


Abstract
Type III CRISPR-Cas effector systems detect foreign RNA triggering DNA and RNA cleavage and synthesizing cyclic oligoadenylate molecules (cA) in their Cas10 subunit. cAs act as a second messenger activating auxiliary nucleases, leading to an indiscriminate RNA degradation that can end in cell dormancy or death. Standalone ring nucleases are CRISPR ancillary proteins which downregulate the strong immune response of Type III systems by degrading cA. These enzymes contain a CRISPR-associated Rossman-fold (CARF) domain, which binds and cleaves the cA molecule. Here, we present the structures of the standalone ring nuclease from Sulfolobus islandicus (Sis) 0811 in its apo and post-catalytic states. This enzyme is composed by a N-terminal CARF and a C-terminal wHTH domain. Sis0811 presents a phosphodiester hydrolysis metal-independent mechanism, which cleaves cA4 rings to generate linear adenylate species, thus reducing the levels of the second messenger and switching off the cell antiviral state. The structural and biochemical analysis revealed the coupling of a cork-screw conformational change with the positioning of key catalytic residues to proceed with cA4 phosphodiester hydrolysis in a non-concerted manner.
INTRODUCTION
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) that associate with CRISPR associated (Cas) proteins, constitute an adaptive immune system in bacteria and archaea against foreign mobile genetic elements (MGEs), such as plasmids and phages (1,2). The CRISPR immune response consists of three stages mediated by a distinct subset of Cas proteins involving adaptation, CRISPR (cr) RNA maturation and interference, concluding with the recognition and cleavage of the target DNA or RNA (3–6). CRISPR systems are divided into two classes and six types based on the interference complex subunit composition, while Class 1 systems interference complexes are built by multi-subunit ribonucleoprotein (RNP) complexes, the Class 2 RNPs are composed by a multidomain protein and the crRNA (7,8).
The Class 1 Type III constitutes a complex CRISPR immune system of particular interest, as its members deploy an intricated response controlled by a multifaceted regulatory pathway to degrade both the mRNA and DNA of the invader (9–11). The large Type III interference complexes build a large filament around the crRNA and they are characterized by the presence of the multidomain Cas10 signature protein, which commonly harbours two active sites: an HD nuclease domain for ssDNA cleavage (10,12–14) and a cyclase domain for cyclic oligoadenylate (cA) synthesis (15–17). The recognition of a target RNA triggers the catalytic activities of the Cas10 subunit, and the Cas10 cyclase domain polymerises ATP into cA species ranging between 3- and 6-AMP subunits (cAn) (15–18). cAn molecules act as a second messenger promoting the activation of CRISPR ancillary nucleases (Csx1/Csm6, Can1/Can2 and NucC families), which are the effector “work horse” of the Type III immune response (15,16,19–21). Once these CRISPR ancillary nucleases are activated, these enzymes degrade both host and invading nucleic acids in the cell, resulting in viral clearance, cell dormancy or cell death (22).
Therefore, the control of cAn levels require a regulatory system that can modulate or stop the activity of the indiscriminate nucleases. Although the CARF domains of some Csm6 proteins have been shown to slowly degrade cA4 or cA6, thereby self-limiting their ribonuclease activity (23–25), a major part of Type III systems include standalone cyclic oligoadenylate degrading enzymes, termed CRISPR ring nucleases (Crn), to fulfil this function. The first two members of this family were described in Sulpholobus solfataricus (Sso), where the CARF-domain proteins Sso1393 and Sso2081, were shown to bind and degrade cA4 using a metal-independent mechanism, thus returning cells to a basal uninfected state (26). Despite this information, no structural details addressing the catalytic mechanism of these regulatory enzymes of the Type III immune response is available.
Here, we show the molecular details of how a standalone ring nuclease degrades cA4 by determining the atomic structures of the Sulfolobus islandicus (Sis) ring nuclease Sis0811 in its apo and post-catalytic form in complex with the two molecules of 5′-OH-ApA-2′,3′-cyclic phosphate generated after cA4 cleavage. We observe that cA4 binding and degradation fires a conformational change transmitting the signal from the CARF domains to the wHTH domains. A structure-function analysis allowed us to provide the first structural insight into the catalytic mechanism of a standalone ring nuclease which regulates the type III CRISPR defence system.
MATERIALS AND METHODS
Construction of expression plasmids
Sis0811 wild type sequence from Sis REY15A and the variants used in this study were synthesized by the Integrated DNA Technology (IDT, USA). The genes were then cloned by In-Fusion HD Cloning Plus (Tanaka) into pET-21 with a C-terminal extension encoding a TEV (Tobacco Etch Virus) protease target site and a 6x His-tag (Histidine tag). All sequences were verified by DNA sequencing of the constructed expression plasmids by Sanger sequencing (LIGHTRUN, GATC or Genewiz).
Purification of ring nucleases
His-tagged Sis0811 full length and all its variants were expressed and purified from Escherichia coli BL21 pRARE cells. Cells were grown in LB media containing ampicillin (1 mM) and chloramphenicol (34 μg/ml) at 37 °C until an OD at 600nm wavelength of 0.7–0.9 was reached. Expression was induced by adding 0.5 mM of isopropyl β-d-1-thiogalactopyranoside (IPTG) at 37 °C during 3h. The cells were harvested and re-suspended in lysis buffer (50 mM HEPES pH 7.5, 2 M NaCl, 5 mM MgCl2) in a ratio of about 10 ml buffer/1 g cells supplemented with 1 protease inhibitor tablet (Roche Diagnostics GmbH), lysozyme, 1 μl benzonaze and 0.5 mM TCEP (tris(2-carboxyethyl) phosphine). Cells were lysed by sonication for 10 min with 10 s on and 20 s off cycle and then cell debris and insoluble particles were removed by centrifugation at 11 000 rpm for 30–45 min (Thermo Fisher Scientific, Multifuge X Pro). The supernatant was diluted to 500 mM NaCl (Dilution buffer: 50 mM HEPES pH 7.5, 5 mM MgCl2) and loaded onto a 5 ml Crude HisTrap FF column (GE Healthcare) equilibrated in buffer A (50 mM HEPES pH 7.5, 5 mM MgCl2, 500 mM NaCl). The elution was performed by a step gradient of buffer B (50 mM HEPES pH 7.5, 5 mM MgCl2, 500 mM NaCl, 500 mM imidazole). Enriched protein fractions were applied onto a 5 ml HiTrap Q HP column (GE Healthcare) equilibrated with buffer A2 (20 mM Tris–HCl pH 8.0, 50 mM NaCl). The protein was eluted with a linear gradient of 0–100% buffer B2 (20 mM Tris–HCl pH 8.0, 1M NaCl). Protein-rich fractions were loaded onto a HiLoad 16/600 75 Superdex column (GE Healthcare) equilibrated in gel filtration buffer GF (25 mM HEPES pH 8.0, 300 mM KCl, 0.5 mM TCEP). The protein fractions were concentrated to ∼10 mg/ml (using 10 kDa MWCO Centriprep Amicon Ultra devices) and aliquots were flash-frozen in liquid nitrogen and subsequently stored at -80°C.
