


Abstract
CstR is a persulfide-sensing member of the functionally diverse copper-sensitive operon repressor (CsoR) superfamily. While CstR regulates the bacterial response to hydrogen sulfide (H2S) and more oxidized reactive sulfur species (RSS) in Gram-positive pathogens, other dithiol-containing CsoR proteins respond to host derived Cu(I) toxicity, sometimes in the same bacterial cytoplasm, but without regulatory crosstalk in cells. It is not clear what prevents this crosstalk, nor the extent to which RSS sensors exhibit specificity over other oxidants. Here, we report a sequence similarity network (SSN) analysis of the entire CsoR superfamily, which together with the first crystallographic structure of a CstR and comprehensive mass spectrometry-based kinetic profiling experiments, reveal new insights into the molecular basis of RSS specificity in CstRs. We find that the more N-terminal cysteine is the attacking Cys in CstR and is far more nucleophilic than in a CsoR. Moreover, our CstR crystal structure is markedly asymmetric and chemical reactivity experiments reveal the functional impact of this asymmetry. Substitution of the Asn wedge between the resolving and the attacking thiol with Ala significantly decreases asymmetry in the crystal structure and markedly impacts the distribution of species, despite adopting the same global structure as the parent repressor. Companion NMR, SAXS and molecular dynamics simulations reveal that the structural and functional asymmetry can be traced to fast internal dynamics of the tetramer. Furthermore, this asymmetry is preserved in all CstRs and with all oxidants tested, giving rise to markedly distinct distributions of crosslinked products. Our exploration of the sequence, structural, and kinetic features that determine oxidant-specificity suggest that the product distribution upon RSS exposure is determined by internal flexibility.
INTRODUCTION
Bacterial pathogens persist in the hostile environment of the infected host as a result of adaptation to host-derived stressors and tolerance to broad-spectrum antibiotics that are used to clear infections. Two broadly recognized anti-bacterial host strategies are the induction of oxidative stress, mediated by a myriad of highly reactive, inorganic oxygen- and nitrogen species (ROS and RNS), and copper (Cu) toxicity (1,2). Bacteria upregulate the biogenesis of hydrogen sulfide (H2S) in response to ROS- and antibiotic stress, which enhances persistence in biofilms or in cells (3,4), while adaptation to Cu poisoning involves cytoplasmic Cu(I) sensing by specialized Cu(I)-sensing metalloregulatory proteins (5–7). Cu(I) sensing turns on a transcriptional program to sequester and/or efflux Cu(I) from the cytoplasm of cells. In order for bacteria to deploy H2S and more oxidized H2S-derived reactive sulfur species (RSS) as infection-relevant antioxidants (8,9), cellular H2S/RSS levels must be tightly regulated (10) to avoid H2S poisoning and elevated inorganic RSS which can be bactericidal at high levels (11,12). RSS-sensing transcriptional regulators have evolved to regulate the expression of RSS biogenesis and clearance enzymes known or projected to reduce cellular loads of H2S, RSS and proteome persulfidation, central to the concept of bacterial sulfide/RSS homeostasis (13–18).
We lack a comprehensive understanding of the chemical specificity and scope of allosteric mechanisms in what we now know are ubiquitous RSS sensors that tightly regulate cellular H2S/RSS levels. It is not clear what prevents crosstalk of RSS with metallosensors in cells, and the extent to which RSS sensors exhibit specificity over other oxidants, with the possible exception of the RSS-sensing repressor SqrR (19). Detailed investigations of paralogs in the copper-sensitive operon repressor (CsoR) superfamily of bacterial transcriptional repressors (6,20,21) present an excellent opportunity to elucidate determinants of biological specificity of closely related proteins (22,23). The founding structurally characterized member of the CsoR family is the Cu(I) sensor initially characterized in Mycobacterium tuberculosis (24). Since that time, several crystallographic structures have become available for CsoR family repressors (25), and each is characterized by a disc-shaped D2-symmetric or pseudosymmetric tetrameric architecture consisting of ≈ 90-residue protomers, and exhibit significant pairwise sequence similarities. The CsoR family is known to detect a wide variety of transition metal- and small molecule inducers and comprises other Cu(I) sensors (26), including a second mycobacterial Cu(I) sensor, RicR (27), as well as the Ni(II)/Co(II) sensor RncR from Escherichia coli (28), the Ni(II) sensor InrS from Synechocystis (29), the formaldehyde-sensing repressor FrmR from E. coli (22,23), and the persulfide sensor CstR, now functionally characterized in Staphylococcus aureus and Enterococcus faecalis (13,14,30). Inducer specificity by CsoR-family repressors was originally described in the context of a simple ‘W-X-Y-Z’ motif using the MtCsoR Cu(I) chelate as a template (24), where the first coordination shell ligands around a subunit-bridging, trigonally coordinated Cu(I) define the X (Cys36), Y (His61’) and Z (Cys65’) residue positions (20) (Figure 1A). While insightful, this simple perspective that four residues fully define inducer specificity across a large protein family, engaging in both distinct coordination chemistries and complex cysteine thiol chemistry, has clear limitations.
Figure 1.

CsoR family repressor tetramer architecture and a cartoon representation of asymmetric crosslinked species. (A) Representation of a generic CsoR-family tetramer (each protomer shaded differently) with the WXYZ motif site shown. (B) SpCstR dimer of dimers architecture with each cysteine thiol represented by an S (right). S30 and S59 represent the thiolate sulfurs of residues Cys30 and Cys59, respectively, that correspond to C31 and C60 in SaCstR. (C) Representation of the different oxidation states that can be captured in a mass spectrometry-based kinetic profiling experiment with each cysteine thiol represented by an S. Red line, covalent crosslink of some kind (either di-, tri- or tetrasulfide bond) between Cys residues on opposite protomers, resolvable by ESI-MS. A more schematic representation of SpCstR or SmCstR (one-half of the tetramer) and used in Figures 7 and 8 is shown in the bottom, with the Cys residues indicated in boxes.
CsoR and CstR from Staphylococcus aureus are CsoR-family paralogs that control distinct physiological processes in the same cytoplasm (30). These proteins share 35% sequence identity and 65% similarity, and each contains two conserved cysteine residues in the X and Z positions. SaCsoR recognizes and coordinates Cu(I) with high affinity (KCu = 1018.1 M−1) through the two cysteine residues, C41 (C36 in MtCsoR) and C70 (C65 in MtCsoR), and the Nδ1 atom of His66 (H61 in MtCsoR) in a trigonal S2N coordination geometry (24,30,31). This results in a decrease in DNA binding affinity, leading to transcriptional derepression of the copper efflux genes copA, a P1B-type ATPase Cu(I) efflux pump, and copZ, a Cu(I) chaperone (24,30). In contrast, S. aureus CstR regulates the expression of the cst operon that encodes enzymes responsible for mediating resistance to sulfide toxicity and reacts with the sulfane sulfur of both inorganic and organic per- and polysulfide sulfide donors, e.g., glutathione persulfide (GSSH), forming di-, tri-, and tetrasulfide linkages between neighboring cysteines on both sides of each dimer pair (C31/C60’ and C31’/C60) (13).
Although CstR, like CsoR, binds Cu(I), Cu(I) binding does not drive dissociation of CstR from cst operator DNA in vitro or in vivo (13,30), as it is missing the allosterically important Cu(I)-ligating histidine in the Y-position. In CstRs, loss of this His weakens Cu(I) affinity such that the intracellular Cu(I) concentration is unlikely to rise to a level required to be bound by CstR, due to intracellular regulation by CsoR (31,32). In bona fide Cu(I) sensors, this His interacts with residues in the second coordination shell including the nearly invariant Tyr35 and Glu81 residues, which in MtCsoR were shown to be energetically linked to Cu(I)-mediated allosteric inhibition of DNA binding (24,31). Thus, fine tuning of Cu(I) affinity and response can fully explain why CstR is insensitive to Cu(I) in cells. However, these prior studies provide no insight into reactant specificity of CstRs or why CsoR fails to sense RSS or other reactive electrophile species in the cell despite of harboring a virtually identical dithiol pair.
In this work, we use a comprehensive sequence similarity network (SSN) analysis of >32,000 sequences to identify three distinct clusters of RSS-sensitive regulators. We validate this analysis by establishing that S. pneumoniae D39 SPD_0073, found in cluster 10 of the SSN analysis, is a bona fide CstR, and present its structure. This structure, which is the first of any CstR, adopts the expected dimer-of-dimers architecture characteristic of the CsoR superfamily (21,24). We find however that while both CsoR (26,33) and CstR proteins have long intersubunit Sγ–Sγ distances, a critical determinant in RSS sensing specificity is a far more nucleophilic cysteine in CstRs. Unexpectedly, kinetically resolved, chemical reactivity profiling experiments reveal that the two intersubunit dithiol sensing sites positioned on opposite sides of each dimer within the tetramer (C30/C59’ and C30’/C59; C31 and C60 in SaCstR) are not equivalent (Figure 1B). One dithiol site is characterized by a significantly faster rate of reactivity, which leads to the formation of an asymmetric singly crosslinked dimer, compared to the symmetric doubly-crosslinked dimer (Figure 1C). NMR experiments and molecular dynamics simulations suggest that this asymmetry can be traced to fast internal dynamics in the dithiol site that arise mainly from two solvent exposed loops. This asymmetry characterizes three SSN cluster 10 CstRs, and occurs with both RSS and simple oxidants, but is largely lost in a ‘wedge’ residue substitution (N55A), despite virtually no impact on the structure of the thiol-reduced CstR. We conclude that the rigidity of the dithiol site permits fine-tuning of the reactivity of the dithiol pair that may ultimately dictate the sensor specificity for different cellular oxidants and reactive sulfur species in cells (19).
MATERIALS AND METHODS
Sequence similarity network (SSN) and genomic neighborhood network (GNN)
The EFI-EST (http://efi.igb.illinois.edu/efi-est/) and EFI-GNT (http://efi.igb.illinois.edu/efi-gnt/) web tools were used to generate SSNs and GNNs, respectively. SSNs were generated using the Option B (Families) option of EFI-EST using the Pfam of SpCstR (PF02583; trans_repr_metal). An alignment score of 28 was used to generate the SSN with no minimum or maximum number of residues, and the final network generated was 80% representative (repnode 80), collapsing sequences of 80% identity over 90% of the sequence into a representative node and was visualized using Cytoscape (http://www.cytoscape.org/). The composite FASTA file containing all unique sequences associated with each cluster of interest was used to generate a multiple sequence alignment (MSA) through Jalview, using ClustalO with default parameters with sequences containing long N-terminal and C-terminal extensions on either side of a core region manually removed to facilitate comparison of sequences. MSAs were then uploaded to Skylign (https://skylign.org/) to generate logo plots associated with each SSN cluster (34). The list of all sequences (Supplementary Table S1A) and sequences used to generate the logo plots (Supplementary Table S1B) are provided, the latter arranged in individual clusters with Uniprot IDs associated with biochemically characterized CsoR-family proteins highlighted in each cluster.
Protein purification
Staphylococcus aureus CsoR and CstR and Enterococcus faecalis CstR were purified from His-tagged constructs to homogeneity in a fully reduced form as previously described (13,14,30). E. coli expression plasmids encoding 6x-His-tagged wild-type, C9A and C9A/N55A S. pneumoniae CstR (locus tag SPD_0073) and S. mitis SVGS_061 CstR (locus tag AXK38_00620) (35) were transformed into BL21(DE3) competent cells and allowed to grow overnight at 37°C. This preculture was used to inoculate LB containing 100 μg/ml ampicillin and allowed to grow at 37°C until an OD of 0.6–0.8 was reached. 0.5 mM IPTG was then added, and the growth allowed to continue overnight at 16°C. Cells were harvested by centrifugation for 20 min, 4°C (3,000 × g), resuspended with 20–30 mL/g wet cells of degassed low imidazole buffer (25 mM Tris, 500 mM NaCl, 20 mM imidazole, 10% glycerol, pH 8.0) and sonicated for a total of 5 min with a 2 s on/8 s off pulse sequence. The lysate was clarified by centrifugation (10 min, 17,000 × g). Polyethyleneimine (PEI) was added to the supernatant at a final concentration of 0.2% (v/v) and allowed to stir for 40 min at 4°C. The resulting fraction was centrifuged (10 min, 17,000 × g) and the soluble fraction subjected to ammonium sulfate precipitation (70% w/v) at 4°C (50 min) and the remaining solution centrifuged (10 min, 17,000 × g) and the pellet used for Ni-NTA affinity chromatography. Low imidazole buffer and high imidazole buffer (25 mM Tris, 500 mM NaCl, 500 mM imidazole, 10% glycerol, pH 8.0) were prepared and degassed and TCEP was added to 2.0 mM. An imidazole gradient was used to elute CstRs, peak fractions collected, subjected to TEV cleavage and dialyzed, centrifuged (10 min, 13,000 rpm) or filtered through 0.45 μm filter and the flow-through fractions from the Ni-NTA pooled and subjected to size exclusion chromatography (Superdex 75; GE, Boston, MA, USA) in degassed G75 buffer (25 mM Tris, 200 mM NaCl, 2 mM EDTA, pH 8.0) and peak fractions were collected and stored at −80°C until use. A typical yield was 25 and 30 mg/L culture for S. pneumoniae and S. mitis CstRs, respectively, and all contained the correct number of reduced cysteines by Ellman's reagent (36). Protein concentrations were determined by UV absorption using the following ϵ280 values: 2,980 M−1 cm−1, for S. pneumoniae, S. mitis and E. faecalis CstRs (14).
