

Abstract
The rare, fatal neurodegenerative disorder Niemann-Pick disease type C1 (NPC1) arises from lysosomal accumulation of unesterified cholesterol and glycosphingolipids. These subcellular pathologies lead to phenotypes of hepatosplenomegaly, neurological degeneration and premature death. The timing and severity of NPC1 clinical presentation is extremely heterogeneous. This study analyzed RNA-Seq data from 42 NPC1 patient-derived, primary fibroblast cell lines to determine transcriptional changes induced by treatment with 2-hydroxypropyl-β-cyclodextrin (HPβCD), a compound currently under investigation in clinical trials. A total of 485 HPβCD-responsive genes were identified. Pathway enrichment analysis of these genes showed significant involvement in cholesterol and lipid biosynthesis. Furthermore, immunohistochemistry of the cerebellum as well as measurements of plasma from Npc1m1N null mice treated with HPβCD and adeno-associated virus gene therapy suggests that one of the identified genes, GPNMB, may serve as a useful biomarker of treatment response in NPC1 disease. Overall, this large NPC1 patient-derived dataset provides a comprehensive foundation for understanding the genomic response to HPβCD treatment.
Introduction
Niemann-Pick disease, type C (NPC) is an autosomal recessive lysosomal storage disorder (LSD) with an incidence between 1 in 120 000 and 150 000 live births (1). The majority of causative mutations (95%) have been identified in the lysosomal transmembrane protein NPC1 (NPC1 disease, OMIM #257220), whereas a much smaller proportion (5%) have been identified in the smaller soluble protein NPC2 [NPC2 disease, OMIM #607625 (1–3)]. Functional impairment of either NPC1 or NPC2 disrupts lysosomal cholesterol transport, resulting in abnormal accumulation of cholesterol and glycosphingolipids within late endosomal/lysosomal (LE/L) structures (4–7).
NPC1 disease has an extremely heterogenous clinical presentation including a variable age of onset that ranges from the pre/perinatal period to adulthood (8). The earliest pre/perinatal onset is usually associated with hepatomegaly or splenomegaly, often progressing to fatal liver disease (9,10) or respiratory complications (11,12). Later onset NPC1 exhibits more insidious neurodegenerative features, including cerebellar-related ataxia and psychiatric manifestations such as schizophrenia-like symptoms or depression (13).
Several investigational therapies for NPC1 are currently being studied (14). Although the glucosyl ceramide synthase inhibitor Miglustat (Zavesca) is approved for use by the European Medical Association and has been correlated with reduced glycosphingolipid levels, stabilized neurological phenotypes and reduced mortality risk in NPC1 (15–17), it has not been approved by the United States Food and Drug Administration (FDA) for the treatment of NPC disease. The heat-shock protein co-inducer arimoclomol has also shown promise in murine studies (18), and is currently part of an NPC clinical trial (NCT02612129, ClinicalTrials.gov). In addition, advances in genome editing as well as improvements in gene therapy techniques hold great promise for the development of effective treatment avenues for NPC1 and other lysosomal storage disorders (19). Multiple studies in the null mouse model, Npc1m1N [also known as Npc1nih (20)], have demonstrated that vectors derived from adeno-associated virus 9 (AAV9) can deliver NPC1 successfully to both the liver and brain, resulting in delayed disease progression and improved lifespan (21–23). However, additional studies are required before gene therapy can safely progress to clinical trials in NPC1 patients. Although these potential treatments represent significant advances in the field, NPC1 remains a fatal and incurable disease, and novel diagnostics and treatments are urgently needed to improve patient care.
Preclinical studies using the Npc1m1N mouse model demonstrated that treatment with the cyclic oligosaccharide derivative 2-hydroxypropyl-β-cyclodextrin (HPβCD) significantly delayed disease progression and extended lifespan (24–29). These promising results have recently translated into the completion of a phase 1-2a clinical trial (NCT01747135, ClinicalTrials.gov) demonstrating intrathecal treatment with HPβCD slowed NPC1 disease progression (30), and have led to a phase 2b-3 clinical trial (NCT02534844, ClinicalTrials.gov). Although HPβCD has been used for many years as an excipient to facilitate drug delivery, the cellular pathways through which HPβCD functions to alleviate NPC1 phenotypes are still not well understood. Several potential models have been suggested to account for the rapid clearance of accumulated cholesterol from treated NPC1 cells (29), including mobilization of cholesterol to the ER, restoration of autophagic flux, exocytosis/endo-lysosomal secretion of lysosomal material and exchange with lipoprotein extracellular acceptors (31–37). In addition, HPβCD treatment has been shown to prevent and reduce artherosclerosis (38,39) as well as provide neuroprotective effects in animal models of neurodegenerative disorders such as Alzheimer disease (40) and Parkinson disease (41). As the usage of HPβCD continues to increase in the clinical setting, further studies are needed to identify the underlying mechanism(s) of action of this drug. Additional work to elucidate the pathways affected by treatment with HPβCD will facilitate identification of novel drugs and biomarkers for NPC with the potential for broader impact among other neurodegenerative disorders.
The absence of diagnostic tools and informative biomarkers is a substantial limitation of current NPC1 treatment (42). Given the clinical heterogeneity of NPC1, an effective biomarker needs to recapitulate disease burden and also correlate with response to disease treatment. Furthermore, a biomarker that can be measured non-invasively with broadly accessible techniques is preferred to minimize patient discomfort and maximize utility across various clinical settings. Although no known biomarkers of NPC1 consistently exhibit all of these characteristics, genomic and lipidomic approaches have identified several promising candidates, many of which are currently employed for patient screening and diagnosis. These include N-palmitoyl-O-phosphocholineserine [previously known as lyso-sphingomyelin-509; (43–46)], cholestane-3β,5α,6β-triol (47–51), 7-ketocholesterol (47–50,52), 24(S)-hydroxycholesterol (30,33) and the bile acid 3β,5α,6β-trihydroxycholanoyl-glycine (53,54). Additional studies have highlighted proteins as potential biomarkers, including transmembrane glycoprotein NMB (encoded by GPNMB, and referred to as GPNMB hereafter), fatty acid binding protein 3, calbindin D, lysozyme, galectin-3 and cathepsins (47,48,55–60). GPNMB encodes a widely expressed type I transmembrane protein that has been implicated in numerous processes, including pigmentary glaucoma, immune response modulation, bone development and mineralization, protection from neuroinflammation, acceleration of wound healing and obesity/obesity-related inflammation [reviewed in (61,62)]. GPNMB is transcriptionally activated by MITF, a member of the Mit/TFE family of transcription factors known to be associated with autophagy and lysosomal biogenesis (63,64), and GPNMB is also a downstream target of the lysosomal transcription factor TFEB (65).
We performed transcriptome analysis of primary fibroblasts derived from 42 NPC1 patients to gain a comprehensive view of genes and biological pathways affected by HPβCD treatment in a large NPC1 patient cohort. Untreated and HPβCD-treated cell lines from the same patient were paired for differential expression analysis to control for patient heterogeneity. Although patients exhibited variability in gene expression responses to treatment, 485 differentially expressed transcripts were identified across the full dataset, including GPNMB as well as many genes related to cholesterol and lipid pathways. Plasma levels of soluble GPNMB protein (sGPNMB) in Npc1m1N/m1N mice directly correlated with both disease progression and phenotype severity in the context of HPβCD treatment. GPNMB protein levels in the cerebellum of Npc1m1N/m1N mice mirrored those of sGPNMB in plasma, with HPβCD treatment lessening the elevated cerebellar levels present in Npc1m1N/m1N mice. In addition, lower plasma sGPNMB levels were identified in Npc1m1N/m1N mice treated with AAV9-mediated NPC1 gene therapy. This study provides both an extensive transcriptomic profile associated with HPβCD treatment in NPC1 patients as well as evidence suggesting plasma GPNMB levels may serve as a useful biomarker in the treatment of NPC1.
Results
Differential gene expression analysis of HPβCD-treated primary fibroblasts from NPC1 patients
Primary skin fibroblasts were collected by skin biopsy from 42 NPC1 patients as part of a natural history study of NPC1 disease at the National Institutes of Health (NCT00344331, Clinical Trials.gov). The fibroblast cells from each individual were treated with 300 μM HPβCD for 24 h, as previously described (32), whereas a second parallel culture from each patient remained untreated. Lysotracker red (Lysotracker), which selectively stains acidic compartments/lysosomes, was used to quantitate the lysosomal storage phenotype in both the treated and untreated cultures from each NPC1 patient (Fig. 1A). We previously demonstrated that methyl-β-cyclodextrin (MβCD) treatment resulted in visible alterations in Lysotracker staining of NPC1 patient fibroblasts (66). Similarly, the HPβCD-treated NPC1 cells showed a significant change in Lysotracker staining in comparison with untreated cells from the same patient (paired t-test P < 0.0001, Supplementary Material, Fig. S1), demonstrating that 24 h of HPβCD treatment in these NPC1 patient-derived cell lines was sufficient to induce changes in a lysosome/cellular phenotype associated with disease.
Figure 1 .