Size-exclusion chromatography–multi-angle light scattering (SEC-MALS)
SEC-MALS experiments were performed using a Dionex (Thermo Scientific) HPLC system connected in-line to a UV detector (Thermo Scientific Dionex Ultimate 3000, MWD-3000), a Wyatt Dawn8+ Heleos 8-angle light-scattering detector and a Wyatt Optilab T-rEX refractive index detector. SEC was performed using a Superdex 200 Increase 10/300 GL column (GE Healthcare) at room temperature in a buffer containing 50 mM bicine pH 8.0, 500 mM KCl and 0.5 mM TCEP. For the analysis, 50 μl of Sis0811, Sis0811Δ268, were injected at 0.75 mg/ml, and 1.00 mg/ml concentrations, respectively, and 0.5 ml/min flow rate. ASTRA (version 6.1.7.17 (RC7)) software was used to collect the data from the UV, refractive index, and light scattering detectors. The weight average molecular masses, Mw, were determined across the elution profile from static LS measurements using ASTRA software and a Zimm model, which relates the amount of scattered light to the weight average molecular weight of the solute, the concentration of the sample, and the square of the refractive index increment (dn/dc) of the sample.
Crystallization
Initial crystallization screening using the full-length version of Sis0811 apo sample was performed at 298 K using the sitting-drop vapor-diffusion method and testing a collection of commercially available crystallization screens. The initial drops consisted of 0.15 μl of protein solution (11.59 mg/ml in 25 mM HEPES pH 8.0, and 300 mM KCl, 0.5 mM TCEP) and 0.15 μl well solution, and were equilibrated against 70 μl of well solution. After 60 days of incubation, the extensive initial screening and optimization only rendered rod-like crystals in 20% PEG 3350, 0.2M KSCN and 0.1 M Bis–Tris pH 8.5. The best crystals grown were systematically providing diffraction data only to 4 Å resolution. To improve the crystallization process, last 54 amino acids, predicted to be disordered, were removed (Sis0811Δ268) and following the same protocol used with the full length, crystallization screening assays were performed by mixing protein solution (9.77 mg/ml in 25 mM HEPES pH 8.0, 300 mM KCl and 0.5 mM TCEP) and commercial crystallization solutions. After 15-20 days of incubation, protein crystals were grown in many screening crystallization conditions and their diffraction quality were checked by synchrotron sources (SLS, Villigen, Switzerland; MAXIV, Lund, Sweden). Best screening diffracting crystals were subsequently scaled up and optimized using a Dragonfly (TTP) screen optimizer yielding triangular-like crystals grown in 30% PEG3000, 0.1 M CHES pH 9.5. Crystals were cryo-protected by adding 20% (v/v) glycerol to the mother liquor before flash-freezing in liquid nitrogen.
Crystallization trials with Sis0811Δ268 and its substrate were initially tried by soaking the apo Sis0811Δ268 crystals into a solution including 200 μM (equimolar ratio) of the commercial ligand cA4 but all crystals were dissolved immediately. Then we performed co-crystallization assays by incubating Sis0811Δ268 with 200 μM of cA4 (1:2 molar ratio) for 30 min at 277 K, and then the reaction was used directly for commercial crystal screening. After optimizing initial hits found in reservoir conditions based on 25% PEG 1500 and 20% PEG 3350, best diffracting crystals were grown in 20% PEG 3350, 0.1M Bis–Tris propane pH 7.5, 0.2M KSCN. Sis0811Δ268(S12A) and Sis0811Δ268(S12G/K169G) crystals were obtained following the same protocol described above for the complex form.
X-ray data collection
All data were collected from frozen crystals at 100 K with EIGER and PILATUS detectors at beamlines PXI and PXIII (SLS, Villigen, Switzerland) and at BioMax (MAX-IV, Lund, Sweden). Data processing and scaling were accomplished using XDS (27) POINTLESS and AIMLESS (28) as implemented in autoPROC (29). Statistics for the crystallographic data and structure solution are summarized in Table 1.
Table 1.
X-ray crystallographic data collection and refinement statistics
|
Sis0811Δ268 Apo |
Sis0811Δ268: 2ApA>p Final product bound form |
Sis0811Δ268(S12A) Apo |
Sis0811Δ268(S12G/K169G) Apo |
|
| Data collection | ||||
| Space group | C2 | P41212 | C2 | C2 |
| Cell dimensions | ||||
| a, b, c (Å) | 77.37, 106.44, 78.44 | 56.76, 56.76, 361.040 | 77.15, 103.95, 77.86 | 76.98, 105.03, 78.36 |
| α, β, γ (°) | 90, 102.92, 90 | 90, 90, 90 | 90, 102.98, 90 | 90.00, 102.89, 90.00 |
| Wavelength | 0.98 | 1.00 | 1.00 | 0.98 |
| Resolution (Å) | 61.73–2.38 (2.43–2.38)a | 61.52–2.85 (2.92–2.85)a | 60.82–2.67 (2.71–2.67)a | 76.38-2.04 (2.07–2.04)a |
| R pim | 0.04 (0.40) | 0.08 (0.66) | 0.17 (0.84) | 0.03 (0.31) |
| CC(1/2) | 0.99 (0.76) | 0.99 (0.37) | 0.95 (0.49) | 0.99 (0.84) |
| Mean I / σI | 15.0 (2.3) | 9.3 (1.1) | 6.7 (2.1) | 18.5 (2.3) |
| Completeness, % | 99.9 (99.5) | 93.5 (79.5) | 98.0 (83.5) | 99.2 (97.6) |
| Redundancy | 7.0 (6.8) | 7.8 (6.9) | 6.3 (4.5) | 7.4 (7.2) |
| Refinement | ||||
| Resolution (Å) | 61.61–2.38 | 48.05–2.85 | 60.90–2.67 | 43.93–2.04 |
| No. reflections | 22 478 | 14 031 | 15 386 | 36 331 |
| R work/Rfree | 0.16/0.22 | 0.25/0.29 | 0.22/0.28 | 0.19/0.24 |
| Molecules by a.u. | 2 | 2 | 2 | 2 |
| No. atoms | ||||
| Protein | 4442 | 4360 | 4441 | 4229 |
| cA4 | 0 | 88 | 0 | 0 |
| Water | 254 | 0 | 3 | 77 |
| Ramachandran | ||||
| Favored (%) | 98.16 | 92.31 | 94.29 | 93.91 |
| Allowed (%) | 1.84 | 7.31 | 4.79 | 4.79 |
| Disallowed (%) | 0.00 | 0.38 | 0.92 | 1.30 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.012 | 0.009 | 0.010 | 0.014 |
| Bond angles (°) | 1.633 | 1.638 | 1.605 | 1.860 |
| PDB code | 7PQ2 | 7PQ3 | 7PQ6 | 7PQA |
aValues in parentheses are for highest-resolution shell. One crystal was used to solve each crystal structure.
