


Abstract
Telomeres are intrinsically difficult-to-replicate region of eukaryotic chromosomes. Telomeric repeat binding factor 2 (TRF2) binds to origin recognition complex (ORC) to facilitate the loading of ORC and the replicative helicase MCM complex onto DNA at telomeres. However, the biological significance of the TRF2–ORC interaction for telomere maintenance remains largely elusive. Here, we employed a TRF2 mutant with mutations in two acidic acid residues (E111A and E112A) that inhibited the TRF2–ORC interaction in human cells. The TRF2 mutant was impaired in ORC recruitment to telomeres and showed increased replication stress-associated telomeric DNA damage and telomere instability. Furthermore, overexpression of an ORC1 fragment (amino acids 244–511), which competitively inhibited the TRF2–ORC interaction, increased telomeric DNA damage under replication stress conditions. Taken together, these findings suggest that TRF2-mediated ORC recruitment contributes to the suppression of telomere instability.
INTRODUCTION
The origin recognition complex (ORC) is composed of 6 subunits (ORC1–6) and binds to replication origins distributed across the eukaryotic genome (1,2). Human ORC binds to origin DNA with no obvious sequence specificity and binding principally depends on the chromatin environment (2–6). ORC-binding sites share several common characteristics, such as the presence of transcriptional start sites with an open chromatin structure, active histone modifications, and CpG islands (3–5). In addition, various chromatin-associated proteins, such as HP1α, dimethylated histone H4 (H4-K20me2), ORCA, and telomeric repeat binding factor 2 (TRF2) (2,6), associate with the ORC complex and act as local ORC recruiters. In late M to G1 phase, ORC, and the additional licensing factors CDC6 and Cdt1, cooperatively promote the loading of minichromosome-maintenance (MCM) complex, a core component of the replicative helicase (1,2,7). During the following S phase, activated cyclin-dependent kinases (Cdks) and Dbf4-dependent kinase (DDK) trigger the initiation of DNA replication. Phosphorylation of MCM is a prerequisite for origin firing, while ORC, CDC6 and Cdt1 are downregulated by phosphorylation to prevent MCM re-loading and DNA re-replication (8,9). Replication stress-induced fork stalling activates MCMs pre-loaded onto dormant origins, promoting origin firing to assist in the completion of replication. Reduction in MCM levels causes DNA breaks, micronuclei formation, and genome instability, eventually leading to cellular senescence, inflammation and increased cancer risk (10–16).
Telomeres are the terminal regions of linear chromosome. In mammals, the chromosome ends form telomere loops (T-loops), protecting DNA ends from detection by DNA damage response sensors (17,18). End-protection is mostly achieved by telomere-specific chromatin-binding proteins that form the shelterin complex, comprised of TRF1, TRF2, RAP1, TIN2, TPP1 and POT1 (18). DNA replication forks are prone to arrest and/or collapse at telomeres, leading to telomere instability, since telomeric higher-order structures and repetitive DNA sequences can interfere with fork progression (6,19–22). In particular, guanine quadruplex (G4 DNA), DNA topological stress, and protective T-loop structures have been shown to lead to telomere instability if left unresolved during S phase (23–27). To facilitate telomere replication, the shelterin complex recruits additional factors to remove such obstacles during DNA replication. For example, TRF2 recruits Apollo, a nuclease that relieves topological stress (28–30); RTEL1 helicase, which dismantles the G4 DNA and the T-loop structure (25,27,31); and SLX4, a multitasking protein involved in the maintenance of telomere stability and the replication stress response (32,33). Overall, a complicated protein network is required to achieve efficient duplication of telomeric DNA tracts.
TRF2 is suggested to play a role in ORC and MCM loading at telomeres. TRF2 directly binds to ORC through the ORC1 subunit (34–36) and RNA interference (RNAi)-mediated TRF2 silencing decreases loading of ORC and MCM onto telomeric DNA (36,37), suggesting that replication origins are assembled at telomeres through the TRF2–ORC interaction. Indeed, DNA combing experiments have demonstrated replication initiation events occurring inside the telomeric tract (38–40). These initiation events may play an important role in telomere maintenance as the persistent arrest of replication forks within a telomere would otherwise result in under replication due to the absence of a converging fork (41). Considering the inherent difficulties associated with telomere replication, these telomeric replication origins may contribute to the complete duplication of telomeric tracts (41).
The biological role of the TRF2–ORC interaction is not fully understood, in part because siRNA-mediated depletion of TRF2 or essential ORC subunits inevitably affects other fundamental functions of these factors; for example, TRF2 knockdown affects telomere protection, while ORC1 knockdown compromises genome-wide DNA replication licensing. In this study, we evaluated the biological relevance of the TRF2–ORC interaction in HeLa cells by two different means: firstly, by using a TRF2 mutant defective in ORC binding, we show that the TRF2–ORC interaction promotes the recruitment of ORC and MCM at telomeres, and may prevent telomere DNA damage and telomere instability under DNA replication stress conditions; secondly, we demonstrate that overexpression of an ORC1 fragment (amino acids 244–511), which binds to TRF2, competitively inhibits ORC recruitment at telomeres and induces the replication stress-associated telomere DNA damage in cells. These results suggest that ORC recruitment by TRF2 underlies formation of telomeric replication origins and telomere stability.
MATERIALS AND METHODS
Cell culture
U2OS, U2OS 2–6-3 (35,42), HEK293T, HeLa, TRF2-edited HeLa clones, and HCT116 cells were maintained in Dulbecco's modified Eagle's medium (Wako) supplemented with 8% fetal calf serum and antibiotics (0.1 mg/ml kanamycin).
Plasmids
pSV40-HA-LacI, pSV40-TRF2-LacI, pSV40-TRF2 (45–244)-LacI, pSV40-TRF2ΔMyb-LacI, pGEX6P-1-TRF2 (45–244), pcDNA3.1-zeo-ORC1-3 × FLAG, pCLMSCV-HA-TRF2, and pCLMSCVhyg-T7-Cdt1 were described previously (35,36,43,44).
pcDNA3.1-zeo-ORC1 (L229A)-3 × FLAG, pcDNA3.1-zeo-ORC1 (D620A)-3 × FLAG, pSV40-TRF2 (45–244/Y73A/G74A)-LacI, pSV40-TRF2 (45–244/V88A/P90A)-LacI, pSV40-TRF2 (45–244/K93A/E94A/H95A/T96A)-LacI, pSV40-TRF2 (45–244/S98A/R102A)-LacI, pSV40-TRF2 (45–244/E111A/E112A)-LacI, pSV40-TRF2 (45–244/S119A/M122A)-LacI, pENTR4-HA-TRF2 (Y73A/G74A), pENTR4-HA-TRF2 (V88A/P90A), pENTR4-HA-TRF2 (K93A/E94A/H95A/T96A), pENTR4-HA-TRF2 (S98A/R102A), pENTR4-HA-TRF2 (E111A/E112A), and pENTR4-HA-TRF2 (S119A/M122A) were produced by oligonucleotide-directed mutagenesis (Quick Change Site-directed Mutagenesis Kit; Stratagene) with the following mutagenic oligonucleotides and their complement.
| cDNA | Mutagenic oligonucleotide |
| ORC1 (L229A) | 5′-CCTACCCATCCTGCTACCCCGCGCGCCAGAAAGAGGC-3′ |
| ORC1 (D620A) | 5′-CGTCCTGCTTGTGGCTGAGCTCGA CC-3′ |
| TRF2 (Y73A/G74A) | 5′-GCGCGCATTTCGCGGATCCCGCGCTGCTGACTTCCGC-3′ |
| TRF2 (V88A/P90A) | 5′-CTCTGCTGGCGCGCGCACTGGGCAAAG-3′ |
| TRF2 (K93A/E94A/ | 5′-GCCCACTGGGCGCTGCAGCTGCCGTGTCCCGCCTGC-3′ |
| H95A/T96A) | |
| TRF2 (S98A/R102A) | 5′-CATACCGTGGCGCGCCTGCTGGCCGTGATGCAG-3′ |
| TRF2 (E111A/E112A) | 5′-GCCGCATTGCAGCCGGCGAAAACCTGGAC-3′ |
| TRF2 (S119A/M122A) | 5′-CCTGGACTGTGCCTTTGATGCGGAAGCTGAGCTGACCCCACTGG-3′ |
A series of pGEX-6P-1 encoding alanine-substitution mutants of TRF2 (45–244) were prepared using the In-Fusion (Clontech) reaction. The cDNAs were amplified by PCR with the primers listed below with a series of pSV40-TRF2 (45–244)-LacI vectors as template DNA. EcoRI- and SalI-digested pGEX-6P-1-TRF2 (45–244) was used as the backbone vector.
| PCR primer | |
| pGEX-6P series | 5′-TCCCCGGAATTCGAGGCCCGCCTGGA AG-3′ |
| 5′-GCTCGAGTCGACCAGAGCCTTTTTG GC-3′ |
pAAVS1-CMV-HA-TRF2 and variants encoding alanine-substitution mutants of TRF2 (used in Figure 2C–E) were prepared using the In-Fusion reaction. The cDNAs were amplified by PCR with the primers listed below with a series of pENTR-HA-TRF2 vectors as template DNA. BglII- and SalI-digested pAAVS1-CMV (provided by Dr. Kanemaki, National Institute of Genetics, Mishima; Addgene # 105924) (45) was used as the backbone vector.
Figure 2.

