

Abstract
Chagas disease is an infection caused by the parasite Trypanosoma cruzi, endemic in Latino America. Leveraging the three-way admixture between Native American (AMR), European (EUR) and African (AFR) populations in Latin Americans, we aimed to better understand the genetic basis of Chagas disease by performing an admixture mapping study in a Colombian population. A two-stage study was conducted, and subjects were classified as seropositive and seronegative for T. cruzi. In stage 1, global and local ancestries were estimated using reference data from the 1000 Genomes Project (1KGP), and local ancestry associations were performed by logistic regression models. The AMR ancestry showed a protective association with Chagas disease within the major histocompatibility complex region [Odds ratio (OR) = 0.74, 95% confidence interval (CI) = 0.66–0.83, lowest P-value = 4.53 × 10−8]. The fine mapping assessment on imputed genotypes combining data from stage 1 and 2 from an independent Colombian cohort, revealed nominally associated variants in high linkage disequilibrium with the top signal (rs2032134, OR = 0.93, 95% CI = 0.90–0.97, P-value = 3.54 × 10−4) in the previously associated locus. To assess ancestry-specific adaptive signals, a selective sweep scan in an AMR reference population from 1KGP together with an in silico functional analysis highlighted the Tripartite Motif family and the human leukocyte antigen genes, with crucial role in the immune response against pathogens. Furthermore, these analyses emphasized the macrophages, neutrophils and eosinophils, as key players in the defense against T. cruzi. This first admixture mapping study in Chagas disease provided novel insights underlying the host immune response in the pathogenesis of this neglected disease.
Introduction
Chagas disease (ICD-10-CM B57) is caused by the protozoan parasite Trypanosoma cruzi and is endemic in Latin American countries (1). This infection affects ~7 million people in endemic and non-endemic areas because of migratory movements (www.who.int/health-topics/chagas-disease#tab=tab_1). The main transmission vectors are members of the Reduviidae family, although other transmission mechanisms such as oral and congenital have been described (2). The infection starts with an acute phase characterized by inflammatory and unspecific clinical symptoms followed by an asymptomatic or chronic phase, the last one with cardiac or digestive involvement (2). In this sense, the critical role played by the host’s innate and adaptive immune responses during the acute phase is well known (2,3).
The differential susceptibility to the infection together with the high exposure to the parasite in endemic areas suggests the implication of the host genetic component in the pathogenesis of the disease (2,4). To elucidate this role, previous candidate gene assessments and genome-wide associations studies (GWAS) have been carried out supporting the existence of this genetic contribution to Chagas disease and to its chronic cardiac form (5–7). Nevertheless, this genetic contribution is complex and our knowledge of it incomplete, as many other genetic polymorphisms are expected to contribute to the risk, including population-specific variants that are not well covered by GWAS (8).
Latin American populations are commonly modeled as a recently admixed population from three main different continental sources: Native Americans (AMR), Europeans (EUR) and Africans (AFR) (9). Specifically, the Colombian population has been described to show one of the most disparate admixture proportions from these three continental ancestries among all Hispanic populations (10,11). Furthermore, the interindividual variation in ancestry proportions has been correlated with the risk of several diseases, including infectious diseases such as malaria (12,13). Functional assessments of prioritized loci suggest that they contain ancestry-specific alleles related to the immune system, proposing that this could be driven by the high exposure to several microbial pathogens (13). This scenario is amenable for admixture mapping studies, which allow the identification of genomic susceptibility regions where affected individuals share their genetic ancestry locally on chromosome regions compared with the non-affected ones. Thus, it allows to scan disease associations with the varying local ancestries in admixed populations (14,15). This approach is complementary to the traditional GWAS, and one of its main advantages is that it requires lower samples sizes for a given statistical power because of the reduced penalty by the multiple testing corrections (14). In Chagas disease, previous studies have assessed global ancestry proportions in Brazilian population, either as a complementary analysis to their main objective (16) or assessing its association with the disease (17), but without evaluating local ancestries.
To better understand the host genetics involved in the risk of T. cruzi infection, we carried out the first admixture mapping study of Chagas disease in the Colombian population.
Results
This study has a two-stage case–control design and samples were classified as seropositive (cases) and seronegative (controls) for parasite antigens. Demographic characteristics are summarized in Table 1. Cases and controls from the stage 1 samples were matched in terms of global ancestries (Supplementary Fig. 1). Local ancestry blocks were estimated for the 471 342 positions corresponding to those of genotyped single nucleotide polymorphisms (SNPs) in stage 1. Local ancestry punctuations per individual were averaged and compared with their global ancestries showing a high correlation among them (r = 0.89–0.96) and indicating consistent global and local ancestry proportions in the population under study. Cases and controls were also matched in terms of local ancestries (Supplementary Table 1), and supervised and unsupervised analysis provided consistent results, reinforcing the selection of the representatives of the parental populations in the ancestry assessment (Supplementary Fig. 2).
Table 1.
Demographical characteristics and sample size of the Colombian collections
| Stage 1 | Stage 2 | |||
|---|---|---|---|---|
| Seropositive | Seronegative | Seropositive | Seronegative | |
| Pre QC sample size | 998 | 659 | 122 | 532 |
| Post QC sample size | 913 | 592 | 122 | 512 |
| Sex, females (%) | 503 (55) | 357 (60) | 65 (53) | 315 (62) |
| Age (mean ± SD) | 62.7 ± 16.2 | 49.0 ± 17.6 | 64.7 ± 11.2 | 50.0 ± 15.6 |
QC, quality controls.
Admixture mapping results based on local ancestry estimations were not inflated because of the presence of population stratification for any of the ancestries after genomic inflation (λ) correction (λAMR = 1.00, λEUR = 1.00, λAFR = 1.00). These results revealed genome-wide significant associations of AMR and EUR ancestries with Chagas disease in the positions of the chromosome 6 region 30 079 993-30 332 160 according to build hg19 (Fig. 1). We observed a lack of association of AFR ancestry with the serological status. These results revealed a differential susceptibility to the infection associated with two of the ancestries in this particular region, where AMR was in higher proportion among seronegative individuals overall, therefore associated with a protective effect. The associated region is located in the major histocompatibility complex (MHC) locus where the leading signal corresponds to rs115833233 [AMR ancestry odds ratio (OR) = 0.74, 95% confidence interval (CI) = 0.66–0.83, P-value = 4.53 × 10−8], which is located in the untranslated region (UTR) of exon 5 in the Tripartite Motif Containing 40 (TRIM40) gene. The association of AMR ancestry was unrelated with the genotypes in that position because conditioning by the allele dosage of the rs115833233 variant did not change the results. Thus, the admixture mapping peak was not explained by the genetic variation of that SNP (Table 2).
