

Abstract
Background
Intratumoral heterogeneity is a hallmark of diffuse gliomas. DNA methylation profiling is an emerging approach in the clinical classification of brain tumors. The goal of this study is to investigate the effects of intratumoral heterogeneity on classification confidence.
Methods
We used neuronavigation to acquire 133 image-guided and spatially separated stereotactic biopsy samples from 16 adult patients with a diffuse glioma (7 IDH-wildtype and 2 IDH-mutant glioblastoma, 6 diffuse astrocytoma, IDH-mutant and 1 oligodendroglioma, IDH-mutant and 1p19q codeleted), which we characterized using DNA methylation arrays. Samples were obtained from regions with and without abnormalities on contrast-enhanced T1-weighted and fluid-attenuated inversion recovery MRI. Methylation profiles were analyzed to devise a 3-dimensional reconstruction of (epi)genetic heterogeneity. Tumor purity was assessed from clonal methylation sites.
Results
Molecular aberrations indicated that tumor was found outside imaging abnormalities, underlining the infiltrative nature of this tumor and the limitations of current routine imaging modalities. We demonstrate that tumor purity is highly variable between samples and explains a substantial part of apparent epigenetic spatial heterogeneity. We observed that DNA methylation subtypes are often, but not always, conserved in space taking tumor purity and prediction accuracy into account.
Conclusion
Our results underscore the infiltrative nature of diffuse gliomas and suggest that DNA methylation subtypes are relatively concordant in this tumor type, although some heterogeneity exists.
Keywords: DNA methylation classification, epigenetics, glioma, imaging, intratumoral heterogeneity
Key Points.
DNA methylation subtype is stable across glioma specimens from the same tumor
Tumor purity is an important consideration for DNA methylation-based glioma classification.
Importance of the Study
Genetic and transcriptional intratumoral heterogeneity is a key feature of diffuse gliomas and thought to drive treatment failure. Epigenetic profiling is an emerging approach used for clinical classification of brain tumors that results in improved alignment of patients and treatments. However, how epigenetic intratumoral heterogeneity may impact the robustness of classification remains unclear. Through DNA methylation profiling of multiple spatially mapped biopsies from diffuse gliomas in 16 adult patients and 11 patients in a validation cohort, our study shows that epigenetic spatial heterogeneity is confounded by the presence of non-tumor cells and the prediction accuracy of the classifier. Taking these confounders into account, DNA methylation-based classification is conserved in space in most patients, including tumor presence outside standard imaging abnormalities. These findings emphasize the importance of tumor purity assessment in DNA methylation studies. The uniformity of DNA methylation-based classification within tumors demonstrates the robustness of this method for classification, corroborating its value for clinical studies and practice.
Diffuse gliomas are the most common malignant brain tumors in adults.1 Patients with a diffuse glioma have a poor prognosis and eventually succumb to treatment failure.2 The diagnosis, treatment, and follow-up of diffuse gliomas rely heavily on imaging,2 with magnetic resonance imaging (MRI) as the current standard. Using contrast-enhanced T1-weighted (T1c) MRI, diffuse gliomas can be divided into enhancing tumors, predominantly glioblastoma, or non-enhancing tumors, predominantly low-grade gliomas (LGG). T1c MRI is used for enhancing and T2/fluid-attenuated inversion recovery (FLAIR) MRI for non-enhancing gliomas.3 However, diffuse glioma infiltration extends beyond the abnormalities detected on standard MRI.4,5 Also, the majority of diffuse gliomas recur directly adjacent to the standard MRI-guided surgical cavity.6 Heterogeneity of tumor cells is a salient feature of diffuse gliomas and thought to be a driver of treatment failure. Treatment exposure may drive the clonal evolution of heterogeneous tumor cell populations, leading to the selection and survival of resistant subpopulations in some gliomas, whereas refractory disease in others may be driven by other factors.7
Numerous studies have looked at genetic and transcriptomic heterogeneity in diffuse glioma. Recent single-cell transcriptome studies have elucidated transcriptional heterogeneity in regulatory programs that converge on the cell cycle or distinct cellular states8,9 while bulk tissue analysis has demonstrated extensive heterogeneity in somatic drivers such as EGFR and PDGFRA10,11 as well as in general somatic alteration burden.12–14
DNA methylation is an epigenetic modification where a methyl group is added to cytosine, most commonly measured in the CpG dinucleotide context. These modifications are of interest to the neuro-oncology field as genome-wide patterns in DNA methylation profiles provide a robust method for disease classification and a viable supplement to traditional histopathology.15,16 Nevertheless, the extent of intratumoral heterogeneity in DNA methylation remains unclear.
To improve our understanding of the epigenetic heterogeneity of diffuse gliomas, we present a comprehensive analysis of DNA methylation of a large number of spatially separated samples taken from regions with and without imaging abnormalities. We devised a 3-dimensional reconstruction of the DNA methylation landscape for each tumor, with particular consideration to the variable ratios of tumor and nonmalignant cells in each sample. These ratios are quantified as tumor purity using a methylation-based metric that, although not being the perfect test due to non-zero levels of tumor purity in nonmalignant samples,17 has proven to be a major confounder in genomic analyses.18 Our analysis underlines the infiltrative nature of gliomas beyond visible tumor boundaries and demonstrates a rather homogeneous DNA methylation landscape across space.
Methods
Sample Acquisition and Study Design
A schematic overview of the study design is given in Figure 1. The exploration cohort consisted of 16 patients with an untreated initial diffuse glioma, treated at the Amsterdam UMC, location VU medical center (VUmc), Amsterdam, the Netherlands. Patients’ characteristics are given in Supplementary Table 1. All patients were participants of the FRONTIER study of which the protocol has been published.19 This protocol was approved by the Medical Ethics Committee of the VUmc and registered in the Dutch National Trial Register (www.trialregister.nl, unique identifier NTR5354). Sampling was performed, using a stereotactic biopsy procedure preceding the craniotomy, to obtain 2 samples of each biopsy location for, respectively, Formalin-Fixed Paraffin-Embedded (FFPE) and Molfix© (patients 1-8) or snap-frozen (patients 9-16) fixation. Samples were obtained from regions with and without abnormalities on T1c and FLAIR MRI in accordance with the study protocol. All procedures were carried out in accordance with the Declaration of Helsinki.20 Written informed consent was obtained from all patients.
Fig. 1.

Graphical overview of the methods. A. Multiple preoperatively planned stereotactic biopsies were taken from each patient tumor. B. Biopsies were acquired in regions in and outside imaging abnormalities. C. Acquired tissue was subject to comprehensive histological (C), molecular (D), and imaging (E) analysis.
The validation cohort comprised 11 patients with 61 FFPE samples from multi-sector sampling of an untreated diffuse glioma treated at the Toronto Western Hospital, Toronto, Canada or UCSF Brain Tumor Center, San Francisco, CA, USA. In addition, 64 FFPE samples from 64 patients without a glioma from the German Cancer Network served as controls.
DNA Isolation
DNA isolation was performed by adding proteinase K and incubating at 56°C using the QIAamp DNA Mini Kit (Qiagen). DNA was quantified using a Qubit Fluorometer (Thermo Fisher). Genomic DNA was bisulfite converted using the QIAamp DNA FFPE Tissue Kit (Qiagen).
DNA Methylation Profiling by Microarray
Data were processed using the minfi packages in R (R Foundation for Statistical Computing, Vienna, Austria). Data from the 450k (IlluminaHumanMethylation450k.ilmn12.hg19) and EPIC platforms (IlluminaHumanMethylationEPICanno.ilm10b2.hg19) were processed separately. Detection P values were calculated for each probe and sample, and samples with an average detection P value >.01 were removed from follow-up analysis. Data were normalized using BMIQ from the wateRmelon package in R. Probes on sex chromosomes and known cross-reactive probes were removed, as were probes mapping to known SNPs and probes with a detection P value >.01. Finally, data from different platforms were merged.
DNA Methylation-Based Classification
Glioma methylation subtype classification was performed using L2-regularized logistic regression using the R package LiblineaR. Classifiers were trained and evaluated on a set of common probes from TCGA glioma samples with known methylation subtypes. The classes LGm6-GBM and PA-like were merged into a single class LGm6-PA as the separation between these classes was based on phenotype. To improve the classification accuracy of samples with low tumor purity, DKFZ controls were added to the classifier as separate classes. DKFZ DNA methylation classification was performed using the molecular neuropathology classification tool (version 11b4).16 Briefly, the software preprocesses, normalizes, and performs batch adjustment on raw DNA methylation data to generate probabilistic estimates for each defined tumor subtype. The tumor subtype with the highest probability was selected as the classification. Samples with a subclassification accuracy lower than 0.5 were assigned to the main DKFZ class in accordance with the Molecular Neuropathology guideline.
Methylation Purity Estimation and Simplicity Score
DNA methylation measurements of tumor purity included the PAMES (Purity Assessment from clonal MEthylation Sites) algorithm and simplicity score.17,21 For the PAMES nonmalignant central nervous system, samples from the German Cancer Research Center (DKFZ) were used as a control. PAMES operates in 3 steps. First, area under the curves are calculated for each probe discriminating between tumor and normal. Second, a selection of the most informative probes is made. Third, tumor purity is calculated on input samples using these probes.
DNA Copy Number Aberrations Inferred From EPIC Microarray
Using the R/Conumee package, copy number aberrations were inferred from the 450k and EPIC array data. Merged data from the control samples were used as a baseline control for all analyses. Genomic data were used to calculate aneuploidy.
Immunohistochemistry and Qualitative Assessment
FFPE samples from the exploration cohort were stained using hematoxylin and eosin (HE) and MIB-1. Two expert neuropathologists independently, blinded for imaging results, assessed the presence or absence of tumor in each sample. Consensus was obtained in case of disagreement. The patient’s histopathological diagnosis was made based on resection material using routine procedures and according to the WHO 2016 criteria.22
Histopathological Analysis of Whole-Slide Scans
Using a Hamamatsu Nanozoomer XR, FFPE slides stained with HE and MIB-1 of each sample were digitalized. The ×40 magnification images were converted to multiple mosaic images using NDPITools software. Cellularity, defined as the number of cells per square millimeter, was calculated with Cellprofiler. Proliferation index, defined as percentage of Ki-67-positive nuclei of all nuclei, was calculated using locally developed software.
