


Abstract
8-Oxo-7,8-dihydro-2′-deoxyguanosine (OG), one of the most common oxidative DNA damages, causes genome instability and is associated with cancer, neurological diseases and aging. In addition, OG and its repair intermediates can regulate gene transcription, and thus play a role in sensing cellular oxidative stress. However, the lack of methods to precisely map OG has hindered the study of its biological roles. Here, we developed a single-nucleotide resolution OG-sequencing method, named CLAPS-seq (Chemical Labeling And Polymerase Stalling Sequencing), to measure the genome-wide distribution of both exogenous and endogenous OGs with high specificity. Our data identified decreased OG occurrence at G-quadruplexes (G4s), in association with underrepresentation of OGs in promoters which have high GC content. Furthermore, we discovered that potential quadruplex sequences (PQSs) were hotspots of OGs, implying a role of non-G4-PQSs in OG-mediated oxidative stress response.
INTRODUCTION
As a constant threat to cellular DNA, oxidative stress can be generated from multiple internal and external sources such as normal metabolism, inflammatory processes and environmental toxins (1,2). Among the >80 different oxidative DNA modifications (3,4), 8-oxo-7,8-dihydro-2′-deoxyguanosine (OG) is one of the most abundant and important lesions since guanine (G) has the lowest redox potential among the four nucleobases (3,5). OGs can induce G to T substitution as DNA polymerases may insert an ‘A’ instead of a ‘C’ opposite an OG base (6). In human cells, OGs and misincorporated As are efficiently removed by DNA glycosylases OGG1 and MUTYH, respectively, generating apurinic/apyrimidinic (AP) sites which are further processed by the base excision repair (BER) pathway (7,8). However, the OG repair intermediates including AP sites and single strand breaks can interfere with DNA replication and other processes and cause genome instability (7,9). Therefore, OGs are relevant to the pathological processes involved in cancers, neurological diseases and aging (2,10). Nonetheless, the steady-state level of cellular OGs, estimated to be about 0.5∼1 lesion per 106 nucleotides (nt) in human cells (11,12), is tightly controlled by the balance between damage formation and repair.
On the other hand, it was reported recently that OGs play an epigenetic role in cellular response to oxidative stress by regulating gene expression through the repair intermediates (13). Indeed, the inhibition of OGG1 suppressed TNFα-induced proinflammatory gene expression and inflammation, suggesting a role of OG and its repair in transcription regulation (14). One well-characterized mechanism is through the excision of OGs to stabilize G-quadruplex (G4) structures (15,16). A G4 is a four-stranded noncanonical DNA secondary structure formed in G-rich regions such as telomeres and promoters (17). G4s are involved in transcriptional regulation and other biological processes including genomic instability, telomere maintenance and DNA replication (17,18). The excision of OG located within a potential quadruplex sequence (PQS) by OGG1 generates AP sites which may drive the switch of a PQS to G4 and recruit APE1 to further stabilize the G4 structure (15,16). G4 in promoter regions and the associated APE1 could regulate gene transcription (13), as demonstrated on selected genes including VEGF-A (16), SRC (19), c-KIT (20), etc. Thus, G4s or PQSs likely serve as a sensor of oxidative damage (17).
As a consequence of the above processes, the precise genomic locations of OGs are expected to impact their biological effects. In this regard, a couple of high-throughput sequencing-based techniques measuring the genomic distribution of OG in yeast, mouse and human genomes were developed recently (21–25). These methods rely on the recognition of OG by an anti-OG antibody (21), selective chemical reaction (22), or OG-specific glycosylases (23–25). Each method has its own advantages and disadvantages (26). These OG sequencing studies demonstrated that OGs were not uniformly distributed across the genome. However, their conclusions were not always consistent with each other (26), possibly due to several common challenges for OG detection. First, OG can be introduced during sample processing such as DNA purification and sonication (12). This incidental damage can be reduced by well-designed protocols and the addition of anti-oxidants, but cannot be completely avoided (11,12). Second, due to the low abundance of OG, captured DNA may contain significant amount of OG-free DNA. However, current methods are unable to distinguish genuine OGs and nonspecific signals. Furthermore, all these methods only reach a resolution of hundreds base pairs except click-code-seq which was applied only to yeast (26).
In order to overcome these obstacles, we developed CLAPS-seq (Chemical Labeling And Polymerase Stalling Sequencing) which captures OGs by an optimized chemical labeling reaction (22,27) and achieves single nucleotide resolution by utilizing the property that high-fidelity DNA polymerase is blocked by the labeled lesion (28). By gently extracting DNA, immediately labeling OGs with biotin, and using streptavidin-based purification, CLAPS-seq minimizes incidental oxidation and greatly reduces background fragments, allowing us to map endogenously formed OGs across the human genome. Specificity was verified by the high ratio of G residues at sequenced damage sites. Exogenous OGs induced by KBrO3 in cellular or naked DNA were also measured to explore the factors affecting OG formation. The results confirm that unexpectedly, genomic regions with the highest GC ratio have relatively low OG levels. Consistent with this observation, OGs are not uniformly distributed across the genome and are depleted at transcription start sites (TSSs). Further analyses suggest this phenomenon is, at least partially, due to the reduced occurrence of OGs at G4s. Surprisingly, non-G4-PQSs, which have G-rich sequences similar to G4s, are hotspots of OGs.
MATERIALS AND METHODS
Biotinylation and purification of OG-containing single-stranded (ss) and double-stranded (ds) DNA substrates
For these studies, we generated single-and double-stranded substrates with and without a uniquely located OG adduct using the oligonucleotides shown in Supplementary Table S1. The labeling reaction was optimized from (22). For ssDNA, 1 pmol of oligo OGT and 5 pmol NOGT were labeled with biotin by incubating with 20 mM of amine-tagged biotin probe (Amine-(PEG)11-biotin, ThermoFisher Scientific, 26136) and 5 mM of potassium hexabromoiridate (K2IrBr6, ThermoFisher Scientific, 012651) in sodium phosphate buffer (100 mM, pH 8.0) for 5 min on ice. The reaction was terminated by passing through a Microspin G50 column (GE Healthcare, 27-5330-01) which was pre-washed with ice-cold deionized water, then labeled oligonucleotides were ethanol precipitated and dissolved in 1× TE buffer (10 mM Tris–Cl pH 8.0, 1 mM EDTA), followed by incubating with 5 μl of Dynabeads MyOne Streptavidin C1 (ThermoFisher Scientific, 65002) in 1× B&W buffer (5 mM Tris–Cl pH 8.0, 0.5 mM EDTA, 1 M NaCl, 0.1% Tween20, 0.1% CA630) at room temperature for 30 min. Then the beads were washed twice with 1× B&W buffer, and the DNA was eluted by incubating with Formamide Elution buffer (98% formamide, 10mM EDTA) at 65°C for 5 min with shaking at 1500 rpm. After ethanol precipitation, products were dissolved in denature loading buffer (98% formamide, 10 mM EDTA and 0.02% bromophenol blue) and separated by 8% denaturing polyacrylamide gel electrophoresis (PAGE). The gel was stained with SYBR gold and visualized by a Gel Imaging System (Clinx Science Instruments Co., Ltd.).
For dsDNA, OGT and NOGT were pre-annealed with the complementary strand OGTB. One pmol of OG-containing dsDNA and 2 pmol of control DNA were subjected to the same procedure as for ssDNA.
Primer extension assay on biotinylated OG-containing template
Ten pmol of OGT was labeled with biotin and captured using 15 μl of Dynabeads MyOne Streptavidin C1 as described above. Then the beads were sequentially washed twice with 1× B&W buffer and one time with 0.1× TE and resuspended in 7.5 μl H2O.
