

Abstract
We have previously established induced pluripotent stem cell (iPSC) models of Huntington’s disease (HD), demonstrating CAG-repeat-expansion-dependent cell biological changes and toxicity. However, the current differentiation protocols are cumbersome and time consuming, making preparation of large quantities of cells for biochemical or screening assays difficult. Here, we report the generation of immortalized striatal precursor neurons (ISPNs) with normal (33) and expanded (180) CAG repeats from HD iPSCs, differentiated to a phenotype resembling medium spiny neurons (MSN), as a proof of principle for a more tractable patient-derived cell model. For immortalization, we used co-expression of the enzymatic component of telomerase hTERT and conditional expression of c-Myc. ISPNs can be propagated as stable adherent cell lines, and rapidly differentiated into highly homogeneous MSN-like cultures within 2 weeks, as demonstrated by immunocytochemical criteria. Differentiated ISPNs recapitulate major HD-related phenotypes of the parental iPSC model, including brain-derived neurotrophic factor (BDNF)-withdrawal-induced cell death that can be rescued by small molecules previously validated in the parental iPSC model. Proteome and RNA-seq analyses demonstrate separation of HD versus control samples by principal component analysis. We identified several networks, pathways, and upstream regulators, also found altered in HD iPSCs, other HD models, and HD patient samples. HD ISPN lines may be useful for studying HD-related cellular pathogenesis, and for use as a platform for HD target identification and screening experimental therapeutics. The described approach for generation of ISPNs from differentiated patient-derived iPSCs could be applied to a larger allelic series of HD cell lines, and to comparable modeling of other genetic disorders.
Introduction
Huntington’s disease (HD) is a fatal, progressive, neurodegenerative disorder caused by a CAG-repeat expansion in the Huntingtin gene (HTT). The disease is characterized by motor abnormalities, cognitive deficits and emotional changes (1,2). HD is recognized as a model disease to study neurodegenerative disorder, since it is caused by a mutation in a single gene and shows correlation between triplet repeat expansion length and age of disease onset (1,3). HD exhibits selective atrophy of the striatum, mainly affecting the caudate and putamen, causing death of up to 95% of GABAergic medium spiny neurons (MSNs) projecting to the globus pallidus and substantia nigra. While the HTT gene mutation resulting in HD was identified in 1993 (4), disease pathogenesis remains incompletely understood and there is currently no disease-modifying therapy available.
HD cell models represent useful tools for understanding the cell biology of the disorder and testing potential therapeutics. While previous HD cell models have provided useful information, most of them have not been derived from patients, or focused on clinically relevant MSNs (5–7). Human embryonic stem cells (hESCs) have been generated from HD-affected embryos (CAG-repeat range from 37 to 51 repeats) (8–10); however, hESCs are hindered by ethical concerns and the relative paucity of HD-related phenotypes (11). In contrast, induced pluripotent stem cells (iPSCs) can be obtained from adult somatic cells, such as skin fibroblasts, by reprogramming with a set of transcription factors into pluripotent cells. Such pluripotent cells can be differentiated into various mature cell types, including neurons, and can be used for modeling human hereditary diseases including neurodegenerative disorders (3,12,13).
The NINDS HD iPSC Consortium has conducted an extensive series of studies on iPSCs generated from HD patients with a range of CAG-repeat expansion lengths, at the neural precursor stage and after differentiation to striatal phenotypes (14–16). The HD iPSCs show patterns of gene and protein expression distinct from controls, and consistent with pathways known to be altered in HD, such as neurodevelopment, cellular transport, proteostasis, GABA and glutamate signaling, axonal guidance and calcium influx, and RNA and cellular metabolism including lower expression of glycolytic enzymes. In addition, the patterns of gene and protein expression correlate with CAG-repeat expansion length. HD iPSCs differentiated into neural cells also exhibit disease-associated phenotypes such as changes in electrophysiology, cell adhesion, and metabolism, including decreased ATP levels. The CAG-repeat expansion in these iPSC lines corresponds with susceptibility to cell stressors (i.e. brain-derived neurotrophic factor (BDNF) withdrawal, glutamate toxicity, oxidative stress and autophagy inhibition) similar to the repeat expansion length dependence in human HD patients. The patient-derived HD iPSC model represents a substantial advance over previous cell models of human genetic disorders [for review see Ref. (17)]. Reversion of HD-associated phenotypes in isogenic cell lines derived by genetic correction of HD iPSCs or HD iPSC-derived neurons treated with mutant allele-specific antisense oligonucleotides (ASOs) confirms the suitability of this model (18–20). Rescue of HD-associated phenotypes in differentiated HD iPSCs by small molecules has also been demonstrated in numerous studies by the HD iPSC Consortium and others (15,16,20–25).
Nevertheless, HD iPSCs still have several significant disadvantages—very complicated differentiation protocols, limited yield of differentiated cells, and substantial variability in cell phenotypes in differentiated cell populations and among different experiments. These disadvantages limit the use of this model for comprehensive biochemical studies, or for development of screenable cell-phenotypic assays.
We hypothesized that conditionally immortalized cell lines—generated by co-expression of hTERT and regulated c-Myc from patient-derived human iPSCs differentiated towards subtype specific striatal precursors in vitro—would recapitulate the main features of the parental iPSC model, including ability to be differentiated to disease-affected cell types, demonstrating disease-associated phenotypes. Here, we report proof of principle for the generation and characterization of a new HD cell model of immortalized striatal precursor neurons (ISPN) derived from differentiated HD and non-disease control human iPSCs. The ISPN lines characterized here may be suitable for studying HD-related cellular pathogenesis and for screening for experimental therapeutics.
Results
Generating human HD iPSC-derived immortalized striatal precursor lines
Immortalization of mouse primary neurons as well as cortical, hippocampal and striatal neurons from human embryonic brains has been successfully used to generate neural cell lines (6,26,27). We hypothesized that similar techniques could be used to immortalize striatal neurons derived by differentiation of human pluripotent cell lines such as HD iPSCs generated and characterized by the HD iPSC Consortium.
Primary neuronal cells can be immortalized using various approaches such as somatic fusion (28) or expression of immortalizing genes/oncogenes including v-myc (29), c-myc (30,31), SV40 large T (6,32–35) and hTERT (36,37). However, overexpression of hTERT alone was not sufficient to immortalize some progenitor cells such as human hematopoietic stem cells, while co-expression of hTERT with human papillomavirus type 16 (HPV16) E6 and E7 oncogenes was reported to immortalize the cells and prevent accumulation of multiple chromosomal abnormalities (38).
To generate ISPN lines, we chosen retroviral transduction of HD and non-disease control iPSCs differentiated into MSN-like phenotypes with vectors encoding c-Myc fused to tamoxifen-sensitive hormone binding domain of the estrogen receptor (ER) (39) and human telomerase reverse transcriptase (hTERT). Regulation of c-Myc-ER expression by 4-hydroxytamoxifen (4-OHT) allows for gene silencing during differentiation by 4-OHT withdrawal and prevents potential disruption of differentiation shown in some cell lines with constitutive expression of immortalizing proto-oncogenes (38,40). Tamoxifen inducible c-Myc-ER, a chimeric construct engineered by fusion of human transcription factor c-Myc to the ER, is shown to efficiently immortalize primary neurons without transformation (26). hTERT is hTERT required for elongation of chromosomal ends and maintenance of genomic integrity (41). Expression of exogenous hTERT in certain normal human somatic cell types stabilizes telomere length and allows for indefinite growth (36,42,43). Using the Consortium differentiation protocol (14) and validated retroviral vectors encoding immortalizing genes c-Myc-ER and hTERT, we developed a scheme for generation of such immortalized lines (Fig. 1).
Figure 1 .
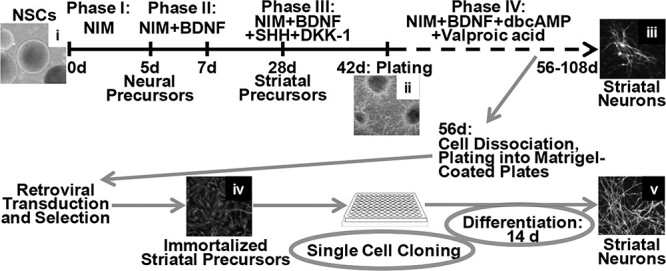
Scheme for generation of immortalized striatal precursor lines. NSCs (also designated as EZ-spheres) were derived from HD and non-disease control iPSCs and differentiated towards striatal neurons for 56 days as described in Materials and Methods. The differentiated cells were dissociated with TrypLE Select solution and plated into Matrigel-coated tissue culture plates. After recovery, the plated cells were transduced with retroviral vectors encoding immortalizing genes and genes for resistance to selective antibiotics and, following the corresponding drug selection, used for single cell cloning. Clones were tested for expression of striatal progenitor markers and differentiation potential. Selected clones were expanded and used for further characterization. Panels show representative images of the following cell cultures: NSCs derived from HD iPSCs (i); NSCs differentiated to the end of Phase III after plating into Matrigel-coated tissue culture plates (ii); NSCs differentiated to MSN-like phenotypes after ICC for MSN markers DARPP32/MAP2 (iii); immortalized striatal precursors after ICC for Nestin (iv); and immortalized striatal precursors differentiated into MSN-like phenotypes after ICC for MSN markers DARPP32/MAP2 (v). NIM—neural induction medium.
We used iPSC lines derived from a healthy donor (33 CAG repeats) and a juvenile onset HD patient (180 CAG repeats) (Table 1) (14)from the same pedigree per the NINDS/Coriell Institute catalog. Neural stem cells (NSCs, also designated as EZ-spheres) derived from HD and control iPSCs were differentiated to an early stage striatal neuron phenotype for 56 days (Fig. 1). The differentiated iPSCs were simultaneously transduced with retroviral vectors encoding c-Myc-ER and hTERT and genes for resistance to selective antibiotics puromycin and neomycin, respectively, followed by drug selection and expansion of surviving cells. Transduction with hTERT alone was not sufficient to immortalize the differentiated iPSCs, as cells did not survive through the selection process for more than 2–3 weeks. In contrast, transduction with c-Myc-ER alone or in combination with hTERT resulted in cell survival in the presence of selective antibiotics for approximately 1 month after transduction, which allowed for expansion of the surviving cells in culture. Therefore, for further characterization we chose cells transduced with both c-Myc-ER and hTERT. C-Myc and hTERT expression in transduced lines was confirmed by Western blotting (Supplementary Material, Fig. S1A).
Table 1.
HD iPSC Consortium cell lines used for generation of ISPN (HD iPSC Consortium, 2012)
| iPSC line | Coriell fibroblasts # | Repeat length | Donor gender | Donor age |
|---|---|---|---|---|
| CTR 33i.8 | GM02183 | 33, 18 | Female | ~ 21 YR |
| HD 180i.5 | GM09197 | 180, 18 | Male | ~ 6 YR |
We also tested two established cell culture media compositions previously used to propagate human immortalized neural precursors [Supplementary Material, Tables S1 and S2, modified from El-Akabawy (44) and Dashinimaev et al. (45)] which we designated as Striatal Cell Medium (SCM) 1 and 2, respectively. Both media compositions were supplemented with 4-hydroxytamoxifen. Cells maintained in SCM-1 did not survive for more than a week, while cells in SCM-2 showed stable survival and growth. SCM-2 medium with minor changes was therefore used in all further experiments. After transduction, we also lowered the oxygen level in the incubator from 21% to 5%, since low oxygen was found to be optimal for human neural precursors (45). Cells that survived over 2 weeks of drug selection were expanded to sub-confluent cultures and used for single-cell cloning performed by serial dilution in 96-well plates.
For further differentiation to MSN-like phenotypes, we used the established HD iPSC differentiation protocol (Fig. 1). After 4 days of induction of differentiation, we replaced the ‘Phase 3’ tamoxifen-free medium with ‘Phase 4’ medium for the following 10 days. The differentiated clones were then screened for the expression of mature neuron and MSN markers, MAP2 and DARPP32, by dot blot analysis (Supplementary Material, Fig. S1B). Clones positive for both markers were expanded, cryopreserved, and used for further characterization. To distinguish our new HD cell model from the parental iPSC model, it was designated ISPN.
ISPNs exhibit a striatal precursor-like phenotype before differentiation and MSN-like phenotype after differentiation
The immortalized cell lines derived from HD iPSCs became proliferative and were grown as homogeneous adherent cultures without any morphological sign of transformation, such as loss of contact inhibition of proliferation, decreased adhesion or change of morphology from polygonal to spherical. The morphology of ISPNs before and after differentiation was similar to striatal precursors and mature neurons, respectively, recapitulating morphological changes during striatal differentiation in human iPSCs and immortalized striatal precursors derived from non-disease human embryonic brains (26,27) (Fig. 2A).