Radiologic Evaluation of Sample Locations
Standard imaging sequences from the patients in the exploration cohort included T1-, T2-, T2/FLAIR, and T1c MRI. For each sample location, the presence of an abnormal signal for each imaging sequence was independently assessed by a neurosurgeon and neurosurgical resident with ample experience in glioma imaging. Consensus was obtained in case of disagreement.
Sample-to-Tumor Surface Distance
Tumors were segmented on FLAIR and, in case of contrast enhancement, also on T1c MRI, using Brainlab Software, by a neurosurgical resident with ample experience in glioma imaging. The segmentations and sample coordinates were exported in 3D T1c MRI space. Sample-to-tumor surface distances were obtained, using Matlab, by calculating the distance between the sample coordinate and the nearest surface coordinate of the tumor segmentation, using FLAIR for non-enhancing and both FLAIR and T1c MRI for enhancing tumors.
Statistical Analysis
Median values with interquartile range were used to describe non-normally distribute data. Mann-Whitney U test was used to compare distributions between subgroups. Correlations were calculated with the Spearman or Pearson correlation and compared using Fisher z transformation. Comparison of percentages between subgroups was performed using Fisher test normalization and scaling of purity measurement modalities was performed by subtracting the mean and dividing by the standard deviation. To compare absolute purity estimates, the normalized and scaled purity measurements were rescaled using the PAMES mean and SD. P values <.05 were considered statistically significant. R (version 3.5.3) was used for all statistical analyses.
Heterogeneity Analysis
Each probe per patient was classified as methylated (B > 0.3) or unmethylated (B < 0.3). A table of all possible pairwise combinations of samples was generated. Each pair of samples was evaluated for heterogeneity by counting the number of identical (homogeneous) probes, the number of differing (heterogeneous) probes, and percentages were subsequently calculated. Each pair was annotated according to the metadata for each sample in the comparison.
For each patient and sample type, we tabulated all possible combinations of any number of samples, iteratively including between 1 and the total number of possible samples. The proportion of heterogeneous and homogeneous probes was calculated when considering each sample in a given set. For each patient/sample type and sample number, we then calculated the mean and standard deviation of the proportion heterogeneous across all sets.
Results
We obtained 133 multi-region image-guided samples from 8 patients (6 glioblastoma, IDH-wildtype, 2 glioblastoma, IDH-mutant) with an enhancing tumor (76 samples) and 8 patients (6 diffuse astrocytoma, IDH-mutant, 1 glioblastoma, IDH-wildtype and 1 oligodendroglioma, IDH-mutant and 1p19q codeleted) with a non-enhancing tumor (47 samples) (Figures 1 and 2). In enhancing gliomas, 12 samples were taken outside both T1c and FLAIR abnormalities (T1c−/FLAIR−), 44 samples outside (T1c−/FLAIR+), and 20 inside T1c abnormalities (T1c+/FLAIR+). In non-enhancing gliomas, 16 samples were taken outside (FLAIR−) and 41 inside FLAIR abnormalities (FLAIR+). The maximum sample-to-tumor surface distance in enhancing tumors was 36.5 mm and 25.6 mm, respectively, assessed with T1c and FLAIR MRI. In non-enhancing tumors, the maximum sample-to-tumor surface distance was 22.5 mm assessed with FLAIR MRI.
Fig. 2.

Overview of 133 samples in 16 patients with initial diffuse glioma. Samples are numbered in order of tumor purity for each patient. First row = IDH status with + representing a mutation, 1p/19q status with + representing a codeletion, and contrast enhancement with + representing the presence of contrast on T1c MRI. Second row = tumor purity assessed with PAMES. Third row = 3D reconstruction of FLAIR (yellow), T1c MRI (red) abnormalities, and sample locations. Due to the 2D representation of 3D object samples behind and outside the abnormalities might appear to be within the abnormalities. Fourth row = Euclidean distance (mm) between sample coordinate and tumor surface assessed with FLAIR (yellow) and T1c MRI (red). Negative values indicate samples obtained within the tumor volume. Fifth row = the presence of abnormalities on FLAIR (top) and T1c MRI (bottom) at the sample location. Sixth row = median cellularity and percentage of MIB1-positive cells of the sample, and final row = consensus assessment of tumor presence by 2 neuropathologists. Abbreviations: FLAIR, fluid-attenuated inversion recovery; IDH, isocitrate dehydrogenase; MRI, magnetic resonance imaging; PAMES, Purity Assessment from clonal MEthylation Sites; T1c, contrast-enhanced T1-weighted.