Effects of the TRF2 mutations on interaction of TRF2 with other TRF2-binding partners. (A) U2OS cells were co-transfected with the indicated expression vectors. At 42 h post-transfection, cells were harvested and subjected to immunoprecipitation with anti-LacI antibody. Immunoprecipitates (IPs) and 3% of the input were separated by SDS-PAGE, followed by immunoblotting with the indicated antibodies. Asterisks indicate the position of full-length LacI-fused proteins. (B) U2OS 2–6-3 cells were co-transfected with the indicated expression vectors for 24 h. Co-localization frequency of FLAG-Apollo and LacI-fused proteins was examined. The values represent the sum scores from at least two biologically independent experiments. a Total number of LacI foci. b Number of LacI foci co-localizing with FLAG-Apollo foci. c Co-localization frequency of FLAG-Apollo foci with LacI foci. d Results of the χ2 test versus HA-LacI. ***P < 0.001. (C) At 42 h post-transfection with the indicated vectors, HCT116 cells were harvested and immunoprecipitated with an anti-RAP1 antibody or normal rabbit IgG (Control IP). IPs and 3% of input were analyzed by immunoblotting with the indicated antibodies. (D) HEK293T cells were transfected with the indicated expression vectors for 42 h, cross-linked with formaldehyde and then solubilized. Soluble fractions were immunoprecipitated with an anti-RTEL1 antibody or normal mouse IgG (Control IP). IPs and 2% of input for RTEL1 or 0.1% of input for HA were analyzed by immunoblotting with the indicated antibodies. (E) At 42 h post-transfection with the indicated vectors, HEK293T cells were harvested and immunoprecipitated with an anti-HA antibody. IPs and 0.5% of input were analyzed by immunoblotting with the indicated antibodies. The signal intensities of the bands were quantitated, and relative SLX4 co-immunoprecipitation (co-IP) values were calculated by normalizing the co-precipitated SLX4 levels to those of the precipitated HA-TRF2. The mean and individual data points from two independent experiments are shown with values with the HA-TRF2 WT set as 1.
| PCR primer | |
| pAAVS1 series | 5′-GGACTCAGATCTGCCACCATGTACC CC-3′ |
| 5′-GGTACCGTCGACTTAGTTCATACCCAGG CG-3′ |
pFLAG-CMV-6a-Apollo was generated as follows: the cDNA was amplified by PCR from the template vector pCMV-SPORT6-Apollo (purchased from Dharmacon, clone# 5001181) using the primers listed below. Amplified cDNA and pFLAG-CMV-6a (SIGMA-ALDRICH) were digested by HindIII and PstI, and ligated using the Takara Ligation Kit (Takara, 6023).
| cDNA | PCR primer |
| Apollo | 5′-CTCAAGCTTGACTCCAACCCTACC-3′ |
| 5′-ATCCTGCAGGTTGTCATTCTGTACTCTCC-3′ |
pFLAG-CMV-6a-SLX4 was generated as follows: the cDNA was amplified from pcDNA-FRT/TO GFP-BTBD12 (provided by Dr. Dario Alessi, University of Dundee, Scotland; clone number DU19216) (46) using the primers listed below. Amplified cDNA and HindIII-digested pFLAG-CMV-6a were subjected to the In-Fusion reaction.
| cDNA | PCR primer |
| SLX4 | 5′-CGACAAGCTCAAGCTTAAACTCAGTGTGAATGAGGCTC-3′ |
| 5′-TCGCGGCCGCAAGCTTCAGTTCCGCTCCACCT-3′ |
pFLAG-CMV-6a-ORC1 was prepared as follows: pBluescript-ORC1 (47) was partially digested by NcoI, and a 5.5 kb fragment was purified by agarose gel electrophoresis and gel extraction. After blunting using the Klenow fragment (Takara, 2140A), the fragment was further digested with NotI and ScaI, and a 2.4 kb ORC1 fragment was purified by agarose gel electrophoresis and gel extraction. pFLAG-CMV-6a was digested with HindIII followed by blunting with the Klenow fragment, and further digested with NotI. After de-phosphorylation of the linear vector using Shrimp Alkaline Phosphatase (Takara, 2660A), the cDNA and the linear vector were mixed and ligated.
pX459-terf2-exon2-1 was generated by inserting a synthesized oligonucleotide into the pX459 vector (a gift from Feng Zhang, Broad Institute, Cambridge, MA; purchased from Addgene, # 62988) (48). To prepare the insert, the oligonucleotides listed below were mixed at final concentration of 4.5 μM each in 1 × annealing buffer [20 mM Tris–HCl pH 7.5, 5 mM MgCl2, 30 mM NaCl and 1 mM dithiothreitol (DTT)]. Mixed oligonucleotides were annealed by incubation at 100ºC for 5 min followed by cooling at room temperature, and then phosphorylated by T4 polynucleotide kinase (Takara, 2021S). pX459 was digested with BbsI. Annealed oligonucleotides and linear vectors were mixed and ligated.
| oligonucleotides for insert DNA |
| 5′-CACCGGGGTTATGCAGTGTCTGTCG-3′ |
| 5′-AAACCGACAGACACTGCATAACCCC-3′ |
pFLAG-CMV-6a-ORC1 (2–511), pFLAG-CMV-6a-ORC1 (2–325), pFLAG-CMV-6a-ORC1 (2–244) and pFLAG-CMV-6a-ORC1 (2–85) were prepared by self-ligation of pFLAG-CMV-6a-ORC1 digested with the following restriction enzymes.
| Restriction enzymes | ||
| ORC1 (2–511) | EcoRI | |
| ORC1 (2–325) | BspT104I * | EcoRV |
| ORC1 (2–244) | BstPI * | EcoRV |
| ORC1 (2–85) | SalI | XhoI (partial digestion) |
*, after digestion, ends were blunted using the Klenow fragment (Takara, 2140A).
pFLAG-CMV-6b-ORC1 (244–511) and pFLAG-CMV-6b-ORC1 (325–511) were prepared as follows: pFLAG-CMV-6a-ORC1 was digested with BstPI [for ORC1 (244–511)] or BspT104I [for ORC1 (325–511)], blunted with the Klenow fragment, and further digested with EcoRI. pFLAG-CMV-6b (SIGMA-ALDRICH) was digested with HindIII, blunted with the Klenow fragment, and further digested with EcoRI. After de-phosphorylation of the linear vector using Shrimp Alkaline Phosphatase, the truncated ORC1 cDNA and the linear vector were mixed and ligated.
pFLAG-CMV-6a-ORC1 (Δ326–510), pFLAG-CMV-6a-ORC1(Δ386–510), pFLAG-CMV-6a-ORC1 (Δ411–510), pFLAG-CMV-6a-ORC1 (Δ446–510) and pFLAG-CMV-6a-ORC1 (Δ411–445) were prepared as follows: two fragments of ORC1 cDNA were amplified by PCR with the primers listed below using pcDNA3.1-zeo-ORC1-3 × FLAG as a template. Amplified cDNA fragments and HindIII-digested pFLAG-CMV-6a were subjected to the In-Fusion or NEBuilder reaction (New England BioLabs, E2621).
| cDNA | Insert | PCR primer |
| ORC1 (Δ326–510) | 1: ORC1 (1-325) | 5′-CGATGACGACAAGCTCATGGCACACTACCCC-3′ |
| 5′-CAAAATTGTAGATGTCTTGGAACGAAGCTGCAATTCG-3′ | ||
| 2: ORC1 (511–861) | 5′-TTCCAAGACATCTACAATTTTG-3′ | |
| 5′-GAATTCGCGGCCGCATTACTCGTCTTTCAGCG-3′ | ||
| ORC1 (Δ386–510) | 1: ORC1 (2–385) | 5′-GATGACGACAAGCTCAAGCTTGCACACTACCCCACAA-3′ |
| 5′-CAAAATTGTAGATGTCTT GGAAGACAGAACTCTTTC TGC-3′ |
||
| 2: ORC1 (511–861) | 5′-TTCCAAGACATCTACAATTTTG-3′ | |
| 5′-TGAATTCGCGGCCGCAAGCTTTACTCGTCTTTCAGCGC-3′ | ||
| ORC1 (Δ411–510) | 1: ORC1 (2–410) | 5′-GATGACGACAAGCTCAAGCTTGCACACTACCCCACAA-3′ |
| 5′-CAAAATTGTAGATGTCTTGGAAAATCTCTTTCTCTTCTTGGTC-3′ | ||
| 2: ORC1 (511–861) | 5′-TTCCAAGACATCTACAATTTTG-3′ | |
| 5′-TGAATTCGCGGCCGCAAGCTTTACTCGTCTTTCAGCGC-3′ | ||
| ORC1 (Δ446–510) | 1: ORC1 (1–445) | 5′-CGATGACGACAAGCTCATGGCACACTACCCC-3′ |
| 5′-CAAAATTGTAGATGTCTTG GAAAGATCGCAGGTTCCTG-3′ |
||
| 2: ORC1 (511–861) | 5′-TTCCAAGACATCTACAATTTTG-3′ | |
| 5′-GAATTCGCGGCCGCATTACTCGTCTTTCAGCG-3′ | ||
| ORC1 (Δ411–445) | 1: ORC1 (2–410) | 5′-GATGACGACAAGCTCAAGCTTGCACACTACCCCACAA-3′ |
| 5′-TGTAAGGATGACTTCAAG GAAATCTCTTTCTCTTCTTGG TC-3′ |
||
| 2: ORC1 (446–861) | 5′-TCCTTGAAGTCATCCTTACATAC-3′ | |
| 5′-TGAATTCGCGGCCGCAAGCTTTACTCGTCTTTCAGCGC-3′ |
The retroviral expression vector pQCXIP-HA-ORC1(244–511) was prepared by In-Fusion reaction. The cDNAs were amplified by PCR with the primers for N-terminal HA-tagging listed below using pcDNA3.1-zeo-ORC1-3 × FLAG as template DNA. NotI-digested pQCXIP (Clontech) was used as the backbone vector.
| cDNA | PCR primer |
| HA-ORC1 (244–511) | 5′-AATTGATCCGCGGCCGCCACCATGTACCCCTACGATGTGCCCGATT |
| ACGCCAACCCTCAGATGTCCCAGC-3′ | |
| 5′-CCTACCGGTGCGGCCTTAGAATTCCTGTTCCCGACAGG-3′ |
As described previously (35,36), our TRF2 cDNA was chemically synthesized with codon optimization and thus some oligonucleotide sequences described here are not comparable to those of cell-derived TRF2 cDNA. The length of our TRF2 oligopeptide is 500 amino acids, which is 42 amino acids shorter at the N-terminus (542 amino acids in total) than that used in studies of the RTEL1-binding regions of TRF2 (25,31). Therefore, the apparent position of residues differ between these studies.
Transfection
For immunofluorescence analysis, expression plasmids (total: 0.56 μg for Figures 1B, 1C, 2B, 5B, 5D; 0.84 μg for Figure 6B) were transiently transfected into 8 × 104 U2OS 2–6–3 cells in 4-well chamber slides using Lipofectamine 2000 reagent (Invitrogen, 11668019) according to the manufacturer's instructions.
Figure 1.

Generation and characterization of a series of TRF2 mutants with alanine-substitutions in the TRFH domain. (A) Crystal structure of the TRF2 TRFH domain (PDB code: 4M7C). Amino acid residues mutated in this study are highlighted and individual TRF2 monomers are depicted with cyan and pink ribbons. The 14 selected residues are marked with blue or red. Top, front view. Bottom, top view. (B) U2OS 2–6-3 cells were transfected with HA-LacI, TRF2 (45–255)-LacI, or alanine-substitution mutant TRF2 (45–244)-LacI (Y73A/G74A, V88A/P90A, K93A/E94A/H95A/T96A, S98A/R102A, E111A/E112A and S119A/M122A; referred to as YG, VP, KEHT, SR, EE and SM, respectively) for 24 h, and then co-immunostained with anti-LacI antibody and anti-ORC1 antibody, followed by DAPI counterstaining. Representative images are shown in the Supplementary Figure S1. Co-localization frequencies of ORC1 with LacI proteins were examined. The values represent the sum scores from two biologically independent experiments. a Total number of LacI foci. b Number of LacI foci co-localizing with ORC1 foci. c Co-localization frequency of ORC1 foci with LacI foci. d Results of the χ2 test versus TRF2 (45–244)-LacI. **P < 0.01; ***P < 0.001; n.s., not significant. (C) U2OS 2–6–3 cells were transfected with the expression vectors as indicated for 24 h, co-immunostained for LacI and ORC2 or ORC3, and analyzed as described in (B). (D) Recombinant GST or a series of GST-TRF2 (45–244) mutant proteins were incubated with HeLa cell extracts, and precipitated with glutathione beads. Precipitates and 2.5% of input were separated by SDS-PAGE, followed by CBB staining or immunoblotting with anti-ORC1 antibody. The signal intensities of ORC1 bands were quantified, and shown with the signal intensities with GST-TRF2 (45–244) WT set as 1. The means and individual data points from two independent experiments are shown. Red asterisks indicate the position of GST-fusion proteins. WT, wild-type. (E) HEK293T cells were co-transfected with FLAG-ORC1 and empty vector or HA-tagged TRF2 proteins, as indicated, for 42 h. After cross-linking with formaldehyde and solubilization, cell lysates were immunoprecipitated with anti-HA antibody. Immunoprecipitates (IPs) and 1% of the input were immunoblotted with the indicated antibodies. The signal intensities of the bands were quantitated, and relative ORC1 co-immunoprecipitation (co-IP) values were calculated by normalizing the co-precipitated ORC1 levels to those of the precipitated HA-TRF2. The mean and individual data points from two independent experiments are shown with values with HA-TRF2 WT set as 1.