Quantitative RT-PCR analysis of sulfide-induced genes in Streptococcus mitis SVGS_061
S. mitis SVGS_061 strain was inoculated from glycerol stocks and grown in 5 ml BHI medium overnight. The overnight culture was pelleted by centrifugation, resuspended in equal volume of BHI and diluted into 30 ml BHI medium with starting OD600 of 0.01. Triplicate cultures were grown to an OD600 of 0.2 aerobically at 37ºC with shaking (200 rpm) in loosely capped 50 mL Falcon tubes, followed by the addition of freshly prepared 0.2 mM Na2S. 5 mL aliquots were withdrawn from the cultures prior to addition of Na2S (t = 0), and at 30 min post addition, and centrifuged for 10 min. Cell pellets were washed with ice-cold PBS, re-centrifuged for 5 min, decanted and stored at –80ºC until analysis by qRT-PCR using the primers specific for the rhdA (locus tag AXK38_00625), coaP (locus tag AXK38_00630) and rhdB (locus tag AXK38_00635) genes, essentially as previously described (13,18).
Ratiometric pulsed-alkylation mass spectrometry (rPA-MS)
Sample preparation for pulsed-alkylation mass spectrometry was adapted from the original report of the technique (37) and optimized for CstR and CsoR. All experiments were carried out anaerobically in a glovebox in a buffer containing 10 mM HEPES and 200 mM NaCl at pH 7.0. CstR, CstR mutants, and CsoR proteins were reacted with a 3-fold molar thiol excess of d5-N-ethylmaleimide (NEM, pulse, Isotech). At discrete time points, 50 μL aliquots were removed and quenched with an equal volume of a solution containing a 900-fold thiol excess of H5-NEM (chase) with 100 mM Tris pH 8.0, and 8 M urea. After a 1 h chase, quenched reactions were removed from the glovebox and precipitated on ice with a final concentration of 12.5% trichloroacetic acid (TCA) for 1.5 h. Precipitated protein was pelleted by centrifugation at 4°C. The supernatant was removed, and the pellet washed twice with ice-cold acetone. The washed pellet was vacuum centrifuged to dryness at 45°C and re-suspended in 10 μL digest buffer (20 mM ammonium bicarbonate, 10% acetonitrile, 50:1 protein:trypsin ratio, pH 8.2). CstRs were digested for 1 h and CsoR for 0.5 h at 37°C. Tryptic digests were quenched with a final concentration of 1% trifluoroacetic acid (TFA) and spotted on a MALDI plate with α-cyano-4-hydroxycinnamic acid (CCA) matrix with a 5:1 matrix:sample ratio for analysis. For examination of the pH-dependence of the reaction, the same experiment was carried out in the following buffers; Mes for pH 6.0–6.5, HEPES for pH 7.0–7.5, and Tris for pH 8.0–9.0.
MALDI-TOF mass spectra were collected and analyzed in triplicate reactions using a Bruker Autoflex III MALDI-TOF mass spectrometer with 200 Hz frequency-tripled Nd:YAG laser (355 nm) and Flex Analysis software (Bruker Daltonics, Billerica, MA, USA). Cysteine-containing peaks were identified by monoisotopic mass (Supplementary Table S3) and resolved as alkylated with d5-NEM (+130.0791 Da) or H5-NEM (+125.0477 Da) with little to no detectable unmodified peptide (data not shown). The theoretical distribution and peak areas were determined using the averagine algorithm (38) and quantified by summing the total peak areas of the full isotopic distribution. Relative peak areas were used to determine the mol fraction of H5-NEM labeled peptide, Θ(H5), as defined by eq. 1. A(H5) and A(d5) correspond to the area (A) of the isotopic distribution of H5-NEM or d5-NEM alkylated peptide, respectively. To obtain the pseudo-first order rate constant of alkylation, k, Θ(H5) was plotted as a function of pulse time, t, and fit to Eq. (2). In some instances, a fit to a sum of two exponentials was used, Eq. (3). The second-order rate constant was obtained by dividing k by the concentration of d5-NEM in the pulse.
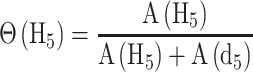 |
(1) |
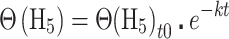 |
(2) |
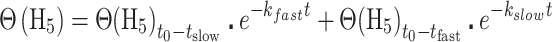 |
(3) |
Fluorescence anisotropy titrations
Double-stranded 5′-fluorescein-labeled DNA constructs were annealed as component single-strands and titrated as previously described using an ISS PC1 Spectrofluorometer (Champaign, IL, USA) equipped with an automatic titrator unit (39). The S. mitis cst operator, 5′-5Fluoro-TACCTCCAAATATACCCTTATGGGGTATATTAAA and the complementary unlabeled strand, was used for all experiments (core cst operator sequence is in bold) (30). 10 nM cst operator dsDNA in 25 mM Tris–HCl, 200 mM NaCl, 2 mM EDTA, 2 mM TCEP, pH 8.0 was titrated with either WT or the indicated CstR mutant (≈12 μM stock). Fluorescein was excited at 495 nm and polarization monitored with a 515 nm cut-off filter in the L-format. Each data point collected was the average and standard deviation of five measurements. Normalized r values for the fractional saturation of cst OP1 was calculated as (robs – r0)/(rcomplex – r0) from 0 to 1 where rcomplex represents the maximum anisotropy obtained and r0 represents free cst OP1 DNA. For titrations not reaching saturation, rcomplex was calculated from the addition of the anisotropy change of reduced CstR to r0 of the corresponding non-saturating CstR. Collected data were fit to a sequential non-dissociable tetramer (CstR4) binding model (13,14,30) using DynaFit (40) assuming a linear relationship between robs and vi and the binding density at the ith addition of titrant (30). The macroscopic binding constant, Ktet, is reported and was determined as Ktet = sqrt(K1•K2) due to high uncertainty in extracting unique Ki values, K1 and K2, due to a strong inverse correlation and little observable sigmoidal behavior in the binding isotherms, as previously described (30). Reported affinities (Ktet) are the average of three independent experiments.
Anaerobic mass spectrometry-based kinetic profiling of CstRs with various oxidants
Protein stocks for these experiments transferred into the argon-filled glove box and buffer exchanged into either 25 mM Tris, 200 mM NaCl, pH 8.0 (for tetrathionate, cystine, H2O2, glutathione disulfide reactions) or 150 mM sodium phosphate pH 7.4, and 2 mM EDTA (for cysteine persulfide) using PD MiniTrap G-25 (GE, Boston, Massachusetts) column, followed by repeated centrifugation (10 min, 13,000 rpm, 6 times). Reaction mixtures contained 60 μM protomer (15 μM tetramer) and a 20-fold molar excess over Cys residues (10-fold over CstR protomer) of each oxidant (1.2 mM) in a total volume of 500 μL Triplicated reactions were performed by withdrawing 50 μL at each time, t (ranging from 30 s to 3 h) and mixed with 50 μL of a 900-fold excess of iodoacetamide (IAM). Prior to initiating the reaction with oxidant, 50 μL was withdrawn in the same way, as t = 0 sample. IAM quenching the reaction and caps (amidomethylates) thiols and per- and polysulfides that form on Cys residues. Quenched reactions were then removed from the glovebox and washed with buffer three times in a 10 kDa mini-concentrator (Millipore-Sigma, Burlington Massachusetts) and 5 μL of each sample was aliquoted into a screwcap vial (Waters, Milford, Massachusetts) with 45 μL of buffer and analyzed by LC–ESI-MS, essentially as previously described (19) using a Waters/Micromass LCT Classic time-of-flight (TOF, Milford, MA, USA) mass spectrometer with a CapLC inlet following chromatography on a 50 μm Agilent BioBasic C4 reverse-phase column (5 μm particle size, 300 Å pore size). A 10 min linear gradient from 10% Solvent A (5% acetonitrile, 95% water, 0.1% formic acid) to 90% Solvent B (95% acetonitrile, 5% water, 0.1% formic acid) was used with the elution monitored at 215 nm. Data were collected and analyzed using MassLynx Software (Waters, Milford, MA).
For data analysis, species with peak intensities >20% that of the most intense feature at every timepoint were picked and the intensities for each peak from triplicate runs were averaged and normalized to determine relative abundance. For dimeric species, a factor of 4 was used to account for the decreased ionization efficiency of the dimer relative to monomeric CstR species, obtained by comparing peak intensities of a CstR in the reduced vs. tetrathionate-oxidized di/di form prepared under identical conditions. There was no significant impact of the nature of the CstR on this intensity ratio (data not shown). The normalized intensities of each of five species that share defined chemical features and kinetic profiles, shown in Supplementary Tables S5–S7 (black circles, red circles, blue squares, green squares and purple squares), were then binned, and a standard calculated, and the resulting progress curves globally analyzed (no weighting function was used) by applying the simplest possible kinetic model, which was a linear model, unless otherwise stated, using Dynafit (40). Schematic representations of these fitting models are shown in each case. Data were fitted using a pseudo-first order reaction, with the concentration of oxidant at a 20-fold excess, and kinetic rate constants compiled in Table 2.
Table 2.
Compilation of all rate constants observed in the thiol kinetic profiling experiments with the indicated oxidanta
| Oxidantb | k i (s−1) | C9A SpCstR | C9A/N55A SpCstR | SmCstR | EfCstR |
|---|---|---|---|---|---|
| Tetrathionate | k 1 | 0.036 ± 0.004 | 0.146 ± 0.004 | 0.0033 ± 0.0002 | n.d.c |
| H2O2 | k 1 | 0.0135 ± 0.0005 | 0.0079 ± 0.0002 | n.d. | n.d. |
| k2 | 0.0079 ± 0.0004 | 0.0069 ± 0.0003 | |||
| Cystine | No reactiond | No reaction | No reaction | No reaction | |
| GSSG | k 1 | 1.35 ± 0.01 × 10−4 | 1.35 ± 0.02 × 10−4 | n.d. | n.d. |
| k2 | 4.7 ± 0.1 × 10−4 | 3.8 ± 0.1 × 10−4 | |||
| k 3 | 1.18 ± 0.03 × 10−4 | 2.7 ± 0.1 × 10−4 | |||
| CysSSH | k 1 | 0.037 ± 0.002 | 0.039 ± 0.002 | 0.044 ± 0.002 | 0.033 ± 0.001 |
| k2 | 0.0014 ± 0.0002 | 0.00062 ± 0.00007 | 1.1 ± 0.1 × 10−5 | 0.00126 ± 0.00004 | |
| k -2 | 0.0016 ± 0.0002 | 0.00024 ± 0.00006 | ≤10−6 | ≤10−6 | |
| k 3 | 4.2 ± 0.6 × 10−5 | 4.2 ± 1.3 × 10−5 | 3 ± 2 × 10−5 | 8.4 ± 2.7 × 10−6 | |
| k 4 | 1.01 ± 0.07 × 10−4 | 6.8 ± 1.5 × 10−5 | 4 ± 2 × 10−5 | 0.02 ± 0.01 e | |
| k -4 | ≤10−6 | ≤10−6 | ≤10−6 | 0.02 ± 0.01 e | |
| k 4’ | ≤10−6 | 0.0012 ± 0.0007 | ≤10−6 | ≤10−6 |
aAll oxidants were present at a 20-fold molar excess of CstR protomer (10-fold excess over Cys thiol). btetrathionate, refer to Supplementary Figure S9; H2O2, refer to Figure 5; cystine, refer to Supplementary Figure S10; GSSG, refer to Figure 7; CysSSH, refer to Figure 6. cn.d., not determined. dNo observable product formation over the course of a 3 h reaction. A sequential kinetic model was used to obtain ki for H2O2 and GSSG, while a reversible bifurcated kinetic model was used for CysSSH. See text for additional details. eFitted bound to the same value according to the final product distribution.
Small angle X-ray scattering (SAXS)
Reduced C9A and C9A/N55A CstR samples were prepared to a final concentration of 1, 3 and 5 mg/mL and data acquired at Argonne National Laboratory (Argonne, Chicago, IL, USA) and analyzed by the Small-Angle X-ray Scattering Core Facility, Frederick National Laboratory for Cancer Research, Frederick, MD, according to published procedures (6). Briefly, the buffer scattering was subtracted from the sample scattering, with modified sample scattering curves merged to generate the scattering curve for further analysis. The scattering data at low q values are used to estimate the radius of gyration (Rg) by the Guinier approximation with the range of qRg < 1.3. Pair distance distribution function (PDDF) plots of each protein were obtained using the distance distribution analysis tool on SAS Data Analysis (Primus). Dmax values were calibrated in order to make the PDDF curve drop smoothly to zero.