Figure 2 .
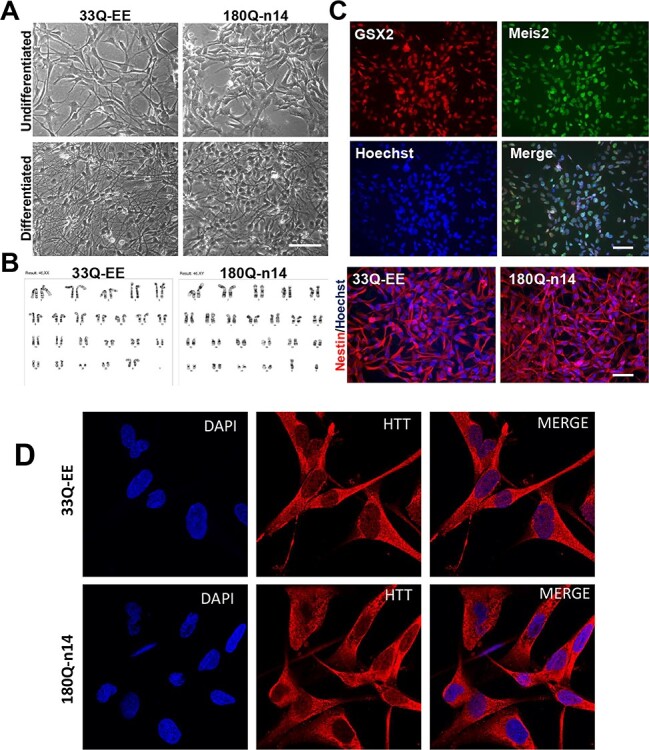
Characterization of selected clones of control and HD ISPNs. (A) Phase-contrast images of non-disease control (33Q, clone EE) and HD (180Q, clone n14) ISPNs demonstrate striatal precursors phenotypes before (undifferentiated) and neural phenotypes after differentiation (differentiated). Differentiation to MSN-like phenotype was performed for 14 days as described in Materials and Methods. Scale bar, 100 μm. (B) G-banding analysis demonstrates normal karyotype of ISPNs from representative karyotype spread images. (C) Immunofluorescence analysis of ISPNs shows expression of striatal precursor markers GSX2 and Meis2 in undifferentiated HD cells (180Q, clone n14) (upper four panels) as well as neural precursor marker Nestin in undifferentiated control (33Q, clone EE) and HD (180Q, clone n14) cells (bottom panels). Scale bars, 100 μm. (D) Immunofluorescence analysis (with MCA2051 Ab) of undifferentiated ISPNs shows expression of HTT predominantly in the cytoplasm of control (33Q, clone EE) and HD (180Q, clone n14) cells. Confocal images.
G-band karyotype analysis showed normal karyotypes in 33Q and 180Q ISPN lines with 46, XX and 46, XY chromosomes, respectively (Fig. 2B and Supplementary Material, Fig. S1D). We also verified the number of CAG-repeats in control and HD ISPN clones by CAG-repeat genotyping. Results confirmed 33/18 CAGs in control ISPN lines and 220/18 CAGs in HD ISPN lines, similar to the parent HD iPSC model (14). (We will still refer to the expanded repeat line as ‘180 CAG’).
To compare our new ISPN model to the parental iPSC model, we characterized selected clones by the expression of striatal precursor markers in the undifferentiated, and on striatal markers in the differentiated state using immunocytochemistry.
Most of the immortalized cells express neuroepithelial marker Nestin and early lateral ganglionic eminence (LGE) markers, homeobox genes GSX2 and Meis2, as well as Ctip2 (BCL11b) and ASCL1 (MASH1) (46). Expression of the neural precursor marker Nestin as well as striatal precursor markers GSX2 and Meis2 was detected in essentially all non-differentiated ISPNs (Fig. 2C and Supplementary Material, Fig. S1C). Our quantification using ImageJ software confirmed that 99.1 ± 1.4% of the total cell populations express both GSX2 and Meis2 and 98.3 ± 1.1% Ctip2 markers. Based on morphological and immunocytochemical analysis, we found that non-differentiated ISPNs display LGE precursor-like phenotypes which reverted from the differentiated neural phenotype of HD iPSCs during immortalization. We suggest that this phenotype resembles the ‘Phase III’ (Fig. 1) stage of iPSC differentiation from the HD iPSC Consortium differentiation protocol (14).
HTT protein expression within cells was also evaluated using immunostaining with an antibody to total HTT (MCA2051, Fig. 2D). In both control and HD cells we observed mostly cytoplasmic with some nuclear HTT localization, consistent with previous reports (47,48).
ISPN lines were then differentiated as described above with minor modifications (Fig. 1). To optimize the ‘Phase 3’ medium composition, we replaced recombinant Sonic Hedgehog with purmorphamine. Purmorphamine is known to significantly upregulate the Hedgehog signaling pathway (GLI1 expression) and enhanced differentiation of the immortalized striatal cell line STROC05 into mature striatal phenotypes (44). We also substituted the recombinant Dickkopf-related protein 1, an inhibitor of the canonical WNT pathway, with IWR-1, a small molecule inhibitor of WNT pathway, that has been shown to promote intermediate progenitor domain specification (49).
Expression of MAP2 and DARPP32 dramatically increased after differentiation, being almost undetectable in the undifferentiated ISPNs (Fig. 3A and Supplementary Material, Fig. S2A). DARPP32 and MAP2 expression levels in ISPNs were further confirmed by Western blotting (Fig. 4A). DARPP32 protein increased from an undetectable level, to detectable at week one and further increased across 3 weeks. DARPP32 induction was significantly greater if the ISPNs were differentiated at normal atmospheric oxygen levels of 21% instead of 5% O2 (Fig. 4A and B). MAP2 protein showed a similar increase; however, we did not observe any oxygen concentration dependency (Fig. 4A). Notably, ISPNs differentiated for 2 weeks demonstrated greater expression of mature neuron and MSN markers compared to parental iPSCs differentiated for over 72 days. Over 90% of the differentiated ISPNs were positive for MAP2a/2b, Ctip2, and DARPP32 (95.2 ± 4.5%) and more than 80% were positive for GABA (89.5 ± 6.7%), while only about 10% of the parental iPSCs were TUJ1/MAP2 positive and 5% were DARPP32/Ctip2 positive (14).
Figure 3 .
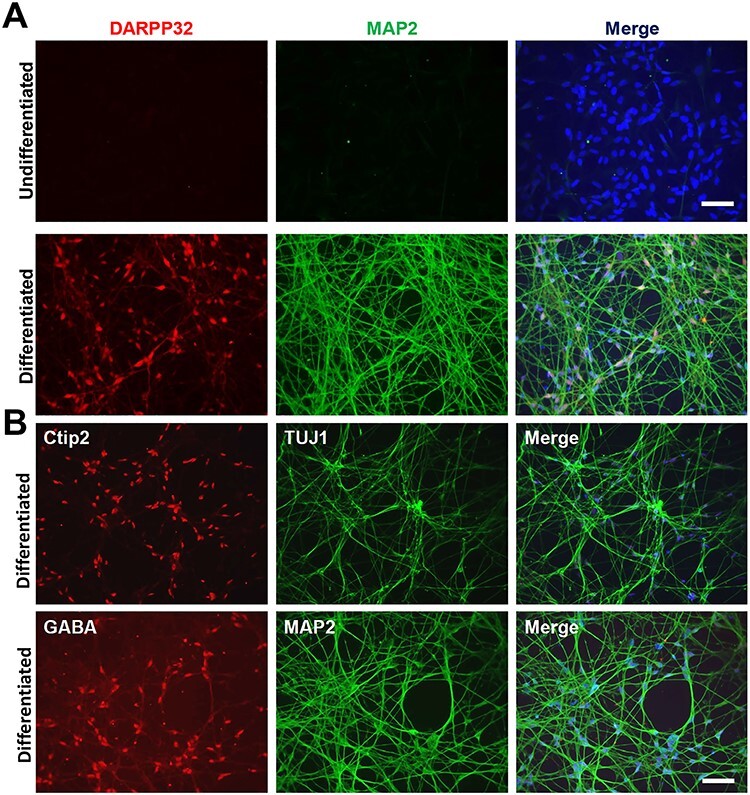
Immunophenotyping of differentiated ISPNs demonstrates MSN-like phenotype of selected clones. (A) Immunofluorescence analysis of ISPNs demonstrates expression of MAP2 (mature neuron marker) and DARPP32 (MSN marker) in differentiated ISPNs (180Q, clone n14) (lower row). No expression of the markers was observed in the undifferentiated ISPNs (upper row). (B) Co-expression of other MSN markers Ctip2 and GABA with neural markers TUJ1 and MAP2 was also observed in the differentiated ISPNs (180Q, clone n14). Nuclei stained with Hoechst. Scale bars, 100 μm.
Figure 4 .
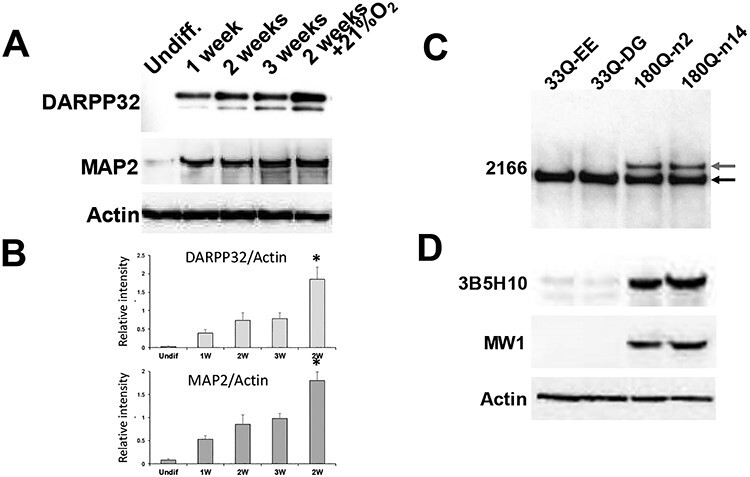
Western blots show expression of MSN-specific genes and proteins in differentiated ISPNs. (A) Western blot for MSN marker DARPP32 and mature neurons marker MAP2 confirms expression of the markers in differentiated versus undifferentiated (Undiff.) ISPNs with gradual increase from week 1 to week 3. Notably, cells differentiated at atmospheric oxygen level (21% O2) show higher levels of the markers on week 2 compared to cells maintained at 5% O2 even for 3 weeks. Actin used as a loading control. (B) Densitometric quantification normalized to actin confirms significant increase of the MSN marker levels in ISPNs differentiated at atmospheric oxygen level. N = 3 independent differentiations. (C) Western blot analysis of ISPNs using anti-HTT MAB2166 antibody (epitope: amino acids 441–455) demonstrates specific expression of mutant HTT (upper band, top red arrow) in HD ISPNs (180Q, clones n2 and n14). Lower band (bottom black arrow) corresponds to normal HTT. (D) Western blots of polyglutamine expression in ISPN lines using two different (3B5H10 and MW1) anti-polyQ antibodies demonstrate mutant HTT in the HD derived lines. Actin used as a loading control.
We confirmed expression of both wild-type and mutant forms of HTT protein by Western blotting using an antibody to total HTT (Fig. 4C). Expression of mHTT in HD ISPNs was confirmed by Western blot using two independent poly-Q recognizing antibodies (Fig. 4D).
Proteome and RNA-seq analysis demonstrate separation of HD and control ISPNs and confirm the upregulation of MSN markers upon differentiation
We compared the proteomes and transcriptomes in non-differentiated and differentiated control and HD cell lines by mass spectrometry (MS) using isobaric mass tags (tandem mass tags, TMTs) and RNA-seq. All experiments were performed on two HD (n2 and n14) and two normal control (EE and DG) ISPN lines. The abundance of over 7000 proteins, identified at 1% false discovery rate (FDR), in non-differentiated and differentiated ISPN samples were compared in each of two separate TMT MS analyses. The data from both TMT runs were combined for downstream analysis. In RNA-seq analysis, 4788 genes were significantly differentially expressed (adjusted P-value < 0.01) between HD and control iPSCs, 5323 genes were differentially expressed between HD and control cells in differentiated MSNs, while 7461 genes were significantly differentially expressed between differentiated and undifferentiated HD cells. Both TMT and RNA-seq analysis demonstrated separation of HD versus control samples by principal component analysis, and greater separation after differentiation (Fig. 5A and B). We searched for the previously reported aberrantly spliced exon-1 HTT1a transcript (50,51) in our RNAseq data. However, it was not detected.
Figure 5 .
