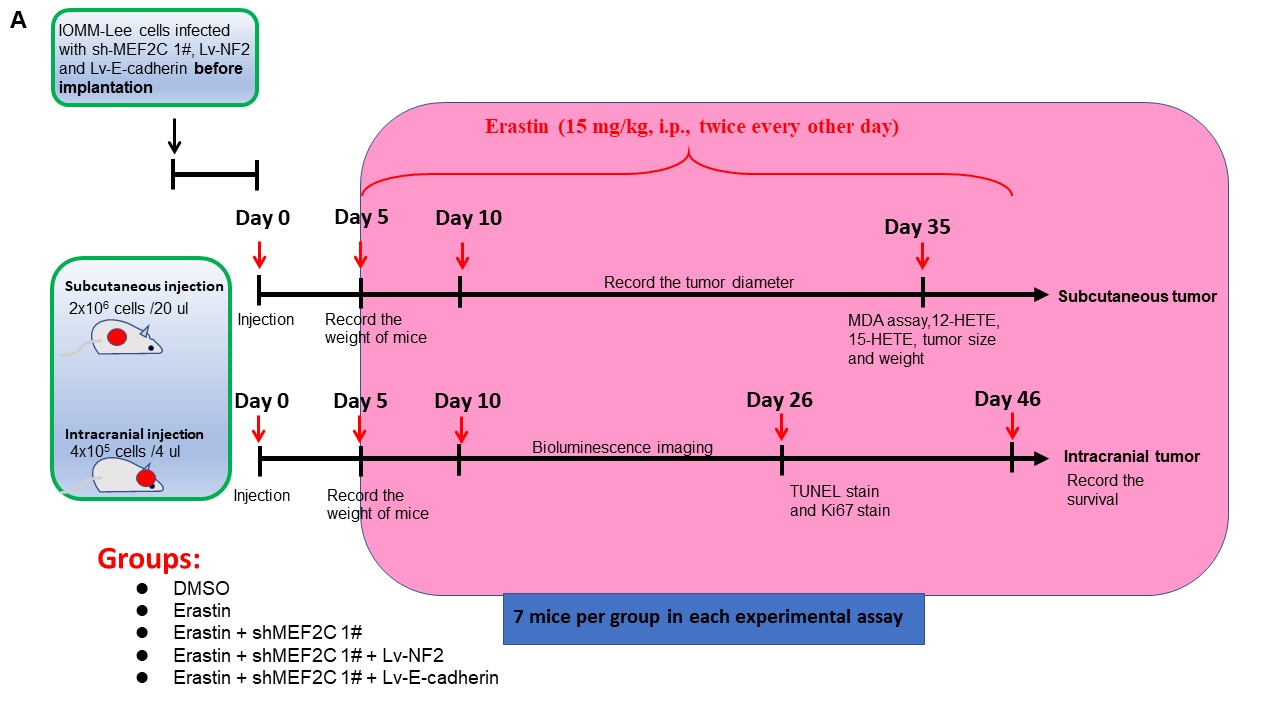
Xenograft Mouse Model and Drug Administration
Animal experiments were approved by the Animal Management Rule of the Chinese Ministry of Health (documentation 55, 2001) and were in accordance with the approved guidelines and the experimental protocol of Nanjing Medical University. See the Supplementary Methods for details.
Mitochondrial Morphology Assays
Transmission electron microscope (TEM) was used to assess mitochondrial morphology. See the Supplementary Methods for details.
Ethics Statement
The studies involving human participants were reviewed and approved by The Research Ethics Committee of Nanjing Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Animal Ethical and Welfare Committee of Nanjing Medical University, and the protocol has been reviewed and approved by the Animal Ethical and Welfare Committee, and the protocol has been reviewed and approved by the Animal Ethical and Welfare Committee.
Statistical Analysis
Data were analyzed with GraphPad 8.0 software and reported as the mean ± standard deviation. An unpaired t test or 1-way analysis of variance (ANOVA) plus Tukey’s test was applied in western blot, fluorescence, immunohistochemical quantification, weight of tumor, and weight of mice in each time point. For the diameter of subcutaneous tumor within continued periods, data of the whole group were analyzed using 2-way ANOVA with repeated measures followed by Tukey post hoc test for comparisons between groups. Significant differences were set at P < .05.
Results
Meningioma With NF2 Inactivation Showed High ACSL4 and Low GPX4 Expression
A total of 35 patients with primary meningiomas were included in this study. Clinical information of the cohort is listed in Supplementary Table 1. NF2 mutation was detected using the Sanger sequencing method. The protein expression level of NF2-encoded protein Merlin was further determined by IHC. To investigate the relationship between NF2 and ferroptosis in meningioma, we used quantitative RT-PCR analysis and demonstrated a significant negative correlation between NF2 and ACSL4 mRNA expression levels, and a positive correlation between NF2 and GPX4 mRNA (Figure 1A–D). Twelve samples (6 NF2 wild type and 6 NF2 mutant) were further analyzed for NF2, GPX4, and ACSL4 protein expression using western blot. The correlation analysis revealed that lower GPX4 and higher ACSL4 expression were significantly more prevalent in the NF2 mutation group (Figure 1E–H). To further confirm the relationship between GPX4, ACSL4, and NF2, these 12 human meningiomas with or without intact NF2 were assessed for GPX4 and ACSL4 protein expression using IHC, revealing that meningiomas with lower or negative NF2 expression correlated with lower GPX4 and higher ACSL4 protein expression (Figure 1I–L). Together, differential expression of ferroptosis modulators GPX4 and ACSL4 suggested that meningioma with inactivated NF2 might be more prone to ferroptosis.
Fig. 1.

The expression of NF2, ACSL4, and GPX4 in human meningioma tissues. (A–D) Quantitative RT-PCR analysis of human meningioma (n = 35) was displayed. (A) A correlation of NF2 and GPX4 mRNA expression (r2 = 0.3374). (B) A correlation of NF2 and ACSL4 mRNA expression (r2 = 0.3454). (C and D) Lower GPX4 was more prevalent in the NF2 loss group, while lower ACSL4 was more prevalent in the NF2 intact group. (E) Western blotting analysis of GPX4, ACSL4, and NF2 protein expression in human meningiomas. GAPDH was used as a control. (F–H) Densitometry quantification of NF2, GPX4, and ACSL4 protein by ImageJ. n = 6. (I) Representative IHC photos of NF2, GPX4, and ACSL4 protein expression. (J–L) IHC image quantification NF2, GPX4, and ACSL4 by ImageJ. n = 6. **P < .01 and ***P < .001. Error bars represent SD. Abbreviations: ACSL4, acyl-CoA synthetase long-chain family member 4; GPX4, glutathione peroxidase 4 (GPX4); IHC, immunohistochemistry; NF2, neurofibromatosis type 2; RT-PCR, reverse transcription polymerase chain reaction.