Crystal structure solution, model building and refinement
Both apo and post-catalytic state structures were solved by molecular replacement method, as implemented in the program PHASER (30). However, while apo structure was solved by using the homologous model from PDB code 3QYF, the cA4 bound form was solved by using the CARF dimer structure from the apo form as a searching model. All the models were then initially subjected to iterative cycles of model building and refinement with PHENIX (31) and Coot (32). Final cycle refinements were performed with REFMAC (33) yielding the refinement and data collection statistics summarized in the Table 1. The Apo, complex, S12A and S12G/K169G final models have an Rwork/Rfree of 16/22, 25/29, 22/28 and 19/24 with 0.00, 0.38, 0.92 and 1.30% of the residues in disallowed regions of the Ramachandran plot, respectively.
The structure of the cA4 bound form was solved by molecular replacement using just the CARF dimer structure from the apo form as a searching model. The quality of the electron density maps provided from this molecular replacement solution allowed us not only to manually retrace the model of the CARF domains but also to build de novo the missing wHTH domains. Figures were generated using PyMOL (34) and ChimeraX (35).
Isothermal titration calorimetry (ITC)
Cyclic tetraadenylate (cA4; catalog number, C 355) was purchased from BIOLOG Life Science Institute (Bremen, Germany). The purity obtained in the synthesis was ≥95% as determined by high performance liquid chromatography (HPLC) and subsequent analysis by mass spectrometry. Prior to ITC experiments both the proteins (Sis0811 and Sis0811Δ268) and the cA4 were extensively dialyzed against ITC buffer, 50 mM HEPES pH 8.0, 300 mM KCl, 0.5 mM TCEP. All ITC experiments were performed on an Auto-iTC200 instrument (Microcal, Malvern Instruments Ltd.) at 25°C. Protein concentrations were determined using a spectrophotometer by measuring the absorbance at 280 nm and applying values for the extinction coefficients computed from the corresponding sequences by the ProtParam program (http://web.expasy.org/protparam/). The cA4 concentration was determined as well by measuring the absorbance at 260 nm and using a extinction coefficient of 54 000 M−1 cm−1. cA4 at approximately 50 or 100 μM concentration was loaded into the syringe and titrated into the calorimetric cell containing the SisRN0811 proteins at ∼5 or 10 μM, respectively. The reference cell was filled with distilled water. In all assays, the titration sequence consisted of a single 0.4 μl injection followed by 19 injections, 2 μl each, with 150 s spacing between injections to ensure that the thermal power returns to the baseline before the next injection. The stirring speed was 750 rpm. Control experiments with the cyclic tetraadenylate injected in the sample cell filled with buffer were carried out under the same experimental conditions. These control experiments showed heats of dilution negligible in all cases. The heats per injection normalized per mole of injectant versus the molar ratio [cA4]/[SisRN0811 variants] were fitted to a single-site model. Data were analysed with MicroCal PEAQ-ITC (version 1.1.0.1262) analysis software (Malvern Instruments Ltd.).
Thin layer chromatography
The Sis0811 homolog Sso1393 has been proposed to cleave cA4 in a consecutive manner where the cA4 substrate is first converted to linear OH-A4>P and then OH-A2>P. To analyse the cA4 cleavage activity of Sis0811 and Sis0811Δ268 purified in this work, the cA4 cleavage products were visualized by TLC. TLC was performed on 10 × 10 cm glass sheets coated with a thin layer of absorbent silica gel containing a green fluorescent indicator that excites at 254nm UV light (Merck Millipore) as the stationary phase and a running buffer containing 30% H2O, 70% ethanol and 0.2 M ammonium bicarbonate as the mobile phase. Reactions of 50 μl containing 40 μM cA4, increasing protein concentration and reaction buffer (20 mM MES pH 6, 5 mM MgCl2) were incubated at 70°C for 60 min in a thermocycler. A “mock” reaction containing no protein but both 40 μM cA2 and 40 μM cA4 was used as a control for each experiment. The reactions were stopped by cooling to 4°C and subsequently performing a phenol extraction to remove the protein from each sample. Phenol extraction was performed by adding an equal volume of phenol to the reaction and vortexing until milky in colour. Samples were then centrifuged for 60 seconds at 7000 rpm to separate the organic and aqueous phase. The cA4 cleavage products remained in the top aqueous phase and 30 μl were carefully extracted to a clean tube, ensuring no phenol was transferred as well. 15 μl of the phenol extracted sample were dotted onto the TLC plate 1 cm from the bottom, 2 μl at a time allowing to dry completely between each application. The plates were dried completely before performing TLC. The dried plates were applied to a previously equilibrated TLC chamber containing 0.5 cm of running buffer at room temperature. The mobile phase was allowed to rise though the stationary phase by capillary action until the solvent front reached about 1 cm from the top. The plates were finally dried before being imaged using UV light of 254 nm. Experiments for each protein were designed with four points of increasing protein concentration and one “mock” reaction as control as previously described. All experiments were performed in triplicates.
Liquid chromatography (LC) ESI-MS
Approximately 20 pmol of the samples were deproteinized as described for TLC, injected into a UPLC system (UltiMate 3000, Dionex) on a Kinetex® EVO C18 reverse phase column (Phenomenex, 2.1 mm × 100 mm, 5.0 μm particle size) and analysed with a microOTOF-Q II mass spectrometer (Bruker Daltonik GmhH) equipped with an electrospray ionization (ESI) source (capillary voltage 4500 V, end plate offset –500 V, nebulizer gas (nitrogen) pressure 2.0 bar, flow 9 l/min and drying gas temperature 200°C). The LC method uses a gradient of A (8.6 mM TEA pH 8.3) and B (8.6 mM TEA pH 8.3, 5% acetonitrile) with a flow rate of 0.2 ml/min as follows: 0–4 min 100% B, 4–6 min 100% B, 6–8 min 0% B. The column temperature was 30°C. Mass data were acquired in negative-ion mode with a scan range m/z 300–1500 and a mass resolving power of 10 000. Data acquisition was done under the control of the module Hystar 3.2-SR 2 from Bruker Compass 1.3 software that integrates both the LC chromatographic separation and MS methods. Data analysis was done with DataAnalysis Version 4.0 SP5 (Bruker Daltonik GmhH). The commercial standards cA2, pApA and cA4 used in this study to identify the mass of the reaction products were purchased from BIOLOG Life Science Institute (Bremen, Germany). cA2 and pApA are isobaric to the reaction products P1 (ApA>p) and P2 (ApAp) but chemically different therefore showing different retention times (Supplementary Figure S5).