For primer extension reaction, 25 pmol of OGT Primer was annealed to biotinylated OGT (bound on streptavidin beads) or non-biotinylated OGT, and extended by KAPA HiFi DNA polymerase (Roche, KM2605) in a thermo cycler (98°C 21s, 68°C 2min). Excess primers were degraded by incubating with 1.5 μl of ExoI (New England Biolabs, M0293S) at 37°C for 15 min. The reaction was ended by adding an equal volume of 1% SDS. Next, for the unlabeled control, the products were purified by phenol/chloroform extraction followed by ethanol precipitation and then dissolved in denature loading buffer. For biotinylated samples, the beads were sequentially washed twice with 1× B&W buffer and incubated with fresh NaOH elution buffer (0.1 M NaOH + 0.1% CA630) at room temperature for 2 min to release extension products from templates. After ethanol precipitation, the extension products were dissolved in denature loading buffer and separated by 10% denaturing PAGE. The gel was stained with SYBR gold and visualized by a Gel Imaging System.
Cell culture and potassium bromate treatment
HeLa-S3 cells were purchased from American Type Culture Collection (ATCC, CCL-2.2) and cultured in Dulbecco's modified Eagle's medium (Gibco, C11995500BT) supplemented with 10% fetal bovine serum (Gibco, 10091-148) and 1% Penicillin–Streptomycin (Gibco, 10378016) at 37°C in a 5% CO2 humidified chamber.
For in vivo potassium bromate (KBrO3) treatment (exogenous-cellular samples), KBrO3 (400 mM stock in 1× PBS, Sigma-Aldrich, 60085) and glutathione (100 mM stock in 1× phosphate buffered saline (PBS), Sigma-Aldrich, V900456) were added to the culture medium to a final concentration of 20 and 2.5 mM, respectively. After incubation at 37°C for 30 min in a 5% CO2 humidified chamber, medium was removed and cells were washed three times with 1× PBS with Ca2+ and Mg2+ (Dulbecco's phosphate buffered saline, Sigma-Aldrich, D8662). Cells were further cultured in fresh medium for 30 min before harvesting, as cellular OG levels kept increasing during this time (data not shown). For in vitro KBrO3 treatment (exogenous-naked samples), genomic DNA from untreated cells was incubated with 1 mM of KBrO3 and 2 mM of glutathione in sodium phosphate buffer (10 mM, pH 7.8) at 37°C for 80 min to achieve a comparable level of OG as the in vivo treatment (Supplementary Figure S1C).
For CLAPS-seq, the endogenous sample had three biological replicates, while KBrO3-treated samples (both exogenous-cellular and exogenous-naked damage) had two biological replicates. The correlations (r) of replicates at 10 kb resolution were shown in Supplementary Figure S2A.
Genomic DNA extraction
HeLa cells from one R150 dish were collected by trypsin (ThermoFisher Scientific, 25300054). Genomic DNA was extracted by 4ml DNAzol (ThermoFisher Scientific, 10503027) with the addition of DTT to a final concentration of 10 mM. DNA pellets were resolved in 300 μl of Alkaline Resolving buffer (8 mM NaOH and 100 μM TEMPO) and quantified by Qubit dsDNA BR Assay kit (ThermoFisher Scientific, Q32853) according to the manufacturer's instruction. Typically, more than 100 μg of genomic DNA was obtained from one dish of cells, and were used for ELISA and/or CLAPS-seq immediately. Notably, all buffers and reagents used in DNA extraction, ELISA and chemical labeling steps were prepared with ultrapure deionized water (Merck, 1.01262.1000) in order to prevent further DNA oxidation during these steps, while DTT and TEMPO were added for the same purpose (11).
Quantification of OG by enzyme-linked immunosorbent assay (ELISA)
Certain amounts of genomic DNA (17–80 μg, depending on the expected OG level of each sample) were applied to the 8-OHdG Assay Preparation Reagent Set (Wako Pure Chemical Industries, Ltd., 292-67801) according to the manufacturer's instruction. Briefly, DNA was denatured by incubating at 98°C for 2 min followed by placing on ice-cold water, then digested by nuclease P1 and alkaline phosphatase sequentially. The digested samples were purified by passing through centrifugal ultrafiltration devices (Amicon Ultra-0.5 10 000 MW, Merck, UFC5010), and only the filtrate containing the digested nucleotides was collected. Subsequently, the amounts of OG were determined by the Highly Sensitive 8-OHdG Check kit (JaICA, KOG-HS10/E) according to the manufacturer's instruction. The absolute concentrations of OG were normalized to total nucleotide content (OG/106 nt) for further analysis. Each sample had three biological replicates.
Selective chemical labeling for CLAPS-seq
Extracted DNA (8–10 μg) was labeled and purified by Microspin G50 columns as described above. Since the incidental/artificial DNA oxidation after labeling reaction would not interfere with the following steps, biotinylated genomic DNA could be stored at -20°C until the next step.
Fragmentation, end-repair and first adaptor ligation
Biotinylated DNA was sheared using a Q800 Sonicator (Qsonica) to generate fragments averaging 600 bp in length. Purified DNA fragments (up to 5 μg) were used for End-repair and dA-tailing by NEBNext DNA Library Prep Kit (New England Biolabs, E6040) following the manufacture's instruction. Subsequently, Ad1 (100 pmol/μg of input DNA, Supplementary Table S1) was ligated to both ends by NEBNext T4 Quick Ligase at 4°C overnight. The ligation products were purified by 0.7× DNA FragSelect XP Magnetic Beads (Smart-lifesciences, S3801) and eluted in 15 μl 0.1× TE.
Affinity purification and primer extension
Each sample was incubated with 5 μl of Dynabeads MyOne Streptavidin C1 beads in 1× binding buffer (0.5 M NaCl, 5 mM MgCl2, 10 mM Tris–Cl pH 8.0, 1 mM EDTA, 0.1% Tween20, 0.1% CA630) in the presence of 10 μg of salmon sperm DNA at 4°C for 1 h. Then the beads were sequentially washed twice with 1× B&W buffer, twice with wash buffer II (100 mM Tris–Cl pH 8.0, 1 mM EDTA, 0.5 M LiCl, 1% CA630, 1% sodium deoxycholate), twice with wash buffer III (1 mM Tris–Cl pH 8.0, 0.1 mM EDTA, 100 mM NaCl, 2% SDS), one time with 1× TE and finally one time with 0.1× TE, followed by resuspending in 6 μl 0.1× TE.
For primer extension reaction, 30 pmol of Extension Primer (Supplementary Table S1) was attached to biotinylated DNA which was still bound on beads and extended by KAPA HiFi DNA polymerase in a thermo cycler (98°C 21s, 68°C 2min). Excess primers were degraded by incubating with 1.5 μl of ExoI at 37°C for 10 min. The reaction was ended by adding an equal volume of 1% SDS. Next, the beads were sequentially washed twice with 1× B&W buffer, twice with wash buffer II and twice with wash buffer III. Extension products were released from templates by incubating with fresh NaOH elution buffer (0.1 M NaOH + 0.1% CA630) at room temperature for 2 min to release the complementary strands. The collected DNA was neutralized by adding 0.5 M acetic acid and purified by ethanol precipitation.
Second adaptor ligation, PCR amplification and high-throughput sequencing
The precipitated DNA was dissolved and subjected to ligation with 40 pmol of Ad2 (Supplementary Table S1) by Instant Stick-end Ligase Master Mix (New England Biolabs, M0370L) at 4°C overnight. The ligation products were purified using 0.8× DNA FragSelect XP Magnetic Beads. Two percent of ligated DNA were used for library quality check while the rest were amplified with NEBNext Multiplex Oligos for Illumina (New England Biolabs, E7335L or E7600S) by NEBNext UltraII Q5 DNA polymerase (New England Biolabs, M0544S) following the manufacturer's instruction to generate sequencing libraries. Qualified libraries were pooled and sequenced on Illumina Hiseq X or NovaSeq platform by Mingma Technologies Company.
Input sequencing
For the input library preparation, unlabeled genomic DNA was sheared by sonication. Purified DNA fragments (5 ng for each sample) were treated with the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, E7103L) according to the manufacturer's instructions. After PCR amplification, input libraries were pooled and sequenced on Illumina Hiseq X by Mingma Technologies Company.