Tumor Purity Accounts for a Considerable Amount of Variation in DNA Methylation Profiles
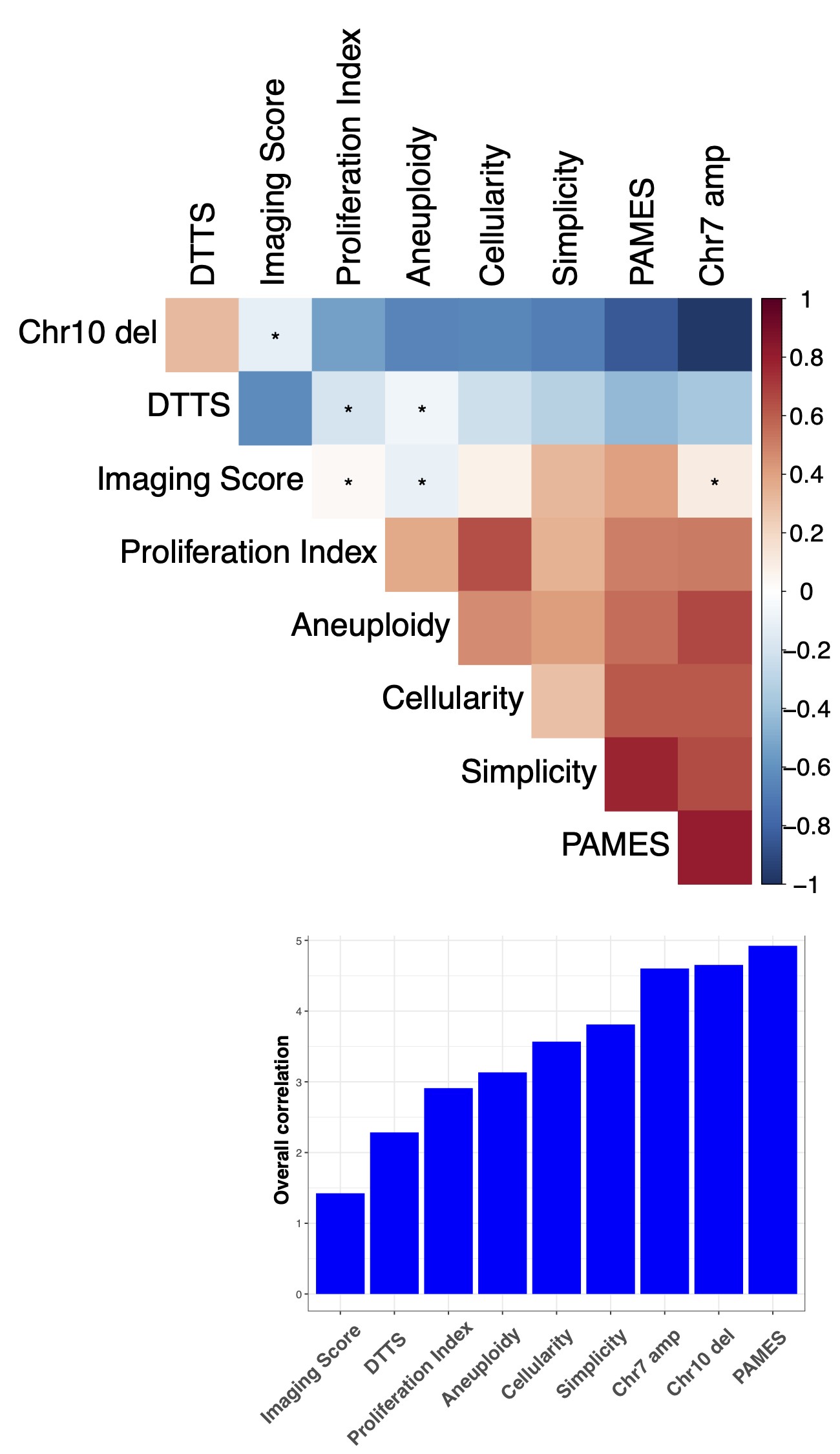
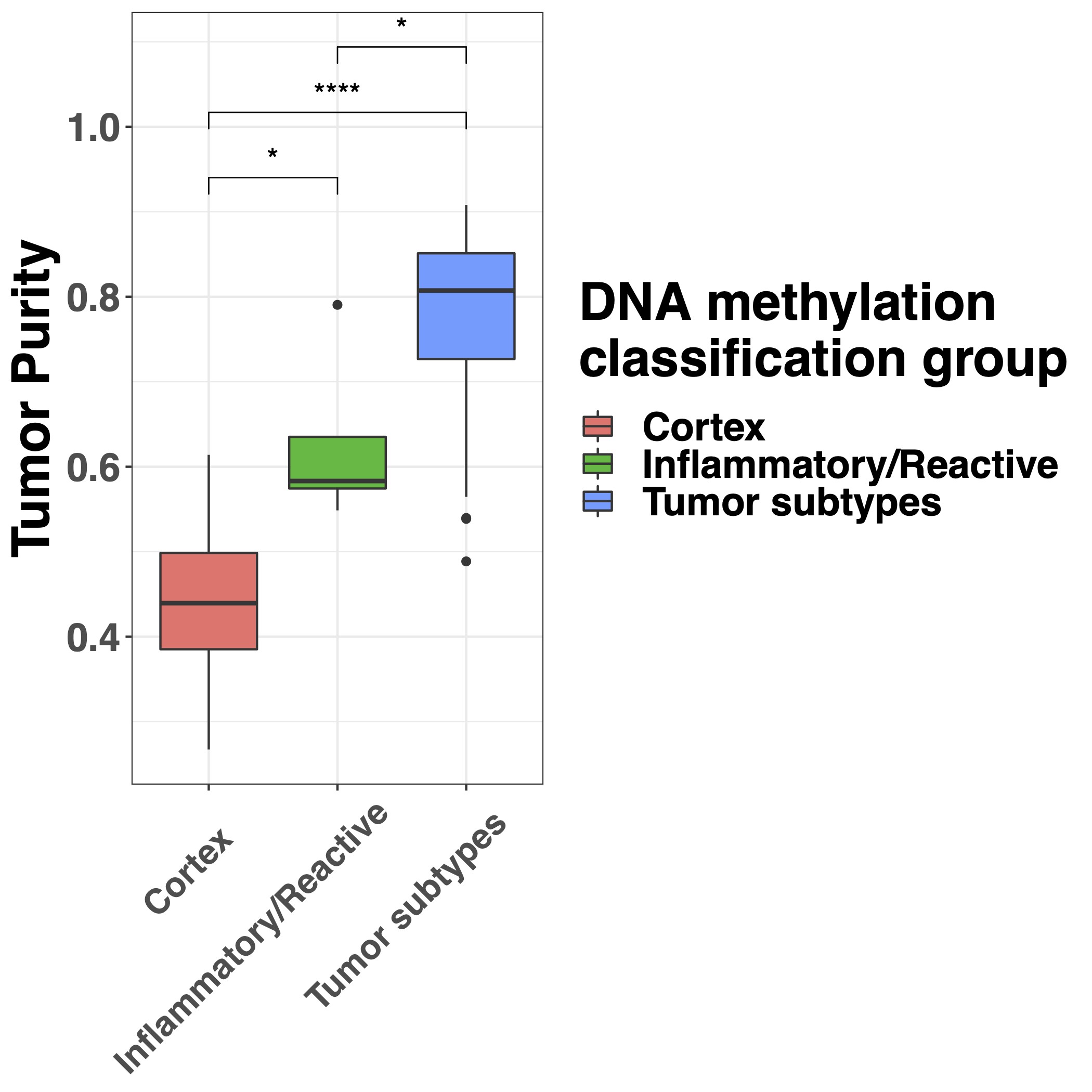
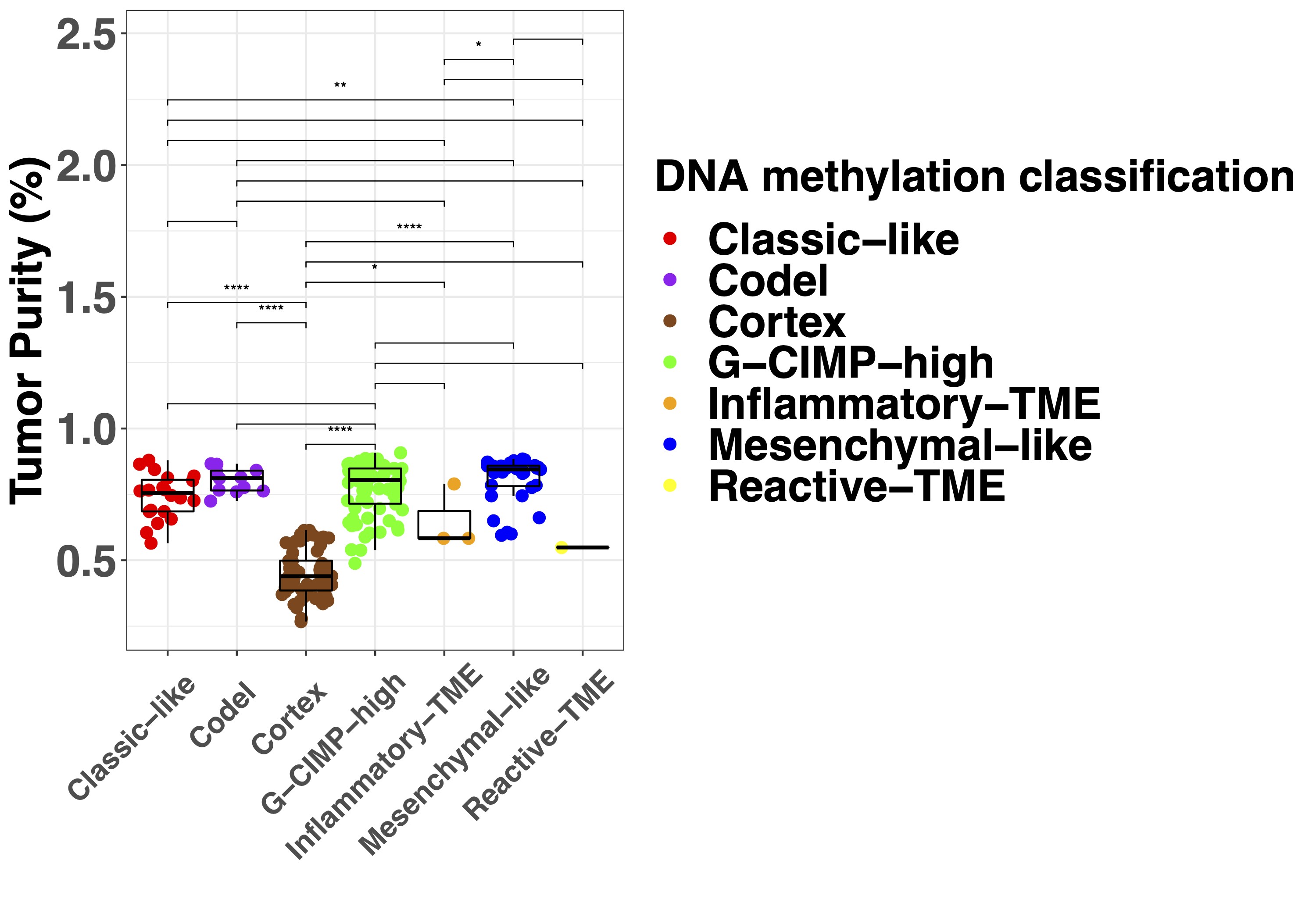
Since nonmalignant cells in a sample influence molecular tumor assessment,21 we sought to quantify tumor purity, defined as the ratio of tumor to nonmalignant cells. We evaluated and compared several methods of tumor purity estimation based on histology, MRI, DNA methylation, and DNA copy number (Supplementary Figure 1A and B). DNA methylation-based purity estimates, PAMES provided the strongest correlations with all other features.17 There was no difference in tumor purity between samples from IDH-mutant and IDH-wildtype tumors (Supplementary Figure 1C). WHO subtype was associated with tumor purity (Kruskal-Wallis P < .001), with the highest tumor purities in oligodendroglioma, IDH-mutant and 1p/19q codeleted and the lowest for glioblastoma, IDH-mutant (Supplementary Figure 1D), most likely due to the known admixture of nonmalignant cells in grade IV diffuse glioma.23 This association between grade and tumor purity is in line with a recent comprehensive analysis of TCGA samples.18
We performed a principal component analysis of the DNA methylation data to elucidate drivers of differences in methylation (194 samples, Figure 3A). Included in the analysis were samples from a second cohort consisting of 61 multi-sector tumor samples from 11 gliomas (5 diffuse astrocytoma, IDH-mutant, 4 glioblastoma, IDH-wildtype, 1 anaplastic astrocytoma, IDH-mutant, and 1 anaplastic oligodendroglioma, IDH-mutant and 1p19q codeleted).16,24,25 The first principal component (percentage of variance 71.2%) separated samples based on IDH status (Figure 3A). The second principal component (percentage of variance 5.8%) was associated with tumor purity, as evidenced by the linear increase in tumor purity and the samples from the control cohort (Figure 3B). These findings indicate that tumor purity accounts for a considerable amount of variation in DNA methylation profiles.
Fig. 3.

Exploration of spatial distribution of DNA methylation-based subtypes. A. Principal component analysis of exploration and validation cohort (samples = 194). B. Correlation between the second principal component and tumor purity, with the correlation calculated with Pearson R.
DNA Methylation Heterogeneity Is a Function of Space
To precisely quantify DNA methylation heterogeneity, we performed pairwise comparisons of binarized methylation values between samples. The vast majority of probes were homogeneously methylated (mean 0.93, range 0.83-1.0) between samples, suggesting that only a small fraction of probes is responsible for all intratumor heterogeneity. Similar trends have been observed in comparisons of nonmalignant samples from the same lineage.26 Unsurprisingly, any 2 samples from different unrelated tumors showed less probes with identical methylation (mean 0.93 ± 0.02) compared to any 2 samples from the same tumor (mean 0.96 ± 0.03). However, this difference was subgroup-dependent. For example, any 2 samples from 2 unrelated IDH-mutant tumors show more similarity on average than any 2 samples from 2 unrelated IDH-wildtype tumors (Figure 4A), likely related to the propensity of (G-CIMP-positive) IDH-mutant tumors to uniformly methylate. As expected, a higher degree of heterogeneity can be observed when comparing samples classified as nonmalignant to samples classified as tumor, based on DNA methylation classification, within the same patient. Any 2 IDH-wildtype tumor samples from the same patient show a comparable degree of heterogeneity when compared to 2 nonmalignant samples from the same patient (Kolmogorov-Smirnov P = 1.0, pink and green dashed lines). In comparison, any 2 IDH-mutant tumor samples from the same patient demonstrate less heterogeneity compared to any 2 nonmalignant samples from the same patient (Kolmogorov-Smirnov P < .001). These findings may reflect the clonal nature and shared ancestry of IDH-mutant tumor cells, whereas specimens are classified as nonmalignant harbor cells from a mixture of lineages.
Fig. 4.

