

Abstract
Charcot–Marie–Tooth (CMT) disease is a neuropathy that lacks effective therapy. CMT patients show degeneration of peripheral nerves, leading to muscle weakness and loss of proprioception. Loss of mitochondrial oxidative phosphorylation proteins and enzymes of the antioxidant response accompany degeneration of nerves in skin biopsies of CMT patients. Herein, we followed a drug-repurposing approach to find drugs in a Food and Drug Administration-approved library that could prevent development of CMT disease in the Gdap1-null mouse model. We found that the antibiotic florfenicol is a mitochondrial uncoupler that prevents the production of reactive oxygen species and activates respiration in human GDAP1-knockdown neuroblastoma cells and in dorsal root ganglion neurons of Gdap1-null mice. Treatment of CMT-affected Gdap1-null mice with florfenicol has no beneficial effect in the course of the disease. However, administration of florfenicol, or the antioxidant MitoQ, to pre-symptomatic GDAP1-null mice prevented weight gain and ameliorated the motor coordination deficiencies that developed in the Gdap1-null mice. Interestingly, both florfenicol and MitoQ halted the decay in mitochondrial and redox proteins in sciatic nerves of Gdap1-null mice, supporting that oxidative damage is implicated in the etiology of the neuropathy. These findings support the development of clinical trials for translation of these drugs for treatment of CMT patients.
Graphical Abstract
Graphical Abstract.

Introduction
Drug repurposing is a strategy to overcome the millionaire costs and decade-long tasks of drug discovery needed to reach a safe treatment for the patients (1). This alternative is especially relevant in the case of rare diseases (RDs) where pharmaceutical investments are scant. Charcot–Marie–Tooth (CMT) disease is a RD and the most common hereditary condition of the peripheral nervous system with an estimated overall population prevalence of 10–28/100 000 in Europe (2). CMT is a heterogeneous genetic disease, which onsets usually in the first two decades of life (3). The symptoms include motor and sensory nerve deficits, loss of proprioception and pinprick sensation and are characterized by distal muscle weakness and atrophy (3,4). Despite the efforts to characterize and treat the disease, CMT lacks an effective drug or advanced therapy to treat the patients (5).
More than 90 genes have been linked to CMT disease and related neuropathies (6). One of them encodes the ganglioside-induced differentiation-associated protein 1 (GDAP1) (7,8). Mutations in GDAP1 gene cause axonal CMT with both recessive (AR-CMT2K) (7) and dominant (CMT2K) inheritance (9), and recessive demyelinating CMT4A (10). GDAP1 is an atypical glutathione S-transferase (GST) (11) of the outer mitochondrial membrane (12), which is also located in both the mitochondria-associated membranes (MAMs) (13) and the mitochondria–lysosome membrane contact sites (14). Mutations in GDAP1 have been associated with abnormal changes in mitochondrial morphology and dynamics (13,15) and with a decreased Ca2+ entry through the store-operated calcium entry (SOCE), which leads to a failure in the stimulation of mitochondrial respiration and the inhibition of mitochondrial ATP production (15,16). The lack of GDAP1 also induces an inflammatory response in the spinal cord and sciatic nerve (17). Furthermore, GDAP1 participates in the defense against oxidative stress (18). In this context, it has been shown that members of the GDAP1 family are responsive and protect against stress associated with increased levels of oxidized glutathione (19). Likewise, GDAP1 mutations lead to defective mitochondrial complex I activity and oxidative stress (20,21). Consistent with these observations, loss of mitochondrial and antioxidant proteins have been reported in skin biopsies of CMT patients (22) and in peripheral nerves of the Gdap1 knock-out mouse (Gdap1-null) (15), further suggesting that mitochondrial function and oxidative stress provide potential targets to treat CMT disease.
Herein, we have performed a screening of 1018 FDA-approved small compounds searching for activators of mitochondrial respiration to be repurposed for the treatment of CMT disease. We found that the antibiotic florfenicol activated mitochondrial respiration in human neuroblastoma SH-SY5Y and GDAP1-silenced SH-SY5Y (G4) cells, and in dorsal root ganglion (DRG) neurons of the Gdap1-null mice. Mechanistically, we show that florfenicol is a mitochondrial uncoupler that prevents the production of reactive oxygen species (ROS) in mitochondria (mtROS) of SH-SY5Y and GDAP1-silenced SH-SY5Y (G4) cells. To verify the efficacy of florfenicol in a preclinical model of CMT disease, we treated Gdap1-null mice before and after development of CMT symptoms and found that florfenicol is an effective treatment only when administered before development of CMT disease. Moreover, aside from increasing mitochondrial respiration by the uncoupling mtROS-preventing activity of florfenicol as a therapeutic strategy for CMT disease, we also ameliorated mtROS production with the antioxidant MitoQ in pre-symptomatic Gdap1-null mice. In vivo, both florfenicol and MitoQ prevented the development of motor deficiencies, the decline in mitochondrial and redox proteins and ameliorated oxidative stress in sciatic nerves of Gdap1-null mice. These results support the use of florfenicol or MitoQ as therapeutic strategies to prevent/minimize the development of symptoms in CMT patients.
Results
Searching for activators of mitochondrial respiration
To identify activators of oxidative phosphorylation (OXPHOS) that could be employed for the treatment of mitochondrial disorders, which are usually ascribed to a deficit of cellular ATP provision, we screened an FDA-approved library of 1018 small compounds that in short-term treatment of 3 h stimulated mitochondrial respiration in HCT116 colon cancer cells (Fig. 1A). We used the oligomycin-sensitive respiration (OSR) as reporter of the drugs’ effect because it represents an estimate of the activity of ATP synthase (23). We identified 139 compounds that enhanced OSR by 20% when compared to cells treated with the vehicle dimethyl sulfoxide (DMSO) (Fig. 1A, Supplementary Material, Table S1). Since our interest at this stage was to find a drug for the treatment of CMT disease, further in-depth investigation of the effect of the activators on cell viability (Fig. 1B) and mitochondrial respiration (Fig. 1C) was carried out using the neuroblastoma SH-SY5Y cell line. Only 86 compounds that surpassed the threshold of cellular viability after 24-h incubation with the drugs (Fig. 1B, Supplementary Material, Table S1) were selected for further analysis of mitochondrial respiration in the SH-SY5Y cell line (Fig. 1C).
Figure 1 .

Florfenicol, methylthiouracil and isosorbide activate mitochondrial respiration of SH-SY5Y neuroblastoma cells. Cells were incubated for 24 h with 1 μm solution of the indicated drugs. (A) One hundred and thirty-nine compounds of a 1018 FDA-approved drug library activate (>20%) OSR of HCT116 colon cancer cells in a 1 μm, 3 h short-term treatment. (B) Viability of SH-SY5Y neuroblastoma cancer cells a 1 μm, 24 h treatment. Eighty-six compounds that had no significant effect on viability (≥75%; in yellow) out of the 139 activators identified were selected for further study. (C) Heat map of the respiration parameters of SH-SY5Y cells treated 24h with 1 μm solution of the 86 selected compounds. The map indicates the OSR, basal (BAS) and maximum (MAX) respiration. The calculated z-score is indicated. (D) Molecular formula of top three mitochondrial activators: florfenicol (blue), methylthiouracil (red) and isosorbide (green). (E) SH-SY5Y cells were treated 1 μm, during 24 h with florfenicol (blue lines and bars), methylthiouracil (red lines and bars) or isosorbide (green lines and bars) or left untreated (black lines and bars) and the respiratory profiles (upper panel) recorded in a Seahorse analyzer. The histograms (bottom panel) show the basal, OSR and maximum oxygen consumption rates (OCR). OL, oligomycin; ROT, rotenone; ANT, antimycin A. Bars indicate the mean ± SEM of three biological replicates. *P < 0.05 when compared to CRL by Student’s t-test.
Florfenicol (Fig. 1D), a fluorinated synthetic analog of thiamphenicol with bacteriostatic activity; methylthiouracil (Fig. 1D), an antithyroid preparation; and isosorbide (Fig. 1D), a heterocyclic compound used to induce the relaxation of smooth muscle fibers in coronary heart disease, were the three drugs that had the strongest stimulatory effect on mitochondrial respiration in the SH-SY5Y cell line (Fig. 1C and E). In fact, treatment of the cells with any of these compounds significantly increased basal, OSR and maximal respiration of the cells (Fig. 1E), showing florfenicol and methylthiouracil a stronger effect than that of isosorbide (Fig. 1E).