Data processing
Paired-end sequencing reads were directly aligned to reference genome hg19 using BWA MEM (version 0.7.17) with default parameters, then Picard MarkDuplicates (version 2.25.0) was used to remove the duplicates. Next, function multiBamSummary in deeptools with default parameters in bins mode was used for counting reads, then Pearson correlation coefficients were calculated using cor, the built-in function of R, to verify reproducibility as well as robustness. The deduped reads in BAM format were further filtered and converted into BED format with Python scripts. The records with the base guanine at 1 nt upstream of the read start (or the end for reads mapped to negative strand) were selected and re-converted into BAM format using the bedtobam function in the bedtools (version 2.29.2). To facilitate downstream analysis, we merged input samples using Picard MergeSamFiles. Lastly, the bamCoverage function in the deeptools (version 3.5.0) (29) was used to generate predicted-OG density BigWig files with the following parameters: ‘–normalizeUsing RPKM’. All downstream analyses only considered common chromosomes including chr1-22 and chrX.
Data normalization
In this study, two methods were used to calculate the relative enrichment of OG. One was called local normalization (1). Within a given interval (e.g. chr1:100–150), the RPKM value of the experimental group was divided by that of the control group within the same interval. The regions where the read count was zero were removed before normalization. This method was used in the analysis of chromatin state and the relationship between OG and GC. The other method was to divide the values of the experimental group by the average value of the control group in a certain interval type. Unless otherwise stated, the second approach was used in this study (2).
 |
(1) |
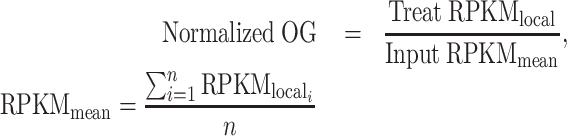 |
(2) |
Background GC content and G content
We made the atlas of background GC and G contents. GC and G contents were assessed at 50 bp bins. We calculated GC and G content and rounded it to a two-digit percentage for each bin. Finally, the GC and G ratios were averaged over bins. For the regions in different chromatin states, we calculated the GC/G content directly by dividing the total counts of GC/G by the total length for each region.
We used the computeGCBias function or a modified version in the deeptools to investigate the relationship between OG and GC content or OG and G content, respectively. In brief, we took random samples from the genome with a window of 1 kb and calculated the GC/G content and normalized OG signal on the positive or negative strands for each window. In this analysis, we only included the regions with GC content 0.2–0.7 or G content 0.1–0.35.
Chromatin state
Chromatin state combined segmentations produced by chromHMM and Segway of HeLa cells were retrieved from UCSC at https://genome.ucsc.edu/cgi-bin/hgTrackUi?g = wgEncodeAwgSegmentation&db = hg19 (30). For a single sample, we counted the number of reads in each chromatin state region, removed regions with zero count in both the experimental and control groups, and then calculated the base-2 logarithm of the normalized OG signal. To compare the relative difference between exogenous-cellular and exogenous-naked samples, we combined biological replicates of each condition and then retained regions that contained reads in both treatment conditions.
Distribution profiles
The protein-coding genes with gene length greater than 10 kb were selected using the gene quantification data of HeLa cells obtained from ENCODE consortium at https://www.encodeproject.org/ under accession number ENCFF622JEX. The OG signals were calculated as mean values in 50bp bins along the genes when metaprofiles were plotted.
R-loops BigWig files were downloaded from GEO under GSE87607 (31). OG profiles and R-loop profiles were produced based on the template strand (TS) or non-template strand (NTS) by Python scripts. G4P-ChIP-seq data and PQSs genome coordinates were derived from GEO under GSE133379 (32). The transcription factor binding sites (TFBS) data, which comprise 109 kinds of TFs, were downloaded from http://bg.upf.edu/group/projects/tfbs/. These binding sites were obtained by matching TF motifs with peak regions of multiple ChIP-seq data from ENCODE (33). DNase I hypersensitive sites (DHS) and corresponding signal files were also from ENCODE under ENCFF775GBM and ENCFF830TNF.
The promoter proximal region was defined as the region of the TSS ±2 kb. Metaprofiles of all grouped regions were achieved by the computeMatrix function in the deeptools with key parameters: ‘–referencePoint center –upstream 3000 –downstream 3000 -bs 50’. Statistical significance of the differences in OG signal levels between them G4+ and G4– TSS surrounding regions were evaluated by one-sided Wilcoxon rank test using 95% confidence interval in R version 3.6.0, and the boxplot was performed using ggplot2. Furthermore, we generated PQS profiles around PQSs within or out of G4P peaks by converting PQSs BED into BigWig format and using deeptools again with the same parameters as above.
IGV snapshot
For aesthetic purposes, we remade the OG signal tracks using makeBW.py with a window of 300 bp and a step size of 10 bp across the genome and computed corresponding OG read density using the following formula:
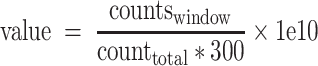 |
RESULTS
Conception and feasibility of CLAPS-seq
In order to sequence OG at single nucleotide resolution, we considered the strategy of Damage-seq which successfully detected the precise positions of UV- and cisplatin-induced DNA adducts by the combination of immunoprecipitation and DNA polymerase stalling (28,34). Unfortunately, in contrast to these damages, OG itself could not block DNA polymerase (7) (see also Figure 1C and Supplementary Figure S1B), while the antibodies might have relatively high cross-reactivity with undamaged dGs (35). A possible solution is conjugating a side-group like biotin to OG which allows the capture by streptavidin-coupled beads and might be able to stop DNA polymerase. Due to the lower redox potential than G, OG can be selectively oxidized by a weak oxidant (e.g. K2IrBr6) and the intermediate can form a covalent adduct with an amine-conjugated biotin (22,27). This chemical labeling reaction was demonstrated to be highly specific and efficient regardless of flanking nucleotides and secondary structures, and has been used for quantification and sequencing of OG (22,27). However, the sequencing method named OG-seq could only map OG at a lower resolution of several hundreds of base pairs (22). We therefore designed the CLAPS-seq (Figure 1A) by optimizing and combining the selective-OG-labeling reaction (22,27) and DNA polymerase stalling strategy (28,34).
Figure 1.

CLAPS-seq method. (A) Schematic of CLAPS-seq. The solid pentacle indicates OG which is labeled with biotin. This labeled OG can block DNA polymerase, generating extension products ending at one nucleotide before OGs (indicated by the hollow pentacle). The blocked extension products are ligated to adaptor 2 (Ad2), PCR-amplified and sequenced to determine the precise locations of OGs. (B) SsDNA containing an OG (OGT) and control ssDNA (NOGT) were biotinylated and purified by streptavidin-coupled beads. Input and pull-down samples were analyzed by denaturing PAGE. Note that the biotinylated-OGT exhibited a shift on the gel (compare lanes 1 and 2). (C) Primer extension by DNA polymerase was performed with the biotinylated or non-biotinylated OGT as templates. Extension products were analyzed by denaturing PAGE. The solid pentacle indicates the position of OG, while the hollow pentacle indicates extension products blocked by biotin-labeled OG. M30 and M31 are markers corresponding to the products ending just before and at the OG site, respectively. M is the degenerated base of A and C which can be incorporated opposite an OG. # indicates a band of undenatured or reannealed dsDNA. The uncropped image is shown in Supplementary Figure S1B. (D) Quality check of representative CLAPS-seq libraries. Two percent of ligation products were amplified by 16 (lanes 1–3) and 19 (lane 4) cycles of PCR. Two necessary reagents for biotinylation, namely potassium hexabromoiridate and amine-tagged biotin probe, were omitted in lanes 1 and 2, respectively. Lanes 3 and 4 show libraries from potassium bromate-treated and untreated HeLa cells, respectively. (E) Base frequencies at sequenced damage sites. The ratios of G were 72.8% in untreated cellular DNA (Endo, endogenous OGs from untreated cells) and 82.4% in potassium bromate-treated sample (Exo-C, exogenous OGs from KBrO3 treated cells.), both of which were much more than 19.4% in the input sample, suggesting CLAPS-seq are specific for both endogenous and exogenous OG damage. Replicates 1 of each condition are shown.
