

Abstract
Understanding the microstructural parameters of amphiphilic copolymers that control the formation and structure of aggregated colloids (e.g., micelles) is essential for the rational design of hierarchically structured systems for applications in nanomedicine, personal care and food formulations. Although many analytical techniques have been employed to study such systems, in this investigation we adopted an integrated approach using non-interfering techniques – diffusion nuclear magnetic resonance (NMR) spectroscopy, dynamic light scattering (DLS) and synchrotron small-angle X-ray scattering (SAXS) – to probe the relationship between the microstructure of poly(ethylene glycol-b-caprolactone) (PEG-b-PCL) copolymers [e.g., block molecular weight (MW) and the mass fraction of PCL (fPCL)] and the structure of their aggregates. Systematic trends in the self-assembly behaviour were determined using a large family of well-defined block copolymers with variable PEG and PCL block lengths (number-average molecular weights (Mn) between 2–10 and 0.5–15 kDa, respectively) and narrow dispersity (Ð < 1.12). For all of the copolymers, a clear transition in the aggregate structure was observed when the hydrophobic fPCL was increased at a constant PEG block Mn, although the nature of this transition is also dependent on the PEG block Mn. Copolymers with low Mn PEG blocks (2 kDa) were observed to transition from unimers and loosely associated unimers to metastable aggregates and finally, to defined cylindrical micelles as the fPCL was increased. In comparison, copolymers with PEG block Mn of between 5 and 10 kDa transitioned from heterogenous metastable aggregates to cylindrical micelles and finally, well-defined ellipsoidal micelles (of decreasing aspect ratios) as the fPCL was increased. In all cases, the diffusion NMR spectroscopy, DLS and synchrotron SAXS results provided complementary information and the grounds for a phase diagram relating copolymer microstructure to aggregation behaviour and structure. Importantly, the absence of commonly depicted spherical micelles has implications for applications where properties may be governed by shape, such as, cellular uptake of nanomedicine formulations.
Keywords: poly(ethylene glycol-b-caprolactone) copolymers, self-assembly, aggregated colloid, micelle, aggregate, diffusion NMR spectroscopy, microstructure, small angle X-ray scattering
Graphical Abstract
1. INTRODUCTION
The development of optimal drug delivery systems is a major area of research in the pharmaceutical industry, and provides the opportunity to improve the solubility, stability, bioavailability and efficacy of many therapeutics.1,2 Colloidal polymer drug delivery systems, such as Genexol-PM, have been established as drug delivery platforms3, 4 and currently there are numerous others in the clinical trial pipeline.5, 6 However, systematic evaluation of the impacts of polymer microstructure on the self-assembly behaviour, dispersity and structural attributes (morphology) of aggregates assembled from particular polymers is needed to further advance the development of tailored colloidal drug delivery systems.7–9 Elucidation of the microstructure-assembly relationship for a given polymer system will allow for the precise and reliable preparation of well-defined aggregates with desirable characteristics for the development of new drug delivery platforms.
The microstructural characteristics of an amphiphilic copolymer (composition, MW of hydrophilic and hydrophobic blocks, and the mass fractions of those blocks) and the employed method of micellisation contribute significantly to the self-assembly process, greatly impacting the thermodynamic state and stability of aggregates within a given solution,10–14, and influencing aggregate morphology, size and distribution.15–17 For instance, the hydrodynamic radius (Rh) and aggregation number (Nagg) of poly(ethylene glycol-b-lactic acid) (PEG-b-PLA) micelles was reported to increase as the weight fraction of the PLA block increased at a constant PEG block Mn, and the morphology remained spherical.18 In comparison, the morphology of poly(styrene-b-acrylic acid) (PS-b-PAA) aggregates was reported to change from spherical to cylindrical to bilayers (vesicles) with the oil-water interfacial curvature decreasing as the weight fraction of the PAA block (fPAA) was decreased.19 At very low fPAA the formation of large colloidal particles (compound micelles) was observed.19 Thus, the chemical composition and microstructural attributes of amphiphilic polymers significantly influence the characteristics of their aggregated colloids.12,19
PEG-b-PCL diblock copolymers have been extensively used to prepare colloidal drug delivery systems as a result of their good biocompatibility, biodegradability and ability to encapsulate a wide variety of therapeutics.20–22 Nevertheless, few systemic studies have investigated the influence of the PEG-b-PCL microstructure on the morphology of their aggregates.23–26 Although valuable in their insights, these studies have either been restrictive in polymer sample size or have only used limited observational techniques to characterise the aggregates. Thus, a systematic appraisal of a relationship between the microstructure and the self-assembly behavior for the PEG-b-PCL system would provide the foundation for the development of tailored drug delivery vectors. A thorough investigation of such a relationship, as with any copolymer system, requires an integrated multi-technique approach to evaluate aggregates that have been produced from a large library of polymers of defined structure.