Potential drug candidates for CMT disease
Mutations in GDAP1 cause axonal recessive (AR-CMT2), axonal dominant (CMT2K) and demyelinating recessive (CMT4A) forms of CMT neuropathy (24). Hence, we next studied the effect of the 87 drugs that activated mitochondrial respiration in GDAP1 knockdown SH-SY5Y cells (G4 cells) (Fig. 2A). The results confirmed that florfenicol, methylthiouracil and isosorbide were also the strongest activators from the screen (Fig. 2B). Indeed, 24-h incubation with the drugs significantly increased basal, OSR and maximal respiration of the GDAP1 knockdown cells (Fig. 2C), revealing marginal differences in potency among the three drugs (Fig. 2C).
Figure 2 .

Florfenicol, methylthiouracil and isosorbide activate mitochondrial respiration of GDAP1-knockdown SH-SY5Y cells and Gdap1−/− DRG neurons. (A) Representative western blot of the expression of GDAP1 in WT SH-SY5Y (WT) and GDAP1-knockdown SH-SY5Y (G4) cells. Tubulin is shown as loading control. (B) Heat map of the respiratory parameters of GDAP1-knockdown G4 cells 24 h-treated with 1 μm solution of the 86 compounds. OSR, basal (BAS) and maximum (MAX) respiration. The calculated z-score is indicated. (C) GDAP1-knockdown G4 cells were treated with 1 μm, during 24 h, florfenicol (blue lines and bars), methylthiouracil (red lines and bars) or isosorbide (green lines and bars) or left untreated (black lines and bars) and the respiratory profiles (upper panel) recorded in a Seahorse analyzer. The histograms (bottom panel) show the basal, OSR and maximum oxygen consumption rates (OCR). OL, oligomycin; ROT, rotenone; ANT, antimycin A. (D) Respiratory profiles (upper panel) and parameters (bottom panel) of DRG neurons from Gdap1−/− mice (blue lines and bars) over DRG neurons of WT mice (black lines and bars) both 24 h-treated with 1 μm florfenicol. (E, F) Respiratory profiles (upper panel) and parameters (bottom panel) of DRG neurons of Gdap1−/− mice were 24 h-treated with 1 μm methylthiouracil (E, red lines and bars) or 1 μm isosorbide (F, green lines and bars) or left untreated (E, F, black lines and bars). Bars indicate the mean ± SEM of three biological replicates. *P < 0.05 when compared to the respective CRL by Student’s t-test.
Postnatal dorsal root ganglia (DRG) neurons from wild-type (WT) and Gdap1-null mice, a mouse model of axonal recessive form of the GDAP1-related CMT (15), were incubated for 24 h with the drugs (Fig. 2D). Only florfenicol stimulated significantly basal, OSR and maximal respiration of DRG neurons from Gdap1-null mice when compared to its effect in DRG neurons of WT mice (Fig. 2D). In fact, the effect of methylthiouracil (Fig. 2E) and isosorbide (Fig. 2F) in the respiratory profiles of DRG neurons from Gdap1-null mice was marginal. These findings suggested that florfenicol could provide a potential drug for the treatment of CMT disease.
Florfenicol is a potent mitochondrial uncoupler that prevents the generation of ROS
Florfenicol is a highly lipophilic broad-spectrum antibiotic used in veterinary medicine with antibacterial effects in both Gram-negative and Gram-positive bacteria because it binds the prokaryotic 50S ribosomal subunit, leading to the inhibition of protein synthesis (25). Florfenicol also binds the mitoribosome and inhibits mitochondrial protein synthesis with concomitant mitochondrial dysfunction at higher concentrations of the drug (70–100 μm) (26). At first sight, it seems unexpected that a low dose of an inhibitor of mitochondrial protein synthesis could result in enhanced cellular respiration. Hence, to explain its positive effect in cellular respiration, we focused on the potential uncoupling effect of the drug because of its lipophilic nature. Indeed, incubation of cells with 1 μm florfenicol promoted a sharp decline in mitochondrial membrane potential (ΔΨm) in both SH-SY5Y WT and G4 cell lines, similarly as when the cells were incubated with the classic mitochondrial uncoupler dinitrophenol (DNP) (Fig. 3A). Interestingly, ΔΨm in G4 cells was significantly augmented when compared to the parental SH-SY5Y WT cells (Fig. 3A). As a result, G4 cells produced more superoxide radical in mitochondria (mtROS), as assessed with the MitoSOX probe than SH-SY5Y WT cells (Fig. 3B). Consistent with the uncoupling effect of florfenicol (Fig. 3A), the drug significantly diminished the production of mtROS in both SH-SY5Y WT and G4 cells (Fig. 3B), to a comparable level of the mitochondrial antioxidant MitoQ (Fig. 3B). Moreover, in contrast with the positive effect observed with 1 μm florfenicol in mitochondrial respiration (Figs 1E and 2C and D), incubation of the cells with 100 μm florfenicol promoted a sharp inhibition of mitochondrial respiration (Supplementary Material, Fig. S1A and B). These results further suggested that the drug effect on the respiratory function of the organelle is independent from its effect on the synthesis of mitochondrial encoded proteins. Overall, these findings thus support that the enhanced mitochondrial respiration promoted by florfenicol stems from its activity as an uncoupling agent and emphasize the relevance that oxidative stress plays in the pathophysiology of CMT disease in agreement with previous reports (20,21).
Figure 3 .

Florfenicol is a mitochondrial uncoupler that prevents the generation of mtROS. (A) The ΔΨm was assessed in TMRM+-stained SH-SY5Y WT and G4 cells treated during 24 h with 1 μm florfenicol (gray bar) or left untreated (black bar). The mitochondrial uncoupler DNP 0.5 mm (red bar) was added to collapse ΔΨm. (B) Mitochondrial ROS production was assessed using 5 μm MitoSOX in SH-SY5Y WT and G4 cells treated during 24 h with 1 μm florfenicol (gray bar), 20 nm MitoQ (yellow bar) or left untreated (black bar). Bars indicate the mean ± SEM of three biological replicates. **P < 0.01 and ##P < 0.01 when compared to CRL and WT, respectively, by Student’s t-test. See also Supplementary Material, Fig. S1.
Florfenicol does not improve motor coordination of CMT-affected GDAP1-null mice
Our first approach to investigate the potency of florfenicol as a repurposed drug for the in vivo treatment of CMT disease was to verify its effect in Gdap1-null mice that have developed the axonal recessive form of CMT disease (Supplementary Material, Fig. S2A) (15). To this aim, 7-month-old WT and Gdap1-null mice were tested for motor and coordination behavior by rotarod test (Supplementary Material, Fig. S2B). Mutant mice had significantly reduced latency to fall in the test when compared to WT mice (Supplementary Material, Fig. S2B), in agreement with previous findings (15). After, Gdap1-null mice were distributed into two groups and treated (FLORF, 0.84 mg/ml) or not (KO) with florfenicol, which was administered in the drinking water for 4 months (Supplementary Material, Fig. S2A). A group of WT mice was also maintained for comparison. After ending drug treatment, motor and coordination behavior was monitored by rotarod test (Supplementary Material, Fig. S2C). No significant differences were observed between non-treated and florfenicol-treated Gdap1-null mice (Supplementary Material, Fig. S2C). Both groups showed significantly reduced latency to fall in the rotarod test when compared to age-matched WT mice (Supplementary Material, Fig. S2C). These results most likely indicate that florfenicol is not able to improve CMT symptoms after developing the disease.
Florfenicol and MitoQ prevent the development of CMT symptoms in Gdap1-null mice
Despite the discouraging findings described above, we reasoned that perhaps florfenicol might prevent or delay the appearance of CMT symptoms. Therefore, florfenicol was administered to Gdap1-null mice from the time of weaning and up to 10 months of age (Fig. 4A). In this experiment, we also included a group of non-treated WT and Gdap1-null mice (Fig. 4A). The reduction of epidermal nerve density is a common neuropathic abnormality observed in skin biopsies of CMT patients (27) that is accompanied by loss of the mitochondrial proteins involved in OXPHOS and scavenging ROS (22). Not surprisingly, fibroblasts of patients with GDAP1 dominant mutations show enhanced ROS production (21), as we have observed in knockdown SH-SY5Y cells (Fig. 3B). Moreover, the peripheral nervous system of Gdap1-null mice has evidence of oxidative stress (19). Therefore, to investigate the potential implication of ROS in the etiology of CMT, we decided to include an additional group of Gdap1-null mice treated with the mitochondrial-targeted antioxidant MitoQ (Fig. 4A) that ameliorates mtROS production (Fig. 3B) without affecting mitochondrial respiration neither in SH-SY5Y WT nor G4 cells (Supplementary Material, Fig. S1A and B).