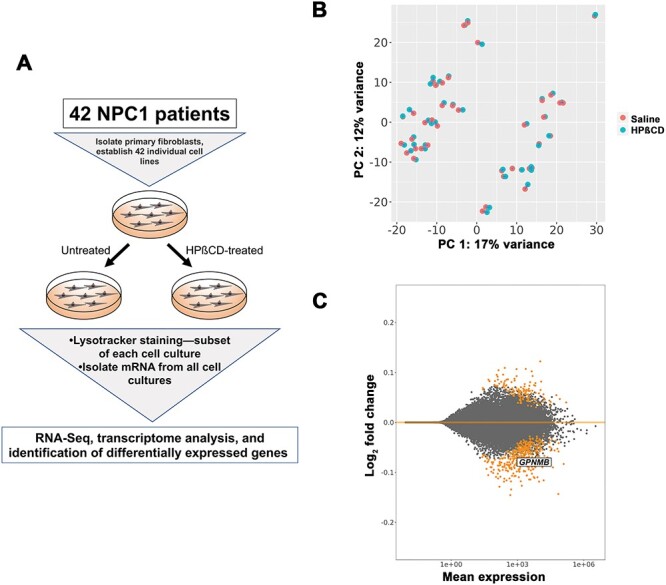
Analysis of differentially expressed genes in NPC1 patient-derived fibroblasts treated with HPβCD. (A) Experimental design. Individual cell lines were established from each patient, then split into cultures that were treated with saline or HPβCD. Lysotracker staining was performed on a subset of cells from each culture. The mRNA from each culture was subjected to RNA-Seq and differential expression analysis, in which pairwise comparisons between saline- and HPβCD-treated samples from the same patient were used. (B) Principal component analysis (PCA) plot showing that the differences between sex (PC 1) are much greater than the difference between saline-treated (orange) and HPβCD-treated (teal) cells. The blue lines connect data points from the same patient. (C) MA plot summarizing results of the differential expression contrast using ‘cell line’ as a blocking factor to control for the patterns observed in (B). Significantly different genes with FDR < 0.1 are shown in orange. Magnitude of the effect (log2 fold change) is on the y-axis, and the normalized read count (averaged across all replicates) is on the x-axis.
RNA-Seq data from all untreated and HPβCD-treated fibroblasts were analyzed to identify differentially expressed genes (DEGs) that were shared across the entire patient dataset (see Materials and methods). Heterogeneity among these individuals is highlighted in a principal components analysis (PCA) plot (Fig. 1B) as well as a clustered heatmap (Supplementary Material, Fig. S2), where the tight clustering of paired HPβCD-treated and untreated samples from each patient indicated that patient origin contributes significantly to the observed variation in gene expression. The separation into two distinct groups along the X-axis by Principal Component 1 was not related to HPβCD treatment but instead attributable to sex-specific differential gene expression (Supplementary Material, Fig. S3), which has been observed in other studies (67–69). Collectively, interpatient variation and sex differences account for larger variability in the dataset than HPβCD treatment. To control for this variability, ‘cell line’ was used as a blocking factor when assessing differential expression. This blocking method allowed the statistical analyses to focus on the specific effect due to HPβCD. Overall, HPβCD induced moderate, statistically significant changes in expression levels of 485 genes (Supplementary Material, Table S1), as illustrated by an MA plot of the log2 fold change in untreated vs. HPβCD treated cells plotted against base mean read counts (Fig. 1C). In addition, HPβCD treatment was associated with a heterogenous response, as the difference in log2 fold changes in gene expression levels between untreated and HPβCD-treated cells varied across our patient cohort (Supplementary Material, Fig. S4).
The majority of the 485 DEGs were downregulated in response to HPβCD treatment (74%, 359 out of 485, Supplementary Material, Table S1). Functional enrichment analysis of the 485 DEGs revealed that the top five canonical pathways were related to cholesterol biosynthesis and the mevalonate pathway, and the top five molecular and cellular functions were associated with cell death and survival, lipid metabolism, small molecule biochemistry, vitamin and mineral metabolism and cellular development [Ingenuity Pathway Analysis (IPA), www.ingenuity.com, Table 1]. The predicted upstream regulators included well-known lipid synthesis regulators, including SREBF1-regulated pathways which were downregulated in the dataset (Supplementary Material, Table S2). These pathways and predicted regulators are consistent with previous studies suggesting that HPβCD treatment of NPC1-deficient cells at lower, therapeutic-level concentrations resulted in mobilization of cholesterol to the ER and downregulation of SREBF1 downstream targets (29,70,71).
Table 1.
Top canonical pathways and molecular and cellular functions from IPA of differentially expressed genes in HPβCD-treated NPC1 fibroblasts
| Top canonical pathways | P-value | Overlap |
|---|---|---|
| Superpathway of cholesterol biosynthesis | 8.61E−17 | 48.3% (14/29) |
| Cholesterol biosynthesis I | 2.97E−11 | 61.5% (8/13) |
| Cholesterol biosynthesis II (via 24,25-dihydrolanosterol) | 2.97E−11 | 61.5% (8/13) |
| Cholesterol biosynthesis III (via desmosterol) | 2.97E−11 | 61.5% (8/13) |
| Superpathway of geranylgeranyldiphosphate biosynthesis I (via mevalonate) | 9.70E−07 | 33.3% (6/18) |
| Top molecular and cellular functions | P-value range | Number of molecules |
| Cell death and survival | 1.08E−04 to 1.94E−18 | 201 |
| Lipid metabolism | 1.59E−04 to 1.20E−13 | 92 |
| Small molecule biochemistry | 1.59E−04 to 1.20E−13 | 92 |
| Vitamin and mineral metabolism | 1.59E−04 to 1.20E−13 | 41 |
| Cellular development | 1.17E−04 to 1.07E−12 | 166 |
Notably, the cholesterol pathway genes SREBF2, HMGCS1, HMGCR and LDLR were downregulated. These results are consistent with reports from previous studies analyzing HPβCD-treated tissues from Npc1m1N/m1N mice (26,72,73), and reflect alterations in cholesterol sensing and synthesis pathways as a result of cholesterol mobilization. Comparison of the 485 DEGs with a previously curated list of 435 human lysosomal genes (74) along with PubMed literature searches found that 37 DEGs encode lysosomal proteins (Supplementary Material, Table S1). Since abnormal autophagy has been shown in NPC1 mutant cells (75–79), the DEGs were also compared with 524 known autophagy-related genes (http://autophagy.lu/index.html). This comparison along with PubMed searches identified 35 autophagy-related genes (Supplementary Material, Table S1). In addition, the 485 DEG list was examined for previously identified downstream targets of TFEB, the master transcription factor known to regulate lysosomal function as well as many aspects of autophagy (65,80,81). Forty-eight TFEB target genes were present in the 485 DEGs, suggesting TFEB-regulated pathways were altered in response to HPβCD treatment (Supplementary Material, Table S1).
Overall, the small number of DEGs suggests that early HPβCD treatment does not have a widespread effect on gene expression, but instead exerts a targeted impact on relevant pathways. Indeed, 218 of the 485 DEGs (45%) had previously published data linking them to lysosomes/endosomes, cholesterol synthesis or signaling pathways, lipid signaling/processing or autophagy (Supplementary Material, Table S1). The perturbation of lysosomal pathways is also highlighted by the presence of 14 genes associated with lysosomal storage disorders within this gene list (Supplementary Material, Table S1). In addition, the lysosomal/mTORC1 regulator FLCN and its interacting partners FNIP1 and FNIP2 all show downregulated expression following HPβCD treatment (82). Many of the remaining genes are indirectly implicated in lysosomal and lipid-related pathways, including 17 genes related to obesity/insulin resistance [such as LPIN1, LPIN2 and ENPP1 (83,84)] as well as PCSK9, which has been suggested as a central regulator of LDLR and may have broad roles in lipid metabolism and cardiovascular health (85,86). Overall, this DEG list representing cell lines from over 40 NPC1 individuals provides the largest transcriptome analysis to date of NPC1 patients, presenting a broad picture of the many lysosomal and lipid/cholesterol pathways that are affected by HPβCD treatment of NPC1 cells.
Decreased sGPNMB levels are associated with HPβCD treatment and AAV gene therapy
GPNMB was one of the DEGs that was downregulated in response to HPβCD treatment (Fig. 1C). GPNMB was previously identified as a potential biomarker for NPC1 disease (59,87) as well as the lysosomal storage disorder Gaucher disease (88–90). In contrast, none of the other previously published biomarker candidates [fatty acid binding protein 3, calbindin D, lysozyme, galectin-3 and cathepsins (47,48,55–60)] showed differential expression in response to HPβCD treatment. GPNMB was also identified as a highly relevant biomarker by IPA analysis (Supplementary Material, Table S3), and a well-characterized GPNMB ELISA assay was available (see Materials and methods). In addition, differential mRNA expression of GPNMB was present in NPC1 patient fibroblasts relative to controls (91–93), and multiple studies found differential expression of Gpnmb mRNA and GPNMB protein in tissues of Npc1m1N/m1N mice in comparison to Npc1+/+ control mice (57,58,87,94,95). Therefore, we chose to further characterize GPNMB expression levels in Npc1m1N/m1N mice undergoing potential therapeutic interventions.