X-ray crystallography
Crystals of C9A SpCstR, selenomethionine (SeMet)-substituted C9A SpCstR, and C9A/N55A SpCstR (6–8 mg/mL) were grown at 20°C using the hanging-drop vapor-diffusion method. Crystals formed under the following conditions: C9A SpCstR (PDB code 7MQ1): 0.1 M MES, pH 5.5, 0.25 M (NH4)2SO4, 15–18% PEG 4000; SeMet-substituted C9A SpCstR (PDB code: 7MQ2): 0.1 M HEPES, pH 7.5, 34–40% PEG 200; C9A/N55A SpCstR (PDB code: 7MQ3): 0.1 M MES, pH 6.0, 50 mM CaCl2, 40–50% PEG 200. Crystals were harvested, cryo-protected in a reservoir solution supplemented with 25% glycerol, except for C9A/N55A SpCstR, and were flash-frozen in liquid nitrogen. Diffraction data were collected at 100 K at the Beamline station 4.2.2 at the Advanced Light Source (Berkeley National Laboratory, CA) and were indexed, integrated, and scaled using XDS (41). A complete dataset of the native C9A SpCstR in space group C2 at ≈2 Å and a redundant dataset of the same variant were collected. (Supplementary Table S5, first and second column at wavelength 1.0 and 1.77 Å, respectively). Despite the excellent anomalous signal for sulfur in the latter dataset, phase estimation using Single Anomalous Diffraction (SAD) was unsuccessful. A third dataset derived from a crystal of a SeMet derivative was obtained in space group P21 (Supplementary Table S5, third column). SAD methods failed again to phase the data; however, molecular replacement (MR) using MORDA (42) was performed. A probable solution was found using PDB 3AAI as search model. Then, phases were improved using MR-SAD methods in PHENIX (43) and the previous dataset obtained at 1.77 Å in space group C2. Four sulfur sites with occupancies 0.81–0.91 were found. A Figure of Merit (FOM) of 0.38 was obtained and an incomplete model with Rwork/Rfree of 0.374/0.434 was built. Successive cycles of automatic building in Autobuild (PHENIX) and manual building in Coot, as well as refinement (PHENIX Refine) led to a complete model. These coordinates were then used as search model for MR to phase datasets of native C9A SpCstR, (Supplementary Table S5, first column), SeMet-substituted C9A SpCstR, with four molecules in the asymmetric unit (Supplementary Table S5, third column) and C9A/N55A SpCstR (Supplementary Table S5, fourth column). Some side chains for several residues in these models were not modeled due to the lack of electron density. The SeMet-substituted C9A SpCstR model obtained at pH 7.5 is characterized by relatively high B-factors (Supplementary Table S5), very weak electron density in the α1–α2 and α2–α3 loops and a loss of helicity in the α3 helix on one of four chains in the asymmetric unit. Thus, Rwork/Rfree is 0.2589/0.2991, where Rfree falls approximately in the 95th percentile as compared to structures of similar resolution. In contrast, the C9A/N55A SpCstR model in space group I 2 2 2 shows an Rwork/Rfree of 0.1998/0.2251 with excellent geometry and no Ramachandran outliers (Supplementary Table S5). However, C9A/N55A SpCstR dataset presented translational pseudo-symmetry, Patterson peaks with length larger than 15 Å and pseudo-translation vector 0.0, 0.5, 0.0 (as defined by Xtriage in Phenix). As a result, the final model Rfree falls in the 80th percentile. All crystallographic data acquisition and refinement statistics are shown in Supplementary Table S5.
NMR spectroscopy
Uniformly 15N, 13C, 2H protein was expressed in E. coli BL21 (DE3) cells, as described above, except in M9 minimal medium containing 1 kg D2O, with 1.0 g of 15NH4Cl and 2 g 13C6,2H-glucose as the sole nitrogen and carbon sources, respectively. Uniformly 15N-labeled protein was expressed in M9 minimal medium containing 1 L H2O, as well as 1.0 g of 15NH4Cl as the sole nitrogen source. NMR samples for backbone assignment contained 0.2–0.6 mM protein, with 25 mM MES pH 5.5, 150 mM NaCl, and 10% (v/v) D2O, with 0.3 mM 2,2-dimethyl-2-silapentanesulfonic acid (DSS) as an internal reference. NMR spectra were recorded at 35°C on a Bruker Avance Neo 600 MHz spectrometer equipped with a cryogenic probe in the METACyt Biomolecular NMR Laboratory at Indiana University, Bloomington. Backbone chemical shifts were assigned for each mutant at pH 5.5 using TROSY versions of the following standard triple-resonance experiments: HNCACB, HNCOCACB and HNCO (44) using non-uniform sampling with Poisson gap schedules (45). Data were collected using Topspin 4.0.7 (Bruker), processed using NMRPipe (46) and istHMS (47), and analyzed using CARA (www.nmr.ch) and NMRFAM-Sparky (48), all on NMRbox (49). Assignments at pH 7.5 were obtained by titration from pH 5.5 through pH 6.0 and pH 6.5. Backbone chemical shift perturbations (CSP) were calculated using 1H and 15N chemical shifts with Δδ = ((ΔδH)2 + 0.2(ΔδN)2)1/2. Chemical shift assignments have been deposited at the BMRB under accession codes 50893 and 50894 for SpCstR C9A and SpCstR C9A N55A, respectively.
Molecular dynamics simulations
Initial coordinates for molecular dynamics simulations for C9A/N55A SpCstR were obtained from reduced crystal structure reported here (PDB code: 7MQ3). In the case of C9A SpCstR, two sets of initial coordinates were considered: an in silico mutation of N55 was performed on the C9A/N55A SpCstR that has both high resolution and resolved N- and C-terminal regions, and the C9A SpCstR crystallographic structure obtained in pH 7.5. In both C9A SpCstR MD simulations, the tetrameric assembly state remains stable throughout the simulations (Supplementary Figure S8), however the in-silico mutant is of remarkable higher quality and was used for the analysis. Crystallization ions, organic molecules and water molecules were not considered part of the initial coordinates of the system.
All molecular dynamics simulations were performed using the Gromacs MD engine (50) using the Amber99SB force field (51). Initial coordinates of the tetrameric protein structures were solvated in an octahedral box of TIP3P water molecules (52) and sodium ions to achieve electroneutrality. All simulations were performed using periodic boundary conditions and Ewald sums to treat long range electrostatic interactions, the SHAKE algorithm (53) to keep bonds involving hydrogen atoms at their equilibrium length, 2 fs time step for the integration of Newton's equations, and the Berendsen thermostat and barostat to control the system temperature and pressure respectively. Equilibration consisted of an energy minimization of the initial structures, followed by a slow, 2 ns long heating up to 300 K (in the NVT ensemble), and slow relaxation of positional restraints to the protein atoms in the NPT ensemble. Production MD runs consisted of 400 ns long trajectories, but we considered 200 ns of equilibrium runs to ensure a valid comparison between authentic C9A/N55A SpCstR and the in-silico C9A SpCstR mutant. Frames were collected at 500 ps intervals, which were subsequently used to analyze the production trajectories. Trajectory analyses were carried out using the CPPTRAJ module of AmberTools (54) and visualization and figures were generated using VMD (55) and Pymol (v2.4.0, Schrödinger, LLC).
RESULTS
Sequence similarity network (SSN) analysis of CsoR-family repressors delineates distinct functional groups
Early work revealed that founding members of the CsoR family, M. tuberculosis copper-sensing repressor (CsoR) (24) and nickel-sensing repressor E. coli RcnR (20,28) were representative of a large superfamily from which CstR ultimately emerged, initially characterized in S. aureus (13,30). In order to understand the sequence relationships between individual members of this very large superfamily of repressors (21), we performed a comprehensive sequence similarity network (SSN) analysis (56) of 32,476 unique PF02583 entries (11,726 sequences that are <90% identical over 80% of the sequence; Supplementary Tables S1A, S2) found in the UniProt database (Figure 2A) with sequence logo representations of SSN cluster sequence conservation (34) shown in Figure 2B. We note that individual SSN clusters represent largely isofunctional groups, as revealed by common sets of regulated genes in the genomic context, as well as by conservation of inducer recognition residues (Supplementary Figure S1). These functional assignments are further supported by at least one functionally characterized member in each cluster. Still, several large SSN clusters (6–9; 9.5% of all PF02583 sequences) remain entirely uncharacterized (Supplementary Figure S2).
Figure 2.

Sequence similarity network (SSN) analysis of the CsoR superfamily of bacterial repressors. (A) Results of an SSN clustering analysis of 32,476 unique sequences belonging to the Pfam PF02583 using genomic enzymology tools and visualized using Cytoscape. Clusters are designated by a number and ranked according to the number of unique sequences (Supplementary Tables S1A, S2), color-coded, and functionally annotated as copper (Cu), nickel/cobalt (Ni/Co), persulfide (RSSH) or formaldehyde (HCHO) sensors, if known. Each node corresponds to sequences that are 80% identical over 80% of the sequence, using an alignment score of 28 (see Materials and Methods). Functionally characterized members in each of nine clusters (1–5, 10, 12, 17, 23) are indicated with species name and trivial name (25). Cluster 10 proteins are the subject of the work presented here. (B) Sequence logo representations of sequence conservation of the Cu(I) and RSSH sensors defined by the indicated cluster of sequences derived from panel A. The residues that coordinate the Cu(I) in CsoRs are marked by the yellow and blue stars, the residue equivalent to C9 in SpCstR (cluster 10) marked by a red star, and the two allosterically important second Cu(I) coordination shell residues in MtCsoR (cluster 23) (86) marked with a green circle. WebLogos for the Ni/Co and other uncharacterized PF02583 sensor clusters are also provided (Supplementary Figure S2A).
Nearly all functionally characterized bona fide CsoR-like Cu sensors with the prominent exception of founding member MtCsoR (24), are grouped in cluster 1 (9,450 sequences), which represents the most diverse group, with SlCsoR (26), MtRicR (27), SaCsoR (30) and BsCsoR (6,57) found in distinct subclusters within SSN cluster 1. Despite this diversity, all subclusters conserve the three cognate Cu binding residues that form a C-H-C (Cys-His-Cys) ‘X-Y-Z’ motif and lack an N-terminal conserved metal binding residue ‘W’ (x-C-H-C, Figure 2). This motif is also present in cluster 23, defined by MtCsoR and found only in actinomycetes, and is characterized by an ≈30-residue, poorly conserved C-terminal tail not found in other CsoRs (Figure 2B). Another cluster of Cu sensors is represented by T. thermophilus CsoR (cluster 17) where one of the Cu(I)-coordinating Cys is replaced by His, defining a x-C-H-H motif (Figure 2B) (58). The assignment of SSN cluster 1, 17 and 23 proteins as bona fide Cu(I) sensors is further supported by a genomic neighborhood network (GNN) analysis of these sequences, which in the vast majority of cases encodes a CopA efflux pump and a CopZ metallochaperone in the immediate vicinity of the csoR gene (Supplementary Figure S1). Sequence similarity and genomic context suggest that the uncharacterized clusters 6 and 7 may also contain Cu sensors (Supplementary Figure S2).
The second largest cluster of sequences is cluster 2 (6,010 unique sequences) and groups the Ni/Co-sensing repressor RcnR, characterized by a H-C-H-H ‘W-X-Y-Z’ motif, and the formaldehyde (HCHO) sensor, FrmR (23,59), characterized by a P-C-H-x ‘W-X-Y-Z’ motif, on opposite ends of the cluster (Figure 2 and Supplementary Figure S2). The fact that these sequences fail to segregate in our SSN analysis is consistent with a high level of pairwise similarity and the relative ease with which inducer specificity can be relaxed or switched in an FrmR by simply reintroducing the His residue that characterize the RcnR subcluster (23) (Supplementary Figure S2). Here, the GNN analysis can be used to predict the function of individual cluster 2 sequences, as an RcnR or FrmR operon contains either a rcnA or an fmrA gene, respectively (Supplementary Figure S1). Clusters 5 and 12 also harbor distinct classes of Ni/Co sensors with a H-C-H-C ‘W-X-Y-Z’ motif, represented by NreA from Bradyrhizobium and Sphingobium spp. (60) and NcrB from Leptospirillum ferriphilum (61) (1836 members) and cyanobacterial InrS (29,62) (372 members), respectively (Supplementary Figure S2). Cluster 8 likely also contains Ni/Co sensors, based on genomic context (Supplementary Figure S2).
Beyond Cu(I), Ni/Co and HCHO, candidate persulfide (RSSH) sensors constitute the next largest subgroup of clusters from the CsoR family of proteins. Cluster 3 corresponds to a significant group (3,175 members) of largely uncharacterized candidate persulfide (RSSH) sensors from actinomycetes, including S. coelicolor CsoR (CstR) (63) and an uncharacterized mycobacterial protein, MtRv1766 (24). Cluster 4 harbors the biochemically characterized RSSH sensor from S. aureus (13), while cluster 10 contains E. faecalis CstR (14). All three candidate RSSH sensor clusters (3, 4 and 10) harbor two conserved Cys, forming a x-C-x-C W-X-Y-Z motif and their functional role is confirmed by a genomic neighborhood that encodes for distinct sulfide or sulfite detoxification pathways previously described (Supplementary Figure S1) (10,14). Overall, our SSN and GNN analyses suggest that the differences in CsoR-family protein function and inducer recognition can be traced to sequence differences beyond known or projected metal binding residues in the context of the ‘W-X-Y-Z’ motif model (20,21), and makes compelling functional predictions for currently uncharacterized CsoR-family SSN clusters (Supplementary Figure S2).