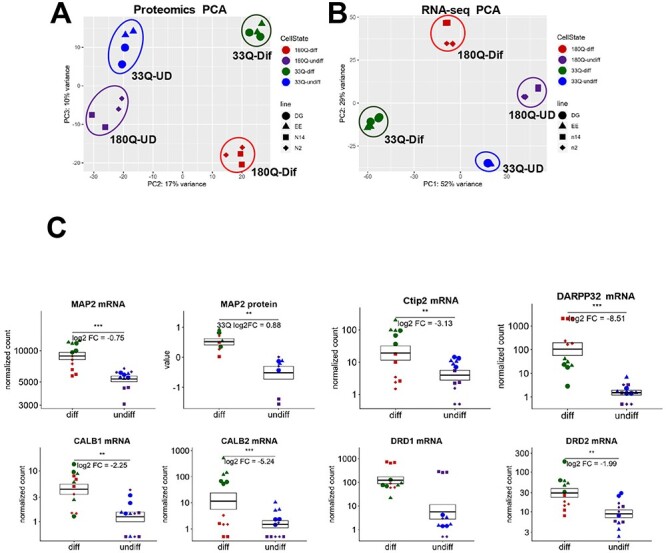
Proteome and RNA-seq analysis demonstrate separation of HD and control ISPNs and confirm the upregulation of MSN markers upon differentiation. (A) Proteomics principal component analysis of log2 normalized protein expression values shows separation of HD (red and purple) versus non-disease control (green and blue) ISPNs along principal component 3. Shapes of points for different lines are shown in graph. (B) RNA-seq principal component analysis of log2 normalized global gene count values demonstrates clear separation between undifferentiated HD and non-disease ISPNs. (C) Upregulation of MSN markers MAP2, DARPP32, Ctip2, Calbindin 1 and 2, and DRD2 but not DRD1 in differentiated ISPNs by Omics analysis. Y-axes for mRNA graphs are on a log scale and represent read counts normalized by size factor. Y-axes for protein graphs represent log2 normalized TMT reporter ion signal intensity values. Colors and shapes of points as in (A, B). *** adjusted P-value <1e-5, ** adjusted P-value < 0.01.
Omics data demonstrate that major MSN markers (MAP2, DARPP32 and Ctip2) are significantly upregulated in ISPNs upon differentiation as was validated by immunocytochemistry (Fig. 5C). mRNA levels of other striatal markers such as Calbindin 1, Calretinin (Calbindin 2) and D1R/D2R were also increased in the differentiated cells (Fig. 5C).
RNA-seq and proteomics results both detected a significant increase of synaptic markers such as GAD1, GAD2, DLG4, GPHN and SLC32A1 after differentiation in non-disease control and HD ISPNs (Supplementary Material, Fig. S3). Notably, levels of major MSN and synaptic markers were similar between differentiated ISPNs and a previously published dataset procured from iPSC-derived neural cells (15), demonstrating the comparability of the two HD cell models (Supplementary Material, Fig. S4). Levels of glial markers including GFAP and PDGF-RA were lower in differentiated ISPN compared to iPSC-derived neural cells (Supplementary Material, Fig. S4), indicating a much more homogeneous MSN-like phenotype.
HD relevant signaling pathways and protein networks are altered in HD compared to control ISPNs
Ingenuity pathway analysis (IPA) of proteomics data identified pathways altered in our HD ISPN model that are similarly altered in other HD models including iPSCs and patient brains (14,15,48). These pathways are involved in endocytosis including caveolar-mediated signaling, and neural development and maturation including GABA receptor, semaphorin and axonal guidance signaling (Fig. 6A and Supplementary Material, Figs S5, S6 and S8).
Figure 6 .
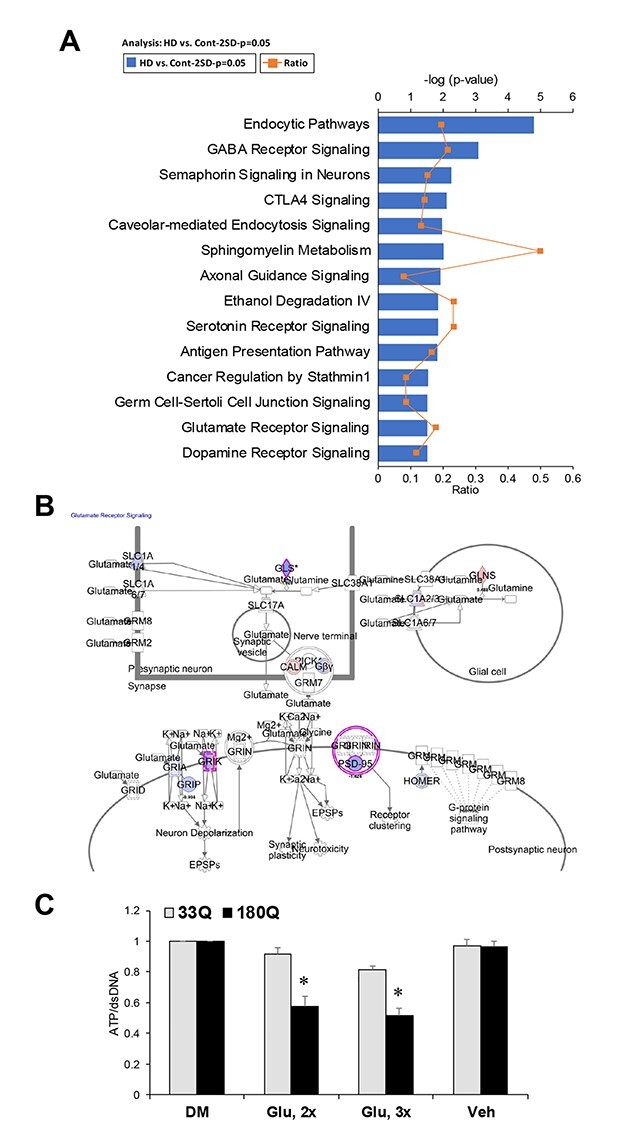
HD relevant signaling pathways are altered in HD compared to control ISPNs. (A) Top IPA pathways showing differential expression in differentiated HD versus control ISPNs in proteomics analysis. (B) Glutamate receptor signaling pathway is altered in HD ISPNs. Proteins found to be downregulated in HD lines are shown in blue and proteins upregulated are in red. (C) ATP levels in differentiated ISPNs after induction of cell toxicity by glutamate pulse treatment. HD (180Q, clone n14) and non-disease control (33Q, clone EE) ISPNs were differentiated in Matrigel coated 96-well assay plates for 2 weeks. Cell viability was measured with CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s protocol. One-way ANOVA: *P < 0.001 versus Vehicle (Veh); DM—complete differentiation medium; 3× – 3 pulses of glutamate toxicity, 2× – 2 pulses of glutamate toxicity.
Previously, we identified several genes in GABA and glutamate signaling pathways downregulated in differentiated HD iPSCs compared to control cells (15). IPA analysis of HD ISPN proteomics data also identified a panel of differentially downregulated proteins in GABA receptor signaling pathway including GABA transporter (GAT1), vesicular GABA transporter (VGAT), GABA-B receptor 2, Adenylyl cyclase 2 and gephyrin (Supplementary Material, Fig. S5) We also found significant alteration of glutamate receptor signaling including downregulation of K-type mitochondrial glutaminase, glutamate receptor 7 and PSD-95 in differentiated HD ISPNs (Fig. 6B). We observed decreased viability of HD ISPNs compared to control cells after induction of cell toxicity by glutamate pulse treatment (Fig. 6C), consistent with previously shown increased glutamate toxicity in HD iPSC-derived neural cultures (14).
In addition, we observed alterations in metabolic pathways such as sphingomyelin metabolism and ethanol degradation, which correlate with dysregulation of metabolism described in HD iPSC derived neural cells and other HD models.
The top altered network identified using IPA of proteomics data was ‘Neurological Disease, Cell-To-Cell Signaling and Interaction, Nervous System Development and Function’ (Supplementary Material, Fig. S6). Notably, most proteins in the network are downregulated in HD ISPNs and one of the central nodes in the network is BDNF, which plays a crucial role in neuronal survival and activity in HD (52,53). Top functional categories altered in differentiated HD ISPNs versus control included behavior, cellular development, cell-to-cell signaling and interaction, tissue development, nervous system development and function, and neurological disease. These categories are similar to those found altered in HD iPSCs (15). Notably, the nervous system development and function category was significantly decreased in HD ISPNs compared to control cells, while neurological disease functional category was significantly increased (Supplementary Material, Table S3). We also identified the ingenuity top toxicity list altered in differentiated HD versus control ISPNs, which includes biogenesis of mitochondria, swelling of mitochondria and glutathione depletion (Supplementary Material, Table S4). Various impairments of mitochondria and bioenergetics are well documented in HD models and patients.
Expression levels of the key regulators of neurodevelopment are altered and can be pharmacologically rescued in differentiated HD ISPNs
Further analysis of proteomics data identified a list of upstream regulators of differential protein expression altered in differentiated HD ISPNs (Supplementary Material, Table S5). The top upstream regulators include REST and BDNF known to be altered in HD.
In agreement with our Omics results for the HD iPSC model (15), REST (a regulator of NeuroD1 expression) was classified by IPA as the top ‘activated upstream regulator’ of differential protein expression in differentiated HD ISPNs with activation Z-score 3.388 (Fig. 7A and Supplementary Material, Table S5) RNA-seq analysis confirmed a significant increase of REST mRNA levels (Fig. 7B). NEUROD1, a key regulator of neurodevelopment altered in the HD iPSC model (15) was also altered in our novel ISPN model. IPA analysis predicted that NEUROD1 is another upstream regulator of differential protein expression with one of lowest activation Z-score: −1.98 (Fig. 7C). RNA-seq analysis confirmed significantly decreased NEUROD1 mRNA levels in differentiated HD ISPN lines (Fig. 7D), as well as ASCL1, one of the pro-neural bHLH factors upstream of NEUROD1 (Fig. 7E). Validation of the Omics results by Western blot confirmed significantly decreased levels of NEUROD1 and its downstream targets Calbindin1 and CAMK IV in HD ISPNs compared to control, and an increase after treatment of differentiated ISPNs with the small molecule Isoxazole-9 (Isx-9) in the absence of BDNF, similar to results obtained using the HD iPSC model (Fig. 7F and G).
Figure 7 .
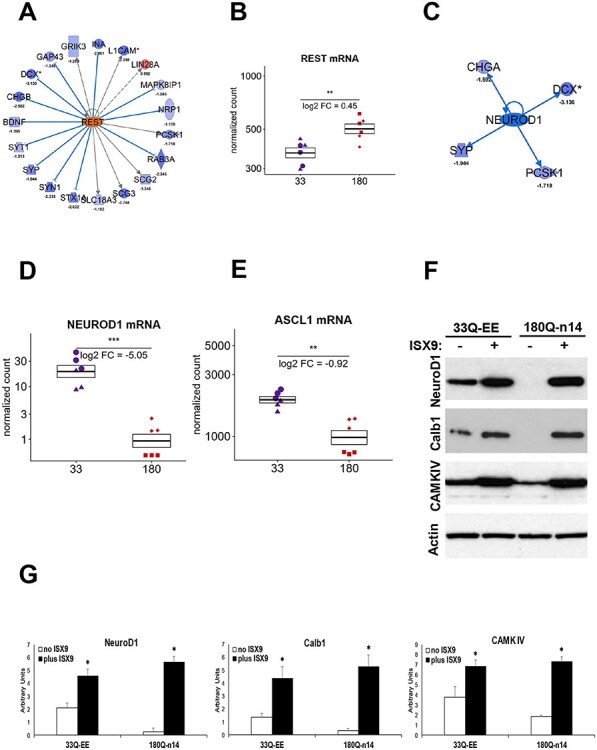
Expression levels of the key regulators of neurodevelopment are altered and can be pharmacologically rescued in differentiated HD ISPNs. (A) IPA pathway tools suggest that REST, a regulator of NEUROD1 expression, is the most activated upstream regulator of the differential protein expression in the differentiated HD ISPNs. (B) RNA-seq analysis confirms significantly increased REST mRNA levels in differentiated HD ISPN lines. Y-axis is on a log scale and represents read counts normalized by size factor. Colors and shapes of points as in Figure 5. *** adjusted P-value < 1e-5, ** adjusted P-value < 0.01. (C) IPA pathway tools suggest that NEUROD1 is one of altered upstream regulators of the differential protein expression in differentiated HD ISPNs (Activation Z-score: −1.980, P-value of overlap: 2.24E-03). (D) RNA-seq analysis confirms significantly decreased NEUROD1 mRNA levels in differentiated HD ISPN lines. Statistics and axes as in (B). (E) Results of RNA-seq analysis demonstrates significantly decreased mRNA levels of ASCL1, one of the pro-neural bHLH factors upstream of NEUROD1, in HD ISPN lines. Stats and axes as in (B). (F) Western blot analysis validates increase of NEUROD1 and its downstream targets Calbindin1 (CALB1) and CAMK IV protein levels after treatment of differentiated ISPNs with 20 μm Isx-9 in the absence of BDNF. (G) Quantification of Western blot densitometry data normalized to actin. Statistical significance between Isx-9 and untreated cells was determined by one-way ANOVA. N = 3, *P < 0.05.