Previously, various observational techniques have been employed to evaluate aggregated colloidal systems, including dynamic and static light scattering (DLS and SLS, respectively),27 nuclear magnetic resonance (NMR) spectroscopy,26,28,29 microscopy,27 and X-ray scattering.30 When used in isolation these techniques rarely provide a complete picture of the dispersity, compositional structure and morphology of aggregates, and in some cases sample preparation may influence the self-assembly process and the resulting structure of the aggregates. For instance, Rajagopal et al. investigated the self-assembly of a large family of PEG-b-PCL block copolymers that analysed aggregate morphology via fluorescence microscopy, which necessitated the inclusion of a dye; a factor known to influence the assembly process.23 Similarly, transmission electron microscopy (TEM) is extensively employed to observe the morphology of PEG-b-PCL aggregates, although concentration and dehydration during sample preparation for TEM may lead to rearrangement, deformation, uncontrolled aggregation and artifacts.24,31 The complementary determination of size, shape, dispersity and Nagg using light scattering techniques or small-angle X-ray scattering (SAXS) is essential in the determination of microstructure-assembly relationships.30,32–34 Although previous studies have used scattering techniques to evaluate aggregated colloids they too have been limited to a small number of block copolymers hindering the establishment of a robust microstructure-assembly relationship.24,33–35
To date, no studies have employed the complementary and non-interfering analytical techniques of diffusion NMR spectroscopy, DLS and Synchrotron SAXS in tandem to accurately define the aggregation behavior, dispersity and morphology of PEG-b-PCL aggregated colloids. Thus, in this study a library of well-defined PEG-b-PCL polymers with varying Mn and fPCL were prepared and their aggregated colloids were thoroughly examined without interference under identical assembly conditions. Each of the techniques provided a means to probe either aggregate composition or morphology in an unperturbed solution state, in situ. Diffusion NMR allowed for the determination of the distribution of self-diffusion coefficients (D) of aggregates, revealing valuable insights into the composition and behaviour of both PEG and PCL blocks in the aggregates. Evaluation of the aggregates by DLS provided an indication of their dispersity and size, whereas synchrotron SAXS provided further insights into the morphology of the aggregates. Thorough multifocal evaluation of the aggregates enabled the accurate determination of the underlying microstructural attributes which contributed to PEG-b-PCL aggregate characteristics.
2. EXPERIMENTAL
2.1. Materials
Stannous octoate (Sn(Oct)2; 92.5–100%), anhydrous toluene (99.8%), deuterated chloroform (CDCl3; 99.8% D), deuterium oxide (D2O; 99.9% D), lithium chloride (LiCl; 99%) and ε-caprolactone (ε-CL; 97%) were purchased from Sigma-Aldrich. ε-CL was dried under vacuum (0.1 mbar) for 1 h prior to use. α-Methoxy-ω-hydroxy PEG with Mn of 2 kDa (PEG2) and 5 kDa (PEG5) were purchased from Sigma-Aldrich, and 10 kDa (PEG10) from Creative PEG Works. PEG2 and PEG5 were dried at 120 °C under vacuum (0.1 mbar) for 1 h prior to use. Analytical grade diethyl ether, chloroform, toluene, ethyl acetate, N,N-dimethylformamide (DMF), acetone and hexane were purchased from Chem-Supply. All reagents were used as received unless otherwise stated. For DLS experiments, ultrapure water with a resistivity of > 18.2 MΩ.cm was obtained from a Sartorius Arium® ultrapure water purification system. For Synchrotron SAXS experiments, ultrapure water with a resistivity of 18.2 MΩ∙cm, interfacial tension of 72.4 mN∙m−1 at 22°C, and less than 4 ppb of total organic carbon was obtained from a Milli-Q Advantage A10 water purification system.
