

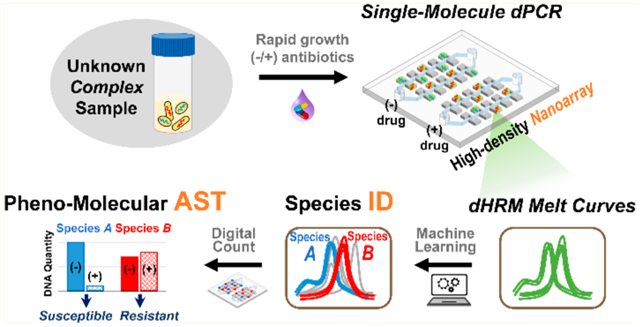
Abstract
Toward combating infectious diseases caused by pathogenic bacteria, there remains an unmet need for diagnostic tools that can broadly identify the causative bacteria and determine their antimicrobial susceptibilities from complex and even polymicrobial samples in a timely manner. To address this need, a microfluidic and machine-learning-based platform that performs broad bacteria identification (ID) and rapid yet reliable antimicrobial susceptibility testing (AST) is developed. Specifically, this platform builds on “pheno–molecular AST”, a strategy that transforms nucleic acid amplification tests (NAATs) into phenotypic AST through quantitative detection of bacterial genomic replication, and utilizes digital polymerase chain reaction (PCR) and digital high-resolution melt (HRM) to quantify and identify bacterial DNA molecules. Bacterial species are identified using integrated experiment–machine learning algorithm via HRM profiles. Digital DNA quantification allows for rapid growth measurement that reflects susceptibility profiles of each bacterial species within only 30 min of antibiotic exposure. As a demonstration, multiple bacterial species and their susceptibility profiles in a spiked-in polymicrobial urine specimen were correctly identified with a total turnaround time of ~4 h. With further development and clinical validation, this platform holds the potential for improving clinical diagnostics and enabling targeted antibiotic treatments.
Graphical Abstract
Infectious diseases inflicted by pathogenic bacteria continue to threaten human health and incur heavy economical burdens. For example, sepsis is the leading cause of deaths in hospitals1,2 and approximately 270 000 Americans die from sepsis each year.3 Similarly, urinary tract infections (UTIs) affect approximately 50% of women at least once in their lifetime and incur >$2 billion in treatment cost in the United States per year.4–8 Adding severity to the problem, recent reports reveal that these bacterial infections (e.g., UTIs9–12 and wound infections13–15) can be polymicrobial, often leading to increased infection severity and poorer patient outcome.16–21 Despite this looming threat, diagnostic methods that rely heavily on bulk bacterial culture and can take several days or even up to weeks to identify the causative bacteria and test their antibiotic susceptibility22 remain as the standard. The lengthy lag to definitive diagnosis results in the common use of broad-spectrum antibiotics, which can lead to poor patient outcome and rampant spread of antimicrobial resistance.23,24 Thus, there remains a critical need for rapid diagnostic tools capable of broad bacteria identification (ID) antimicrobial susceptibility testing (AST) to combat infectious diseases.25
Nucleic acid amplification tests (NAATs) provide an effective foundation for accelerating bacteria ID.26–32 Specifically, employing polymerase chain reaction (PCR) for bacteria ID—now a turnkey process—can be achieved either via a multiplexed assay with a panel of bacterial species-specific primers or probes (as employed in commercial platforms such FilmArray from BioFire Diagnostics, BD Max from Becton Dickinson Diagnostics, and GeneXpert from Cepheid) or via a universal assay with a pair of panbacteria primers coupled with post-PCR analysis techniques such as high-resolution melt (HRM). In particular, the combination of universal PCR and HRM has received significant research attention33–41 due to its capacity for broad-based detection and potential for broad bacterial ID beyond the restriction of primer panels, once a database of bacterial species-specific melt curves is built. In contrast to the maturity of bacteria ID, incorporating NAATs in AST remains a work in progress. This is because, except for a few well-established markers (e.g., mecA42–44 and vanA and vanB45,46), resistance genes cannot reliably predict susceptibility to a particular antibiotic.47,48 Therefore, new NAAT-based methods that not only enable broad bacteria ID but also provide reliable AST information must be developed.
“Pheno–molecular AST” is an emerging approach that combines reliable growth-based (i.e., phenotypic) AST with quantitative, nucleic-acids-based molecular detection of bacteria.22,49–59 In pheno–molecular AST, bacteria are first briefly incubated in the presence and absence of antibiotics. The amounts of bacterial nucleic acids—serving as surrogates of bacterial growths—between antibiotic-treated samples and no-antibiotic controls are then quantitatively detected and compared to reveal antibiotic susceptibilities. To date, researchers have developed “single-organism” pheno–molecular AST assays based on real-time PCR,49 digital PCR,54 and microfluidic digital LAMP.55 Notably, Schoepp et al.54,55 leveraged the digital-level DNA quantification to shorten the antibiotic exposure time to 15–30 min, though they only focused on Escherichia coli as the target organism. Meanwhile, we and others50,56,57 have combined pheno–molecular AST with real-time, quantitative universal PCR–HRM to enable broad bacteria ID, though these bulk-based assays have a limited ability of analyzing polymicrobial samples because composite melt curves from multiple bacteria species cannot be easily decoupled and resolved. Currently, however, no platform can simultaneously achieve pheno–molecular AST, broad bacteria ID, and analysis of polymicrobial samples. This limitation must be overcome to further advance pheno–molecular AST toward clinical applications.
In response, we have developed the first universal digital PCR and HRM (dPCR–HRM) platform for performing broad bacteria ID and rapid pheno–molecular AST toward clinical diagnosis of infectious diseases. At the center of our platform is the nanoarray—a microfluidic device that we have engineered to perform dPCR–HRM upon digitizing single bacterial DNA molecules. In doing so, our nanoarray not only facilitates precise quantification of bacterial DNA molecules but also ensures that each melt curve in the device is generated from a single bacterial species, which allows us to identify individual bacterial species even from polymicrobial samples. We have also developed a machine-learning-based algorithm for the analysis of digital HRM (dHRM) profile from each bacterial DNA to achieve species identification and accurate quantification. Antibiotic susceptibility of each species can thus be determined based on digital counts of DNA that reflects bacterial growth under antibiotic exposure. The quantitative precision of our dPCR–HRM platform allows for rapid antibiotic exposure time in as little as 30 min. Finally, to illustrate the potential of our platform, we correctly identified both gentamicin-sensitive E. coli and gentamicin-resistant Staphylococcus aureus in a polymicrobial urine sample in ~4 h.
EXPERIMENTAL SECTION
Design of Nanoarray Devices.
The nanoarray is composed of three separated but identical modules. Each module has an inlet and an outlet to facilitate sample loading and partitioning and houses a total of 5040 nanowells of 125 μm in length × 100 μm in width × 80 μm in height (making 1 nL in volume/nanowell, Figure S1). Importantly, the array area of the device is designed to have an “ultrathin” profile60 to minimize sample evaporation during thermocycling and to reduce background fluorescence. Loading and digitization of PCR mixtures in nanoarrays were achieved via vacuum-assisted loading and oil-driven digitization.60,61
Overview of Bacteria ID and Pheno–Molecular AST in the Nanoarray.
To perform digital-based bacterial ID/AST in nanoarrays, we began with sample preparation where a bacterial sample was divided into two portions: one portion was added with the antibiotic of interest, while the other portion was without antibiotics and served as the no-drug control. Both portions were then briefly incubated and followed by DNA extraction. Extracted DNA was mixed with PCR mixture and loaded into nanoarrays to perform dPCR–HRM. Finally, the results were analyzed to achieve bacterial ID and AST (Figure S2). Each of these steps is described in detail in the Supplementary Methods section of the Supporting Information.