IPA pathway tools suggest that the down-regulated BDNF could be an upstream regulator of the differential protein expression in differentiated HD ISPNs (activation Z-score − 2.653, Fig. 8A and Supplementary Material, Table S5). RNA-seq analysis confirmed significantly decreased BDNF mRNA levels in HD ISPN lines (Fig. 8B). To investigate whether BDNF withdrawal from the cell culture medium can induce cell death in HD ISPNs that can be rescued with small molecules, we used a drug previously validated in HD iPSC model, Isx-9 and a nuclear condensation cell death assay (14,15). We observed a 3-fold increase in death of differentiated HD ISPNs after BDNF withdrawal for 48 h, while control cells were not significantly affected. Addition of Isx-9 to the BDNF-free differentiation medium ameliorated toxicity in the HD ISPNs (Fig. 8C), similar to what has been shown for HD iPSCs and other HD models (14).
Figure 8 .
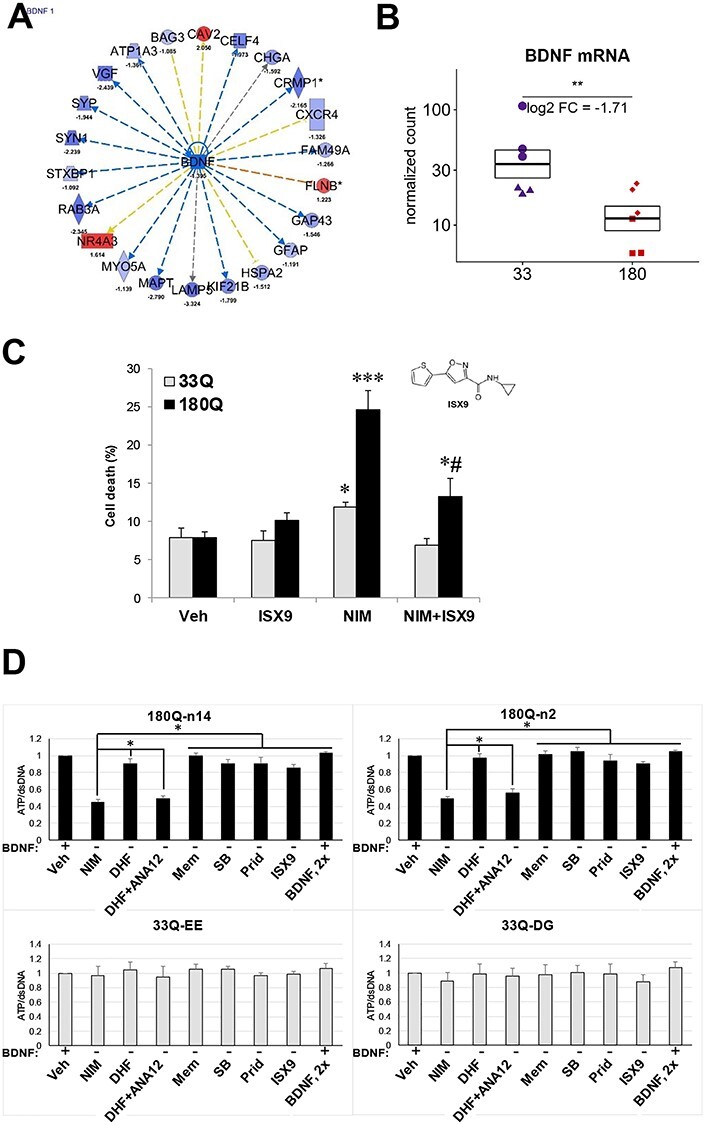
CellTiter-Glo cell viability assay can be used for 96-well format cell toxicity rescue experiments with differentiated HD ISPNs. (A) Analysis of proteomics data using IPA suggest that BDNF is one of the most inhibited upstream regulators of the differential protein expression in differentiated HD ISPN cells. (B) Results of RNA-seq analysis confirm significantly decreased BDNF mRNA levels in HD ISPN lines. Y-axis is on a log scale and represents read counts normalized by size factor. Colors and shapes of points as in Figure 5. *** adjusted P-value < 1e-5, ** adjusted P-value <0.01. (C) Rescue of BDNF withdrawal induced cell toxicity using Isx-9. Immortalized striatal precursors were differentiated in 24-well plates for 2 weeks and treated with 20 μm Isx-9 for 48 h. Percentage of dead cells measured by nuclei condensation assay. *P < 0.05 versus Ctrl 33Q; ***P < 0.001 versus Ctrl 33Q; #P < 0.05 versus NIM 180Q. Automated picture acquisition was performed using a Zeiss Axiovert 200 inverted microscope and nuclear intensity measured using Volocity. (D) Immortalized striatal precursor cells HD (180Q, clones n14 and n2) and non-disease controls (33Q, clones EE and DG) were differentiated in Matrigel coated 96-well assay plates for 2 weeks. Cell viability was measured with CellTiter-Glo luminescent cell viability assay and double-stranded DNA amount was then measured using the QuantiFluor dsDNA fluorescent system in the same plate for normalization (both from Promega) according to the manufacturer’s protocol. BDNF withdrawal was performed for 24 h. Prior to measurements. Small molecules were added to the BDNF-free media for all 24 h. NIM – BDNF free NIM; vehicle was in complete differentiation medium (Veh, DMSO); Small Molecules: 7,8-Dihydroxyflavone (DHF; TrkB receptor agonist, 1 μm); ANA-12 (TrkB receptor antagonist, 10 μm); Memantine (Mem; NMDA antagonist, 10 μm); SB 239063 (SB; p38 MAPK inhibitor, 200 nm); Pridopidine (Prid; Sigma1R agonist, 1 μm); Isx-9 (20 μm); BDNF used at 2× concentration (40 ng/ml) compared to differentiation media. One-way ANOVA: *P < 0.01 (n = 3).
We used a BDNF withdrawal model of neuronal death to develop a screening platform using our new HD ISPNs (Fig. 8D). The assay was performed in a 96-well plate format using a CellTiter-Glo luminescent cell viability assay (Promega). HD and control ISPNs were differentiated for 2 weeks before being transferred into BDNF-free neural induction medium (NIM); ATP levels were determined after 24 h. We tested rescue of cell death using small molecules previously validated in the HD iPSC model such as memantine (NMDA receptor antagonist), SB 239063 (p38 MAPK inhibitor) (20) and Isx-9 (15). We also tested the BDNF receptor TrkB agonist, 7,8-Dihydroxyflavone (7,8-DHF), known to activate TrkB in primary neurons cultures (54). BDNF was used as a positive control at 2× concentration (40 ng/ml) compared to differentiation media. Our results demonstrated that all the tested small molecules ameliorated BDNF-withdrawal induced toxicity in HD ISPNs, and rescue with 7,8-DHF can be blocked by TrkB receptor antagonist ANA-12 (Fig. 8D).
Pridopidine (a sigma-1 receptor agonist) protection of neurons from mHTT toxicity via the sigma-1 receptor was recently shown in mouse primary neurons and human HD iPSCs (25). Similar protection was observed in our ISPN model and this protection was blocked by the sigma-1R antagonist NE-100 (Supplementary Material, Fig. S7).
Discussion
The recent development of patient-derived models is a substantial advance for cell models of HD and other genetic diseases. However, developing iPSC-based platforms for biochemical studies and therapeutic screenings has been hindered by heterogeneity, long differentiation times and complicated methods of propagation of iPSC models. We present here a proof-of principle for the development of a novel model of HD patient-derived ISPNs which ameliorates some of these disadvantages. Our HD ISPN model recapitulates key features of the parental cell lines but is experimentally more tractable. Key properties of the HD ISPN model versus the parental iPSC model are as summarized in Table 2.
Table 2.
Comparison of HD iPSC and ISPN cell models
| HD iPSCs | ISPNs | |
|---|---|---|
| Starting cell culture | iPSC clones or neural precursors (e.g. ‘EZ-spheres’ in suspension) | Homogeneous culture of adherent cells |
| Differentiation time | 8 weeks | 2 weeks |
| Heterogeneity of differentiated population | 5–10% of DARPP32/Ctip2 positive cells; up to 40–50% glia | over 90% of DARPP32/Ctip2 positive cells |
| Expansion and modification | Complicated methods of propagation; difficult to subclone | Easy to expand adherent cell lines; relatively easy to derive lines by single-cell cloning followed by genetic or other modifications |
| Generation of material for biochemical characterization | Restricted amount of material after differentiation, usually requiring ICC or single-cell molecular analysis | Can generate large amounts of material applicable for biochemical analysis including Omics and Western blotting, and for screening |
Differentiated ISPNs exhibit morphology similar to striatal projection neurons and show expression of many of the same mature neuron and MSN markers as the parental HD iPSCs. HD ISPNs demonstrate much broader expression of DARPP32, Ctip2 and GABA in over 90% of differentiated cells versus 5–10% in the iPSC model. We are also currently optimizing our experiments for further functional maturation of the ISPNs, such as synapse formation and electrophysiological recordings.
To further elucidate whether HD ISPNs recapitulate major features of HD iPSCs, we performed RNA-seq and proteomics pathways analysis (using IPA), and compared results to previously reported Omics analysis of HD iPSCs (14). We observed a significant overlap of signaling pathways, upstream regulators, and functions altered in our differentiated HD ISPNs and iPSCs. Many of these dysregulated pathways have been previously implicated in HD.
Pathways identified as altered in HD ISPNs included endocytic pathways, consistent with early studies of biopsies from postmortem HD patient brains, which showed a dramatic increase in endosome–lysosome-like organelles and HTT-positive tubulovesicular structures in remaining striatal neurons (55,56). Alterations in caveolar-mediated endocytosis signaling in HD ISPNs correlate with reported inhibition of endocytosis in mHTT-expressing primary striatal neurons through a non-clathrin caveolar-related pathway, suggesting a potentially novel mechanism for HD neurotoxicity (57).
Alterations of GABAergic neurotransmission are well documented in HD patient brains and rodent models. These include changes in GABA levels and synthesis, abnormal receptor subunit compositions and distribution of GABAA receptors, and aberrant GABAA receptor-mediated signaling (58,59). GABA receptor signaling is also one of the top IPA canonical pathways showing differential gene expression in differentiated HD iPSCs (15). Hence, changes in GABA receptor signaling in our HD ISPN model correlate well with data from HD patients, iPSCs and other models.
Axonal guidance signaling was previously found altered in the HD iPSC model (14,15) and the closely related semaphorin signaling pathway is significantly perturbed in differentiated HD ISPNs. It has been reported that anti-semaphorin 4D (SEMA4D) immunotherapy ameliorates neuropathology and some cognitive impairment in YAC128 mice, consistent with the idea that semaphorins may represent a therapeutic target in HD (60). The recently concluded SIGNAL trial of Pepinemab (conducted by Vaccinex), a humanized monoclonal antibody to SEMA4D, yielded promising results.
Changes in CTLA4 and antigen presentation pathways identified in our ISPN model suggest possible immune dysregulation involved in HD pathogenesis since CTLA4 is a crucial immune regulator (61). Identification of changes in two metabolic pathways, sphingomyelin metabolism and ethanol degradation, confirms the alteration of metabolism that has been described in HD iPSCs (16). Dysfunction in serotonin and dopamine receptor signaling in HD ISPNs is also well documented in HD (62–64).
Alterations in Stathmin1, a microtubule destabilizer involved in cell cycle progression, segregation of chromosomes, clonogenicity, cell motility and survival (65), is a new observation for HD, and could be either relevant to chromosomal instability in HD cells (11) or a result from the immortalization process. Changes in germ cell-Sertoli cell junction signaling could be relevant to reported sperm and testicular degeneration in HD transgenic animal model through coordinated interactions between Sertoli cells and germ cells (66,67).