The nucleophilicity of the N-terminal Cys dictates RSS selectivity over metal ions
Our SSN analysis identifies three Cu(I)-sensing clusters and three RSS-sensing clusters of CsoR-family proteins. Clusters 1, 3, 4, 10 and 23 all share two conserved cysteine residues irrespective of their cognate inducers (Cu or RSS) (Figure 2). These patterns identified in our SSN analysis raise the question of whether differences in cysteine reactivity (nucleophilicity) can explain the inducer selectivity of the dithiol pair for either Cu(I) ions or RSS. We performed pulsed alkylation mass spectrometry experiments (37,39) with the neutral alkylating reagent, N-ethylmaleimide (NEM) at pH 7.0 under anaerobic conditions to directly compare the chemical reactivities of the cysteines in CstR relative to a Cu(I)-sensing CsoR isolated from the same organism, Staphylococcus aureus (Sa) (clusters 1 versus 4; Figure 2B). Briefly, each protein was reacted with a pulse of deuterium-labeled ‘heavy’ NEM (d5-NEM) for a variable time, t, then chased with a vast excess of ‘light’ NEM (H5-NEM) to alkylate any unreacted cysteine residues. Proteins were then digested with trypsin and analyzed by MALDI-TOF mass spectrometry (Figure 3). All observed and expected d5- and H5-NEM derivatized peptide masses are shown in Supplementary Table S3, with the increase in molar fraction of ‘heavy’-labeled peptide modeled by a single or double exponential function (Supplementary Table S4; Supplementary Figure S3A, B).
Figure 3.
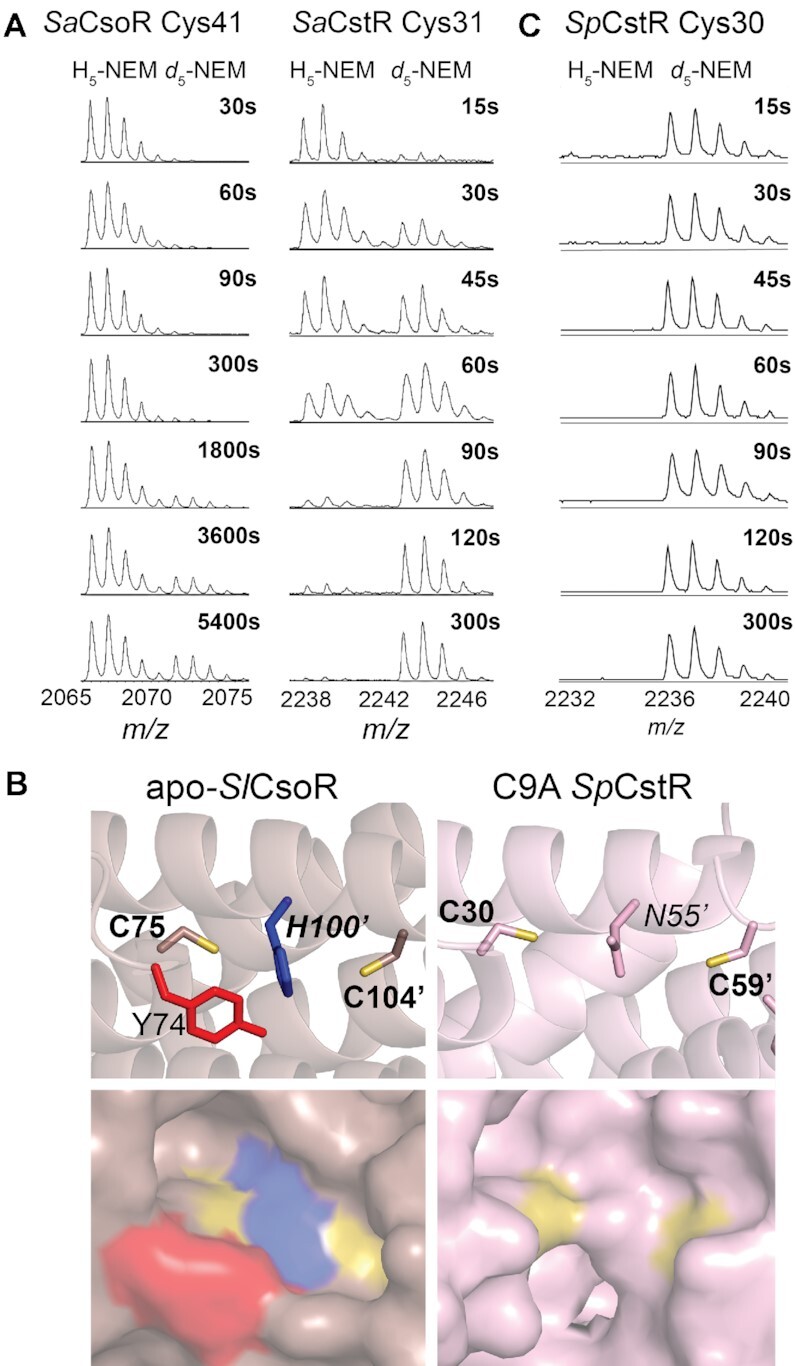
Selected regions of a series of MALDI-TOF mass spectra obtained by rPA-MS of (A) wild-type SaCsoR (left) and SaCstR (right) showing only the peptide containing the more N-terminal Cys in each case. The d5-NEM pulse time is indicated on each panel, with the H5-NEM and d5-NEM isotopic mass distributions shown, with expected and measured monoisotopic masses (38) shown for each tryptic peptide shown compiled in Supplementary Table S3. Representative MALDI-TOF spectral regions for Cys9, and Cys59 of SpCstR are provided (Supplementary Figure S3). The data in panel were analyzed using single or double exponential model (see Supplementary Figure S3) with the kinetic parameters compiled in Supplementary Table S4. (B) Structures of the dithiol regulatory sites shown for the Cu(I) sensor Streptomyces lividans (Sl) CsoR in the apo-state (33) (left) and the persulfide C9A SpCstR (right) (this work). Residues of interest are shown. (C) Selected region of a series of MALDI-TOF mass spectra obtained by rPA-MS of wild-type SpCstR.
We find that the reactivity of the more N-terminal Cys in SaCsoR (C41) is very slow (Figure 3A) when compared to the more C-terminal Cys (C70) (Supplementary Table S4), which is characterized by a nucleophilicity largely as expected for a solvent-exposed thiolate. Indeed, inspection of a structure of apo-CsoR from S. lividans reveals that while C104 (C70 in SaCsoR) is on the edge of a tetramer in a solvent exposed helix, C75 (C41 in SaCsoR) is boxed into a cage-like structure formed by the Cu-ligating His100’ (H66’ in SaCsoR; analogous to N55 in SpCstR) and the allosteric residue Y74 (Y40 in SaCsoR), that are conserved among all Cu(I) sensing clusters (Figures 2 and 3B, left) (31). This is also the case for TtCsoR in the apo-state, where C41 is found in the bottom of a cavity, while C66 is located on a solvent-exposed loop that is not resolved in this structure. These observations suggest that a restriction of solvent accessibility by the N-terminal cysteine attenuates its reactivity and thus enforces Cu(I) specificity.
In striking contrast, in SaCstR the N-terminal Cys (C31) is more reactive than the C-terminal Cys (C60) (Figure 3A; Supplementary Table S4) and a comparison of reactivities of the N-terminal Cys in SaCstR vs. SaCsoR reveals a 50-fold difference in the second order rate constant, a trend that persists over a wide range of pH values (6.0–9.5) (Supplementary Figure S3). Indeed, these data suggest that Cys31 in SaCstR has a pKa of ≈8.6, or similar to that of a free thiol, 8.5–9.0 (64,65), while Cys41 in SaCsoR is elevated to ≈9.3 or higher, consistent with its decreased reactivity over the entire pH range (Supplementary Figure S3). Furthermore, this relative trend of cysteine nucleophilicity is common among other RSS-sensors found in cluster 10, where only the more N-terminal of the two conserved cysteines is invariant. The previously characterized EfCstR (14) and two candidate CstRs from Streptococcal spp (vide infra) exhibit an even higher reactivity of the N-terminal Cys than SaCstR (Figure 3C, Supplementary Table S4), with SmCstR nearly too fast to measure. These findings reveal that the microenvironment near C41 in CsoR makes deprotonation less favorable than in a CstR, thus providing a mechanistic rationale for a lack of transcriptional regulatory crosstalk between CstR and CsoR sensors in the S. aureus cytoplasm. This also suggests that CstRs employ a highly reactive N-terminal attacking Cys that readily reacts with electrophiles, such as RSS and possibly resolves forming a bridge with a resolving C-terminal Cys (30). Moreover, these findings make the structural prediction that the N-terminal cysteine of a CstR is more solvent accessible than the analogous residue in a Cu(I)-sensing protein.
Streptococcus pneumoniae D39 SPD_0073 is a CstR
In order to identify new CstR proteins that would serve as structural targets to test the predictions from our pulsed alkylation experiments, we took advantage of our SSN analysis to make functional predictions for individual protein nodes contained within a single SSN cluster (66). We turned our attention to a single cluster 10 node, encoded by a gene designated locus tag SPD_0073 in the serotype 2 pneumococcal strain D39 (67). SPD_0073 possesses significant sequence similarity to the previously described CstR from E. faecalis (cluster 10; Figure 2A) but is more distantly related to S. aureus CstR, and not nearby other genes that suggest a role in RSS sensing and metabolism (Figure 4A). This prompted a broader search for other cluster 10 candidate CstRs, which are closely related to SPD_0073. We identified a candidate CstR encoded in mitis group oral streptococci closely related to S. pneumoniae (68) that is known to encounter fluctuations in H2S in periodontal biofilms (69). The operon bears strong resemblance to the sulfide- and RSS-inducible cst-like operon that we characterized in E. faecalis, encoding two single-domain sulfurtransferases or rhodaneses, RhdA and RhdB, and a coenzyme A persulfide reductase (Figure 4A; Supplementary Figure S1) (14) Those operons are found in most strains of S. constellatus, S. anginosus, S. cristatus, S. gallolyticus and S. mitis, and also harbor a consensus CstR binding sequence in the operator-promoter region upstream of the rhdB gene (30). These features make the prediction that these operons would be inducible by Na2S or RSS (14).
Figure 4.

S. pneumoniae (Sp) D39 SPD_0073 encodes a bona fide persulfide-sensing CstR. (A) Genomic neighborhood and location of a candidate CstR-regulated operator-promoter site around selected streptococcal cstR genes (cluster 10; Figure 1A) investigated here. The DNA sequence of the core Sm cst operator is shown (30). (B) Induction of the expression of genes in S. mitis SVGS_061 by exogenous sulfide, as measured by qRT-PCR. AXK38_00625 (rhdA; see panel A); AXK38_00630 (coaP); AXK38_00635 (rhdB). (C) Left, SmCstR (locus tag AXK38_00620) binds to the S. mitis SVGS061 cst operator-containing DNA (black circles) and is inhibited by pre-treatment with CysSSH (cyan triangles, >90% persulfidated species according to the data in Figure 8) which is reversible upon addition of TCEP (cyan time course, inset). Right, representative DNA binding curves obtained for reduced (black circles) or tetrathionate-treated (purple triangles, >90% symmetric disulfide species according to the data in Supplementary Figure S9), C9A SpCstR on the S. mitis SVGS061 cst operator-containing DNA. Black open circles, C9A SpCstR binding to an AdcR operator (87), representative of non-specific binding. The continuous lines drawn through each data set represent the results of a fit to a two-tetramer binding model (Supplementary Figure S4B) (30), with parameters complied in Table 1. (D) Bar chart representation of the kinetics of Sm cst operator binding, dissociation by tetrathionate and subsequent reduction by TCEP by various SpCstR dithiol site mutants in a C9R SpCstR parent background, from left to right. The arrows labeled DNA CstR and DNA are the anisotropies associated with the CstR-bound and free DNAs. 1 min, anisotropy 1 min after addition of tetrathionate; eq, equilibrium anisotropy value.
To test this prediction, we focused on S. mitis SVGS_061, isolated from the bloodstream of an infected cancer patient (35). The S. mitis CstR (AXK38_00620) is 95% identical to SPD_0073 (Supplementary Figure S4A) and a comparison of the genomic neighborhoods around putative cstR-encoded genes suggests a precise genomic deletion in D39, relative to the S. mitis strain (Figure 4A) (67). We used qRT-PCR to assess induction of the candidate CstR-regulated genes (AXK38_00625, rhdA; AXK38_00630; coaP; AXK38_00635, rhdB) before and 30 min after addition of 0.2 mM Na2S (Figure 4B). We observe robust sulfide-inducible expression, consistent with the designation of S. mitis CstR as a bona fide persulfide sensor analogous to the enterococcal and staphylococcal systems (13,14).
We confirmed the sulfide-induced regulation by studying DNA binding affinities of recombinant wild-type S. mitis CstR and the C9A S. pneumoniae SPD_0073 (this mutation has no impact on DNA binding and was introduced to avoid undesirable oxidative chemistry at this non-conserved Cys; Supplementary Figure S4A). We carried out quantitative fluorescence anisotropy-based DNA binding experiments with a duplex harboring the S. mitis cst operator and analyzed with a two-tetramer binding model (Figure 4A; Supplementary Figure S4B) (57,70). These experiments show that the average tetramer binding affinity (Ktet) is virtually identical for both proteins (Figure 4C; Table 1) and is significantly weakened, in a reversible fashion, upon incubation with RSS or a reactive disulfide-inducing electrophile, tetrathionate (Figure 4C, D), provided that C30, and to lesser degree C59, is present. A van’t Hoff analysis of the DNA-binding thermodynamics reveals that assembly of two CstR tetramers on the DNA operator is largely driven by a favorable enthalpy change, consistent with previous studies of a Streptomyces ssp. CsoR (70) and a modest electrostatic component (Supplementary Figure S4C, D). These studies establish that S. pneumoniae SPD_0073 exhibits DNA binding properties and regulation consistent with that of a CstR, which we denote SpCstR.