Protein domain analysis
A HHPred (36) search was performed with 6 CARF domain sequences: Sis0811, Sis0455, Sso1393, Sso2081, SisCsx1 and TonCsm6. A multiple sequence alignment was performed using MUSCLE (37) and submitted to HHpred for sequence and further structural homology comparison. Additionally, a DALI (38) search was performed with the structure of the CARF domain of Sis0811 in order to attempt to discover new structural homologies. Ring Nuclease activity information was found in the literature on nine CARF domain containing proteins. Information about these structures is summarized in Supplementary Table S1. Thereafter, a multiple structure alignment of CARF domains was performed using mTM-align (39).
RESULTS
Crystal structure of Sis0811
Sis0811 is composed by an N-terminal CRISPR-Cas associated Rossmann Fold (CARF) domain (residues 1–199) connected to a winged helix-turn-helix (wHTH) domain (residues 208-268) and a C-terminal tail (residues 269–322), which is predicted to be disordered (Figure 1A, Supplementary Figure S1). Sis0811 crystals did not provide diffraction-quality data better than 4 Å resolution. To circumvent that problem, we designed a truncated version (Sis0811Δ268), removing the C-terminal 54 residues. The Sis0811Δ268 yielded high-quality crystals and the structure was solved at 2.38 Å resolution by molecular replacement (Figure 1B, Material and Methods, Table 1).
Figure 1.

Structural, biophysical and cleavage activity characterization of the ring nuclease Sis0811. (A) Domain organization of full-length ring nuclease Sis0811. Dash lines represent the sequence fragment removed to obtain the truncated construct Sis0811Δ268. (B) Cartoon models (each domain is coloured differently) of the dimeric apo Sis0811Δ268 crystal structure rotated 90 degrees between them along its 2-fold axis. (C) Sis0811 and Sis0811Δ268 substrate binding assays by ITC. Affinities and thermodynamic values of Sis0811, Sis0811Δ268 binding events to cA4 inferred from ITC measurements performed at 25°C. Gibbs free energy (ΔG), enthalpy (ΔH), entropy (–TΔS), equilibrium dissociation constant (KD) are shown. The protein-cyclic oligonucleotide interaction affinity is defined by the Gibbs energy for binding ΔG = –RT ln KA = RT ln KD. The errors are the standard deviation of three independent experiments. (D) Sis0811 and Sis0811Δ268 substrate cleavage assays analysed by TLC. The panel represents the percentage of the final reaction product (2 ApA>p) generated by SisRN0811 and Sis0811Δ268. (E) TLC cleavage assay experiment. Arrow reflects the sample migration direction.
The structure of the apo Sis0811Δ268 monomers within the asymmetric unit interact parallelly along the 2-fold axis to form a dimer (Figure 1B), which correlates with the dimeric assembly observed in solution from SEC-MALS experiments (Supplementary Figure S2). Thus, the CARF domains shape the catalytic pocket at their interface of the dimer assembly, while wHTH domains are oppositely oriented along the 2-fold dimer axis (Figure 1B). Hence, the 2-fold axis bisects the cA4 catalytic pocket and the wHTH domains in the dimer. As in other CARF domains, its core is a 6-stranded Rossmann-like fold, with the core β5 and β6 strands forming a ß-hairpin. By comparison with its structural homologue Sso1393 in its apo form, where K168 and a S11 of each monomer has been proposed to be involved in its catalytic activity (26), the equivalent residues in Sis0811Δ268, K169 and S12, display the same configuration, respectively (Supplementary Figure S3A and B). The conserved lysine residues are present at the very bottom of the catalytic pocket, parallel to each other. The ϵ-ammonium group of the lysine residue is surrounded by five α-carboxyl groups from the opposite chain, most likely assisting in the stabilization of the dimer and creating a positively charged environment. The conserved serine residues are located to each side of the catalytic pocket ∼8 Å from the bottom of the pocket and the carboxyl group faces away from the catalytic pocket (Supplementary Figure S3A and B). The N-terminal α-helixes are positioned symmetrically across each other forming the catalytic pocket. β-sheets in the centre of the protein angle the C-terminal α-helixes so that the chains cross each other in a ‘ribbon-like’ fashion. Modelling the electrostatic potential reveals a positively charged pocket at the CARF dimer interface, pointing the cavity candidate of the negatively charged cA4 substrate binding (Supplementary Figure S3B).
Next, we analysed whether the deletion of the C-terminal tail affected cA4 binding and cleavage. We performed ITC binding assays with the truncated and full-length versions of SisRN0811. The experiments show that both versions of the enzyme displayed a similar KD of 9.1 and 5.9 nM for the full length and the truncated form respectively, suggesting that the deletion of the C-terminal tail does not affect cA4 binding substantially (Figure 1C). These minor differences are also observed in the catalytic activity of the enzyme. We monitored the enzymatic reaction quantifying the di-AMP reaction products of the cA4 degradation after separation using TLC (Figure 1D and E, Materials and Methods). The truncated version of Sis0811 (Sis0811Δ268) cleaved cA4 slightly more efficiently than the full-length (Figure 1D and E), suggesting that the removal of the C-terminal tail could facilitate the entrance of the substrate.
Structure of the Sis0811: 2ApA>p post-catalytic complex
To understand cA4 interaction and degradation mechanism by Sis0811, we attempted to obtain the crystal structure of Sis0811Δ268 in complex with its substrate cA4. Thus, we initially performed soaking experiments of Sis0811Δ268 crystals into its mother liquor solution including cA4 in an equimolar ratio. However, once the apo crystals were soaked into the cA4 containing solution, they were immediately dissolved suggesting that the interaction of cA4 with the ring nuclease dismantled the crystal lattice, indicating a possible conformational change upon cA4 binding. A similar behaviour was observed with the full-length protein. Hence, we decided to perform co-crystallization assays by incubating the ring nuclease and its cA4 substrate prior to the crystallization trials to obtain the structure of the post-catalytic state. Following this strategy, we obtained new crystals under different conditions, which diffracted to 2.7 Å resolution. The post-catalytic state structure was determined by molecular replacement using the CARF domain of the apo structure as a model (Figure 2A, Supplementary Figure S4A, Table 1, Materials and Methods). The quality of the 2FoFc and omit electron density maps unambiguously revealed the presence of the cleaved cyclic oligoadenylate at the catalytic pocket in a post-catalytic state (Figure 2B and C, Supplementary Figure S4A).