RESULTS AND DISCUSSION
Assay Overview.
We have developed a streamlined, dPCR–HRM-based workflow for performing broad bacteria ID and pheno–molecular AST from complex samples. We begin by evenly dividing the sample into two aliquots, exposing one aliquot to an antibiotic and the other to no antibiotics (i.e., no-drug control), and incubating both aliquots at 37 °C for 30 min (Figure 1, step 1). Following incubation, bacterial DNA from both aliquots are extracted in parallel, diluted (if necessary), mixed with universal PCR mixture, and loaded into two independent modules on a nanoarray device composed of thousands of nanoliter reaction wells (nanowells) (Figure 1, step 2). The use of limiting dilutions with nanoarrays leads to digitization of DNA molecules in the nanowells for dPCR and dHRM and that each melt curve is generated from a single bacterial species. In doing so, even multiple species in heterogeneous samples can be individually and independently enumerated. Next, we place the device onto our custom thermal–optical platform60 (Figure S3), which performs thermocycling for dPCR and temperature ramping for dHRM, and acquires fluorescence images of the entire nanoarray at all temperature increments during dHRM. These “temperature-lapse” fluorescence images are then compiled to generate digital melt curves for all nanowells within the device in parallel. Each digital melt curve in a nanowell is analyzed via our in-house, machine-learning-based melt curve identification algorithm to match to the species-specific melt curves in our digital melt curve database, thereby identifying the bacterial species present in the nanowell (Figure 1, step 3, ID). The total number of bacterial species-specific DNA molecules from each aliquot can then be accurately counted. Finally, antibiotic susceptibility of each bacterial species is determined by comparing the number of DNA molecules of the drug-treated aliquot with the no-drug control aliquot, where significantly fewer DNA counts indicate susceptibility to the antibiotic while comparable DNA counts indicate resistance to the antibiotic (Figure 1, step 3, AST).
Figure 1.
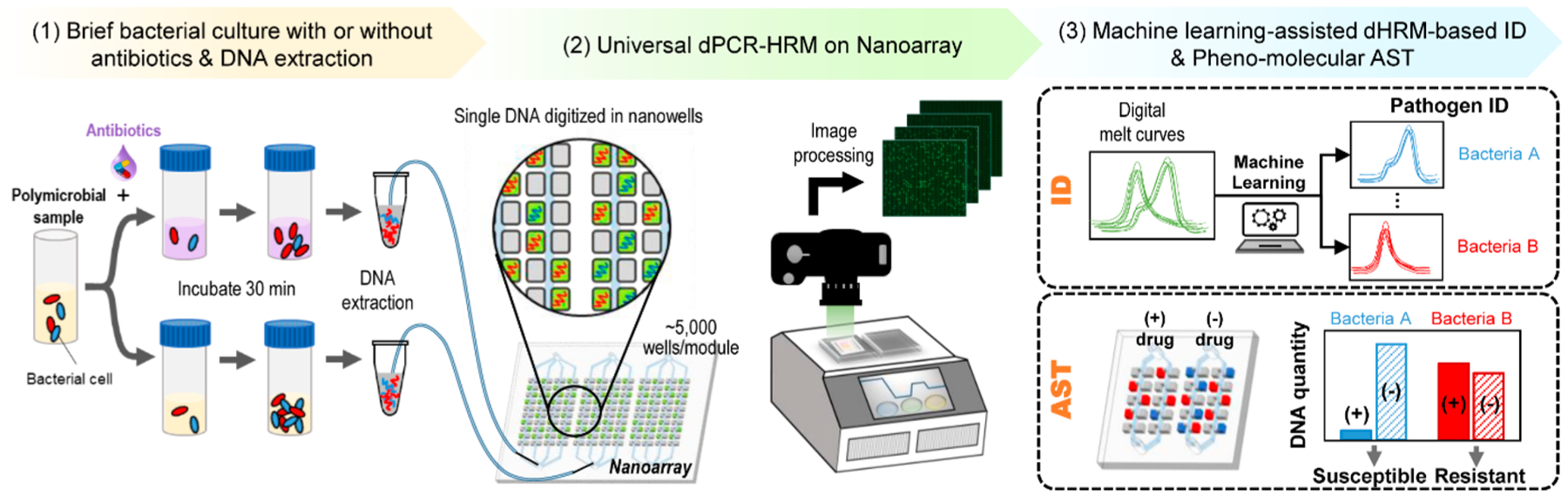
Overview of digital PCR and melt platform with machine-learning-assisted algorithm for rapid bacteria ID and pheno–molecular AST. The streamlined workflow begins with (1) briefly incubating evenly divided bacterial sample aliquots with and without antibiotics and extracting bacterial DNA from both aliquots. (2) Bacterial DNA from each aliquot is then mixed with a universal PCR mixture that contains panbacteria primers and Evagreen dye, loaded into separate modules of a nanoarray device, and placed on a thermal–optical platform to perform dPCR–HRM. (3) After dPCR–HRM, fluorescence intensities within all nanowells are extracted from these “temperature-lapse” fluorescence images to generate digital melt curves for all nanowells. The digital melt curves are analyzed by a machine-learning-based melt curve identification algorithm to identify the bacterial species. Next, the number of species-specific digital melt curves is enumerated to quantify the DNA copy number for each bacterial species in each aliquot. Finally, the comparison between the DNA copy numbers of the two aliquots reveals the antibiotic susceptibility.
Validation of dPCR–HRM in the Nanoarray and Construction of the Digital Melt Curve Database.
An enabling element of our platform is the nanoarray—a microfluidic device that we have engineered to rapidly and efficiently digitize single bacterial DNA molecules and reliably perform dPCR–HRM. Our nanoarray features three identical but independent modules; each of these high-density modules houses 5040 1 nL nanowells (Figure 2A). The three parallel modules within a single device allow us to analyze both drug-treated and no-drug AST conditions while performing a control dPCR–HRM with a well-characterized DNA sample. Within the nanoarray, we have implemented a universal dPCR–HRM assay with PCR primers that hybridize to conserved regions (i.e., consistent among all bacteria) flanking the V1–V6 hypervariable region33,62–66 (i.e., unique to each bacterial species) in the 16S rRNA gene and Evagreen dye that facilitates HRM analysis. We first validated our universal dPCR–HRM assay in the nanoarray using E. coli genomic DNA as the target. In this initial validation, we loaded the target DNA and PCR mixture into a module of a nanoarray device at a “digital concentration” such that only some of nanowells would be filled with a single copy of E. coli DNA. After dPCR, we indeed observed many “negative” nanowells that showed weak green fluorescence signal comparable to only surrounding background and channels, indicating that no dPCR occurred in these nanowells as they contained no target DNA (Figure 2B, left). Importantly, we also detected a number of “positive” nanowells that exhibited strong green fluorescence, suggesting that a single copy of the target DNA was digitized and amplified in each of these nanowells. Subsequently during dHRM, temperature-lapse fluorescence images of the entire module revealed that fluorescence signals in all positive nanowells decreased as the temperature ramped up, indicating that DNA amplicons in these nanowells became increasingly melted (Figure 2B). After extracting fluorescence intensities within nanowells from the temperature-lapse fluorescence images (Figure S4), we obtained hundreds of high-resolution digital melt curves for E. coli (Figure 2C, E. coli, blue). These E. coli digital melt curves closely resembled each other, indicating they originated from the same bacterial species. Importantly, they also resembled the melt curve of E. coli obtained from a benchtop PCR–HRM (Figure S5). These results provide strong validation for our universal dPCR–HRM in the nanoarray.