Table 1.
Summary of DNA binding parameters for SpCstR and SmCstRa
| Protein | Treatmentb | DNA operator | K tet (M−1) |
|---|---|---|---|
| C9A SpCstR | None | S. mitis cst | 2.2 ± 0.2 × 108 |
| SmCstR | None | S. mitis cst | 1.4 ± 0.2 × 108 |
| C9A SpCstR | None | Sp adcA | 4.9 ± 0.1 × 105 |
| C9A SpCstR | Tetrathionate | S. mitis cst | <3.0 × 105 |
| SmCstR | CysSSH | S. mitis cst | <6.3 × 105 |
aConditions: 10 nM cst operator dsDNA in 25 mM Tris–HCl, 200 mM NaCl, 2 mM EDTA, 2 mM TCEP, pH 8.0, 25.0ºC. The DNA binding data were fit to a two-tetramer DNA binding model (see Materials and Methods) and reflect the results of triplicate titrations. bNone, dithiol-reduced protein.
The crystallographic structure of C9A SpCstR reveals a more accessible N-terminal Cys and functional asymmetry that is confirmed by cysteine reactivity experiments
Having validated the SSN-predicted functions of SpCstR and SmCstR, we turned to crystallography to further understand the structural basis of RSS-sensing specificity. We solved the crystallographic structure of reduced C9A SpCstR at pH 7.5 from 34% PEG200 to 2.3 Å resolution. This first crystal structure of a CstR shows the anticipated dimer-of-dimers pseudo-tetrameric architecture (Figure 5A; Supplementary Table S5 for structure statistics), with each protomer harboring three helices (α1–α3). This is compatible with our small angle X-ray scattering (SAXS) and NMR spectroscopy analysis that indicates that SpCstR adopts a homogeneous tetrameric assembly state at pH 7.5 (Supplementary Figures S5 and S6). Despite the relatively low resolution, the structure reveals a microenvironment around the N-terminal Cys that is far less sterically hindered when compared to structures of apo-CsoR (Figure 3B). These structural data are fully compatible with the chemical reactivity data above (Figure 3C). Moreover, two Cys side chains in the C9A SpCstR tetramer maintain long Sγ–Sγ interprotomer distances (7.0–8.4 Å, Figure 5A) suggesting the possibility that this is a common feature of dithiol-harboring CsoR family proteins irrespective of their cognate inducer.
Figure 5.

Ribbon and electrostatic surface representations of the crystallographic structures of homotetrameric C9A SpCstR (A, B, respectively) at pH 7.5 and C9A/N55A SpCstR (D, E), at pH 6.0. See Supplementary Table S5 for structure statistics. In panels A and D, each of the protomers are shaded white, blue, grey and light blue. The side chains of C30, N55 (A55) and C59 are presented in sphere representation, with CPK coloring and Sγatoms yellow. Interprotomer C30 Sγ-C59’ Sγ distances are indicated. In C9A/N55A SpCstR, the Sγ atom of C59’ occupies two rotamer positions. (C) and (F), results of molecular dynamics (MD) simulations obtained for C9A SpCstR derived from in silico mutation of C9A/N55A SpCstR to C9A SpCstR as the starting structure (C) or authentic C9A/N55A SpCstR as starting structure (F). The temporal displacements of the Cys side chains (yellow) and N55 (C) or A55 (F) over the course of these simulations are shown.
The C9A SpCstR tetramer is highly asymmetric (with an asymmetric unit of 4 protomers), with one dimer laterally offset relative to the other, and a wide range of C30 Sγ–C59’ Sγ distances, but with the side chain of N55 generally wedged between the two Cys side chains (Figure 5A). Surprisingly, the orientations of the C-terminal α3 helices in each protomer at the tetramer interface vary widely in this model (Supplementary Figure S5A). Small angle x-ray scattering (SAXS) analysis reveals that C9A SpCstR adopts a homogeneous tetrameric assembly state at pH 7.5 (Supplementary Figure S5B), while NMR spectroscopy shows that any deviations from a D2-symmetric tetrameric structure at this pH must occur on a fast timescale, since only one set of crosspeaks is observed in the 1H–15N TROSY spectra (Figure 6A and Supplementary Figure S6, right panel). Moreover, an additional structure solved at pH 5.5 to a resolution of 2.0 Å (Supplementary Table S5) adopts a distinct assembly state (hexameric) in the crystal lattice, accommodated by significant displacements of the α3 helix (Supplementary Figure S5D, top). The difficulties obtaining a high-resolution structure of the tetrameric assembly are also reflected in our molecular dynamics simulations of C9A SpCstR at pH 7.5, where the interface between protomers appears to have significant mobility and does not necessarily reach an equilibrium state (Supplementary Figure S8). These data show that packing of the dimer-dimer interface is somewhat plastic in C9A SpCstR. Altogether our data suggest the differences between protomers are not necessarily strictly structural as could be inferred from the crystal structures alone; indeed, the crystallography may have trapped a structurally asymmetric state that reflects local differences in solution state dynamics.
Figure 6.

Solution NMR analysis of C9A/N55A SpCstR vs. the parent C9A SpCstR. (A)15N,1H TROSY spectra of reduced C9A SpCstR at pH 5.5 (blue contours) (left) compared to the same spectrum of C9A/N55A SpCstR at pH 5.5 (cyan contours) (right), with both spectra overlaid (middle), acquired at 35 ºC. (B) 15N and 1HN chemical shift perturbation versus residue number, with these changes painted onto the structure of C9A/N55A SpCstR (inset). (C) A comparison of the predicted helical propensities (range 0, no propensity; 1, high propensity) of C9A/N55A (red) versus the parent C9A SpCstR (black), based on chemical shifts analysis by TALOSN (72). (D) TALOSN-predicted backbone order parameter (S2) for C9A/N55A (red) versus the parent C9A SpCstR (black).
In order to explore the functional impact of this asymmetry on chemical reactivity, we implemented a mass spectrometry-based kinetic profiling approach. Purified protein was anaerobically exposed to each oxidant for a variable time t, followed by quenching with excess iodoacetamide (IAM, +57 amu), and mass determination by ESI-MS. The strong oxidant tetrathionate reacts quantitatively within the first ≈1 min with no evidence of intermediates (Supplementary Figure S9), forming quantitatively doubly disulfide-crosslinked dimer which is DNA binding-incompetent (Figure 4). In contrast, the more weakly reactive oxidants hydrogen peroxide (H2O2) and glutathione disulfide (GSSG) show three and four major species, respectively (Figure 7A; Supplementary Table S6). In addition to the two end-states, fully reduced/capped monomer (black) and doubly disulfide-crosslinked dimer (denoted di/di; blue), we observe a transiently populated, on-pathway intermediate dimer species in which one side of the dimer is closed with a disulfide, with the other side remaining reduced (or sulfenylated (71)), and subject to capping by IAM, or in the case of GSSG, singly- or doubly-glutathionylated (Figure 7B). The differences in the rate of formation of the first disulfide bond in the dimer unit relative to that of the second on the other side of the dimer constitute direct evidence for asymmetry of reactivity in the SpCstR dimer-of-dimers architecture. This observation is consistent with an asymmetric reduced tetramer where the asymmetry may arise from the internal dynamics and the plasticity between protomers (Supplementary Figure S6). Furthermore, given the long distance between the two thiols in the crystal, closing of one dithiol site may introduce strain on the opposite site of the dimer that inhibits the subsequent closure as a result of this asymmetry in other oxidation states.
Figure 7.

Thiol reactivity kinetic profiling experiments with non-sulfur-based oxidants, hydrogen peroxide (H2O2) and glutathione disulfide. (A) (top) Overall workflow with a schematic non-covalent ‘dimer’ representation of SpCstR shown with the Cys residues indicated. (bottom) Representative regions of the ESI-MS data obtained for a 10 min (left) or 60 min (right) reaction of GSSG with C9A/N55A SpCstR. The symbols correspond to the state harboring distinct modifications on one or both thiols as shown in panel (B). Supplementary Tables S5-S6 lists expected and observed masses for each of these species. (C) Representative kinetic profiling experiments obtained for C9A SpCstR (left) and C9A/N55A SpCstR (right) and H2O2. (D) Kinetic scheme used to analyze these data with ki compiled in Table 2, and defined by the continuous lines (dashed line, symmetric limit, k1=k2, and dotted line, 2-fold asymmetry, k1= 2k2, in panel (C). (E) Representative kinetic profiling experiments obtained for C9A SpCstR (left) and C9A/N55A SpCstR (right) and GSSG. (F) Kinetic scheme used to analyze these data with ki are compiled in Table 2, and defined by the continuous lines (dashed line, symmetric limit, k1=k2; dotted line, two-fold asymmetry, k1= 2k2, and dashed-dotted line 4-fold asymmetry, k1= 4k2, in panel E.
An N55A substitution diminishes asymmetry of reactivity in SpCstRs
Our results thus far suggest that C9A SpCstR harnesses a highly reactive N-terminal attacking Cys capable of forming a disulfide with a resolving C-terminal Cys in which the Sγ atoms are separated by 7.0–8.4 Å; formation of that disulfide, in turn, may further enhance the asymmetry observed in the reduced state and inhibits, to some extent, the formation of a symmetrically disulfide-crosslinked (di/di) dimer. We reasoned that N55, which replaces the Cu(I)-coordinating His in Cu(I)-sensing CsoRs (Figure 2B), might enforce this asymmetry of reactivity, given its positioning as a ‘wedge’ residue between the cysteines. To test this, we solved the crystallographic structure of C9A/N55A SpCstR to 1.4 Å at pH 6.0 (Supplementary Table S5) and investigated its dynamics and kinetics of reactivity toward various oxidants. The structure reveals a highly symmetric tetrameric architecture (Figure 5D), characterized by an asymmetric unit of 1 protomer, in striking contrast to the parent C9A SpCstR structure, and confirmed as adopting a tetrameric assembly state in solution by SAXS analysis (Supplementary Figure S5C). Interestingly, all the other structural features remain unaltered relative to C9A SpCstR. Long Sγ–Sγ interprotomer distances (8.6 Å) in two rotamer populations of C59 are observed, as is an unbroken α2 helix, despite the loss of the N55 side chain. We next turned to our mass spectrometry-based kinetic profiling to interrogate the effect of this N55A cavity mutation on chemical reactivity and functional asymmetry. We find that the N55A substitution reduces the asymmetry in chemical reactivity with both GSSG and H2O2 (Figure 7, Table 2).
Our crystallography data provide residue-specific information on the differences in asymmetry between the two variants of SpCstR, and our kinetic profiling data demonstrate that the differences in asymmetry are preserved in the solution state but provide no details on which structural motifs are responsible. To explore the residue-specific structural and dynamic impact of the N55A mutation in solution, we turned to NMR and molecular dynamics. NMR backbone (1HN, 13Cα, 13Cβ, 13C’ and 15NN) resonance assignments for both C9A and C9A/N55A SpCstRs at pH 5.5 reveal that the N55A substitution induces only local perturbations in the α2 helix, propagating to the structurally adjacent region of the C-terminal α3 helix (Figure 6A, B). Analysis of the chemical shifts with TALOS-N (72) shows that the secondary structures of the two variants are identical and fully consistent with C9A/N55A SpCstR structure (Figure 6C). In contrast, a preliminary comparative analysis of the 1H,15N TROSY spectra of the disulfide crosslinked states of C9A and C9A/N55A SpCstRs reveals far more differences than similarities (Supplementary Figure S7A), consistent with globally distinct tetrameric structures. Chemical shift-based predictions of sub-ns backbone disorder for both variants indicate that the N- and C-termini and interhelical loops are flexible, while α1 and most of α2 are quite rigid, α3 is well-structured but less rigid than the other helices. Close inspection shows that the α2- α3 loop is longer and more flexible than the α1–α2 loop, and although the α2 helix is unbroken by the mutation, the C-terminal region around N55 is relatively flexible, even approaching the flexibility of α3 in the case of the N55A mutation (Figure 6D). Experimental heteronuclear NOE measurements largely confirm these predictions, showing that the α2–α3 loop is the most flexible region of the body of the protein, and that α3 helix is the most flexible helix. These experimental data suggest increased order around the site of the N55A mutation, but increased flexibility in the N-terminal region of the α2–α3 loop (Supplementary Figure S7B). These data suggest that the α2–α3 loop is likely responsible for the solution state asymmetry in the C9A SpCstR, although we cannot measure differences in flexibility between protomers directly, because they have the same chemical shifts. We propose that this loop may have a different flexibility in each protomer in the C9A SpCstR tetramer giving rise to asymmetry in reactivity and an asymmetric plasticity in the α3 helix that is captured in the crystal, while the flexibility of all four α2–α3 loops is fairly similar and on average more flexible in the C9A/N55A SpCstR tetramer.