Figure 3.

Selective sweep scan results for Native Americans (AMR) from the 1000 Genomes Project. Y and X axes represent the iSAFE scores and hg19 genomic positions, respectively. (A) iSAFE scores within the MHC region (chr6:28477895-33 389 603). Top ranking variants included in the significant admixture mapping region are highlighted in red (chr6:30079993-30 332 160). (B) Zoom in of the admixture mapping significant region, where the top 25 variants according to their iSAFE score are represented. Genetic variants are depicted in colored dots to reflect its LD with the variant with the highest iSAFE score based on pairwise r2 values in AMR population. Their dot size correlates to their composite variant to genes score (V2G) from Open Targets Genetics (https://genetics.opentargets.org/).
Table 2.
Joint single nucleotide polymorphism-ancestry analysis in the discovery stage
| Factor | OR (95% CI) | P-value |
|---|---|---|
| AMR ancestry | 0.74 (0.66–0.83) | 4.53 × 10−08 |
| Allele dosage rs115833233 | 1.00 (0.89–1.13) | 0.99 |
| AMR ancestry (conditioned on rs115833233) | 0.73 (0.65–0.82) | 2.15 × 10−08 |
AMR, Native American ancestry; CI, confidence interval; OR, odds ratio.
Figure 1.
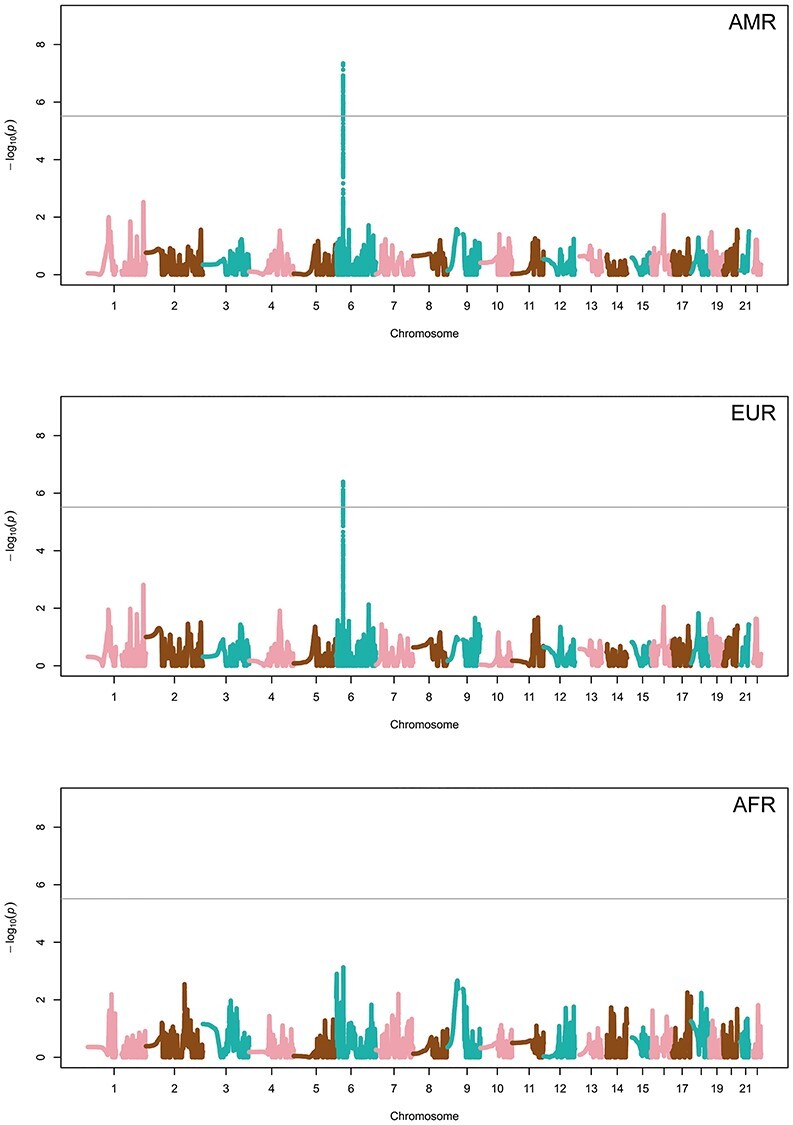
Manhattan plots of the admixture mapping results based on local ancestry estimates of Native American (AMR; op), European (EUR; middle) and African (AFR bottom). Y and X axes refer to the –log10 transformed P-values and hg19 positions in chromosomes, respectively. The horizontal line indicates the significance threshold (P-value = 3.07 × 10−6).
To identify potential variants explaining the result, imputed genotype data from the significant admixture mapping region were assessed in the stage 1 samples and in independent samples from stage 2. This analysis identified 23 variants in high linkage disequilibrium (LD, r2 > 0.8) with nominal significance and consistent direction of effects in the two stages (Table 3). The leading variant was rs2032134, located intergenic to the Ribonuclease P/MRP Subunit P21 (RPP21) and TRIM39 genes (OR for the C allele = 0.93, 95% CI = 0.90–0.97, P-value = 3.54 × 10−4) (Fig. 2). We confirmed this association was dependent on the AMR ancestry because adjusting the models by the ancestry score resulted not significant (OR for the C allele = 0.99, 95% CI = 0.95–1.03, P-value = 0.766). In addition, we confirmed that the C allele was associated with local AMR ancestry as stratifying cases and controls by this ancestry proportion, the allele frequency was increased among carriers of local AMR ancestry in cases and controls in comparison with individuals bearing other ancestries in that position (Supplementary Fig. 3). Functional analysis showed that rs2032134 is an expression-quantitative trait loci (eQTL) for different human leukocyte antigen (HLA) members in different tissues, most significantly for the HLA-C in whole blood (P-value = 5.1 × 10−35) according to eQTLGen. Evidence of long-distance interactions between rs2032134 and another member of the Tripartite Motif (TRIM) family, TRIM31, was observed in macrophages and neutrophils (18), as well as in lymphoblastoid cell lines (19). Further functional assessments for rs2032134 and its best LD-proxies are summarized in Table 4.