Figure 2.
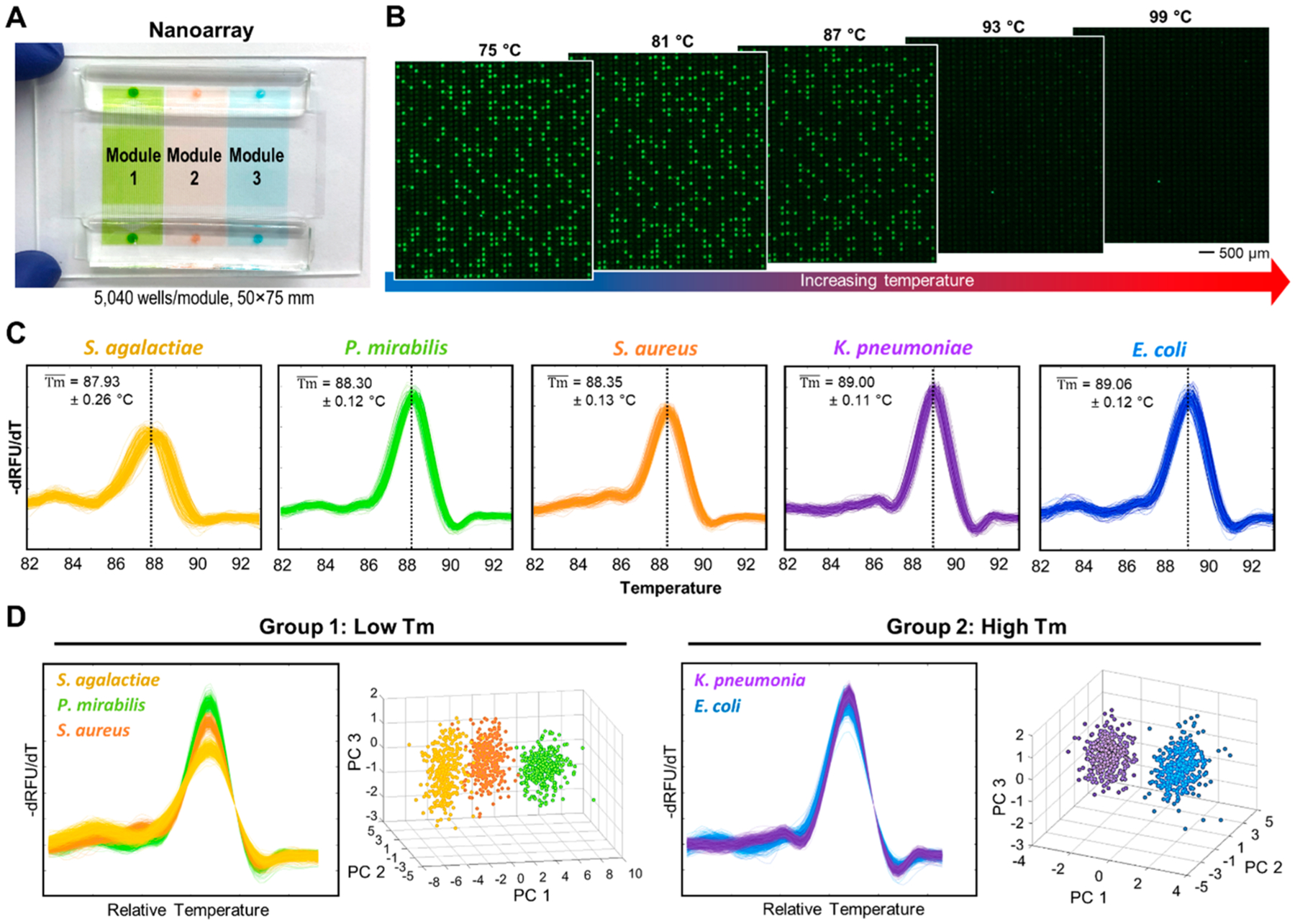
Bacteria ID via dPCR–HRM in the nanoarray and machine-learning-assisted digital melt curve identification. (A) Each nanoarray device contains three independent modules; each houses 5040 1 nL nanowells. (B) During dHRM, double-stranded dPCR products in these strongly fluorescent, positive wells become increasingly melted as temperature increases, resulting in decreasing fluorescence intensities in these nanowells. (C) A total of 320 digital melt curves from five species of bacteria commonly found in urinary tract infections—S. agalactiae, P. mirabilis, S. aureus, K. pneumoniae, and E. coli—are collected to build a digital melt curve database toward broad bacteria ID. (D) To achieve reliable bacteria ID, both the melting temperature (Tm) and the shape of the digital melt curves are used for analysis. On the basis of Tm, our digital melt curve database is divided into the low-Tm group with S. agalactiae, P. mirabilis, and S. aureus and the high-Tm group with K. pneumoniae and E. coli. Within each Tm group, digital melt curves are aligned to a single point to facilitate shape-based digital melt curve analysis before a one-versus-one support vector machine algorithm is used to compare species-specific melt curve shapes and identify bacterial species. Principle component analysis using the first three principle components (i.e., PC1, PC2, and PC3) is performed to visualize that the digital melt curves from each species indeed cluster into distinguishable populations.
We then performed dPCR–HRM in our nanoarray to detect and generate digital melt curves for four additional bacteria, thereby building a digital melt curve database with five common UTI bacterial species toward bacteria identification. As expected, our universal dPCR–HRM successfully detected genomic DNA from Streptococcus agalactiae (ATCC 13813), Proteus mirabilis (ATCC 12453), S. aureus (ATCC 29213), and Klebsiella pneumoniae (ATCC BAA-1705) (Figure S6) and produced hundreds of digital melt curves for each species (Figure 2C) that exhibit similar profiles as their respective benchtop melt curves (Figure S5). For all five species, the digital melt curves universally exhibited the single main peak profile—an indication that the melt curves were derived from single species. Importantly, however, the digital melt curves of each species differed from those of the other four species in their melting temperature (Tm) and their shape. For example, the digital melt curves from E. coli (Figure 2C, blue) and P. mirabilis (Figure 2C, green) showed similar shapes but differed by nearly 0.8 °C in average Tm. The digital melt curves from S. aureus (Figure 2C, orange) displayed a distinctive ramp leading to the main peak. The digital melt curves from S. agalactiae (Figure 2C, yellow) exhibited a small bump before a low main peak. Finally, the digital melt curves from K. pneumoniae (Figure 2C, purple) had a small but noticeable drop immediately before the main peak. These differences form the basis for achieving bacteria ID based on their unique melt curves.
Machine-Learning-Assisted Algorithm for Digital Melt Curve and Bacteria ID.
Having built our digital melt curve database, we next developed a new identification algorithm for distinguishing and identifying these species-specific digital melt curves. As part of our algorithm, we first concurrently perform in our three-module devices dPCR–HRM for two samples of interest (e.g., antibiotic-treated sample and no-antibiotic control) and purified genomic DNA of E. coli, whose digital melt curves serve as in situ references. These reference digital melt curves were used to mitigate undesirable experiment-to-experiment variations, enabling us to compare digital melt curves that were obtained from different nanoarray devices through separate experiments. Specifically, we use differences in Tm between the digital melt curves of interest and the reference digital melt curves (i.e., relative Tm) to classify the digital melt curves of interest into groups that contain a subset of the bacterial species within the database (Figure 2D and Figure S7). Next, we normalize the area under the curve of all digital melt curves, and then align them to a single point to facilitate the identification of digital melt curves of interest based on their shapes. Finally, using an adapted in-house developed digital melt curve classification tool based on one-versus-one support vector machine (ovoSVM) algorithm,33,57,67 the shape of each digital melt curve of interest is compared to the digital melt curves in the Tm-classified group to identify the bacterial species represented by the digital melt curve of interest.