To ensure the integrity of this approach, we first validated that the labeling reaction was specific for OG using synthesized single-stranded (ss) and double-stranded (ds) DNA containing a uniquely located OG (Figure 1B and Supplementary Figure S1A). More importantly, the primer extension assay indicated that high-fidelity DNA polymerase completely and precisely stalled just before the biotin-labeled OG but not before the non-biotinylated OG (Figure 1C and Supplementary Figure S1B), indicating the feasibility to detect the exact position of OG by biotin-labeling and the high-fidelity DNA polymerase.
Measure OG distribution in HeLa cells by CLAPS-seq
We next applied CLAPS-seq to HeLa cells to detect endogenous and chemically-induced OG. We first extracted more than 100 μg of genomic DNA by DNAzol reagent for the purpose of minimizing incidental oxidation during sample processing. It was reported that the extent of incidental oxidation was negatively correlated with the amount of obtained DNA if it was less than 100 μg (36), while the use of DNAzol could reduce spurious OGs (37). As confirmed by ELISA, the DNAzol-extracted DNA possessed a low level (<1/106 nt) of endogenous OGs (Supplementary Figure S1C) which was comparable with reported values (11). Biotin-labeling was performed immediately after DNA extraction, to limit the possibility of oxidation before labeling. Notably, incidental oxidation after biotinylation would not interfere with subsequent affinity purification, thus should have no effect on the final sequencing results. After chemical labeling, genomic DNA was sheared and ligated to the first adaptor as described previously (28). Then fragments conjugated to biotin were purified with streptavidin-coupled beads. The biotin-streptavidin interaction is so strong that we could use harsh wash condition to remove unspecific binding and perform on-bead primer extension without eluting DNA from the beads. Extension products were dissociated from templates by alkaline denaturation and ligated to the second adaptor, followed by PCR amplification to obtain libraries for sequencing (Figure 1A).
A quality check of CLAPS-seq libraries shows that both selective oxidant and biotin-probe are essential for producing sequencing libraries, and the yields of libraries are in accord with OG levels in the samples (Figure 1D, Supplementary Figure S1C), indicating CLAPS-seq is specific for OG damage. As in Damage-seq (28), the OG site should be the nucleotide adjacent to the 5′ end of Read 1 mapped to the genome. As shown in Figure 1E and Supplementary Figure S2B, G is highly enriched at sequenced damage sites in all samples. It is noteworthy that the ratio of Gs at sequenced damage sites provides a simple way to evaluate the specificity of CLAPS-seq. The average G ratio in the human genome is about 20% (38), in other words, G : (A + C + T) ratio is about 20%/(1–20%) = 0.25, which could be loosely thought of as the ratio of unspecific G to (A + C + T) at sequenced damage sites, then the ratios of unspecific and specific Gs in all Gs can be calculated. For instance, if the G ratio is 70% and the sum of A + C + T is 30%, the unspecific Gs should be about 30% × 0.25 = 7.5%, therefore the specific Gs (from OGs) should occupy about 1–7.5/70 ≈ 89% of all Gs at sequenced damage sites. Indeed, the ratio of G at sequenced damage sites ranged from nearly 70% to more than 82% in all CLAPS-seq samples, thus about 89% to 95% of Gs at sequenced damage sites were from genuine OGs, demonstrating that CLAPS-seq is able to specifically detect both exogenous and endogenous OGs at single nucleotide resolution.
Local sequence context of OGs
The susceptibility of Gs to be oxidized is determined by their ionization potential, i.e. Gs with lower ionization potential are easier to be oxidized, and vice versa. Theoretical studies revealed that the ionization potential of G bases would be affected by neighboring bases. The 5′ G in GG dinucleotides has the lowest ionization potential, while Gs in the context of NGT and NGC have apparently higher ionization potential (Supplementary Figure S3A) (39,40). In order to investigate the effect of flanking bases, the relative frequencies of each tri-nucleotide with an OG in the middle were calculated (Figure 2A and Supplementary Figure S3B-F). Consistent with theoretical calculation, NGG was enriched while NGT was depleted in all samples. However, the extents of enrichment and depletion were more obvious in KBrO3-treated samples, and NGC was only depleted in KBrO3-treated samples. Notably, the 5′ adjacent base of OG was also enriched for G and depleted for C, to a much less extent. The neighboring nucleotides on both sides of OG preferred G, suggesting the relative OG level should be elevated in G-rich regions. However, as shown in Figure 2B and Supplementary Figure S4A, OG frequency peaked in the regions of ∼50% GC content and decreased drastically when GC content increased to 60–70% in all samples. A similar pattern was reported by other groups (24,41). As the correlation between G frequency in ssDNA and GC content in corresponding dsDNA is relatively weak in G-rich regions (Supplementary Figure S4C), we calculated OG level as a function of G frequency in ssDNA and found that OG level was highest in regions with ∼27% G and declined slightly when the G frequency kept increasing (Figure 2C and Supplementary Figure S4B). Therefore, the strand-specific information provided by CLAPS-seq revealed a more accurate correlation between OG frequency and local sequence context. These results suggest that local sequence context had complicated impacts on OG distribution.
Figure 2.

Local sequence context of OGs. (A) The relative frequencies (log2 OG/Input) of all 16 tri-nucleotides having a central OG (for CLAPS-seq) or G (for input). (B, C) The relationship between normalized OG signal and (B) GC content in double strands or (C) G content in single strand. The shadow represents mean ± S.E.M. Replicates 1 of endogenous (left panels) and exogenous-cellular samples (right panels) are shown.
The influence of chromatin states and genome accessibility on OG distribution
Genomic distribution of endogenous OGs is determined by damage formation and repair, both of which may be affected by chromatin organization, while the pattern of KBrO3-induced cellular OGs at an early time point should be mainly attributed to damage formation since only a small portion of OGs have been repaired. We analyzed the distribution of endogenous and exogenous cellular OGs (Exo-C) and found that the most open regions, annotated as ‘TSS’, have the highest GC content and lowest OG level among seven chromatin states (Figure 3A and B and Supplementary Figure S5A-C). Similar patterns observed between endogenous and exogenous damage suggest the low level of OGs in ‘TSS’ state is caused by limited damage formation. Intriguingly, KBrO3-treated naked DNA (Exo-N) which are not impacted by chromatin compaction show similar OG local sequence features (relative tri-nucleotides frequencies and local GC/G content, Supplementary Figure S2B and S3E-F) and distribution among chromatin states (Figure 3C and D and Supplementary Figure S5D) with Exo-C, indicating the suppressed damage formation in ‘TSS’ regions should be attributed to DNA itself, i.e. specific sequence and secondary structures, which are examined below.
Figure 3.

OG distribution across different chromatin states. (A–C) Relative OG levels and corresponding GC contents (blue) of 7 chromatin states in (A) Endo, (B) Exo-C and (C) Exo-N samples. Replicates 1 are shown. (D) The ratios of Exo-C OGs to Exo-N OGs in seven chromatin states. Biological replicates of each condition were combined for analysis.
Another factor that may influence OG distribution is genome accessibility which can be measured by DNase-seq. Thus, we checked OG distributions around DNase I hypersensitive sites (DHSs). Since OGs were strongly decreased in the ‘TSS’ state (Figure 3), DHSs were separated into two classes based on their distance from TSSs. For promoter-distal DHSs (DHSs >2 kb from TSSs), endogenous OG signals peaked at the center and dropped moderately at flanking regions, largely consistent with the GC content (Figure 4A and Supplementary Figure S6A and B). In contrast, for the GC-enriched promoter-proximal DHS regions (DHSs within the TSSs ± 2 kb regions), endogenous OGs decreased broadly with a weak peak at the center (Figure 4B and Supplementary Figure S6A and B). As the pattern of endogenous OGs was determined by both damage formation and repair, we explored the impact of genome accessibility on damage formation by comparing OG distribution in KBrO3-treated cellular and naked DNA which should be hardly affected by repair. Surprisingly, the pattern of OGs in Exo-N samples was similar to endogenous damage, whereas the peaks at the centers of both proximal and distal DHSs were lost in Exo-C samples, suggesting the inhibition of OG formation in vivo at the center of DHSs (Figure 4C and D and Supplementary Figure S6C and D). It was reported that the binding of transcription factors (TFs) at DHS centers could inhibit the repair of UV-induced DNA lesions due to the reduced accessibility to the protein-bound DNA (33). Thus we hypothesized that TF binding also inhibit OG formation at DHS centers by blocking the access of oxidants.