Table 3.
Association testing results and allele frequencies on imputed data for the admixture mapping associated region
| Stage 1 | Stage 2 | Meta-analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP_ID(*EA) | CHR:BP | EA frequency (cases/controls) | OR (95% CI) | P-value | EA frequency (cases/controls) | OR (95% CI) | P-value | OR (95% CI) | P-value |
| rs2032134*C | 6:30360509 | 0.81/0.86 | 0.95 (0.90–0.99) | 2.57 × 10−2 | 0.75/0.84 | 0.92 (0.87–0.97) | 3.67 × 10−3 | 0.93 (0.90–0.97) | 3.54 × 10−4 |
EA, effect allele; BP, base pair; CHR, chromosome; CI, confidence interval; OR, odds ratio; SNP, single nucleotide polymorphism.
Figure 2.

Regional plots of the results from admixture (top) and fine mapping (bottom) analyses, showing the −log10 transformed P-value in the y-axis and the hg19 genomic position in the x-axis. Top: Region centered on the significant variants for the AMR ancestry association. The horizontal line indicates the significance threshold (P-value = 3.07 × 10−6). Estimated recombination rates (light blue line) are plotted on the right y-axis. Bottom: Meta-analysis results from stages 1 and 2 within the admixture mapping significant region (chr6:30079993-30 332 160) and its proxies (r2 > 0.8) indicating the associated variant with the lowest significance in the region (rs2032134). The results for the remaining single nucleotide polymorphisms (SNPs) are color coded to reflect their degree of LD with the indicated SNP based on pairwise r2 values in AMR. The horizontal lines indicate the significant (solid line, P-value = 3.23 × 10−4) and suggestive thresholds (broken line, P-value = 6.46 × 10−3).
Table 4.
In silico functional assessment of the fine mapping and selective sweep analyses top variants
| Chr | SNP | Function | Nearest gene | eQTLsa | C-HiC genes | PheWAS |
|---|---|---|---|---|---|---|
| 6 | rs2032134 | Intergenic | RPP21/TRIM39 | HLA-C, HCG18, HLA-E, HLA-G, TRIM10 | TRIM31 in GM12878b, macrophagesc and neutrophilsc | - |
| 6 | rs9261440 | Intergenic | TRIM31/TRIM40 | HLA-F, HLA-G, HLA-A, HLA-L, TRIM26, TRIM10 | TRIM10 in GM12878b | Hematological measurement, eosinphil count |
eGENES in whole blood.
Lymphoblastoid cell line from Mifsud et al.
Macrophages and neutrophil from Javierre et al.
Queried databases were Open Targets Genetics, eQTLGen, HaploReg and Capture HiC Plotter.
Chr, chromosome; C-HiC, capture Hi-C; eQTL, expression quantitative trait loci; PheWAS, phenome-wide association study.
In order to assess adaptive signals within the significant admixture mapping region, we performed a selective sweep analysis in this region using a reference population. From this analysis, we found that the iSAFE scores were high; with the top-ranking variants corresponding to positions 30 087–30 102 Mb (iSAFE score ≥ 0.331). The top 25 variants ranked by the iSAFE score mapped within (n = 5; only rs28400887 was coding but synonymous) or nearby (n = 20) the TRIM31 gene, with the furthest SNP located at 21.1 Kb from the gene (Fig. 3). Given the excellent performance of iSAFE in prioritizing the most likely favored variant in 94% of the times (20), we performed a functional assessment of the variant with top scoring (rs9261440; iSAFE score = 0.340) assuming it to be the variant driving the selective sweep. According to Open Targets Genetics portal, rs9261440 is an eQTL for some HLA class I genes in whole blood, where the most significant were HLA-F (P-value = 5.4 × 10−143), HLA-G (P-value = 2.4 × 10−280) and HLA-A (P-value = 2.4 × 10−156), and also for the TRIM family member TRIM26 (P-value = 7.9 × 10−79). Additionally, HaploReg indicates that this variant is an eQTL of HLA-L and TRIM10 in whole blood (21). In agreement with this, phenome-wide association studies (PheWAS) data from Open Targets Genetics indicates that this variant associates with hematological measurements, particularly with the eosinophil count in the UK Biobank (P-value = 7.6 × 10−43).
Discussion
We used an admixture mapping strategy to leverage varying local genomic ancestries in Colombians to identify loci associated with differential susceptibility to Chagas disease. We found significant associations of AMR and EUR ancestries within the MHC locus with the development of the infection, pointing out to the role of the immune response on the disease risk. Additionally, fine mapping assessments and a selective sweep scan of this region prioritized variants with potential functional implications in the disease and highlighting how powerful this strategy is for identifying regions that had been previously overlooked in other genomic studies.
Several studies in bioarchaeological material confirmed that the American trypanosomiasis already existed long before European settlement when ancestral populations domesticated plants and animals in the process of sedentarization. This provided the vector with food availability and a more rapid domiciliation (22,23). Host–parasite co-evolution is considered one of the most important generators of biological diversity in the genome, as is the case of selective sweeps at loci with functional role in their interaction (24). It occurs when parasites trigger host adaptations, which will lead the parasite to adapt again to this new environment in their hosts (24). This co-evolution has been described for T. cruzi (3,25) and, based on the observed protective association of the AMR ancestry, one can speculate that the long-term exposure of ancestral populations with larger proportions of AMR ancestry may have provided a more efficient immune response to the parasite, as previously hypothesized, with an adequate defense against pathogens that are endemic to the New World (13). This response could be responsible for parasite clearance, precluding the establishment of a serological response. This ancestry-specific effective response against parasites and other infectious agents has been previously hypothesized to explain the enrichment of African alleles within the HLA-B locus in a Colombian population (12). Another study correlated the EUR and AFR ancestries with differential immune response to bacterial infections and hypothesized that this could be related to the differential pathogen exposure of each population and different selective pressures after the human migrations out of Africa (26). Local adaptations have shaped human genetic variation together with drift and migrations, and admixed populations are likely to have a larger number of genetic variants that have functional effects (13,27). Therefore, it is particularly striking that the selective sweep scan within the significant admixture mapping region revealed variants associated with eosinophil counts because eosinophils have cytotoxic functions to fight parasitic infections, and along with macrophages, monocytes and neutrophils, are the innate immune cells responsible for the control of the initial infection by T. cruzi (28,29). Interestingly, blood cell traits have been reported to differ among ancestries and are subjected to different selective pressures (30).