NF2 Inhibition Promoted Ferroptosis-Related Cytotoxicity and Lipid Peroxidation
To explore the effect of NF2 on ferroptosis, we used 2 meningioma cell lines, CH157 (NF2 mutant) and IOMM-Lee (NF2 intact),18 as well as the primary meningioma cells PM3 (Supplementary Figure 1A). CH157 and IOMM-Lee cells have a high number of copy number alterations, consistent with the genetic profiles of human malignant meningioma.18 Regarding genetic alterations relevant to meningioma, both cell lines have TERT promoter mutation C228T, while CH157 has loss of chromosome 22 and IOMM-Lee has loss of chromosome 9p21.3 that harbors CDNK2A.18 Sanger sequencing of PM3 cells identified c.731delA frame-shift mutation in NF2 (Supplementary Figure 1B). Erastin, the specific ferroptosis inducer, was used to trigger ferroptosis in the 3 types of meningioma cells. All meningioma cells responded to ferroptosis, since with the increasing concentration of Erastin, LDH release was induced in the 3 cell lines. However, LDH release was more easily induced from CH157 and PM3 at lower concentrations of Erastin (Supplementary Figure 2A and B). In contrast, higher concentrations of Erastin were needed to cause significantly increased LDH release from IOMM-Lee cells (Supplementary Figure 2C). Based on these results, we selected Erastin at the concentration of 6 µM, with which significant LDH release was induced in CH157 and PM3 but not in IOMM-Lee cells, in the following experiments. The differing NF2 status in IOMM-Lee, CH157, and PM3 made these cells suitable to explore the relationship between NF2 and ferroptosis.
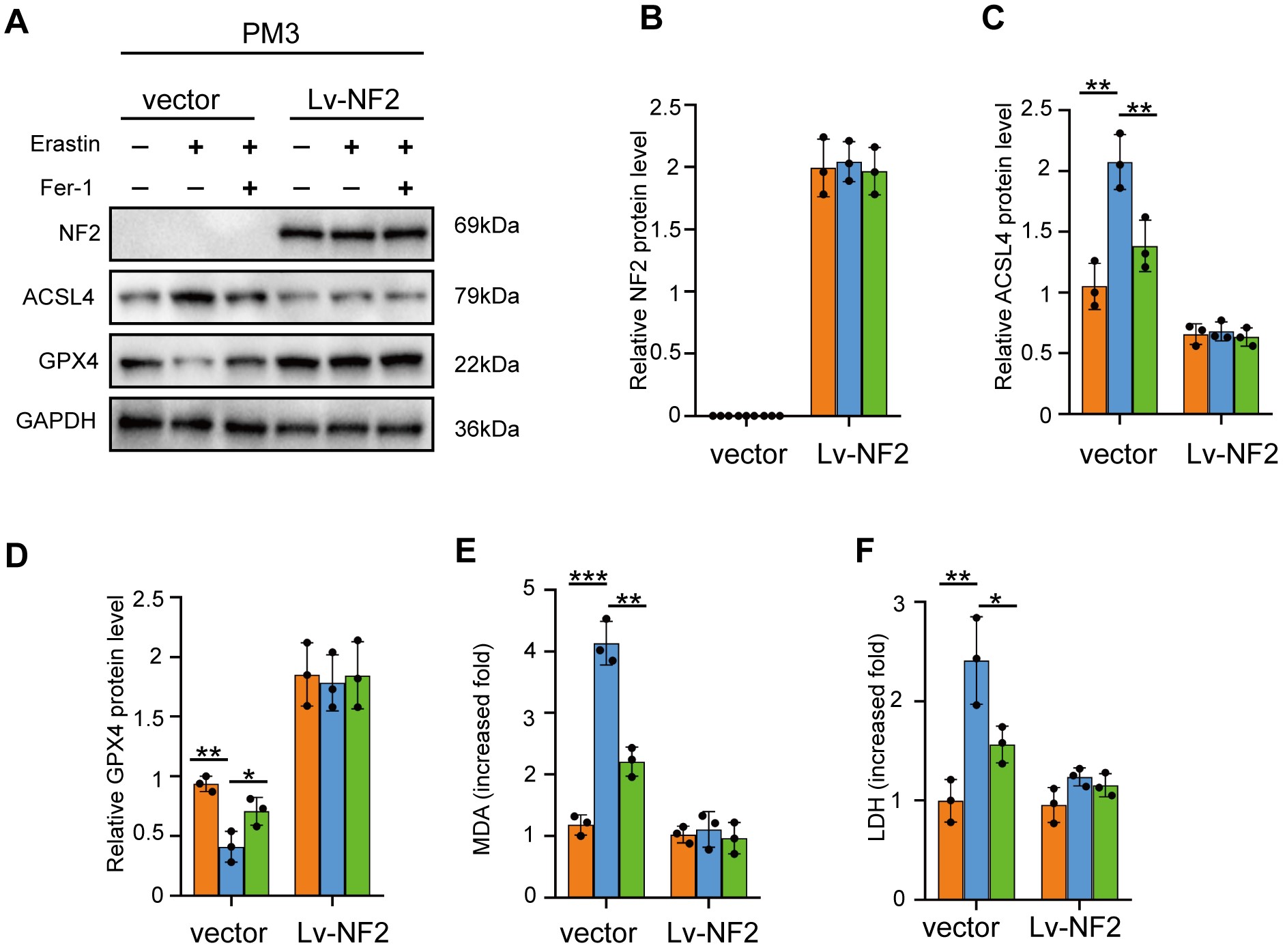
We first tested the effect of NF2 on ferroptosis in CH157 and PM3 using Erastin and the specific ferroptosis inhibitor, Ferrostain-1 (Fer-1). 6 µM Erastin enhanced ACSL4 expression and reduced GPX4 in CH157 and PM3, which was alleviated by Fer-1. However, exogenous overexpression of wild-type NF2 abrogated the changes induced by Erastin and Fer-1 (Figure 2A–D; Supplementary Figure 3A–D). Increased lipid peroxidation measured by malonaldehyde (MDA) and level of LDH were found in CH157 and PM3 cells treated with Erastin, which was limited by Fer-1. However, no changes were observed in CH157 and PM3 overexpressing wild-type NF2 (Figure 2E and F; Supplementary Figure 3E and F). The effect of NF2 on ferroptosis was also tested in IOMM-Lee with intact NF2. We generated NF2 knockdown using 2 independent shRNA sequences in IOMM-Lee cells (Supplementary Figure 4A and B). 6 µM Erastin did not significantly change ACSL4 and GPX4 expression in shCtrl IOMM-Lee cells. However, when NF2 was silenced, Erastin induced ACSL4 upregulation and GPX4 downregulation, which were alleviated by Fer-1 (Figure 2G–J). Similarly, increased MDA and LDH were significantly triggered by Erastin in NF2-knockdown IOMM-Lee cells, but not in shCtrl cells (Figure 2K and L). Together, these results suggest that loss of NF2 might sensitize meningioma to ferroptosis-induced cytotoxicity.
Fig. 2.