We next turned to molecular dynamics simulations to further validate this model for dynamic asymmetry. Our simulations of C9A/N55A SpCstR reveal a highly symmetric conformational ensemble where the α2–α3 loop is indeed the most dynamic loop, as reflected by the high RMSF in all protomers (Supplementary Figure S8A). Moreover, an in silico A55-to-N55 substitution introduced into C9A/N55A SpCstR reintroduces asymmetry by restricting the dynamics of the α2–α3 loop in two of the four protomers (namely A and C, Supplementary Figure S8A). This asymmetry is also illustrated at the level of the two dithiol sites on opposite ends of the tetramer within a dimer, as reflected by the low spread of visited coordinates of the Sγ atoms in half of the sites in C9A (Figure 5C), while the spread is comparable in the C9A/N55A tetramer (Figure 5F). These C9A SpCstR simulations are remarkably reminiscent of the structure of the formaldehyde sensor FrmR with formaldehyde adducts at half of the dithiol sites (Figure 5C) (22). Overall, these MD simulations as well as the NMR and SAXS data from C9A and C9A/N55A SpCstRs support the idea that functional asymmetry arises from fast internal dynamics in the loops that may impact packing in the dithiol sensing sites on opposite sides of the SpCstR tetramer, an extreme example of which is simply trapped in our highly asymmetric structure of C9A SpCstR (Figure 5A).
Distribution of crosslinked products upon reaction with thiol persulfides varies among cluster 10 CstRs
We next wished to determine if these specific features of dynamic and functional asymmetry observed with SpCstR can be extended to other cluster 10, RSS-sensing repressors and to other biologically relevant oxidants. We therefore implemented a mass spectrometry-based kinetic profiling approach (Figure 8A) to elucidate the mechanism and rates of product formation for three cluster 10 CstRs (SpCstRs, SmCstR and E. faecalis CstR) toward cysteine persulfide (CysSSH) (8,14,18,73), in an effort to understand if the asymmetry observed in SpCstR plays a role in fine-tunning thiol reactivity towards the cognate inducer (19). CysSSH is generated in situ by anaerobic incubation of cystine with an excess Na2S, forming equimolar CysSH and CysSSH, with remaining HS− and cystine, the latter of which does not react with CstR thiols (Supplementary Figure S10) and is used immediately.
Figure 8.

Cys persulfide (CysSSH) reactivity kinetic profiling experiments with cluster 10 CstRs. (A) Overall workflow. A schematic noncovalent ‘dimer’ representation of SpCstR or SmCstR (one-half of the tetramer) is shown, with the Cys residues indicated. For EfCstR, Cys residues corresponding to C30 and C59 in SpCstR are C32 and C61, respectively. Representative regions of the ESI-MS data obtained for a 10 min (B) or 60 min (C) reaction of CysSSH with C9A/N55A SpCstR. In each case, the monomer region is shown at left, and the crosslinked dimer region is shown at right. The symbols correspond to the generalized structures shown in panel D, with numbers within each symbol corresponding to the ith state harboring n distinct + 32 species distributed among one or both thiols (isomers cannot be resolved using this method): i= 1, n= 1; i= 2, n= 2; i= 3, n= 3; i= 4, n= 4. Supplementary Table S8 lists expected and observed masses for each of these species. (C) Global analysis of these kinetic profiles obtained for C9A SpCstR, C9A/N55A SpCstR, SmCstR and EfCstR (left to right) with CysSSH, using the generalized kinetic scheme in panel D. The species are colored according to the key shown in panel B, with the sum of all ith species grouped into each data point. The continuous lines drawn through each dataset reflect the results of global fitting to the minimal bifurcated model shown in panel D, with individual ki compiled in Table 2. (D) Generalized kinetic scheme used to analyze the reaction profiles obtained with persulfide donors. Continuous lines represent the processes that were determined in all profiles, whereas dashed lines represent the processes that could only be determined for some of the proteins. Rate constants are compiled in Table 2.
As expected from previous work, the dithiol persulfide-sensing site in CstRs is capable of forming a variety of protomer crosslinked oxidized products upon persulfide treatment in a way that depends on the incubation time (Figure 8A) (13). Five groups of reaction products are needed to fully describe the kinetic data (Figure 8B, Supplementary Table S8) and were fit to a kinetic scheme in which a thiol-derivatized per- and polysulfide protomeric intermediate (red) containing up to three sulfur additions capped by IAM and comprising more than 70% of species at early time points, is irreversibly formed from the reduced and capped monomer (black), defined by k1 (Figure 8D). This gives rise to three groups of covalently crosslinked species, which we denote as closed/open (purple), a di/di (blue) species introduced above, and a new closed/closed (green) dimer (see Figure 8B) that harbors C30–C59’ tri- and/or tetrasulfide crosslinks (Figure 8C, green) (13). The kinetic data for C9A SpCstR were globally fitted using a linear (unbranched) model that incorporates formation of the closed/closed (green) product from the closed/open intermediate (purple, Figure 8C, left). Also, since the derivatized monomer is not completely consumed, a rate constant for its formation from the closed/open intermediate was introduced (k-2, Table 2). Although the formation of closed/closed products are favored in C9A SpCstR with respect to di/di species (k4 > k3), the formation of these species is slow. This is not unexpected given the kinetic barrier to formation of the symmetric di/di product with other oxidants (Figure 7).
While treatment of C9A SpCstR with CysSSH gives a mixture of products with different degrees of asymmetric connectivity, C9A/N55A SpCstR yields nearly quantitative formation of a more symmetric closed/closed product (green). The time-dependence of the production of the closed/closed product is best modelled with the formation of these species from the polysulfidated monomer (red) with no accumulation of closed/open intermediates (purple). Notably, numerical simulations with different rate constants of formation of a more symmetric closed/closed product from the polysulfidated monomer (k4’, Table 2, Figure 8C, dashed and dotted lines) are capable of explaining the differences in the kinetic profiles between the C9A and C9A/N55A SpCstRs nearly entirely. This suggests that the loss of the bulky N55 side chain removes a kinetic block to the formation of higher order tri- and tetrasulfide crosslinked dimers within the CstR tetramer, supporting the idea that asymmetry determines not only the kinetic profile but also the final product distribution where more symmetric reduced tetramers form faster more symmetric products.
Remarkably, the reactivity profile observed for SmCstR collapses to one highly populated (≈90%) species, the polysulfidated monomer (red), with successive sulfur additions (up to three) presumably added to C30 (Figure 8C, Supplementary Table S8). This modification appears to be regulatory for DNA binding (Figure 4C). A very small amount of asymmetric closed/open species is formed at long time points, with the ‘open’ site again harboring mixed disulfide linkages to cysteine. We see no evidence for the formation of di/di, di/tri and other closed/closed species over a 3 h reaction. Analysis using the same kinetic scheme (Figure 8D) reveals a significant decrease in the rate constant for the formation of linkages between the C30 and C59’ (k2, Table 2, Figure 6C, dotted lines). These kinetic profiling data with SmCstR appear to represent an extreme case of the reactivity profile of C9A SpCstR (Figure 8C) except that no closed/closed species are formed. It is interesting to note that this product distribution appears to resemble that obtained previously with a cluster 3 CstR (63). These kinetic profiling data are compatible with a more significant kinetic block for the formation of higher order tri- and tetrasulfide crosslinked dimers within the CstR tetramer for SmCstR. Indeed, the SmCstR tetrathionate reactivity profile confirms that this protein can access a closed/closed conformation but does so significantly more slowly relative to SpCstRs (Supplementary Figure S9; Table 2). This kinetic block renders the resolving Cys unreactive, and provides a rationale for the comparatively low level of conservation of this residue in cluster 10 CstRs, suggesting that this extreme case of asymmetry may be more prevalent in cluster 10 than in other CstR clusters (Figure 2B).
It is interesting to note that the differences in product distributions between SmCstR and SpCstR originate from just four amino acid substitutions, one of which is the non-conserved C9 in SpCstR (Supplementary Figure S4A). Of the remaining three amino acid differences between SmCstR and SpCstR, G26, derived from a non-conservative E26G substitution, is most conspicuous (Supplementary Figure S4A). G26 terminates the α1 helix in SpCstR and defines a point of increased backbone motional disorder on the sub-ns timescale by NMR (Supplementary Figure S7). This Gly is positioned above the attacking Cys, C30, and thus may markedly impact its range of chemical reactivity in comparing these two closely related CstRs (Figure 5A). Indeed, MD simulations of an in silico G26E substitution mutant reveal that E26 engages in several salt bridges with neighboring residues that may inhibit formation of di- and polysulfide crosslinks in SmCstR (Supplementary Figure S8C).
The reactivity profile of a third cluster 10 CstR, EfCstR (14), on the other hand, bears close resemblance to that of C9A/N55A SpCstR except that the relative concentrations of the closed/open species is higher, accumulating to ≈ 40% of all species (purple, Figure 8C), with correspondingly less closed/closed species formed (green, Figure 8C). This kinetic profile can be modelled considering a fast equilibrium between closed/open and closed/closed species (defined by k4 and k-4) (red). This fast interconversion and the relatively high kinetic constants suggest that EfCstR is even more flexible and less kinetically trapped than C9A/N55A SpCstR. This observation also suggests that the E26, which is present in both SmCstR (kinetically trapped) and EfCstR (fast interconversion from open to closed), is not necessarily sufficient to create a kinetic block, and other residues that differ between SmCstR and EfCstR might be responsible for the dramatic change in reactivity (Supplementary Figure S4A). Altogether our kinetic profiling experiments with CysSSH suggest that dynamical asymmetry and kinetic barriers to the formation of interprotomer linkages can be tuned with minimal sequence alterations; this in turn significantly impacts the product distribution while retaining the fundamental property of connecting cysteine reactivity to conformational change associated with release from DNA.
DISCUSSION
In this work, we present new insights into the structure and chemical reactivity of organic persulfide sensors in the CsoR superfamily of bacterial repressors (10,13). We have defined the structure and sequence features that distinguish a Cu(I)-sensing CsoR from a persulfide-sensing CstR, focusing on characterized proteins from S. aureus, a human pathogen known to encode both of these dithiol CsoRs with no evidence of crosstalk in cells (30). Our SSN and GNN analyses allow us to identify three clusters of putative persulfide sensors, from which we were able to functionally and structurally characterize, for the first time, a CstR, representative of closely related persulfide sensors from the lactic acid bacteria (14). These structures and comprehensive chemical reactivity data, specifically for C9A SpCstR, suggest the presence of high kinetic barriers toward the formation of symmetrically crosslinked states. Further, this asymmetry can be traced to internal dynamics that can be fine-tuned by minimal sequence perturbations in what is rather limited sequence diversity within a single cluster of protein sequences.
Our structures of SpCstR in the dithiol-reduced and DNA-binding competent state, when placed in the context of conserved sequence features of the cluster 10 family repressors, reveal characteristics that are both common to, and distinct from, Cu(I)-sensing CsoRs of known structure. SpCstR features a pair of symmetry-related ion pairs across the α1–α1’ dimer interface, E14-R18’, and an additional ion pair in the α1–α2 loop, R28–D32, which surrounds C30, and thus may help orient the attacking thiol in CstR; both features are widespread in the CsoR superfamily (24). A prominent stripe of positive electrostatic potential crosses one face of the tetramer, over the positively charged ‘hole’ of the tetramer (R84 and K85 are highly dynamic and not visible in our structure) and includes superfamily-invariant residues, K11 and R12 of the RXXK/R sequence (X, any amino acid) in α1 helix (C9-X-K11-R12 in SpCstR) (24,70,74), Q36 (70) and R46 in the α2 helix and the C-terminal K83-R84-K85 motif (74,75), all known to play energetically important roles in DNA binding (Figure 5). It is interesting to note that these DNA binding residues are near the dynamic loops and could be affected by the plasticity of the interfaces between the promoters in the tetramer, which may be general a feature of the CsoR superfamily. A major point of departure between CstR and CsoR is the intrinsic reactivity of the N-terminal Cys which is far higher in a CstR and appears readily explained by the differences in microenvironment in apo-SlCsoR (33) vs. reduced SpCstR. This 50-fold difference between second order rate constants in SaCstR and SaCsoR could be defined by the intracellular concentration range of these species within which inducer selectivity must have evolved. This difference in nucleophilicity does not negatively impact Cu(I) binding in Cu(I)-sensing CsoR. Indeed, close approach of Cu(I) to the Cu(I) sensing site in CsoR would result in the concerted formation of the two strong Cu(I) coordination bonds to the Cys and His, which upon recruitment of the more distal C-terminal Cys into the first coordination shell, breaks the helical sense of the α2 helix, enforcing an allosteric switch that ultimately drives CsoR off the DNA (6,33,76).
In addition to understanding what distinguishes a Cu(I) sensor from an RSS sensor, the extent to which an RSS sensor is specific for these oxidants over others remains the subject of debate (77,78). It has recently been shown that R. capsulatus SqrR and BigR from an animal pathogen, which adopt entirely distinct structures from that of CstR beyond a common dithiol persulfide sensing site (15,16,19) can distinguish RSS from ROS by means of structural ‘frustration’ of the disulfide form (19). In striking contrast, CstRs react rapidly with H2O2, but at a ≈3–5-fold slower rate than observed with CysSSH. Despite this reactivity in vitro, it has been shown that H2O2 added to the outside of S. aureus cells does not induce the cst operon (13). Nevertheless, more recent work reveals that a range of potent oxidants, including polysulfane compounds, are capable of inducing transcription of CstR-regulated genes (12,79). Thus, the persulfide specificity of a CstR may well be lower than for SqrR/BigR, but the primary physiological (versus pathophysiological) inducer is likely to be reactive sulfur species.