Spatial heterogeneity of genome-wide methylomes. A. Empirical cumulative density function (ECDF) curves reflecting similarity (homogeneity) across all pairwise combinations of samples. Comparisons were separated based on whether they involved 2 samples from the same patient (intra-patient) or between 2 patients (interpatient) and based on whether the 2 samples spanned one or multiple sample types. B. Line plot showing the cumulative homogeneity associated with additional samples taken from the same tumor. Lines were colored by dataset, tumor and normal samples were separated, and tumor samples were further separated into IDH-mutant and IDH-wildtype. C. Scatter plot of the relation between distance and methylation heterogeneity. Distance is the Cartesian distance in mm between 2 samples and methylation heterogeneity as described above. Correlation is calculated with Pearson R.
To assess the impact of additional samples on tumor heterogeneity, we calculated the percentage of identical probes pooling any number of samples per patient, separating samples classified as tumor and nonmalignant (Figure 4B). The majority of heterogeneity was captured by the first 2 samples per patient. Although additional samples further contributed to overall heterogeneity, the change in heterogeneity decreased with each additional sample. Next, we investigated the relation between sample-to-sample distance and heterogeneity. These were positively correlated for both nonmalignant and tumor samples (Figure 4C). These results suggest that heterogeneity is a function of space and that increased physical separation between cells increases cell-to-cell heterogeneity.
DNA Methylation-Based Classification Is Highly Conserved in Space After Adjusting for Tumor Purity and Classification Ambiguity
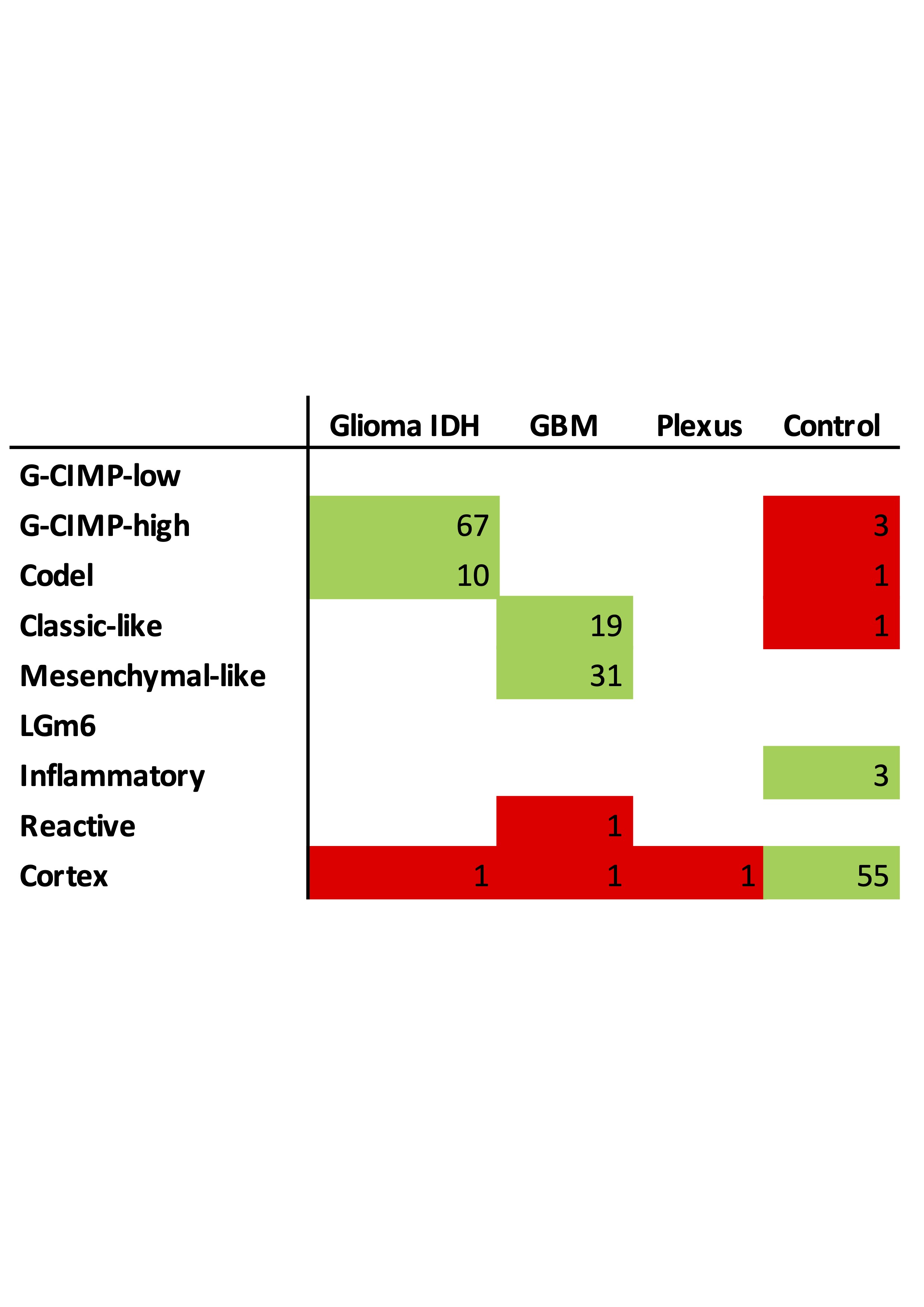
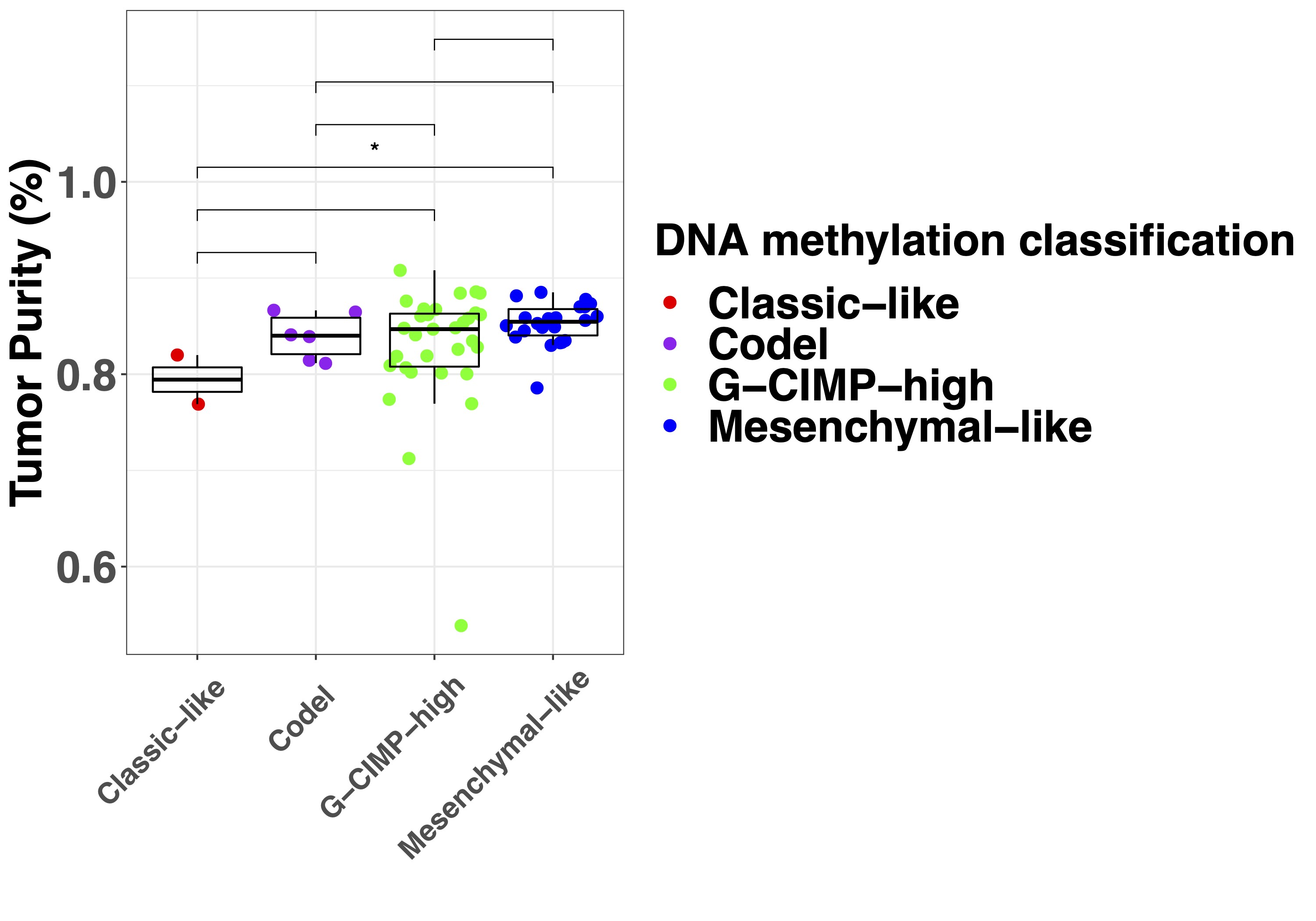
To establish the relationship between DNA methylation-based classification and tumor purity, we inferred sample subtypes based on 2 previous published classifiers (Supplementary Figure 2A),15,16 which showed a large conformity for classification families (4.6% discordance, Supplementary Figure 2B) and slightly lower conformity for family subtypes (12.4% discordance, Supplementary Figure 2C). As expected, samples with a low tumor purity were assigned a control subtype whereas high tumor purity samples were assigned a tumor subtype when assessed by the TCGA classifier (Supplementary Figure 2D). The differences in subtype assignment and its relation to tumor purity were clearly captured by the principal component analysis (Figure 3A). There were no significant differences between the tumor purity of the different tumor subtypes in the exploration dataset (Supplementary Figure 2E) while in the validation dataset (Supplementary Figure 2F), the tumor purity was lower in the Classical-like compared to Mesenchymal-like subtype, which are methylation subtypes based on unsupervised hierarchical clustering with a majority of, respectively, Classical and Mesenchymal expression subtypes as described by Verhaak et al.27 To explore the heterogeneity of tumor subtypes within a tumor, we analyzed which tumor subtypes were recognized within each patient across the core and validation dataset. The majority of patients (24 of 27) did not show heterogeneity in tumor subtype as assessed by the TCGA classifier (Figure 5). In 3 patients (Toronto-02, VUmc-05, and VUmc-17) with TCGA tumor subtype heterogeneity, the 1 (VUmc-05, VUmc-17) or 2 (Toronto-02) discordant samples were the lowest purity tumor sample identifying tumor purity as the confounding factor for the found heterogeneity (Supplementary Figure 3A). Also, the prediction accuracy of tumor samples was lower in patients with subtype heterogeneity (mean prediction accuracy 71.0%) than without (mean 94.9%, P < .001).