NF2 loss sensitized meningioma cells to Erastin-induced ferroptosis. CH157 (A–F) and IOMM-Lee (G–L) cells transduced with control (Vector) and NF2 expression/silencing lentivirus vector (Lv-NF2/sh-NF2) were stimulated with 6 µM Erastin for 24 hours to trigger ferroptosis. Fer-1 (3 µM for 24 hours) was used to inhibit ferroptosis. (A–D) Western blot assays and densitometry quantification for NF2, ACSL4, and GPX4. n = 3 experiments. GAPDH was used as a control. (E) Lipid peroxidation tested by MDA assays. n = 5 experiments. (F) Cytotoxicity tested by LDH assays. n = 5 experiments. (G–J) Western blot assays and densitometry quantification for NF2, ACSL4, and GPX4. n = 3 experiments. (K) Lipid peroxidation tested by MDA assays. n = 5 experiments. (L) Cytotoxicity tested by LDH assays. n = 5 experiments. *P < .05, **P < .01, and ***P < .001. Error bars represent SD. Abbreviations: ACSL4, acyl-CoA synthetase long-chain family member 4; GPX4, glutathione peroxidase 4; NF2, neurofibromatosis type 2.
E-Cadherin Induced Cell Density-Dependent Inhibition of Ferroptosis
We next sought other factors than NF2 that influence cell fates after exposure to Erastin. We found distinct characteristics in cell growth of the 3 cell lines; IOMM-Lee cells showed the tendency to form locally high-density clusters while CH157 and PM3 cells showed dispersed distribution. A previous study reporting that cell density affects ferroptosis in mesothelioma cells,12 prompted us to examine whether cell density had a similar influence in meningioma.
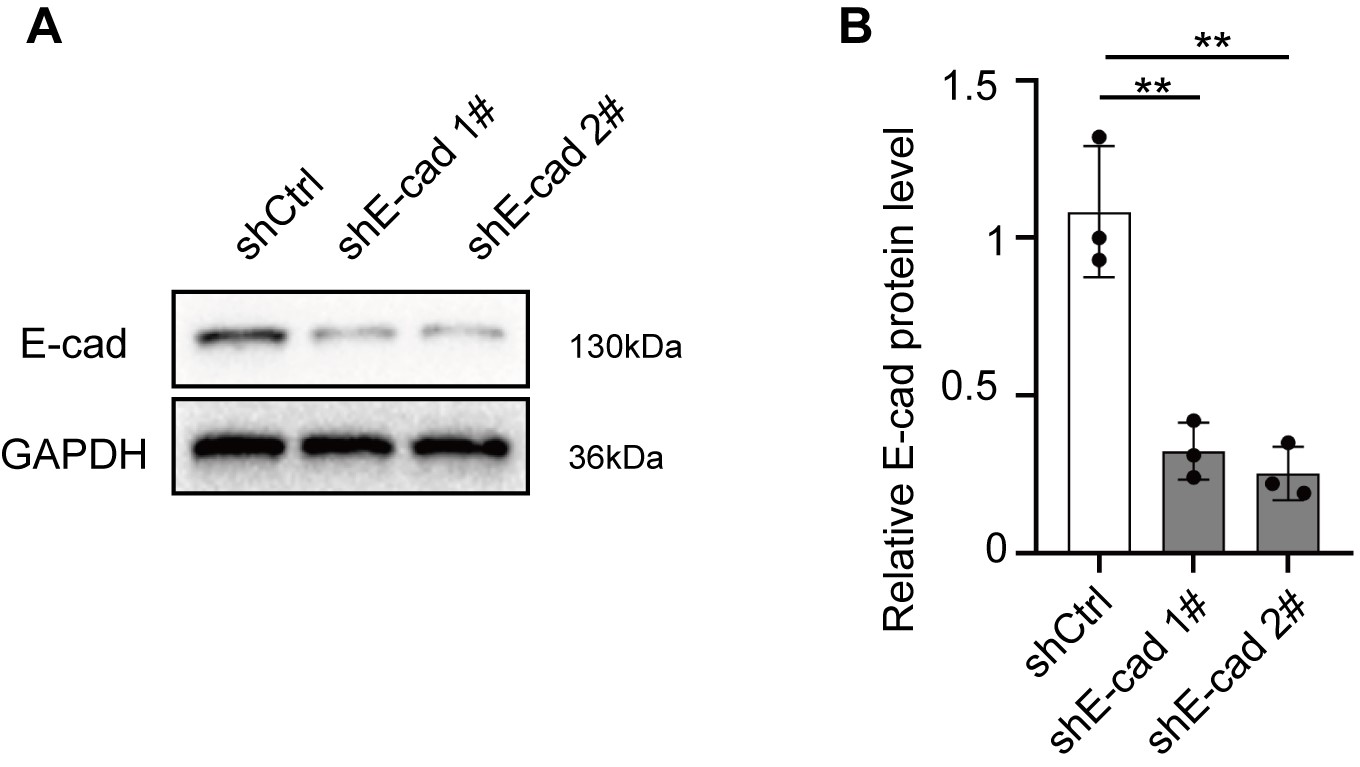
The level of lipid peroxidation (reactive oxygen species [ROS]) assayed by C11-BODIPY (581/591) gradually declined with the increasing cell density of CH157, IOMM-Lee, and PM3 cells (Figure 3A and B; Supplementary Figure 5A and B), suggesting that ferroptosis induced by Erastin was dependent on cell density. Generally, cell density and cell-cell adhesion modulated the expression of E-cadherin that mediates interactions between cells.12,19 Western blot assay showed that the expression of E-cadherin was upregulated as meningioma cell density increased (Figure 3C and D; Supplementary Figure 5C and D). Immunofluorescence assays showed similar results (Figure 3E–G; Supplementary Figure 5E and F). To ask whether E-cadherin inhibited lipid peroxidation and ferroptosis when cell density was high, we downregulated E-cadherin in IOMM-Lee cells (Supplementary Figure 6A and B). In shCtrl IOMM-Lee cells grown at high density, Erastin up to 11 µM was not cytotoxic (Figure 3H), with no change in ACSL4 and GPX4 levels after Erastin treatment. However, in IOMM-Lee cells expressing shE-cadherin, Erastin induced ACSL4 and lowered GPX4, which was inhibited by Fer-1 (Figure 3I–L). Similarly, Erastin promoted lipid peroxidation and cytotoxicity in shE-cadherin-transfected cells, but not in shCtrl cells, cultured at high density, which was alleviated by Fer-1 (Figure 3M and N). In order to eliminate the effect of NF2, we also tested the relationship between E-cadherin and ferroptosis in NF2-knockdown IOMM-Lee cells and PM3 cells at high density. shNF2 IOMM-Lee cells were more sensitive than the parental cells and the maximum nontoxic concentration with high density was 2 μM (Supplementary Figure 7A). Low dose (2 μM) Erastin stimulation of shNF2 IOMM-Lee did not significantly change ACSL4 and GPX4 levels. However, upregulated ACSL4 and downregulated GPX4 were found after additional E-cadherin knockdown, which was blocked by Fer-1 (Supplementary Figure 7B–E). Furthermore, levels of LDH and MDA were increased in NF2/E-cadherin double knockdown cells even when 2 µM Erastin was used (Supplementary Figure 7F and G). Similar results were obtained with PM3 at high density. 6 µM Erastin was the maximum nontoxic concentration (Supplementary Figure 8A) and did not change the level of ACSL4 and GPX4 (Supplementary Figure 8B–E). Knockdown of E-cadherin induced a significant increase of ACSL4 but a decrease of GPX4. Levels of MDA and LDH were also enhanced when 6 µM Erastin was exposed to PM3 cells at high density (Supplementary Figure 8F and G). Therefore, deficient E-cadherin promoted the sensitivity to ferroptosis in meningioma cells independent of the effect of NF2.