Several genes of the TRIM family mapped within the significant admixture mapping region, and the top variant was located in the TRIM40 gene (Fig. 2). More importantly, the variants prioritized both by the fine mapping and the selective sweep scan are also eQTLs of TRIM genes in whole blood. The TRIM family is E3 ubiquitin kinases that play an important role in immune signaling pathways, and the expansion of this multigene family suggests their key regulatory role during the immune response against pathogens (31,32). When the pathogens are recognized by the immune system through the pattern-recognition receptors, several immune responses are initiated, including the production of interferons (IFNs), leading to the expression of TRIM proteins, among others (31). The upregulation of TRIM genes in response to IFN-γ has been reported in human monocytes and macrophages (33). The expression of IFN-γ is induced by interleukin 18 (IL-18), which is a pro-inflammatory cytokine mainly produced by macrophages (3). Remarkably, genetic variants of the gene encoding IL-18 have been associated with T. cruzi infection in previous candidate gene studies and showed suggestive association in a recently published GWAS (7,34,35).
The fact that the top variant from the fine mapping analysis mapped in an intergenic region, made us speculate on its potential relation with nearby genes through an in silico functional analysis. This strategy suggested the significant correlation of this variant with the HLA-C and HLA-G expression in whole blood as eQTLs. HLA genes are well-known for their role in the modulation of the immune response during T. cruzi infection (6). A previous study associated the HLA-C*03 allele with the susceptibility to the chronic cardiac form of the disease in a Venezuelan population (36). Regarding the HLA-G, several alleles from the HLA-G 3′UTR region were tested in a Brazilian population, reporting evidence of association with different clinical forms of the disease (37). Additionally, lower gene expression and plasma concentrations of this gene have been described in the chronic phase of T. cruzi and Plasmodium falciparum infections (38). Further long-distance chromosome interaction analysis of this variant revealed a significant interaction with the promoter of TRIM31 in macrophages, neutrophils and the lymphoblastoid cell line GM12878. This gene is a member of the TRIM family that acts as a regulator of the NLR pyrin domain-containing 3 (NLRP3) expression (39). The NLRP3 is an inflammasome component that is well-known to be activated by molecular patterns associated with pathogens. The activation of the inflammasome is crucial for the control of intracellular protozoan parasitic infections, like T. cruzi and the production of nitric oxide during its acute infection in mouse models (40,41). Interestingly, TRIM31 has been also prioritized by the selective sweep scan as a locus under putative positive selection in the AMR population.
There are some major limitations in our study that need to be taken into consideration. One of those limitations relates to the identification of the causal agent of the selective signal. The local adaptive signal maps within the MHC locus, which is a well-recognized target of selection. However, the causal factor underlying the selective sweep has been identified only in few instances in humans, such as skin pigmentation (42), lactase persistence (43) and adaptation to altitude (44). Therefore, we cannot infer nor guarantee that T. cruzi is the causal factor driving this putative adaptive signal. Another limitation is the limited statistical power, especially in the fine mapping, where we only had 80% power to detect variants with an allele frequency >20% and minimum effect allele of 1.4, which is fairly larger than expected for common variants in complex traits (8). Larger sample sizes would improve the statistical power of the study. Another limitation refers to the analyzed genetic variants, as only common SNPs were assessed in the fine mapping, and structural or less frequent genetic variants underlying the admixture mapping signal of Chagas susceptibility remain unexplored. Further insights concerning these types of genetic variants may be assessed by next-generation sequencing approaches. Moreover, despite the advantages of admixture mapping studies for identifying susceptibility loci, we are not in the position to warrantee if the results are generalizable to other populations with AMR ancestry, considering as well the variability of the infectious agent throughout the American continent. Finally, the scarce representation of AMR populations in reference databases reduces the precision to provide the exact functional implication of the associated variants, given the differences in LD structure among populations as well as other potential phenotypic features in different cell types.
To our knowledge, this is the first admixture mapping analysis carried out in Chagas disease in the Colombian population. This assessment allows us to associate the AMR local ancestry at the MHC locus with protection from Chagas disease and highlights the role of the immune response during the acute phase of the infection.
Material and Methods
Ethical considerations
The protocols used in the study followed the Declaration of Helsinki principles and informed consent was obtained from all individuals included in the study design. The Industrial University of Santander and Cardiovascular Foundation (Colombia) Ethics Committee approved this study (Act No. 15/2005).
Study population and genotyping
All donors were recruited by the health care system from the Industrial University of Santander and Cardiovascular Foundation in the provinces of Guanentina, Comunera and Garcia Rovira, which are the provinces with the highest prevalence of Chagas disease in the Santander department in Colombia (45). We used a two-stage case–control design where individuals were classified as seropositive (cases) or seronegative (controls) for T. cruzi antigens according to an indirect hemagglutination commercial test (Chagatest, Wiener, Argentina) and enzyme-linked immunosorbent assays (Test ELISA Chagas III, Grupo Bios, Chile; Chagas ELISA IgG + IgM, Vircell, Spain). Samples from stage 1 comprised 1576 individuals (933 classified as cases and 643 as controls) as described elsewhere (46). Further sample recruitment composed stage 2, including 654 independent samples (122 cases and 532 controls) according to the same classification criteria. Genomic DNA isolation of blood samples was performed using the QIAamp Midi DNA Kit (QIAGEN, Germany) following manufacturer’s recommendations.
All samples were genotyped with the Global Screening Array Platform (Illumina Inc., San Diego, CA, USA) as described elsewhere (7). As part of the quality controls (QCs) of genotyped data, individuals and variants with missing genotype rate > 5%, SNPs with different call rates between cases and controls (P-value < 0.05), and SNPs with large deviations from Hardy–Weinberg equilibrium (HWE) in the control group (P-value ≤ 1 × 10−6) was removed. QCs were performed using PLINK v.1.9 (47).