Fig. 5.

Overview of DNA methylation subtypes. Patients of both the exploration and validation cohort are ordered according to their cohort and DNA methylation subtype. Both TCGA and DKFZ subtypes and classification probabilities are given. Intratumoral heterogeneity of DNA methylation subtypes is represented as not present (−), possible present (+/−), or present (+).
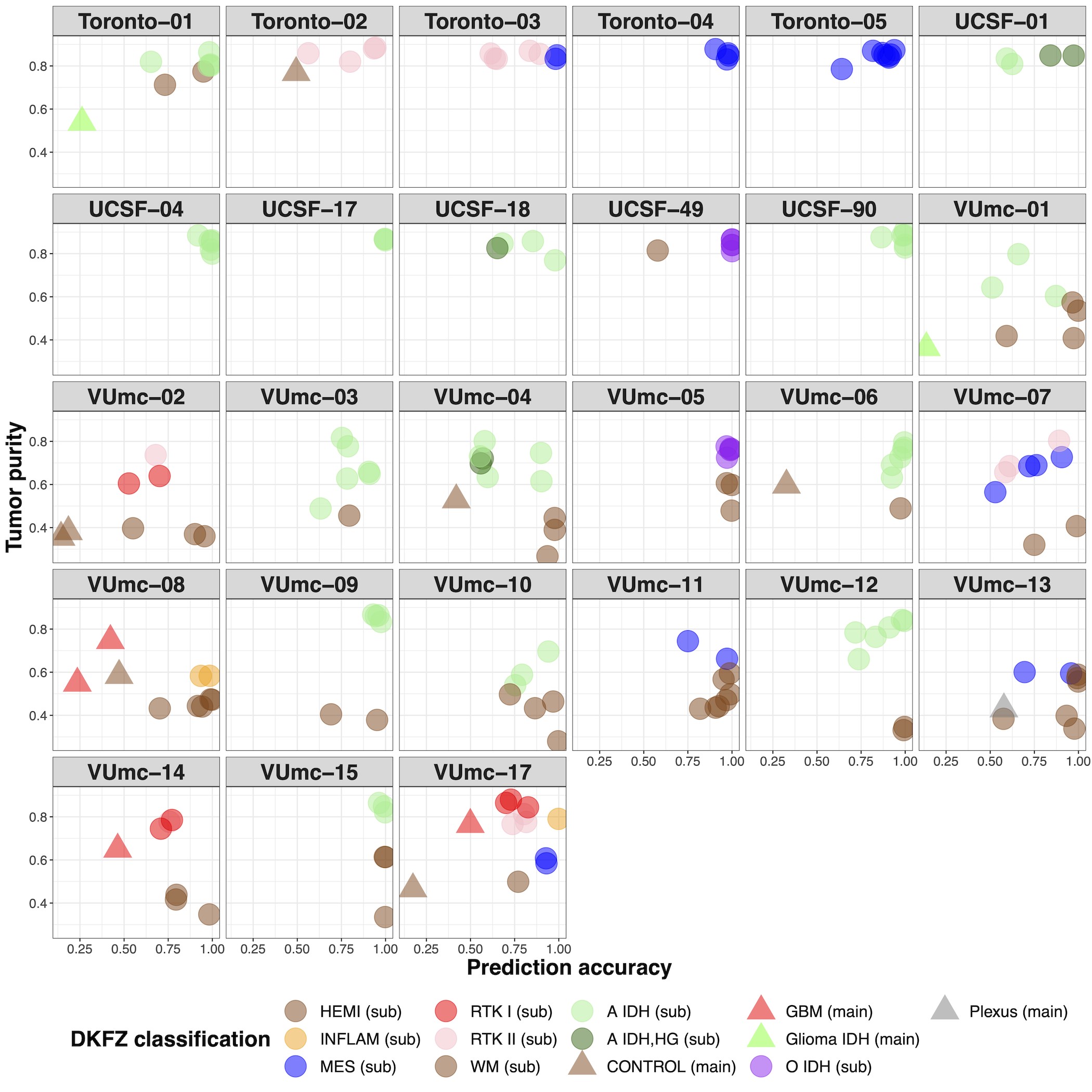
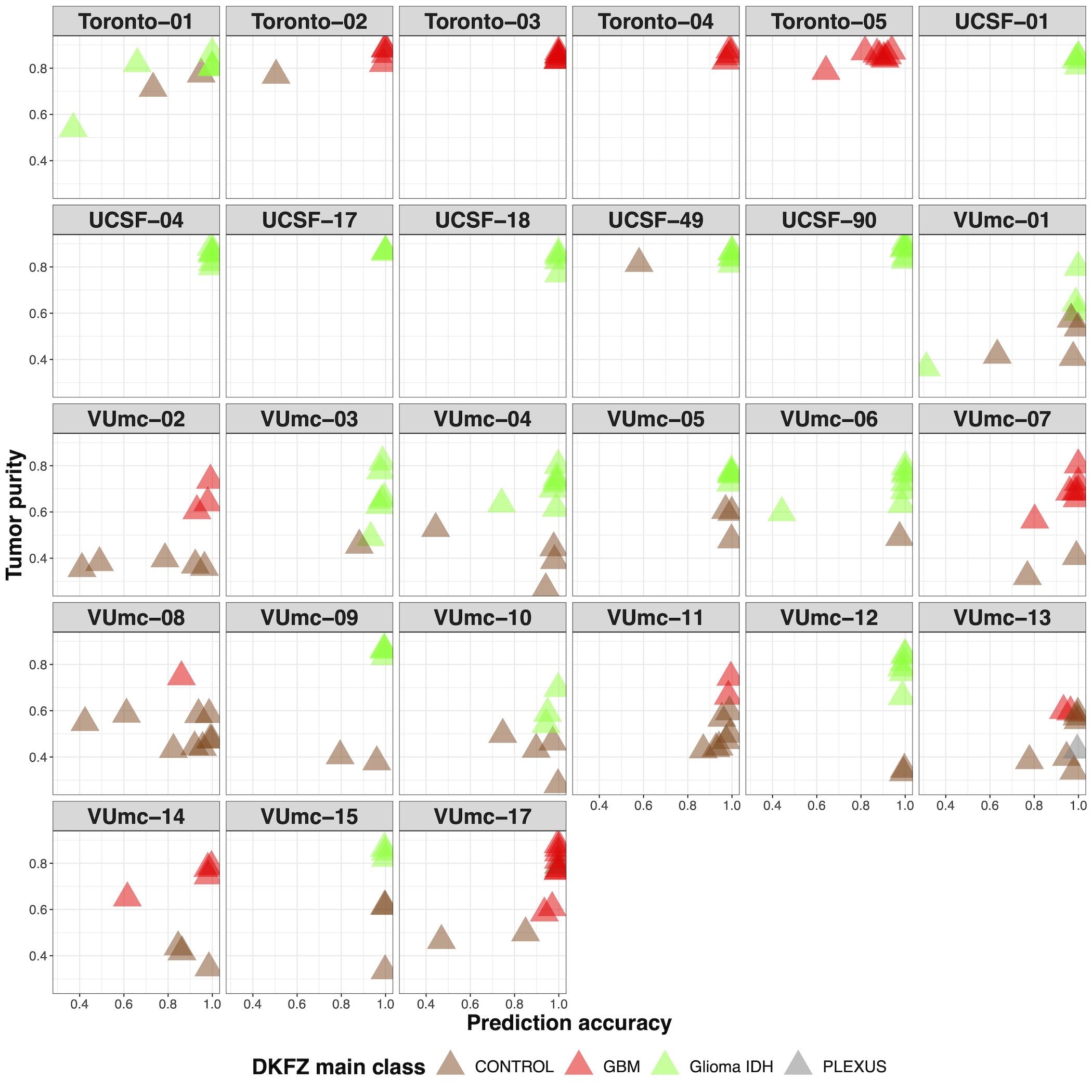
When we classified samples according to the DKFZ classifier, no heterogeneity was found for the main classes (Supplementary Figure 3B and C). Heterogeneity of main class subtypes, so-called family members, was found in 8 patients (VUmc-02, VUmc-04, VUmc-07, VUmc-14, VUmc-17, Toronto-03, UCSF-01, and UCSF-18). Both tumor purity and prediction accuracy were lower in tumor samples of these 8 patients with subtype heterogeneity (mean tumor purity 75.9 ± 0.09 and mean prediction accuracy 73.4 ± 14.9) compared to tumor samples of patients without heterogeneity (mean 78.1 ± 10.9, P = .046 and 87.8 ± 18.0, P < .001, respectively), suggesting tumor purity and prediction accuracy as potential confounding factors for the observed heterogeneity in these patients (Supplementary Figure 3D). In 2 patients (VUmc-02 and VUmc-17), the discordant samples were the lowest tumor purity, although the absolute difference in tumor purity was small. In 3 patients (Toronto-03, UCSF-01, and UCSF-18), the discordant samples were the lowest prediction accuracy, although the absolute difference in prediction accuracy was small except for patient UCSF-01. Three patients (VUmc-04, VUmc-07, and VUmc-14) showed variable tumor purity and prediction accuracy of the tumor samples suggesting true heterogeneity. To validate the possible heterogeneity in these patients, we evaluated the histological and imaging data that was available for the exploration cohort. The only difference we found between subtypes was higher cellularity (P = .016) and more frequent contrast enhancement on T1c (P < .001) in A IDH, HG samples compared to A IDH samples (Figure 6A). This confirmed the true heterogeneity in patient VUmc-04, with both A IDH, HG samples having the highest cellularity and showing contrast enhancement on T1c (Figure 6B). In the 4 glioblastoma patients with multiple subtypes, neither histology nor imaging could confirm or reject the subtype heterogeneity (Figure 6C). Therefore, we conclude there is true DNA methylation subtype heterogeneity in patient VUmc-04, based on DNA methylation, histology, and imaging data, and patients VUmc-07 and VUmc-14, based on DNA methylation data. Based on all available data, it remains unclear if there is true heterogeneity in the remaining 5 patients. Although not statistically significant, all true heterogeneous patients had a glioblastoma (chi-squared P = .17), yet heterogeneity was not related to IDH status (chi-squared P = .73). Overall in our cohort, a low frequency of true spatial heterogeneity of DNA methylation-based subtypes was observed (14%, 3 of the 22 patients), when taking tumor purity and prediction accuracy into account, yet possible heterogeneity cannot be ruled out in another 5 (19%) patients.