Fig. 3.

Cell density-related E-cadherin upregulation had anti-ferroptosis effects. (A and B) 9 µM Erastin for 24 hours was used to trigger ferroptosis in CH157 and IOMM-Lee cells at low, normal, and high density, respectively. (I–N) IOMM-Lee cells at a high density were stimulated by 11 µM Erastin for 24 hours. Fer-1 (3 µM for 24 hours) was used to inhibit ferroptosis. (A and B) Lipid peroxidation was detected by C11-BODIPY (581/591) in CH157 and IOMM-Lee cells. Mean fluorescence intensity was analyzed by Image J. n = 5 experiments. Scale bar is 50 µm. (C and D) E-cadherin protein expression tested by western blot. n = 3 experiments. (E–G) Immunofluorescence assay for E-cadherin. n = 5 experiments. Scale bar is 50 µm. (H) Cytotoxicity tested by LDH assays. n = 4 experiments. (I–L) Western blot and densitometry quantification for E-cadherin, ACSL4, and GPX4. GAPDH was used as a loading control. n = 5 experiments. (M) Lipid peroxidation tested by MDA assays. n = 5 experiments. (N) Cytotoxicity tested by LDH assays. n = 5 experiments. *P < .05, **P < .01, and ***P < .001. Error bars represent SD. Abbreviations: ACSL4, acyl-CoA synthetase long-chain family member 4; GPX4, glutathione peroxidase 4.
MEF2C Promoted Both NF2 and E-Cadherin Transcription
Our results showed that NF2 and E-cadherin both limited meningioma cells’ sensitivity to Erastin-induced ferroptosis. We speculated whether NF2 and E-cadherin (CDH1) genes were regulated by the same transcription factor. We searched potential transcription factors regulating expression of both NF2 and CDH1 in the JASPAR 2020. Based on relative scores shown, we screened out the top 10 transcription factors (Supplementary Table 4), and only MEF2C emerged as the transcription factor common for NF2 and CDH1 (Figure 4A). MEF2C was a member of the family of transcription factors MEF2 and was initially described in embryogenesis regulating tissue-specific genes. It was also expressed in many types of adult cells, including neuronal and endothelial cells20 and associated with tumor progression.21 We here studied whether MEF2C regulated transcription of NF2 and CDH1 in meningioma.
Fig. 4.

MEF2C promoted the expression of both NF2 and E-cadherin. (A) MEF2C was screened out from top10 transcription factors regulating NF2 and E-cadherin (CDH1) expression (JASPAR 2020). (B and C) Dual-luciferase reporter gene assays for NF2 and E-cadherin promoters. MEF2C increased luciferase activity. n = 3 experiments. (D and E) Western blot and densitometry quantification for MEF2C, NF2, and E-cadherin. GAPDH was used as a control. n = 3 experiments. (F) ChIP assays. (G) Structure drawing for wild type (WT) and mutated sequences of NF2 and E-cadherin (CDH1) promoters. Luciferase reporter gene assays were used to verify MEF2C-binding sites. n = 3 experiments. **P < .01 and ***P < .001. Error bars represent SD. Abbreviations: MEF2C, myocyte enhancer factor 2C; NF2, neurofibromatosis type 2.
In IOMM-Lee cells, dual-luciferase reporter assay indicated MEF2C overexpression promoted the transcription of NF2 and E-cadherin (Figure 4B and C). MEF2C also increased the protein expressions of NF2 and E-cadherin (Figure 4D and E). Moreover, ChIP assay demonstrated that MEF2C can directly bind to the specific region of the NF2 and E-cadherin promoters in IOMM-Lee (Figure 4F). We also conducted dual-luciferase reporter assays using constructs driven by promoter sequences containing mutations in the putative binding sites (Figure 4G). Mutations in sequence 1 of the NF2 promoter and those in sequences 1 and 2 of the CDH1 promoter diminished the promoter activity (Figure 4G), providing results consistent with the ChIP assay. Together, these findings indicate that MEF2C is a common transcription factor driving expressions of both NF2 and E-cadherin genes.
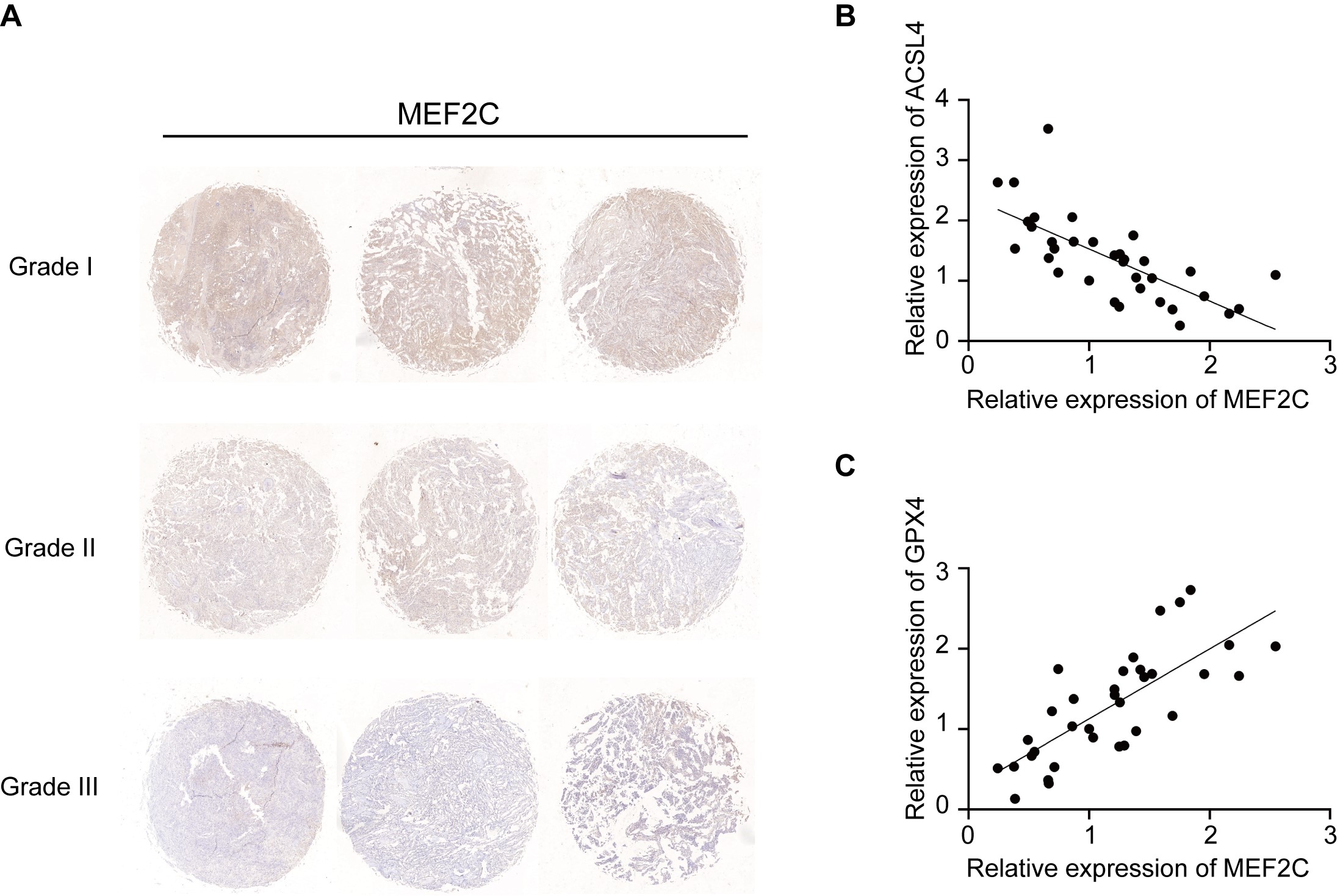
Next, we tested the expression of MEF2C in human meningioma tissues. The levels of MEF2C protein declined as the malignancy grade increased (Supplementary Figure 9A). A significant negative correlation between MEF2C and ACSL4 protein expression levels and a positive correlation between MEF2C and GPX4 protein were found (Supplementary Figure 9B and C). These data suggest loss of MEF2C to be a clinically applicable biomarker of meningioma vulnerability to ferroptosis.