Admixture mapping analysis
Reference data from the 1KGP Phase 3 (48) was used as representatives of the parental populations in the downstream stage 1 global and local ancestry assessments. Briefly, we used data from AMR (n = 85), EUR (n = 503) and AFR (n = 504). Regarding the AMR population, only Peruvian from Lima (PEL) were included because this population has been described to have the highest proportion of AMR ancestry after the Maya population (49). All EUR subpopulations were taken into account (Utah residents, Finnish, British, Iberian and Toscani populations). In the case of AFR, both the African Caribbean from Barbados, and individuals with African ancestry from Southwest US were excluded as they are populations with recent admixture events (50). Using PLINK, we intersected the autosomal SNPs from stage 1 genotyped data (post QC 493 271 SNPs) with those from the 1KGP dataset, removing from the latter those variants with missing genotype rate >5% or with large deviations from HWE (P-value ≤ 1 × 10−6) in at least one population. After the intersection of datasets and QCs, a total of 471 342 SNPs were kept for further analysis.
Global ancestry assessment
Individual global ancestries were obtained with ADMIXTURE v1.3.0 (51), which calculates average ancestry proportions across the genome. We estimated the best number of ancestral populations (K) using a 10-time cross-validation with random seeds. The best fitting was obtained for K = 3, i.e. assuming 3 ancestral populations distinguishing AMR, EUR and AFR ancestries.
Local ancestry assessment and association testing
Local ancestries were estimated with ELAI v1.0 (52) assuming a three-way admixture and 10 generations since the last admixture event as has been indicated by previous studies (9,53). As QC steps, a correlation was calculated for individual global and averaged local ancestries across the genome using R v3.6.1. Principal components were calculated using PLINK and plotted along with those from the representative parental populations from 1KGP.
For the admixture mapping, the three local ancestries by separate were tested for case–control association using the EMMAX mixed model (54) implemented in EPACTS (https://genome.sph.umich.edu/wiki/EPACTS). As related individuals were considered, EMMAX calculates the kinship matrix to include it as a covariate in the model, in addition to age and sex. We controlled for the type I error rate by calculating the λ using an in-house script for R v3.6.1. Significance was adjusted by the number of ancestry blocks across the genome and the number of generations since the admixture using the R package STEAM (9). Based on that, the significance threshold for the admixture mapping study was established at P-value < 3.07 × 10−6, similar to estimates that have been declared in independent studies in Latin American populations (9).
Fine mapping
A fine mapping assessment was performed to elucidate the most likely genetic variant(s) underlying the admixture mapping study results combining data from the two stages. Briefly, genotypic data from stage 2 were subjected to the same QCs that were used in stage 1. After this, imputation of both stages was performed with the Michigan Imputation Server using the admixed American population from 1KGP phase 3 as reference panel. Imputed variants were filtered by their minor allele frequency (MAF) and the imputation quality metric Rsq. Those variants satisfying both a MAF > 1% and Rsq > 0.3 were kept for the study. Association testing of imputed allele dosages was performed in both stages by separate using the EMMAX mixed model implemented in EPACTS, including age, sex and the kinship matrix as covariates. Summary-level statistics of each stage were meta-analyzed using METASOFT v2.0 (55). A random- or fixed-effect size meta-analysis was selected for each variant based on the results for the Cochran’s Q-test of heterogeneity. Significant and suggestive thresholds for the assessed region were established at P-value < 3.23 × 10−4 and P-value < 6.46 × 10−3, respectively, according to the estimates of GEC software (56) based on the LD structure from the stage 1 samples. In order to confirm the association of the specific allele with the local AMR ancestry, samples were stratified according to their ancestry punctuations and allele frequencies were recalculated in cases and controls separately. The statistical power of the fine mapping analysis was estimated using the Power Calculator for Two Stage Association Studies software CaTS (57).
Selective sweep analysis
Given the high rate of adaptive signals in the genome triggered by parasites, and that admixture serves as a mechanism driving adaptive evolution in humans (13,24), we used iSAFE (20) v1.0.4 to provide evidence of a selective sweep embedded in the admixture mapping region and to pinpoint the most likely favored variant. iSAFE exploits the evolutionary contributions hidden in the flanking regions surrounding the region under selection to provide a ranking of variants (iSAFE-score) based on their contribution to the overall signal of selection. For the analysis, we used 1KGP data from unrelated subjects from PEL population (n = 77) and from a random selection of 10% Yoruba individuals drawn from the reference (n = 91), which represents a non-target or outgroup population. iSAFE was executed enabling the IgnoreGaps flag and the default MaxFreq value (0.95). Ancestral fasta sequences for Homo sapiens (GRCh37) were downloaded from ENSEMBL release 75 (http://ftp.ensembl.org/pub/release-75/fasta/ancestral_alleles/).
In silico functional analysis
In silico functional analyses were performed to assess the biological consequences of the leading associated variants in the fine mapping and of those variants prioritized in the selective sweep analysis. The Open Targets Genetics portal (https://genetics.opentargets.org/) was used for functional annotation, to assess trait associations based on PheWAS and to retrieve the evidence of eQTLs in relevant tissues based on eQTLGen database (https://www.eqtlgen.org/). Additionally, long-distance physical interactions and regulatory genomic regions were considered using Capture HiC Plotter (58) and HaploReg v4.1 (59).
Supplementary Material
Acknowledgements
We thank all the patients who participated in this study and the Medical team from Colombia for the sample recruitment. This research is part of the doctoral degree awarded to D.C.M., within the Biomedicine program from the University of Granada entitled ‘Bases moleculares de la enfermedad de Chagas: Integrando Genómica, Transcriptómica y Epigenómica’. Conflict of Interest statement. None declared.
Contributor Information
Desiré Casares-Marfil, Cell Biology and Immunology Department, Institute of Parasitology and Biomedicine López-Neyra, CSIC, Granada, P.C. 18016, Spain.
Beatriz Guillen-Guio, Research Unit, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, P.C. 38010, Spain.
Jose M Lorenzo-Salazar, Genomics Division, Instituto Tecnológico y de Energías Renovables (ITER), Santa Cruz de Tenerife, P.C. 38600, Spain.