In this context, it is interesting to speculate on the extent to which a relaxed degree of oxidant specificity may impact bacterial physiology. Cluster 10 proteins are derived nearly exclusively from the order Lactobacillales or lactic acid bacteria that are generally non-respiring and catalase-negative, acid-tolerant, low GC-content Gram-positive bacteria that meet their energy needs via carbohydrate fermentation with the production of lactic acid as a waste product. These organisms lack a TCA cycle and an electron transport chain that can generate significant endogenous ROS. Some Streptococcus spp., e.g., Streptococcus mutans, are important components of dental plaque, a biofilm that forms on the surface of teeth, and NADH oxidase is capable of reducing any O2 to water, regenerating NAD+ and redox homeostasis, while limiting exposure to other reactive oxygen species (80). As a result, the apparent inability of CstR to readily distinguish H2O2 from RSS in these species is unlikely to be physiologically important, even in microaerobic conditions. However, this likely is not the case for Streptococcus pneumoniae, a facultative anaerobe that is uniquely capable of respiring on oxygen by employing pyruvate oxidase (spxB), and to a lesser extent, lactate oxidase (lctO). These enzymes collectively convert lactic acid to acetyl-phosphate via pyruvate, to generate copious H2O2 that is used to limit microbial competitors during the colonization of the respiratory tract (81,82) and induce host cell damage in the presence of host-derived nitric oxide (83). It is interesting to note that S. pneumoniae is unique among cluster 10 organisms in that it has clearly lost the RSS sensing and detoxification response (see Figure 4 and Supplementary Figure S1) found in other lactic acid bacteria, such as S. mitis and E. faecalis. This is consistent with the idea that RSS would not be expected to accumulate in S. pneumoniae, due to high levels of endogenous H2O2 (8).
A striking finding is that the kinetic barrier to the formation of symmetric di-, tri- or tetrasulfide crosslinks dictates the product distribution of CstRs upon treatment with a biologically relevant persulfide donor, cysteine persulfide. The relative impact of this kinetic barrier appears to be determined by the number of accessible states of the Sγ atoms in the dithiol sensing site, which is tunable by minimal sequence changes. The flexibility of this site determines the probability of resolving a polysulfide built on the attacking cysteine and, thus, the relative abundance of species harboring di-, tri- or tetrasulfide linkages. An extreme case is SmCstR, where even long incubations with CysSSH give rise to only small amounts of the closed/open species, below 5% (Figure 8). This finding is consistent with a strong structural asymmetry in a ‘half-the-sites’ reacted dimer. Interestingly, the crystal structure of the CsoR-family formaldehyde-sensing repressor FrmR is characterized by the same tetrameric asymmetry, with one site ‘closed’ by a methylene bridge initiated by Michael addition of formaldehyde to the sole attacking Cys (equivalent to C30 in SpCstR), and the other sensing site in the same dimer unreacted (22). No reaction kinetics or product distributions were presented in that work, but this finding is consistent with the kinetic profiling we present here for CstRs and with the asymmetric tetramer structure of C9A CstR at pH 7.5, in which the two dimers are strongly offset relative to one another, in striking contrast to the C9A/N55A SpCstR structure (Figure 5). Based on these results and those others, we suggest that the microenvironment around each cysteine thiol and the dynamic flexibility of those sites gives rise to a spectrum of derivatized and crosslinked species that is far more complex than anticipated on the basis of other sulfur-nitrogen and sulfur-sulfur bridges found in other allosteric redox switches in regulatory proteins and enzymes (19,22,84,85). Ongoing studies are directed toward elucidating the persulfide switching mechanism in CstRs, and the extent to which this diversity in thiol reactivity impacts bacterial physiology.
DATA AVAILABILITY
The coordinates and structural factors have been submitted to the Protein Data Bank with accession codes 7MQ1 (C9A SpCstR), 7MQ2 (C9A SeMet SpCstR) and 7MQ3 (C9A/N55A SpCstR). Backbone Chemical shift assignments have been deposited at the BMRB under accession codes 50893 and 50894 for C9A and C9A/N55A SpCstRs, respectively. All data are available in the manuscript, with strains available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Giedroc and Capdevila laboratories for helpful comments on the manuscript. We gratefully acknowledge use of the Macromolecular Crystallography Facility and J. Trinidad for his assistance in the Laboratory for Biological Mass Spectrometry at IU- Bloomington. We thank J. Nix for his assistance during X-ray data collection at beamline 4.2.2, ALS and Prof. Sam Shelbourne (MD Anderson, UT-Houston) for his gift of the S. mitis strain used in these studies. We thank H. Wu of the METACyt Biomolecular NMR Laboratory for his help with NMR data acquisition. We thank Dr. Lixin Fan of the Small-Angle X-ray Scattering Core Facility, National Cancer Institute, Frederick, MD for acquiring the SAXS data. We thank the INQUIMAE-DQIAyQF cluster (FCEN, UBA) for providing computational resources.
Contributor Information
Joseph N Fakhoury, Department of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405-7102, USA.
Yifan Zhang, Department of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405-7102, USA; Department of Molecular and Cellular Biochemistry, Indiana University, 212 S. Hawthorne Drive, Bloomington, IN 47405 USA.
Katherine A Edmonds, Department of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405-7102, USA.
Mauro Bringas, Fundación Instituto Leloir, Av. Patricias Argentinas 435, Buenos Aires C1405BWE, Argentina.
Justin L Luebke, Department of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405-7102, USA.
Giovanni Gonzalez-Gutierrez, Department of Molecular and Cellular Biochemistry, Indiana University, 212 S. Hawthorne Drive, Bloomington, IN 47405 USA.
Daiana A Capdevila, Department of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405-7102, USA; Fundación Instituto Leloir, Av. Patricias Argentinas 435, Buenos Aires C1405BWE, Argentina.
David P Giedroc, Department of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405-7102, USA; Department of Molecular and Cellular Biochemistry, Indiana University, 212 S. Hawthorne Drive, Bloomington, IN 47405 USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health [R35 GM118157 to D.P.G.]; Bunge & Born, Argentina; Williams Foundations; MinCyT Argentina [PICT 2019-00011 to D.A.C.]; D.A.C. is Staff Member from CONICET, Argentina; M.B. is supported by a postdoctoral fellowship provided by CONICET, Argentina. Funding for open access charge: US National Institutes of Health [R35 GM118157 to D.P.G.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Fu Y., Chang F.M., Giedroc D.P.. Copper transport and trafficking at the host-bacterial pathogen interface. Acc. Chem. Res. 2014; 47:3605–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diaz-Ochoa V.E., Lam D., Lee C.S., Klaus S., Behnsen J., Liu J.Z., Chim N., Nuccio S.P., Rathi S.G., Mastroianni J.R.et al.. Salmonella mitigates oxidative stress and thrives in the inflamed gut by evading calprotectin-mediated manganese sequestration. Cell Host Microbe. 2016; 19:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shatalin K., Nuthanakanti A., Kaushik A., Shishov D., Peselis A., Shamovsky I., Pani B., Lechpammer M., Vasilyev N., Shatalina E.et al.. Inhibitors of bacterial H2S biogenesis targeting antibiotic resistance and tolerance. Science. 2021; 372:1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mishra R., Kohli S., Malhotra N., Bandyopadhyay P., Mehta M., Munshi M., Adiga V., Ahuja V.K., Shandil R.K., Rajmani R.S.et al.. Targeting redox heterogeneity to counteract drug tolerance in replicating Mycobacterium tuberculosis. Sci. Transl. Med. 2019; 11:eaaw6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Changela A. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003; 301:1383–1387. [DOI] [PubMed] [Google Scholar]
- 6. Chang F.M., Coyne H.J., Cubillas C., Vinuesa P., Fang X., Ma Z., Ma D., Helmann J.D., Garcia-de Los Santos A., Wang Y.X.et al.. Cu(I)-mediated allosteric switching in a copper-sensing operon repressor (CsoR). J. Biol. Chem. 2014; 289:19204–19217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Achard M.E., Stafford S.L., Bokil N.J., Chartres J., Bernhardt P.V., Schembri M.A., Sweet M.J., McEwan A.G.. Copper redistribution in murine macrophages in response to Salmonella infection. Biochem. J. 2012; 444:51–57. [DOI] [PubMed] [Google Scholar]
- 8. Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T.et al.. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li K., Xin Y., Xuan G., Zhao R., Liu H., Xia Y., Xun L.. Escherichia coli uses separate enzymes to produce H2S and reactive sulfane sulfur from L-cysteine. Front. Microbiol. 2019; 10:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walsh B.J.C., Giedroc D.P.. H2S and reactive sulfur signaling at the host-bacterial pathogen interface. J. Biol. Chem. 2020; 295:13150–13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu Z., Qiu Z., Liu Q., Huang Y., Li D., Shen X., Fan K., Xi J., Gu Y., Tang Y.et al.. Converting organosulfur compounds to inorganic polysulfides against resistant bacterial infections. Nat. Commun. 2018; 9:3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chi B.K., Huyen N.T.T., Loi V.V., Gruhlke M.C.H., Schaffer M., Mader U., Maass S., Becher D., Bernhardt J., Arbach M.et al.. The disulfide stress response and protein S-thioallylation caused by allicin and diallyl polysulfanes in Bacillus subtilis as revealed by transcriptomics and proteomics. Antioxidants (Basel). 2019; 8:E605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luebke J.L., Shen J., Bruce K.E., Kehl-Fie T.E., Peng H., Skaar E.P., Giedroc D.P.. The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol. 2014; 94:1343–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen J., Walsh B.J.C., Flores-Mireles A.L., Peng H., Zhang Y., Zhang Y., Trinidad J.C., Hultgren S.J., Giedroc D.P.. Hydrogen sulfide sensing through reactive sulfur species (RSS) and nitroxyl (HNO) in Enterococcus faecalis. ACS Chem. Biol. 2018; 13:1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimizu T., Shen J., Fang M., Zhang Y., Hori K., Trinidad J.C., Bauer C.E., Giedroc D.P., Masuda S.. Sulfide-responsive transcriptional repressor SqrR functions as a master regulator of sulfide-dependent photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2355–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guimarães B.G., Barbosa R.L., Soprano A.S., Campos B.M., De Souza T.a., Tonoli C.C.C., Leme A.F.P., Murakami M.T., Benedetti C.E.. Plant pathogenic bacteria utilize biofilm growth-associated repressor (BigR), a novel winged-helix redox switch, to control hydrogen sulfide detoxification under hypoxia. J. Biol. Chem. 2011; 286:26148–26157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H., Li J., Lu C., Xia Y., Xin Y., Liu H., Xun L., Liu H.. FisR activates sigma54-dependent transcription of sulfide-oxidizing genes in Cupriavidus pinatubonensis JMP134. Mol. Microbiol. 2017; 105:373–384. [DOI] [PubMed] [Google Scholar]
- 18. Walsh B.J.C., Wang J., Edmonds K.A., Palmer L.D., Zhang Y., Trinidad J.C., Skaar E.P., Giedroc D.P.. The response of Acinetobacter baumannii to hydrogen sulfide reveals two independent persulfide-sensing systems and a connection to biofilm regulation. mBio. 2020; 11:e01254-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Capdevila D.A., Walsh B.J.C., Zhang Y., Dietrich C., Gonzalez-Gutierrez G., Giedroc D.P.. Structural basis for persulfide-sensing specificity in a transcriptional regulator. Nat. Chem. Biol. 2021; 17:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iwig J.S., Leitch S., Herbst R.W., Maroney M.J., Chivers P.T.. Ni(II) and Co(II) sensing by Escherichia coli RcnR. J. Am. Chem. Soc. 2008; 130:7592–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Higgins K.A., Giedroc D. Insights into protein allostery in the CsoR/RcnR family of transcriptional repressors. Chem. Lett. 2014; 43:20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Denby K.J., Iwig J., Bisson C., Westwood J., Rolfe M.D., Sedelnikova S.E., Higgins K., Maroney M.J., Baker P.J., Chivers P.T.et al.. The mechanism of a formaldehyde-sensing transcriptional regulator. Sci. Rep. 2016; 6:38879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Osman D., Piergentili C., Chen J., Sayer L.N., Uson I., Huggins T.G., Robinson N.J., Pohl E.. The effectors and sensory sites of formaldehyde-responsive regulator FrmR and metal-sensing variant. J. Biol. Chem. 2016; 291:19502–19516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu T., Ramesh A., Ma Z., Ward S.K., Zhang L., George G.N., Talaat A.M., Sacchettini J.C., Giedroc D.P.. CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat. Chem. Biol. 2007; 3:60–68. [DOI] [PubMed] [Google Scholar]
- 25. Barchi E., Musiani F.. Molecular modelling of the Ni(II)-responsive Synechocystis PCC 6803 transcriptional regulator InrS in the metal bound form. Inorganics. 2019; 7:76. [Google Scholar]