Figure 4.
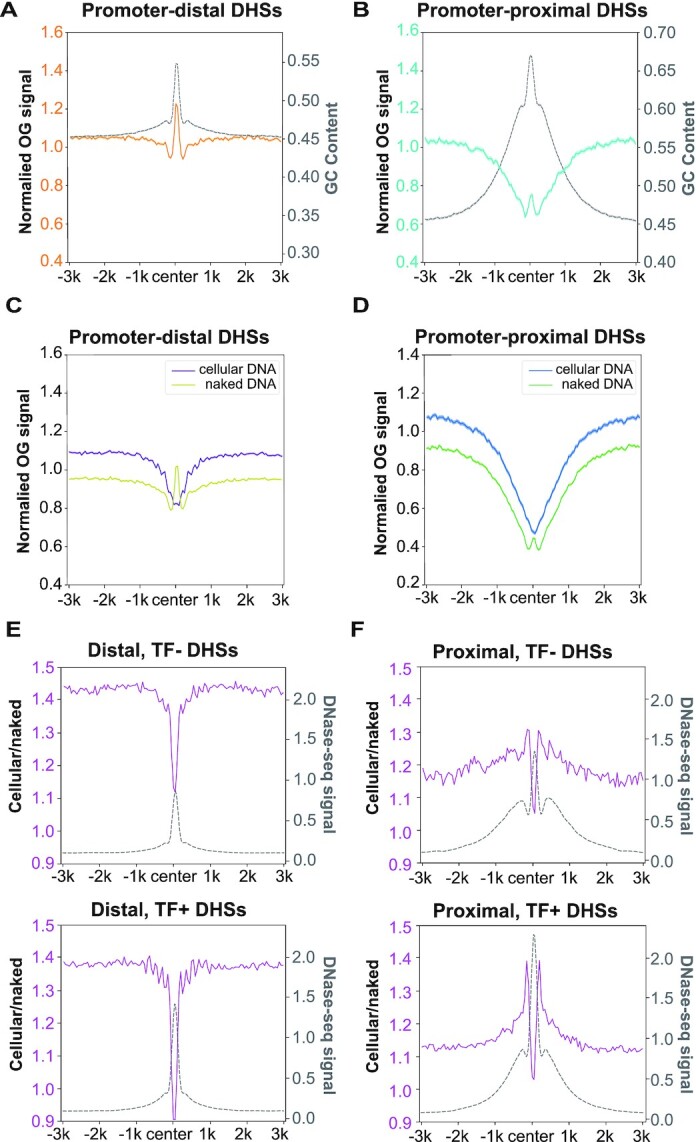
The influence of genome accessibility on OG distribution. (A, B) Relative OG levels of endogenous OGs centred in (A) promoter-distal DHSs (n = 69 327) and (B) promoter-proximal DHSs (n = 26 391) were plotted alongside the corresponding GC contents (gray). (C, D) Relative OG levels of Exo-C and Exo-N samples centred in (C) promoter-distal DHSs and (D) promoter-proximal DHSs. For A–D, replicates 1 are shown and the shadow represents mean ± S.E.M. (E, F) The ratios of Exo-C OGs to Exo-N OGs centred in (E) promoter-distal DHSs and (F) promoter-proximal DHSs alongside the relative DNase-seq signals (gray). Top panels (TF–): DHSs non-overlapped with 109 ENCODE-mapped TFBS (n = 31 253 for distal, n = 6527 for proximal); bottom panels (TF+): DHSs overlapped with 109 ENCODE-mapped TFBS (n = 38 074 for distal, n = 19 864 for proximal). For E and F, biological replicates of each condition were combined for analysis.
To test TFs inhibition hypothesis, both proximal and distal DHSs were further divided into TF+ and TF– DHSs based on HeLa cell ChIP-seq data of 109 common TFs from ENCODE project (42). As shown in Figure 4E and F, the dip of Exo-C/Exo-N ratio at DHS centers was more prominent for TF + DHSs, consistent with our hypothesis. A weaker suppression at TF- DHS centers were also observed, which might be explained by the fact that the peaks of ENCODE ChIP-seq data could not cover all transcription factor binding sites. Alternatively, other events occurring at DHSs, e.g. bidirectional transcription at promoters and enhancers, might also affect OG distribution. Furthermore, the Exo-C/Exo-N ratio was raised in the flanking regions of proximal DHSs (two peaks around DHS centers in Figure 4F) but not distal ones (Figure 4E), implying the stimulation of OG formation in those regions. Coincidentally, the flanking regions of proximal DHSs had remarkably higher DNase-seq signal than distal DHSs (Figure 4E and F), suggesting that the higher accessibility of these regions could allow the attack of oxidant to DNA and generate more OGs. Altogether, our results indicated that OG formation would be affected by genome accessibility.
Reduced levels of OG at TSSs and G4s
In light of the non-homogenous distribution of OG, we looked into OG distribution along individual genes. The screenshots of VEGFA and SESN2 genes clearly show less OGs around the TSS under all conditions (top panels of Figure 5A and Supplementary Figure S7A). Metagene analysis supports the same conclusion that OGs are depleted around TSSs. In addition, a weaker drop is also seen near the TES (Figure 5B and Supplementary Figure S7B-E). However, this could be explained by the dip of G content near TESs. In contrast, the TSS-flanking regions are enriched in G residues (Figure 5C). Therefore, we investigated OG formation in TSS-flanking regions in more detail. As shown in Figure 5D and Supplementary Figure S7F–I, the non-template strands (NTS) have more OGs than template strands (TS) in the downstream regions of TSSs, coinciding with the G frequencies in the same regions (Figure 5E). More importantly, OGs on each strand show a nadir whose center is located on each side of TSSs (Figure 5D and Supplementary Figure S7F–I). As similar patterns are seen in all samples including endogenous damage and exogenous damage on cellular and naked DNA, we hypothesized that it is caused by DNA secondary structures. R-loop and G4 are two common secondary structures enriched around TSSs, both of which exhibit two peaks which virtually overlap with the valleys of OGs (Figure 5F) (31,32). However, since the peaks of R-loops on both strands have few overlaps and are more asymmetric than the patterns of OGs and G4s, we focused on the association between G4s and OGs. The overlapping of G4 signals and OG depletion near TSSs was also observed in individual genes (bottom panels of Figure 5A and Supplementary Figure S7A). Intriguingly, the depletion of OGs near the TSS of VEGFA gene is mainly at the region downstream of TSS, which is slightly different from the OG meta-profile but coincident with the peaks of G4P-ChIP-seq that characterized G4s by an artificial G4 probe (G4P) (Figure 5A, bottom panel). G4 structures in fact have been reported to inhibit OG formation in vitro (43). Therefore, G4 is a candidate responsible for the depletion of OG around TSSs.
Figure 5.

OG distribution along genes. (A) Top panel, genome browser screenshots of strand-specific OG distributions around the VEGFA gene. Bottom panel, zoomed-in screenshots of OG distributions in the TSS surrounding region alongside G4P peaks and PQSs. Replicates 1 are presented. The G4 distribution measured by G4P-ChIP-seq and strand-specific PQS data were obtained from (32). (B) Metaprofiles of OGs on both strands of all analyzed genes and 2 kb upstream and downstream. Replicates 1 are presented. (C) G content profile of both strands over the same gene loci. (D) Metaprofiles of OGs of both strands around TSSs. Results from replicates 1 are presented. (E) G content profile of both strands around TSSs. For B–E, TS: template strand (blue); NTS: non-template strand (red). The shadow represents mean ± S.E.M. (F) G4P-ChIP-seq (black) (32) and RR-ChIP-seq (blue and red for two strands) (31) signals around TSSs. RR-ChIP-seq mapped the RNA strands of R-loops.