Figure 4 .
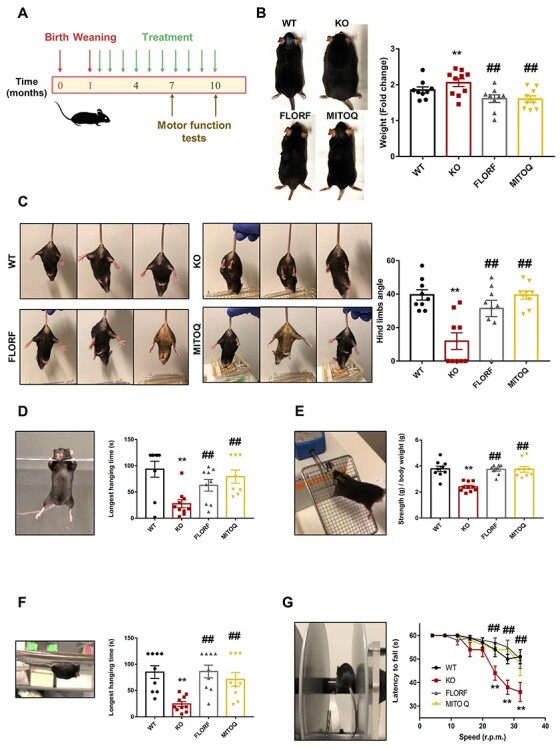
Florfenicol and MitoQ prevent motor function disabilities of Gdap1−/− mice. (A) Graphical scheme showing the course of mice treatment. After weaning (1 month), florfenicol (0.84 mg/ml) and MitoQ (0.14 mg/ml) were administered in the drinking water of mice. Motor function tests were run at 7 and 10 months of age. (B–G) WT mice (n = 9, black line bar and dots), untreated Gdap1−/− mice (KO, n = 10, red line bar and dots), florfenicol-treated Gdap1−/− mice (FLORF, n = 9, gray line bar and dots) and MitoQ-treated Gdap1−/− mice (MITOQ, n = 9, yellow line bar and dots). (B) Representative images of mice at 10 months of age. Histograms show the quantification of the fold change in mice weight during the treatments. (C) Representative images of 10-month-old mice suspended by the tail. WT, florfenicol- and MitoQ-treated Gdap1−/− mice show the characteristic response trying to escape by splaying the hind limbs away from the trunk of its body. In contrast, hind limbs of non-treated Gdap1−/− mice are held closed to the trunk in an abnormal dystonic posture. Histograms show the quantification of the angle form by the right hind limb and the body axis. (D) Representative image of two limbs hanging test. Histograms show the quantification of the longest time that the mice exhibited sustained limb tension. (E) Representative image of grip force test. Histograms show the quantification of mice strength (g) over body weight (g). (F) Representative image of four limbs hanging test. Histograms show the quantification of the longest time that the mice exhibited sustained limb tension. (G) Representative image of rotarod test. The graph shows the quantification of the mice latency to fall (s) at 4, 8, 12, 16, 20, 24, 28 and 32 r.p.m. of speed. Bars indicate the mean ± SEM of the n above indicated. **P < 0.01 when compared to WT mice and ##P < 0.01 when compared to KO mice by Student’s t-test. See also Supplementary Material Figs S2 and S3.
At 7 months of age, we noted that Gdap1-null mice showed a significant increase in body weight when compared to WT (Supplementary Material, Fig. S3A). In contrast, both florfenicol and MitoQ prevented the increase in body weight of Gdap1-null mice (Supplementary Material, Fig. S3A). Motor coordination of the four groups of mice under study, as assessed with the rotarod at midterm of the experiment (7 months), revealed no significant differences between them (Supplementary Material, Fig. S3B).
At 10 months of age, non-treated Gdap1-null mice also showed a relevant increase in body weight when compared to WT mice (Fig. 4B). Both florfenicol and MitoQ treatments significantly diminished the body weight gain of Gdap1-null mice (Fig. 4B). GDAP1-related CMT promotes weakness and wasting of feet and hands leading to pronounced disability of the patients. Hind-limb clasping is a marker of disease progression in a number of mouse models of neurodegeneration (28). Consistent with previous results (15), we observed abnormal hind-limb clasping reflex in non-treated Gdap1-null mice, as assessed by the angle formed between the right hind limb and the body axis when compared to WT mice (Fig. 4C). Remarkably, treatment of mice with florfenicol or MitoQ significantly improved the hind-limb clasping reflex of the animals (Fig. 4C), suggesting that both treatments improved CMT symptoms.
Determination of the forelimbs muscle strength and maximum strength performed by the forelimbs were assessed by the hanging (Fig. 4D; Supplementary Material, Videos S1 and S2) and grip force (Fig. 4E) tests, respectively. Both the longest hanging time (Fig. 4D; Supplementary Material, Videos S1 and S2) and minimal holding impulse (Supplementary Material, Fig. S3C) were significantly reduced in non-treated Gdap1-null when compared to WT mice. Treatment of mice with florfenicol or MitoQ significantly improved the muscle strength of the forelimbs (Fig. 4D; Supplementary Material, Fig. S3C and Supplementary Material, Videos S1 and S2). Similar findings were obtained in the grip force test (Fig. 4E), supporting that both florfenicol and MitoQ treatment prevented the wasting of muscles in the forelimbs of Gdap1-null mice.
The muscle strength of the four limbs was determined by the ability of mice to oppose their weight making use of the four limbs (Fig. 4F; Supplementary Material, Videos S3 and S4). Both the longest hanging time (Fig. 4F; Supplementary Material, Videos S3 and S4) and minimal holding impulse (Supplementary Material, Fig. S3D) were significantly reduced in non-treated Gdap1-null when compared to WT mice. Treatment of mice with florfenicol or MitoQ significantly improved the muscle strength of the four limbs (Fig. 4F; Supplementary Material, Fig. S3C and Supplementary Material, Videos S3 and S4), further supporting that both treatments prevented muscle wasting of the limbs in Gdap1-null mice.
Finally, motor coordination behavior was assessed by rotarod test (Fig. 4G). Consistent with the results of previous tests, the latency to fall was significantly reduced in Gdap1-null mice (Fig. 4G). Remarkably, treatment of Gdap1-null mice with either florfenicol or MitoQ abolished these differences (Fig. 4G), strongly supporting that florfenicol or mitochondrial ROS scavenging from early life prevents the appearance of the deleterious muscle wasting phenotype of the GDAP1-CMT mouse model.
Florfenicol and MitoQ prevent deterioration of mitochondrial and redox proteomes in sciatic nerves of Gdap1-null mice
We have already reported that electrophysiological analysis of the nerves in 5-month Gdap1-null mice showed a significant reduction of compound muscle action potential (CMAP) amplitude obtained for distal (at the ankle) and proximal (at the hip) stimulation, indicative for an axonal neuropathy (15). In morphological studies, we did not detect any significant reduction in the axonal number correlating with the observed reduction in CMAP amplitude in 5-month-old mice (15). We reasoned that prevention of CMT symptoms was enough evidence of the electrophysiological functionality of peripheral nerves. However, we carried out their analysis at the molecular level because sciatic nerves of Gdap1-null mice showed a reduction of proteins of energy metabolism and of the antioxidant response (15), in agreement with the partial loss of these proteins in skin biopsies of CMT1A patients as a result of the injury of epidermal nerve terminals (22). Hence, we next explored the effect of florfenicol and MitoQ in the expression of selected proteins of energy metabolism and of the oxidative stress response in tissue extracts derived from sciatic nerves, skeletal muscle, liver and cerebellum of Gdap1-null mice (Figs 5 and 6; Supplementary Material, Figs S5–S7). For this purpose, we used a quantitative high-throughput reverse phase protein array (RPPA) approach (29,30) (Supplementary Material, Fig. S4). In agreement with previous observations (15), the peripheral nerves of GDAP1-null mice showed significant loss of the enzymes involved in the oxidation of glucose and mitochondrial respiration (LDHA, PDH, IDH1 and CORE2) and of the antioxidant response (GR, SOD1, catalase and SOD2) when compared to WT mice (Fig. 5). Consistent with this last observation, the oxidative modification of cellular proteins by MDA and 4HNE was significantly increased in Gdap1-null mice (Fig. 5). Other mitochondrial proteins showed no relevant changes (Fig. 5).
Figure 5 .