Using a total of 1600 digital melt curves—320 from each of the five bacterial species in our database—we illustrate the outcomes of the key steps and demonstrate the overall performance of our machine-learning-assisted algorithm in bacteria identification. In our database, classification of our digital melt curves based on relative Tm results in the low-Tm group with S. agalactiae, P. mirabilis, and S. aureus and the high-Tm group with K. pneumoniae and E. coli (Figure 2D). To show that the digital melt curves within each group have distinct shapes, we performed principle component analysis and indeed observed distinct S. agalactiae, P. mirabilis, and S. aureus clusters in the low-Tm group results (Figure 2D, left) and distinct K. pneumoniae and E. coli clusters in the high-Tm group (Figure 2D, right). Notably, our principle component analysis shows that, when put in a single group, the digital melt curves of the five species are not fully distinguished (Figure S8). The analysis therefore confirms the importance of relative Tm-based classification toward robust identification. Finally, we iteratively performed 320 leave-one-out cross-validation experiments to validate our identification algorithm. That is, for each bacterial species, we trained our algorithm using 319 digital melt curves, and then challenged our algorithm to identify the last digital melt curve, and repeated until all 320 digital melt curves had been tested. Our identification algorithm correctly identified 98.5% of the 1600 digital melt curves, illustrating the effectiveness of our machine-learning-assisted algorithm in the identification of digital melt curves, and hence, bacterial species.
Bacterial DNA Quantification in the Nanoarray.
Precise quantification of DNA molecules is essential for pheno–molecular AST. To this end, our nanoarray supports digital quantification of DNA molecules through both dPCR alone and dPCR–HRM. Notably, our dPCR–HRM adds the dHRM-based verification step that can remove false-positives from the counting results (Figure S9). To demonstrate quantification of DNA molecules at concentrations relevant to our subsequent pheno–molecular AST in the nanoarray, we performed dPCR–HRM for three dilutions of purified E. coli genomic DNA at estimated 0.01, 0.1, and 1 genomic copy per nanowell (Figure 3A), Using dPCR–HRM for analysis, we counted 5.3%, 39.5%, and 99.2% positives for these three dilutions (Figure 3B), comparable to the 5.4%, 40.1%, and 99.3% positives for these three dilutions that we quantified via dPCR alone (Figure S10).
Figure 3.
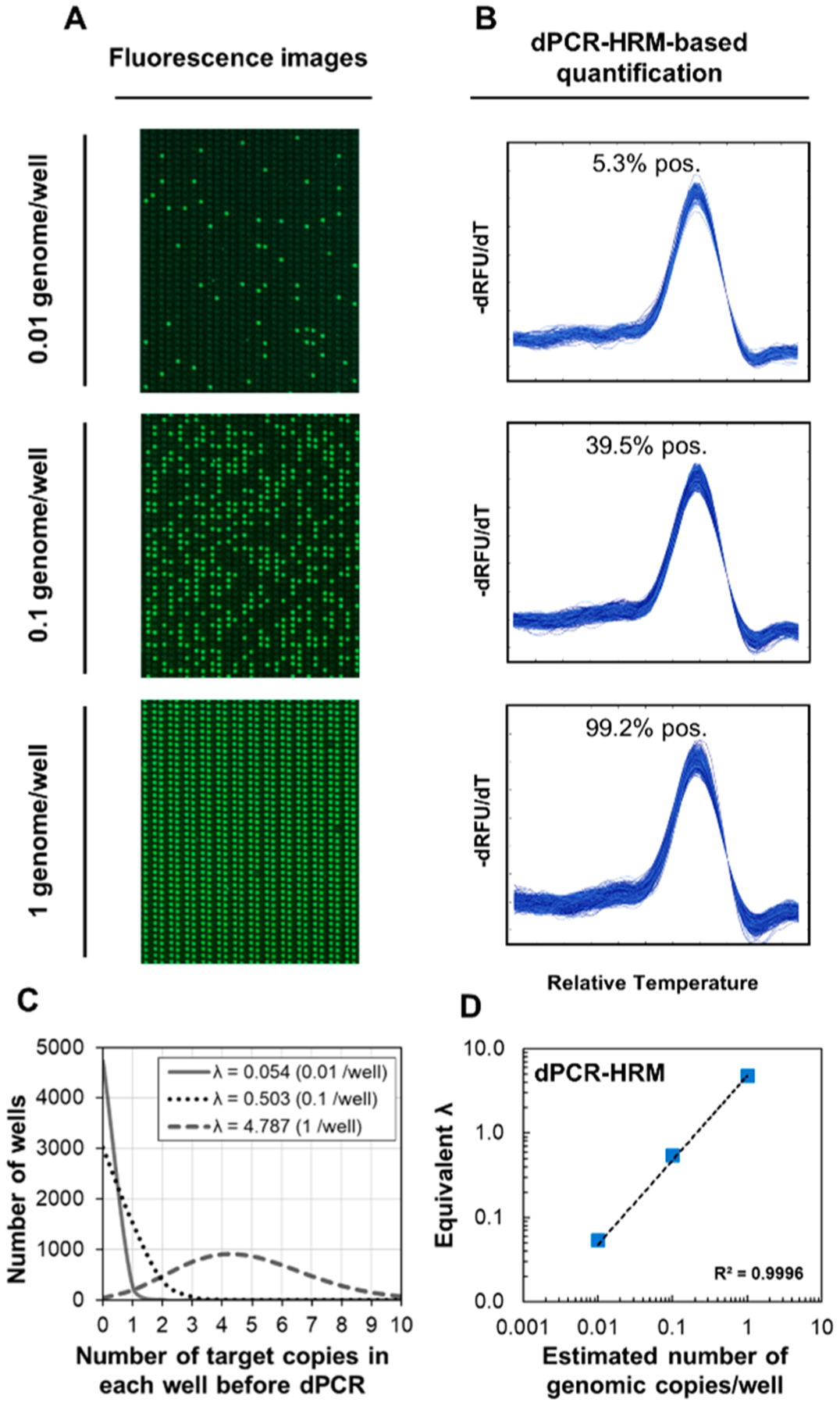
Digital bacterial DNA quantification in the nanoarray. (A) Fluorescence images after performing dPCR for three concentrations of E. coli genomic DNA are shown. (B) dPCR–HRM-based quantification relies on counting the number of correctly identified digital melt curves, which can enhance the assay specificity. (C) The percentage of positive nanowells from of each concentration can be used to calculate the corresponding mean occupancy (λ) via Poisson distribution. (D) A linear relationship between the estimated number of genomic copy per nanowell and λ can be observed from these three input concentrations quantified via dPCR–HRM.
Consistent with standard digital-based quantification methods, we can calculate the quantity of our E. coli target based on the percent positive for each dilution. This calculation corrects for positive nanowells that contain more than one copy of the target. Specifically, we applied Poisson distribution to calculate the mean occupancy of DNA per nanowell, λ, using eq 1
| (1) |
where p represents the percent positives. Hereafter, we used λ as our standard quantitation metric. Using the percent positives from dPCR–HRM, the λʼs were 0.054, 0.503, and 4.787 for the three dilutions. We can also calculate the probability distribution of the number of DNA molecules per nanowell (k) that would result from λ of each dilution calculated from our dPCR–HRM results using eq 2 (Figure 3C, based on a total of 5000 wells):
| (2) |
More importantly, we observed strong linear relationships between λʼs and the input genomic copy numbers (Figure 3D). The excellent linearity provides strong support for precise quantification of DNA molecules in the nanoarray.
dPCR–HRM-Based Pheno–Molecular AST in the Nanoarray.