The top two upstream regulators identified by IPA, REST and CREB1, were shown to modulate neurotoxicity in HD (68,69) and predicted via weighted gene correlation network analysis (WGCNA) of HD datasets as upstream regulators highly correlated to disease stage (70). REST is a master regulator of neurogenesis and neurodevelopment. Its elevated levels may be responsible for reduced neural maturation of HD iPSCs and contribute to unbalanced generation of GABAergic and glutamatergic neurons. REST was reported as the most activated upstream regulator of the differential gene expression in the HD iPSC derived neurons, which again demonstrates the great correlation in global Omics profiles between HD iPSC and ISPN models (15). In previous studies of HD iPSC lines, the HD iPSC Consortium reported decreased expression of one of the REST target genes NEUROD1 (a proneuronal basic helix–loop–helix gene, that regulates neurodevelopment and adult neurogenesis), as well as altered expression of several genes that regulate NEUROD1 expression including ASCL and REST (15). The Consortium also established that mHTT-related phenotypes could be modulated in HD iPSC neurons by a small molecule, isoxazole-9 (Isx-9), which targets several of these dysregulated genes. In particular, Isx-9 was shown to rescue BDNF-withdrawal induced toxicity in differentiated HD iPSCs specifically through upregulation of NEUROD1 levels. Our ISPN model recapitulates altered levels of NEUROD1, CALB1 and CAMK4 as well as the effects of Isx-9 on induction of these neurodevelopmental regulators and on cell viability.
BDNF is another upstream regulator identified by IPA in HD ISPNs with decreased gene expression levels in HD versus control cells. Decreased BDNF levels in HD brains and cell models are well documented and considered one of the most important contributors to loss of striatal neurons (52,53). BDNF signaling alterations was reported in differentiated HD iPSCs (15). Notably, increased expression of REST is implicated in dysregulation of BDNF: in HD, REST translocates from the cytoplasm to nucleus resulting in BDNF transcription repression in neurons (68). BDNF withdrawal causes reduced cell viability of differentiated HD iPSCs in a CAG-repeat-dependent manner that can be rescued by various small molecules (14,15,20,25). We observed similar protective effects of these compounds in our HD ISPN model. In addition, we tested the pro-survival effect of a small molecule Tropomyosin receptor kinase B (TrkB) agonist 7,8-dihydroxyflavone (7,8-DHF). 7,8-DHF is shown to protect neurons in the N171-82Q transgenic HD mouse model while significantly improving motor deficits, ameliorating brain atrophy, and extending survival (71). 7,8-DHF treatment in primary striatal cultures from R6/1 HD mice induces phosphorylation of TrkB at tyrosine 816 and activation of the downstream PLCγ1 pathway. Chronic administration of 7,8-DHF delays motor deficits in R6/1 mice (72). Our results show that 7,8-DHF and BDNF itself can rescue BDNF-withdrawal induced toxicity in HD ISPNs, demonstrating that the rescue of HD toxicity with 7,8-DHF is applicable to an HD patient derived model.
Overcoming iPSC disadvantages in our novel ISPN model allows to generate larger amounts of material that can be used for molecular and biochemical analysis, including identification of post-translational modifications (PTMs) in mHTT. PTMs can contribute to HD onset age, and relevant kinases are being considered as HD therapeutic targets (73,74). Furthermore, we demonstrate that our HD ISPN model is a suitable screening platform for experimental therapeutics in a 96-well format, using quantitative readout methods like the CellTiter-Glo luminescent cell viability assay. However, more work is needed to determine optimal tests (phenotypic readouts) and analytical endpoints, and validate the assay parameters including range, specificity, accuracy, precision, linearity and robustness (5). In addition to cell viability/toxicity assays, candidate endpoints could include, among others, assays for mitochondrial dysfunction (Supplementary Material, Table S4).
We are currently developing more ISPN lines with an allelic series of repeat lengths. We anticipate studying the pathologic pathways related to HD in the context of differences in gender and genetic background by using this cell model. As a separate approach, isogenic lines carrying varying CAG repeat lengths in the first exon of HTT for ISPNs will be useful for investigating CAG repeat length related mechanisms. Currently, the CAG size of our original 180Q ISPN line has expanded to around 220, indicating somatic expansion in our cells. Thus, we anticipate the ISPN model cane also be used for somatic CAG repeat instability studies in the future.
Taken together, our results demonstrate that our new HD ISPN model recapitulates some key disease-related phenotypes of the parental iPSC model, and it will be amenable to the studies of pathogenesis, and screening for experimental therapeutics. We believe our approach also represents a novel paradigm for generating tractable cell models of other genetic disorders.
Materials and Methods
Cell culture and differentiation of iPSC derived NSC lines
NSCs (also designated as EZ-spheres) were derived from human HD (180Q, clone 180i.5) and non-disease control (33Q, clone 33i.8) iPSCs as described previously (14). Briefly, iPSCs were converted into non-adherent NSCs by lifting iPSC colonies with Collagenase IV solution (ThermoFisher), followed by propagation in Stemline NSC medium (Sigma-Aldrich) supplemented with heparin (5 μg/ml, Sigma-Aldrich, cat # H3149) and high concentrations of EGF and FGF-2 (100 ng/ml, both from Peprotech). NSCs were maintained as spherical aggregates in polyhema-coated flasks to prevent attachment and split weekly by mechanical chopping as described before (40). Differentiation of NSCs towards striatal neurons was performed according to the EZ-sphere differentiation protocol (14,75) for 56 days prior to viral transduction. First, the NSCs were transferred into NIM composed of DMEM/F12 (ThermoFisher-Invitrogen) supplemented with 25 μg/ml Insulin (cat # I3536), 50 μg/ml Transferrin (cat # T1147), 100 μm Putrescine (cat # P5780), 20 nm Progesterone (cat # P6149), and 30 nm Selenium chloride (cat # 323527) (all from Sigma-Aldrich) for 5 days (Phase I) and then, BDNF (20 ng/ml; Peprotech) was added for 2 days (Phase II). For the following 21 days, cells were differentiated in NIM with 20 ng/ml BDNF, 200 ng/ml rhSHH (Peprotech) and 100 ng/ml DKK1 (Peprotech) to promote a rostral forebrain fate (Phase III). Finally, the cells were matured into MSN-like phenotypes in NIM supplemented with 20 ng/ml BDNF, dibutyryl cyclic AMP (dbcAMP, 0.5 mm; Sigma-Aldrich, cat # D0627) and valproic acid (VPA, 0.5 mm; Sigma-Aldrich, cat # P4543) for 14 days (Phase IV). The differentiation media were half-changed three times per week.
Generation of retroviral stocks and transduction of differentiated iPS cells
Viral stocks were prepared by transfection of Phoenix-AMPHO cells (ATCC, CRL3213) with pBabe-puro-myc-ER [a gift from Dr Wafik El-Deiry (Addgene plasmid # 19128; http://n2t.net/addgene:19128; RRID: Addgene_19128)] or pBabe-neo-hTERT [a gift from Dr Bob Weinberg (Addgene plasmid # 1774; http://n2t.net/addgene:1774; RRID: Addgene _1774)] plasmid DNA using LipofectAMINE 2000 reagent (ThermoFisher Scientific, cat #11668027) followed by concentration of retroviral particles from conditioned media using Retro-X Concentrator (Clontech, cat # 631455) according to the manufacturer’s protocol. Transfection efficiency of approximately 90% was verified by in parallel transfection with pBabe-GFP (a gift from Dr William Hahn, Addgene plasmid # 10668; http://n2t.net/addgene:10668; RRID: Addgene_10668) plasmid DNA. The average viral titer measured by QuickTiter Retrovirus Quantitation Kit (Cell Biolabs Inc., VPK-120) was ~108 per ml.
For generation of immortalized striatal neurons, non-disease control and HD iPSCs were differentiated for 56 days, dissociated with TrypLE Select solution (ThermoFisher-Invitrogen), and re-plated in Striatal Cell Medium-2 (SCM2, for composition see Supplementary Material, Table S2) into 6-well plates pre-coated for 1 h with Matrigel (0.167 mg/ml DMEM; Corning). After 48 h, the cells were transduced or co-transduced with the fresh or frozen viral stocks diluted with fresh SCM2 1:10. Polybrene (Hexadimethrine bromide, Sigma-Aldrich, #H9268, 6 mg/ml stock solution made in sterile water) was added to a concentration of 6 μg/ml. After overnight incubation, the medium was changed to fresh SCM2 for 48 h then, to SCM2 containing selective antibiotics 0.2 μg/ml Puromicin dihydrocholoride (ThermoFisher Scientific) and/or 50 μg/ml Geneticin (ThermoFisher Scientific). Cells were maintained in the media with selective antibiotics until selection was complete, and sub-confluent cultures were split using TrypLE Select solution for single-cell cloning and frozen stock preparation.
Single-cell cloning of ISPNs
ISPN single-cell cloning was performed according to a Cell Cloning by Serial Dilution in 96 Well Plates protocol (Corning). After the first split of non-disease control or HD ISPNs following drug selection, the cells were re-plated from one well of a 6-well plate into Matrigel coated 96-well plates using the serial dilution technique and maintained in medium prepared by mixing of equal volumes of fresh SCM2 and SCM2 conditioned medium from non-disease control ISPNs culture. Wells of the 96-well plate containing single-cell clones were monitored daily, and cells were split into two new 96-well plates after reaching sub-confluent density. One of these two plates was used for induction of differentiation followed by dot-blot analysis of striatal marker expression and another one for further propagation, Western blot validation of c-myc and hTERT transduction, and preparation of frozen cell stocks.
Immortalized striatal precursors propagation and freezing
For propagation, selected ISPN clones were cultured in Matrigel-coated 6-well plates with SCM2 medium at 37°C, 5% CO2 and 5% O2 atmosphere incubator. The cells were split approximately twice per week after reaching sub-confluent density. First, cells were treated with TrypLE solution for 10 min at 37°C. Then, the cells were harvested with DMEM +0.02% BSA into 15 ml tube and counted with a hemocytometer. After spinning at 150 g for 5 min at room temperature, cell pellets were re-suspended in SCM2 and plated at a density of 2 × 105 per well in Matrigel coated 6-well plates. SCM2 was changed every 2–3 days. For freezing, cells were harvested as for propagation and cell pellets were re-suspended in SCM2 plus 10% DMSO (Sigma-Aldrich) to a density of 5 × 105 to 106 cells per ml. Cryovials with 1 ml cell suspension per vial were placed into a −80°C freezer in an isopropyl alcohol chamber for 24 h. The frozen vials were then transferred for long-term storage into a liquid nitrogen tank.
Karyotyping and CAG-repeat genotyping
Karyotyping of live cells was performed by Cell Line Genetics laboratories, Madison, WI. Cytogenetic analysis was performed on 20 G-banded metaphase cells for each tested cell line.
Genomic DNA was collected from cells using the Qiagen DNeasy Blood and Tissue Kit (cat # 69504) per the manufacturer’s protocol. Genotyping was performed by Laragen Inc. (Culver City, CA), while the remaining DNA was frozen as stocks at −80°C.
Immortalized striatal precursors differentiation
To induce striatal differentiation sub-confluent monolayer ISPN cells were grown in Phase 3 medium on Matrigel-coated plates at a density of 3 × 104 cell/cm2. The cells were maintained 4 days in modified ‘Phase-3 medium’ (14) composed as NIM supplemented with 20 ng/ml BDNF (Peprotech), 0.67 uM purmorphamine (Stemgent), and either 100 ng/ml Dickkopf-related protein 1 (DKK-1) (Peprotech) or 1.5 μm IWR-1 (Sigma-Aldrich). Then, the cells were transferred for 10 days into ‘Phase-4 medium’: NIM with 20 ng/ml BDNF (Peprotech), 0.5 mm valproic acid (VPA) (Sigma-Aldrich) and 0.5 mm dibutyryl cyclic adenosine monophosphate (dbcAMP) (Sigma-Aldrich).
Proteomics
Protein samples preparation
Quantitative proteomics samples of two HD and two control undifferentiated and differentiated ISPN lines were prepared and characterized in parallel cultures according to the previously described protocol (73,76). Briefly, cell pellets were lysed in 2% SDS/1 mm EDTA/1 mm PMSF in dH2O at room temperature and sonicated 2–3 times with 22% power input and 5 s intervals to reduce viscosity of the lysate. The lysates were diluted with water and protein amounts were determined via BCA assay (Pierce) to normalize protein levels between samples (100 μg protein/108 μl each). For additional QC, aliquots of the prepared material were fractionated on SDS-PAGE and stained with Coomassie blue protein stain to ensure the lack of protein degradation during the procedure. Two independent sets of protein samples were used in parallel for the proteome analysis.