MEF2C Inhibited Ferroptosis by Upregulating NF2 and E-Cadherin
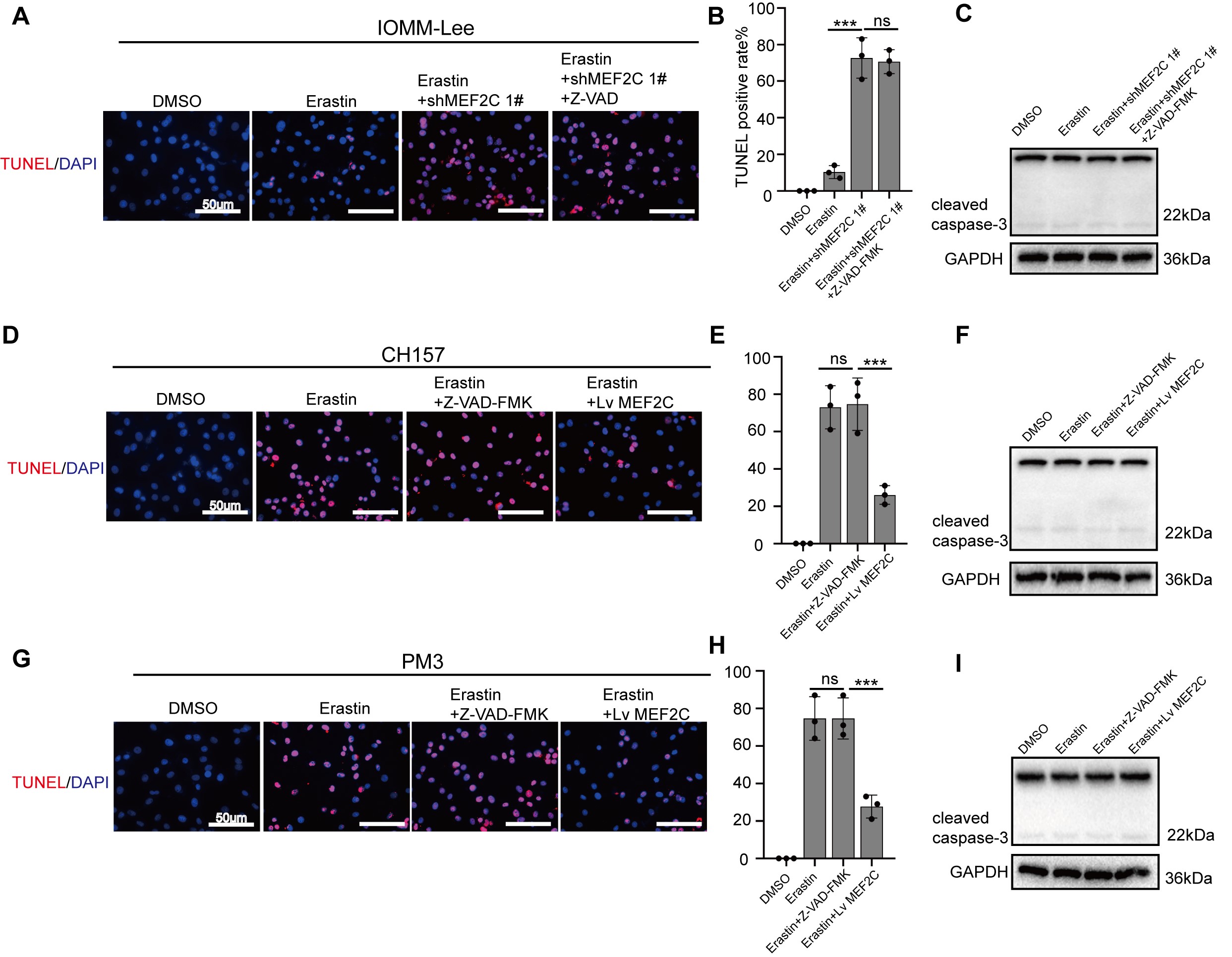
We next examined the potential role of MEF2C in ferroptosis. MEF2C was silenced efficiently by shMEF2C sequences (Supplementary Figure 10A and B). TUNEL assay revealed that 6 µM Erastin had little effect in IOMM-Lee cells, but profoundly induced cell death in MEF2C-knockdown cells, which was not reversed by Z-VAD-FMK, a specific inhibitor of caspase-mediated apoptosis (Supplementary Figure 11A and B). In CH157 and PM3 cells, overexpression of MEF2C reduced the fraction of TUNEL-positive cells post-Erastin exposure, while Z-VAD-FMK did not (Supplementary Figure 11D, E, G, and H). In addition, Erastin treatment of IOMM-Lee (MEF2C-knockdown), CH157, or PM3 cells did not induce cleaved caspase-3 despite its cytotoxic effects (Supplementary Figure 11C, F, and I). These results indicate that caspase-dependent apoptosis does not play a significant role in Erastin-induced meningioma death that is accompanied by TUNEL positivity.