Figure 2.

Structural analysis of the ring nuclease SiRe-0811 in complex with its post-catalytic reaction product. (A) Cartoon models of the dimeric Sis0811Δ268 crystal structure in complex with 2 ApA>p (post-catalytic reaction product from cA4 substrate).(B) 2Fo – Fc map at the substrate binding pocket superimposed onto its corresponding refined structure. Map displayed at 2.0σ contour value. (C) Fo – Fc omit map at the substrate binding pocket superimposed onto its corresponding 2 ApA>p post-catalytic structure. FoFc omit map is displayed at 4.0σ contour value. (D) LC–MS analysis of SiRe-0811 reaction products. Upper panel: extracted ion chromatograms for m/z 657 (±0.5) (violet trace; ApA>p–1; retention time 5 min) and m/z 675 (±0.5) (orange trace; ApAp-1; retention time 6.4 min). Lower panel: extracted ion chromatogram for m/z 1315 (±0.5) (cyan trace; cA4-1; retention time 6.8 min). * Symbol means that the hydrolysis of P1 at the cyclic phosphate could generate a P2 product having the phosphate group either at 2′ or 3′.
Collectively, the apo and post-catalytic structure of the Sis0811Δ268, including the reaction product bound, provide a detailed glimpse on the conformational changes of the enzyme and the key catalytic residues involved in cA4 binding and cleavage of the canonical metal-independent standalone ring nucleases (Figure 1B, Figure 2) (26,40).
cA4 degradation
RNase and DNase metal independent nucleases generate 2′3′cyclic phosphate intermediates (41). Although there are differences among their active pockets, they share common feaures, such as the fact that water appears to be excluded from the active site, the 2′-OH of the ribose or a protein side chain serve as the nucleophile and the highly negatively charged pentacovalent intermediate is neutralized by positively charged side chains (41). A cleavage mechanism was proposed in a previous study combining the docking of a cA4 molecule in the apo structure of Sso1393 (PDB:3QYF), a Sis0811 homologue, with biochemical assays (26). In this mechanism the 2′-OH- of the ribose would initate the nucleophilic atack leaving the 5′-OH of the 2-AMP product unprotected and a 2′,3′-cyclyc phosphate. Our post-catalytic state 2FoFc and omit electron density maps support this hypothesis since the reaction product structure could be fitted containing a 5′-OH and the 2′,3′-cyclic phosphate termini (2 ApA>p, Figure 2B and C). The identification of this reaction product was confirmed by mass spectrometry analysis (Figure 2D, Supplementary Figure S5).
cA4 triggers a conformational change in SisRN0811
The binding and cleavage of cA4 in the cataytic pocket seems to induce a large conformational change in the protein (Figure 3, Supplementary Video S1). The two monomers undergo a corkscrew movement closing the CARF domains on the substrate while the wHTH domains separate. The CARF domains fold inwards from the apo to the cA4 cleaved state, trapping the reaction product 2 ApA>p into the positively charged environment of the catalytic pocket. The corresponding wHTH domains turn 30° in counter-clockwise manner, like a ‘cork-screw’, around the 2-fold axis (Figure 3, Supplementary Video S1).
Figure 3.

Conformational change of the ring nuclease Sis0811 from its apo form to its state in complex with its product reaction. (A) Cartoon/surface models comparing the cA4 non-bound (upper panels) and its product complex forms (bottom panels). Coloured arrows represent the conformational change path of each corresponding domain from apo structure towards the product reaction bound form. (B) Zoom view at the substrate binding pocket within the apo dimer Sis0811Δ268 and (C) in complex with its post-catalytic reaction product, depicting their key interacting residues. Each monomer is coloured differently and key side chains residues from each monomer interacting with the 2 ApA>p product (white sticks) are depicted accordingly in sticks.
The comparison of the active site between the apo and post-cleavage structures indicates the key residues which are repositioned in the conformational change (Figure 3B-C). Among them K169 seems to play an important role in the relocation of the side chains to favour catalysis. The comparison suggests that the correponding 2′-OH of the ribose could be the responsible of the nucleophilic attack, while the K169 residues in each monomer could be involved in the reaction intermediate stabilisation and S12 would stabilize the 2′,3′cyclic phosphate after cleavage (Figure 3C, Supplementary Figure S4A). During this conformational change between the apo and post-catalytic states, S12 and S12′ undergo a concerted change in their side chain orientation, moving from their configuration opposite to the catalytic pocket to engage with the 2′,3′cyclic termini after the hydrolysis reaction (Figure 3B and C, Supplementary Figure S3A, Supplementary Figure S4A, Supplementary Video 1). The closed state conformation induced by the substrate cleavage was stabilized by the generation of new interactions at the interface of the CARF domains highlighting the creation of two interchain salt bridge interactions: E193-R37′ and E193′-R37. Q192-Q192′ is also involved in the stabilization of the R37–R37′ by polar interactions in both monomers while its side chain interacts with A1 and A1′ by stacking with their adenine rings (Supplementary Figure S4B). Apart from the two catalytic residues, this structure revealed a network of interactions that were involved in substrate binding. Y189 from each monomer is interacting with the non-cleaved phosphates, while additional residues are involved in binding and positioning the adenines. Hence, N16–N16′ and D75–D75′ interact with the A1–A1′ adenines while T98–T98′, T100–T100′, S103–S103′, Y191–Y191′ interact with A2–A2′ adenines (Figure 3C).
The conserved S12 and K169 are essential for cA4 degradation
Based on the structural information we generated several mutants (S12A, S12G, K169G and S12G/K169G) to investigate their role in catalysis. We attempted to crystallize these mutants with the ligand in order to trap some intermediates of the cleavage reaction. We succeeded in crystallizing and solving the structures of the S12A and S12G/K169G mutants. However, although crystallization assays were performed at protein: cA4 molar ratio of 1:2 (Materials and Methods), the second messenger was not visualized at the catalytic pocket and the structure of the mutants is like the wild type apo form (Table 1, Methods).