Mass tag Labeling
Protein extracts were reduced with 10 μl of 200 mm TCEP then alkylated with 10 μl or 375 mm iodoacetamide in the dark for 30 min and TCA/Acetone precipitated (100 μg). Protein pellets were resolubilized in 100 μl of 100 mm triethyl ammonium bicarbonate (TEAB) and digested overnight at 37°C by adding 10 μg of Trypsin/LysC mixture (V5071, Promega) in 100 mm TEAB. Individual samples (100 μg) were labeled with a unique isobaric mass tag reagent (TMT 10-plex, ThermoFisher Scientific) according to the manufacturer instructions. Both pairing and labeling order of TMT reagent and peptide sample were randomized. Briefly, TMT-10 plex reagents (0.8 μg vials) were allowed to come to room temperature before adding 41 μl of anhydrous acetonitrile, then briefly vortexed and centrifuged. The entire TMT reagent vial was added to the 100 μg peptide sample and reacted at room temperature for 1 h. 5% hydroxylamine (8 μl) was then added to quench the reaction. All TMT labeled samples were combined and vacuum centrifuged to dryness. The combined samples of TMT labeled peptides was resuspended in 10 mm TEAB and separated into 84 fractions using a 0–90% acetonitrile gradient in 10 mm TEAB on a 5 μm C18 Waters XBridge column using an Agilent 1200 capillary HPLC in normal flow mode and Agilent 1260 micro-fraction collector. The 84 fractions were concentrated into 24 fractions by combining all odd rows into 12 fractions and all even rows into another 12 fractions.
Tandem mass spectrometry
Peptide fractions were resuspended in 20 μl 2% acetonitrile in 0.1% formic acid and analyzed by reverse phase liquid chromatography coupled to tandem mass spectrometry. Peptides were separated on a 75 μm × 150 mm ProntoSIL-120-5-C18 H column (3 μm, 120 Å (BISCHOFF), www.bischoff-chrom.com) using 2–90% acetonitrile gradient at 300 nl/min over 90 min on a EasyLC nanoLC 1000 (ThermoFisher Scientific). Eluting peptides were sprayed through 1 μm emitter tip (New Objective, www.newobjective.com) at 2.0 kV directly into a Q-Exactive Plus (QE Plus, ThermoFisher Scientific) mass spectrometer. Survey scans (full ms) were acquired from 350 to 1700 m/z with data-dependent monitoring of up to 15 peptide masses (precursor ions), each individually isolated in a 1.2 Da window and fragmented using HCD activation collision energy 32 and 30 s dynamic exclusion. Precursor and the fragment ions were analyzed at resolutions 70 000 and 35 000, respectively, with automatic gain control (AGC) target values at 3e6 with 50 ms maximum injection time (IT) and 1e5 with 200 ms maximum IT, respectively.
Peptide identification and quantification
Isotopically resolved masses in precursor (MS) and fragmentation (MS/MS) spectra were processor in Proteome Discoverer (PD) software (v1.4, ThermoFisher Scientific). Spectra were extracted and deconvoluted using both MS2 processor and Xtract. All data were searched using Mascot (2.5.1; www.matrixscience.com) against the Refseq 2015_Complete database (57 479 sequences for taxonomy Homo sapiens). The following criteria were set for all database searches: sample’s species; trypsin as the enzyme, allowing one missed cleavage; N-terminal TMT label as fixed modifications; TMT label on lysine, methionine oxidation, asparagine, and glutamine deamidation, and cysteine carbamidomethylation as variable modifications. Peptide identifications from Mascot searches were filtered at 1% false discovery rate (FDR) confidence threshold, based on a concatenated decoy database search, using the Proteome Discoverer. Proteome Discoverer uses only the peptide identifications with the highest Mascot score for the same peptide matched spectrum from the different extraction methods.
Protein quantification and statistical analysis
The peptides identified with a confidence threshold 1% FDR as described above were considered for analysis. Reporter ions from MS/MS spectra with isolation interferences greater or equal to 30% were excluded from further analysis. Protein relative abundances were determined from the normalized log2 values of the above spectra. In brief, reporter ion intensities were first transformed into log2 notation and for each sample the median value for each peptide was taken to represent that peptide. The values of all samples were then quantile normalized to minimize technical variation, such as differential amounts of material loading, prior to fold change analysis.
Samples were compared based on their number of HD repeats using the two-tailed one-way ANOVA t-test using the Partek Genomics Suite v6.6 platform. This provided each protein’s relative expression, as fold change, and that fold change’s statistical significance as P-value. Those proteins whose log2 fold changes differed by greater than two standard deviations (2SD) from the mean of zero were deemed to be significantly changed for purposes of downstream functional analysis by QIAGEN Ingenuity Pathway Analysis (IPA). Proteomic raw data and search results can be downloaded from ‘figshare’ database using the following link: https://doi.org/10.6084/m9.figshare.14781480
RNA-seq analysis
RNA-seq library preparation
RNA-sequencing was performed on two HD (n2 and n14) and two normal control (EE and DG) ISPN lines. Each line was evaluated both before and after differentiation into MSN-like phenotypes. Three separately generated sets of samples were obtained for each line and differentiation state, totaling 24 samples. RNA was extracted from each sample using the GenElute™ Universal Total RNA Purification Kit (MilliporeSigma). RIN values were obtained for the extracted RNA samples using the Agilent 2000 Bioanalyzer. Samples with a RIN > 8 were taken forward to sequencing. RNA-Seq was performed by the University of Iowa Genomics Division using manufacturer recommended protocols. Initially, 500 ng of DNase I-treated total RNA was used to enrich for polyA containing transcripts using oligo(dT) primers bound to beads. The enriched RNA pool was then fragmented, converted to cDNA, and ligated to sequencing adaptors containing indexes using the Illumina TruSeq stranded mRNA library preparation kit (Cat. #RS-122-2101, Illumina, Inc., San Diego, CA). The molar concentrations of the indexed libraries were measured using the Agilent 2100Bioanalyzer (Agilent Technologies, Santa Clara, CA) and combined equally into pools for sequencing. The concentration of the pools was measured using the Illumina Library Quantification Kit (KAPA Biosystems, Wilmington, MA) and sequenced on the Illumina HiSeq 4000 genome sequencer using 150 bp paired-end SBS chemistry.
RNA-seq data analysis
FASTQ files for paired-end RNA-seq data were aligned to the hg19 genome using Tophat 2.1.1 running Bowtie2 (v2.1.0) with default parameters. Reads were assigned to genes using the Transcription Database object provided in the R library TxDb.Hsapiens.UCSC.hg19.knownGene. Differential genes between HD and control samples were calculated with DESeq2 (v1.18.1). Genes with no reads across all samples were eliminated from analysis, as well as genes on the X or Y chromosome. Genes with adjusted P-value less than 0.01 were considered differentially expressed.
The FASTQ RNAseq data can be downloaded from the link: http://dnacore454.healthcare.uiiowa.edu/20160727-0041_Davidson_607 (Username: davidson, password:6lswewdavidson). Samples key can be found in Supplementary Material, Table S6.
Western blot and dot blot analysis
Cells were lysed with RIPA buffer (MilliporeSigma, cat# R0278) supplemented with 1% Triton X100 (MilliporeSigma, cat # 93443) and 0.5% Protease Inhibitor Cocktail (Set III, Calbiochem, now a part of MilliporeSigma, cat # 539134). Then, samples were sonicated 3 times for 5–10 s on ice and clarified from cell debris by centrifugation at 14 000 rpm in a desktop Eppendorf centrifuge at +4°C for 15 min. Protein concentrations were determined using Pierce BCA Protein Assay Kit (ThermoFisher Scientific, cat # 23225), according to the manufacturer’s directions. Approximately 15 μg of protein was loaded into NuPAGE 4–12% Bis-Tris protein gels (ThermoFisher Scientific, cat # NP0321BOX), separated with 200 V for approximately 1 h, and electrotransferred to PVDF membrane in the XCell II Blot Module transfer (ThermoFisher Scientific) for 90 min at 30 V. The membrane was blocked in 5% non-fat dry milk in TBS buffer, pH 7.4 (Quality Biological, cat # 351-086-101) plus 0.1% Tween 20 (Sigma-Aldrich) for 1 h at room temperature and then, exposed to primary antibodies with shaking overnight in a cold room. The HTT-specific antibody 2166 (1:1000, EMDMillipore MAB2166) was used to detect HTT protein in the control and HD lines. The expanded polyglutamine-specific antibodies 3B5H10 (1:20 000, Sigma-Aldrich, cat # P1874) and MW1 (1:10 000, DSHB) were used to detect mutant HTT in the HD lines. Antibodies for detection of DARPP32 (1:250, sc-11365) and MAP2 (1:1000, cat # MAB3418) were from Santa Cruz biotechnology and EMDMillipore, respectively. Antibodies for detection of NEUROD1 (1:1000, NeuroD (D90G12) rabbit mAb: cat # 7019), Calbindin 1 (1:1500, Calbindin (D1I4Q) XP rabbit mAb: cat # 13176), and CAMK IV (1:1000, rabbit, cat # 4032) were purchased from Cell Signaling Technology. Equal loading was verified by Western blotting for actin (β-actin: 1:10 000, mouse, Sigma-Aldrich, clone AC-15, cat # A5441). Densitometry was performed using ImageJ software and statistical analysis was performed using one-way ANOVA for three independent experiments.
For dot-blot analysis, ISPNs differentiated in 96-well plates were lysed on wet ice with 15 μl per well of the same lysis buffer as for Western blotting and 5 μl of each lysate were transferred with a pipette tip to nitrocellulose membrane. The membranes were air-dried and processed with primary and secondary antibodies as for Western blotting.
Immunofluorescence
For immunophenotyping, ISPNs were differentiated in 24-well plates on Matrigel coated 12 mm coverslips, fixed with 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature, permeabilized with PBS containing 0.5% Triton X-100 (Sigma-Aldrich) for 10 min, washed with PBS, and blocked with 10% Normal Goat Serum (NGS, Sigma-Aldrich) in PBS for 1 h. Then, cells were co-incubated overnight at +4°C in PBS containing 10% NGS and a combination of two primary antibodies—one of mouse and one of rabbit origin. The list of primary antibodies includes GSX2 (1:250, rabbit polyclonal, LSBio, cat # LS-C31550), Meis2 (1:250, mouse, clone 63-T, Santa Cruz Biotechnology, cat # sc-81986), Nestin (1:500, mouse, clone 10c2, Santa Cruz biotech, cat # sc-23927), MAP2 (1:500, mouse, clone AP20, EMD Millipore, cat # MAB3418), Tuj1 (1:1000, neuron-specific class III beta-tubulin, mouse, Neuromics, cat # MO15013), DARPP32 (1:150, rabbit polyclonal, Santa Cruz Biotechnology, cat # sc-11365), Bcl11b/Ctip2 (1:200, rabbit mAb D6F1, Cell Signaling Technology, cat # 12120S),GABA (1:500, rabbit, Sigma-Aldrich, cat # A2052) or HTT (1:200, mouse MCA2051 Bio-Rad). The next morning, cells were rinsed three times with PBS and co-incubated for 1 h with goat anti-mouse Alexa488-conjugated (1:1000, Cell Signaling Technology, cat #4408) and goat anti-rabbit Alexa555-conjugated (1:1000, Cell Signaling Technology, cat #4413) antibodies in 10% NGS in PBS. Nuclei were stained with 0.2 μg/ml Hoechst 33342 (Sigma Aldrich) in PBS and after washing twice in PBS, coverslips with fixed cells were mounted onto slides with FluorSave reagent (MilliporeSigma-Calbiochem, cat # 345789) and imaged using a Zeiss Axiovert 100 fluorescence microscope. Quantification of cells positive for subtype specific markers was performed using ImageJ software.
Cell viability and cell death assays
BDNF withdrawal
BDNF withdrawal to induce cell toxicity was performed as described before for HD iPSCs with some changes (14,20). Briefly, ISPNs were differentiated toward a striatal fate for 14 days and then washed once with either DMEM/F12 or NIM before being transferred into basic NIM for 24 or 48 h. NIM was not supplemented with dbcAMP and VPA because they increase endogenous BDNF transcription but are not critical for cell survival (77). NIM with 40 ng/ml BDNF was used as a positive control.
Nuclear condensation cell death assay
Cell toxicity was measured by nuclear condensation assay per our published protocol (14,15,24). To rescue from BDNF withdrawal induced toxicity, cultures of differentiated HD and control ISPNs were supplemented with Isx-9 (20 μm, Tocris), Pridopidine (0.1 μm or 1 μm, a kind gift from TEVA Pharmaceutical Industries), 3PPP (1 μm, Sigma-Aldrich), or 40 ng/ml BDNF. After 48 h of treatment, cells were fixed with 4% PFA and stained for 15–20 min with 0.2 μg/ml Hoechst 33342 (Sigma-Aldrich) in PBS. Automated picture acquisition was performed on a Zeiss Axiovert 200 fluorescence inverted microscope at 10× objective, and automated quantification of nuclear intensity was performed using Volocity (Perkin Elmer). Cells were considered dead when their average nuclear intensity was more than twice the average intensity of nuclei in untreated cells.