In NF2-mutant CH157 cells, MEF2C overexpression inhibited Erastin (6 µM)-triggered ferroptotic ACSL4 increase and GPX4 decrease, which was counteracted by E-cadherin knockdown (Figure 5A–E; Supplementary Figure 12A–E). The enhanced lipid peroxidation and cytotoxicity in response to Erastin were mitigated by MEF2C overexpression. E-cadherin knockdown recovered the levels of MDA and LDH diminished by MEF2C overexpression (Figure 5F and G; Supplementary Figure 12F and G).
Fig. 5.

Silencing of MEF2C promoted ferroptosis by downregulating NF2 and E-cadherin. CH157 (A–G) and IOMM-Lee (H–P) cells were stimulated by 6 µM Erastin for 24 hours to trigger ferroptosis. (A–E) Western blot and densitometry quantification for MEF2C (B), E-cadherin (C), ACSL4 (D), and GPX4 (E). GAPDH was used as a control. (F) Lipid peroxidation tested by MDA assays. n = 3 experiments. (G) Cytotoxicity tested by LDH assays. n = 3 experiments. (H–M) Western blot and densitometry quantification for MEF2C (I), E-cadherin (J), NF2 (K), ACSL4 (L), GPX4 (M), and P-YAP (S-127) (N). GAPDH was used as a control. n = 3 experiments. (O) Lipid peroxidation tested by MDA assays. n = 3 experiments. (P) Cytotoxicity tested by LDH assays. n = 3 experiments. *P < .05, **P < .01, and ***P < .001. Error bars represent SD. Abbreviations: ACSL4, acyl-CoA synthetase long-chain family member 4; GPX4, glutathione peroxidase 4; MEF2C, myocyte enhancer factor 2C; NF2, neurofibromatosis type 2.
Next, we studied the relationship between MEF2C and NF2 or E-cadherin in NF2 wild-type IOMM-Lee. shRNA knockdown of MEF2C caused an increase of ACSL4 and a decrease of GPX4 when 6 µM Erastin was used, which was alleviated by forced expression of NF2 or E-cadherin (Figure 5H–M). Previous studies showed that NF2-YAP signaling was involved in the induction of ferroptosis in cancer.12 We found a marked decrease of P-YAP (S-127) in Erastin-treated, MEF2C-silenced IOMM-Lee cells, which was reversed by overexpression of NF2 or E-cadherin, connecting YAP activity to ferroptosis (Figure 5H and N). 6 µM Erastin did not simulate lipid peroxidation and cytotoxicity in cells with intact MEF2C, but both were observed when MEF2C was silenced (Figure 5O and P). These Erastin-induced increases of MDA and LDH in MEF2C depleted IOMM-Lee cells were blocked when NF2 or E-cadherin was overexpressed. Therefore, MEF2C mediated the anti-ferroptotic effect via upregulating NF2 and E-cadherin.
MEF2C Silencing Enhanced Ferroptosis-Mediated Inhibition of Meningioma Growth In Vivo
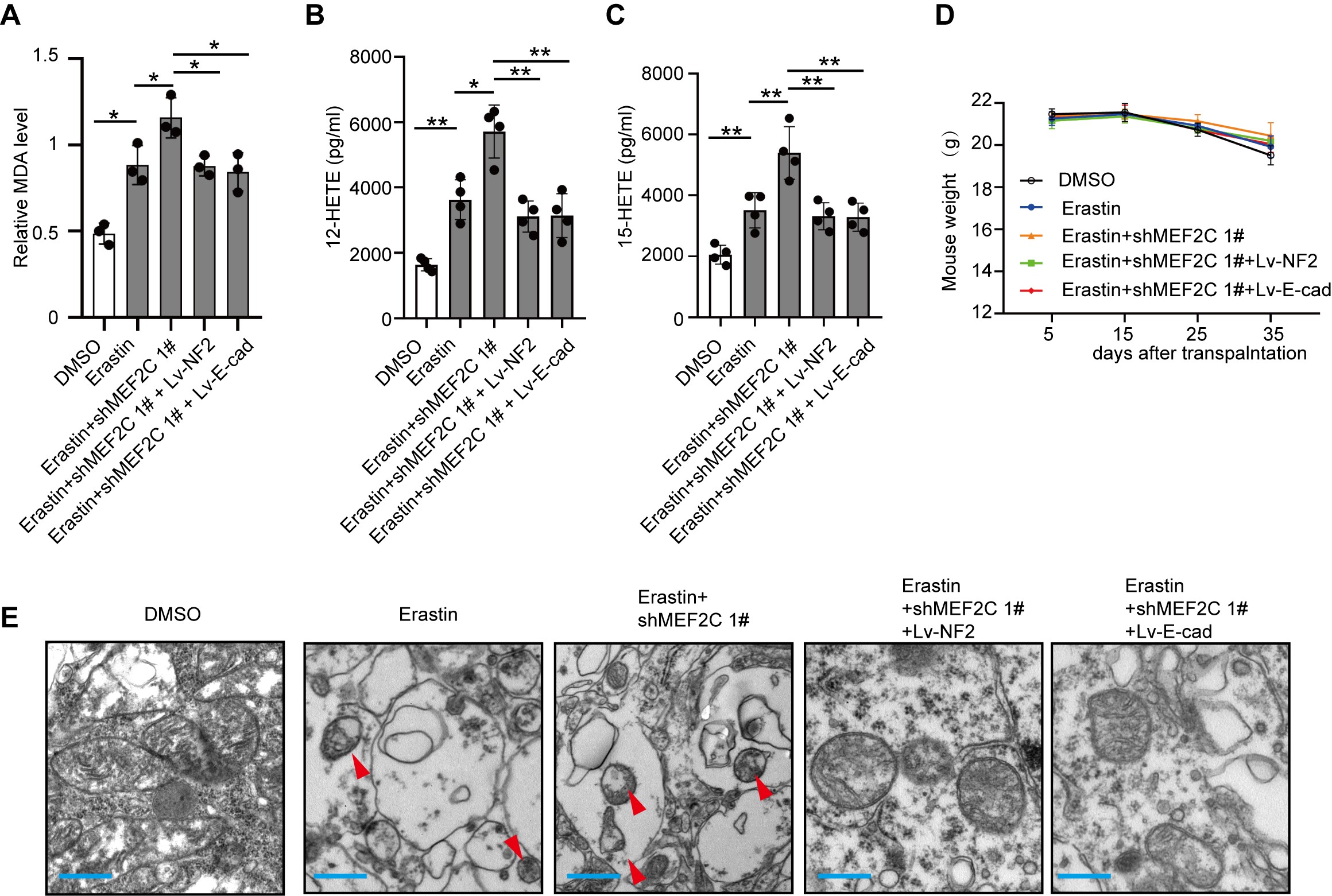
Our results showed that silencing of MEF2C can sensitize meningioma cells to ferroptosis, by downregulating NF2 and E-cadherin. We next performed in vivo studies to assess the therapeutic effects of Erastin and MEF2C knockdown (Supplementary Figure 13A). Subcutaneous transplantation of IOMM-Lee cells in nude mice led to the establishment of visible tumors at about 10 days after transplantation. Treatment with Erastin slowed tumor growth rate, and tumor inhibition was more prominent when Erastin was combined with MEF2C-knockdown. NF2 or E-cadherin overexpression restored the growth of Erastin-treated shMEF2C 1# IOMM-Lee, to the degree of Erastin-treated parental IOMM-Lee tumors (Figure 6A). At 35 days after transplantation, meningiomas were harvested and weighed (Figure 6B and C), confirming the findings of tumor growth measurement. MDA analysis of tumors showed that Erastin administration increased the level of lipid peroxidation, which was further elevated by MEF2C knockdown. Overexpression of NF2 or E-cadherin inhibited this upregulation (Supplementary Figure 14A). MEF2C knockdown also promoted increased 12-HETE (12-hydroxyeicosatetraenoic acid) and 15-HETE (15-hydroxyeicosatetraenoic acid), the 2 metabolites produced during lipid peroxidation.22 Elevated NF2 or E-cadherin reduced the production of 12-HETE and 15-HETE (Supplementary Figure 14B and C). Overall, there was a slow decline of weight loss but was no difference between groups at the 35th day (Supplementary Figure 14D). Since ferroptosis can be accompanied by mitochondrial injury,23 we tested the morphology of mitochondria in transplanted meningioma tumors using TEM (Supplementary Figure 14E). Mitochondrial shrinking, outer mitochondrial membrane (OMM) ruptures and the formation of light vacuoles, likely related to the characteristic mitochondria collapse caused by ferroptosis,6,8 were found in the Erastin group and Erastin + shMEF2C 1# group. Overexpression of NF2 or E-cadherin alleviated these changes of mitochondrial morphology.