A comparison of the active sites with the wild type shows that the S12A mutation did not alter substantially the configuration and polarity of the substrate binding pocket substantially. However, in the case of the S12G/K169G mutant, the active site is clearly distorted as shown by the absence of a clear electron density map between residues 166–169 (Figure 4A). The K169G mutation removes the strong interchain associations of both K169, thus affecting the configuration and the electrostatic potential of the substrate binding site (Supplementary Figure S6). To further analyse the role of these catalytic residues, we performed activity assays incubating the S12G, K169G and S12G/K169G mutants with the substrate cA4. The reaction mixture was analysed by mass spectrometry revealing the degradation products (Figure 4B, Supplementary Figure S5, Materials and Methods). Five different species were detected: the substrate cA4, P1 (ApA>P), P2 (hydrolysis product of P1, ApAp), P3 (intermediate reaction product, ApApApA>p) and P4 (hydrolysis of P3, ApApApAp). The assay confirmed that the individual S12G, K169G mutants and the S12G/K169G combination strongly affect the degradation of cA4, abolishing the production of the final product P1 (ApA>p) (Figure 4B). The major compound found in the assay for the K169G and S12G/K169G mutants is cA4, while in the case of the S12G variant the intermediate product P3 (ApApApA>p) was detected (Figure 4B), suggesting that cleavage of the cA4 phosphodiesters to generate P1 does not proceed in a concerted manner.
Figure 4.

Sis0811 reaction product analysis from the designed mutants. (A) 2Fo – Fc map at the substrate binding pocket of the Sis0811Δ268 (S12A) and Sis0811Δ268 (S12G/K169G) mutants. (B) LC–MS analysis of the reaction products obtained from Sis0811Δ268 (S12G), Sis0811Δ268 (K169G) and Sis0811Δ268 (S12G/K169G) mutants compared to the wild type. Ion chromatograms extracted for m/z 657 (±0.5) (ApA>p–1, cA4–2, ApApApA>p–2; retention times 5, 6.8 and 7.1 min), m/z 675 (±0.5) (ApAp -1; retention time 6.4 min), m/z 1315 (±0.5) (cA4–1, ApApApA>p–1; retention times 6.8 and 7.1 min) and m/z 1333 (±0.5) (ApApApAp–1; retention time 7.3 min). Chemical formula of the reaction species is depicted. The asterisk symbol (*) means that the hydrolysis of P1/P3 at the cyclic phosphate could generate a P2/P4 product having the phosphate group either at 2′ or 3′.
Comparison of cA4 cleavage properties by different CARF domains
Some CARF-containing proteins bind and cleave cA molecules, while others just bind cA. A general description of the cA cleaving mechanism by CARF domains is elusive because of the different set of residues involved (40). With this aim, we compiled a group of CARF-containing protein structures with and without cA cleavage catalytic activity and the general fold of the CARF domains were compared using the TM-align web server (39). The general fold is very well conserved (Supplementary Figure S7), and from the TM-scores calculated there are no differences that allow the prediction of which of the domains are capable of cA4 catalysis (Supplementary Table S1). For example, Sis0811 has a higher similarity to certain CARF domains that do not cleave cA, such as Can2 or Card1, than to those which display catalytic activity, i.e, Thermococcus onnurineus (Ton) Csm6 or Enterococcus italicus (Ei) Csm6. Therefore, the presence of certain residues, which provide the reactant groups, in the CARF scaffold is the key factor to allow cA cleavage. A comparison of SisRN0811 with the structure of TonCsm6 (PDB: 6O6Y) (Figure 5), which to our knowledge is the only structure of a CARF containing protein in the post-catalytic state, shows that Sis0811-K169 and TonCsm6-N135 share the role of stabilizing the reaction intermediate, while Sis0811-S12 and TonCsm6-W14 are the residues involved in stabilizing the product of the cleavage reaction (Figure 5A-B). However, the differences in the catalytic sites generate a rather different electrostatic potential in the active centre, conferring different enzymatic properties to these two enzymes who cleave cA4 generating the same product (Figure 5C-D).
Figure 5.
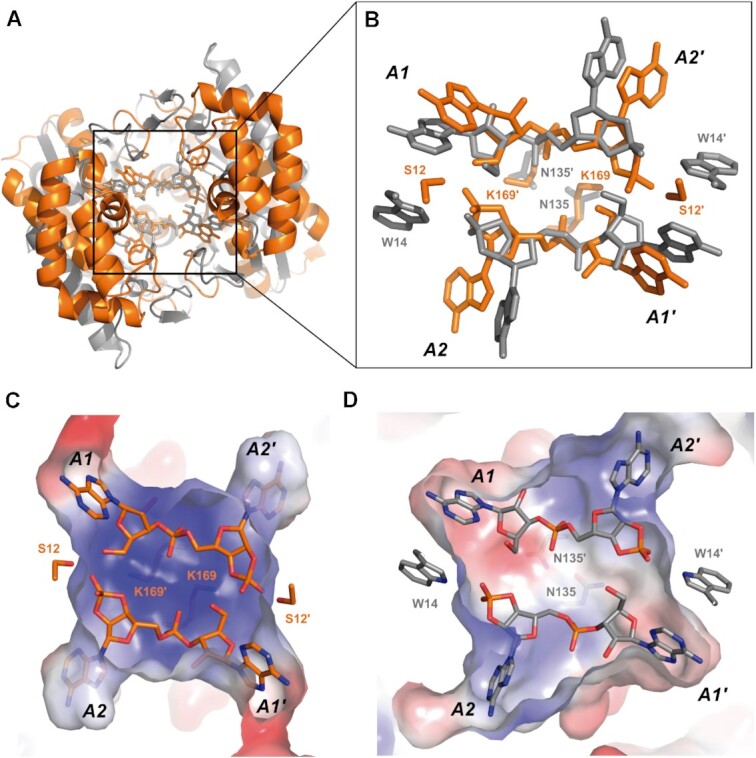
Sis0811:2ApA>p–TonCsm6:2ApA>p complex comparison. (A) Top view of the CARF structural alignment from Sis0811:2ApA>p (orange) and TonCsm6:2ApA>p complexes (grey). (B) Zoom view of the key residues involved in catalysis of both complexes. (C) Electrostatic potential representation of the active site cavity in Sis0811:2ApA>p and (D) TonCsm6:2ApA>p post-catalytic complexes.
DISCUSSION
The regulation of type III CRISPR-Cas systems utilizes specific nucleases to deactivate the signal unleashed by cyclic oligonucleotides and stop the indiscriminate RNA degradation by Csx1/Csm6 nucleases. In this study we combine structural and biochemical approaches showing how a stand-alone ring nuclease degrades a cA4 molecule. The apo and post-cleavage crystal structures provide a detailed view of the phosphodiester hydrolysis mechanism of Sis0811. The dimeric Sis0811 undergoes a conformational change that resembles a corkscrew and triggers cA4 degradation, thereby decreasing the levels of this second messenger in the cell. Although we could not capture the intermediate state where the substrate is bound before catalysis, and therefore our structural data cannot distinguish whether the conformational change is caused by the ligand binding or its cleavage; our previous analysis in SisCsx1 suggests that most likely the binding of the cA4 molecule is sufficient to induce a conformational change in the CARF domain, which positions the key residues involved in phosphodiester hydrolysis (42).