To determine if GPNMB protein levels correlated with the presentation and progression of NPC1 disease phenotypes in mutant mice, plasma levels of sGPNMB protein were quantified by ELISA in Npc1m1N/m1N mice over the time course of disease. Npc1m1N/m1N mutant mice and Npc1+/+ controls received serial injections of either saline or HPβCD from postnatal day (P7) through 9 weeks of age (which is the natural end-stage of disease), and plasma was serially collected from weeks 3–9 to measure sGPNMB levels (Fig. 2A). HPβCD treatment was associated with strikingly decreased lipid accumulation in the liver of Npc1m1N/m1N mice (9-week old) compared with age-matched, saline-injected Npc1m1N/m1N mice, as demonstrated by reduced CD68-positive foamy macrophages with increased lipid storage in the liver (Fig. 2B, Supplementary Material, Fig. S5). This is consistent with the expected phenotypic improvements from HPβCD treatment (26,28,72). Interestingly, immunohistochemistry detected notable increases in GPNMB protein in the cerebellum of 9-week-old saline-injected Npc1m1N/m1N mice compared with Npc1+/+ (Fig. 2C, left two panels). HPβCD treatment of Npc1m1N/m1N mice greatly reduced these high GPNMB levels to levels approaching those seen in Npc1+/+ mice (Fig. 2C). Furthermore, increased sGPNMB levels were present in saline-treated Npc1m1N/m1N mice from weeks 7 to 9 (Fig. 2D, Supplementary Material, Fig. S6). HPβCD-treated Npc1m1N/m1N mice did not show increased sGPNMB, and instead maintained consistent sGPNMB levels from weeks 3 to 9 that were not significantly different from Npc1+/+ mice (Fig. 2D and Supplementary Material, Fig. S6). These results show that sGPNMB acts as a robust biomarker of disease progression in this animal model.
Figure 2 .
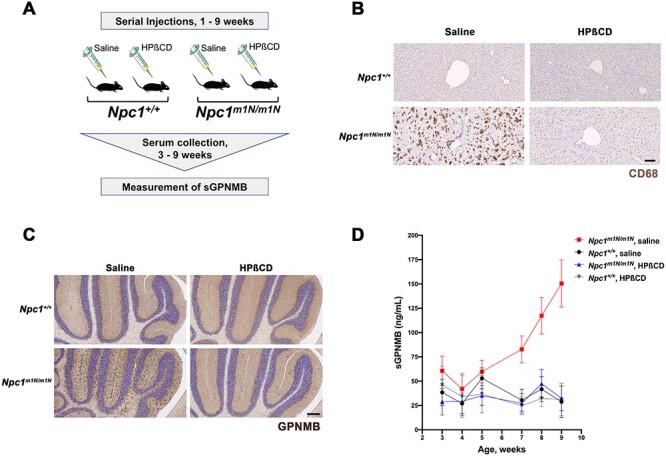
GPNMB levels are increased in Npc1m1N/m1N mutant mice and reduced by HPβCD treatment. (A) Experimental design. Mice received serial injections of HPβCD between 1 and 9 weeks of age, and serial plasma collection was performed for sGPNMB measurement by ELISA. (B) CD68 immunohistochemistry (brown staining) of liver sections demonstrated that the notable increase in foam cells characteristic of Npc1m1N/m1N mutant mice (lower left panel) is greatly reduced by HPβCD treatment (lower right panel). Saline-injected and HPβCD-treated Npc1+/+ controls are shown for comparison (upper left and right panels, respectively). Scale bar = 100 μm. (C) Immunostaining of midline sagittal sections of the cerebellum showed that elevated levels of GPNMB protein were present in 9-week-old saline-injected Npc1m1N/m1N mice (dark brown staining, lower left panel) when compared with the GPNMB levels in saline-injected Npc1+/+control mice (upper left panel). HPβCD-treated Npc1m1N/m1N mice (lower right panel) showed lower GPNMB expression in the cerebellum when compared with saline-injected Npc1m1N/m1N mice. HPβCD treatment of Npc1+/+ controls (upper right panel) did not affect GPNMB expression. Scale bar = 200 μm. (D) Levels of sGPNMB in plasma rose in untreated Npc1m1N/m1N mutant mice and were significantly different from Npc1+/+ controls. In contrast, sGPNMB levels remained low in HPβCD-treated mice and were not statistically different from those of Npc1+/+ mice. The mice shown in (D) were divided into two cohorts; see Supplementary Material, Fig. S6 for detailed, repeated measures data on individual mice, as well as a description of statistical analyses.
To further explore sGPNMB as a potential biomarker, plasma was collected at 9 weeks from Npc1m1N/m1N mutant mice treated with an AAV9 gene therapy vector, AAV9.EF1a(s).hNPC1 (Fig. 3A), previously described by our group to improve lifespan and other NPC1 phenotypes (21). Systemic treatment with AAV9.EF1a(s).hNPC1 reduced the foam cells and abnormal lipid storage present in the liver (Fig. 3B), consistent with gene therapy treatment improving disease-related phenotypes. Furthermore, these AAV9.EF1a(s).hNPC1-treated mice exhibited significantly lower plasma sGPNMB levels than saline-injected Npc1m1N/m1N control mice (Fig. 3C). Taken together with the results in HPβCD-treated Npc1m1N/m1N mice, this suggests that measurement of sGPNMB levels can be used to monitor the biological response to different potential therapeutics in a preclinical setting using this NPC1 animal model.
Figure 3 .
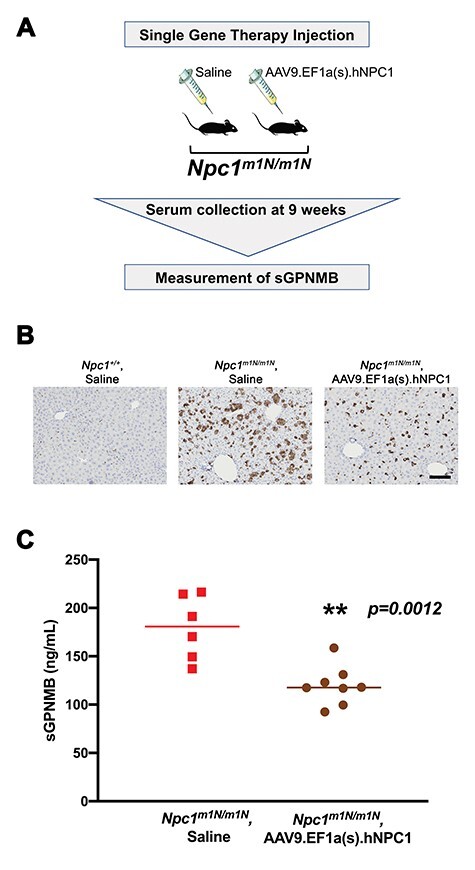
Reduced sGPNMB levels are present in Npc1m1N/m1N mutant mice that received AAV9.EF1a(s).hNPC1 gene therapy treatment. (A) Experimental design. Mice received a single injection of saline or AAV9.EF1a(s).hNPC1 at weaning, and plasma was collected at 9 weeks of age. (B) CD68 staining of liver sections demonstrated that the increased foam cells characteristic of Npc1m1N/m1N mutant mice (middle panel, saline-treated) are reduced by gene therapy treatment (right panel). Saline-treated Npc1+/+ controls are shown for comparison. Scale bar = 100 μm. (C) Mice receiving gene therapy exhibited significantly lower sGPNMB levels at 9 weeks of age in comparison to age-matched, saline-injected Npc1m1N/m1N mutant mice (P = 0.012, t-test).
Discussion
Many of the complex cellular pathways regulating lipid and cholesterol homeostasis throughout the cell are centrally focused on the lysosome (96–100). The lysosomal localization of NPC1 protein and its crucial role in cholesterol transport suggests that these signaling pathways may be affected in NPC1 mutant cells, and thus knowledge of the genomics underlying these pathways could be used to develop targeted treatments in NPC1 disease. However, genomic-based studies on NPC1 disease have faced many obstacles, including the limited patient numbers inherent in studying a rare disease as well as the high levels of clinical and genetic heterogeneity observed among NPC1 patients. To address these issues, this study generated comprehensive RNA-Seq data on paired, untreated and HPβCD-treated fibroblasts from 42 NPC1 patients with diverse NPC1 mutations (detailed in Supplementary Material, Table S4), making it the largest published transcriptome analysis of NPC1 patients to date. The larger size of this dataset compared with previous efforts has now allowed a broad, genome-wide view of shared, significant gene expression changes across multiple patients with varied NPC1 mutations and genetic backgrounds.
Our analysis suggests that initial HPβCD treatment induces moderate and specific changes in gene expression, as opposed to widespread alteration of the transcriptional landscape. These expression changes correlate with previous data showing that cholesterol biosynthesis pathways are affected in conjunction with SREBP2 downstream targets. A recent study examining Npc1m1N/m1N mutant mice 24 h after HPβCD treatment found that suppressed sterol synthesis was a primary response in liver and spleen (101), which correlates with our fibroblast data indicating downregulation of cholesterol synthesis pathway genes at 24 h post-treatment. In addition, our study found that out of 628 previously identified TFEB target genes (65,80,81), 48 were altered in response to HPβCD treatment, suggesting enrichment of these targets in our dataset. These data correlate with a recent study on NPC1 mutant fibroblasts which reported that TFEB upregulation and activation of TFEB-governed CLEAR network genes is associated with increased autophagy and lysosomal movement to the ER in response to cyclodextrin treatment (102).