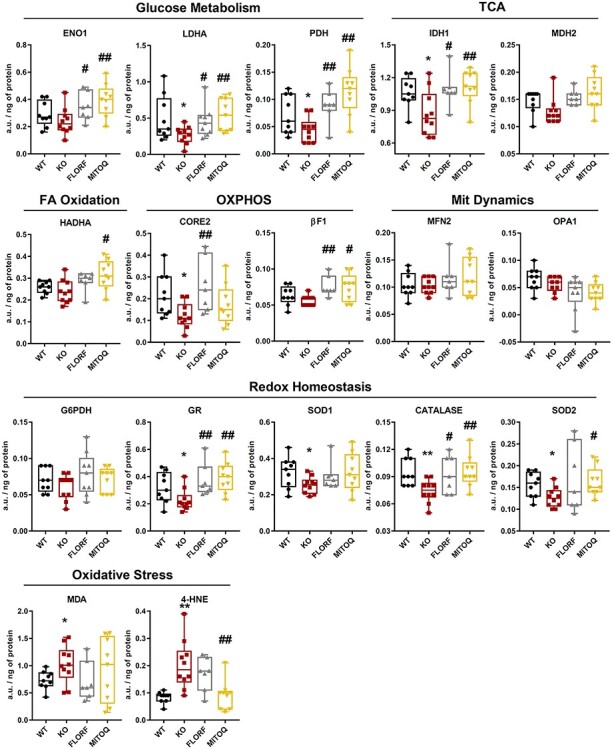
Florfenicol and MitoQ prevent the metabolic and redox deficiencies observed in sciatic nerves of GDAP1 −/− mice. Sciatic nerve extracts derived from WT mice (black boxed) (n = 9), non-treated Gdap1−/− mice (KO, red boxed) (n = 10) and Gdap1−/−-treated mice with florfenicol (FLORF, grey boxed) (n = 9) or MitoQ (MITOQ, yellow boxed) (n = 9) were spotted onto the arrays and processed. The arrays were developed with the indicated primary antibodies and the quantity of protein in the extracts expressed in arbitrary units/ng of protein using as standard the linear plot of the C2C12 cell line (Supplementary Material, Fig. S3). Box plots represent 25th–75th percentiles with the median value in the middle line and with all data represented from minimal to maximal values of the expression level of the proteins. Enzymes of glucose metabolism (ENO1, LDHA, PDH), TCA cycle (IDH1, MDH2), fatty acid oxidation (HADHA), OXPHOS (CORE2, β-F1-ATPase), mitochondrial dynamics (MFN2, OPA1), redox homeostasis (G6PDH, GR, SOD1, Catalase, SOD2) and oxidative stress (MDA, 4HNE) are presented. Bars indicate the mean ± SEM of three replicates of the n above indicated. * and ** indicates a P < 0.05 and P < 0.001 when compared to WT mice, respectively; # and ##, P < 0.05 and P < 0.001 when compared to Gdap1−/− mice, respectively, by Student’s t-test. See also Supplementary Material, Fig. S3.
Figure 6 .

Muscle, liver and cerebellum of GDAP1 −/− mice show marginal effect in the protein content of metabolic proteins. (A) Scheme summarizing significant changes in protein expression levels [decreased (blue) or increased (red)] in sciatic nerve, muscle, liver and cerebellum of non-treated Gdap1−/− mice (KO) (n = 10) and Gdap1−/−-treated mice with florfenicol (FLORF) (n = 9) or MitoQ (MITOQ) (n = 9) when compared to WT mice (n = 9) as compiled in Fig. 4; Supplementary Material, Figs S4–S6. (B) Sciatic nerve extracts derived from WT mice (black boxed) (n = 9), non-treated Gdap1−/− mice (KO, red boxed) (n = 10) and Gdap1−/−-treated mice with florfenicol (FLORF, grey boxed) (n = 9) or MitoQ (MITOQ, yellow boxed) (n = 9) were spotted onto the arrays and processed. The arrays were developed with the indicated primary antibodies and the quantity of protein in the extracts expressed in arbitrary units/ng of protein using as standard the linear plot of the C2C12 cell line (Supplementary Material, Fig. S3). Box plots represent 25th–75th percentiles with the median value in the middle line and with all data represented from minimal to maximal values of the expression level of the proteins. Additional mitochondrial proteins of OXPHOS (NADH9, COXIV), mitochondrial dynamics (MFN1) and structure (HSP60) are presented. Bars indicate the mean ± SEM of three replicates of the n above indicated. *P < 0.05 when compared to WT mice; #P < 0.05 when compared to Gdap1−/− mice by Student’s t-test. See also Supplementary Material, Figs S4–S7.
Remarkably, both florfenicol and MitoQ treatments significantly prevented and/or increased the expression of a large number of proteins of energy metabolism (ENO1, LDHA, PDH, IDH, CORE2, βF1) and of the antioxidant response (GR, catalase and SOD2) in the sciatic nerves of GDAP1-null mice (Fig. 5). Both treatments partially ameliorated oxidative damage of nerve proteins of the Gdap1-null mice, although only MitoQ reached statistical significance in 4HNE modifications (Fig. 5). These results strongly support at the molecular level that the bioenergetic and redox compromise that onsets in Gdap1-null mice during development could be prevented by treatment with the antibiotic florfenicol or the mitochondrial-targeted antioxidant MitoQ.
It should be noted that except for mitofusin 2 (MFN2) in muscle (Fig. 6A; Supplementary Material, Fig. S5), ENO1 and SOD1 in liver (Fig. 6A; Supplementary Material, Fig. S6) and none in cerebellum (Fig. 6A; Supplementary Material, Fig. S7), ablation of the Gdap1 gene had marginal effect in the tissue content of many of the proteins investigated. Likewise, both florfenicol and MitoQ had negligible impact on the content of the proteins in these tissues (Fig. 6A; Supplementary Material, Figs S5–S7). It should be noted the reduction of SOD2 observed in the liver and cerebellum of Gdap1-null mice in response to the antioxidant MitoQ (Fig. 6A; Supplementary Material, Figs S5–S7).
The quantification of additional mitochondrial proteins in sciatic nerves, such as NADH9 and COXIV of the respiratory chain, and of mitochondrial dynamics (MFN1) and structure (HSP60) further confirmed the specific deleterious effect of the ablation of the Gdap1 gene in proteins involved in mitochondrial energy production when compared to proteins involved in other mitochondrial functions (Fig. 6B). In addition, these findings, further confirmed the positive effect of both treatments in preventing the loss of the enzymes of energy metabolism in Gdap1-null mice (Fig. 6B). Overall, the findings indicate that both florfenicol and MitoQ are two drugs that prevent the development of CMT symptoms by ameliorating the dysfunction of energy metabolism and redox homeostasis in peripheral nerves of a mouse model of CMT disease.
Discussion
Multiple genetic alterations result in the degeneration of the peripheral nerves and death of neurons that lead to muscle weakness and atrophy in CMT patients (27,31). These genetic alterations are expressed at the protein level by the loss of mitochondrial enzymes involved in OXPHOS and ROS scavenging (22). Despite efforts to find targets and drugs for the treatment of CMT disease to date, and although different mouse models of the disease are already available (15,32), no clinical benefits have been advanced limiting current treatments to supportive care of the patients (5). Since the duplication of the PMP22 gene is causative for the most prevalent form CMT1A (33), most of the strategies developed look for the downregulation of PMP22 gene. In this context, PXT3003 has been shown to be safe and tolerable in a phase II trial in CMT1A patients (34) (NCT01401257). Potential treatments for other subtypes of CMT disease are scant, and among them, HDCA6 inhibitors have shown correction of the axonal transport deficits in CMT2 (HSPB1) (35) and CMT2D (36) mouse models. Moreover, agonists of MFN2 promote mitochondrial fusion and ameliorate mitochondrial trafficking deficits in cultured mouse neurons containing Mfn2R94G and Mfn2T105M mutations (37). Therefore, treatments that target molecular pathways to stop the onset and progression of the disease are imperative to overcome CMT symptoms in patients.