The capacity for digitally detecting even minute increases in the quantity of DNA molecules via dPCR–HRM in the nanoarray enables brief antibiotic exposure and consequently rapid pheno–molecular AST. To demonstrate this concept, we incubated gentamicin-susceptible and gentamicin-resistant strains of E. coli and S. aureus with gentamicin (a commonly used intravenous antibiotic for infections) at either 0 or 4 μg/mL (the low end of the clinical breakpoint for resistance/susceptibility) for 30 min before quantifying the DNA molecules from each case via dPCR–HRM in the nanoarray. For the gentamicin-susceptible E. coli strain, λ of 0.120 (i.e., 0.120 λ) and significantly lower 0.042 λ were measured for the no-gentamicin control and the gentamicin-treated sample, respectively, suggesting that gentamicin indeed inhibited the growth of this susceptible E. coli (Figure 4A). Conversely, for the gentamicin-resistant E. coli strain, comparable 0.037 λ and 0.033 λ were observed from the no-gentamicin control and the gentamicin-treated sample, respectively, indicating that gentamicin could not inhibit the growth of this resistant E. coli (Figure 4B). By taking the ratio between λ of the drug-treated sample and λ of the no-drug control (i.e., eq 3)
| (3) |
we calculate the “growth ratio” to concisely represent bacterial susceptibility and resistance to drugs. Here, the growth ratios of the gentamicin-susceptible E. coli strain and the gentamicin-resistant E. coli strain were 0.418 ± 0.100 and 0.985 ± 0.124, respectively (Figure 4C). Similarly, for gentamicin-susceptible S. aureus, we measured 0.140 λ and substantially lower 0.026 λ from the no-gentamicin control and the gentamicin-treated sample, respectively (Figure 4D). On the contrary, for gentamicin-resistant S. aureus, we observed similar 0.037 λ and 0.046 λ from the no-gentamicin control and the gentamicin-treated sample, respectively (Figure 4E). The growth ratios of the gentamicin-susceptible S. aureus and the gentamicin-resistant S. aureus were 0.217 ± 0.043 and 1.282 ± 0.024 (Figure 4F). The clearly distinguishable growth ratios demonstrate that, when coupling pheno–molecular AST with dPCR–HRM in the nanoarray, even 30 min of exposure was sufficient for completing the AST that effectively ascertained the susceptibility or resistance of E. coli and S. aureus strains to gentamicin. Of note, to the best of our knowledge, this is the first demonstration of rapid pheno–molecular AST with S. aureus with only 30 min of antibiotic exposure.
Figure 4.
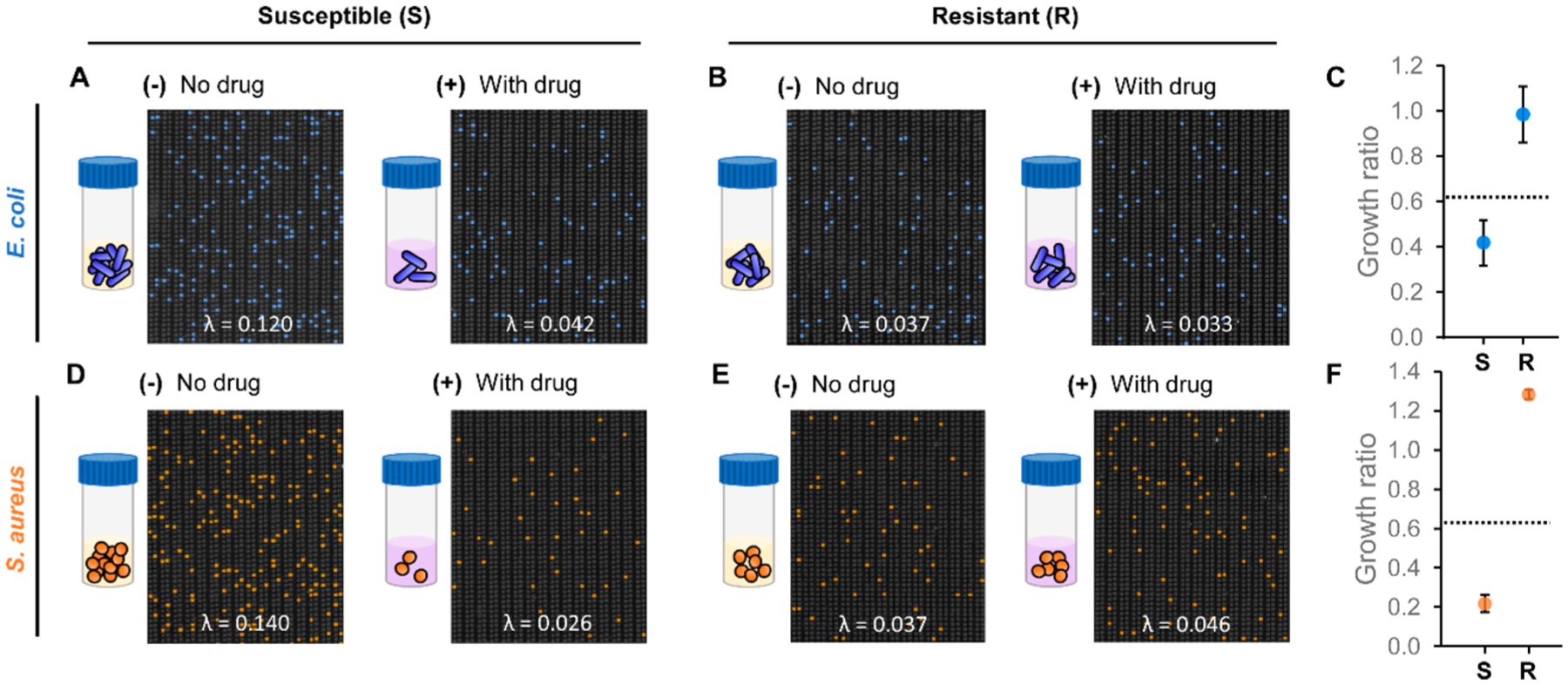
Characterization of pheno–molecular AST with dPCR–HRM-based quantitative analysis in the nanoarray. After 30 min of incubation (A) for susceptible E. coli, significantly more E. coli DNA is measured from the no-gentamicin control aliquot than the gentamicin-treated sample aliquot. (B) For resistant E. coli, comparable amounts of E. coli DNA are measured from both aliquots. (C) The “growth ratios” of susceptible E. coli and resistant E. coli are calculated from the ratio between λ of the gentamicin-treated sample and λ of the no-gentamicin control from duplicate experiments. In the growth ratio plot, the dashed line represents the susceptibility threshold for determining whether the bacteria strain is susceptible (S) or resistant (R) to gentamicin. Similar results are observed for S. aureus in panels D and E. (F) The growth ratios from duplicate experiments of susceptible S. aureus and resistant S. aureus correctly fall below and above the susceptibility threshold.
On the basis of the growth ratios, we established a “susceptibility threshold” for distinguishing resistance from susceptibility for subsequent AST experiments. Our results show that both gentamicin-resistant strains of E. coli and S. aureus both had growth ratios of ~1, whereas both susceptible strains had growth ratios of <1, indicating that gentamicin could effectively prevent their growth. We therefore set the susceptibility threshold that is applicable for both species by subtracting 3 standard deviations from the mean of the growth ratio for the gentamicin-resistant E. coli and obtained 0.614 as our susceptibility threshold. In subsequent experiments, bacteria samples with growth ratios below the susceptibility threshold will be identified as susceptible. Conversely, bacteria samples with growth ratios above the susceptibility threshold will be called resistant.