Fig. 6.

Histology and imaging of patients with DNA methylation subtype heterogeneity. A. Boxplots of cellularity (upper) and proliferation index (middle) and barplot of T1c MRI contrast enhancement (lower) for the DKFZ subtypes MES (blue), RTK I (red), RTK II (pink), A IDH (light green), and A IDH, HG (dark green) of all patients from the exploration cohort. Statistical significance is given using the Mann-Whitney U test for the continuous data (Histology) and the chi-squared test for the frequency data (Imaging), with * representing P < .05. B. Representative examples of the histological and radiological images of the patient VUmc-04: HE-stained histology slide of a sample classified as A IDH (S3) and A IDH, HG (S9) showing the higher cellularity in the A IDH, HG sample. Sagittal T1c MRI images of the same samples showing contrast enhancement in the A IDH, HG, but not in the A, IDH sample. C. Barplot of cellularity (upper), proliferation index (middle), and contrast enhancement on T1c MRI (lower) of the tumor samples of glioblastoma patients of the exploration cohort with subtype heterogeneity. Abbreviations: HE, hematoxylin and eosin; MRI, magnetic resonance imaging; T1c, contrast-enhanced T1-weighted.
Since O6-methylguanine-DNA methyl-transferase (MGMT) promoter methylation status is an important prognostic and predictive marker we assessed its spatial distribution. Heterogeneity was found in 30% of patients and was not related to tumor purity (Pearson correlation P = .19), IDH status, histology or DNA methylation classification heterogeneity (chi-squared P = 1.0, P = .76, and P = .78, respectively).
DNA Methylation Abnormalities Extend Beyond Standard MRI Boundaries
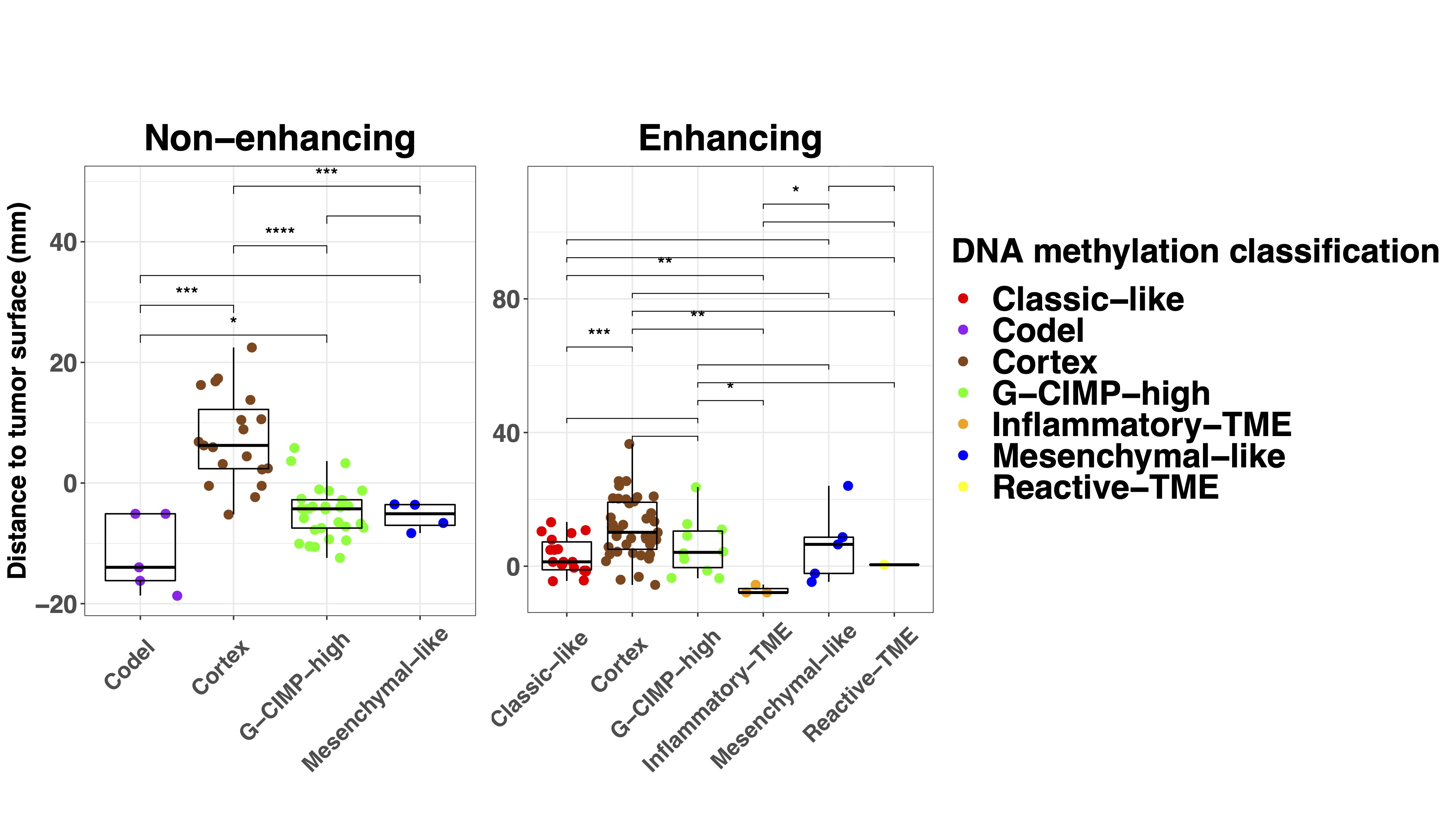
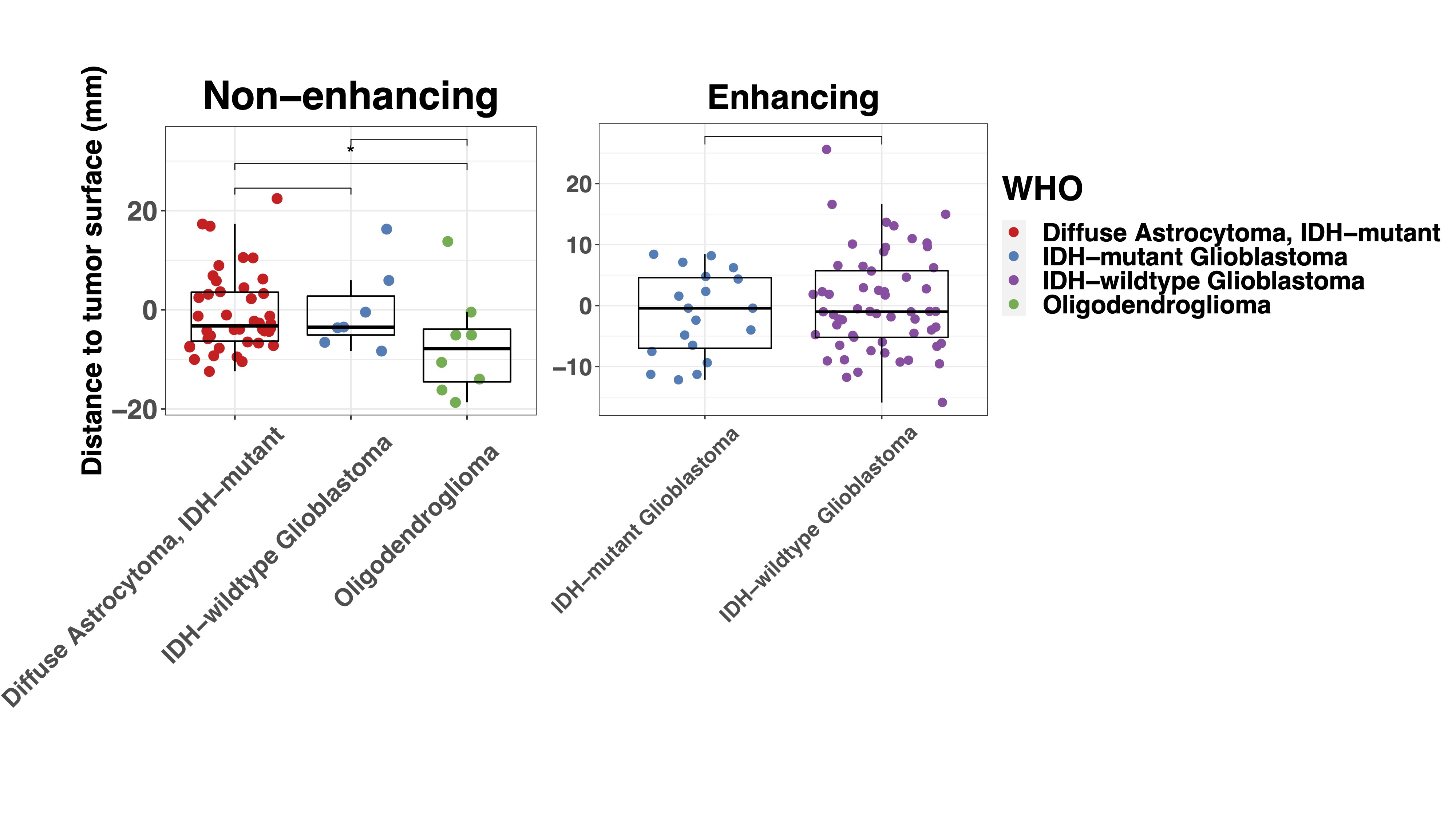
To understand the spatial distribution of glioma infiltration, we analyzed the correlation of tumor purity and subtypes with the distance to the tumor surface assessed with T1c for enhancing and FLAIR MRI for non-enhancing tumors. As expected, the distance of samples to the tumor surface showed a linear relationship with tumor purity (Supplementary Figure 4A). In non-enhancing tumor, samples classified as Cortex were found further away from the radiological tumor boundaries than all the other subtypes (Supplementary Figure 4B). Yet, in enhancing tumor, this difference was not found, possibly indicating a more diffuse infiltration pattern of enhancing tumors. When evaluating the infiltration pattern in the histological subgroups, no difference was found except that distance to tumor surface in 1 oligodendroglioma (median −7.9 mm, IQR −14.5 to −3.9 mm) was lower than in IDH-mutant diffuse astrocytoma (−3.2 mm, −6.3 to 3.6 mm, P = .047) (Supplementary Figure 4C).