Héctor Rodríguez-Pérez, Research Unit, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, P.C. 38010, Spain.
Martin Kerick, Cell Biology and Immunology Department, Institute of Parasitology and Biomedicine López-Neyra, CSIC, Granada, P.C. 18016, Spain.
Mayra A Jaimes-Campos, Grupo de Inmunología y Epidemiología Molecular, Escuela de Microbiología, Universidad Industrial de Santander, Bucaramanga, P.C. 680006, Colombia.
Martha L Díaz, Grupo de Inmunología y Epidemiología Molecular, Escuela de Microbiología, Universidad Industrial de Santander, Bucaramanga, P.C. 680006, Colombia.
Elkyn Estupiñán, Cell Biology and Immunology Department, Institute of Parasitology and Biomedicine López-Neyra, CSIC, Granada, P.C. 18016, Spain; Grupo de Inmunología y Epidemiología Molecular, Escuela de Microbiología, Universidad Industrial de Santander, Bucaramanga, P.C. 680006, Colombia.
Luis E Echeverría, Heart Failure and Heart Transplant Clinic, Fundación Cardiovascular de Colombia, Floridablanca, P.C. 681004, Colombia.
Clara I González, Grupo de Inmunología y Epidemiología Molecular, Escuela de Microbiología, Universidad Industrial de Santander, Bucaramanga, P.C. 680006, Colombia.
Javier Martín, Cell Biology and Immunology Department, Institute of Parasitology and Biomedicine López-Neyra, CSIC, Granada, P.C. 18016, Spain.
Carlos Flores, Research Unit, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, P.C. 38010, Spain; Genomics Division, Instituto Tecnológico y de Energías Renovables (ITER), Santa Cruz de Tenerife, P.C. 38600, Spain; CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, P.C. 28029, Spain.
Marialbert Acosta-Herrera, Cell Biology and Immunology Department, Institute of Parasitology and Biomedicine López-Neyra, CSIC, Granada, P.C. 18016, Spain.
Funding
Red Iberoamericana de medicina genómica en enfermedad de Chagas (217RT0524)—Programa Iberoamericano de Ciencia y Tecnología para el Desarrollo; Ministerio de Ciencia e Innovación (RTC-2017-6471-1; AEI/FEDER, UE); Instituto de Salud Carlos III (FI18/00230); European Regional Development Funds ‘A way of making Europe’ from the European Union; Cabildo Insular de Tenerife (CGIEU0000219140); agreement with Instituto Tecnológico y de Energías Renovables to strengthen scientific and technological education, training, research, development and innovation in Genomics, Personalized Medicine and Biotechnology (OA17/008); and Ministerio de Ciencia e Innovación. Juan de la Cierva fellowship (IJC2018-035131-I to M.A.H.).
Author Contributions
Conceptualization: M.A.H., C.F. resources: M.A.J.C., M.L.D., E.E., L.E.E., C.I.G., J.M., C.F. Formal analysis: D.C.M., B.G.G., H.R.P., J.M.L.S., M.K., C.F., M.A.H. Software: H.R.P., J.M.L.S. Writing—original draft preparation: D.C.M., B.G.G., C.F., M.A.H. Review and editing: D.C.M., B.G.G., J.M.L.S., H.R.P., M.A.J.C., M.L.D., E.E., L.E.E., C.I.G., J.M., C.F., M.A.H.
Data Availability
All relevant data could be found in the Figshare repository DOI:10.6084/m9.figshare.c.5370824.
References
- 1. Bern, C. (2015) Chagas' disease. N. Engl. J. Med., 373, 456–466. [DOI] [PubMed] [Google Scholar]
- 2. Perez-Molina, J.A. and Molina, I. (2018) Chagas disease. Lancet, 391, 82–94. [DOI] [PubMed] [Google Scholar]
- 3. Acevedo, G.R., Girard, M.C. and Gomez, K.A. (2018) The unsolved jigsaw puzzle of the immune response in Chagas disease. Front. Immunol., 9, 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chevillard, C., Nunes, J.P.S., Frade, A.F., Almeida, R.R., Pandey, R.P., Nascimento, M.S., Kalil, J. and Cunha-Neto, E. (2018) Disease tolerance and pathogen resistance genes may underlie Trypanosoma cruzi persistence and differential progression to Chagas disease cardiomyopathy. Front. Immunol., 9, 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cunha-Neto, E. and Chevillard, C. (2014) Chagas disease cardiomyopathy: immunopathology and genetics. Mediat. Inflamm., 2014, 683230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Acosta-Herrera, M., Strauss, M., Casares-Marfil, D., Martin, J. and Network, C.G.C.Y.T.E.D. (2019) Genomic medicine in Chagas disease. Acta Trop., 197, 105062. [DOI] [PubMed] [Google Scholar]
- 7. Casares-Marfil, D., Strauss, M., Bosch-Nicolau, P., Lo Presti, M.S., Molina, I., Chevillard, C., Cunha-Neto, E., Sabino, E., Ribeiro, A.L., Gonzalez, C.I. et al. (2021) A genome-wide association study identifies novel susceptibility loci in chronic Chagas cardiomyopathy. Clin. Infect. Dis., ciab090. 10.1093/cid/ciab090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bomba, L., Walter, K. and Soranzo, N. (2017) The impact of rare and low-frequency genetic variants in common disease. Genome Biol., 18, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grinde, K.E., Brown, L.A., Reiner, A.P., Thornton, T.A. and Browning, S.R. (2019) Genome-wide significance thresholds for admixture mapping studies. Am. J. Hum. Genet., 104, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruiz-Linares, A., Adhikari, K., Acuna-Alonzo, V., Quinto-Sanchez, M., Jaramillo, C., Arias, W., Fuentes, M., Pizarro, M., Everardo, P., de Avila, F. et al. (2014) Admixture in Latin America: geographic structure, phenotypic diversity and self-perception of ancestry based on 7,342 individuals. PLoS Genet., 10, e1004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Norris, E.T., Wang, L., Conley, A.B., Rishishwar, L., Marino-Ramirez, L., Valderrama-Aguirre, A. and Jordan, I.K. (2018) Genetic ancestry, admixture and health determinants in Latin America. BMC Genomics, 19, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rishishwar, L., Conley, A.B., Wigington, C.H., Wang, L., Valderrama-Aguirre, A. and Jordan, I.K. (2015) Ancestry, admixture and fitness in Colombian genomes. Sci. Rep., 5, 12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Norris, E.T., Rishishwar, L., Chande, A.T., Conley, A.B., Ye, K., Valderrama-Aguirre, A. and Jordan, I.K. (2020) Admixture-enabled selection for rapid adaptive evolution in the Americas. Genome Biol., 21, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shriner, D. (2017) Overview of Admixture mapping. Curr. Protoc. Hum. Genet., 94, 1 23 21-21 23 28. [DOI] [PubMed] [Google Scholar]