CellTiter-Glo luminescent cell viability assay and rescue of BDNF withdrawal
The cell viability assay was performed as described before for HD iPSCs with modifications (15,16). HD and control ISPNs were plated into Matrigel coated 96-well plates at a density of 104 cells/well in ‘Phase-3 medium’ and differentiated to striatal fate for 14 days including transfer to ‘Phase-4 medium’ after 4 days of differentiation. BDNF withdrawal was performed for 24 h. Cell viability was determined using the CellTiter-Glo luminescent cell viability assay (Promega, cat # G7571) according to manufacturer’s instructions. Luminescence was measured on a SpectraMax 340PC microplate reader (Molecular Devices) and the values were normalized to the amount of double-stranded DNA per well determined by QuantiFluor® dsDNA System (Promega, cat # E2670) according to the manufacturer’s instructions. Statistical analysis was performed using one-way ANOVA for at least three independent experiments. For rescue experiments, 7,8-Dihydroxyflavone (7,8-DHF, 1 μm, a kind gift from Dr Keqiang Ye, Emory University School of Medicine, Atlanta, GA), Memantine (10 μm, Sigma-Aldrich), SB239063 (200 nm, Sigma-Aldrich), Pridopidine (1 μm, a kind gift from TEVA Pharmaceutical Industries), Isx-9 (20 μm, Tocris) or 3PPP (1 μm, Sigma-Aldrich) small molecules were supplemented into NIM without BDNF for the full 24 h of BDNF withdrawal. ANA-12 (10 μm, TrkB receptor antagonist, IC50 of the high-affinity TrkB receptor sites, 45.6 ± 6.7 nm, and low-affinity sites, 41.1 ± 21.7 μm; (78), Sigma-Aldrich) or NE-100 (1 μm, Sigma-1 receptor antagonist, IC50 = 4.16 nm, (79), Sigma-Aldrich) were used to block 7,8-DHF or Sigma-1 receptor agonists, Pridopidine and 3PPP, induced rescue, respectively. BDNF was used for rescue of cell toxicity at 2× concentration (40 ng/ml) compared to differentiation media.
Statistical analysis
Multiple statistical models were used during the analysis as detailed in specific sections of Materials and Methods, Figure legends, and below. Principal component analysis was used to assess variance in proteomic and RNA-seq analysis between non-differentiated and differentiated or HD and non-disease control cells using the prcomp function in the R stats package.
Western blot results (densitometry) were normalized to actin. Statistical significance was determined by one-way ANOVA.
CellTiter-Glo luminescent cell viability assay (Promega) results were normalized to amount of dsDNA per well measured by QuantiFluor dsDNA fluorescent system (Promega). Statistical significance was determined by one-way ANOVA.
One-way ANOVA was performed using the results of at least three independent experiments at VassarStats statistical computation web site (http://vassarstats.net/).
Cell toxicity studies using nuclear condensation assay were performed blind with a coding system. The placement of the treatments on the plates was randomized with each experiment. All statistics are shown for independent experiments. No power analysis was done a priori for these experiments since we had performed power analysis for similar conditions previously (14). Four wells per condition were used per experiment and the experiments were reproduced at least 3 times each. A total of 5000–10 000 cells were quantified per condition per experiment. Statistical analysis was performed using ANOVA with a Bonferroni post-hoc test.
Supplementary Material
Acknowledgements
We are grateful to Lily Zhang for technical support.
The aliquots of ISPNs with 33 or 180 CAG repeats at early passage numbers are available from our lab upon request.
Conflict of Interest statement. The authors have no conflicts of interest.
Contributor Information
Sergey S Akimov, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Mali Jiang, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Amanda J Kedaigle, Department of Biological Engineering, Computational and Systems Biology Graduate Program, Massachusetts Institute of Technology, Cambridge, MA 02142, USA.
Nicolas Arbez, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Leonard O Marque, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Chelsy R Eddings, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Paul T Ranum, The Department of Pathology and Laboratory Medicine, The University of Pennsylvania, The Raymond G Perelman Center for Cellular and Molecular Therapeutics, The Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Emma Whelan, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Anthony Tang, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Ronald Wang, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Lauren R DeVine, Mass Spectrometry and Proteomics Facility, Department of Biological Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Conover C Talbot, Jr, The Johns Hopkins School of Medicine, Institute for Basic Biomedical Sciences, Baltimore, MD 21205, USA.
Robert N Cole, Mass Spectrometry and Proteomics Facility, Department of Biological Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Tamara Ratovitski, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Beverly L Davidson, The Department of Pathology and Laboratory Medicine, The University of Pennsylvania, The Raymond G Perelman Center for Cellular and Molecular Therapeutics, The Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA; The Department of Pathology and Laboratory Medicine, The University of Pennsylvania, Philadelphia, PA 19104, USA.
Ernest Fraenkel, Department of Biological Engineering, Computational and Systems Biology Graduate Program, Massachusetts Institute of Technology, Cambridge, MA 02142, USA.
Christopher A Ross, Division of Neurobiology, Department of Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA; Department of Neurology, Neuroscience and Pharmacology, The Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Funding
National Institute of Health (R21NS083365, 2013, R21NS104320-01A1, 2018, R01NS086452 to C.A.R.); NIH/NIGMS Biotechnology Training Program (T32 GM008334 to A.J.K.); Hopkins Post-baccalaureate Research Education Program (PREP) (NIH R25GM109441 to C.R.E., R01 NS089076 to E.F.).
References
- 1. Ross, C.A. and Tabrizi, S.J. (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol., 10, 83–98. [DOI] [PubMed] [Google Scholar]
- 2. Ross, C.A., Aylward, E.H., Wild, E.J., Langbehn, D.R., Long, J.D., Warner, J.H., Scahill, R.I., Leavitt, B.R., Stout, J.C., Paulsen, J.S. et al. (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol., 10, 204–216. [DOI] [PubMed] [Google Scholar]
- 3. Ross, C.A. and Akimov, S.S. (2014) Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum. Mol. Genet., 23, R17–R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes (1993) The Huntington's disease collaborative research group. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 5. Schmidt, B.Z., Lehmann, M., Gutbier, S., Nembo, E., Noel, S., Smirnova, L., Forsby, A., Hescheler, J., Avci, H.X., Hartung, T. et al. (2017) In vitro acute and developmental neurotoxicity screening: an overview of cellular platforms and high-throughput technical possibilities. Arch. Toxicol., 91, 1–33. [DOI] [PubMed] [Google Scholar]
- 6. Trettel, F., Rigamonti, D., Hilditch-Maguire, P., Wheeler, V.C., Sharp, A.H., Persichetti, F., Cattaneo, E. and Mac Donald, M.E. (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet., 9, 2799–2809. [DOI] [PubMed] [Google Scholar]
- 7. Vonsattel, J.P. (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol., 115, 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verlinsky, Y., Strelchenko, N., Kukharenko, V., Rechitsky, S., Verlinsky, O., Galat, V. and Kuliev, A. (2005) Human embryonic stem cell lines with genetic disorders. Reprod. BioMed. Online, 10, 105–110. [DOI] [PubMed] [Google Scholar]
- 9. Niclis, J., Trounson, A.O., Dottori, M., Ellisdon, A., Bottomley, S.P., Verlinsky, Y. and Cram, D. (2009) Human embryonic stem cell models of Huntington disease. Reprod. BioMed. Online, 19, 106–113. [DOI] [PubMed] [Google Scholar]
- 10. Bradley, C.K., Scott, H.A., Chami, O., Peura, T.T., Dumevska, B., Schmidt, U. and Stojanov, T. (2011) Derivation of Huntington's disease-affected human embryonic stem cell lines. Stem Cells Dev., 20, 495–502. [DOI] [PubMed] [Google Scholar]
- 11. Ruzo, A., Croft, G.F., Metzger, J.J., Galgoczi, S., Gerber, L.J., Pellegrini, C., Wang, H., Jr., Fenner, M., Tse, S., Marks, A. et al. (2018) Chromosomal instability during neurogenesis in Huntington's disease. Development, 145. [DOI] [PubMed] [Google Scholar]
- 12. Takahashi, K. and Yamanaka, S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126, 663–676. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K. and Yamanaka, S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 14. Consortium, H.D.i. (2012) Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell, 11, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Consortium, H.D.i. (2017) Developmental alterations in Huntington's disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci., 20, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kedaigle, A.J., Fraenkel, E., Atwal, R.S., Wu, M., Gusella, J.F., Mac Donald, M.E., Kaye, J.A., Finkbeiner, S., Mattis, V.B., Tom, C.M. et al. (2020) Bioenergetic deficits in Huntington's disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum. Mol. Genet., 29, 1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tousley, A. and Kegel-Gleason, K.B. (2016) Induced pluripotent stem cells in Huntington's disease research: progress and opportunity. J. Huntingtons Dis., 5, 99–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. An, M.C., Zhang, N., Scott, G., Montoro, D., Wittkop, T., Mooney, S., Melov, S. and Ellerby, L.M. (2012) Genetic correction of Huntington's disease phenotypes in induced pluripotent stem cells. Cell Stem Cell, 11, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu, X., Tay, Y., Sim, B., Yoon, S.I., Huang, Y., Ooi, J., Utami, K.H., Ziaei, A., Ng, B., Radulescu, C. et al. (2017) Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep., 8, 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mattis, V.B., Tom, C., Akimov, S., Saeedian, J., Ostergaard, M.E., Southwell, A.L., Doty, C.N., Ornelas, L., Sahabian, A., Lenaeus, L. et al. (2015) HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet., 24, 3257–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu, X.H., Mattis, V.B., Wang, N., Al-Ramahi, I., van den Berg, N., Fratantoni, S.A., Waldvogel, H., Greiner, E., Osmand, A., Elzein, K. et al. (2014) Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington's disease. Sci. Transl. Med., 6, 268ra178. [DOI] [PubMed] [Google Scholar]
- 22. Dickey, A.S., Pineda, V.V., Tsunemi, T., Liu, P.P., Miranda, H.C., Gilmore-Hall, S.K., Lomas, N., Sampat, K.R., Buttgereit, A., Torres, M.J. et al. (2016) PPAR-delta is repressed in Huntington's disease, is required for normal neuronal function and can be targeted therapeutically. Nat. Med., 22, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dickey, A.S., Sanchez, D.N., Arreola, M., Sampat, K.R., Fan, W., Arbez, N., Akimov, S., Van Kanegan, M.J., Ohnishi, K., Gilmore-Hall, S.K. et al. (2017) PPARdelta activation by bexarotene promotes neuroprotection by restoring bioenergetic and quality control homeostasis. Sci. Transl. Med., 9, eaal2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arbez, N., Roby, E., Akimov, S., Eddings, C., Ren, M., Wang, X. and Ross, C.A. (2019) Cysteamine protects neurons from mutant huntingtin toxicity. J. Huntingtons Di.s, 8, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eddings, C.R., Arbez, N., Akimov, S., Geva, M., Hayden, M.R. and Ross, C.A. (2019) Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor. Neurobiol. Dis., 129, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pollock, K., Stroemer, P., Patel, S., Stevanato, L., Hope, A., Miljan, E., Dong, Z., Hodges, H., Price, J. and Sinden, J.D. (2006) A conditionally immortal clonal stem cell line from human cortical neuroepithelium for the treatment of ischemic stroke. Exp. Neurol., 199, 143–155. [DOI] [PubMed] [Google Scholar]
- 27. Johansson, S., Price, J. and Modo, M. (2008) Effect of inflammatory cytokines on major histocompatibility complex expression and differentiation of human neural stem/progenitor cells. Stem Cells, 26, 2444–2454. [DOI] [PubMed] [Google Scholar]
- 28. Choi, H.K., Won, L.A., Kontur, P.J., Hammond, D.N., Fox, A.P., Wainer, B.H., Hoffmann, P.C. and Heller, A. (1991) Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res., 552, 67–76. [DOI] [PubMed] [Google Scholar]