Bacteria ID and Pheno–Molecular AST from Complex Samples.
By performing dPCR–HRM in the nanoarray, our platform offers unique capacity for analyzing complex samples—samples that may contain multiple species of bacteria in a clinically relevant sample matrix—without additional sample processing or species isolation steps. For initial demonstration, we used the nanoarray to identify and enumerate a polymicrobial sample that contains two bacterial species. This mock polymicrobial sample was generated by mixing S. aureus with P. mirabilis at equivalent amounts. After dPCR–HRM and species identification using our identification algorithm, 0.011 λ were identified as S. aureus and relatively similar amount of 0.012 λ were identified as P. mirabilis (Figure 5A). This result evidently supports that our nanoarray is capable of simultaneously identifying multiple bacterial species in a single sample.
Figure 5.
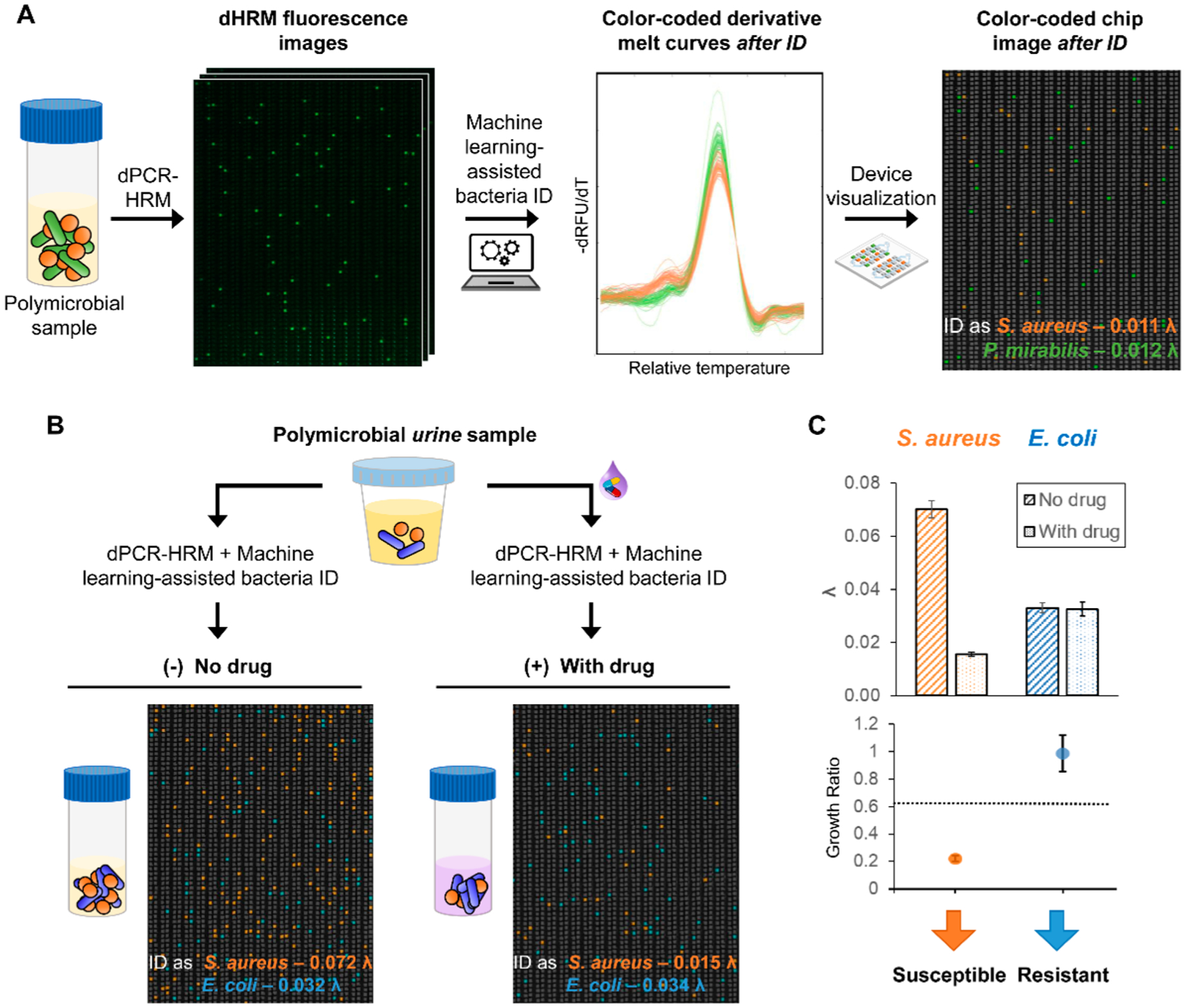
Bacteria ID and pheno–molecular AST from complex samples in the nanoarray. (A) Equal concentrations of S. aureus and P. mirabilis spiked in the same sample are both correctly identified and enumerated by performing dPCR–HRM and machine-learning-assisted bacteria ID. (B) Samples containing 10% urine with spiked-in gentamicin-susceptible S. aureus and gentamicin-resistant E. coli are used for testing. Following pheno–molecular AST, dPCR–HRM, and machine-learning-assisted bacteria ID, color-coded nanoarray images clearly show that not only are both species in the sample identified, but more S. aureus DNA is detected in the no-gentamicin control and comparable amounts of E. coli DNA are detected in both samples. (C) The corresponding λʼs and growth ratios confirm the identity and the gentamicin susceptibility for both gentamicin-susceptible S. aureus and gentamicin-resistant E. coli.
As the final demonstration of our method, we identified two species of bacteria and determined their susceptibilities to gentamicin directly from a simulated polymicrobial urine sample. Here, we created the simulated polymicrobial urine sample by spiking both gentamicin-susceptible S. aureus and gentamicin-resistant E. coli in culture medium laced with 10% culture-negative (i.e., bacteria-free) urine sample (Figure 5B). After 30 min of gentamicin exposure, in a representative no-gentamicin control, 0.072 λ were identified as S. aureus and 0.032 λ were identified as E. coli. On the other hand, from the corresponding gentamicin-treated sample, we identified 0.015 λ as S. aureus and 0.034 λ as E. coli (Figure 5B, representative color-coded nanoarray devices are shown; S. aureus—orange, E. coli—blue). Similarly, from duplicate experiments, growth ratios were calculated to be 0.224 ± 0.022 and 0.991 ± 0.133 for S. aureus and E. coli, respectively (Figure 5C). By comparing these ratios with the susceptibility threshold previously determined for these two bacterial species, S. aureus was determined to be gentamicin-susceptible while E. coli was determined to be gentamicin-resistant—both in accordance with what we expected. These results demonstrate the use of the nanoarray for simultaneously identifying multiple bacterial species and performing AST, even in the presence of urine.