Figure 5.
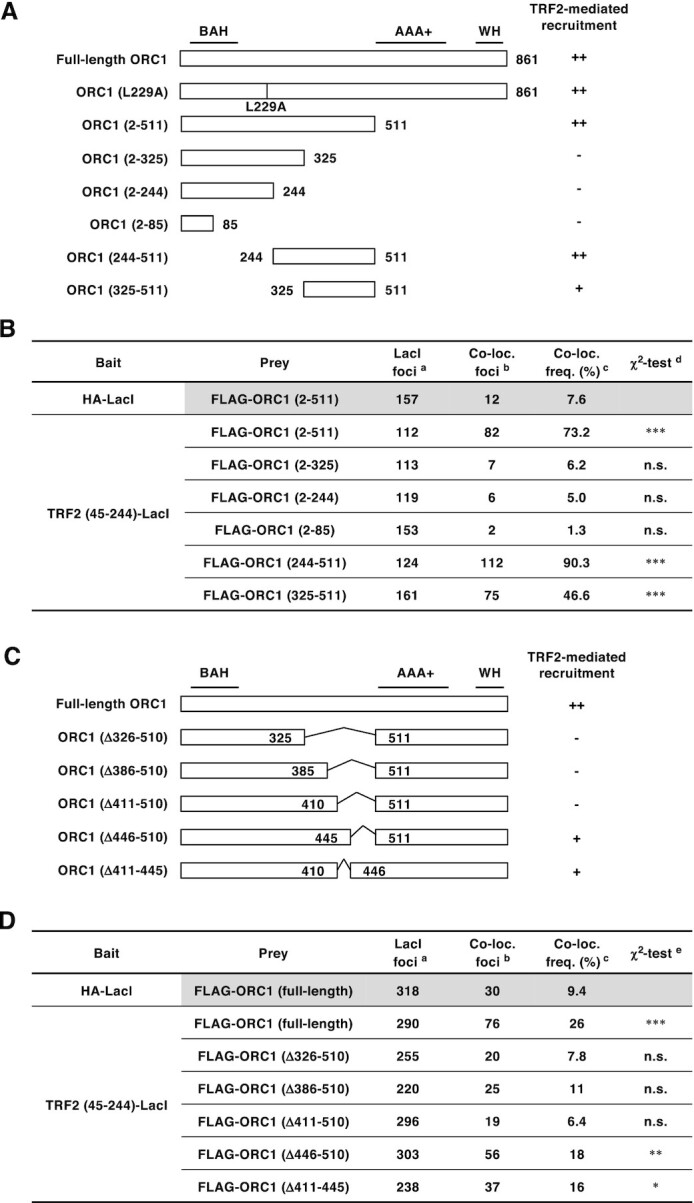
ORC1 amino acids 411–510 are required for efficient ORC1 recruitment by TRF2. (A, C) Schematics of ORC1 mutants used in this study. Domain information is shown at the top. A summary of their ability to bind TRF2 is shown on the right (see Figure 5B, D and Supplementary Figure S4 for detail). BAH, bromo adjacent homology domain; AAA+, ATPase associated with diverse cellular activities domain; WH, winged helix domain. (B, D) Summary of the co-localization frequencies of ORC1 mutants with TRF2 (45–244)-LacI. U2OS 2–6-3 cells were co-transfected with the indicated ORC1 and LacI expression vectors. Co-localization frequency was examined as in Figure 1B. The values represent the sum score from at least two biologically independent experiments. a Total number of LacI foci. b Number of LacI foci co-localizing with FLAG foci. c Co-localization frequency of FLAG foci with LacI foci. d Results of the χ2 test vs. co-localization of HA-LacI and FLAG-ORC1 (2–511). e Results of the χ2 test vs. co-localization of HA-LacI and FLAG-ORC1 (full-length). *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant.
Figure 6.

Overexpression of ORC1 (244–511) fragment competitively inhibits TRF2–ORC1 binding, but has little effect on the genome-wide replication activity of ORC1. (A) HEK293T cells were co-transfected with the indicated expression vectors (equal amounts of each plasmid) for 42 h. After cross-linking with formaldehyde and solubilization, cell lysates were immunoprecipitated with anti-LacI antibody or normal mouse IgG (Control). IPs and 1% of the input were immunoblotted with the anti-FLAG or anti-LacI antibody. The data are representatives of two independent experiments. (B) U2OS 2–6–3 cells were co-transfected with the indicated expression vectors (equal amounts of each plasmid) for 24 h, then double-immunostained with the indicated antibodies, followed by DAPI staining. Co-localization frequencies of FLAG-ORC1 foci with LacI foci were examined. The values represent the sum score of two biologically independent experiments. a Total number of LacI foci. b Number of LacI foci co-localizing with FLAG-ORC1 foci. c Co-localization frequency of FLAG-ORC1 foci with LacI foci. d Results of the χ2 test versus co-localization of FLAG-ORC1 and TRF2 (45–244)-LacI in the absence of HA-ORC1 (244–511). ***P < 0.001. (C–E) HEK293T cells were co-transfected with mixture of the expression vectors (equal amounts of each plasmid) or their empty vectors (-) as indicated for 48 h. (C) Whole-cell lysates were subjected to immunoblotting with the indicated antibodies. CBB serves as a loading control. (D) DNA content was analyzed by flow cytometry. After excluding the sub-G1 fraction, the percentages of re-replicated cells (DNA content greater than 4N) were calculated (shown on the right). (E) The relative abundance of re-replicated cells as a proportion of control cells. The means and SDs from three biologically independent experiments were shown. ***P < 0.001; n.s., not significant (Tukey–Kramer test).
For immunoprecipitation, ChIP and re-replication analysis, transfection was performed with PEImax reagent (Polysciences, 24765), as previously described (49). Detailed transfection conditions used in the experiments are listed below. For ChIP analysis (Figure 3E, F and Figure 7A, B), the medium was changed 6 h after transfection.
Figure 3.

ORC recruitment to telomeres is impaired in HeLa TRF2 EE clones. (A) The sequences of single stranded oligo DNA (ssODN) used to establish TRF2-edited HeLa clones. Top, reference sequence of the target site. Middle, ssODN used for the TRF2 WT clone. Bottom, ssODN used for TRF2 EE clones. Introduced mutations are shown in red. The PAM sequence required for Cas9 recognition is underlined. Codons corresponding to the E111/E112 residues are marked with a dotted underline. The newly introduced NruI site is marked with a dashed underline. (B) Restriction fragment length polymorphism analysis of the HeLa clones. The target site was amplified from genomic DNA derived from each cell line by PCR. Amplicons were incubated with or without NruI restriction enzyme and then separated by agarose gel electrophoresis, followed by EtBr staining. When the target genes were edited, NruI treatment produced 427 bp and 209 bp fragments (frag. 1 and frag. 2, respectively) from the 636 bp PCR product (*). (C) Summary of the sequencing of the target sites in the TERF2 gene of each cell line. For detail, see Material and Methods. Eleven colonies from each clone were analyzed. (D) Whole cell extracts were separated by SDS-PAGE, and then analyzed by immunoblotting with anti-TRF2 antibody or by CBB staining (as a loading control). The signal intensities of TRF2 and CBB bands were quantified, and the TRF2/CBB signal ratio is shown relative to that of parental HeLa cells. The means ± SDs are shown (n = 3). (E, F) At 48 h post-transfection with empty vector or ORC1-3 × FLAG expression vector, cells were subjected to ChIP with an anti-FLAG antibody. (E) Purified DNA from input and immunoprecipitates was analyzed by qPCR with primer pairs amplifying either a telomeric sequence or the LMNB2 replication origin. Relative specific ChIP values were calculated as follows: (Telomeric DNA as a % of input from ORC1-3 × FLAG ChIP − Telomeric DNA as a % of input from empty vector ChIP) / (LMNB2 origin DNA as a % of input from ORC1-3 × FLAG ChIP − LMNB2 origin DNA as a % of input from empty vector ChIP). The means ± SDs are shown (n = 9). * P < 0.05; ***, P < 0.001 (two-tailed Student's t-test). (F) Lysates were separated by SDS-PAGE and then analyzed by immunoblotting with anti-FLAG antibody and by CBB staining (as a loading control). (G) Cells were harvested and stained with propidium iodide to allow cell cycle analysis by flow cytometry. The means ± SDs are shown (n = 3). (H, I) Cells were subjected to ChIP assay with control IgG or anti-MCM7 antibody. (H) Purified DNAs from input and immunoprecipitates were analyzed by qPCR with a primer pair amplifying a telomeric sequence. Results are shown as the percent of input DNA. The means ± SDs are shown (n = 4). *P < 0.05; n.s., not significant (two-tailed Student's t-test). (I) Left, the total amounts of co-precipitated DNA and input DNA were analyzed using SYBR Gold staining and UV photography. Right, results are shown as the percent of input DNA. The means ± SDs are shown (n = 4). n.s., not significant (two-tailed Student's t-test).
Figure 7.