Our structural data provide experimental evidence of a previously proposed mechanism for Sso1393 (26) (Figure 6). The 2′OH of the ribose will initate the nucleophilic atack on the phosphate, subsequently K169 seem to stabilize the pentacovalent phosphorous formed in the transition state, and the 2′-3′cyclic phosphate will be stabilized by the S12 OH group, which could previously position the 2′-OH of the ribose for the nucleophilic attack. The cyclic 2′,3′-cyclic phosphate can be destabilized and disrupted over time (41), as it has been observed in our assays some P2 product (ApAp) can be detected (Figure 4B). In addition, our analysis of the reaction products suggest that the cleavage reaction of the phosphodiester bonds does not occur in a concerted manner, as the linear P3 product (ApApApA>p) is formed in the case of the S12G mutant. This product was also observed in TonCsm6 before ApA>p was detected (23), thereby resembling the cleavage observed in Sis0811. Therefore, the data indicates that the role of S12 in stabilizing the linear ApApApA>p product could be an important stage before the second hydrolysis proceeds and 2 molecules of ApA>p are generated. Future studies trapping non-cleaved cA4 and linear intermediates may help to solve this point.
Figure 6.

Model of Sis0811 catalytic mechanism. The S12 position the 2′-OH of a ribose to initate the nucleophilic atack on the corresponding phosphate and subsequently K169′ stabilize the pentavalent phosphorous formed in the transition state. Then, the 2′-3′cyclic phosphate is stabilized by the S12 OH group, producing the intermediate reaction product P3 (ApApApA>p). Next, the other cleavable phosphodiester bond is attacked by the 2′-OH of the ribose which is close to S12′ that positions it. Thus, K169 stabilize the pentacovalent phosphorous formed in the transition state and the cyclic 2′,3′-cyclic phosphate is stabilised by S12′ OH group generating two molecules of P1 (ApA>p) as final reaction product.
Unfortunately, the structural comparison of the CARF domains does not provide clear insights to distinguish those that can degrade cA from others that are unable of cleaving the second messenger. The current data suggest that the degradation of cA is a mechanism to control the catalytic activity of Csm6/Csx1 RNAses. However, in Csm6 proteins containing a CARF domain that hydrolizes the cyclic compound, such as TonCsm6 and EiCsm6, the cleavage of the second messenger is slow, and the control of the catalytic activity seems to be accomplished by downregulating the RNAse activity of the HEPN domain by eliminating the activating effect of the ligand binding on the CARF. On the other hand, the CARF domains of the Csx1 family of auxiliary RNAses, such as SisCsx1 or Sulfolobus solfataricus (Sso) Csx1, which also contain HEPN domains, do not cleave cAs (26,42) and seem to be dependent on other CARF containing proteins, the stand-alone ring nucleases, to downregulate their RNA degradation. The CARF domains of these stand-alone ring nucleases seem to degrade the compounds more swiftly than those with catalytic activity in Csm6 proteins (23,25,26,42).
Collectively, our analysis indicates that the CARF domain in these CRISPR systems seems to have evolved as a protein module to recognise cA, then the recognition module has diversified its properties. As an appropriate control of the cA levels is crucial for the cell, the differences observed in the catalytic activities and regulatory mechanisms are likely a product of the large diversity of strategies developed during evolution to adapt this signalling systems to the habitats of these organisms.
Some members of the standalone Crn1 subfamily are composed by only the CARF domain, while others, such as Sis0811 and Sso1393 contain a C-terminal wHTH domain fused to it (43). As we have shown in this study the wHTH domain is not involved in catalysis. Although they do not contribute to the dimerization of Sis0811, they seem to be important to undergo the conformational movement which triggers activity. In addition, S. islandicus REY15A contains a second ring nuclease, Sis0455, which does not include an wHTH domain and displays a higher cA4 degradation activity than Sis0811 (42). In addition, the Sis0811Δ268 mutant also displays a slightly higher activity than the wild type (Figure 1D). These observations lead us to speculate with the possibility that the wHTH of this stand-alone nuclease may downregulate its activity.
A structural homology search performed in the DALI server using the wHTH domain rendered many DNA binding proteins, including transcriptional repressors and activators, with substantial structural homology (38) (between 1.9 and 3.0 Å for the 65 residues of the wHTH domain). Sis0811 wHTH domain matches with transcriptional regulator domains from the MarR (Multiple antibiotic resistance regulator) transcriptional regulator family (RMSD of 2.2Å). This observation led us to speculate whether the wHTH domain and the conformational change upon cA4 binding and degradation, could be related to a functional DNA binding event, by which the degradation of cA4 may be linked to the activation or repression of certain genes involved in the bacterial immune defence system. We performed a dsDNA docking analysis in Sis0811 using structures that contained structurally similar DNA bound wHTH domains. However, the arrangement of the Sis0811 wHTH dimer in our structures does not support the docking of dsDNA molecules. Consequently, with the current data, the role of the wHTH domains in Sis0811 seems to be only structural regulating the interplay of the protein scaffold to proceed with the degradation of cA4. Further studies are needed to fully explore and confirm this hypothesis.
DATA AVAILABILITY
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 7PQ2, 7PQ3, 7PQ6 and 7PQA accordingly. All other data are available from the corresponding author upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank SLS and MAX-IV synchrotrons for help during X-ray diffraction data collection. Data processing has been performed at the Computerome, the Danish National Computer for Life Sciences. GM is a member of the Integrative Structural Biology Cluster (ISBUC) at the University of Copenhagen.
Notes
Present address: Stefano Stella, Twelve Bio ApS, Ole Maaløes Vej 3, Copenhagen, 2200, Denmark.
Contributor Information
Rafael Molina, Structural Molecular Biology Group, Novo Nordisk Foundation Centre for Protein Research, Faculty of Health and Medical Sciences University of Copenhagen, Blegdamsvej 3-B, Copenhagen, 2200, Denmark.
Anne Louise Grøn Jensen, Structural Molecular Biology Group, Novo Nordisk Foundation Centre for Protein Research, Faculty of Health and Medical Sciences University of Copenhagen, Blegdamsvej 3-B, Copenhagen, 2200, Denmark.
Javier Marchena-Hurtado, Structural Molecular Biology Group, Novo Nordisk Foundation Centre for Protein Research, Faculty of Health and Medical Sciences University of Copenhagen, Blegdamsvej 3-B, Copenhagen, 2200, Denmark.
Blanca López-Méndez, The Novo Nordisk Foundation Center for Protein Research, Protein Structure & Function Programme, Faculty of Health and Medical Sciences, University of Copenhagen, Blegdamsvej 3B, 2200 Copenhagen, Denmark.