In order to test this possibility, OG patterns around TSSs with or without G4P peaks were compared. As shown in Figure 6A and B and Supplementary Figure S8A and B, there are significantly fewer OGs around TSSs with G4P peaks than those without G4P peaks in all samples, although both types of TSSs are G-rich (Supplementary Figure S8C). Next, OGs around promoter-distal G4s were checked to exclude the impact of other factors at TSSs. Interestingly, OGs are reduced at both promoter-proximal G4s and promoter-distal G4s under all conditions despite their different GC contents (Figure 6C and Supplementary Figure S8D and E). Therefore, OGs are depleted at G4s, which should be, at least in part, responsible for the reduced OGs around TSSs.
Figure 6.

G4 structure reduces OG occurrence. (A) Metaprofiles of OG around TSSs with G4P peaks (G4+, n = 7173) and without G4P peaks (G4–, n = 7140). The shadow represents mean ± S.E.M. (B) Quantification of OGs in G4+ and G4– TSS surrounding regions (±500 bp). Endo, P value < 2.2e–88; Exo-C, P value < 3.5e–251; Exo-N, P value < 5.0e–228. (C) Normalized OG signals around promoter-proximal G4P peaks (n = 12 311) and promoter-distal G4P peaks (n = 5476). (D) OG distributions around PQSs within G4P peaks (G4-PQSs, n = 31 623) and out of G4P peaks (non-G4-PQSs, n = 1 474 730). Replicates 1 are presented for A–D. (E) The profiles of PQSs around G4-PQSs and non-G4-PQSs. (F) The model illustrating the relationship between OG formation and G4 regulation. G4s can prevent OG formation to protect genome stability. On the other hand, non-G4-PQSs are hotspots of OGs which can stimulate the transformation of duplex to G4 and regulate downstream transcription through OGG1 and APE1.
Furthermore, we wondered whether the decrease of OG at G4P peaks was due to the secondary structure or underlying sequence, i.e. PQS. Accordingly, OG distributions around PQSs overlapping (G4-PQSs) or non-overlapping (non-G4-PQSs) with G4P peaks were compared. In striking contrast to G4-PQSs, there was a remarkable peak of OGs at non-G4-PQSs (Figure 6D and Supplementary Figure S8F–I), indicating that the G4 structure, not the underlying sequence, is the factor responsible for OG depletion. It is worth noting that OGs form a sharp peak at non-G4-PQSs while suppression of OGs around G4-PQSs spread to a wide range. This might be explained by the fact that one G4P peak might contain more than one PQS, as there are more G4-PQSs than G4P peaks (31 623 versus 17 787) in HeLa cells (32). Therefore, we plotted PQSs around G4- and non-G4-PQSs and found that the patterns of surrounding PQSs coincide with the patterns of OGs around G4- or non-G4-PQSs, respectively (Figure 6E). In conclusion, these results reveal that non-G4-PQSs are hotspots of OG due to their G-rich property, while the G4-PQSs, which are also G-rich (Supplementary Figure S8J), are protected from this damage.
DISCUSSION
Comparison of CLAPS-seq and other OG sequencing methods
A unique challenge for OG sequencing is the incidental oxidation after sample collection. Besides choosing a proper DNA extraction protocol (37), acquiring enough DNA (36) and adding anti-oxidant (11), reducing processing steps and time before damage recognition or labeling can help minimize this effect in theory. In this respect, CLAPS-seq and OGG1-AP-seq (24) performed damage recognition immediately after DNA extraction by directly labeling with biotin, or converting OGs to AP sites which were then biotinylated with ARP, respectively, thus these methods should suffer from less incidental oxidation. In contrast, OG-seq (22), click-code-seq (25), enTRAP-seq (23) and OxiDIP-seq (21) all sheared DNA before damage recognition/labeling. OxiDIP-seq had an extra heat-denaturing step (5 min at 95 °C) before immunoprecipitation (21). These manipulations can induce additional oxidative damage and compromise final results.
Similar to other sequencing techniques, specificity and resolution are two key parameters for OG sequencing. Specificity is particularly important due to the low abundance of OG (1). For CLAPS-seq, selective biotinylation was demonstrated to be efficient on OG independent of flanking nucleotides and secondary structures, but not on undamaged dG, 8-oxo-2′-deoxyadenosine, 5-hydroxy-2′-deoxyuridine or 5-hydroxy-2′-deoxycytidine (22,27). Nonetheless, each method including CLAPS-seq might inevitably capture some non-specific DNA by unspecific binding, side reaction, etc. Moreover, as claimed by Poetsch et al. (24), peak calling might not be appropriate for OG sequencing, because OGs occur widely across the genome with few hot spots. Therefore, it is difficult to assess and compare the specificity of low-resolution methods. Although other single nucleotide resolution sequencing methods for AP sites (44) and single strand breaks (45) could also be used for OGs if combined with OGG1 or FPG (and APE1), to the best of our knowledge, only click-code-seq had detected OGs at single-nucleotide resolution in yeast (25). Notably, click-code-seq used a G-derivate nucleotide analog to fill the gap generated by FPG and APE1, thus the detected damage sites were nearly exclusively Gs (25). Thus, reads generated of click-code-seq from OGs and nonspecific dGs (from unspecific reaction or pre-exist AP sites or nicks) could not be distinguished or estimated, so the ratio of specific Gs could not be calculated as described above. In summary, CLAPS-seq can map endogenous levels of genomic OGs at single-nucleotide resolution in human cells with evaluable high specificity and minimal incidental oxidation. The properties of recent OG sequencing methods including CLAPS-seq are summarized in Supplementary Table S2. It is worth noting that differences in incidental oxidation, specificity and resolution might be responsible for the discrepancies between OG sequencing studies.
The impacts of neighboring nucleotides on OG formation
Theoretical calculation suggested that the 5′ G of a G stretch has a lower ionization potential due to π-stacking, thus it should more readily be oxidized (39,40). Wu et al. (25) and we showed this preference in yeast and human cells, respectively. Furthermore, we find that endogenous and KBrO3-induced OGs had different features for neighboring nucleotides. The exogenous damage showed stronger bias than endogenous damage, and the relative frequency of NGC decreased to a similar level of NGT for exogenous damage (Figure 2A and Supplementary Figure S3B-F), which was consistent with the similar higher ionization potential of NGC and NGT (40). The different patterns between endogenous and exogenous damage may be attributed to the different sources of oxidant such that endogenous OGs are mainly caused by ROS like HO• and CO3•– (2,46) whereas exogenous OGs are induced by KBrO3 through a different mechanism (47). An alternative explanation is that the repair system (mainly BER through OGG1 in human cells) may remove OGs with different efficiency according to the sequence context (48). Further experiments are needed to clarify these possibilities.
Impacts of chromatin states and genome accessibility on OG distribution
The genomic distribution of endogenous OGs is controlled by the balance between damage formation and repair (28,34). We find that the distributions of endogenous and exogenous OGs were similar in different chromatin states (Figure 3 and Supplementary Figure S5), arguing that any impact of chromatin organization on OG repair was not much stronger than that on damage formation. More experiments are required to explore how chromatin compaction affects OG repair.