Overexpression of an ORC1 (244–511) fragment inhibits ORC- and MCM-binding to telomeres and exacerbates the accumulation of 53BP1 at telomeres upon replication stress. (A, B) Control and HA-ORC1 (244–511)-overexpressing HeLa cells were transfected with empty vector or ORC1-3 × FLAG expression vector. At 48 h post-transfection, cells were subjected to ChIP with anti-FLAG antibody. (A) Cell lysates for ChIP assay were subjected to immunoblotting with the indicated antibodies. CBB serves as a loading control. (B) Purified DNAs from input and IPs were analyzed by qPCR with a primer pair amplifying a telomeric sequence or the LMNB2 replication origin. Relative specific ChIP values were calculated as described in Figure 3E. The means ± SDs are shown (n = 6). **P < 0.01 (two-tailed Student's t-test). (C, D) Cells were subjected to the ChIP assay with control IgG or anti-MCM7 antibody. (C) Purified DNAs from input and IPs were analyzed by qPCR with a primer pair amplifying a telomeric sequence. Results are shown as the percent of input DNA. The means ± SDs are shown (n = 6). ***P < 0.001 (two-tailed Student's t-test). (D) Left, the total amounts of co-precipitated DNA and input DNA were analyzed using SYBR Gold staining and UV photography. Right, the means ± SDs are shown (n = 2). (E) Cells were treated with 0.1 mM hydroxyurea (HU) for 16 h. Telomere foci co-localizing with 53BP1 were analyzed as in Figure 4A and B. The frequency of cells carrying ≥5 TIFs was scored. The means ± SDs are shown (n = 3). One hundred cells were analyzed for each sample. The sum scores are used for statistical analyses. *P < 0.05 (Fisher's exact test).
| Experiment | Cells | Total DNA | Cellular density |
| Figure 1E | HEK293T | 12 μg | 4 × 106/10 cm dish |
| Figure 2A | U2OS | 2 μg | 5 × 105/6 cm dish |
| Figure 2C | HCT116 | 4 μg | 1 × 106/6 cm dish |
| Figure 2D | HEK293T | 8 μg | 6 × 106/10 cm dish |
| Figure 2E | HEK293T | 3 μg | 3 × 106/6 cm dish |
| Figure 3E and F | TRF2-edited HeLa clones | 19 μg | 3.8 × 106/15 cm dish |
| Figure 6A | HEK293T | 18 μg | 6 × 106/10 cm dish |
| Figure 6C-E | HEK293T | 4 μg | 2 × 106/6 cm dish |
| Figure 7A and B | HeLa expressing ORC1 (244–511) | 7.5 μg | 1.5 × 106/10 cm dish |
Immunofluorescence staining
For Figure 1B and C, co-staining of endogenous ORC1, ORC2 and ORC3 with LacI were performed as described previously (35). Briefly, cells were washed with PBS (Wako, 048-29805), fixed with chilled 100% methanol for 10 min, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and then immunostained.
For the staining of FLAG-Apollo, and FLAG-ORC1 truncation mutants (Figures 2B and 5B), cells were fixed with 3.7% formaldehyde (Nacalai Tesque, 16223–55) in PBS. For the staining of full-length FLAG-ORC1 and FLAG-ORC deletion mutants (Figures 5D and 6B), cells were fixed with chilled 100% methanol for 10 min, and permeabilized with 0.1% Triton X-100 in PBS for 10 min. The staining of 53BP1 was performed after peptide nucleic acid (PNA)-fluorescence in situ hybridization (FISH) (Figures 4A, B and 7E). Cells were then incubated with primary antibodies in PBS supplemented with 1% fetal calf serum for 1 h at room temperature for 53BP1 staining or 2 h at 37ºC for LacI co-staining, followed by incubation with secondary antibody for 1 h at room temperature and 0.2 μg/ml 4,6-diamidino-2-phenylindole (DAPI) in PBS for 15 min at room temperature. Cells were washed three times after each staining with PBS for 5 min at room temperature, and finally mounted in Fluoro-KEEPER Antifade Reagent (Nacalai Tesque, 12593-64) and stored at 4ºC.
Figure 4.

Telomere instability in HeLa TRF2 EE clones upon DNA replication stress. (A, B) Cells treated with 0.1 mM hydroxyurea (HU) for 16 h were stained with PNA FISH using a telomere probe (TelC, red), followed by immunostaining with anti-53BP1 antibody (green) and DAPI counterstaining (blue). (A) Representative images; telomere foci co-localizing with 53BP1 (53BP1 TIFs) are indicated by yellow arrowheads. Scale bar, 10 μm. (B) The frequencies of cells carrying ≥ 10 53BP1 TIFs were quantified. The means ± SDs are shown (n = 2). One hundred cells were analyzed for each sample. The sum scores are used for statistical analyses. n.s., not significant; *P < 0.05; **P < 0.01 (Fisher's exact test). (C, D) Cells treated with 0.1 mM HU for 72 h were stained with PNA-FISH using a telomere probe (red), followed by DAPI counterstaining (cyan). (C) Representative images; telomere-containing micronuclei are indicated by yellow arrowheads. Scale bar, 10 μm. (D) The number of nuclei and telomere-containing micronuclei were counted, and the ratio of telomere-containing micronuclei/nucleus was calculated. The means ± SDs are shown (n = 3). At least 82 cells were analyzed for each sample. *P < 0.05 (two-tailed Student's t-test).
Microscopic analysis was performed with the KEYENCE BZ-9000 or BZ-X700 fluorescence microscope. Microscopic images were acquired by single image captures (5–20 fields for each sample) using the BZ-X viewer software (KEYENCE). For calculation of co-localization frequencies, at least 31 nuclei with a single prominent LacI focus were scored for each experiment/condition. Co-localization frequency of foci of each protein with the LacI foci was then manually scored. A summary of the co-localization frequency calculated from multiple biologically independent experiments is shown in the figures. The values are sum scores from at least two independent experiments performed for each condition.
Immunoprecipitation
For Figure 2A, C and E, cells were lysed in 500 mM NaCl NET gel buffer (500 mM NaCl, 0.1% Triton X-100, 50 mM Tris–HCl pH 7.4, 1 mM EDTA and 1 mM dithiothreitol). The lysates were diluted with 0 M NaCl NET gel buffer to a final NaCl concentration of 150 mM and then subjected to immunoprecipitation with 2 μg antibody. Bound proteins were eluted in 1× sample buffer (62.5 mM Tris–HCl pH 6.8, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, 0.01% bromo- phenol blue).
For Figures 1E, 2D and 6A, transfected HEK293T cells were cross-linked with 1% formaldehyde and then subjected to immunoprecipitation as described previously (35) using 2 μg (Figure 2D) or 4 μg (Figures 1E and 6A) of antibody.
GST pull-down assay
GST-TRF2 (45–244) and its variants encoding the alanine-substitution mutants of TRF2 were bacterially expressed and purified as described previously (35).
HeLa cells were lysed in 500 mM NaCl NET gel buffer supplemented with protease inhibitors and phosphatase inhibitors. The lysates were diluted with 0 M NaCl NET gel buffer to a final NaCl concentration of 150 mM. GST-fused proteins were bound to glutathione Sepharose beads, and the beads were incubated with the lysates. After washing four times with 150 mM NaCl NET gel buffer, the bound proteins were eluted and subjected to SDS-PAGE followed by immunoblotting.
Immunoblotting and antibodies
Immunoblotting was performed as described previously (43). Coomassie brilliant blue (CBB) staining was performed with the Rapid Stain CBB Kit (Nacalai Tesque, 30035-14). Quantification was performed with the LumiVision Analyzer (for immunoblots, AISIN Seiki) or ImageJ software (for CBB, NIH).
Preparation of polyclonal rabbit antibodies against human ORC1, ORC2, Cdt1 and MCM7 was described previously (36). Rabbit anti-LacI antibody was obtained by immunizing rabbits with a bacterially produced His-T7-LacI protein.
Other antibodies used in this study are as follows: LacI (clone 9A5, Merck, 05-503), ORC1 (Santa Cruz, ac-23887), ORC3 (Clone 1D6, Santa Cruz), HA-tag (clone 3F10, Roche), HA-tag (COVANCE MMS-101R), FLAG-tag (clone M2, SIGMA-ALDRICH), FLAG-tag (Thermo, PA1-984B), RAP1 (Bethyl, A300-306A), RTEL1 (Novus, NBP2-22360), SLX4 (Novus, NBP1-28680), TRF2 (Merck, 4A794), 53BP1 (Novus, NB100-904), rabbit normal IgG (DAKO, X0903), mouse normal IgG (Southern Biotech, 0107-01), rat normal IgG (MBL, M080-3), horseradish-peroxidase (HRP)-conjugated anti-mouse IgG (H + L) (Invitrogen, 61-6520 or ROCKLAND, 18-8817-33), HRP-conjugated anti-rabbit IgG (H + L) (Invitrogen, 65-6120 or ROCKLAND,18–8816-33), HRP-conjugated anti-rat IgG (H + L) (Zymed, 62–9520), CF488A-conjugated goat anti-rabbit IgG (Biotium, 20019), CF594-conjugated donkey anti-rabbit IgG (Biotium 20152), CF594-conjugated goat anti-mouse IgG (Biotium, 20111), Alexa488-conjugated goat anti-mouse IgG (Invitrogen, A11029).
Cas9-based gene-editing
Cas9-based gene-editing was performed by transient transduction of Cas9/guide RNA and single stranded oligonucleotide DNA (ssODN) (48). HeLa cells cultured in 6 well plates (1 × 105/well) were co-transfected with 2 μg pX459-terf2-exon2-1 and 2 μg ssODN using Lipofectamine 2000 reagent. ssODN carries the following sequence: ssODN WT, 5′- AGAGAAGAACACAAAAATAGCCATACCTAAATTTTCCCCTTCTTCAATTCGCGACAGACACTGCATAACCCGCAGCAATCGGGACACGGT-3′; ssODN EE, 5′-AGAGAAGAACACAAAAATAGCCATACCTAAATTTTCCCCTGCTGCAATTCGCGACAGACACTGCATAACCCGCAGCAATCGGGACACGGT-3′.
After puromycin (0.5 μg/ml) selection for 3 days, cells were subjected to limiting dilution. Aliquots of the obtained clones were re-seeded into 96-well plates, and clones were screened by RFLP analysis. Positive clones were re-subjected to further limiting dilution. After the second selection, confirmed positive clones by RFLP analysis were used for subsequent experiments.
RFLP analysis
For screening of HeLa clones, cells were seeded into 96-well plate and rinsed with PBS once after reaching confluence. Forty-five microliters of 50 mM NaOH were then added to each well, and the cells were lysed in a 90–95ºC water bath. Cell lysates were directly added to the PCR reaction mix (Toyobo, KOD-201).
For Figure 3B, genomic DNA was prepared as follows: cells were lysed with SDS and treated with RNase A (Invitrogen) and proteinase K (Roche, 3115887001). Genomic DNA was precipitated by isopropanol (Nacalai tesque, 29113–95). Purified genomic DNA was dissolved in TE (10 mM Tris–HCl pH 8.0 and 1 mM EDTA) and fragmented by incubation at 70ºC for 2–4 h.
The following primers were used to amplify the target site in TERF2 gene exon 2: 5′-GGACTTCAGACAGATCCGGG-3′ and 5′- CTCCTCAGATACGAGTGGCAAG-3′. Amplified DNA was incubated with or without NruI and separated by agarose gel electrophoresis.
Sequencing of the target locus of the TERF2 gene
The target sites in the TERF2 gene exon2 were amplified as described above. For TA cloning, the PCR products were 1000-fold diluted and re-amplified with Taq polymerase (Bioacademia, 02-001) and the same primer pair. The products were purified by agarose gel extraction, cloned into T-Vector pMD20 (Takara, 3270), and sequenced (Thermo, 4337454). Eleven colonies from each clone were analyzed.