Drug repurposing is a strategy that could accelerate the translation of active molecules for the treatment of CMT disease. Since mitochondrial dysfunction and oxidative stress are two hallmarks of CMT disease (22), which are also reproduced in the Gdap1-null mouse model (15), we have screened a library of small compounds to search for activators of mitochondrial respiration to be tested in vivo in the preclinical model of the disease (15). We have discovered that the antibiotic florfenicol is a mitochondrial uncoupler that prevents mtROS generation and increases mitochondrial respiration in GDAP1-silenced SH-SY5Y cells, and DRG neurons of Gdap1-null mice. When florfenicol is administered from early life to Gdap1-null mice, it prevents the development of motor deficiencies and other symptoms of the CMT neuropathy. Florfenicol also prevents the loss of mitochondrial proteins involved in metabolic and redox functions of peripheral sciatic nerves. Interestingly, treatment of Gdap1-null mice with the mitochondrial-targeted antioxidant MitoQ mimicked at the functional and molecular levels the beneficial effects of florfenicol, strongly supporting the implication of oxidative damage of metabolic proteins in the etiology of CMT neuropathy. Overall, we advance two drugs that by either ameliorating (florfenicol) or scavenging (MitoQ) mtROS production prevent the development of CMT disease in a preclinical mouse model. We support that the two drugs hold great promise to be repurposed for the treatment of these patients. Unfortunately, florfenicol does not revert motor deficiencies when administered to already symptomatic CMT mice, which may be due to the ROS-mediated progressive accumulation of oxidative damage in neurons. It is likely that the oxidative damage cannot be amended to recover functionality, neither the neurons can be replaced because they are post-mitotic cells. In this line, it has been suggested that failure of the antioxidant MitoQ in relieving the symptoms of Parkinson’s disease resulted because of the advanced oxidative damage in the patients enrolled in the clinical trial (38).
Weight gain is a serious issue in CMT patients because weakened muscles make it difficult for them to keep active. In agreement with this, we observed that Gdap1-null mice showed a progressive increase in body weight when compared to WT mice due to the accumulation of visceral fat. Interestingly, Gdap1-null mice treated with florfenicol or MitoQ prevented the body weight increase in these animals, presumably because avoiding muscle weakness allowed the mice to exercise as controls. Indeed, mice with a restrained mitochondrial function in skeletal muscle present higher body weight and visceral adipose tissue than WT mice (39). Of note, weight in these mice can be reestablished by treatment with edaravone, a potent antioxidant and activator of mitochondrial respiration (39), supporting the importance of preserving mitochondrial and redox function to prevent muscle weakness.
The doses of florfenicol used to inhibit mitochondrial protein synthesis and function (26) are much higher than the ones used in this study. Hence, we support that the beneficial effect of florfenicol results from its lipophilic nature and uncoupler activity that, on the one hand, enhances mitochondrial respiration and, on the other, ameliorates mtROS production. In fact, long-term in vivo treatment with florfenicol prevents the downregulation of mitochondrial metabolic and redox proteins in peripheral nerves. Overall, we postulate that the long-term beneficial effect of florfenicol in GDAP1-null mice results from preventing oxidative damage to metabolic and redox proteins in peripheral nerves what results in less deterioration of motor functions.
Deterioration of the cellular and mitochondrial antioxidant response is a consistent feature observed in different human genetic disorders that affect mitochondrial activity (30). Consistently, we showed that Gdap1-null mice present a specific deficiency in metabolic and redox proteins in sciatic nerves, which leads to an increase oxidative damage and development of CMT symptoms. Remarkably, long-term treatment with MitoQ prevents the molecular deterioration and oxidative damage observed and the development of CMT symptoms in Gdap1-null mice, strongly supporting oxidative damage as a major contributing factor in the etiology of the disease. MitoQ is a mitochondrial targeted antioxidant that consists of a quinone moiety linked to a triphenylphosphonium by a 10-carbon alkyl chain (40). MitoQ has no toxic effects in long-term treatments administered to mice in drinking water and is commercialized as a dietary supplement for humans (41). Hence, we support that MitoQ administration early at the stage of diagnosis to CMT patients might contribute to prevent the loss of mitochondrial and antioxidant proteins observed in peripheral nerves of mildly affected CMT1A patients (22). This treatment might contribute to halt the development of CMT symptoms. In this context, it has been shown that Coenzyme Q10 treatment—which plays a role in antioxidant responses—partially resolved visual impairment in hereditary motor sensory neuropathy related to CMT2A (42). MitoQ has been shown to decrease blood pressure in old human subjects (43). Moreover, MitoQ administration reduces oxidative stress and mitochondrial dysfunction in a mouse model of multiple sclerosis (44) and a phase II clinical trial in multiple sclerosis patients to overcome fatigue is already ongoing (NCT03166800). Since ROS production is a common feature in different mitochondrial disorders, including MFN2-related CMT forms, we suggest that florfenicol or MitoQ deserve to be investigated for the treatment of OXPHOS and redox-related pathologies in appropriate preclinical mouse models of these disorders.
In conclusion, we highlight a drug repurposing strategy to find drugs for the treatment of CMT disease and emphasize the potential clinical relevance of florfenicol and MitoQ for the treatment of these patients as positively exemplified at the molecular and functional levels in the Gdap1-null mouse model of the disease. We further suggest that several of the plasma metabolites previously identified as potential biomarkers of CMT1A disease (22) could help the follow-up of the response to these drugs in future clinical trials to be developed in CMT patients.
Materials and Methods
Cell lines
Cells were cultured in an incubator at 37°C with a controlled atmosphere of ambient air 10% CO2. Human colorectal carcinoma HCT116 cells were grown in McCoy’s 5A media supplemented with 10% fetal bovine serum (FBS). Human neuroblastoma WT and GDAP1 knockdown (G4) (13) SH-SY5Y cells were grown in DMEM-F12 media supplemented with 10% FBS. For stable control of G4 SH-SY5Y cells, 2 μg/ml puromycin was added to the medium, to maintain the selection. To preserve cell lines, aliquots of 5 million cell were gradually frozen (14 h at −20°C, 24 h at −70°C and then they were kept in liquid nitrogen) in 1 ml culture medium with 10% DMSO. The unfreezing was carried out at 37°C immediately diluting them in culture medium.
DRG neuronal cultures
DRGs were obtained from newborn (postnatal days 0–2) WT and Gdap1 knockout (Gdap1−/−) mice according to (45). Ganglia were digested in collagenase (Worthington, USA) and trypsin (Sigma-Aldrich, St. Louis, MO), dissociated by trituration and plated in DMEM-F12 containing 50 ng/ml NGF (Tebu-Bio, Le-Perray-en-Yvelines, France), 10% FBS and 5 ng/ml of aphidicolin (A.G. Scientific, San Diego, CA). DRG neurons were seeded on poly-l-lysine and laminin (1 μg/ml)-coated (Sigma-Aldrich, St. Louis, MO) plates and used for experimentation between 2 and 4 DIV.
Drug library screening
The effects of compounds from a 1018 FDA-approved Drug Library (Selleckchem, Texas, USA) on mitochondrial respiratory parameters were determined in XFe96 Seahorse flux analyzer (Agilent Technologies, California, USA) as described in (23). In primary screenings, the effect was assessed in 35 000 HCT116 colon cancer cells after 1 μm short-term incubation (3 h) of the drugs on the basal and OSR using 10 mm glucose, 1 mm pyruvate and 2 mm glutamine to identify inhibitors and activators of mitochondrial respiration. Inhibitors (23) and activators (this study) were, respectively, defined when the effect of the drugs promoted a 40% decrease or activated 20% mitochondrial respiration when compared to the cells incubated only with the vehicle. To validate the potential hits in the appropriate cellular models, compounds that positively affected respiration were used for secondary screenings in 35 000 cells of SH-SY5Y WT and G4 cell lines. In the secondary screening, the effect of the drugs was assessed in triplicate assays after 1 μm incubation of the drugs for 24 h using 10 mm glucose, 1 mm pyruvate and 2 mm glutamine. The effect on mitochondrial respiration of the three confirmed hits, florfenicol, isosorbide and methylthiouracil, was further assessed in a XF24 Seahorse flux analyzer (Agilent Technologies). WT and Gdap1−/− DRGs were seeded at a density of 1 × 105 cells/well. Bioenergetics characterization of cultured DRGs was determined through sequential addition of 6 μm oligomycin, 0.25 mm 2,4-dinitrophenol (DNP) and 1 μm antimycin A/1 μm rotenone.