Our results also revealed the involvement of lysosomal and autophagy genes in the HPβCD response of NPC1 mutant cells. Recent reports have shown the importance of autophagy in NPC1 disease biology, suggesting future studies of autophagy and NPC1 disease may be warranted (75–79). Overall, the 485 genes identified in this study expand our knowledge of cholesterol- and lipid-related pathways affected by HPβCD treatment in the context of NPC1 mutations, and also may highlight novel biomarkers to track disease progression and efficacy of interventions. This publicly available dataset will provide a robust foundation for future analyses on the genetics of NPC1 as well as the effects of HPβCD on NPC1 patients, including perturbations in basal lysosomal functions and autophagy pathways.
Of note, some RNA-Seq results reported here do not correlate with previous analyses. For example, the downregulation of the autophagy protein SQSTM1 in our dataset agreed with results from human neurons with reduced NPC1 expression generated from embryonic stem cells (77), but contrasts with data showing HPβCD treatment increased SQSTM1 levels in Npc1 mutant cells and also in Alzheimer disease model mice (78,102,103). These differences may reflect differing experimental conditions, including effective HPβCD concentrations and/or length of treatment, thus each study may measure very different phases of cellular response to HPβCD. The 24 h timepoint after 300 μM HPβCD treatment used in our study likely measures conditions where NPC1 mutant cells are still sensing excess unesterified cholesterol that has been released from lysosomes. Future studies examining a time course of the transcriptional response to HPβCD will be useful to resolve these questions and capture a clearer understanding of the cellular changes in NPC1 mutant cells in response to HPβCD.
NPC1 disease presentation is extremely heterogenous, with broad phenotypic variations across patients, and even in patients with the same NPC1 mutation (10,104–106). Our large transcriptome analysis revealed that this striking variability is also present at the mRNA expression level. These results could be explained by the presence of genetic variants elsewhere in the genome that are capable of modifying the NPC1 phenotype (107), including HPβCD treatment response. Of note, differences in treatment response have also been reported in the phase 1/2a clinical trial, as a subset of patients, ‘responders’, had a better response to HPβCD treatment compared with ‘non-responders’ (30). This variability emphasizes the need for a reliable biomarker that could consistently measure disease hallmarks in spite of such complexity.
Our study supports GPNMB as a robust NPC1 biomarker, both for disease progression and to measure response to therapeutic treatments in both peripheral and neuronal tissues. Our studies found that GPNMB expression is significantly altered in NPC1 fibroblasts in response to HPβCD treatment across a large panel of patients that are heterogeneous in both NPC1 mutation type and gene expression profiles, thus indicating that GPNMB has the potential to act as a biomarker across a highly variable patient cohort. The significant changes in sGPNMB levels identified in both HPβCD-treated as well as gene therapy-treated Npc1m1N/m1N mutant mice demonstrated that sGPNMB level alterations are not solely limited to HPβCD treatment but also appear to correlate with disease severity and therapeutic efficacy. Furthermore, overexpression of GPNMB protein in the cerebellum of Npc1m1N/m1N mutant mice was reduced with HPβCD treatment, indicating systemic HPβCD administration is able to exert changes in the brain. These results also suggest changes in gene expression of primary fibroblast cells could act as surrogates of disease severity and treatment response in plasma and the CNS. Importantly, these sGPNMB alterations are present in plasma and can be easily assayed by ELISA, therefore this molecule could be readily evaluated as a biomarker with minimal patient discomfort.
The molecular details of GPNMB function in the setting of NPC1 remain uncharacterized. Previously, GPNMB was reported as overexpressed in NPC1 in comparison to normal cells/tissues and suggested as a potential disease biomarker (59). Broader studies of expression variation in NPC1 correlate with these results, revealing that GPNMB mRNA or protein expression is altered by NPC1 mutations in 6 out of 8 studies from NPC1 patients and mouse models (57,58,91–95,108). GPNMB occurs as a transmembrane protein localized to endosomes/lysosomes and melanosomes, but it also can act as a secreted, soluble factor following cleavage and release of its extracellular portion (109–112). GPNMB is implicated in lysosomal function and autophagy, and it is a downstream target of TFEB, a master regulator of numerous lysosomal genes (65,111,113). Of note, GPNMB is overexpressed in another lysosomal storage disorder, Gaucher disease, and its expression level correlates with phenotype severity (88–90,114). In addition, GPNMB has been linked to several diseases and processes, including neuro-protective properties in Parkinson disease and Alzheimer disease (and related animal models), regulation of cell differentiation and modulation of inflammation (61,62,115–123).
The slowly progressing nature of many rare diseases such as NPC1 adds complexity to the already difficult process of therapeutic development. Emphasis in recent years has been placed on biomarker discovery and development. Biomarkers present opportunities to not only track disease progression and understand genotype–phenotype correlations, but also determine or monitor efficacy of therapeutic compounds. A validated biomarker can be used as a surrogate endpoint for clinical trials, which can be especially useful for diseases such as NPC1 where clinically significant decline occurs over many years. Herein, we show the utility of GPNMB as a treatment-related biomarker for both HPβCD and gene therapy in NPC1 murine studies. Our results along with corroborating work from multiple groups suggest continued development of GPNMB is warranted, including further studies to validate that both plasma- and CSF-derived GPNMB can act as a biomarker in NPC1 patients.
Materials and Methods
Cell culture, RNA isolation, Lysotracker staining and HPβCD treatment
Primary skin fibroblasts from NPC1 patients were obtained with consent as part of a natural history study of NPC1 disease at the National Institutes of Health (NCT0034433, ClinicalTrials.gov) approved by the Eunice Kennedy Shriver National Institute of Health Institutional Review Board. A diverse range of NPC1 mutations were present across the patient cohort (Supplementary Material, Table S4). Cell lines were cultured in DMEM (Invitrogen, Waltham, MA, USA, Catalog no. 11995-040) supplemented with 10% fetal bovine serum in a humidified incubator with 5% CO2 at 37°C. A parallel set of cells from each line was also treated with 300 μM of 2-hydroxypropyl-ß-cyclodextrin (HPßCD; Sigma-Aldrich, St. Louis, MO, USA, Catalog no. C0926) for 24 h. Prior to isolation of RNA for sequencing, a portion of the culture was stained with Lysotracker-red (Lysotracker, Invitrogen, catalog no. L7528).
Levels of Lysotracker were analyzed by fluorescence-activated cell sorter (FACS) analysis as previously published (66). This was done coincidently with cells being harvested for RNA isolation with TRIzol reagent (ThermoFisher, Waltham, MA, USA, catalog no. 15596018) and purified with Qiagen RNA Easy Mini Columns (Qiagen, Germantown, MD, USA, Catalog no. 74104). RNA quality was assessed using a Bioanalyzer (Angilent, Inc., Santa Clara, CA, USA).
RNA sequencing and analysis
RNA sequencing (RNA-Seq) was performed at the NIH Intramural Sequencing Center (NISC) using a HiSeq400 instrument (Illumina, San Diego, CA, USA). Poly-A selected stranded mRNA libraries were constructed from 0.51 μg total RNA using the Illumina TruSeq Stranded mRNA Sample Prep Kit, version 2 according to manufacturer’s instructions except where noted. The resulting cDNA was fragmented using a Covaris E210. Amplification was performed using 10 cycles to minimize the risk of over-amplification. Unique barcode adapters were applied to each library. Libraries were pooled for sequencing. The pooled libraries were sequenced on multiple lanes of a HiSeq 4000 to achieve a minimum of 42 million 76 base read pairs. The data were processed using RTA version 1.18.54 and CASAVA 1.8.2. Sequence data will be made available through dbGaP, study accession number phs002392.v1.p1.
Demultiplexed 76-bp paired-end reads were aligned to the GRCh38 human reference using HISAT2 v2.10 (124). Reads were then counted in genes with the featureCounts program of the subread package v1.6.4 using the GENCODE release 28 annotations (125). For the PCA and clustered heatmaps (Fig. 1B, and Supplementary Material, Figs S2, S3 and S4) we used counts normalized with the DESeq2 v1.22.1 variance stabilizing transform (126). These analyses indicated the majority of the cell lines were clustered into pre/post-treatment pairs. One sample pair (NPC2) behaved differently, with the pre-treatment sample clustering into its own top-level cluster, suggesting this sample was an outlier (Supplementary Material, Fig. S2), and thus it was removed from the pre-post treatment contrast. Differential expression was performed using raw counts provided to DESeq2. The primary contrast presented here used cell line as a blocking factor. That is, it used the model ‘~treatment + cell line’ and extracted the contrast only for the treatment effect. A gene was considered differentially expressed if the false discovery rate (FDR) was <0.1, as recommended by DESeq2 parameters (126). The MA plot shows the shrunken log2 fold change calculated by DESeq2 using the ‘normal’ method.