2.2. Characterisation
Nuclear magnetic resonance (NMR) spectroscopy
One-dimensional NMR spectroscopy experiments were performed on a 600 MHz Bruker Avance III HD spectrometer (Bruker BioSpin). For proton (1H) NMR spectroscopy of the polymers a sample concentration of 5–30 mg/mL in CDCl3 or D2O was used. The Mn values of the PCL blocks of the copolymers were calculated from the ratio between the integral values of the methylene protons adjacent to the ester group in the PCL repeat unit (δH 4.06 ppm) and the methylene protons in the PEG repeat unit (δH 3.65 ppm) from spectra acquired in CDCl3, referencing to the Mn values of PEG provided by the manufacturers.
Diffusion NMR measurements
Diffusion NMR spectroscopy experiments were performed with PEG and PEG-b-PCL copolymer solutions (1 mg/mL, 500 µL) in D2O, using 5 mm NMR tubes (Aldrich, ColorSpec). The D of the polymers were measured using 1H pulsed gradient stimulated echo (PGStE) NMR experiments at 600 MHz with a 14.09 T superconducting vertical bore magnet, a Micro5 Imaging Probe, a 5 mm radio frequency coil, and a Diff30 (11.7 T/m maximum) gradient set.36 PGStE experiments were performed in the Diff module of TopSpin 3.2 (Bruker) and used sinusoidal gradient pulse shapes with pulse duration of δ = 1.58 ms. The time between the leading edges of the pulses (Δ) defines the observation time of the diffusion measurement and was set to 100 ms. The repetition time was 10 s. For each experiment, 32 gradient points were acquired with 16 averages and the gradient amplitude, g, varied linearly to 11.67 T/m. Experiments were performed at a core temperature of 25 °C. Attenuation of spectrally resolved PEG and PCL signals was modeled as arising from a lognormal distribution of diffusion coefficients.37,38 Refer to Supplementary Information (SI) for further details on the processing parameters employed in diffusion NMR analysis, and representative signal attenuation and model fits (SI, Figure S1).
Gel permeation chromatography (GPC)
GPC was performed on a Prominence Liquid Chromatography system (Shimadzu) equipped with a RID-10A refractive index detector and three Shimadzu Shim-pack columns in series (GPC-8025D, GPC-805D and GPC-80MD) at 60 °C. 0.05 M LiCl in DMF was used as the mobile phase at a flow rate of 1.0 mL/min. Sample concentrations of 10 mg/mL were used with an injection volume of 50 µL. MW characteristics were determined with reference to a conventional calibration with PEG standards (Polymer Standards Service GmbH; molar mass at peak maximum (Mp) = 194 to 969 kDa).
Dynamic light scattering (DLS) experiments
DLS was conducted using a Malvern Zetasizer NANO ZS (Malvern Instruments) equipped with a 4 mW He–Ne laser operating at a wavelength of 633 nm. Samples were prepared in ultrapure water and analysed at an angle of 173 ° using a quartz cuvette. The polydispersity index (PDI) values were determined from the correlograms using the Zetasizer 7.11 software (Malvern Instruments). All DLS experiments were conducted in triplicate for three independent repeats to calculate the average values and standard deviation of PDI from the correlation analysis.