Protein extraction and western blot analysis
Cells were resuspended in lysis buffer containing 25 mm HEPES, 2.5 mm EDTA, 1% Triton X-100 supplemented with protease and phosphatase inhibitor cocktails. Lysates were clarified by centrifugation at 11 000×g for 15 min. The resulting supernatants were fractionated on SDS-PAGE and transferred onto nitrocellulose membranes for immunoblot analysis. Protein concentrations were determined using Bradford reagent (Bio-Rad protein assay). The primary antibodies used were anti-GDAP1 (1:500, Abnova, Taipei, Taiwan), anti-tubulin (1:1000, Sigma, Darmstadt, Germany), anti-ATF4 (1:500, Cell Signaling, Massachussets, USA), anti-p-EIF2α (1:500, Cell Signaling), anti-eIF2α (1:500, SantaCruz, Texas, USA), anti-HSP70 (1:500, SantaCruz), anti-HSP60 (1:2000, Stressgene SPA-807, Canada), anti-GAPDH (1:20 000, Abcam, Massachusetts, USA). Peroxidase conjugated anti-mouse or anti-rabbit IgGs (Promega, 1:3000, Wisconsin, USA) were diluted in 5% non-fat dried milk in Tris-buffered saline (TBS) with 1% Tween 20 and used as secondary antibodies. The Novex® ECL (Thermo Fisher Scientific, Massachusetts, USA) system was used to visualize the bands.
Cell viability
Cell viability of SH-SY5Y cells after 24-h treatment with 1 μm of the selected compounds was assessed using the calcein AM probe (Invitrogen, Massachusetts, USA). 10 000 cells were seeded in 200 μl medium into a 96-well black plate with clear flat bottom and treated with the compounds. DMSO treatment was also added as control. The calcein solution was prepared by dissolving 50 μg of calcein in 125 μl DMSO and adding 25 μl–10 ml sterile phosphate-buffered saline (PBS). After 24 h, 100 μl of calcein solution were added to each well and, immediately after, Ex/Em = 485/530 nm fluorescence was recorded in a FLUOstar Omega (BMG LABTECH, Ortenberg, Germany) luminometer.
Mitochondrial membrane potential and ROS production
Cells (3 × 105) were incubated with 0.5 μm TMRM+ in 300 μl of FACS buffer (1 mm EDTA, 2% FBS in PBS) and processed for flow cytometry to determine ΔΨm with 1 μm florfenicol, 0.5 mm DNP or left untreated (46). For the determination of the mitochondrial production of superoxide (mtROS), 3 × 105 cells were incubated with 5 μm MitoSoX, resuspended in 300 μl of FACS buffer and processed for flow cytometry with 1 μm florfenicol, 20 nm MitoQ or left untreated. Cells were analyzed in a BD FACScan (46). For each analysis, 10 000 events were recorded. Data were analyzed in FlowJo software v10.6.2.
Animals
Mice were housed in the Animal Facility of the CBMSO with a 12-h light/12-h dark cycle and temperature of 18–23°C with 40–60% humidity. The Gdap1 knockout (Gdap1−/−) mice were previously generated and characterized (15). The Ethics Committee of Animal Experimentation (PROEX 233/19 and PROEX 352/15) and the Institutional Review Board of UAM (CEI 52-961) approved the project. Florfenicol (0.84 mg/ml) (47) and MitoQ (0.14 mg/ml) (48) treatments were administered in drinking water.
Determination of the hind-limb clasping reflex
To quantify the hind-limb clasping reflex, mice were suspended from the tip of the tail and the natural position taken by every mouse was recorded taking a picture of the animal. Then, in a blinded analysis, the angle form between the right hind limb and the longitudinal body axis of the mouse was determined using a protractor.
Assessment of mouse motor function: rotarod test
Motor performance and balance were tested using an accelerating rotarod. Each mouse underwent for 2 days the same procedure. The first day was used to train the mice in one session of 2 min, walking at 4 r.p.m. Mice that fell off after completing 2 min walking within 10 min were discarded from the experiment. The test sessions were run on day 2. During the test, the speed of the rotarod was accelerated from 4, 8, 12, 16, 20, 24, 28 to 32 r.p.m. The mice spent 1 min running at each speed. Their latency was measured. Mice that fell off six times within 60 s were discarded from the experiment.
Grip force/strength test
The grip force test was used to measure the maximum strength that could be performed by a mouse with its forelimbs by taking advantage of the animal’s tendency to grasp to surfaces. One mouse at a time was let to grasp the metallic bars and then was gently pulled away until its grasp is broken. The tests were run by the same investigator (C.N.-T.) who placed her elbow on top of the bench, and at the same distance from the apparatus, to exert the minimum force required to break the grasp. The pulling was performed sufficiently slowly to permit the mouse to build up a resistance against it. The test was repeated five times per mouse, with at least 1 min elapsing between each of the five determinations per animal (49).
Hanging test: two limbs
The forelimbs muscle strength was measured by monitoring the ability of mice to exhibit sustained limb tension to oppose their weight. Mice were placed in a 2 mm thick metal bar at 35 cm of a padded surface and time until falling was recorded. The test ended after a hanging time of 2 min was achieved or otherwise after three sessions. Longest hanging time (s) and minimal holding impulse (body mass × hang time) were calculated (50).
Hanging test: four limbs
The four limbs muscle strength was measured by monitoring the ability of mice to exhibit sustained limb tension to oppose their weight. Mice were placed in a wire grid at 35 cm of a padded surface and time until falling was recorded. The test ended after a hanging time of 2 min was achieved or otherwise after three sessions. Longest hanging time (s) and minimal holding impulse (body mass × hang time) were calculated (50).
Protein extraction from mouse tissues
For protein extraction, tissue sections were homogenized in the OMNI Bead Ruptor 24 bead mill homogenizer (Fisher Scientific, 15515799, Massachusetts, USA) in T-PER Tissue Protein Extraction Reagent (Cat. No. 78510, Thermo Scientific, Ins. Madrid, Spain,) in a 1:8 (w/v) ratio and further freeze thawed three times in liquid nitrogen. The protein concentration was determined with the Bradford reagent (Cat. No. 5000001, Bio-Rad, Inc. Madrid, Spain) using BSA as standard.
Printing and processing of RPPAs
Protein extracts from tissue biopsies were diluted with T-PER to a final protein concentration of 2 μg/μl. Before printing, samples were diluted with PBS to a final protein concentration of 0.5 μg/μl. Serially diluted protein extracts (0–1.5 μg/μl) derived from C2C12 mouse myoblast and HCT116 human colocarcinoma cells were also prepared to assess printing quality and the linear response of protein recognition by the antibodies used (30). Standard curves of BSA (0–2 μg/μl) and mouse IgGs (1–30 ng/μl) were also prepared as internal negative and positive controls, respectively.
Approximately, 1.5 nl of each sample was spotted in quintuplicated or triplicate onto nitrocellulose-coated glass slides (ONCYTE® SuperNOVA 8 pad—Grace Bio-Labs, 705118, Oregon, USA) using a iTWO-300P pico system printer (M2-Automation, Inc., Idaho, USA) equipped with a Piezo Driven Micro-Dispenser (PDMD) 30–150 pl at constant chamber humidity (RH 52%) and temperature (16°C), and plate temperature (10°C).
After printing, arrays were allowed to dry and further blocked in Super G Blocking Buffer (Cat. No. 10501, Grace Biolabs, Madrid, Spain). After, the arrays were incubated overnight at 4°C with the indicated dilutions of the primary antibodies listed in Supplementary Material, Table S2. After incubation, the arrays were washed with PBS-T and further incubated with goat anti-mouse or goat anti-rabbit highly cross-adsorbed antibodies conjugated with CF™ 647 (Sigma Aldrich, SAB4600183–SAB4600185; 1:500, Madrid, Spain). Pads incubated directly with secondary antibodies and with 0.0001% Fast Green FCF (Cat. No. F7252, Sigma Aldrich, Madrid, Spain) were used to evaluate potential unspecific binding to non-masked mouse IgGs and the total protein amount present in the spotted samples, respectively.
Microarrays were scanned using Typhoon 9410 scanner (GE Healthcare, Inc. Madrid, Spain). The mean fluorescent intensity of the spots was measured using GenePix® Pro 7 (USA) and normalized relative to the protein amount contained in the sample obtained from the FCF stained pad. After quantification, the relative fluorescent intensity was converted into arbitrary units of expressed protein/ng of protein in the extract using as standard the linear plot of the C2C12 cell line (Supplementary Material, Fig. S3) (29,51).
Statistics
The results shown are means ± SEM. Statistical analysis was performed by Student’s t-test. Statistical tests were two-sided at the 5% level of significance. Statistical analyses were performed using Excel Microsoft 365 and GraphPad Prism 7. With the exception of the hind-limb clasping reflex, the results were not evaluated blind to treatment.
Supplementary Material
Acknowledgements
We thank Dolores Lopez for her technical assistance in the mouse motor behavior experiments.
Conflict of Interest Statement. M.P.M. consults for Antipodean Pharmaceutical Inc., which is commercializing MitoQ and is the inventor of Patent US 6331532 B1, dated December18, 2001 entitled ‘Mitochondrially targeted antioxidants’. C.N.-T., J.S., F.P. and J.M.C. are inventors of UAM/CIBERER/HSJD Spanish Patents for the treatment of CMT disease. The rest of authors declare no competing interests.