CONCLUSIONS
We have developed the first microfluidic-based universal dPCR and machine-learning-assisted dHRM analysis platform that enables broad bacteria ID and rapid pheno–molecular AST for addressing critical unmet needs in the clinical diagnosis of infectious diseases. We first engineered the nanoarray device and performed dPCR–HRM to broadly detect five common UTI species based on their 16S rRNA gene and generate hundreds of melt curves for each bacterial species in parallel, which have been stored in our digital melt curve database. We subsequently created our machine-learning-based melt curve classification algorithm and used it in tandem with reference melt curves generated directly in each nanoarray device to achieve bacteria ID. We have also shown the capability of the nanoarray in measuring the concentrations of E. coli genomic DNA across three titrations. Precise quantification of bacterial DNA in the nanoarray allowed us to accurately determine susceptibility profiles of gentamicin-susceptible and gentamicin-resistant strains of E. coli and S. aureus via pheno–molecular AST with as little as 30 min of exposure to gentamicin. Leveraging our platform’s unique capability in polymicrobial detection, we analyzed a spiked, polymicrobial urine sample and correctly identified the gentamicin-sensitive E. coli and the gentamicin-resistant S. aureus that were both present in the sample with a turnaround time of ~4 h. These results illustrate the potential of our platform as a comprehensive and rapid diagnostic method for infectious diseases.
We envision to continue improving the performance of our platform. Specifically, although we have introduced several technical and conceptual advances, including polymicrobial detection, in situ reference digital melt curves, high-throughput digital melt curve generation and training, and new digital melt curve classification algorithm, it is imperative to continue simplifying and accelerating our assay. To this end, we can expand the dynamic range of the nanoarray by increasing the number and/or the volume of the nanowells to facilitate direct analysis of bacterial DNA without dilution. This strategy could be particularly useful for the diagnosis of UTIs, for which the relevant bacteria concentrations range from 104 to 107 CFU/mL. Moreover, we can implement a rapid PCR assay in the nanoarray to shorten the turnaround time. We must also continue expanding the number of bacterial species by including other pathogenic species and also common flora in our digital melt curve database to broaden our capacity of bacteria ID. Similarly, for pheno–molecular AST, we must test our platform against bacteria with various growth rates, as well as antibiotics with different mechanisms and at their respective minimum inhibitory concentrations (MICs). Finally, our platform offers unique capacity for diagnosing polymicrobial infections, which present an increasingly relevant clinical challenge, especially for UTIs.10,12,17,18,68,69 For example, up to one in three elderly UTI patients are polymicrobial68 and up to 31% of catheter-associated UTIs especially with long-term catheters are polymicrobial.10,69 We therefore envision testing the performance of our platform in diagnosing polymicrobial infections with different organisms and different input concentrations from urine and potentially other types of clinical samples. Taken together, we believe that our platform, when fully developed, will become a useful diagnostic tool for rapid pathogen identification and antibiotic susceptibility testing from complex specimens, thereby facilitating precision-directed therapy and reducing the spread of antibiotic resistance in the future.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Kathleen E. Mach and Professor Joseph C. Liao from the Veterans Affairs Palo Alto Health Care System for providing the urine specimen and Aniruddha Kaushik for his help in coordinating. We also thank Dr. Helena C. Zec, Dr. Wei Liu, and Dr. Liben Chen for the initial design of the microfluidic devices and the imaging system. This research is supported by the National Institutes of Health [Grant Nos. R01AI117032, R01AI137272, R01AI138978]. K.H. is financially supported through a Hartwell Postdoctoral Fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b02344.
Nanoarray device fabrication, schematic of bacterial ID/AST using the nanoarray digital melt, thermal–optical setup for dHRM analysis, melt curve acquisition and data processing, benchtop- and nanoarray-generated melt curves comparison, fluorescence images of all bacterial species for generating digital melt curve database, dHRM-based bacteria identification using the nanoarray digital melt, principal component analysis of digital melt curves from five bacterial species, no-template control experiment on the nanoarray, dPCR-based quantification in the nanoarray, and supplementary methods (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Jabaley CS; Blum JM; Groff RF; O’Reilly-Shah VN Crit. Care 2018, 22 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Reinhart K; Daniels R; Kissoon N; Machado FR; Schachter RD; Finfer SN Engl. J. Med 2017, 377 (5), 414–417. [DOI] [PubMed] [Google Scholar]
- (3).Rhee C; Dantes R; Epstein L; Murphy DJ; Seymour CW; Iwashyna TJ; Kadri SS; Angus DC; Danner RL; Fiore AE; et al. JAMA 2017, 318 (13), 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brown P; Ki M; Foxman B PharmacoEconomics 2005, 23, 1123–1142. [DOI] [PubMed] [Google Scholar]
- (5).Foxman B; Barlow R; D’Arcy H; Gillespie B; Sobel JD Ann. Epidemiol 2000, 10 (8), 509–515. [DOI] [PubMed] [Google Scholar]
- (6).Dielubanza EJ; Schaeffer AJ Med. Clin. North Am 2011, 95 (1), 27–41. [DOI] [PubMed] [Google Scholar]
- (7).Barber AE; Norton JP; Spivak AM; Mulvey MA Clin. Infect. Dis 2013, 57 (5), 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Simmering JE; Tang F; Cavanaugh JE; Polgreen LA; Polgreen PM Open Forum Infect. Dis 2017, 4 (1), ofw281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Croxall G; Weston V; Joseph S; Manning G; Cheetham P; McNally AJ Med. Microbiol 2011, 60 (1), 102–109. [DOI] [PubMed] [Google Scholar]
- (10).Armbruster CE; Smith SN; Johnson AO; DeOrnellas V; Eaton KA; Yep A; Mody L; Wu W; Mobley HLT Infect. Immun 2017, 85 (2), e00808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kline KA; Schwartz DJ; Gilbert NM; Hultgren SJ; Lewis AL Infect. Immun 2012, 80 (12), 4186–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wolfe AJ; Toh E; Shibata N; Rong R; Kenton K; FitzGerald MP; Mueller ER; Schreckenberger P; Dong Q; Nelson DE; et al. J. Clin. Microbiol 2012, 50 (4), 1376–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bertesteanu S; Triaridis S; Stankovic M; Lazar V; Chifiriuc MC; Vlad M; Grigore R Int. J. Pharm 2014, 463 (2), 119–126. [DOI] [PubMed] [Google Scholar]
- (14).Peters BM; Jabra-Rizk MA; O’May GA; Costerton JW; Shirtliff ME Clin. Microbiol. Rev 2012, 25 (1), 193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bowler PG; Duerden BI; Armstrong DG Clin. Microbiol. Rev 2001, 14 (2), 244–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Short FL; Murdoch SL; Ryan RP Trends Microbiol 2014, 22 (9), 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Brogden KA; Guthmiller JM; Taylor CE Lancet 2005, 365 (9455), 253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tay WH; Chong KKL; Kline KA J. Mol. Biol 2016, 428 (17), 3355–3371. [DOI] [PubMed] [Google Scholar]
- (19).Griffiths EC; Pedersen AB; Fenton A; Petchey OL J. Infect 2011, 63 (3), 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Stacy A; McNally L; Darch SE; Brown SP; Whiteley M Nat. Rev. Microbiol 2016, 14 (2), 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang J; Foxman B; Mody L; Snitkin ES Proc. Natl. Acad. Sci. U. S. A 2017, 114, 10467–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Davenport M; Mach KE; Shortliffe LMD; Banaei N; Wang T-H; Liao JC Nat. Rev. Urol 2017, 14 (5), 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wise R; Hart T; Cars O; Streulens M; Helmuth R; Huovinen P; Sprenger M BMJ 1998, 317 (7159), 609–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013. https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf (accessed September 9, 2019).
- (25).The White House. National Strategy for Combating Antibiotic-Resistant Bacteria, 2015. https://www.cdc.gov/drugresistance/pdf/national_action_plan_for_combating_antibotic-resistant_bacteria.pdf (accessed September 9, 2019).