Small-angle X-ray scattering (SAXS) experiments
SAXS measurements were performed on the SAXS/WAXS beamline at the Australian Nuclear Science and Technology Organisation (ANSTO), Australian Synchrotron, Melbourne.39 Samples were loaded into glass capillaries with wall thickness of 0.01 mm and a nominal external diameter of 1.5 mm (Charles Supper), before being placed into a custom built thermostated capillary holder. The temperature of the capillary holder was controlled by a circulating water bath and set to 37 °C. The temperature during the SAXS experiments was monitored using a thermocouple, which was inserted into one of the empty capillaries. Two detector configurations were used: (i) photon energy = 15.1 keV (wavelength, λ = 0.821 Å) with a sample-to detector distance of 1350 mm, and (ii) photon energy = 11.0 keV (λ = 1.127 Å) with a sample-to detector distance of 7398 mm. 2D scattering patterns were recorded for each sample using a Dectris Pilatus 2M detector and the data were reduced to profiles of scattered X-ray intensity [(I(Q)] versus the magnitude of the scattering vector Q [= (4π/λ)sinθ, 2θ = scattering angle] using the in-house developed software Scatterbrain. Data from the two detector configurations were stitched together into a single continuous scattering profile with a Q range from 1.9 × 10−3 to 2.0 × 10−1 Å−1 using the IRENA small-angle scattering macros (release 2.68) within the IgorPro software package (version 7.0.8.1).40 These scattering profiles were subsequently analysed using the SASView small angle scattering data analysis package (version 5.0).41 Full descriptions of the form factor modelling are given in the SI and parameters that are pertinent to describing the shape and size of the polymer aggregates are provided in the results and discussion section (vide infra). Given the small variations between capillaries (i.e., the thickness of the capillary walls and their inner diameters), and hence the variation in the pathlength in the X-ray beam, the scattered X-ray intensity was not placed on an absolute scale as the absolute scattering volume was not known precisely. Only the shape of the particles in solution was therefore analysed, not their absolute scattering power (scattering length density).
2.3. Methods
Synthesis of PEG-b-PCL block copolymers
PEG-b-PCL block copolymers were synthesised via ring-opening polymerisation (ROP) of ε-caprolactone from PEG macroinitiators (Mn = 2, 5 and 10 kDa) using Sn(Oct)2 as the catalyst. Slightly different methods were used for each of the macroinitiators as described in greater detail below. The block copolymers are referred to as PEGxPCLy copolymers where the subscripts x and y refer to the Mn (kDa) of the respective blocks.
For the PEG2PCLy block copolymer series, PEG2 macroinitiator (1.5 g, 0.75 mmol) was weighed into a Schlenk flask and dried in vacuo (0.1 mbar) at 120 °C for 1 h before back-filling with nitrogen (99.999% purity, BOC). The flask was placed in a preheated oil bath at 115 °C, and ε-caprolactone (8.8 mL, 79 mmol) and Sn(Oct)2 (0.12 mL, 0.38 mmol) were added consecutively under an atmosphere of nitrogen to provide a monomer:initiator:catalyst ([M]:[I]:[cat]) mole ratio of 210:2:1. PEG2PCL0.4, PEG2PCL1.1, PEG2PCL1.8 and PEG2PCL4.0 were prepared using polymerisation times of 40, 60, 120 and 230 min, respectively. The crude reaction mixture was cooled to ambient temperature, diluted in chloroform (4 mL) and precipitated into diethyl ether (40 mL). The precipitated polymer was collected via centrifugation (6000 rpm, 15 min). This precipitation procedure was repeated three times to remove all unreacted monomer and catalyst. The polymer was then dried in vacuo (0.01 mbar) for 72 h and stored at ambient temperature, protected from light. 1H NMR spectra and GPC differential refractive index (DRI) chromatograms of the polymers are provided in the SI, Figures S2 and S3, respectively.
For the PEG5PCLy block copolymer series, PEG5 macroinitiator (1.5 g, 0.30 mmol) was weighed into a Schlenk flask and dried in vacuo at 120 °C for 1 h before back-filling with nitrogen. The flask was placed in a preheated oil bath at 120 °C, and ε-caprolactone (3.5 mL, 32 mmol) and Sn(Oct)2 (0.049 mL, 0.15 mmol) were added consecutively under an atmosphere of nitrogen to provide a [M]:[I]:[cat] mole ratio of 210:2:1. PEG5PCL0.6, PEG5PCL1.3, PEG5PCL2.3, PEG5PCL4.2 and PEG5PCL9.4 were prepared using polymerisation times of 2, 6, 10, 24 and 50 min, respectively. The crude reaction mixture was cooled to ambient temperature, diluted in chloroform (4 mL) and precipitated into 30% v/v ethyl acetate in diethyl ether (40 mL). The polymer was collected via centrifugation (6000 rpm, 15 min). This precipitation procedure was repeated three times and the polymers were then dried in vacuo (0.01 mbar) for 72 h and stored at ambient temperature, protected from light. 1H NMR spectra and GPC DRI chromatograms of the polymers are provided in the SI, Figures S4 and S5, respectively.