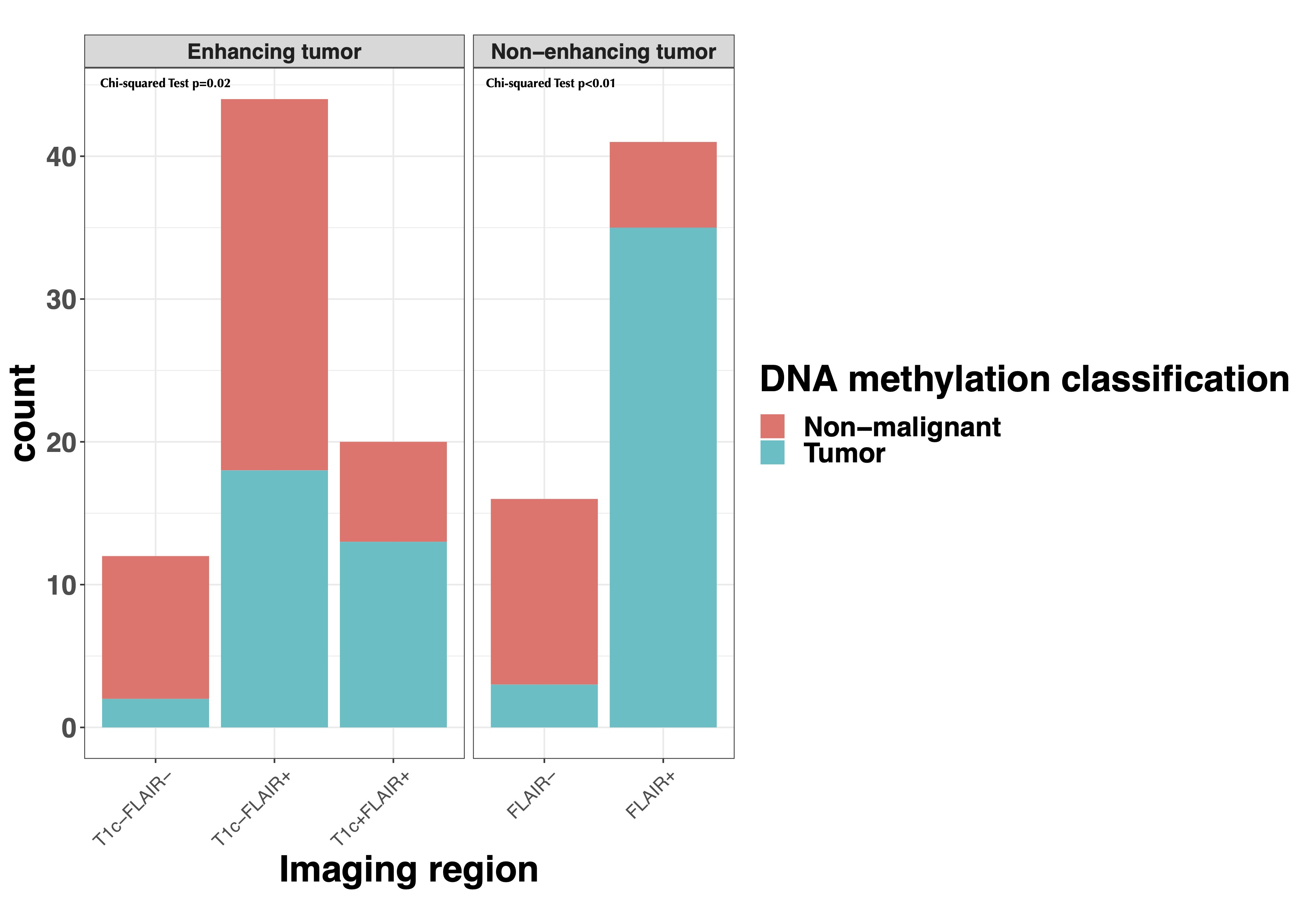
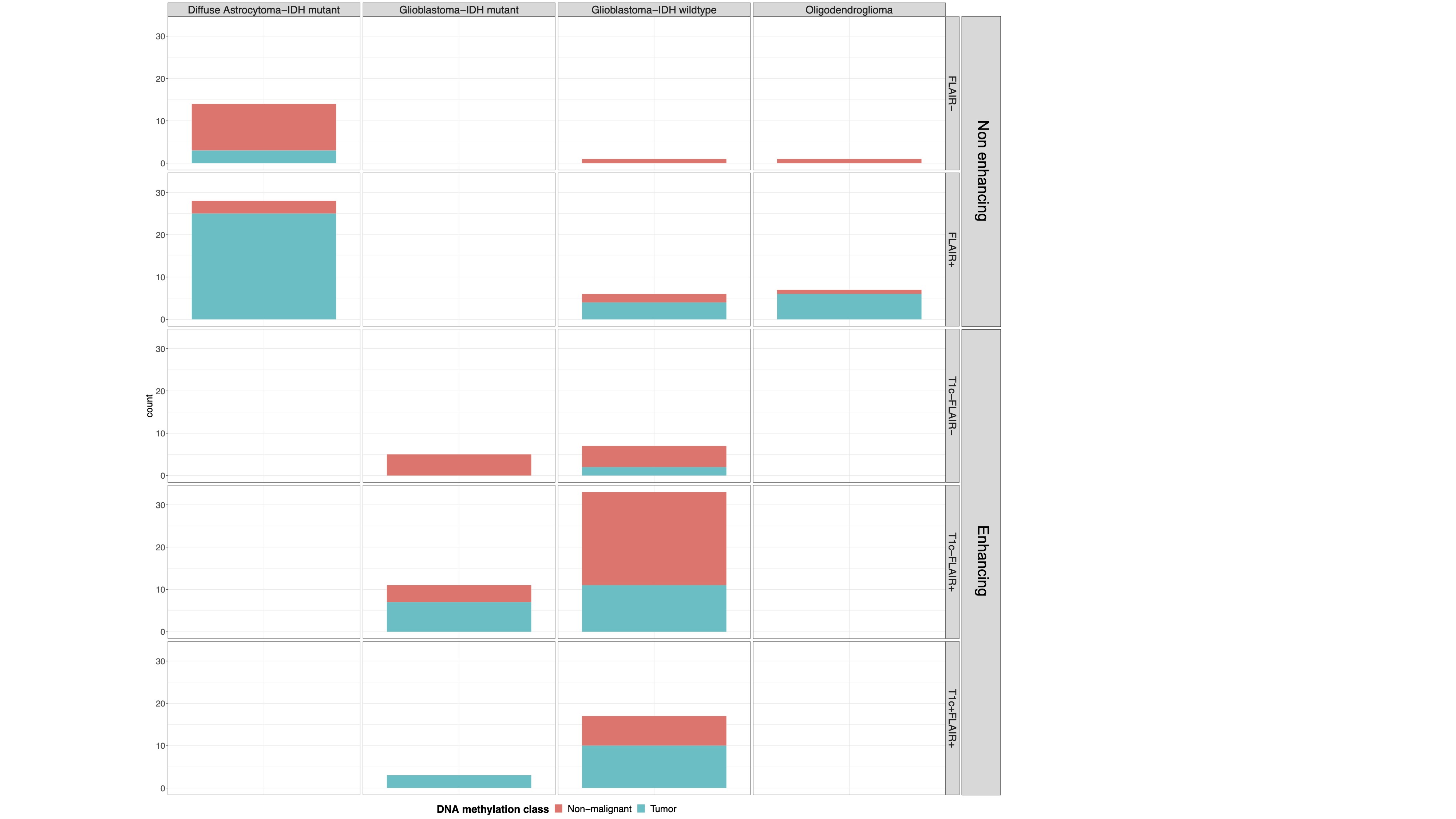
As anticipated, in enhancing tumors the T1c+/FLAIR+ region showed the highest tumor purity, followed by the T1c−/FLAIR+ and T1c−/FLAIR− (Supplementary Figure 5A). In non-enhancing tumors, tumor purity was higher in the FLAIR+ than the FLAIR− region. Interestingly, samples taken from regions outside standard imaging abnormalities (FLAIR− in non-enhancing and T1c−/FLAIR− and T1c−/FLAIR+ in enhancing tumors) showed a tumor subtype in 36% and 17% of enhancing and non-enhancing gliomas, respectively (Supplementary Figure 5B). Conversely, samples taken from within the standard imaging abnormalities showed a nonmalignant subtype in 35% of enhancing tumors, which is most likely due to necrosis in these samples. In non-enhancing tumors, 15% of samples within the FLAIR+ region showed a nonmalignant subtype. Tumor presence, assessed as the ratio of tumor and nonmalignant TCGA subtypes, in the different imaging regions was comparable between histological subgroups (all chi-squared tests P > .05) (Supplementary Figure 5C). Samples with tumor subtypes were found up to 24 mm outside imaging abnormalities. These findings support the diffusely infiltrative nature of these tumors and corroborate the notion that standard MRI does not capture the true extent of diffuse glioma infiltration.