Contributor Information
Cristina Nuevo-Tapioles, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Instituto de Investigación Hospital 12 de Octubre, 28041 Madrid, Spain.
Fulvio Santacatterina, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Instituto de Investigación Hospital 12 de Octubre, 28041 Madrid, Spain.
Brenda Sánchez-Garrido, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Instituto de Investigación Hospital 12 de Octubre, 28041 Madrid, Spain.
Cristina Núñez de Arenas, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Instituto de Investigación Hospital 12 de Octubre, 28041 Madrid, Spain.
Adrián Robledo-Bérgamo, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain.
Paula Martínez-Valero, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain.
Lara Cantarero, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Laboratorio de Neurogenética y Medicina Molecular - IPER, Institut de Recerca Sant Joan de Déu, 08950 Barcelona, Spain.
Beatriz Pardo, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain.
Janet Hoenicka, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Laboratorio de Neurogenética y Medicina Molecular - IPER, Institut de Recerca Sant Joan de Déu, 08950 Barcelona, Spain.
Michael P Murphy, Medical Research Council Mitochondrial Biology Unit, Wellcome Trust/MRC Building, University of Cambridge, Cambridge CB2 0XY, UK.
Jorgina Satrústegui, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain.
Francesc Palau, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Laboratorio de Neurogenética y Medicina Molecular - IPER, Institut de Recerca Sant Joan de Déu, 08950 Barcelona, Spain; Department of Genetic and Molecular Medicine - IPER, Hospital Sant Joan de Déu, 08950 Barcelona, Spain; Clinic Institute of Medicine and Dermatology (ICMiD), Hospital Clínic, 09036 Barcelona, Spain; Division of Pediatrics, School of Medicine and Health Sciences, University of Barcelona, 08036 Barcelona, Spain.
José M Cuezva, Departamento de Biología Molecular, Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain; Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain; Instituto de Investigación Hospital 12 de Octubre, 28041 Madrid, Spain.
Author contributions
C.N.-T., F.S., J.S., F.P. and J.M.C. designed experiments and interpreted results. C.N.-T., F.S., B.S.-G., C.N.d.A., A.R.-B., L.C. and P.M.-V. performed experiments. B.P. and J.H. assisted in mouse experiments. M.P.M. contributed to the design and supplied MitoQ. J.S., F.P. and J.M.C. secured funding and C.N.-T. and J.M.C. wrote the paper. All authors read, contributed to and approved the final manuscript.
Funding
ACCI (grant ER18TRL713 to F.P., J.S. and J.M.S.) from CIBERER-Instituto de Salud Carlos III, Spain; Ministerio de Economía y Competitividad (MINECO) (SAF2016-75916-R and PID2019-108674RB-100 to J.M.C, and SAF2015-66625-R to F.P.), Spain.
References
- 1. Sleire, L., Forde, H.E., Netland, I.A., Leiss, L., Skeie, B.S. and Enger, P.O. (2017) Drug repurposing in cancer. Pharmacol. Res., 124, 74–91. [DOI] [PubMed] [Google Scholar]
- 2. Pareyson, D., Saveri, P. and Pisciotta, C. (2017) New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol., 30, 471–480. [DOI] [PubMed] [Google Scholar]
- 3. Pareyson, D. and Solari, A. (2009) Charcot-Marie-Tooth disease type 1A: is ascorbic acid effective? Lancet Neurol., 8, 1075–1077. [DOI] [PubMed] [Google Scholar]
- 4. Rossor, A.M., Liu, C.H., Petzold, A., Malaspina, A., Laura, M., Greensmith, L. and Reilly, M.M. (2016) Plasma neurofilament heavy chain is not a useful biomarker in Charcot-Marie-Tooth disease. Muscle Nerve, 53, 972–975. [DOI] [PubMed] [Google Scholar]
- 5. Boutary, S., Echaniz-Laguna, A., Adams, D., Loisel-Duwattez, J., Schumacher, M., Massaad, C. and Massaad-Massade, L. (2021) Treating PMP22 gene duplication-related Charcot-Marie-Tooth disease: the past, the present and the future. Transl. Res., 227, 100–111. [DOI] [PubMed] [Google Scholar]
- 6. Rossor, A.M., Tomaselli, P.J. and Reilly, M.M. (2016) Recent advances in the genetic neuropathies. Curr. Opin. Neurol., 29, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cuesta, A., Pedrola, L., Sevilla, T., Garcia-Planells, J., Chumillas, M.J., Mayordomo, F., LeGuern, E., Marin, I., Vilchez, J.J. and Palau, F. (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet., 30, 22–25. [DOI] [PubMed] [Google Scholar]
- 8. Rzepnikowska, W. and Kochanski, A. (2018) A role for the GDAP1 gene in the molecular pathogenesis of Charcot-Marie-Tooth disease. Acta Neurobiol. Exp. (Wars), 78, 1–13. [PubMed] [Google Scholar]
- 9. Claramunt, R., Pedrola, L., Sevilla, T., López de Munain, A., Berciano, J., Cuesta, A., Sánchez-Navarro, B., Millán, J.M., Saifi, G.M., Lupski, J.R. et al. (2005) Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet., 42, 358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baxter, R.V., Ben Othmane, K., Rochelle, J.M., Stajich, J.E., Hulette, C., Dew-Knight, S., Hentati, F., Ben Hamida, M., Bel, S., Stenger, J.E. et al. (2002) Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet., 30, 21–22. [DOI] [PubMed] [Google Scholar]
- 11. Marco, A., Cuesta, A., Pedrola, L., Palau, F. and Marin, I. (2004) Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol. Biol. Evol., 21, 176–187. [DOI] [PubMed] [Google Scholar]
- 12. Pedrola, L., Espert, A., Wu, X., Claramunt, R., Shy, M.E. and Palau, F. (2005) GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet., 14, 1087–1094. [DOI] [PubMed] [Google Scholar]
- 13. Pla-Martin, D., Rueda, C.B., Estela, A., Sanchez-Piris, M., Gonzalez-Sanchez, P., Traba, J., de la Fuente, S., Scorrano, L., Renau-Piqueras, J., Alvarez, J. et al. (2013) Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis., 55, 140–151. [DOI] [PubMed] [Google Scholar]
- 14. Cantarero, L., Juárez-Escoto, E., Civera-Tregón, A., Rodríguez-Sanz, M., Roldán, M., Benítez, R., Hoenicka, J. and Palau, F. (2021) Mitochondrial-lysosome membrane contact sites are defective in GDAP1-related Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet., 29, 3589–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barneo-Muñoz, M., Juárez, P., Civera-Tregón, A., Yndriago, L., Pla-Martin, D., Zenker, J., Cuevas-Martín, C., Estela, A., Sánchez-Aragó, M., Forteza-Vila, J. et al. (2015) Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of Charcot-Marie-Tooth neuropathy. PLoS Genet., 11, e1005115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez-Sanchez, P., Pla-Martin, D., Martinez-Valero, P., Rueda, C.B., Calpena, E., Del Arco, A., Palau, F. and Satrustegui, J. (2017) CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca(2+) entry-stimulated respiration. Sci. Rep., 7, 42993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandez-Lizarbe, S., Civera-Tregon, A., Cantarero, L., Herrer, I., Juarez, P., Hoenicka, J. and Palau, F. (2019) Neuroinflammation in the pathogenesis of axonal Charcot-Marie-Tooth disease caused by lack of GDAP1. Exp. Neurol., 320, 113004. [DOI] [PubMed] [Google Scholar]
- 18. Noack, R., Frede, S., Albrecht, P., Henke, N., Pfeiffer, A., Knoll, K., Dehmel, T., Meyer Zu Horste, G., Stettner, M., Kieseier, B.C. et al. (2012) Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet., 21, 150–162. [DOI] [PubMed] [Google Scholar]
- 19. Niemann, A., Huber, N., Wagner, K.M., Somandin, C., Horn, M., Lebrun-Julien, F., Angst, B., Pereira, J.A., Halfter, H., Welzl, H. et al. (2014) The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain, 137, 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cassereau, J., Chevrollier, A., Gueguen, N., Malinge, M.C., Letournel, F., Nicolas, G., Richard, L., Ferre, M., Verny, C., Dubas, F. et al. (2009) Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics, 10, 145–150. [DOI] [PubMed] [Google Scholar]