The uneven OG distribution around DHSs indicated genome accessibility could affect OG formation. The valley of Exo-C to Exo-N ratio at DHS centers suggests a lower accessibility which might be, at least partially, caused by TF binding. Indeed, it was reported that TF binding could suppress cisplatin-induced damage (28). Alternatively, depleted OGs in treated cellular DNA might be due to enhanced repair at DHS centers. However, if the enhanced repair was the real reason, reduced damage at DHS centers should also be observed in endogenous samples as repair was continuously operating in untreated cells. In fact, small peaks but not dips are seen for endogenous DNA damage at DHS centers, implying the absence of enhanced repair in this case. Furthermore, elevated Exo-C to Exo-N ratio was seen in the promoter-proximal DHS-flanking regions (about ±100–500 bp from DHS centers) which have moderate DHS signals, and should be more accessible than more distant regions. If the repair was the determinant for exogenous cellular OG distribution, a reversed pattern of Exo-C/Exo-N ratio would be observed in these promoter-proximal DHS-flanking regions, as high accessibility can stimulate repair. Therefore, the distribution of Exo-C damage was mainly determined by damage formation rather than repair. In summary, these results suggest that high genome accessibility could stimulate damage formation in oxidant-treated cells.
Depletion of OGs at G-quadruplex sites
The most conspicuous feature of CLAPS-seq profiles is the valley at promoters, which was also reported by Poetsch et al. and Wu et al. (24,25). Although OxiDIP-seq revealed enrichment of OGs at promoters, analysis at higher resolution uncovered a dip within a narrow region around TSSs where the GC content is higher than adjacent regions, generating a bimodal pattern (41). OG-seq reported an increase of OGs at promoters (22), however, reanalysis of the data by Poetsch et al. and Gorini et al. exhibited opposite conclusions (24,41). Therefore, OG sequencing by different methods all revealed that TSSs regions that possess extremely high GC content have fewer OGs than flanking regions. More importantly, we observed the same pattern in KBrO3-treated naked DNA, excluding the impacts of many cellular factors such as repair, transcription and protein binding. Among DNA secondary structures and modifications which might be kept during DNA extraction, we focused on G4 because it overlaps closely with TSSs and its distribution pattern around TSSs is complementary to that of OGs. It was reported that G4 structures are stable and have a slow unfolding kinetics in vitro (49). Although whether G4 structures changed during our DNA extraction and in vitro oxidation steps was unclear, we did observe the depletion of OGs in G4s but not non-G4-PQSs in treated naked DNA.
Besides the complementary distribution between G4s and OGs around TSSs, the effect of G4 on OG distribution was further evidenced by two observations: (i) TSSs without G4 had more OGs; 2) OGs were also depleted at promoter-distal G4s. Moreover, in HeLa cells about half of TSSs have G4P peaks in their flanking regions. Therefore, G4s appear to play an important role in the low level of OGs around TSSs. The offset of OG nadirs on two strands may be ascribed to the split peaks of G4s, however, it is unknown whether the distribution of G4s around TSSs are strand asymmetric, because the data of G4P-ChIP-seq lacks strand information (32). On the other hand, as another common DNA secondary structure at promoters, R-loops may also be involved in the low level of OGs around TSSs. Although R-loops have a different pattern than G4s and OGs, G4s and R-loops usually colocalize around TSSs as both of them are positively correlated with transcription (31,32,50). Therefore, it is difficult to distinguish the contributions of these two secondary structures.
It is worth noting that some studies reported an enrichment of OGs at G4s in general (22,24,41). However, this is not contradictory to our conclusion, since they all used ‘predicted’ G4 sites which have the potential to form G4, in other words, some kind of PQSs for analysis. Recent G4 in situ capturing and sequencing (G4P-ChIP-seq) data we adopted presented an in vivo G4 pattern distinct from PQSs (32). More importantly, we observed an increase of OGs at non-G4-PQSs, while in sharp contrast G4s were devoid of OG. Considering the depletion of OGs at TSSs in those studies and extensive overlap of G4s and promoters, it is reasonable to suspect that OGs were also depleted at G4-PQSs in their results. However, we could not test this hypothesis as G4 distribution is cell type specific, and G4P-ChIP-seq was not performed in cell lines or organs used in those studies (32).
G4 has been proposed as a sensor of cellular oxidation stress since the repair intermediate of OGs, i.e. AP sites, can stabilize G4 structure and regulate downstream gene expression (15,16). However, it is dangerous for cells to use OG which is a mutagenic modification as a regulator, especially at G4 sites, since OGG1 can only efficiently recognize OGs paired with dC in dsDNA (8). Indeed, OG can destabilize G4 and lead to the duplex structure in vitro (51,52). Therefore, it is hypothesized that OG could induce the transformation of G4 to duplex, then it could be incised by OGG1, and the resulting AP would be bound by APE1 and promote the formation of G4 (15,16). Our results show that G residues within G4 structures are relatively poor substrates for OG formation. On the other hand, non-G4-PQSs are hotspots of OG, which can be recognized and incised by OGG1. The resulting AP site can stimulate the formation of G4 and regulate downstream transcription. In other words, non-G4-PQSs, but not the existing G4s, might play the role of a sensor in response to oxidation pressure (Figure 6F). More experiments are required for exploring the relationship among OG, G4 and transcription.
In conclusion, our results provide a new view of how to coordinate the epigenetic role and mutagenic property of OG at G4 sites, indicating that CLAPS-seq is a powerful tool for studying the biological role of OG. Moreover, CLAPS-seq has the potential to be applied to other lesions like AP sites by choosing different labeling reactions.
DATA AVAILABILITY
Sequencing data have been submitted to the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE181312. The codes are publicly available at https://github.com/Jyyin333/CLAPS-seq.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Hongzhou Gu (Fudan University) and Dr Feilong Meng (University of Chinese Academy of Sciences) for careful reading of the manuscript and valuable suggestions.
Contributor Information
Jiao An, Shanghai Fifth People's Hospital, Fudan University, and Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
Mengdie Yin, Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
Jiayong Yin, Institute of Pediatrics and Department of Hematology and Oncology, Children's Hospital of Fudan University, and Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
Sizhong Wu, Shanghai Fifth People's Hospital, Fudan University, and Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
Christopher P Selby, Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina, Chapel Hill, NC 27599-7260, USA.
Yanyan Yang, Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina, Chapel Hill, NC 27599-7260, USA.
Aziz Sancar, Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina, Chapel Hill, NC 27599-7260, USA.
Guo-Liang Xu, Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
Maoxiang Qian, Institute of Pediatrics and Department of Hematology and Oncology, Children's Hospital of Fudan University, and Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
Jinchuan Hu, Shanghai Fifth People's Hospital, Fudan University, and Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism (Ministry of Science and Technology), Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China (NSFC) [21807013, 31870804, 81973997, 82170157]; Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning [2019]; innovative research team of high-level local university in Shanghai (to M.Q. and J.H.); Shanghai Outstanding Young Talent Program [2019 to J.H.]; National Institutes of Health [GM118102, ES033414 to A.S.]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of Interest. None declared.