Stefano Stella, Structural Molecular Biology Group, Novo Nordisk Foundation Centre for Protein Research, Faculty of Health and Medical Sciences University of Copenhagen, Blegdamsvej 3-B, Copenhagen, 2200, Denmark.
Guillermo Montoya, Structural Molecular Biology Group, Novo Nordisk Foundation Centre for Protein Research, Faculty of Health and Medical Sciences University of Copenhagen, Blegdamsvej 3-B, Copenhagen, 2200, Denmark; The Novo Nordisk Foundation Center for Protein Research, Protein Structure & Function Programme, Faculty of Health and Medical Sciences, University of Copenhagen, Blegdamsvej 3B, 2200 Copenhagen, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Novo Nordisk Foundation Center for Protein Research is supported financially by the Novo Nordisk Foundation Core grant [NNF14CC0001]; Distinguished Investigator [NNF18OC0055061 to G.M.]. Funding for open access charge: Novo Nordisk Foundation Core [NNF14CC0001].
Conflict of interest statement. G.M. and S.S. are co-founders of Twelve BIO.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709. [DOI] [PubMed] [Google Scholar]
- 2. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E.v, van der Oost J.. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016; 353:aad5147. [DOI] [PubMed] [Google Scholar]
- 4. Amitai G., Sorek R.. CRISPR–Cas adaptation: insights into the mechanism of action. Nat. Rev. Microbiol. 2016; 14:67–76. [DOI] [PubMed] [Google Scholar]
- 5. Jackson S.A., McKenzie R.E., Fagerlund R.D., Kieper S.N., Fineran P.C., Brouns S.J.J.. CRISPR-Cas: adapting to change. Science. 2017; 356:eaal5056. [DOI] [PubMed] [Google Scholar]
- 6. Barrangou R., Horvath P.. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2017; 2:17092. [DOI] [PubMed] [Google Scholar]
- 7. Koonin E.v, Makarova K.S., Zhang F.. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017; 37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P.et al.. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Molina R., Sofos N., Montoya G.. Structural basis of CRISPR-Cas Type III prokaryotic defence systems. Curr. Opin. Struct. Biol. 2020; 65:119–129. [DOI] [PubMed] [Google Scholar]
- 10. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA cleavage during Type III CRISPR-Cas immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng W., Feng M., Feng X., Liang Y.X., She Q.. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015; 43:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR–Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Estrella M.A., Kuo F.-T., Bailey S.. RNA-activated DNA cleavage by the Type III-B CRISPR–Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas Č., Siksnys V.. Spatiotemporal control of Type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 15. Kazlauskiene M., Kostiuk G., Venclovas Č., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017; 357:605. [DOI] [PubMed] [Google Scholar]
- 16. Niewoehner O., Garcia-Doval C., Rostøl J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:543–548. [DOI] [PubMed] [Google Scholar]
- 17. Rouillon C., Athukoralage J.S., Graham S., Grüschow S., White M.F.. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. eLife. 2018; 7:e36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nasef M., Muffly M.C., Beckman A.B., Rowe S.J., Walker F.C., Hatoum-Aslan A., Dunkle J.A.. Regulation of cyclic oligoadenylate synthesis by the Staphylococcus epidermidis Cas10-Csm complex. RNA. 2019; 25:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McMahon S.A., Zhu W., Graham S., Rambo R., White M.F., Gloster T.M.. Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nat. Commun. 2020; 11:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu W., Mcquarrie S., Gr S., Mcmahon S.A., Graham S., Gloster T.M., White M.F.. The CRISPR ancillary effector Can2 is adual-specificity nuclease potentiating type III CRISPR defence. Nucleic Acids Res. 2021; 49:2777–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lau R.K., Ye Q., Patel L., Berg K.R., Mathews I.T., Watrous J.D., Whiteley A.T., Lowey B., Mekalanos J.J., Kranzusch P.J.et al.. Structure and mechanism of a cyclic trinucleotide-activated bacterial endonuclease mediating bacteriophage immunity. Mol. Cell. 2020; 77:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rostøl J.T., Marraffini L.A.. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR–Cas immunity. Nat. Microbiol. 2019; 4:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia N., Jones R., Yang G., Ouerfelli O., Patel D.J.. CRISPR-Cas III-A Csm6 CARF domain is a ring nuclease triggering stepwise cA4 cleavage with ApA>p formation terminating RNase activity. Mol. Cell. 2019; 75:944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Athukoralage J.S., Graham S., Grüschow S., Rouillon C., White M.F.. A Type III CRISPR ancillary ribonuclease degrades its cyclic oligoadenylate activator. J. Mol. Biol. 2019; 431:2894–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia-Doval C., Schwede F., Berk C., Rostøl J.T., Niewoehner O., Tejero O., Hall J., Marraffini L.A., Jinek M., Jakob T.et al.. Activation and self-inactivation mechanisms of the cyclic oligoadenylate-dependent CRISPR ribonuclease Csm6. Nature Communications. 2020; 11:1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Athukoralage J.S., Rouillon C., Graham S., Grueschow S., White M.F.. Ring nucleases deactivate Type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature. 2018; 562:277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W. XDS. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution?. Acta Crystallogr. D. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G.. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D. 2011; 67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P.D., Afonine P.v., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 33. Winn M.D., Murshudov G.N., Papiz M.Z.. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods in Enzymology. 2003; 374:Academic Press; 300–321. [DOI] [PubMed] [Google Scholar]
- 34. Schrödinger L. The PyMOL molecular graphics system. Version 2.0 Schrödinger, LLC. 2015; [Google Scholar]
- 35. Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E.. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zimmermann L., Stephens A., Nam S.Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A.N., Alva V.. A completely reimplemented MPI bioinformatics toolkit with a new hhpred server at its core. J. Mol. Biol. 2018; 430:2237–2243. [DOI] [PubMed] [Google Scholar]
- 37. Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004; 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holm L. DALI and the persistence of protein shape. Protein Sci. 2020; 29:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y., Skolnick J.. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005; 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Makarova K.S., Timinskas A., Wolf Y.I., Gussow A.B., Siksnys V., Venclovas Č., Koonin E.v.. Evolutionary and functional classification of the CARF domain superfamily, key sensors in prokaryotic antivirus defense. Nucleic Acids Res. 2020; 48:8828–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011; 44:1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Molina R., Stella S., Feng M., Sofos N., Jauniskis V., Pozdnyakova I., López-méndez B., She Q., Montoya G.. Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nat. Commun. 2019; 10:4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Athukoralage J.S., White M.F.. Cyclic oligoadenylate signalling and regulation by ring nucleases during type III CRISPR defence. RNA. 2021; 27:855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 7PQ2, 7PQ3, 7PQ6 and 7PQA accordingly. All other data are available from the corresponding author upon reasonable request.