- 15. Gignoux, C.R., Torgerson, D.G., Pino-Yanes, M., Uricchio, L.H., Galanter, J., Roth, L.A., Eng, C., Hu, D., Nguyen, E.A., Huntsman, S. et al. (2019) An admixture mapping meta-analysis implicates genetic variation at 18q21 with asthma susceptibility in Latinos. J. Allergy Clin. Immunol., 143, 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng, X., Sabino, E.C., Cunha-Neto, E., Ribeiro, A.L., Ianni, B., Mady, C., Busch, M.P., Seielstad, M. and REDSII Chagas Study Group from the NHLBI Retrovirus Epidemiology Donor Study-II Component International (2013) Genome wide association study (GWAS) of Chagas cardiomyopathy in Trypanosoma cruzi seropositive subjects. PLoS One, 8, e79629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lima-Costa, M.F., Macinko, J., Mambrini, J.V., Peixoto, S.V., Pereira, A.C., Tarazona-Santos, E. and Ribeiro, A.L. (2016) Genomic African and Native American ancestry and Chagas disease: the Bambui (Brazil) Epigen cohort study of aging. PLoS Negl. Trop. Dis., 10, e0004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Javierre, B.M., Burren, O.S., Wilder, S.P., Kreuzhuber, R., Hill, S.M., Sewitz, S., Cairns, J., Wingett, S.W., Varnai, C., Thiecke, M.J. et al. (2016) Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell, 167, 1369, e1319–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mifsud, B., Tavares-Cadete, F., Young, A.N., Sugar, R., Schoenfelder, S., Ferreira, L., Wingett, S.W., Andrews, S., Grey, W., Ewels, P.A. et al. (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet., 47, 598–606. [DOI] [PubMed] [Google Scholar]
- 20. Akbari, A., Vitti, J.J., Iranmehr, A., Bakhtiari, M., Sabeti, P.C., Mirarab, S. and Bafna, V. (2018) Identifying the favored mutation in a positive selective sweep. Nat. Methods, 15, 279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Westra, H.J., Peters, M.J., Esko, T., Yaghootkar, H., Schurmann, C., Kettunen, J., Christiansen, M.W., Fairfax, B.P., Schramm, K., Powell, J.E. et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet., 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aufderheide, A.C., Salo, W., Madden, M., Streitz, J., Buikstra, J., Guhl, F., Arriaza, B., Renier, C., Wittmers, L.E., Jr., Fornaciari, G. et al. (2004) A 9,000-year record of Chagas' disease. Proc. Natl. Acad. Sci. U. S. A., 101, 2034–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lidani, K.C.F., Andrade, F.A., Bavia, L., Damasceno, F.S., Beltrame, M.H., Messias-Reason, I.J. and Sandri, T.L. (2019) Chagas disease: from discovery to a worldwide health problem. Front. Public Health, 7, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ebert, D. and Fields, P.D. (2020) Host-parasite co-evolution and its genomic signature. Nat. Rev. Genet., 21, 754–768. [DOI] [PubMed] [Google Scholar]
- 25. Flores-Ferrer, A., Marcou, O., Waleckx, E., Dumonteil, E. and Gourbiere, S. (2018) Evolutionary ecology of Chagas disease; what do we know and what do we need? Evol. Appl., 11, 470–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nedelec, Y., Sanz, J., Baharian, G., Szpiech, Z.A., Pacis, A., Dumaine, A., Grenier, J.C., Freiman, A., Sams, A.J., Hebert, S. et al. (2016) Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell, 167, 657, e621–669. [DOI] [PubMed] [Google Scholar]
- 27. Seldin, M.F., Pasaniuc, B. and Price, A.L. (2011) New approaches to disease mapping in admixed populations. Nat. Rev. Genet., 12, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosenberg, H.F., Dyer, K.D. and Foster, P.S. (2013) Eosinophils: changing perspectives in health and disease. Nat. Rev. Immunol., 13, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sousa-Rocha, D., Thomaz-Tobias, M., Diniz, L.F., Souza, P.S., Pinge-Filho, P. and Toledo, K.A. (2015) Trypanosoma cruzi and its soluble antigens induce NET release by stimulating toll-like receptors. PLoS One, 10, e0139569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen, M.H., Raffield, L.M., Mousas, A., Sakaue, S., Huffman, J.E., Moscati, A., Trivedi, B., Jiang, T., Akbari, P., Vuckovic, D. et al. (2020) Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell, 182, 1198, e1114–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ozato, K., Shin, D.M., Chang, T.H. and Morse, H.C., 3rd. (2008) TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol., 8, 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang, J., Villar, V.A.M., Rozyyev, S., Jose, P.A. and Zeng, C. (2019) The emerging role of sorting nexins in cardiovascular diseases. Clin. Sci. (Lond.), 133, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carthagena, L., Bergamaschi, A., Luna, J.M., David, A., Uchil, P.D., Margottin-Goguet, F., Mothes, W., Hazan, U., Transy, C., Pancino, G. et al. (2009) Human TRIM gene expression in response to interferons. PLoS One, 4, e4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leon Rodriguez, D.A., Carmona, F.D., Echeverria, L.E., Gonzalez, C.I. and Martin, J. (2016) IL18 gene variants influence the susceptibility to Chagas disease. PLoS Negl. Trop. Dis., 10, e0004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strauss, M., Acosta-Herrera, M., Alcaraz, A., Casares-Marfil, D., Bosch-Nicolau, P., Lo Presti, M.S., Molina, I., Gonzalez, C.I., Chagas Genetics, C.N. and Martin, J. (2019) Association of IL18 genetic polymorphisms with Chagas disease in Latin American populations. PLoS Negl. Trop. Dis., 13, e0007859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Layrisse, Z., Fernandez, M.T., Montagnani, S., Matos, M., Balbas, O., Herrera, F., Colorado, I.A., Catalioti, F. and Acquatella, H. (2000) HLA-C(*)03 is a risk factor for cardiomyopathy in Chagas disease. Hum. Immunol., 61, 925–929. [DOI] [PubMed] [Google Scholar]