REFERENCES
- 1. Cadet J., Davies K.J.A., Medeiros M.H., Di Mascio P., Wagner J.R.. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med. 2017; 107:13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yu Y., Cui Y., Niedernhofer L.J., Wang Y.. Occurrence, biological consequences, and human health relevance of oxidative stress-induced DNA damage. Chem. Res. Toxicol. 2016; 29:2008–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cadet J., Douki T., Gasparutto D., Ravanat J.L.. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat. Res. 2003; 531:5–23. [DOI] [PubMed] [Google Scholar]
- 4. Kuznetsova A.A., Knorre D.G., Fedorova O.S.. Oxidation of DNA and its components with reactive oxygen species. Russ. Chem. Rev. 2009; 78:659–678. [Google Scholar]
- 5. Steenken S., Jovanovic S.V.. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997; 119:617–618. [Google Scholar]
- 6. Shibutani S., Takeshita M., Grollman A.P.. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991; 349:431–434. [DOI] [PubMed] [Google Scholar]
- 7. Gorini F., Scala G., Cooke M.S., Majello B., Amente S.. Towards a comprehensive view of 8-oxo-7,8-dihydro-2′-deoxyguanosine: highlighting the intertwined roles of DNA damage and epigenetics in genomic instability. DNA Repair (Amst.). 2021; 97:103027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dizdaroglu M., Coskun E., Jaruga P.. Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutat. Res. Rev. Mutat. Res. 2017; 771:99–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abbotts R., Wilson D.M.. Coordination of DNA single strand break repair. Free Radical. Biol. Med. 2017; 107:228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J., Yandava C.N.et al.. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018; 359:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. European Standards Committee on Oxidative DNA Damage (ESCODD) Measurement of DNA oxidation in human cells by chromatographic and enzymic methods. Free Radic. Biol. Med. 2003; 34:1089–1099. [DOI] [PubMed] [Google Scholar]
- 12. Ravanat J.L., Douki T., Duez P., Gremaud E., Herbert K., Hofer T., Lasserre L., Saint-Pierre C., Favier A., Cadet J.. Cellular background level of 8-oxo-7,8-dihydro-2′-deoxyguanosine: an isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis. 2002; 23:1911–1918. [DOI] [PubMed] [Google Scholar]
- 13. Fleming A.M., Burrows C.J.. Interplay of guanine oxidation and G-quadruplex folding in gene promoters. J. Am. Chem. Soc. 2020; 142:1115–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Visnes T., Cazares-Korner A., Hao W., Wallner O., Masuyer G., Loseva O., Mortusewicz O., Wiita E., Sarno A., Manoilov A.et al.. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science. 2018; 362:834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roychoudhury S., Pramanik S., Harris H.L., Tarpley M., Sarkar A., Spagnol G., Sorgen P.L., Chowdhury D., Band V., Klinkebiel D.et al.. Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:11409–11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fleming A.M., Ding Y., Burrows C.J.. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spiegel J., Adhikari S., Balasubramanian S.. The structure and function of DNA G-quadruplexes. Trends Chem. 2020; 2:123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rhodes D., Lipps H.J.. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015; 43:8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rodriguez R., Miller K.M., Forment J.V., Bradshaw C.R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S.P.. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012; 8:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei D., Parkinson G.N., Reszka A.P., Neidle S.. Crystal structure of a c-kit promoter quadruplex reveals the structural role of metal ions and water molecules in maintaining loop conformation. Nucleic Acids Res. 2012; 40:4691–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amente S., Di Palo G., Scala G., Castrignano T., Gorini F., Cocozza S., Moresano A., Pucci P., Ma B., Stepanov I.et al.. Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019; 47:221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ding Y., Fleming A.M., Burrows C.J.. Sequencing the mouse genome for the oxidatively modified base 8-oxo-7,8-dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017; 139:2569–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang Y., Zou P.. Genome-wide mapping of oxidative DNA damage via engineering of 8-oxoguanine DNA glycosylase. Biochemistry. 2020; 59:85–89. [DOI] [PubMed] [Google Scholar]
- 24. Poetsch A.R., Boulton S.J., Luscombe N.M.. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 2018; 19:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu J., McKeague M., Sturla S.J.. Nucleotide-resolution genome-wide mapping of oxidative DNA damage by click-code-Seq. J. Am. Chem. Soc. 2018; 140:9783–9787. [DOI] [PubMed] [Google Scholar]
- 26. Poetsch A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020; 18:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xue L., Greenberg M.M.. Facile quantification of lesions derived from 2′-deoxyguanosine in DNA. J. Am. Chem. Soc. 2007; 129:7010–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu J., Lieb J.D., Sancar A., Adar S.. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:11507–11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoffman M.M., Ernst J., Wilder S.P., Kundaje A., Harris R.S., Libbrecht M., Giardine B., Ellenbogen P.M., Bilmes J.A., Birney E.et al.. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013; 41:827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan-Wong S.M., Dhir S., Proudfoot N.J.. R-loops promote antisense transcription across the mammalian genome. Mol. Cell. 2019; 76:600–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng K.W., Zhang J.Y., He Y.D., Gong J.Y., Wen C.J., Chen J.N., Hao Y.H., Zhao Y., Tan Z.. Detection of genomic G-quadruplexes in living cells using a small artificial protein. Nucleic Acids Res. 2020; 48:11706–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sabarinathan R., Mularoni L., Deu-Pons J., Gonzalez-Perez A., Lopez-Bigas N.. Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature. 2016; 532:264–267. [DOI] [PubMed] [Google Scholar]
- 34. Hu J., Adebali O., Adar S., Sancar A.. Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yin B., Whyatt R.M., Perera F.P., Randall M.C., Cooper T.B., Santella R.M.. Determination of 8-hydroxydeoxyguanosine by an immunoaffinity chromatography-monoclonal antibody-based ELISA. Free Radic. Biol. Med. 1995; 18:1023–1032. [DOI] [PubMed] [Google Scholar]
- 36. Badouard C., Menezo Y., Panteix G., Ravanat J.L., Douki T., Cadet J., Favier A.. Determination of new types of DNA lesions in human sperm. Zygote. 2008; 16:9–13. [DOI] [PubMed] [Google Scholar]
- 37. Mangal D., Vudathala D., Park J.H., Lee S.H., Penning T.M., Blair I.A.. Analysis of 7,8-dihydro-8-oxo-2′-deoxyguanosine in cellular DNA during oxidative stress. Chem. Res. Toxicol. 2009; 22:788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piovesan A., Pelleri M.C., Antonaros F., Strippoli P., Caracausi M., Vitale L.. On the length, weight and GC content of the human genome. BMC Res Notes. 2019; 12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugiyama H., Saito I.. Theoretical studies of GC-specific photocleavage of DNA via electron transfer: Significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA. J. Am. Chem. Soc. 1996; 118:7063–7068. [Google Scholar]
- 40. Senthilkumar K., Grozema F.C., Guerra C.F., Bickelhaupt F.M., Siebbeles L.D.A.. Mapping the sites for selective oxidation of guanines in DNA. J. Am. Chem. Soc. 2003; 125:13658–13659. [DOI] [PubMed] [Google Scholar]
- 41. Gorini F., Scala G., Di Palo G., Dellino G.I., Cocozza S., Pelicci P.G., Lania L., Majello B., Amente S.. The genomic landscape of 8-oxodG reveals enrichment at specific inherently fragile promoters. Nucleic. Acids. Res. 2020; 48:4309–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sabarinathan R., Mularoni L., Deu-Pons J., Gonzalez-Perez A., Lopez-Bigas N.. Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature. 2016; 532:264–267. [DOI] [PubMed] [Google Scholar]
- 43. Merta T.J., Geacintov N.E., Shafirovich V.. Generation of 8-oxo-7,8-dihydroguanine in G-Quadruplexes Models of Human Telomere Sequences by One-electron Oxidation. Photochem. Photobiol. 2019; 95:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu Z.J., Cuesta S.M., van Delft P., Balasubramanian S.. Sequencing abasic sites in DNA at single-nucleotide resolution. Nat. Chem. 2019; 11:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zilio N., Ulrich H.D.. Exploring the SSBreakome: genome-wide mapping of DNA single-strand breaks by next-generation sequencing. FEBS J. 2021; 288:3948–3961. [DOI] [PubMed] [Google Scholar]
- 46. Halliwell B., Adhikary A., Dingfelder M., Dizdaroglu M.. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 2021; 50:8355–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawanishi S., Murata M.. Mechanism of DNA damage induced by bromate differs from general types of oxidative stress. Toxicology. 2006; 221:172–178. [DOI] [PubMed] [Google Scholar]
- 48. Allgayer J., Kitsera N., von der Lippen C., Epe B., Khobta A.. Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence. Nucleic Acids Res. 2013; 41:8559–8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lane A.N., Chaires J.B., Gray R.D., Trent J.O.. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008; 36:5482–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miglietta G., Russo M., Capranico G.. G-quadruplex-R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020; 48:11942–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bielskute S., Plavec J., Podbevsek P.. Impact of oxidative lesions on the human telomeric G-quadruplex. J. Am. Chem. Soc. 2019; 141:2594–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee H.T., Sanford S., Paul T., Choe J., Bose A., Opresko P.L., Myong S.. Position-dependent effect of guanine base damage and mutations on telomeric G-quadruplex and telomerase extension. Biochemistry. 2020; 59:2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been submitted to the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE181312. The codes are publicly available at https://github.com/Jyyin333/CLAPS-seq.