Pathway analysis was performed with Ingenuity pathway analysis software (Qiagen, Germantown, MD, USA) using the 485 DEG set. Literature searches were performed in PubMed for all 485 genes, using each gene name as a search term paired with ‘lysosome’, ‘autophagy’, ‘lipids’, ‘cholesterol’, ‘NPC’ or ‘cyclodextrin’.
Colony management and genotype identification
Colonies were maintained in a specific pathogen free AAALAC-approved facility by following the standard protocol of the Institutional Animal Care and Use Committee from the National Human Genome Research Institute (NHGRI). Mating between heterozygous BALB/cNctr-Npc1m1N/J mice (Jackson Laboratories, Bar Harbor, ME, USA, stock number: 003092) generated Npc1m1N/m1N mutant offspring. Mice were housed between two and five adult mice per cage (regardless of their genotype). DNA for genotype analysis (20) was extracted from tail biopsies at P10 and purified using a Gentra Puregene Mouse Tail Kit (Qiagen, Germantown, MD, USA).
Mouse HPβCD, AVV treatment and plasma extraction
Mice received serial subcutaneous injections of either saline or HPβCD starting at postnatal day (P7) and continued to receive injections every other day from P7 to P21 as previously described in (127). From P21 onward, injections were administered three times per week. Plasma was collected serially by retro-orbital bleeding, but collection alternated between two cohorts of mice in each treatment group (see Supplementary Material, Fig. S6), so that blood collection was performed on each mouse every other week and blood volume drawn based on body weight as per NIH ACUC guidelines.
AAV gene therapy [AAV9.EF1a(s).hNPC1] was performed as previously described (21) with a dose of 4.3 × 1012 gene copy (GC) per mouse. Plasma collection was performed at 9 weeks for each group.
Tissue histology and immunohistochemistry
Liver histology for both Hematoxylin and eosin stain (H&E) and anti-CD68 were performed as previously described (107). Immunohistochemistry for GPNMB expression in cerebellum was performed by Histoserv (Germantown, MD, USA) using mouse osteoactivin/GPNMB antibody following manufacturer’s instructions (catalog # AF2330, R&D systems, Minnesota USA) with biotin-conjugated anti-mouse IgG as a secondary antibody.
sGPBMB measurement and analysis
Soluble GPNMB levels (sGPNMB) were measured from plasma samples in triplicates (1:50 dilution) by ELISA (catalog # DY2330, R&D systems, Minneapolis, MN, USA) as described per manufacturer. The aspiration and washing steps were performed using an automatic microplate washer and dispenser (405 TS, BioTek, Winooski, VT, USA). The optical density was measured at 450 nm using a microplate reader (Epoch 2, BioTek, Winooski, VT, USA). Interpolated concentrations were reported in ng/ml.
Statistical analysis
All statistical analyses were performed using Prism software (GraphPad).
Members of the NISC comparative sequencing program
Beatrice B. Barnabas, Gerard G. Bouffard, Shelis Y. Brooks, Holly Coleman, Lyudmila Dekhtyar, Xiaobin Guan, Joel Han, Shi-ling Ho, Richelle Legaspi, Quino L. Maduro, Catherine A. Masiello, Jennifer C. McDowell, Casandra Montemayor, James C. Mullikin, Morgan Park, Nancy L. Riebow, Karen Schandler, Brian Schmidt, Christina Sison, Raymond Smith, Sirintorn Stantripop, James W. Thomas, Pamela J. Thomas, Meghana Vemulapalli and Alice C. Young.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank members of the Pavan lab for helpful input and discussions. F.M.P. is a Wellcome Trust Investigator in Science and a Royal Society Wolfson Research Merit Award holder. C.D.D. received support from the Hide & Seek Foundation and Dana’s Angels Research Trust (both part of Support Of Accelerated Research for Niemann-Pick C). This study was also supported by the Ara Parseghian Medical Research Fund and Niemann-Pick Canada.
Conflict of Interest statement. The authors declare no competing or financial interests.
Contributor Information
Jorge L Rodriguez-Gil, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA; Medical Scientist Training Program, University of Wisconsin-Madison, School of Medicine and Public Health, Madison, WI 53705, USA.
Laura L Baxter, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Dawn E Watkins-Chow, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Nicholas L Johnson, Bioinformatics and Scientific Programming Core, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA.
Cristin D Davidson, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Steven R Carlson, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Arturo A Incao, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
NISC Comparative Sequencing Program, National Institutes of Health Intramural Sequencing Center, Bethesda, MD 20892, USA.
Kerri L Wallom, Department of Pharmacology, University of Oxford, Oxford OX1 3QT, UK.
Nicole Y Farhat, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA.
Frances M Platt, Department of Pharmacology, University of Oxford, Oxford OX1 3QT, UK.
Ryan K Dale, Bioinformatics and Scientific Programming Core, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA.
Forbes D Porter, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA.
William J Pavan, Genomics, Development and Disease Section, Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Funding
This research was supported by the National Institutes of Health Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development [ZIA HD008989] and the National Human Genome Research Institute [1ZIAHG000068-15]. J.L.R.-G. is supported by an NHGRI Intramural Research Training Award, the NIH Oxford-Cambridge Scholars Program, and the Medical Scientist Training Program from the University of Wisconsin-Madison School of Medicine and Public Health, [3T32GM008692].
References
- 1. Vanier, M.T. and Millat, G. (2003) Niemann-Pick disease type C. Clin. Genet., 64, 269–281. [DOI] [PubMed] [Google Scholar]
- 2. Carstea, E.D., Morris, J.A., Coleman, K.G., Loftus, S.K., Zhang, D., Cummings, C., Gu, J., Rosenfeld, M.A., Pavan, W.J., Krizman, D.B. et al. (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science, 277, 228–231. [DOI] [PubMed] [Google Scholar]
- 3. Naureckiene, S., Sleat, D.E., Lackland, H., Fensom, A., Vanier, M.T., Wattiaux, R., Jadot, M. and Lobel, P. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science, 290, 2298–2301. [DOI] [PubMed] [Google Scholar]
- 4. Infante, R.E., Wang, M.L., Radhakrishnan, A., Kwon, H.J., Brown, M.S. and Goldstein, J.L. (2008) NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. PNAS, 105, 15287–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vanier, M.T. (2015) Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis., 38, 187–199. [DOI] [PubMed] [Google Scholar]
- 6. Cologna, S.M. and Rosenhouse-Dantsker, A. (2019) Insights into the molecular mechanisms of cholesterol binding to the NPC1 and NPC2 proteins. Adv. Exp. Med. Biol., 1135, 139–160. [DOI] [PubMed] [Google Scholar]
- 7. Pfeffer, S.R. (2019) NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem., 294, 1706–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vanier, M.T. (2013) Niemann-Pick diseases. Handb. Clin. Neurol., 113, 1717–1721. [DOI] [PubMed] [Google Scholar]
- 9. Colin, E., Barth, M., Boussion, F., Latour, P., Piguet-Lacroix, G., Guichet, A., Ziegler, A., Triau, S., Loisel, D., Sentilhes, L. et al. (2016) In utero diagnosis of Niemann-Pick type C in the absence of family history. JIMD Rep., 28, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vanier, M.T. (2010) Niemann-Pick disease type C. Orphanet. J. Rare. Dis., 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gumus, E., Haliloglu, G., Karhan, A.N., Demir, H., Gurakan, F., Topcu, M. and Yuce, A. (2017) Niemann-Pick disease type C in the newborn period: a single-center experience. Eur. J. Pediatr., 176, 1669–1676. [DOI] [PubMed] [Google Scholar]
- 12. Staretz-Chacham, O., Aviram, M., Morag, I., Goldbart, A. and Hershkovitz, E. (2018) Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatr., 177, 1609–1615. [DOI] [PubMed] [Google Scholar]
- 13. Geberhiwot, T., Moro, A., Dardis, A., Ramaswami, U., Sirrs, S., Marfa, M.P., Vanier, M.T., Walterfang, M., Bolton, S., Dawson, C. et al. (2018) Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet. J. Rare. Dis., 13, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pallottini, V. and Pfrieger, F.W. (2020) Understanding and treating Niemann-Pick type C disease: models matter. Int. J. Mol. Sci., 21, 8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stein, V.M., Crooks, A., Ding, W., Prociuk, M., O'Donnell, P., Bryan, C., Sikora, T., Dingemanse, J., Vanier, M.T., Walkley, S.U. et al. (2012) Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J. Neuropathol. Exp. Neurol., 71, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fecarotta, S., Romano, A., Casa Della, R., Del Giudice, E., Bruschini, D., Mansi, G., Bembi, B., Dardis, A., Fiumara, A., Di Rocco, M. et al. (2015) Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet. J. Rare. Dis., 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patterson, M.C., Garver, W.S., Giugliani, R., Imrie, J., Jahnova, H., Meaney, F.J., Nadjar, Y., Vanier, M.T., Moneuse, P., Morand, O., et al. (2020) Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: a large retrospective observational study. J. Inherit. Metab. Dis., 43, 1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kirkegaard, T., Gray, J., Priestman, D.A., Wallom, K.-L., Atkins, J., Olsen, O.D., Klein, A., Drndarski, S., Petersen, N.H.T., Ingemann, L. et al. (2016) Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med., 8, 355ra118–355ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Platt, F.M., d'Azzo, A., Davidson, B.L., Neufeld, E.F. and Tifft, C.J. (2018) Lysosomal storage diseases. Nat. Rev. Dis. Primers, 4, 27–25. [DOI] [PubMed] [Google Scholar]