Fig. 6.

Erastin and MEF2C silencing inhibited meningioma growth in vivo. Starting on day 5, Erastin was given to treat subcutaneous and orthotopic meningiomas. (A) Diameter of subcutaneous IOMM-Lee tumors was measured. Erastin was given at 15 mg/kg, i.p., twice every other day. n = 7 mice/group. (B) Image of subcutaneous tumors. (C) Tumor weight. (D and E) Tumor formation was assessed using bioluminescence imaging on days 10, 18, and 26 days after implantation. (F and G) IHC assay for Ki-67. Scale bar is 50 µm. n = 3/group. (H and I) TUNEL assay. Scale bar is 100 µm. n = 3/group. (J) Kaplan-Meier analysis of animal survival. n = 7 mice/group. (K) Weight of mice during the experiment. n = 7 mice/group. *P < .05, **P < .01, and ***P < .001. Error bars represent SD. Abbreviations: IHC, immunohistochemistry; MEF2C, myocyte enhancer factor 2C.
We next studied the orthotopic meningioma model of IOMM-Lee-Fluc. In vivo bioluminescence imaging of tumor volume showed that Erastin treatment reduced the size of intracranial tumors. Intracranial meningiomas with MEF2C knockdown responded better to Erastin, which were reversed by overexpression of NF2 and E-cadherin (Figure 6D and E). Erastin inhibited the proliferation of IOMM-Lee cells (labeled by Ki-67) in vivo, which was further inhibited in the sh-MEF2C plus Erastin group. Overexpression of NF2 or E-cadherin abrogated the effect of MEF2C knockdown on inhibiting proliferation (Figure 6F and G). TUNEL assays revealed that Erastin induced cell death in IOMM-Lee meningioma cells in vivo. MEF2C knockdown intensified Erastin-induced meningioma cell death, however, NF2 or E-cadherin upregulation limited this effect (Figure 6H and I). Importantly, Erastin administration extended survival time in this orthotopic meningioma xenograft model, which was further increased when Erastin was combined with MEF2C knockdown. NF2 or E-cadherin overexpression inhibited the effect of MEF2C knockdown and reduced the survival time (Figure 6J). Orthotopic meningioma development significantly decreased mouse weight, which was alleviated by Erastin administration (Figure 6K).
Discussion
In this study, we examined ferroptosis-related molecular mechanisms in meningioma, with the goal of identifying possible molecular targets for meningioma therapies. NF2 is the most commonly mutated gene in meningioma as up to 60% of sporadic meningioma cases exhibit inactivation of NF2 by somatic mutation, epigenetic inactivation, or allelic loss of chr22q.24 We demonstrate that meningioma cells with absent or lower level of NF2 are more susceptible to ferroptosis. We also found that Erastin-induced ferroptosis was dependent on the cell density. E-cadherin-related high cell density and cell-cell connections had anti-ferroptotic effects. E-cadherin knockdown triggered cell death when exposed to Erastin regardless of the cell density, which was consistent with prior reports with other cancer types.12 Interestingly, previous studies indicated that tumor-suppressive effects of NF2 were in part mediated indirectly through its membrane organization of proteins (ie, CD44, epidermal growth factor receptor, layilin) and cell-to-cell adhesion.25,26
Notably, we found that NF2 and E-cadherin gene transcription was both regulated by the same transcription factor, MEF2C. MEF2C increased NF2 and E-cadherin and limited Erastin-induced ferroptosis in meningioma, suggesting MEF2C as the transcription factor determining the meningioma fate in response to Erastin. Our work revealed that inhibition of MEF2C sensitized meningioma to Erastin-triggered ferroptosis, and nominated MEF2C as a potential molecular target in ferroptosis-related meningioma therapy. A member of the MEF2 family of transcription factors, MEF2C plays an important role in the development of organs such as the heart27 and the nervous system.28 The function of MEF2C in cancer is context-dependent as it can act as an oncogene in T-cell acute lymphoblastic leukemia29 and promote chemoresistance in hepatic cancer cells30 or serve as a tumor suppressor in soft tissue sarcoma.31 Our finding that MEF2C positively regulates NF2 and E-cadherin expression supports the notion that MEF2C inhibits ferroptosis in meningioma. To the best of our knowledge, the current work is the first to mechanistically connect MEF2C to ferroptosis.
Recently, cancer therapy strategies exploiting ferroptosis have been emerging. Several clinical drugs have been found to be able to induce ferroptosis in cancer cells. Erastin-induced ferroptosis in mouse tumor xenografts by depleting glutathione and inactivating GPX4.7 Sorafenib, an inhibitor of oncogenic kinases, induced ferroptosis in hepatocellular carcinoma cells.32 Anti-inflammatory drug Sulfasalazine inhibited the glutamate transporter xCT and induced ferroptotic cell death in glioma cells.33 Antimalarial drug Artemisinin specifically induced ROS- and lysosomal iron-dependent cell death in pancreatic ductal adenocarcinoma cells.34 Other small molecule inducers of ferroptosis include siramesine (a lysosome disrupting agent), lapatinib (a tyrosine kinase inhibitor),35 BAY 87-2243, a potent inhibitor of NADH-coenzyme Q oxidoreductase (complex I),36 and 1,2-dioxolane (FINO2),37 and have a potential clinical utility as ferroptosis-based cancer therapeutics.38 Clinically, Erastin analog PRLX93936 has been in a phase 1/2 clinical trial for multiple myeloma (NCT01695590).