- 26. Dwarakanath S., Chaplin A.K., Hough M.A., Rigali S., Vijgenboom E., Worrall J.A.. Response to copper stress in Streptomyces lividans extends beyond genes under direct control of a copper-sensitive operon repressor protein (CsoR). J. Biol. Chem. 2012; 287:17833–17847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Festa R.A., Jones M.B., Butler-Wu S., Sinsimer D., Gerads R., Bishai W.R., Peterson S.N., Darwin K.H.. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol. Microbiol. 2011; 79:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwig J.S., Rowe J.L., Chivers P.T.. Nickel homeostasis in Escherichia coli - The rcnR-rcnA efflux pathway and its linkage to NikR function. Mol. Microbiol. 2006; 62:252–262. [DOI] [PubMed] [Google Scholar]
- 29. Foster A.W., Patterson C.J., Pernil R., Hess C.R., Robinson N.J.. Cytosolic Ni(II) sensor in cyanobacterium: nickel detection follows nickel affinity across four families of metal sensors. J. Biol. Chem. 2012; 287:12142–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grossoehme N., Kehl-Fie T.E., Ma Z., Adams K.W., Cowart D.M., Scott R.A., Skaar E.P., Giedroc D.P.. Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J. Biol. Chem. 2011; 286:13522–13531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimmermann M., Xiao Z., Cobbett C.S., Wedd A.G.. Metal specificities of Arabidopsis zinc and copper transport proteins match the relative, but not the absolute, affinities of their N-terminal domains. Chem. Commun. (Camb.). 2009; 42:6364–6366. [DOI] [PubMed] [Google Scholar]
- 32. Osman D., Martini M.A., Foster A.W., Chen J., Scott A.J.P., Morton R.J., Steed J.W., Lurie-Luke E., Huggins T.G., Lawrence A.D.et al.. Bacterial sensors define intracellular free energies for correct enzyme metalation. Nat. Chem. Biol. 2019; 15:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Porto T.V., Hough M.A., Worrall J.A.R.. Structural insights into conformational switching in the copper metalloregulator CsoR from Streptomyces lividans. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2015; 71:1872–1878. [DOI] [PubMed] [Google Scholar]
- 34. Wheeler T.J., Clements J., Finn R.D.. Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics. 2014; 15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Petrosyan V., Holder M., Ajami N.J., Petrosino J.F., Sahasrabhojane P., Thompson E.J., Kalia A., Shelburne S.A.. Complete genome sequence of Streptococcus mitis strain SVGS_061 isolated from a neutropenic patient with viridans group Streptococcal shock syndrome. Genome Announc. 2016; 4:e00259-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. VanZile M.L., Chen X., Giedroc D.P.. Structural characterization of distinct alpha3N and alpha5 metal sites in the cyanobacterial zinc sensor SmtB. Biochemistry. 2002; 41:9765–9775. [DOI] [PubMed] [Google Scholar]
- 37. Apuy J.L., Chen X., Russell D.H., Baldwin T.O., Giedroc D.P.. Ratiometric pulsed alkylation/mass spectrometry of the cysteine pairs in individual zinc fingers of MRE-binding transcription factor-1 (MTF-1) as a probe of zinc chelate stability. Biochemistry. 2001; 40:15164–15175. [DOI] [PubMed] [Google Scholar]
- 38. Senko M.W., Beu S.C., McLaffertycor F.W.. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 1995; 6:229–233. [DOI] [PubMed] [Google Scholar]
- 39. Ray A., Edmonds K.A., Palmer L.D., Skaar E.P., Giedroc D.P.. Staphylococcus aureus glucose-induced biofilm accessory protein A (GbaA) is a monothiol-dependent electrophile sensor. Biochemistry. 2020; 59:2882–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996; 237:260–273. [DOI] [PubMed] [Google Scholar]
- 41. Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993; 26:795–800. [Google Scholar]
- 42. Vagin A., Lebedev A.. MoRDa, an automatic molecular replacement pipeline. Acta Crystallogr. Sect. A: Found. Adv. 2015; 71:s19. [Google Scholar]
- 43. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D: Biol Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Salzmann M., Pervushin K., Wider G., Senn H., Wuthrich K.. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:13585–13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hyberts S.G., Takeuchi K., Wagner G.. Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J. Am. Chem. Soc. 2010; 132:2145–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 47. Hyberts S.G., Milbradt A.G., Wagner A.B., Arthanari H., Wagner G.. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson gap scheduling. J. Biomol. NMR. 2012; 52:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maciejewski M.W., Schuyler A.D., Gryk M.R., Moraru II, Romero P.R., Ulrich E.L., Eghbalnia H.R., Livny M., Delaglio F., Hoch J.C. NMRbox: a resource for biomolecular NMR computation. Biophys. J. 2017; 112:1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abraham M.J., Murtola T., Schulz R., Pall S., Smith J.C., Hess B., Lindahl E.. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1-2:19–25. [Google Scholar]
- 51. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C.. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jorgensen W. L., Chandrasekhar J., Madura J.D.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926. [Google Scholar]
- 53. Ryckaert J.-P., Ciccotti G., Berendsen H.J.C.. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977; 23:327–341. [Google Scholar]
- 54. Salomon-Ferrer R., Case D.A., Walker R.C.. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013; 3:198–210. [Google Scholar]
- 55. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 56. Zallot R., Oberg N., Gerlt J.A.. The EFI web resource for genomic enzymology tools: leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry. 2019; 58:4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ma Z., Cowart D.M., Scott R.A., Giedroc D.P.. Molecular insights into the metal selectivity of the copper(I)-sensing repressor CsoR from Bacillus subtilis. Biochemistry. 2009; 48:3325–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sakamoto K., Agari Y., Agari K., Kuramitsu S., Shinkai A.. Structural and functional characterization of the transcriptional repressor CsoR from Thermus thermophilus HB8. Microbiology. 2010; 156:1993–2005. [DOI] [PubMed] [Google Scholar]
- 59. Herring C.D., Blattner F.R.. Global transcriptional effects of a suppressor tRNA and the inactivation of the regulator frmR. J. Bacteriol. 2004; 186:6714–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Manzari C., Chiara M., Costanza A., Leoni C., Volpicella M., Picardi E., D’Erchia A.M., Placido A., Trotta M., Horner D.S.et al.. Draft genome sequence of Sphingobium sp. strain ba1, resistant to kanamycin and nickel ions. FEMS Microbiol. Lett. 2014; 361:8–9. [DOI] [PubMed] [Google Scholar]
- 61. Zhu T., Tian J., Zhang S., Wu N., Fan Y.. Identification of the transcriptional regulator NcrB in the nickel resistance determinant of Leptospirillum ferriphilum UBK03. PLoS One. 2011; 6:e17367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Foster A.W., Pernil R., Patterson C.J., Scott A.J., Palsson L.O., Pal R., Cummins I., Chivers P.T., Pohl E., Robinson N.J.. A tight tunable range for Ni(II) sensing and buffering in cells. Nat. Chem. Biol. 2017; 13:409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lu T., Cao Q., Pang X., Xia Y., Xun L., Liu H.. Sulfane sulfur-activated actinorhodin production and sporulation is maintained by a natural gene circuit in Streptomyces coelicolor. Microb. Biotechnol. 2020; 13:1917–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reddie K.G., Carroll K.S.. Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 2008; 12:746–754. [DOI] [PubMed] [Google Scholar]
- 65. Benchoam D., Semelak J.A., Cuevasanta E., Mastrogiovanni M., Grassano J.S., Ferrer-Sueta G., Zeida A., Trujillo M., Moller M.N., Estrin D.A.et al.. Acidity and nucleophilic reactivity of glutathione persulfide. J. Biol. Chem. 2020; 295:15466–15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zallot R., Oberg N.O., Gerlt J.A.. Democratized’ genomic enzymology web tools for functional assignment. Curr. Opin. Chem. Biol. 2018; 47:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lanie J.A., Ng W.L., Kazmierczak K.M., Andrzejewski T.M., Davidsen T.M., Wayne K.J., Tettelin H., Glass J.I., Winkler M.E.. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 2007; 189:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Doern C.D., Burnham C.A.. It's not easy being green: the viridans group streptococci, with a focus on pediatric clinical manifestations. J. Clin. Microbiol. 2010; 48:3829–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ooi X.J., Tan K.S.. Reduced glutathione mediates resistance to H2S toxicity in oral Streptococci. Appl. Environ. Microbiol. 2016; 82:2078–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tan B.G., Vijgenboom E., Worrall J.A.. Conformational and thermodynamic hallmarks of DNA operator site specificity in the copper sensitive operon repressor from Streptomyces lividans. Nucleic Acids Res. 2014; 42:1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reisz J.A., Bechtold E., King S.B., Poole L.B., Furdui C.M.. Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 2013; 280:6150–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shen Y., Bax A.. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR. 2013; 56:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peng H., Shen J., Edmonds K.A., Luebke J.L., Hickey A.K., Palmer L.D., Chang F.J., Bruce K.A., Kehl-Fie T.E., Skaar E.P.et al.. Sulfide homeostasis and nitroxyl intersect via formation of reactive sulfur species in Staphylococcus aureus. mSphere. 2017; 2:e00082-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chang F.M., Martin J.E., Giedroc D.P.. Electrostatic occlusion and quaternary structural ion pairing are key determinants of Cu(I)-mediated allostery in the copper-sensing operon repressor (CsoR). Biochemistry. 2015; 54:2463–2472. [DOI] [PubMed] [Google Scholar]
- 75. Chang F.M., Lauber M.A., Running W.E., Reilly J.P., Giedroc D.P.. Ratiometric pulse-chase amidination mass spectrometry as a probe of biomolecular complex formation. Anal. Chem. 2011; 83:9092–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Coyne H.J. 3rd, Giedroc D.P.. Backbone resonance assignments of the homotetrameric (48 kD) copper sensor CsoR from Geobacillus thermodenitrificans in the apo- and Cu(I)-bound states: insights into copper-mediated allostery. Biomol. NMR Assign. 2013; 7:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen H., Hu J., Chen P.R., Lan L., Li Z., Hicks L.M., Dinner A.R., He C.. The Pseudomonas aeruginosa multidrug efflux regulator MexR uses an oxidation-sensing mechanism. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:13586–13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xuan G., Lu C., Xu H., Chen Z., Li K., Liu H., Xia Y., Xun L.. Sulfane sulfur is an intrinsic signal activating MexR-regulated antibiotic resistance in Pseudomonas aeruginosa. Mol. Microbiol. 2020; 114:1038–1048. [DOI] [PubMed] [Google Scholar]
- 79. Fritsch V.N., Loi V.V., Busche T., Sommer A., Tedin K., Nurnberg D.J., Kalinowski J., Bernhardt J., Fulde M., Antelmann H.. The MarR-type repressor MhqR confers quinone and antimicrobial resistance in Staphylococcus aureus. Antioxid. Redox. Signal. 2019; 31:1235–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Baker J.L., Derr A.M., Karuppaiah K., MacGilvray M.E., Kajfasz J.K., Faustoferri R.C., Rivera-Ramos I., Bitoun J.P., Lemos J.A., Wen Z.T.et al.. Streptococcus mutans NADH oxidase lies at the intersection of overlapping regulons controlled by oxygen and NAD+ levels. J. Bacteriol. 2014; 196:2166–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pericone C.D., Park S., Imlay J.A., Weiser J.N.. Factors contributing to hydrogen peroxide resistance in Streptococcus pneumoniae include pyruvate oxidase (SpxB) and avoidance of the toxic effects of the fenton reaction. J. Bacteriol. 2003; 185:6815–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lisher J.P., Tsui H.T., Ramos-Montanez S., Hentchel K.L., Martin J.E., Trinidad J.C., Winkler M.E., Giedroc D.P.. Biological and chemical adaptation to endogenous hydrogen peroxide production in Streptococcus pneumoniae D39. mSphere. 2017; 2:e00291-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hoffmann O., Zweigner J., Smith S.H., Freyer D., Mahrhofer C., Dagand E., Tuomanen E.I., Weber J.R.. Interplay of pneumococcal hydrogen peroxide and host-derived nitric oxide. Infect. Immun. 2006; 74:5058–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhu R., Zhang G., Jing M., Han Y., Li J., Zhao J., Li Y., Chen P.R.. Genetically encoded formaldehyde sensors inspired by a protein intra-helical crosslinking reaction. Nat. Commun. 2021; 12:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gray M.J., Li Y., Leichert L.I., Xu Z., Jakob U.. Does the transcription factor NemR use a regulatory sulfenamide bond to sense bleach. Antioxid. Redox. Signal. 2015; 23:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ma Z., Cowart D.M., Ward B.P., Arnold R.J., DiMarchi R.D., Zhang L., George G.N., Scott R.A., Giedroc D.P.. Unnatural amino acid substitution as a probe of the allosteric coupling pathway in a mycobacterial Cu(I) sensor. J. Am. Chem. Soc. 2009; 131:18044–18045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Reyes-Caballero H., Guerra A.J., Jacobsen F.E., Kazmierczak K.M., Cowart D., Koppolu U.M., Scott R.A., Winkler M.E., Giedroc D.P.. The metalloregulatory zinc site in Streptococcus pneumoniae AdcR, a zinc-activated MarR family repressor. J. Mol. Biol. 2010; 403:197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structural factors have been submitted to the Protein Data Bank with accession codes 7MQ1 (C9A SpCstR), 7MQ2 (C9A SeMet SpCstR) and 7MQ3 (C9A/N55A SpCstR). Backbone Chemical shift assignments have been deposited at the BMRB under accession codes 50893 and 50894 for C9A and C9A/N55A SpCstRs, respectively. All data are available in the manuscript, with strains available upon request.