Chromatin immunoprecipitation and quantitative real-time PCR (qPCR) analysis
Chromatin immunoprecipitation was performed as described previously (50). For qPCR analysis, SYBR Premix Ex Taq II (Takara, RR081A) or TB Green Premix Ex Taq II (Takara, RR820A) was used according to the manufacturer's instructions. PCR reactions were performed using the CFX96 Touch Real-Time PCR Detection System (BIO-RAD). For detection of the telomere sequences and LMNB2 replication origin region, the primer sequences and qPCR cycling parameters were as described previously (35,51,52). We have done the q-PCR experiments according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (53). MIQE checklist was shown in Supplementary Table S1. The total amount of co-precipitated DNA was quantified using SYBR Gold staining (Invitrogen, S11494) (49). SYBR Gold signals were captured using cooled-CCD camera detection systems (LumiVision Imager, AISIN Seiki) within the linear range, and band intensities were quantitated using LumiVision Analyzer.
Fluorescence-activated cell sorting (FACS)
Cells were trypsinized and suspended in PBS supplemented with 0.1% Triton X-100 and 10 μg/ml RNase A. After staining with propidium iodide (40 μg/ml), cell cycle distribution was examined using a FACS Verse flow cytometer (BD Bioscience) and ModiFit LT (Verity Software House). For Figure 6 D and E, re-replication was measured as described previously (54). Dots with higher FL2-W signals, which result from aggregated cells and cell debris, were excluded from measurements of re-replication.
PNA-FISH: fluorescence in situ hybridization with peptide nucleic acid probes
After rinsing with PBS, cells were fixed with 3.7% formaldehyde (for Figures 4A, B and 7E) or chilled 100% MeOH (for Figure 4C and D). For formaldehyde fixation, cells were permeabilized in PBS containing 0.1% TX-100. Cells were dehydrated by incubating in serially diluted EtOH (70%, 95%, and 100%). After drying cells completely, hybridizing solution [70% formamide, 9 mM Tris-HCl pH 7.5, 1% skimmed milk, and 200 nM Cy5-TelC (Panagene, F1003)] was added to each well. After placing coverslips onto slides, they were baked at 80ºC for 5 min, followed by incubation at room temperature overnight. After removing coverslips, cells were washed once with PBS, twice with washing solution (70% formamide and 9 mM Tris-HCl pH 7.5) at room temperature for 15 min, and three times with PBS. For co-staining with 53BP1, immunofluorescence staining was performed after PNA-FISH.
Establishment of HeLa cells stably expressing HA-ORC1 (244-511)
HeLa cells were infected with the recombinant retroviruses encoding HA-ORC1(244–511) or an empty vector, as described previously (54). Infected cells were selected with puromycin (0.5 μg/ml) and subjected to assays without cloning.
Data presentation and statistical analysis
Unless otherwise stated, quantitative data are represented as the mean ± SD of three or more independent experiments. The number of experiments was chosen according to the standards of the field. The statistical analyses used are defined in each figure legend. For Figures 1B, 1C, 2B, 4B, 5B, D, 6B and 7E and Supplementary Figure S4, the sum scores of two or more independent experiments were used for the statistical analyses. Individual values of each experiment are provided as Source data files. For the t-tests, data distribution was assumed to be normal but this was not formally tested. Tukey–Kramer multiple comparison test was performed after examination by one-way ANOVA. P-values <0.05 were considered statistically significant. For qualitative data and semi-quantitative data, a representative image from multiple independent experiments is shown; for all such figures, essentially the same results were obtained in the multiple independent experiments.
RESULTS
TRF2 E111/E112 residues are required for efficient recruitment of ORC but not for interactions with other binding partners
To evaluate the TRF2–ORC interaction we sought to identify a TRF2 mutant defective only in its interaction with ORC. We previously reported that the dimerization of TRF2 is required for the recruitment of ORC (35). Using the lacO-LacI protein tethering assay in U2OS 2–6-3 cells containing an array of 256 lacO repeats on chromosome 1 (42), we have also shown that the TRFH (TRF homology) domain of TRF2 (amino acids 45–244) fused to LacI is sufficient for ORC and MCM recruitment to this lacO array (35). TRF2 V52D and N53P mutations in the TRFH domain impairs TRF2 dimer formation and ORC recruitment by TRF2 (35). However, the TRF2 (V52D/N53P) mutant is not suitable as a specific ORC interaction mutant because TRF2 dimerization is critical for its other functions, including telomere binding. To identify a specific TRF2–ORC interaction mutant, residues predicted to be required for binding to ORC1 were identified based on the crystal structure of the TRF2 TRFH domain (PDB code 4M7C) and the previously published data described above (Figure 1A). TRF2 residues were selected according to the following three criteria: (i) Since TRF2 dimerization is required for ORC1 binding and recruitment, we speculated that ORC1 recognizes a specific structure of the TRFH dimer created around the interface of the two monomers; hence, the residues selected were within helix 2 to helix 3 of the TRFH located in proximity to the dimer interface; (ii) selected residues were exposed on the surface of the protein; (iii) selected residues were not conserved in TRF1, since TRF1 is unable to recruit ORC (35). Fourteen residues (Y73, G74, V88, P90, K93, E94, H95, T96, S98, R102, E111, E112, S119 and M122) met these criteria (Figure 1A), and six TRF2 mutants were generated by substituting the selected residues with alanine (Y73A/G74A, V88A/P90A, K93A/E94A/H95A/T96A, S98A/R102A, E111A/E112A and S119A/M122A; referred to as YG, VP, KEHT, SR, EE and SM, respectively).
The ability of the mutants to recruit ORC was examined using the lacO-LacI assay (Figure 1B, C, and Supplementary Figure S1). The results clearly showed that TRF2 (45–244) EE-LacI was defective in recruiting ORC1, ORC2, and ORC3 to the lacO array. We further confirmed that the EE mutations impair the TRF2–ORC1 interaction in GST pull-down assay using purified GST-TRF2 and HeLa cell lysates or in co-immunoprecipitation assay using HEK293T cells (Figure 1D and E). In addition, YG, VP, KEHT, and SM mutants showed partially decreased co-localization with ORC1 (Figure 1B), suggesting that these residues are also involved in the interaction with ORC. Immunoprecipitation assays demonstrated that all the prepared mutants of TRF2 (45–244)-LacI can bind to HA-TRF2 with similar affinity to that of wild-type TRF2 (45–244)-LacI (Figure 2A), indicating that these mutations do not affect dimerization. TRF2 functions as a hub protein that directly binds to telomeres and recruits various factors required for telomere maintenance. For example, a component of shelterin complex, RAP1, binds to telomeres concomitant with TRF2 (55,56). In addition, several factors such as Apollo, RTEL1 and SLX4 access telomeres by binding to the TRF2 TRFH domain, thereby facilitating telomeric DNA replication (6,20). Therefore, we next examined whether our TRF2 mutants were proficient in binding to these partners (Figure 2B–E). YG, VP, EE and SM mutants recruited FLAG-Apollo to the lacO array to a similar extent as wild-type TRF2 (Figure 2B). These mutants could also bind to RAP1 (Figure 2C) and RTEL1 (Figure 2D) in immunoprecipitation assays. On the other hand, the EE mutant showed decreased but detectable binding to SLX4 in immunoprecipitation (Figure 2E). Collectively, these results suggest that TRF2 EE is a TRF2 mutant primarily defective in ORC recruitment.
Recruitment of ORC and MCM to telomeres is inhibited by the TRF2 E111A/E112A mutation in HeLa cells
To evaluate the effects of substitution of endogenous wild-type TRF2 with the TRF2 EE mutant, we established HeLa clones carrying EE mutations in the TERF2 gene by gene-editing using CRISPR-Cas9 and ssODN encoding the EE mutations and a diagnostic NruI restriction site (Figure 3A-D). Gene-editing with an ssODN that introduced the NruI site alone was performed in parallel to establish a control clone (Figure 3A, ssODN WT). Restriction fragment length polymorphism (RFLP) analysis showed that TRF2 WT 7–1, TRF2 EE 4–3 and TRF2 EE 7–7 clones have at least one TERF2 allele carrying the mutated sequences with the newly introduced NruI site (Figure 3B). The target sites in the TERF2 gene exon 2 were amplified and sequenced, as described in Materials and Methods, and no wild-type sequences or in-frame insertions/deletions were detected (Figure 3C). In all TRF2-edited clones, TRF2 protein levels were decreased to 20–40% of that in the parental HeLa cell line (Figure 3D). It is possible that this is caused by frameshift mutations in other TRF2 alleles. Furthermore, it is possible that the EE mutations partially impair the stability of TRF2 protein. To examine whether TRF2 EE affects the recruitment of ORC to telomeres, ChIP-qPCR assays to examine telomere-specific chromatin loading were performed using an anti-FLAG antibody in TRF2-edited clones expressing ORC1-3 × FLAG (Figure 3E and Supplementary Figure S2), and telomere DNA enrichment relative to the LMNB2 replication origin (52,57) was calculated (Figure 3E). ORC1 binding to telomeres was decreased to approximately 60% in the WT clone compared with the parental HeLa cells. This decrease may be associated with the reduced TRF2 protein levels in the WT clone. In the two EE clones, ORC1 binding to telomeres was decreased 4-fold compared with the WT clone. Even after normalizing to the TRF2 protein levels, ORC1 recruitment to telomeres appeared to be reduced in the two EE clones (Supplementary Figure S2B). This impairment was not attributable to the expression level of ORC1-3 × FLAG (Figure 3F) or to cell cycle distribution (Figure 3G). We next examined whether MCM recruitment to telomeres was affected in the TRF2-edited clones using MCM7 ChIP-qPCR. MCM7 binding to telomeres was significantly decreased in the two EE clones when compared with the WT clone (Figure 3H), while MCM7 DNA binding in general was unaffected (Figure 3I). These data suggest that TRF2–ORC binding may be critical for the recruitment of ORC and MCM to telomeres.
Telomere maintenance is impaired in HeLa TRF2 E111A/E112A clones under DNA replication stress conditions
The phenotype of TRF2-edited clones was analyzed with particular focus on telomere maintenance under DNA replication stress conditions. First, we investigated telomere DNA damage. After exposing cells to 0.1 mM hydroxyurea (HU), telomeres and 53BP1, a marker for DNA double-strand breaks (58), were visualized by FISH with a PNA probe targeting a telomere-specific sequence, and immunofluorescence staining, respectively (Figure 4A and B). In the absence of HU-induced replication stress, the number of telomere dysfunction-induced foci (TIFs), detected as telomeric foci co-localizing with 53BP1 (59,60), increased in the WT clone (Figure 4B, Parental HeLa/DDW vs. WT 7–1/DDW). This increase is possibly related to the decreased TRF2 protein level in WT 7–1 (see Figure 3D), which may lead to insufficient telomere protection (61,62). In the absence of HU, the number of endogenous TIFs in the two EE clones were comparable to the WT clone, suggesting that there was no increase in the basal level of telomere damage in the EE clones. Importantly, HU treatment increased the number of TIFs in the two EE clones but not in the WT clone, indicating a defect in the response to HU-induced telomere replication stress in the EE clones.