- 37. Dias, F.C., Mendes-Junior, C.T., Silva, M.C., Tristao, F.S., Dellalibera-Joviliano, R., Moreau, P., Soares, E.G., Menezes, J.G., Schmidt, A., Dantas, R.O. et al. (2015) Human leucocyte antigen-G (HLA-G) and its murine functional homolog Qa2 in the Trypanosoma cruzi infection. Mediat. Inflamm., 2015, 595829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dias, F.C., Castelli, E.C., Collares, C.V., Moreau, P. and Donadi, E.A. (2015) The role of HLA-G molecule and HLA-G gene polymorphisms in tumors, viral hepatitis, and parasitic diseases. Front. Immunol., 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song, H., Liu, B., Huai, W., Yu, Z., Wang, W., Zhao, J., Han, L., Jiang, G., Zhang, L., Gao, C. et al. (2016) The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun., 7, 13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carvalho, C.M., Silverio, J.C., da Silva, A.A., Pereira, I.R., Coelho, J.M., Britto, C.C., Moreira, O.C., Marchevsky, R.S., Xavier, S.S., Gazzinelli, R.T. et al. (2012) Inducible nitric oxide synthase in heart tissue and nitric oxide in serum of Trypanosoma cruzi-infected rhesus monkeys: association with heart injury. PLoS Negl. Trop. Dis., 6, e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paroli, A.F., Gonzalez, P.V., Diaz-Lujan, C., Onofrio, L.I., Arocena, A., Cano, R.C., Carrera-Silva, E.A. and Gea, S. (2018) NLRP3 Inflammasome and Caspase-1/11 pathway orchestrate different outcomes in the host protection against Trypanosoma cruzi acute infection. Front. Immunol., 9, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McNamara, M.E., Rossi, V., Slater, T.S., Rogers, C.S., Ducrest, A.L., Dubey, S. and Roulin, A. (2021) Decoding the evolution of melanin in vertebrates. Trends Ecol. Evol., 36, 430–443. [DOI] [PubMed] [Google Scholar]
- 43. Burger, J., Link, V., Blocher, J., Schulz, A., Sell, C., Pochon, Z., Diekmann, Y., Zegarac, A., Hofmanova, Z., Winkelbach, L. et al. (2020) Low prevalence of lactase persistence in bronze age Europe indicates ongoing strong selection over the last 3,000 years. Curr. Biol.: CB, 30, 4307, e4313–4315. [DOI] [PubMed] [Google Scholar]
- 44. Bigham, A.W. (2016) Genetics of human origin and evolution: high-altitude adaptations. Curr. Opin. Genet. Dev., 41, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olivera, M.J., Fory, J.A., Porras, J.F. and Buitrago, G. (2019) Prevalence of Chagas disease in Colombia: a systematic review and meta-analysis. PLoS One, 14, e0210156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leon Rodriguez, D.A., Acosta-Herrera, M., Carmona, F.D., Dolade, N., Vargas, S., Echeverria, L.E., Gonzalez, C.I. and Martin, J. (2018) Comprehensive analysis of three TYK2 gene variants in the susceptibility to Chagas disease infection and cardiomyopathy. PLoS One, 13, e0190591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang, C.C., Chow, C.C., Tellier, L.C., Vattikuti, S., Purcell, S.M. and Lee, J.J. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Genomes Project, C., Abecasis, G.R., Auton, A., Brooks, L.D., DePristo, M.A., Durbin, R.M., Handsaker, R.E., Kang, H.M., Marth, G.T. and McVean, G.A. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ongaro, L., Scliar, M.O., Flores, R., Raveane, A., Marnetto, D., Sarno, S., Gnecchi-Ruscone, G.A., Alarcon-Riquelme, M.E., Patin, E., Wangkumhang, P. et al. (2019) The genomic impact of European colonization of the Americas. Curr. Biol.: CB, 29, 3974, e3974–e3986. [DOI] [PubMed] [Google Scholar]
- 50. Mathias, R.A., Taub, M.A., Gignoux, C.R., Fu, W., Musharoff, S., O'Connor, T.D., Vergara, C., Torgerson, D.G., Pino-Yanes, M., Shringarpure, S.S. et al. (2016) A continuum of admixture in the western hemisphere revealed by the African diaspora genome. Nat. Commun., 7, 12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alexander, D.H., Novembre, J. and Lange, K. (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res., 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guan, Y. (2014) Detecting structure of haplotypes and local ancestry. Genetics, 196, 625–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bryc, K., Velez, C., Karafet, T., Moreno-Estrada, A., Reynolds, A., Auton, A., Hammer, M., Bustamante, C.D. and Ostrer, H. (2010) Colloquium paper: genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc. Natl. Acad. Sci. U. S. A., 107(Suppl 2), 8954–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kang, H.M., S.J., Service S, S.K, Zaitlen, N.A., Kong, S.Y., Freimer, N.B., Sabatti, C. and Eskin, E. (2010) Variance component model to account for sample structure in genome-wide association studies. Nat. Genet., 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Han, B. and Eskin, E. (2011) Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet., 88, 586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li, M.X., Yeung, J.M., Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet., 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Skol, A.D., Scott, L.J., Abecasis, G.R. and Boehnke, M. (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet., 38, 209–213. [DOI] [PubMed] [Google Scholar]
- 58. Schofield, E.C., Carver, T., Achuthan, P., Freire-Pritchett, P., Spivakov, M., Todd, J.A. and Burren, O.S. (2016) CHiCP: a web-based tool for the integrative and interactive visualization of promoter capture Hi-C datasets. Bioinformatics, 32, 2511–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ward, L.D. and Kellis, M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data could be found in the Figshare repository DOI:10.6084/m9.figshare.c.5370824.