The dose of Erastin (15 mg/kg, i.p., twice every other day) we used herein was lower compared with prior published studies that used 20 mg/kg, i.p., twice every other day in gastric cancer,39 20 mg/kg i.v. twice daily every other day for HeLa-derived subcutaneous tumor,40 and 50 mg/kg to treat subcutaneous lung cancer xenografts tumors. The significant anti-tumor effects and the lack of adverse events of the lower dose Erastin in both flank and intracranial models provided experimental evidence that induction of ferroptosis is a suitable anti-meningioma strategy. Furthermore, inhibition of MEF2C elevated the sensitivity of meningioma to Erastin-induced ferroptosis, resulting in profound tumor inhibition and greater survival extension. Given the emergence of a variety of ferroptosis inducers, it is of interest to screen those clinical candidates for efficacy and safety in preclinical models of NF2-mutant meningioma. Development of selective inhibitors of MEF2C will open up a potential to expand the application of ferroptosis inducers to a broader spectrum of meningioma.
In summary, Erastin-induced ferroptosis represents a promising molecular strategy to target meningiomas. Ferroptosis induction is particularly efficacious for tumors with NF2 loss, which is clinically important given the role of NF2 loss in tumor progression and relapse. Our discovery that MEF2C-mediated upregulation of NF2 and E-cadherin impairs meningioma sensitivity to ferroptosis identifies MEF2C as a potential therapeutic target to enhance meningioma in response to ferroptosis-inducing therapy.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This study was funded by grants from National Natural Science Foundation of China (Grant No. 81972153 to J.J. and 81772674 to Y.G.), Jiangsu Province’s Key Discipline of Medicine (Grant No. XK201117), Shanghai Sailing Program (20YF1403900 to L.H.), and Meningioma Mommas (to H.W.).
Conflict of interest statement. The authors declare no competing financial interests.
Authorship statement. Designed research: J.J. and Y.G.; Performed research: Z.B., Y.Y., L.H., C.L., and D.W.; Data analysis and interpretation: Z.B., and L.H.; Manuscript preparation: Z.B., L.H., and H.W.
References
- 1. Ostrom QT, Patil N, Cioffi G, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(12 Suppl 2):iv1–iv96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 3. Hua L, Zhu H, Li J, et al. Prognostic value of estrogen receptor in WHO Grade III meningioma: a long-term follow-up study from a single institution. J Neurosurg. 2018;128(6):1698–1706. [DOI] [PubMed] [Google Scholar]
- 4. Wang D, Sun S, Hua L, et al. Prognostic model that predicts benefits of adjuvant radiotherapy in patients with high grade meningioma. Front Oncol. 2020;10:568079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu H, Bi WL, Aizer A, et al. Efficacy of adjuvant radiotherapy for atypical and anaplastic meningioma. Cancer Med. 2019;8(1):13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35(6):830–849. [DOI] [PubMed] [Google Scholar]
- 10. Williams EA, Santagata S, Wakimoto H, et al. Distinct genomic subclasses of high-grade/progressive meningiomas: NF2-associated, NF2-exclusive, and NF2-agnostic. Acta Neuropathol Commun. 2020;8(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deng J, Hua L, Han T, et al. The CREB-binding protein inhibitor ICG-001: a promising therapeutic strategy in sporadic meningioma with NF2 mutations. Neurooncol Adv. 2020;2(1):vdz055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu J, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin H, Patel S, Affleck VS, et al. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017;19(1):43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song LR, Li D, Weng JC, et al. MicroRNA-195 functions as a tumor suppressor by directly targeting fatty acid synthase in malignant meningioma. World Neurosurg. 2020;136:e355–e364. [DOI] [PubMed] [Google Scholar]
- 16. Xu X, Bao Z, Liu Y, et al. PBX3/MEK/ERK1/2/LIN28/let-7b positive feedback loop enhances mesenchymal phenotype to promote glioblastoma migration and invasion. J Exp Clin Cancer Res. 2018;37(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tu Y, Xie P, Du X, et al. S100A11 functions as novel oncogene in glioblastoma via S100A11/ANXA2/NF-κB positive feedback loop. J Cell Mol Med. 2019;23(10):6907–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mei Y, Bi WL, Greenwald NF, et al. Genomic profile of human meningioma cell lines. PLoS One. 2017;12(5):e0178322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65(23):3756–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong C, Yang XZ, Zhang CY, et al. Myocyte enhancer factor 2C and its directly-interacting proteins: a review. Prog Biophys Mol Biol. 2017;126:22–30. [DOI] [PubMed] [Google Scholar]
- 21. Bai XL, Zhang Q, Ye LY, et al. Myocyte enhancer factor 2C regulation of hepatocellular carcinoma via vascular endothelial growth factor and Wnt/β-catenin signaling. Oncogene. 2015;34(31):4089–4097. [DOI] [PubMed] [Google Scholar]
- 22. Li Y, Feng D, Wang Z, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26(11):2284–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou J, Jin Y, Lei Y, et al. Ferroptosis is regulated by mitochondria in neurodegenerative diseases. Neurodegener Dis. 2020;20(1):20–34. [DOI] [PubMed] [Google Scholar]
- 24. Ruttledge MH, Sarrazin J, Rangaratnam S, et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet. 1994;6(2):180–184. [DOI] [PubMed] [Google Scholar]
- 25. Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373(9679):1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Curto M, Cole BK, Lallemand D, et al. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177(5):893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim HJ, Oh HJ, Park JS, et al. Direct conversion of human dermal fibroblasts into cardiomyocyte-like cells using CiCMC nanogels coupled with cardiac transcription factors and a nucleoside drug. Adv Sci (Weinh). 2020;7(7):1901818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li M, Santpere G, Imamura Kawasawa Y, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Homminga I, Pieters R, Langerak AW, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19(4):484–497. [DOI] [PubMed] [Google Scholar]
- 30. Zhang H, Liu W, Wang Z, et al. MEF2C promotes gefitinib resistance in hepatic cancer cells through regulating MIG6 transcription. Tumori. 2018;104(3):221–231. [DOI] [PubMed] [Google Scholar]
- 31. Di Giorgio E, Clocchiatti A, Piccinin S, et al. MEF2 is a converging hub for histone deacetylase 4 and phosphatidylinositol 3-kinase/Akt-induced transformation. Mol Cell Biol. 2013;33(22):4473–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lachaier E, Louandre C, Godin C, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34(11):6417–6422. [PubMed] [Google Scholar]
- 33. Sehm T, Fan Z, Ghoochani A, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. 2016;7(24):36021–36033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ooko E, Saeed ME, Kadioglu O, et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22(11):1045–1054. [DOI] [PubMed] [Google Scholar]
- 35. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schockel L, Glasauer A, Basit F, et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abrams RP, Carroll WL, Woerpel KA. Five-membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem Biol. 2016;11(5):1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen Z, Song J, Yung BC, et al. Emerging strategies of cancer therapy based on ferroptosis. Adv Mater. 2018;30(12):e1704007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hao S, Yu J, He W, et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia. 2017;19(12):1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.