Defective DNA replication leads to micronuclei formation (63,64); therefore, we next measured the number of micronuclei containing telomeric FISH signals (telomere-containing micronuclei) as an index of telomere instability (65) (Figure 4C, D). In the absence of HU, the frequency of telomere-containing micronuclei did not differ between the cell lines tested. However, HU induced significantly more telomere-containing micronuclei in EE clones than the WT clone (approximately 2-fold). By contrast, the number of micronuclei that do not contain telomeric FISH signals (telomere-free micronuclei) was comparable among the cell lines even in the presence of HU (Supplementary Figure S3). Taken together, these results suggest that TRF2-mediated recruitment of ORC to telomeres contributes to the maintenance of telomere stability under DNA replication stress conditions.
A broad region of ORC1 including amino acids 411–510 is required for the recruitment by TRF2
As an alternative approach to evaluate the physiological significance of the TRF2–ORC interaction, we searched for a specific ORC1 mutant specifically defective for TRF2 binding alone. Several proteins that bind to the TRFH domain of TRF2, such as Apollo and SLX4, share a TRF2 binding motif (F/Y/HxLxP) (28,32). The leucine residues in the binding motifs of Apollo and SLX4 are important for their interaction with TRF2 (28,32). This short motif is similarly found in human ORC1 at amino acid 227–231 (HTLTP). However, an ORC1 L229A mutant could be recruited by TRF2 in the lacO-LacI assay (Supplementary Figure S4), suggesting that, unlike Apollo and SLX4, this motif in ORC1 may not be involved in TRF2 binding.
Next, we generated a series of ORC1 truncation mutants (Figure 5A) and examined their recruitment by TRF2 (45–244)-LacI (Figure 5B). Transiently-expressed FLAG-ORC1 (2–511) frequently co-localized with TRF2 (45–244)-LacI (73%), indicating that the C-terminal AAA+ (ATPases associated with diverse cellular activities) and WH (winged-helix) domains are dispensable for binding to TRF2. By contrast, three further ORC1 truncation mutants (2–325, 2–244, and 2–85) were not recruited by TRF2 (45–244)-LacI, suggesting that a region spanning amino acid 326–511 of ORC1 is required for TRF2 binding. In addition, ORC1 (244–511), but not ORC1 (325–511), co-localized more frequently with TRF2 (45–244)-LacI (∼90% and ∼46%, respectively) than ORC1 (2–511) (73%). These data suggest that ORC1 efficiently binds to TRF2 via a relatively broad region that includes amino acids 244–511. This is consistent with a previous report that GST-ORC1 (201–511) interacts with TRF2 in HeLa cell extracts (34).
To further narrow the region required for TRF2 binding, we analyzed the recruitment of full-length ORC1 mutants lacking amino acids within the 326–510 region (Figure 5C and D). TRF2 (45–244)-LacI recruited N-terminal FLAG-tagged ORC1 (full-length) at a significantly higher frequency (26%) than the control HA-LacI (9%). However, ORC1 (Δ326–510), ORC1 (Δ386–510), and ORC1 (Δ411–510) co-localized with TRF2 (45–244)-LacI at low frequencies, indicating that amino acids 411–510 of ORC1 are necessary for the recruitment by TRF2. By contrast, ORC1 (Δ446–510) and ORC1 (Δ411–445) partially retained its ability to bind to TRF2. These results suggest that multiple residues distributed across the 411–510 region are responsible for binding to TRF2. Because the loss of such a broad region is likely to have adverse effects on ORC1 stability and function, we considered ORC1 (Δ411–510) to be an unsuitable candidate for a specific ORC1 mutant defective only in TRF2 binding.
Overexpression of an ORC1 (244–511) fragment competitively inhibits TRF2–ORC binding but does not affect the function of ORC in the induction of DNA re-replication
Since the TRF2 binding sites reside within amino acids 244 and 511 of ORC1, we examined whether overexpression of an ORC1 (244–511) construct could competitively inhibit the TRF2–ORC interaction (Figure 6). As shown in Figure 6A and Supplementary Figure S5, FLAG-ORC1 (244–511) bound to TRF2 and competitively inhibited co-immunoprecipitation of full-length ORC1 with TRF2. In addition, overexpression of HA-ORC1 (244–511) impaired the TRF2 (45–244)-LacI-mediated recruitment of ORC1 to the lacO array (Figure 6B). On the other hand, co-immunoprecipitation of RAP1 and SLX4 with TRF2 is not affected by overexpression of FLAG-ORC1 (244–511) (Supplementary Figure S6). Next, we examined whether ORC1 (244–511) overexpression affects the general ORC function during global DNA replication. Although MCM loading is tightly regulated to prevent DNA re-replication, simultaneous deregulation of licensing factors (Cdt1 + ORC1 or Cdt1 + CDC6) can induce re-replication in HEK293T cells, which is detected by FACS as DNA content higher than 4N (54). We used this assay to examine the genome-wide functions of ORC in the presence of FLAG-ORC1 (244–511) (Figure 6C–E). Consistent with a previous report (54), co-expression of Cdt1 + ORC1 induced significant re-replication (∼two-fold higher than that induced by expression of Cdt1 alone) (Figure 6C–E). This re-replication was dependent on ORC1 ATPase activity, as a mutation in the Walker B motif (D620A) abolished the induction of re-replication (Supplementary Figure S7). Importantly, re-replication was induced by Cdt1 + ORC1 to a similar extent in the presence and absence of FLAG-ORC1 (244–511) (Figure 6C–E). These results suggest that overexpression of ORC1 (244–511) may specifically inhibit TRF2–ORC interaction with little effect on the genome-wide replication activity of ORC.
Overexpression of ORC1 (244–511) compromises ORC and MCM recruitment to telomeres and allows telomeric DNA damage to accumulate under replication stress conditions
To investigate whether the overexpression of ORC1 (244–511) inhibits ORC binding to telomeres, HeLa cells stably overexpressing HA-ORC1 (244–511) were established by retroviral infection (Supplementary Figure S8A). Overexpression of HA-ORC1 (244–511) did not affect cell proliferation or cell cycle distribution (Supplementary Figure S8B and C). When ORC1-3 × FLAG was transiently expressed in these cells to perform anti-FLAG ChIP analysis (Figure 7A), ORC1-3 × FLAG binding at telomeres was significantly reduced upon overexpression of HA-ORC1 (244–511) (Figure 7B and Supplementary Figure S9). Furthermore, overexpression of HA-ORC1 (244–511) significantly decreased MCM loading at telomeres (Figure 7C), while the total DNA co-precipitated with an anti-MCM7 antibody was unchanged (Figure 7D). These results suggest that HA-ORC1 (244–511) specifically impairs telomere binding of ORC and MCM. Using telomere-FISH and immunostaining of 53BP1, we next examined telomeric DNA damage upon overexpression of HA-ORC1 (244–511) (Figure 7E). In the absence of exogenous replication stress (Figure 7E; DDW), levels of spontaneous TIF-positive cells were low in both the control and HA-ORC1 (244–511)-overexpressing cells. When treated with 0.1 mM HU, cells overexpressing HA-ORC1 (244–511) showed a significant increase in TIF-positive cells compared with the control (Figure 7E; HU). These results further support the notion that TRF2-mediated ORC binding to telomeres contributes to telomere stability upon DNA replication stress.
DISCUSSION
In this study, we inhibited the TRF2–ORC interaction using the TRF2 E111A/E112A mutant, which is primarily defective for ORC1 binding (Figures 1 and 2). In HeLa TRF2 E111A/E112A knock-in clones, the recruitment of ORC and MCM to telomeres was reduced, and replication-stress-induced telomere instability was significantly enhanced (Figures 3 and 4). Because the TRF2 E111A/E112A mutations may partially affect the protein stability and the binding to SLX4, we cannot exclude the possibility that these also contribute to telomere phenotypes observed in the HeLa EE clones. In addition, potential effects of the mutant TRF2 on chromatin structure could also contribute to the impaired telomere stability. As an alternative experimental system, we identified an ORC1 fragment (244–511), which competitively inhibits the TRF2–ORC interaction without inhibiting TRF2 interaction with SLX4 and RAP1. When this fragment was overexpressed in HeLa cells, levels of telomere-bound ORC and MCM were reduced and replication stress-induced telomere DNA damage was enhanced (Figures 6 and 7). These two specific and complementary methods to inhibit the TRF2–ORC interaction avoid the limitations of simple TRF2 knockdown studies (36,37), which result in the deprotection of telomere ends and prevent TRF2-binding factors from accessing telomeres. Our findings indicate a critical role for the TRF2–ORC interaction in the recruitment of ORC and MCM to telomeres and in the maintenance of telomere stability when cells experience replication stress. On the other hand, our results show that inhibition of TRF2–ORC interaction may have no significant impact on telomere stability under unperturbed conditions in HeLa cells. It has been reported that replication initiation events can be detected in long telomeres ranging from 25 to 50 kb but rarely occur in short telomeres (38,39). In HeLa cells, telomeres usually range from 5 to 15 kb in length (66). Further investigation using cell lines harboring longer telomeres is needed to clarify the role of TRF2–ORC interaction during unperturbed telomere replication. It has recently been demonstrated that overexpression of a TRF2 ΔB mutant, an ORC-binding mutant that lacks the N-terminal basic domain (34,37), decreases ORC recruitment to telomeres and leads to telomere fragility in LOX human melanoma cells (40). However, overexpression of TRF2 ΔB also induces cell cycle arrest, senescence, and rapid loss of telomeres (37,40,62,67,68). These phenotypes may be a consequence of the multiple roles the TRF2 basic domain has been shown to play in preventing homologous recombination-mediated telomere deletion (67–70), promoting the replication of heterochromatin (71,72), and mediating ORC recruitment at telomeres.
The dimerization of the TRF2 TRFH domain is required for the TRF2–ORC interaction (35). Moreover, the TRF2 E111/E112 residues are critical for ORC recruitment (Figure 1). Our data suggest that the SLX4-binding region of TRF2 overlaps with the ORC1-binding region within the TRFH domain, yet the TRF2 E111A/E112A mutant partially retains its ability to interact with SLX4 (Figure 2E). In addition, the Y73, G74, V88, P90, K93, E94, H95, T96, S119 and M122 residues are also involved in ORC recruitment. The involvement of these residues, which are widely distributed, suggests that TRF2 and ORC1 interact through a broad binding interface. Indeed, we found that, although ORC1 (325–511) is sufficient for TRF2 binding, ORC1 (244–511) co-localizes with TRF2 with a 2-fold higher frequency (Figure 5). A flexible linker sequence is present between the N-terminal BAH (bromo adjacent homology) domain and the C-terminal AAA + domain of ORC1 (Supplementary Figure S10). This region is predicted to be a conserved, intrinsically disordered region (Supplementary Figure S10) (73). Therefore, multiple residues in the ORC1 244–511 region may form a flexible binding interface for the interaction with TRF2.