- (26).Hill J; Beriwal S; Chandra I; Paul VK; Kapil A; Singh T; Wadowsky RM; Singh V; Goyal A; Jahnukainen T; et al. J. Clin. Microbiol 2008, 46 (8), 2800–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Caliendo AM Clin. Infect. Dis 2011, 52 (suppl_4), S326–S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lucignano B; Ranno S; Liesenfeld O; Pizzorno B; Putignani L; Bernaschi P; Menichella DJ Clin. Microbiol 2011, 49 (6), 2252–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Fouad AF; Barry J; Caimano M; Clawson M; Zhu Q; Carver R; Hazlett K; Radolf JD J. Clin. Microbiol 2002, 40 (9), 3223–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dong J; Xu Q; Li CC; Zhang CY Chem. Commun 2019, 55 (17), 2457–2460. [DOI] [PubMed] [Google Scholar]
- (31).Matsuki T; Watanabe K; Fujimoto J; Miyamoto Y; Takada T; Matsumoto K; Oyaizu H; Tanaka R Appl. Environ. Microbiol 2002, 68 (11), 5445–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Váradi L; Luo JL; Hibbs DE; Perry JD; Anderson RJ; Orenga S; Groundwater PW Chem. Soc. Rev 2017, 46 (16), 4818–4832. [DOI] [PubMed] [Google Scholar]
- (33).Fraley SI; Athamanolap P; Masek BJ; Hardick J; Carroll KC; Hsieh YH; Rothman RE; Gaydos CA; Wang TH; Yang S Sci. Rep 2016, 6, 19218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yang S; Ramachandran P; Rothman R; Hsieh YH; Hardick A; Won H; Kecojevic A; Jackman J; Gaydos CJ Clin. Microbiol 2009, 47 (7), 2252–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Perng CL; Chen HY; Chiueh TS; Wang WY; Huang CT; Sun JR J. Med. Microbiol 2012, 61 (7), 944–951. [DOI] [PubMed] [Google Scholar]
- (36).Cheng JC; Huang CL; Lin CC; Chen CC; Chang YC; Chang SS; Tseng CP Clin. Chem 2006, 52 (11), 1997–2004. [DOI] [PubMed] [Google Scholar]
- (37).Andini N; Wang B; Athamanolap P; Hardick J; Masek BJ; Thair S; Hu A; Avornu G; Peterson S; Cogill S; Rothman RE; Carroll KC; Gaydos CA; Wang JT-H; Batzoglou S; Yang S Sci. Rep 2017, 7, 42097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Clifford RJ; Milillo M; Prestwood J; Quintero R; Zurawski DV; Kwak YI; Waterman PE; Lesho EP; Mc Gann P PLoS One 2012, 7 (11), e48558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Masek BJ; Hardick J; Won H; Yang S; Hsieh YH; Rothman RE; Gaydos CA J. Mol. Diagn 2014, 16 (2), 261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Velez DO; Mack H; Jupe J; Hawker S; Kulkarni N; Hedayatnia B; Zhang Y; Lawrence S; Fraley SI Sci. Rep 2017, 7, 42326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tong SYC; Giffard PM J. Clin. Microbiol 2012, 50 (11), 3418–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Francois P; Pittet D; Bento M; Pepey B; Vaudaux P; Lew D; Schrenzel JJ Clin. Microbiol 2003, 41 (1), 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Espy MJ; Uhl JR; Sloan LM; Buckwalter SP; Jones MF; Vetter EA; Yao JDC; Wengenack NL; Rosenblatt JE; Cockerill FR; et al. Clin. Microbiol. Rev 2006, 19 (1), 165–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Huletsky A; Giroux R; Rossbach V; Gagnon M; Vaillancourt M; Bernier M; Gagnon F; Truchon K; Bastien M; Picard FJ; et al. Society 2004, 42 (5), 1875–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Arthur M; Quintiliani RJ Antimicrob. Agents Chemother 2001, 45 (2), 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kristich CJ; Rice LB; Arias CA Enterococcal Infection—Treatment and Antibiotic Resistance. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet]; Gilmore MS, Clewell DB, Shankar N, Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, 2014. [PubMed] [Google Scholar]
- (47).Bard JD; Lee F Clin. Microbiol. Newsl 2018, 40 (11), 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Davies J; Davies D Microbiol. Mol. Biol. Rev 2010, 74 (3), 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chen L; Shin DJ; Zheng S; Melendez JH; Gaydos C; Wang TH ACS Infect. Dis 2018, 4 (9), 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Waldeisen JR; Wang T; Mitra D; Lee LP PLoS One 2011, 6 (12), e28528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Barczak AK; Gomez JE; Kaufmann BB; Hinson ER; Cosimi L; Borowsky ML; Onderdonk AB; Stanley SA; Kaur D; Bryant KF; et al. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (16), 6217–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mezger A; Gullberg E; Göransson J; Zorzet A; Herthnek D; Tano E; Nilsson M; Andersson DI J. Clin. Microbiol 2015, 53 (2), 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Altobelli E; Mohan R; Mach KE; Sin MLY; Anikst V; Buscarini M; Wong PK; Gau V; Banaei N; Liao JC Eur. Urol. Focus 2017, 3 (2–3), 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Schoepp NG; Khorosheva EM; Schlappi TS; Curtis MS; Humphries RM; Hindler JA; Ismagilov RF Angew. Chem., Int. Ed 2016, 55 (33), 9557–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Schoepp NG; Schlappi TS; Curtis MS; Butkovich SS; Miller S; Humphries RM; Ismagilov RF Sci. Transl. Med 2017, 9 (410), eaal3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Andini N; Hu A; Zhou L; Cogill S; Wang TH; Wittwer CT; Yang S Clin. Chem 2018, 64 (10), 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Athamanolap P; Hsieh K; Chen L; Yang S; Wang T-H Anal. Chem 2017, 89 (21), 11529–11536. [DOI] [PubMed] [Google Scholar]
- (58).Rolain JM; Mallet MN; Fournier PE; Raoult DJ Antimicrob. Chemother 2004, 54 (2), 538–541. [DOI] [PubMed] [Google Scholar]
- (59).Beuving J; Verbon A; Gronthoud FA; Stobberingh EE; Wolffs PFG PLoS One 2011, 6 (12), e27689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).O’Keefe CM; Pisanic TR; Zec H; Overman MJ; Herman JG; Wang TH Sci. Adv 2018, 4 (9), eaat6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hsieh K; Zec HC; Chen L; Kaushik AM; Mach KE; Liao JC; Wang T-H Anal. Chem 2018, 90 (15), 9449–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yarza P; Yilmaz P; Pruesse E; Glöckner FO; Ludwig W; Schleifer KH; Whitman WB; Euzéby J; Amann R; Rosselló-Móra R Nat. Rev. Microbiol 2014, 12 (9), 635. [DOI] [PubMed] [Google Scholar]
- (63).Clarridge JE Clin. Microbiol. Rev 2004, 17 (4), 840–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Srinivasan R; Karaoz U; Volegova M; MacKichan J; Kato-Maeda M; Miller S; Nadarajan R; Brodie EL; Lynch SV PLoS One 2015, 10 (2), e0117617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Chakravorty S; Helb D; Burday M; Connell N; Alland DJ Microbiol. Methods 2007, 69 (2), 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Laurent FJ; Provost F; Boiron PJ Clin. Microbiol 1999, 37 (1), 99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Athamanolap P; Parekh V; Fraley SI; Agarwal V; Shin DJ; Jacobs MA; Wang TH; Yang S PLoS One 2014, 9 (9), e109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ronald A Am. J. Med 2002, 113 (1), 14–19. [Google Scholar]
- (69).Armbruster CE; Smith SN; Yep A; Mobley HLT J. Infect. Dis 2014, 209 (10), 1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.