- 29. Snyder, E.Y., Deitcher, D.L., Walsh, C., Arnold-Aldea, S., Hartwieg, E.A. and Cepko, C.L. (1992) Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell, 68, 33–51. [DOI] [PubMed] [Google Scholar]
- 30. Colucci-D'Amato, G.L., Tino, A., Pernas-Alonso, R., ffrench-Mullen, J.M. and di Porzio, U. (1999) Neuronal and glial properties coexist in a novel mouse CNS immortalized cell line. Exp. Cell Res., 252, 383–391. [DOI] [PubMed] [Google Scholar]
- 31. Miljan, E.A., Hines, S.J., Pande, P., Corteling, R.L., Hicks, C., Zbarsky, V., Umachandran, M., Sowinski, P., Richardson, S., Tang, E. et al. (2009) Implantation of c-mycER TAM immortalized human mesencephalic-derived clonal cell lines ameliorates behavior dysfunction in a rat model of Parkinson's disease. Stem Cells Dev., 18, 307–319. [DOI] [PubMed] [Google Scholar]
- 32. Prasad, K.N., Carvalho, E., Kentroti, S., Edwards-Prasad, J., Freed, C. and Vernadakis, A. (1994) Establishment and characterization of immortalized clonal cell lines from fetal rat mesencephalic tissue. In Vitro Cell. Dev. Biol. Anim., 30A, 596–603. [DOI] [PubMed] [Google Scholar]
- 33. Truckenmiller, M.E., Tornatore, C., Wright, R.D., Dillon-Carter, O., Meiners, S., Geller, H.M. and Freed, W.J. (1998) A truncated SV40 large T antigen lacking the p53 binding domain overcomes p53-induced growth arrest and immortalizes primary mesencephalic cells. Cell Tissue Res., 291, 175–189. [DOI] [PubMed] [Google Scholar]
- 34. Cattaneo, E. and Conti, L. (1998) Generation and characterization of embryonic striatal conditionally immortalized ST14A cells. J. Neurosci. Res., 53, 223–234. [DOI] [PubMed] [Google Scholar]
- 35. Alwin Prem Anand, A., Gowri Sankar, S. and Kokila Vani, V. (2012) Immortalization of neuronal progenitors using SV40 large T antigen and differentiation towards dopaminergic neurons. J. Cell. Mol. Med., 16, 2592–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roy, N.S., Nakano, T., Keyoung, H.M., Windrem, M., Rashbaum, W.K., Alonso, M.L., Kang, J., Peng, W., Carpenter, M.K., Lin, J. et al. (2004) Telomerase immortalization of neuronally restricted progenitor cells derived from the human fetal spinal cord. Nat. Biotechnol., 22, 297–305. [DOI] [PubMed] [Google Scholar]
- 37. Roy, N.S., Cleren, C., Singh, S.K., Yang, L., Beal, M.F. and Goldman, S.A. (2006) Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med., 12, 1259–1268. [DOI] [PubMed] [Google Scholar]
- 38. Akimov, S.S., Ramezani, A., Hawley, T.S. and Hawley, R.G. (2005) Bypass of senescence, immortalization, and transformation of human hematopoietic progenitor cells. Stem Cells, 23, 1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ricci, M.S., Jin, Z., Dews, M., Yu, D., Thomas-Tikhonenko, A., Dicker, D.T. and El-Deiry, W.S. (2004) Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol. Cell. Biol., 24, 8541–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svendsen, C.N., ter Borg, M.G., Armstrong, R.J., Rosser, A.E., Chandran, S., Ostenfeld, T. and Caldwell, M.A. (1998) A new method for the rapid and long term growth of human neural precursor cells. J. Neurosci. Methods, 85, 141–152. [DOI] [PubMed] [Google Scholar]
- 41. Smogorzewska, A. and de Lange, T. (2004) Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem., 73, 177–208. [DOI] [PubMed] [Google Scholar]
- 42. Bodnar, A.G., Ouellette, M., Frolkis, M., Holt, S.E., Chiu, C.P., Morin, G.B., Harley, C.B., Shay, J.W., Lichtsteiner, S. and Wright, W.E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 43. Simonsen, J.L., Rosada, C., Serakinci, N., Justesen, J., Stenderup, K., Rattan, S.I., Jensen, T.G. and Kassem, M. (2002) Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat. Biotechnol., 20, 592–596. [DOI] [PubMed] [Google Scholar]
- 44. El-Akabawy, G., Medina, L.M., Jeffries, A., Price, J. and Modo, M. (2011) Purmorphamine increases DARPP-32 differentiation in human striatal neural stem cells through the Hedgehog pathway. Stem Cells Dev., 20, 1873–1887. [DOI] [PubMed] [Google Scholar]
- 45. Dashinimaev, E.B., Vishnyakova, K.S., Popov, K.V. and Yegorov, Y.E. (2008) Stable culture of hTERT-transduced human embryonic neural stem cells holds all the features of primary culture. Electronic J. Biol., 4, 93–97. [Google Scholar]
- 46. Ma, L., Hu, B., Liu, Y., Vermilyea, S.C., Liu, H., Gao, L., Sun, Y., Zhang, X. and Zhang, S.C. (2012) Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell, 10, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hung, C.L., Maiuri, T., Bowie, L.E., Gotesman, R., Son, S., Falcone, M., Giordano, J.V., Gillis, T., Mattis, V., Lau, T. et al. (2018) A patient-derived cellular model for Huntington's disease reveals phenotypes at clinically relevant CAG lengths. Mol. Biol. Cell, 29, 2809–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ratovitski, T., Chaerkady, R., Kammers, K., Stewart, J.C., Zavala, A., Pletnikova, O., Troncoso, J.C., Rudnicki, D.D., Margolis, R.L., Cole, R.N. et al. (2016) Quantitative proteomic analysis reveals similarities between Huntington's disease (HD) and Huntington's disease-like 2 (HDL2) human brains. J. Proteome Res., 15, 3266–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Telezhkin, V., Schnell, C., Yarova, P., Yung, S., Cope, E., Hughes, A., Thompson, B.A., Sanders, P., Geater, C., Hancock, J.M. et al. (2016) Forced cell cycle exit and modulation of GABAA, CREB, and GSK3beta signaling promote functional maturation of induced pluripotent stem cell-derived neurons. Am. J. Physiol. Cell Physiol., 310, C520–C541. [DOI] [PubMed] [Google Scholar]
- 50. Neueder, A., Landles, C., Ghosh, R., Howland, D., Myers, R.H., Faull, R.L.M., Tabrizi, S.J. and Bates, G.P. (2017) The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington's disease patients. Sci. Rep., 7, 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sathasivam, K., Neueder, A., Gipson, T.A., Landles, C., Benjamin, A.C., Bondulich, M.K., Smith, D.L., Faull, R.L., Roos, R.A., Howland, D. et al. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A., 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zuccato, C., Liber, D., Ramos, C., Tarditi, A., Rigamonti, D., Tartari, M., Valenza, M. and Cattaneo, E. (2005) Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol. Res., 52, 133–139. [DOI] [PubMed] [Google Scholar]
- 53. Zuccato, C. and Cattaneo, E. (2007) Role of brain-derived neurotrophic factor in Huntington's disease. Prog. Neurobiol., 81, 294–330. [DOI] [PubMed] [Google Scholar]
- 54. Liu, X., Obianyo, O., Chan, C.B., Huang, J., Xue, S., Yang, J.J., Zeng, F., Goodman, M. and Ye, K. (2014) Biochemical and biophysical investigation of the brain-derived neurotrophic factor mimetic 7,8-dihydroxyflavone in the binding and activation of the TrkB receptor. J. Biol. Chem., 289, 27571–27584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tellez-Nagel, I., Johnson, A.B. and Terry, R.D. (1974) Studies on brain biopsies of patients with Huntington's chorea. J. Neuropathol. Exp. Neurol., 33, 308–332. [DOI] [PubMed] [Google Scholar]
- 56. Sapp, E., Schwarz, C., Chase, K., Bhide, P.G., Young, A.B., Penney, J., Vonsattel, J.P., Aronin, N. and DiFiglia, M. (1997) Huntingtin localization in brains of normal and Huntington's disease patients. Ann. Neurol., 42, 604–612. [DOI] [PubMed] [Google Scholar]
- 57. Trushina, E., Singh, R.D., Dyer, R.B., Cao, S., Shah, V.H., Parton, R.G., Pagano, R.E. and McMurray, C.T. (2006) Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum. Mol. Genet., 15, 3578–3591. [DOI] [PubMed] [Google Scholar]
- 58. Garret, M., Du, Z., Chazalon, M., Cho, Y.H. and Baufreton, J. (2018) Alteration of GABAergic neurotransmission in Huntington's disease. CNS Neurosci. Ther., 24, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hsu, Y.T., Chang, Y.G. and Chern, Y. (2018) Insights into GABAAergic system alteration in Huntington's disease. Open Biol., 8, 180165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Southwell, A.L., Franciosi, S., Villanueva, E.B., Xie, Y., Winter, L.A., Veeraraghavan, J., Jonason, A., Felczak, B., Zhang, W., Kovalik, V. et al. (2015) Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis., 76, 46–56. [DOI] [PubMed] [Google Scholar]
- 61. Walker, L.S. and Sansom, D.M. (2015) Confusing signals: recent progress in CTLA-4 biology. Trends Immunol., 36, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cross, A.J., Reynolds, G.P., Hewitt, L.M. and Slater, P. (1986) Brain serotonin receptors in Huntington's disease. Neurochem. Int., 9, 431–435. [DOI] [PubMed] [Google Scholar]
- 63. Pang, T.Y., Du, X., Zajac, M.S., Howard, M.L. and Hannan, A.J. (2009) Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington's disease. Hum. Mol. Genet., 18, 753–766. [DOI] [PubMed] [Google Scholar]
- 64. Cepeda, C., Murphy, K.P., Parent, M. and Levine, M.S. (2014) The role of dopamine in Huntington's disease. Prog. Brain Res., 211, 235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Machado-Neto, J.A., Saad, S.T. and Traina, F. (2014) Stathmin 1 in normal and malignant hematopoiesis. BMB Rep., 47, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Syed, V., Gomez, E. and Hecht, N.B. (1999) mRNAs encoding a von Ebner's-like protein and the Huntington disease protein are induced in rat male germ cells by Sertoli cells. J. Biol. Chem., 274, 10737–10742. [DOI] [PubMed] [Google Scholar]
- 67. Macakova, M., Bohuslavova, B., Vochozkova, P., Pavlok, A., Sedlackova, M., Vidinska, D., Vochyanova, K., Liskova, I., Valekova, I., Baxa, M. et al. (2016) Mutated huntingtin causes testicular pathology in transgenic minipig boars. Neurodegener. Dis., 16, 245–259. [DOI] [PubMed] [Google Scholar]
- 68. Buckley, N.J., Johnson, R., Zuccato, C., Bithell, A. and Cattaneo, E. (2010) The role of REST in transcriptional and epigenetic dysregulation in Huntington's disease. Neurobiol. Dis., 39, 28–39. [DOI] [PubMed] [Google Scholar]
- 69. Steffan, J.S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y.Z., Gohler, H., Wanker, E.E., Bates, G.P., Housman, D.E. and Thompson, L.M. (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA, 97, 6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Neueder, A. and Bates, G.P. (2014) A common gene expression signature in Huntington's disease patient brain regions. BMC Med. Genet., 7, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang, M., Peng, Q., Liu, X., Jin, J., Hou, Z., Zhang, J., Mori, S., Ross, C.A., Ye, K. and Duan, W. (2013) Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington's disease. Hum. Mol. Genet., 22, 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Garcia-Diaz Barriga, G., Giralt, A., Anglada-Huguet, M., Gaja-Capdevila, N., Orlandi, J.G., Soriano, J., Canals, J.M. and Alberch, J. (2017) 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington's disease mouse model through specific activation of the PLCgamma1 pathway. Hum. Mol. Genet., 26, 3144–3160. [DOI] [PubMed] [Google Scholar]
- 73. Ratovitski, T., O'Meally, R.N., Jiang, M., Chaerkady, R., Chighladze, E., Stewart, J.C., Wang, X., Arbez, N., Roby, E., Alexandris, A. et al. (2017) Post-translational modifications (PTMs), identified on endogenous huntingtin, cluster within proteolytic domains between HEAT repeats. J. Proteome Res., 16, 2692–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Arbez, N., Ratovitski, T., Roby, E., Chighladze, E., Stewart, J.C., Ren, M., Wang, X., Lavery, D.J. and Ross, C.A. (2017) Post-translational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J. Biol. Chem., 292, 19238–19249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ebert, A.D., Shelley, B.C., Hurley, A.M., Onorati, M., Castiglioni, V., Patitucci, T.N., Svendsen, S.P., Mattis, V.B., McGivern, J.V., Schwab, A.J. et al. (2013) EZ spheres: a stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res., 10, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wisniewski, J.R., Zougman, A., Nagaraj, N. and Mann, M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- 77. Pruunsild, P., Sepp, M., Orav, E., Koppel, I. and Timmusk, T. (2011) Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J. Neurosci., 31, 3295–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cazorla, M., Premont, J., Mann, A., Girard, N., Kellendonk, C. and Rognan, D. (2011) Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest., 121, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Okuyama, S., Imagawa, Y., Ogawa, S., Araki, H., Ajima, A., Tanaka, M., Muramatsu, M., Nakazato, A., Yamaguchi, K., Yoshida, M. et al. (1993) NE-100, a novel sigma receptor ligand: in vivo tests. Life Sci., 53, PL285–PL290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.