Our results further reveal that TRF2 Y73A/G74A, V88A/P90A, E111A/E112A and S119A/M122A mutations have no obvious effect on TRFH dimerization or the ability of TRF2 to bind Apollo, RAP1 or RETL1 (Figure 2A-D). TRF2 F120 is particularly important for the interaction with Apollo and SLX4 through their shared F/Y/HxLxP motif (28,32). RTEL1 binds to both TRF2 amino acids 64–83 and 312–341, with I79 playing a particularly crucial role in RTEL1 binding (25,31). A RAP1-binding motif is found at the TRF2 275–316 region (74). The present data are in line with these findings. Although the TRF2 S119A/M122A mutations are in the proximity of F120, this mutant interacts with Apollo and SLX4, but not ORC1, suggesting that the TRF2 residues responsible for the bindings are different among these factors.
ChIP analysis revealed that ORC and MCM loading at telomeres is reduced by the overexpression of ORC1 (244–511) in HeLa cells (Figure 7). Importantly, ORC1 (244–511) overexpression did not affect genome-wide DNA binding of MCM (Figure 7D). Moreover, re-replication induced by co-expression of Cdt1 + ORC1 was not compromised by overexpression of ORC1 (244–511). These results suggest that ORC1 (244–511) overexpression selectively impairs the telomere binding activity of ORC. However, it remains possible that the overexpression of ORC1 (244–511) affects particular replication origins in other genomic regions such as pericentromeric heterochromatin, where TRF2 has been suggested to promote ORC recruitment (72). This issue should be addressed in future studies. The formation of the ORC complex should be unaffected by ORC1 (244–511) overexpression since the ORC1 N-terminal region (amino acids 1–470) is not essential for ORC complex formation (75). The BAH domain of ORC1 recognizes histone H4-K20me2 and regulates the loading of ORC onto replication origins, while mutations in this domain are implicated in the etiology of Meier-Gorlin syndrome (76–78). The ORC1 244–511 region is situated away from the BAH domain and is unlikely to affect BAH domain-mediated binding to replication origins. However, ORC1 (244–511) overexpression could have additional impact, besides interference with TRF2 interaction, on cell cycle-dependent regulation of ORC loading on replication origins because the ORC1 244–511 region is involved in ORC1–ORC1 self-interaction and interactions with a number of regulatory factors, such as CDC6 and protein phosphatase 1 (79). In addition, the ORC1 244–511 region overlaps with basic patches and G-rich RNA/ssDNA binding regions (80–82) (Supplementary Figure S10) thought to be involved in the interaction with origin DNA. It is therefore also possible that the binding of DNA through these regions of ORC1 is affected by ORC1 (244–511) overexpression. Future studies are required to evaluate the contribution of these regions to ORC recruitment to the G-rich telomere repeat sequence.
Dormant replication origins are critical for complete duplication of the genome (41,83). A paucity of backup replication origins results in chromosome instability (10–12,15). It has been reported that dormant telomeric origins are activated by replication stress to complete telomere replication (40). Although whether telomere replication is actually impaired due to the defects in ORC recruitment should be determined in future studies, our findings support the view that TRF2-mediated ORC and MCM recruitment plays a pivotal role in maintaining telomere stability through the formation of dormant telomeric origins that fire when replication forks stall or collapse.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Tohru Kiyono (National Cancer Center EPOC, Kashiwa), Masato Kanemaki (National Institute of Genetics, Mishima), Yoko Katsuki (Kyoto University, Kyoto), Minoru Takata (Kyoto University, Kyoto), Hilary McLauchlan (University of Dundee, Scotland), and John Rouse (University of Dundee, Scotland) for plasmids. We thank Toshiki Tsurimoto (Kyushu University, Fukuoka), Tsutomu Katayama (Kyushu University, Fukuoka), and Hironori Kawakami (Sanyo-Onoda City University, Sanyo-Onoda) for discussion and comment. We appreciate technical support from the Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences.
Notes
Present address: Kazumasa Yoshida. Department of Cell Biology, Faculty of Medicine, Fukuoka University, Fukuoka 814-0180, Japan; Central Research Institute for Advanced Molecular Medicine, Fukuoka University, Fukuoka 814-0180, Japan.
Contributor Information
Mitsunori Higa, Department of Cellular Biochemistry, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Yukihiro Matsuda, Department of Cellular Biochemistry, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Jumpei Fujii, Department of Cellular Biochemistry, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Nozomi Sugimoto, Department of Cellular Biochemistry, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Kazumasa Yoshida, Department of Cellular Biochemistry, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Masatoshi Fujita, Department of Cellular Biochemistry, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science [KAKENHI grant number JP17K15065 to K.Y.]; M.H. was supported by a JSPS research fellowship [JP18J11443]. Funding for open access charge: Publication charge will be funded from the university.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bleichert F. Mechanisms of replication origin licensing: a structural perspective. Curr. Opin. Struct. Biol. 2019; 59:195–204. [DOI] [PubMed] [Google Scholar]
- 2. Parker M.W., Botchan M.R., Berger J.M.. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2017; 52:107–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dellino G.I., Cittaro D., Piccioni R., Luzi L., Banfi S., Segalla S., Cesaroni M., Mendoza-Maldonado R., Giacca M., Pelicci P.G.. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013; 23:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miotto B., Ji Z., Struhl K.. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E4810–E4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Picard F., Cadoret J.C., Audit B., Arneodo A., Alberti A., Battail C., Duret L., Prioleau M.N.. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet. 2014; 10:e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Higa M., Fujita M., Yoshida K.. DNA replication origins and fork progression at mammalian telomeres. Genes (Basel). 2017; 8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sugimoto N., Fujita M.. Molecular mechanism for chromatin regulation during MCM loading in mammalian cells. Advances in Experimental Medicine and Biology. 2017; 61–78. [DOI] [PubMed] [Google Scholar]
- 8. Fujita M. Cdt1 revisited: complex and tight regulation during the cell cycle and consequences of deregulation in mammalian cells. Cell Div. 2006; 1:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hills S.A., Diffley J.F.X.. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014; 24:R435–R444. [DOI] [PubMed] [Google Scholar]
- 10. Chuang C.H., Wallace M.D., Abratte C., Southard T., Schimenti J.C.. Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet. 2010; 6:e1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ibarra A., Schwob E., Méndez J.. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawabata T., Luebben S.W., Yamaguchi S., Ilves I., Matise I., Buske T., Botchan M.R., Shima N.. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell. 2011; 41:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kunnev D., Rusiniak M.E., Kudla A., Freeland A., Cady G.K., Pruitt S.C.. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene. 2010; 29:3630–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McNairn A.J., Chuang C.H., Bloom J.C., Wallace M.D., Schimenti J.C.. Female-biased embryonic death from inflammation induced by genomic instability. Nature. 2019; 567:105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shima N., Alcaraz A., Liachko I., Buske T.R., Andrews C.A., Munroe R.J., Hartford S.A., Tye B.K., Schimenti J.C.. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007; 39:93–98. [DOI] [PubMed] [Google Scholar]
- 16. Sedlackova H., Rask M.B., Gupta R., Choudhary C., Somyajit K., Lukas J.. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nature. 2020; 587:297–302. [DOI] [PubMed] [Google Scholar]
- 17. Van Ly D., Low R.R.J., Frölich S., Bartolec T.K., Kafer G.R., Pickett H.A., Gaus K., Cesare A.J.. Telomere loop dynamics in chromosome end protection. Mol. Cell. 2018; 71:510–525. [DOI] [PubMed] [Google Scholar]
- 18. de Lange T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018; 52:223–247. [DOI] [PubMed] [Google Scholar]
- 19. Gilson E., Géli V.. How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 2007; 8:825–838. [DOI] [PubMed] [Google Scholar]
- 20. Martínez P., Blasco M.A.. Replicating through telomeres: a means to an end. Trends Biochem. Sci. 2015; 40:504–515. [DOI] [PubMed] [Google Scholar]
- 21. Pfeiffer V., Lingner J.. Replication of telomeres and the regulation of telomerase. Cold Spring Harb. Perspect. Biol. 2013; 5:a010405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stroik S., Hendrickson E.A.. Telomere replication—when the going gets tough. DNA Repair (Amst.). 2020; 94:102875. [DOI] [PubMed] [Google Scholar]
- 23. Fouché N., Özgür S., Roy D., Griffith J.D.. Replication fork regression in repetitive DNAs. Nucleic. Acids. Res. 2006; 34:6044–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martínez P., Thanasoula M., Muñoz P., Liao C., Tejera A., McNees C., Flores J.M., Fernández-Capetillo O., Tarsounas M., Blasco M.A.. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009; 23:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarek G., Vannier J., Panier S., Petrini J.H.J., Boulton S.J.. TRF2 recruits RTEL1 to telomeres in S phase to promote T-Loop unwinding. Mol. Cell. 2015; 57:622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T.. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009; 138:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vannier J.B., Pavicic-Kaltenbrunner V., Petalcorin M.I.R., Ding H., Boulton S.J.. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012; 149:795–806. [DOI] [PubMed] [Google Scholar]
- 28. Chen Y., Yang Y., van Overbeek M., Donigian J.R., Baciu P., de Lange T., Lei M.. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008; 319:1092–1096. [DOI] [PubMed] [Google Scholar]
- 29. van Overbeek M., de Lange T.. Apollo, an Artemis-related nuclease, interacts with TRF2 and protects human telomeres in S phase. Curr. Biol. 2006; 16:1295–1302. [DOI] [PubMed] [Google Scholar]
- 30. Ye J., Lenain C., Bauwens S., Rizzo A., Saint-Léger A., Poulet A., Benarroch D., Magdinier F., Morere J., Amiard S.et al.. TRF2 and apollo cooperate with topoisomerase 2α to protect human telomeres from replicative damage. Cell. 2010; 142:230–242. [DOI] [PubMed] [Google Scholar]
- 31. Sarek G., Kotsantis P., Ruis P., Van Ly D., Margalef P., Borel V., Zheng X.F., Flynn H.R., Snijders A.P., Chowdhury D.et al.. CDK phosphorylation of TRF2 controls t-loop dynamics during the cell cycle. Nature. 2019; 575:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wan B., Yin J., Horvath K., Sarkar J., Chen Y., Wu J., Wan K., Lu J., Gu P., Yu E.Y.et al.. SLX4 assembles a telomere maintenance toolkit by bridging multiple endonucleases with telomeres. Cell Rep. 2013; 4:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guervilly J.H., Gaillard P.H.. SLX4: multitasking to maintain genome stability. Crit. Rev. Biochem. Mol. Biol. 2018; 53:475–514. [DOI] [PubMed] [Google Scholar]