Discussion
This study represents a comprehensive analysis of spatially separated samples in diffuse glioma. The combination of histological, radiological, and DNA methylation data enabled us to explore the spatial contexts of tumor purity, epigenetic molecular subtypes, and tumor heterogeneity. Our study demonstrates that in most tumors, molecular subtypes are stable and homogeneous after considering tumor purity. Moreover, gliomas are diffusely infiltrative tumors, and our data clearly show that they indeed extend beyond the tumor boundaries found on MRI. Finally, in our study, the extent of heterogeneity in tumor samples was predominantly equal to or less than in nonmalignant samples.
Information on the spatial heterogeneity of epigenetic molecular subtypes in the literature is limited. A recent study reported intratumor DNA methylation-based subtype heterogeneity in 5 of the 12 glioblastomas in their cohort.28 We were unable to confirm this extent of heterogeneity in our study. The differences may be explained by our approach to account for tumor purity prior to determining intratumoral epigenetic subtype classification. The non-purity–related heterogeneity that was found could be explained by focal malignant progression in patients with an IDH-mutant astrocytoma, while cellular state plasticity, as described by Neftel et al,8 might be an explanation for the coexistence of both mesenchymal and RTK II subtypes in one of the IDH-wildtype glioblastoma patients. Although technically difficult,29 future single-cell DNA methylation profiling can be expected to drastically increase our understanding of DNA methylation classification heterogeneity. MGMT promoter methylation status proved to be more heterogeneous than DNA methylation subtypes, which is most likely due to the use of 2 probes for the MGMT status compared to ~1000 probes for the DNA methylation subtype.
We observed that samples obtained outside standard imaging abnormalities, on FLAIR in non-enhancing and on T1c MRI in enhancing gliomas, displayed similar epigenetic molecular subtypes as the samples from the tumor core. The fraction of tumor cells per specimen varied between the different MRI regions, with lower fractions in regions outside of imaging abnormalities. The presence of tumor tissue outside standard imaging abnormalities is well known.4,5 Our results suggest that spatial imaging heterogeneity in glioma is associated with tumor purity and not with epigenetic heterogeneity, although heterogeneity at the genomic and mutational level as cause cannot be excluded and has recently been reported for progressive IDH-mutant astrocytoma.30 This was confirmed by the strong correlation between tumor purity and the imaging score. Our observations imply that a viable part of the tumor, especially in IDH-wildtype glioblastomas, is left behind after resection of standard imaging abnormalities.
A limitation of this study is the difficulty of assessing the true tumor purity since there is no gold standard. Still, we have compared many tumor purity metrics and choose the most representative of those. Another limitation is the evaluation of tumor heterogeneity by bulk DNA methylation analysis instead of single-cell analysis, although correlating imaging data with single-cell analysis might prove difficult due to the different levels of detail. Finally, the use of T1c and FLAIR MRI for the assessment of tumor to sample distance is a limitation, since these modalities are known to be less accurate for the detection of diffuse glioma infiltration.31 However, they are still the current standard imaging and therefore represent the daily practice of glioma treatment.
In conclusion, we demonstrate that DNA methylation subtypes in diffuse glioma show little intratumoral heterogeneity and are uniform across the different imaging regions, underscoring the diffuse infiltrative nature of this disease and the robustness of DNA methylation subtypes.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Zoe Reifsnyder from the Jackson Laboratory Creative Team for assistance in graphic design.
Funding
This work was supported by Cancer Center Amsterdam Support Grant 2012-2-05, Cancer Center Support Grant P30CA034196; and NIH/NCA R01 CA190121, R01 CA237208, NIH/NINDS R21 NS114873, Department of Defense W81XWH1910246 (R.G.W.V.). N.V. is the recipient of a Dutch Cancer Society Fellowship (OAA/H1/VU 2015-7502) and a Niels Stensen Fellowship. F.P.B. is supported by the JAX Scholar program and the National Cancer Institute (K99 CA226387). K.C.J. is the recipient of an American Cancer Society Fellowship (130984-PF-17-141-01-DMC). F.B. is supported by the NIHR Biomedical Research Centre at UCLH.
Conflict of interest statement. R.G.W.V. is a co-founder of Boundless Bio, Inc. F.P.B. has performed consulting for Celgene. S.D. receives research funding from Alkermes and has received research funding from Medicenna (completed 12/2019), is a consultant for XPan Medical, and is on the advisory board for the Subcortical Surgery Group.
Authorship statement. Study concept and design: O.S.H., F.B., P.J.W.P., J.C.R., W.P.V., P.W., P.C.W.H., and R.G.W.V. Acquisition, analysis, or interpretation of data: N.V., F.P.B., K.J.A., K.C.J., T.K., M.M.Y., O.S.H., A.A.L., F.B., P.J.W.P., J.C.R., A.J.M.R., J.A.M.B., R.B., M.D.T., S.D., J.F.C., W.P.V., P.W., P.C.W.H., and R.G.W.V. Drafting of the manuscript: N.V. and F.P.B. Critical revision of the manuscript for important intellectual content: K.J.A., K.C.J., P.J.W.P., J.C.R., W.P.V., P.W., P.C.W.H., and R.G.W.V. Statistical analysis: N.V. and F.P.B. Obtained funding: N.V., P.C.W.H., and R.G.W.V. Study supervision: P.C.W.H. and R.G.W.V.
References
- 1. Ho VK, Reijneveld JC, Enting RH, et al. ; Dutch Society for Neuro-Oncology (LWNO) . Changing incidence and improved survival of gliomas. Eur J Cancer. 2014;50(13):2309–2318. [DOI] [PubMed] [Google Scholar]
- 2. Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–446. [DOI] [PubMed] [Google Scholar]
- 3. Ellingson BM, Bendszus M, Boxerman J, et al. ; Jumpstarting Brain Tumor Drug Development Coalition Imaging Standardization Steering Committee . Consensus recommendations for a standardized Brain Tumor Imaging Protocol in clinical trials. Neuro Oncol. 2015;17(9):1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pallud J, Varlet P, Devaux B, et al. . Diffuse low-grade oligodendrogliomas extend beyond MRI-defined abnormalities. Neurology. 2010;74(21):1724–1731. [DOI] [PubMed] [Google Scholar]
- 5. Kelly PJ, Daumas-Duport C, Kispert DB, Kall BA, Scheithauer BW, Illig JJ. Imaging-based stereotaxic serial biopsies in untreated intracranial glial neoplasms. J Neurosurg. 1987;66(6):865–874. [DOI] [PubMed] [Google Scholar]
- 6. Petrecca K, Guiot MC, Panet-Raymond V, Souhami L. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J Neurooncol. 2013;111(1):19–23. [DOI] [PubMed] [Google Scholar]
- 7. Barthel FP, Johnson KC, Varn FS, et al. ; GLASS Consortium . Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576(7785):112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neftel C, Laffy J, Filbin MG, et al. . An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019; 178(4):835–849 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venteicher AS, Tirosh I, Hebert C, et al. . Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355(6332):1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Snuderl M, Fazlollahi L, Le LP, et al. . Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. [DOI] [PubMed] [Google Scholar]
- 11. Szerlip NJ, Pedraza A, Chakravarty D, et al. . Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012;109(8):3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sottoriva A, Spiteri I, Piccirillo SG, et al. . Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki H, Aoki K, Chiba K, et al. . Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458–468. [DOI] [PubMed] [Google Scholar]
- 14. Kim H, Zheng S, Amini SS, et al. . Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25(3):316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ceccarelli M, Barthel FP, Malta TM, et al. ; TCGA Research Network . Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Capper D, Jones DTW, Sill M, et al. . DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benelli M, Romagnoli D, Demichelis F. Tumor purity quantification by clonal DNA methylation signatures. Bioinformatics. 2018;34(10):1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verburg N, Pouwels PJ, Boellaard R, et al. . Accurate delineation of glioma infiltration by advanced PET/MR neuro-imaging (FRONTIER Study): a diagnostic study protocol. Neurosurgery. 2016;79(4):535–540. [DOI] [PubMed] [Google Scholar]
- 20. Nathanson V. Revising the Declaration of Helsinki. BMJ. 2013;346:f2837. [DOI] [PubMed] [Google Scholar]
- 21. Wang Q, Hu B, Hu X, et al. . Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017; 32(1):42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Louis DN, Perry A, Reifenberger G, et al. . The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 23. Darmanis S, Sloan SA, Croote D, et al. . Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017;21(5):1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mazor T, Pankov A, Johnson BE, et al. . DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell. 2015;28(3):307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morrissy AS, Cavalli FMG, Remke M, et al. . Spatial heterogeneity in medulloblastoma. Nat Genet. 2017;49(5):780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ziller MJ, Gu H, Müller F, et al. . Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500(7463):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network . Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wenger A, Ferreyra Vega S, Kling T, Bontell TO, Jakola AS, Carén H. Intratumor DNA methylation heterogeneity in glioblastoma: implications for DNA methylation-based classification. Neuro Oncol. 2019;21(5):616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson KC, Anderson KJ, Courtois ET, et al. . Single-cell multimodal glioma analyses reveal epigenetic regulators of cellular plasticity and environmental stress response. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jonsson P, Lin AL, Young RJ, et al. . Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin Cancer Res. 2019;25(18):5537–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verburg N, Koopman T, Yaqub MM, et al. . Improved detection of diffuse glioma infiltration with imaging combinations: a diagnostic accuracy study. Neuro Oncol. 2020;22(3):412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.