- 20. Loftus, S.K., Morris, J.A., Carstea, E.D., Gu, J.Z., Cummings, C., Brown, A., Ellison, J., Ohno, K., Rosenfeld, M.A., Tagle, D.A. et al. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science, 277, 232–235. [DOI] [PubMed] [Google Scholar]
- 21. Chandler, R.J., Williams, I.M., Gibson, A.L., Davidson, C.D., Incao, A.A., Hubbard, B.T., Porter, F.D., Pavan, W.J. and Venditti, C.P. (2017) Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum. Mol. Genet., 26, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xie, C., Gong, X.-M., Luo, J., Li, B.-L. and Song, B.-L. (2017) AAV9-NPC1 significantly ameliorates Purkinje cell death and behavioral abnormalities in mouse NPC disease. J. Lipid Res., 58, 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hughes, M.P., Smith, D.A., Morris, L., Fletcher, C., Colaco, A., Huebecker, M., Tordo, J., Palomar, N., Massaro, G., Henckaerts, E. et al. (2018) AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann-Pick type C1 disease. Hum. Mol. Genet., 27, 3079–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Camargo, F., Erickson, R.P., Garver, W.S., Hossain, G.S., Carbone, P.N., Heidenreich, R.A. and Blanchard, J. (2001) Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci., 70, 131–142. [DOI] [PubMed] [Google Scholar]
- 25. Liu, B., Li, H., Repa, J.J., Turley, S.D. and Dietschy, J.M. (2008) Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res., 49, 663–669. [DOI] [PubMed] [Google Scholar]
- 26. Liu, B., Turley, S.D., Burns, D.K., Miller, A.M., Repa, J.J. and Dietschy, J.M. (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the NPC 1−/− mouse. PNAS, 106, 2377–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davidson, C.D., Ali, N.F., Micsenyi, M.C., Stephney, G., Renault, S., Dobrenis, K., Ory, D.S., Vanier, M.T. and Walkley, S.U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One, 4, e 6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramirez, C.M., Liu, B., Taylor, A.M., Repa, J.J., Burns, D.K., Weinberg, A.G., Turley, S.D. and Dietschy, J.M. (2010) Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr. Res., 68, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peake, K.B. and Vance, J.E. (2012) Normalization of cholesterol homeostasis by 2-hydroxypropyl-β-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem., 287, 9290–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ory, D.S., Ottinger, E.A., Farhat, N.Y., King, K.A., Jiang, X., Weissfeld, L., Berry-Kravis, E., Davidson, C.D., Bianconi, S., Keener, L.A. et al. (2017) Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Lancet, 390, 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen, F.W., Li, C. and Ioannou, Y.A. (2010) Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One, 5, e15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenbaum, A.I., Zhang, G., Warren, J.D. and Maxfield, F.R. (2010) Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. PNAS, 107, 5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tortelli, B., Fujiwara, H., Bagel, J.H., Zhang, J., Sidhu, R., Jiang, X., Yanjanin, N.M., Shankar, R.K., Carillo-Carasco, N., Heiss, J. et al. (2014) Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1). Hum. Mol. Genet., 23, 6022–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Demais, V., Barthélémy, A., Perraut, M., Ungerer, N., Keime, C., Reibel, S. and Pfrieger, F.W. (2016) Reversal of pathologic lipid accumulation in NPC1-deficient neurons by drug-promoted release of LAMP1-coated lamellar inclusions. J. Neurosci., 36, 8012–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dai, S., Dulcey, A.E., Hu, X., Wassif, C.A., Porter, F.D., Austin, C.P., Ory, D.S., Marugan, J. and Zheng, W. (2017) Methyl-β-cyclodextrin restores impaired autophagy flux in Niemann-Pick C1-deficient cells through activation of AMPK. Autophagy, 13, 1435–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vacca, F., Vossio, S., Mercier, V., Moreau, D., Johnson, S., Scott, C.C., Montoya, J.P., Moniatte, M. and Gruenberg, J. (2019) Cyclodextrin triggers MCOLN1-dependent endo-lysosome secretion in Niemann-Pick type C cells. J. Lipid Res., 60, 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feltes, M., Gale, S.E., Moores, S., Ory, D.S. and Schaffer, J.E. (2020) Monitoring the itinerary of lysosomal cholesterol in Niemann-Pick type C1-deficient cells after cyclodextrin treatment. J. Lipid Res., 61, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coisne, C., Tilloy, S., Monflier, E., Wils, D., Fenart, L. and Gosselet, F. (2016) Cyclodextrins as emerging therapeutic tools in the treatment of cholesterol-associated vascular and neurodegenerative diseases. Molecules, 21, 1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zimmer, S., Grebe, A., Bakke, S.S., Bode, N., Halvorsen, B., Ulas, T., Skjelland, M., De Nardo, D., Labzin, L.I., Kerksiek, A. et al. (2016) Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci. Transl. Med., 8, 333ra50–333ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yao, J., Ho, D., Calingasan, N.Y., Pipalia, N.H., Lin, M.T. and Beal, M.F. (2012) Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J. Exp. Med., 209, 2501–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kilpatrick, K., Zeng, Y., Hancock, T. and Segatori, L. (2015) Genetic and chemical activation of TFEB mediates clearance of aggregated α-synuclein. PLoS One, 10, e0120819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sitarska, D. and Ługowska, A. (2019) Laboratory diagnosis of the Niemann-Pick type C disease: an inherited neurodegenerative disorder of cholesterol metabolism. Metab. Brain Dis., 34, 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giese, A.-K., Mascher, H., Grittner, U., Eichler, S., Kramp, G., Lukas, J., Vruchte Te, D., Eisa Al, N., Cortina-Borja, M., Porter, F.D. et al. (2015) A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet. J. Rare. Dis., 10, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuchar, L., Sikora, J., Gulinello, M.E., Poupetova, H., Lugowska, A., Malinova, V., Jahnova, H., Asfaw, B. and Ledvinova, J. (2017) Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem., 525, 73–77. [DOI] [PubMed] [Google Scholar]
- 45. Pettazzoni, M., Froissart, R., Pagan, C., Vanier, M.T., Ruet, S., Latour, P., Guffon, N., Fouilhoux, A., Germain, D.P., Levade, T. et al. (2017) LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: a novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS One, 12, e0181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sidhu, R., Kell, P., Dietzen, D.J., Farhat, N.Y., Do, A.N.D., Porter, F.D., Berry-Kravis, E., Vite, C.H., Reunert, J., Marquardt, T. et al. (2020) Application of N-palmitoyl-O-phosphocholineserine for diagnosis and assessment of response to treatment in Niemann-Pick type C disease. Mol. Genet. Metab., 129, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Porter, F.D., Scherrer, D.E., Lanier, M.H., Langmade, S.J., Molugu, V., Gale, S.E., Olzeski, D., Sidhu, R., Dietzen, D.J., Fu, R. et al. (2010) Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med., 2, 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang, X., Sidhu, R., Porter, F.D., Yanjanin, N.M., Speak, A.O., Vruchte te, D.T., Platt, F.M., Fujiwara, H., Scherrer, D.E., Zhang, J. et al. (2011) A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res., 52, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boenzi, S., Deodato, F., Taurisano, R., Martinelli, D., Verrigni, D., Carrozzo, R., Bertini, E., Pastore, A., Dionisi-Vici, C. and Johnson, D.W. (2014) A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease. Clin. Chim. Acta, 437, 93–100. [DOI] [PubMed] [Google Scholar]
- 50. Romanello, M., Zampieri, S., Bortolotti, N., Deroma, L., Sechi, A., Fiumara, A., Parini, R., Borroni, B., Brancati, F., Bruni, A. et al. (2016) Comprehensive evaluation of plasma 7-Ketocholesterol and Cholestan-3β,5α,6β-Triol in an Italian cohort of patients affected by Niemann-Pick disease due to NPC1 and SMPD1 mutations. Clin. Chim. Acta, 455, 39–45. [DOI] [PubMed] [Google Scholar]
- 51. Kannenberg, F., Nofer, J.-R., Schulte, E., Reunert, J., Marquardt, T. and Fobker, M. (2017) Determination of serum cholestane-3β,5α,6β-triol by gas chromatography-mass spectrometry for identification of Niemann-Pick type C (NPC) disease. J. Steroid Biochem. Mol. Biol., 169, 54–60. [DOI] [PubMed] [Google Scholar]