- 34. Atanasiu C., Deng Z., Wiedmer A., Norseen J., Lieberman P.M.. ORC binding to TRF2 stimulates OriP replication. EMBO Rep. 2006; 7:716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Higa M., Kushiyama T., Kurashige S., Kohmon D., Enokitani K., Iwahori S., Sugimoto N., Yoshida K., Fujita M.. TRF2 recruits ORC through TRFH domain dimerization. Biochim. Biophys. Acta - Mol. Cell Res. 2017; 1864:191–201. [DOI] [PubMed] [Google Scholar]
- 36. Tatsumi Y., Ezura K., Yoshida K., Yugawa T., Narisawa-Saito M., Kiyono T., Ohta S., Obuse C., Fujita M.. Involvement of human ORC and TRF2 in pre-replication complex assembly at telomeres. Genes Cells. 2008; 13:1045–1059. [DOI] [PubMed] [Google Scholar]
- 37. Deng Z., Dheekollu J., Broccoli D., Dutta A., Lieberman P.M.. The origin recognition complex localizes to telomere repeats and prevents telomere-circle formation. Curr. Biol. 2007; 17:1989–1995. [DOI] [PubMed] [Google Scholar]
- 38. Drosopoulos W.C., Kosiyatrakul S.T., Schildkraut C.L.. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J. Cell Biol. 2015; 210:191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Drosopoulos W.C., Kosiyatrakul S.T., Yan Z., Calderano S.G., Schildkraut C.L.. Human telomeres replicate using chromosomespecific, rather than universal, replication programs. J. Cell Biol. 2012; 197:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drosopoulos W.C., Deng Z., Twayana S., Kosiyatrakul S.T., Vladimirova O., Lieberman P.M., Schildkraut C.L.. TRF2 mediates replication initiation within human telomeres to prevent telomere dysfunction. Cell Rep. 2020; 33:108379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alver R.C., Chadha G.S., Blow J.J.. The contribution of dormant origins to genome stability: From cell biology to human genetics. DNA Repair (Amst.). 2014; 19:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janicki S.M., Tsukamoto T., Salghetti S.E., Tansey W.P., Sachidanandam R., Prasanth K.V, Ried T., Shav-Tal Y., Bertrand E., Singer R.H.et al.. From silencing to gene expression: Real-time analysis in single cells. Cell. 2004; 116:683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sugimoto N., Kitabayashi I., Osano S., Tatsumi Y., Yugawa T., Narisawa-Saito M., Matsukage A., Kiyono T., Fujita M.. Identification of novel human Cdt1-binding proteins by a proteomics approach: Pproteolytic regulation by APC/CCdh1. Mol. Biol. Cell. 2008; 19:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tatsumi Y., Ohta S., Kimura H., Tsurimoto T., Obuse C.. The ORC1 cycle in human cells. I. Cell cycle-regulated oscillation of human ORC1. J. Biol. Chem. 2003; 278:41528–41534. [DOI] [PubMed] [Google Scholar]
- 45. Okumura M., Natsume T., Kanemaki M.T., Kiyomitsu T.. Dynein–dynactin–NuMA clusters generate cortical spindle-pulling forces as a multiarm ensemble. Elife. 2018; 7:e36559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Muñoz I.M., Hain K., Déclais A.C., Gardiner M., Toh G.W., Sanchez-Pulido L., Heuckmann J.M., Toth R., Macartney T., Eppink B.et al.. Coordination of Structure-Specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell. 2009; 35:116–127. [DOI] [PubMed] [Google Scholar]
- 47. Fujita M., Ishimi Y., Nakamura H., Kiyono T., Tsurumi T.. Nuclear organization of DNA replication initiation proteins in mammalian cells. J. Biol. Chem. 2002; 277:10354–10361. [DOI] [PubMed] [Google Scholar]
- 48. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sugimoto N., Maehara K., Yoshida K., Yasukouchi S., Osano S., Watanabe S., Aizawa M., Yugawa T., Kiyono T., Kurumizaka H.et al.. Cdt1-binding protein GRWD1 is a novel histone-binding protein that facilitates MCM loading through its influence on chromatin architecture. Nucleic Acids Res. 2015; 43:5898–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ishimoto R., Tsuzuki Y., Matsumura T., Kurashige S., Enokitani K., Narimatsu K., Higa M., Sugimoto N., Yoshida K., Fujita M.. SLX4–XPF mediates DNA damage responses to replication stress induced by DNA–protein interactions. J. Cell Biol. 2021; 220:e202003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cawthon R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002; 30:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sugimoto N., Yugawa T., Iizuka M., Kiyono T., Fujita M.. Chromatin remodeler sucrose nonfermenting 2 homolog (SNF2H) is recruited onto DNA replication origins through interaction with Cdc10 protein-dependent transcript 1 (Cdt1) and promotes pre-replication complex formation. J. Biol. Chem. 2011; 286:39200–39210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L.et al.. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009; 55:611–622. [DOI] [PubMed] [Google Scholar]
- 54. Sugimoto N., Yoshida K., Tatsumi Y., Yugawa T., Narisawa-Saito M., Waga S., Kiyono T., Fujita M.. Redundant and differential regulation of multiple licensing factors ensures prevention of re-replication in normal human cells. J. Cell Sci. 2009; 122:1184–1191. [DOI] [PubMed] [Google Scholar]
- 55. Li B., Oestreich S., De Lange T.. Identification of human Rap1: implications for telomere evolution. Cell. 2000; 101:471–483. [DOI] [PubMed] [Google Scholar]
- 56. Sfeir A., Kabir S., van Overbeek M., Celli G.B., de Lange T.. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage Signal. Science (80-.). 2010; 327:1657–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giacca M., Zentilin L., Norio P., Diviacco S., Dimitrova D., Contreas G., Biamonti G., Perini G., Weighardt F., Riva S.et al.. Fine mapping of a replication origin of human DNA. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:7119–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 59. Takai H., Smogorzewska A., De Lange T.. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003; 13:1549–1556. [DOI] [PubMed] [Google Scholar]
- 60. d’Adda di Fagagna F., Reaper P.M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N.P., Jackson S.P.. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003; 426:194–198. [DOI] [PubMed] [Google Scholar]
- 61. Celli G.B., de Lange T.. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005; 7:712–718. [DOI] [PubMed] [Google Scholar]
- 62. van Steensel B., Smogorzewska A., De Lange T.. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998; 92:401–413. [DOI] [PubMed] [Google Scholar]
- 63. Ying S., Minocherhomji S., Chan K.L., Palmai-Pallag T., Chu W.K., Wass T., Mankouri H.W., Liu Y., Hickson I.D.. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013; 15:1001–1007. [DOI] [PubMed] [Google Scholar]
- 64. Chan K.L., Palmai-Pallag T., Ying S., Hickson I.D.. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009; 11:753–760. [DOI] [PubMed] [Google Scholar]
- 65. Lindberg H.K., Falck G.C.M., Järventaus H., Norppa H.. Characterization of chromosomes and chromosomal fragments in human lymphocyte micronuclei by telomeric and centromeric FISH. Mutagenesis. 2008; 23:371–376. [DOI] [PubMed] [Google Scholar]
- 66. Cristofari G., Lingner J.. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 2006; 25:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang R.C., Smogorzewska A., De Lange T.. Homologous recombination generates t-loop-sized deletions at human telomeres. Cell. 2004; 119:355–368. [DOI] [PubMed] [Google Scholar]
- 68. Saint-Léger A., Koelblen M., Civitelli L., Bah A., Djerbi N., Giraud-Panis M.J., Londonõ-Vallejo A., Ascenzioni F., Gilson E.. The basic N-terminal domain of TRF2 limits recombination endonuclease action at human telomeres. Cell Cycle. 2014; 13:2469–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rai R., Chen Y., Lei M., Chang S.. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun. 2016; 7:10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Poulet A., Buisson R., Faivre-Moskalenko C., Koelblen M., Amiard S., Montel F., Cuesta-Lopez S., Bornet O., Guerlesquin F., Godet T.et al.. TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J. 2009; 28:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mendez-Bermudez A., Lototska L., Bauwens S., Giraud-Panis M.J., Croce O., Jamet K., Irizar A., Mowinckel M., Koundrioukoff S., Nottet N.et al.. Genome-wide control of heterochromatin replication by the telomere capping protein TRF2. Mol. Cell. 2018; 70:449–461. [DOI] [PubMed] [Google Scholar]
- 72. Bauwens S., Lototska L., Koundrioukoff S., Debatisse M., Ye J., Gilson E., Mendez-Bermudez A.. The telomeric protein TRF2 regulates replication origin activity within pericentromeric heterochromatin. Life. 2021; 11:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Parker M.W., Bell M., Mir M., Kao J.A., Darzacq X., Botchan M.R., Berger J.M.. A new class of disordered elements controls DNA replication through initiator self-assembly. Elife. 2019; 8:e48562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen Y., Rai R., Zhou Z.R., Kanoh J., Ribeyre C., Yang Y., Zheng H., Damay P., Wang F., Tsujii H.et al.. A conserved motif within RAP1 has diversified roles in telomere protection and regulation in different organisms. Nat. Struct. Mol. Biol. 2011; 18:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tocilj A., On K.F., Yuan Z., Sun J., Elkayam E., Li H., Stillman B., Joshua-Tor L.. Structure of the active form of human origin recognition complex and its ATPase motor module. Elife. 2017; 6:e20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Beck D.B., Burton A., Oda H., Ziegler-Birling C., Torres-Padilla M.E., Reinberg D.. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev. 2012; 26:2580–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bicknell L.S., Bongers E.M.H.F., Leitch A., Brown S., Schoots J., Harley M.E., Aftimos S., Al-Aama J.Y., Bober M., Brown P.A.J.et al.. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat. Genet. 2011; 43:356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kuo A.J., Song J., Cheung P., Ishibe-Murakami S., Yamazoe S., Chen J.K., Patel D.J., Gozani O.. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012; 484:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hossain M., Bhalla K., Stillman B.. Multiple, short protein binding motifs in ORC1 and CDC6 control the initiation of DNA replication. Mol. Cell. 2021; 81:1951–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bleichert F., Leitner A., Aebersold R., Botchan M.R., Berger J.M.. Conformational control and DNA-binding mechanism of the metazoan origin recognition complex. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E5906–E5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li N., Lam W.H., Zhai Y., Cheng J., Cheng E., Zhao Y., Gao N., Tye B.K.. Structure of the origin recognition complex bound to DNA replication origin. Nature. 2018; 559:217–222. [DOI] [PubMed] [Google Scholar]
- 82. Hoshina S., Yura K., Teranishi H., Kiyasu N., Tominaga A., Kadoma H., Nakatsuka A., Kunichika T., Obuse C., Waga S.. Human origin recognition complex binds preferentially to G-quadruplex-preferable RNA and single-stranded DNA. J. Biol. Chem. 2013; 288:30161–30171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Moreno A., Carrington J.T., Albergante L., Mamun M.Al, Haagensen E.J., Komseli E.S., Gorgoulis V.G., Newman T.J., Blow J.J.. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E5757–E5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.