- 21. Cassereau, J., Chevrollier, A., Codron, P., Goizet, C., Gueguen, N., Verny, C., Reynier, P., Bonneau, D., Lenaers, G. and Procaccio, V. (2020) Oxidative stress contributes differentially to the pathophysiology of Charcot-Marie-Tooth disease type 2K. Exp. Neurol., 323, 113069. [DOI] [PubMed] [Google Scholar]
- 22. Soldevilla, B., Cuevas-Martin, C., Ibanez, C., Santacatterina, F., Alberti, M.A., Simo, C., Casasnovas, C., Marquez-Infante, C., Sevilla, T., Pascual, S.I. et al. (2017) Plasma metabolome and skin proteins in Charcot-Marie-Tooth 1A patients. PLoS One, 12, e0178376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nuevo-Tapioles, C., Santacatterina, F., Stamatakis, K., Nunez de Arenas, C., Gomez de Cedron, M., Formentini, L. and Cuezva, J.M. (2020) Coordinate beta-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun., 11, 3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gonzalez-Sanchez, P., Satrustegui, J., Palau, F. and Del Arco, A. (2019) Calcium deregulation and mitochondrial bioenergetics in GDAP1-related CMT disease. Int. J. Mol. Sci., 20, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Syriopoulou, V.P., Harding, A.L., Goldmann, D.A. and Smith, A.L. (1981) In vitro antibacterial activity of fluorinated analogs of chloramphenicol and thiamphenicol. Antimicrob. Agents Chemother., 19, 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu, D., Cao, S., Zhang, G., Xiao, Y., Liu, S. and Shang, Y. (2017) Florfenicol-induced mitochondrial dysfunction suppresses cell proliferation and autophagy in fibroblasts. Sci. Rep., 7, 13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pareyson, D. and Marchesi, C. (2009) Natural history and treatment of peripheral inherited neuropathies. Adv. Exp. Med. Biol., 652, 207–224. [DOI] [PubMed] [Google Scholar]
- 28. Guyenet, S.J., Furrer, S.A., Damian, V.M., Baughan, T.D., La Spada, A.R. and Garden, G.A. (2010) A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp., 39, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Santacatterina, F., Chamorro, M., de Arenas, C., Navarro, C., Martín, M.A., Cuezva, J.M. and Sánchez-Aragó, M. (2015) Quantitative analysis of proteins of metabolism by reverse phase protein microarrays identifies potential biomarkers of rare neuromuscular diseases. J. Transl. Med., 13, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Santacatterina, F., Torresano, L., Nunez-Salgado, A., Esparza-Molto, P.B., Olive, M., Gallardo, E., Garcia-Arumi, E., Blazquez, A., Gonzalez-Quintana, A., Martin, M.A. et al. (2018) Different mitochondrial genetic defects exhibit the same protein signature of metabolism in skeletal muscle of PEO and MELAS patients: a role for oxidative stress. Free Radic. Biol. Med., 126, 235–248. [DOI] [PubMed] [Google Scholar]
- 31. Pareyson, D. and Marchesi, C. (2009) Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol., 8, 654–667. [DOI] [PubMed] [Google Scholar]
- 32. Morena, J., Gupta, A. and Hoyle, J.C. (2019) Charcot-Marie-Tooth: from molecules to therapy. Int. J. Mol. Sci., 20, 3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nelis, E., Van Broeckhoven, C., De Jonghe, P., Lofgren, A., Vandenberghe, A., Latour, P., Le Guern, E., Brice, A., Mostacciuolo, M.L., Schiavon, F. et al. (1996) Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur. J. Hum. Genet., 4, 25–33. [DOI] [PubMed] [Google Scholar]
- 34. Attarian, S., Vallat, J.M., Magy, L., Funalot, B., Gonnaud, P.M., Lacour, A., Pereon, Y., Dubourg, O., Pouget, J., Micallef, J. et al. (2016) Erratum to: an exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet J. Rare Dis., 11, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. d'Ydewalle, C., Krishnan, J., Chiheb, D.M., Van Damme, P., Irobi, J., Kozikowski, A.P., Vanden Berghe, P., Timmerman, V., Robberecht, W. and Van Den Bosch, L. (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med., 17, 968–974. [DOI] [PubMed] [Google Scholar]
- 36. Mo, Z., Zhao, X., Liu, H., Hu, Q., Chen, X.Q., Pham, J., Wei, N., Liu, Z., Zhou, J., Burgess, R.W. et al. (2018) Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy. Nat. Commun., 9, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rocha, A.G., Franco, A., Krezel, A.M., Rumsey, J.M., Alberti, J.M., Knight, W.C., Biris, N., Zacharioudakis, E., Janetka, J.W., Baloh, R.H. et al. (2018) MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science, 360, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Snow, B.J., Rolfe, F.L., Lockhart, M.M., Frampton, C.M., O'Sullivan, J.D., Fung, V., Smith, R.A., Murphy, M.P., Taylor, K.M. and Protect Study Group (2010) A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord., 25, 1670–1674. [DOI] [PubMed] [Google Scholar]
- 39. Sanchez-Gonzalez, C., Nuevo-Tapioles, C., Herrero Martin, J.C., Pereira, M.P., Serrano Sanz, S., Ramirez de Molina, A., Cuezva, J.M. and Formentini, L. (2020) Dysfunctional oxidative phosphorylation shunts branched-chain amino acid catabolism onto lipogenesis in skeletal muscle. EMBO J., 39, e103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith, R.A. and Murphy, M.P. (2010) Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci., 1201, 96–103. [DOI] [PubMed] [Google Scholar]
- 41. Smith, R.A., Porteous, C.M., Gane, A.M. and Murphy, M.P. (2003) Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U. S. A., 100, 5407–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takahashi, R., Ikeda, T., Hamaguchi, A., Iwasa, K. and Yamada, M. (2012) Coenzyme Q10 therapy in hereditary motor sensory neuropathy type VI with novel mitofusin 2 mutation. Intern. Med., 51, 791–793. [DOI] [PubMed] [Google Scholar]
- 43. Rossman, M.J., Santos-Parker, J.R., Steward, C.A.C., Bispham, N.Z., Cuevas, L.M., Rosenberg, H.L., Woodward, K.A., Chonchol, M., Gioscia-Ryan, R.A., Murphy, M.P. et al. (2018) Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension, 71, 1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mao, P., Manczak, M., Shirendeb, U.P. and Reddy, P.H. (2013) MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta, 1832, 2322–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cabrera, J.R., Viejo-Borbolla, A., Martinez-Martin, N., Blanco, S., Wandosell, F. and Alcami, A. (2015) Secreted herpes simplex virus-2 glycoprotein G modifies NGF-TrkA signaling to attract free nerve endings to the site of infection. PLoS Pathog., 11, e1004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Formentini, L., Sánchez-Aragó, M., Sánchez-Cenizo, L. and Cuezva, J.M. (2012) The mitochondrial ATPase inhibitory factor 1 (IF1) triggers a ROS-mediated retrograde pro-survival and proliferative response. Mol. Cell, 45, 731–742. [DOI] [PubMed] [Google Scholar]
- 47. Shuang, G., Yu, S., Weixiao, G., Dacheng, W., Zhichao, Z., Jing, L. and Xuming, D. (2011) Immunosuppressive activity of florfenicol on the immune responses in mice. Immunol. Investig., 40, 356–366. [DOI] [PubMed] [Google Scholar]
- 48. Formentini, L., Santacatterina, F., Nunez de Arenas, C., Stamatakis, K., Lopez-Martinez, D., Logan, A., Fresno, M., Smits, R., Murphy, M.P. and Cuezva, J.M. (2017) Mitochondrial ROS production protects the intestine from inflammation through functional M2 macrophage polarization. Cell Rep., 19, 1202–1213. [DOI] [PubMed] [Google Scholar]
- 49. Smith, J.P., Hicks, P.S., Ortiz, L.R., Martinez, M.J. and Mandler, R.N. (1995) Quantitative measurement of muscle strength in the mouse. J. Neurosci. Methods, 62, 15–19. [DOI] [PubMed] [Google Scholar]
- 50. Aartsma-Rus, A. and van Putten, M. (2014) Assessing functional performance in the mdx mouse model. J. Vis. Exp., 85, 51303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Santacatterina, F., Sanchez-Arago, M., Catalan-Garcia, M., Garrabou, G., de Arenas, C.N., Grau, J.M., Cardellach, F. and Cuezva, J.M. (2017) Pyruvate kinase M2 and the mitochondrial ATPase inhibitory factor 1 provide novel biomarkers of dermatomyositis: a metabolic link to oncogenesis. J. Transl. Med., 15, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.