- 52. Zhang, H., Wang, Y., Lin, N., Yang, R., Qiu, W., Han, L., Ye, J. and Gu, X. (2014) Diagnosis of Niemann-Pick disease type C with 7-ketocholesterol screening followed by NPC1/NPC2 gene mutation confirmation in Chinese patients. Orphanet. J. Rare. Dis., 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jiang, X., Sidhu, R., Mydock-McGrane, L., Hsu, F.-F., Covey, D.F., Scherrer, D.E., Earley, B., Gale, S.E., Farhat, N.Y., Porter, F.D. et al. (2016) Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci. Transl. Med., 8, 337ra63–337ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mazzacuva, F., Mills, P., Mills, K., Camuzeaux, S., Gissen, P., Nicoli, E.-R., Wassif, C., Vruchte, Te, D., Porter, F.D., Maekawa, M. et al. (2016) Identification of novel bile acids as biomarkers for the early diagnosis of Niemann-Pick C disease. FEBS Lett., 590, 1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alam, M.S., Getz, M., Yi, S., Kurkewich, J., Safeukui, I. and Haldar, K. (2014) Plasma signature of neurological disease in the monogenetic disorder Niemann pick type C. J. Biol. Chem., 12, 8051–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cologna, S.M., Jiang, X.-S., Backlund, P.S., Cluzeau, C.V.M., Dail, M.K., Yanjanin, N.M., Siebel, S., Toth, C.L., Jun, H.-S., Wassif, C.A. et al. (2012) Quantitative proteomic analysis of niemann-pick disease, type c1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS One, 7, e47845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alam, M.S., Getz, M., Safeukui, I., Yi, S., Tamez, P., Shin, J., Velázquez, P. and Haldar, K. (2012) Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS One, 7, e48273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cluzeau, C.V.M., Watkins-Chow, D.E., Fu, R., Borate, B., Yanjanin, N., Dail, M.K., Davidson, C.D., Walkley, S.U., Ory, D.S., Wassif, C.A. et al. (2012) Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet., 21, 3632–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marques, A.R.A., Gabriel, T.L., Aten, J., van Roomen, C.P.A.A., Ottenhoff, R., Claessen, N., Alfonso, P., Irún, P., Giraldo, P., Aerts, J.M.F.G. et al. (2016) Gpnmb is a potential marker for the visceral pathology in Niemann-Pick type C disease. PLoS One, 11, e0147208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bradbury, A., Bagel, J., Sampson, M., Farhat, N., Ding, W., Swain, G., Prociuk, M., O'Donnell, P., Drobatz, K., Gurda, B. et al. (2016) Cerebrospinal fluid Calbindin D concentration as a biomarker of cerebellar disease progression in Niemann-Pick type C1 disease. J. Pharmacol. Exp. Ther., 358, 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Budge, K.M., Neal, M.L., Richardson, J.R. and Safadi, F.F. (2018) Glycoprotein NMB: an emerging role in neurodegenerative disease. Mol. Neurobiol., 55, 5167–5176. [DOI] [PubMed] [Google Scholar]
- 62. Tsou, P.-S. and Sawalha, A.H. (2020) Glycoprotein nonmetastatic melanoma protein B: a key mediator and an emerging therapeutic target in autoimmune diseases. FASEB J., 34, 8810–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martina, J.A., Diab, H.I., Li, H. and Puertollano, R. (2014) Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. Life Sci., 71, 2483–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gabriel, T.L., Tol, M.J., Ottenhof, R., van Roomen, C., Aten, J., Claessen, N., Hooibrink, B., de Weijer, B., Serlie, M.J., Argmann, C. et al. (2014) Lysosomal stress in obese adipose tissue macrophages contributes to MITF-dependent Gpnmb induction. Diabetes, 63, 3310–3323. [DOI] [PubMed] [Google Scholar]
- 65. Carey, K.L., Paulus, G.L.C., Wang, L., Balce, D.R., Luo, J.W., Bergman, P., Ferder, I.C., Kong, L., Renaud, N., Singh, S. et al. (2020) TFEB transcriptional responses reveal negative feedback by BHLHE40 and BHLHE41. Cell Rep., 33, 108371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rodriguez-Gil, J.L., Larson, D.M., Wassif, C.A., Yanjanin, N.M., Anderson, S.M., Kirby, M.R., Trivedi, N.S., Porter, F.D. and Pavan, W.J. (2013) A somatic cell defect is associated with the onset of neurological symptoms in a lysosomal storage disease. Mol. Genet. Metab., 110, 188–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kaisers, W., Boukamp, P., Stark, H.-J., Schwender, H., Tigges, J., Krutmann, J. and Schaal, H. (2017) Age, gender and UV-exposition related effects on gene expression in in vivo aged short term cultivated human dermal fibroblasts. PLoS One, 12, e0175657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gershoni, M. and Pietrokovski, S. (2017) The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol., 15, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kassam, I., Wu, Y., Yang, J., Visscher, P.M. and McRae, A.F. (2019) Tissue-specific sex differences in human gene expression. Hum. Mol. Genet., 28, 2976–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Abi-Mosleh, L., Infante, R.E., Radhakrishnan, A., Goldstein, J.L. and Brown, M.S. (2009) Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. PNAS, 106, 19316–19321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vance, J.E. and Karten, B. (2014) Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J. Lipid Res., 55, 1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu, B., Ramirez, C.M., Miller, A.M., Repa, J.J., Turley, S.D. and Dietschy, J.M. (2010) Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res., 51, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Taylor, A.M., Liu, B., Mari, Y., Liu, B. and Repa, J.J. (2012) Cyclodextrin mediates rapid changes in lipid balance in Npc 1−/− mice without carrying cholesterol through the bloodstream. J. Lipid Res., 53, 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brozzi, A., Urbanelli, L., Germain, P.L., Magini, A. and Emiliani, C. (2013) hLGDB: a database of human lysosomal genes and their regulation. Database (Oxford), doi: 10.1093/database/bat024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pacheco, C.D., Kunkel, R. and Lieberman, A.P. (2007) Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet., 16, 1495–1503. [DOI] [PubMed] [Google Scholar]
- 76. Elrick, M.J., Yu, T., Chung, C. and Lieberman, A.P. (2012) Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum. Mol. Genet., 21, 4876–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ordonez, M.P., Roberts, E.A., Kidwell, C.U., Yuan, S.H., Plaisted, W.C. and Goldstein, L.S.B. (2012) Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet., 21, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sarkar, S., Carroll, B., Buganim, Y., Maetzel, D., Ng, A.H.M., Cassady, J.P., Cohen, M.A., Chakraborty, S., Wang, H., Spooner, E. et al. (2013) Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep., 5, 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maetzel, D., Sarkar, S., Wang, H., Abi-Mosleh, L., Xu, P., Cheng, A.W., Gao, Q., Mitalipova, M. and Jaenisch, R. (2014) Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick type C patient-specific iPS cells. Stem Cell Reports, 2, 866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Settembre, C. and Ballabio, A. (2014) Lysosome: regulator of lipid degradation pathways. Trends Cell Biol., 24, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Palmieri, M., Impey, S., Kang, H., di Ronza, A., Pelz, C., Sardiello, M. and Ballabio, A. (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet., 20, 3852–3866. [DOI] [PubMed] [Google Scholar]
- 82. de Martín Garrido, N. and Aylett, C.H.S. (2020) Nutrient Signaling and lysosome positioning crosstalk through a multifunctional protein, Folliculin. Front Cell Dev. Biol., 8, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Reue, K. and Zhang, P. (2008) The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Lett., 582, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Maddux, B.A. and Goldfine, I.D. (2000) Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes, 49, 13–19. [DOI] [PubMed] [Google Scholar]
- 85. Spolitu, S., Dai, W., Zadroga, J.A. and Ozcan, L. (2019) Proprotein convertase subtilisin/kexin type 9 and lipid metabolism. Curr. Opin. Lipidol., 30, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Katzmann, J.L., Gouni-Berthold, I. and Laufs, U. (2020) PCSK9 inhibition: insights from clinical trials and future prospects. Front. Physiol., 11, 595819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fukaura, M., Ishitsuka, Y., Shirakawa, S., Ushihama, N., Yamada, Y., Kondo, Y., Takeo, T., Nakagata, N., Motoyama, K., Higashi, T. et al. (2021) Intracerebroventricular treatment with 2-hydroxypropyl-β-cyclodextrin decreased cerebellar and hepatic glycoprotein nonmetastatic melanoma protein B (GPNMB) expression in Niemann-Pick disease type C model mice. Int. J. Mol. Sci., 22, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zigdon, H., Savidor, A., Levin, Y., Meshcheriakova, A., Schiffmann, R. and Futerman, A.H. (2015) Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS One, 10, e0120194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kramer, G., Wegdam, W., Donker-Koopman, W., Ottenhoff, R., Gaspar, P., Verhoek, M., Nelson, J., Gabriel, T., Kallemeijn, W., Boot, R.G. et al. (2016) Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio., 6, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Murugesan, V., Liu, J., Yang, R., Lin, H., Lischuk, A., Pastores, G., Zhang, X., Chuang, W.-L. and Mistry, P.K. (2018) Validating glycoprotein non-metastatic melanoma B (gp NMB, osteoactivin), a new biomarker of Gaucher disease. Blood Cells Mol. Dis., 68, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Reddy, J.V., Ganley, I.G. and Pfeffer, S.R. (2006) Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS One, 1, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. De Windt, A., Rai, M., Kytömäki, L., Thelen, K.M., Lütjohann, D., Bernier, L., Davignon, J., Soini, J., Pandolfo, M. and Laaksonen, R. (2007) Gene set enrichment analyses revealed several affected pathways in Niemann-Pick disease type C fibroblasts. DNA Cell Biol., 26, 665–671. [DOI] [PubMed] [Google Scholar]
- 93. Hetmańczyk-Sawicka, K., Iwanicka-Nowicka, R., Fogtman, A., Cieśla, J., Włodarski, P., Żyżyńska-Granica, B., Filocamo, M., Dardis, A., Peruzzo, P., Bednarska-Makaruk, M. et al. (2020) Changes in global gene expression indicate disordered autophagy, apoptosis and inflammatory processes and downregulation of cytoskeletal signalling and neuronal development in patients with Niemann-Pick C disease. Neurogenetics, 21, 105–119. [DOI] [PubMed] [Google Scholar]
- 94. Vázquez, M.C., del Pozo, T., Robledo, F.A., Carrasco, G., Pavez, L., Olivares, F., González, M. and Zanlungo, S. (2011) Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PLoS One, 6, e28777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Martin, K.B., Williams, I.M., Cluzeau, C.V., Cougnoux, A., Dale, R.K., Iben, J.R., Cawley, N.X., Wassif, C.A. and Porter, F.D. (2019) Identification of novel pathways associated with patterned cerebellar Purkinje neuron degeneration in Niemann-Pick disease, type C1. Int. J. Mol. Sci., 21, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thelen, A.M. and Zoncu, R. (2017) Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol., 27, 833–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ballabio, A. and Bonifacino, J.S. (2020) Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev. Mol. Cell Biol., 21, 101–118. [DOI] [PubMed] [Google Scholar]
- 98. Luo, J., Yang, H. and Song, B.-L. (2020) Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol., 21, 225–245. [DOI] [PubMed] [Google Scholar]
- 99. Meng, Y., Heybrock, S., Neculai, D. and Saftig, P. (2020) Cholesterol handling in lysosomes and beyond. Trends Cell Biol., 30, 452–466. [DOI] [PubMed] [Google Scholar]
- 100. Shin, H.R. and Zoncu, R. (2020) The lysosome at the intersection of cellular growth and destruction. Dev. Cell, 54, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ramirez, C.M., Taylor, A.M., Lopez, A.M., Repa, J.J. and Turley, S.D. (2020) Delineation of metabolic responses of Npc 1−/−nih mice lacking the cholesterol-esterifying enzyme SOAT2 to acute treatment with 2-hydroxypropyl-β-cyclodextrin. Steroids, 164, 108725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Singhal, A., Krystofiak, E.S., Jerome, W.G. and Song, B. (2020) 2-Hydroxypropyl-gamma-cyclodextrin overcomes NPC1 deficiency by enhancing lysosome-ER association and autophagy. Sci. Rep., 10, 8663–8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yang, D.-S., Stavrides, P., Kumar, A., Jiang, Y., Mohan, P.S., Ohno, M., Dobrenis, K., Davidson, C.D., Saito, M., Pawlik, M. et al. (2017) Cyclodextrin has conflicting actions on autophagy flux in vivo in brains of normal and Alzheimer model mice. Hum. Mol. Genet., 26, 843–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vanier,M.T., Wenger, D.A., Comly, M.E., Rousson, R., Brady, R.O. and Pentchev, P.G. (1988) Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin. Genet., 33, 331–348. [DOI] [PubMed] [Google Scholar]
- 105.Vanier,M.T., Rodriguez-Lafrasse, C., Rousson, R., Gazzah, N., Juge, M.C., Pentchev, P.G., Revol, A. and Louisot, P. (1991) Type C Niemann-Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta , 1096, 328–337. [DOI] [PubMed] [Google Scholar]
- 106. Walterfang, M., Fietz, M., Abel, L., Bowman, E., Mocellin, R. and Velakoulis, D. (2009) Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J. Inherit. Metab. Dis., 32, S221–S226. [DOI] [PubMed] [Google Scholar]
- 107. Rodriguez-Gil, J.L., Watkins-Chow, D.E., Baxter, L.L., Elliot, G., Harper, U.L., Wincovitch, S.M., Wedel, J.C., Incao, A.A., Huebecker, M., Boehm, F.J. et al. (2020) Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann-Pick disease type C1. Dis. Model. Mech., 13, dmm. 042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Singhal, A., Szente, L., Hildreth, J.E.K. and Song, B. (2018) Hydroxypropyl-beta and -gamma cyclodextrins rescue cholesterol accumulation in Niemann-Pick C1 mutant cell via lysosome-associated membrane protein 1. Cell Death Dis., 9, 1019–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ripoll, V.M., Meadows, N.A., Raggatt, L.-J., Chang, M.K., Pettit, A.R., Cassady, A.I. and Hume, D.A. (2008) Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene, 413, 32–41. [DOI] [PubMed] [Google Scholar]
- 110. Tomihari, M., Hwang, S.-H., Chung, J.-S., Cruz, P.D. and Ariizumi, K. (2009) Gpnmb is a melanosome-associated glycoprotein that contributes to melanocyte/keratinocyte adhesion in a RGD-dependent fashion. Exp. Dermatol., 18, 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Li, B., Castano, A.P., Hudson, T.E., Nowlin, B.T., Lin, S.-L., Bonventre, J.V., Swanson, K.D. and Duffield, J.S. (2010) The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J., 24, 4767–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hoashi, T., Sato, S., Yamaguchi, Y., Passeron, T., Tamaki, K. and Hearing, V.J. (2010) Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. FASEB J., 24, 1616–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Calcagnì, A., kors, L., Verschuren, E., De Cegli, R., Zampelli, N., Nusco, E., Confalonieri, S., Bertalot, G., Pece, S., Settembre, C. et al. (2016) Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. Elife, 5, e17047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. van der Lienden, M.J.C., Gaspar, P., Boot, R., Aerts, J.M.F.G. and van Eijk, M. (2018) Glycoprotein non-metastatic protein B: an emerging biomarker for lysosomal dysfunction in macrophages. Int. J. Mol. Sci., 20, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Abdelmagid, S.M., Belcher, J.Y., Moussa, F.M., Lababidi, S.L., Sondag, G.R., Novak, K.M., Sanyurah, A.S., Frara, N.A., Razmpour, R., Del Carpio-Cano, F.E. et al. (2014) Mutation in osteoactivin decreases bone formation in vivo and osteoblast differentiation in vitro. Am. J. Pathol., 184, 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kanaan, N.M., Collier, T.J., Cole-Strauss, A., Grabinski, T., Mattingly, Z.R., Winn, M.E., Steece-Collier, K., Sortwell, C.E., Manfredsson, F.P. and Lipton, J.W. (2015) The longitudinal transcriptomic response of the substantia nigra to intrastriatal 6-hydroxydopamine reveals significant upregulation of regeneration-associated genes. PLoS One, 10, e0127768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Srinivasan, K., Friedman, B.A., Larson, J.L., Lauffer, B.E., Goldstein, L.D., Appling, L.L., Borneo, J., Poon, C., Ho, T., Cai, F. et al. (2016) Untangling the brain's neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun., 7, 11295–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Murthy, M.N., Blauwendraat, C., UKBEC, Guelfi, S., IPDGC, Hardy, J., Lewis, P.A. and Trabzuni, D. (2017) Increased brain expression of GPNMB is associated with genome wide significant risk for Parkinson's disease on chromosome 7p15.3. Neurogenetics, 18, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Noda, Y., Tsuruma, K., Takata, M., Ishisaka, M., Tanaka, H., Nakano, Y., Nagahara, Y., Shimazawa, M. and Hara, H. (2017) GPNMB induces BiP expression by enhancing splicing of BiP pre-mRNA during the endoplasmic reticulum stress response. Sci. Rep., 7, 12160–12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Neal, M.L., Boyle, A.M., Budge, K.M., Safadi, F.F. and Richardson, J.R. (2018) The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflammation, 15, 73–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Moloney, E.B., Moskites, A., Ferrari, E.J., Isacson, O. and Hallett, P.J. (2018) The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson's disease patients and increases after lysosomal stress. Neurobiol. Dis., 120, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Budge, K.M., Neal, M.L., Richardson, J.R. and Safadi, F.F. (2020) Transgenic overexpression of GPNMB protects against MPTP-induced neurodegeneration. Mol. Neurobiol., 57, 2920–2933. [DOI] [PubMed] [Google Scholar]
- 123. Hüttenrauch, M., Ogorek, I., Klafki, H., Otto, M., Stadelmann, C., Weggen, S., Wiltfang, J. and Wirths, O. (2018) Glycoprotein NMB: a novel Alzheimer's disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun., 6, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kim, D., Paggi, J.M., Park, C., Bennett, C. and Salzberg, S.L. (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol., 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liao, Y., Smyth, G.K. and Shi, W. (2014) Feature counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 126. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Davidson, C.D., Fishman, Y.I., Puskás, I., Szemán, J., Sohajda, T., McCauliff, L.A., Sikora, J., Storch, J., Vanier, M.T., Szente, L., et al. (2016) Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol, 3, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.