For the PEG10PCLy block copolymer series, PEG10 macroinitiator (1.0 g, 0.10 mmol) was weighed into a Schlenk flask and purged with nitrogen. The Schlenk flask was then placed in an oil bath at 120 °C. Anhydrous toluene (5 mL), ε-caprolactone (4.3 mL, 39 mmol) and Sn(Oct)2 (0.064 mL, 0.20 mmol) were added consecutively under an atmosphere of nitrogen to provide a [M]:[I]:[cat] mole ratio of 395:1:2. PEG10PCL3.2, PEG10PCL7.9, PEG10PCL10.7, and PEG10PCL14.9 were prepared using polymerisation times of 30, 135, 210 and 300 min, respectively. The crude reaction mixture was cooled to ambient temperature, diluted in chloroform (4 mL) and precipitated into diethyl ether (40 mL). The polymer was collected via centrifugation (6000 rpm, 15 min). This precipitation procedure was repeated three times and the polymers were then dried in vacuo (0.01 mbar) for 72 h and stored at ambient temperature, protected from light. 1H NMR spectra and GPC DRI chromatograms of the polymers are provided in the SI, Figures S6 and S7, respectively.
Self-assembly of PEG-b-PCL copolymers in water
Self-assembly of PEG-b-PCL copolymers in ultrapure water or D2O was conducted using a solvent evaporation technique. Polymer solutions were prepared in acetone (1 mL, 1 mg/mL) in vials (4 mL) and 1 mL of D2O (for NMR experiments) or ultrapure water (for DLS and synchrotron SAXS experiments) was then added dropwise. The vials were placed in a pre-heated block heater at 60 °C and the solvent level was checked periodically until the volume was ~ 1 mL. The vials were then placed in a vacuum desiccator (0.1 mbar) for 1 h to remove the residual organic solvent, and the solvent volume was readjusted with D2O or ultrapure water on a mass basis taking into consideration the density of the solvent to ensure a polymer concentration of 1 mg/mL. Complete removal of the organic solvent was confirmed by 1H NMR spectroscopy that revealed the absence of acetone resonances that would be expected at δH 2.22 ppm (SI, Figure S8). All solutions were prepared fresh prior to analysis. In this study, polymer concentrations of 1 mg/mL were used to minimise secondary aggregate formation and to avoid microscopic averaging effects during NMR diffusion experiments, whilst still being sufficient to generate a strong NMR signal.38 In addition, it is important to note that the concentration used (1 mg/mL) was above the critical micelle concentration (CMC) (< 50 µg/mL) reported for PEG-b-PCL copolymers.27,33,42
3. RESULTS AND DISCUSSION
3.1. Synthesis and characterisation of PEG-b-PCL block copolymers
The PEG-b-PCL block copolymers were synthesised via ROP of ε-caprolactone from PEG macroinitiators (Mn = 2, 5 and 10 kDa) using Sn(Oct)2 as the catalyst, as has previously been reported.43 A library of copolymers was prepared by varying the degree of polymerisation (DP) of both blocks to allow for the effect of the microstructure on their self-assembly to be studied in detail. The block copolymers are herein referred to as PEGxPCLy copolymers where the subscript x and y refer to the Mn (kDa) of the respective blocks. The copolymers were characterised using 1H NMR spectroscopy to determine their Mn (Figure 1 and SI, Figures S2, S4, and S6) and GPC to determine their dispersity (Ð) (SI, Figures S3, S5, and S7), and the results are summarised in Table 1.
Figure 1:
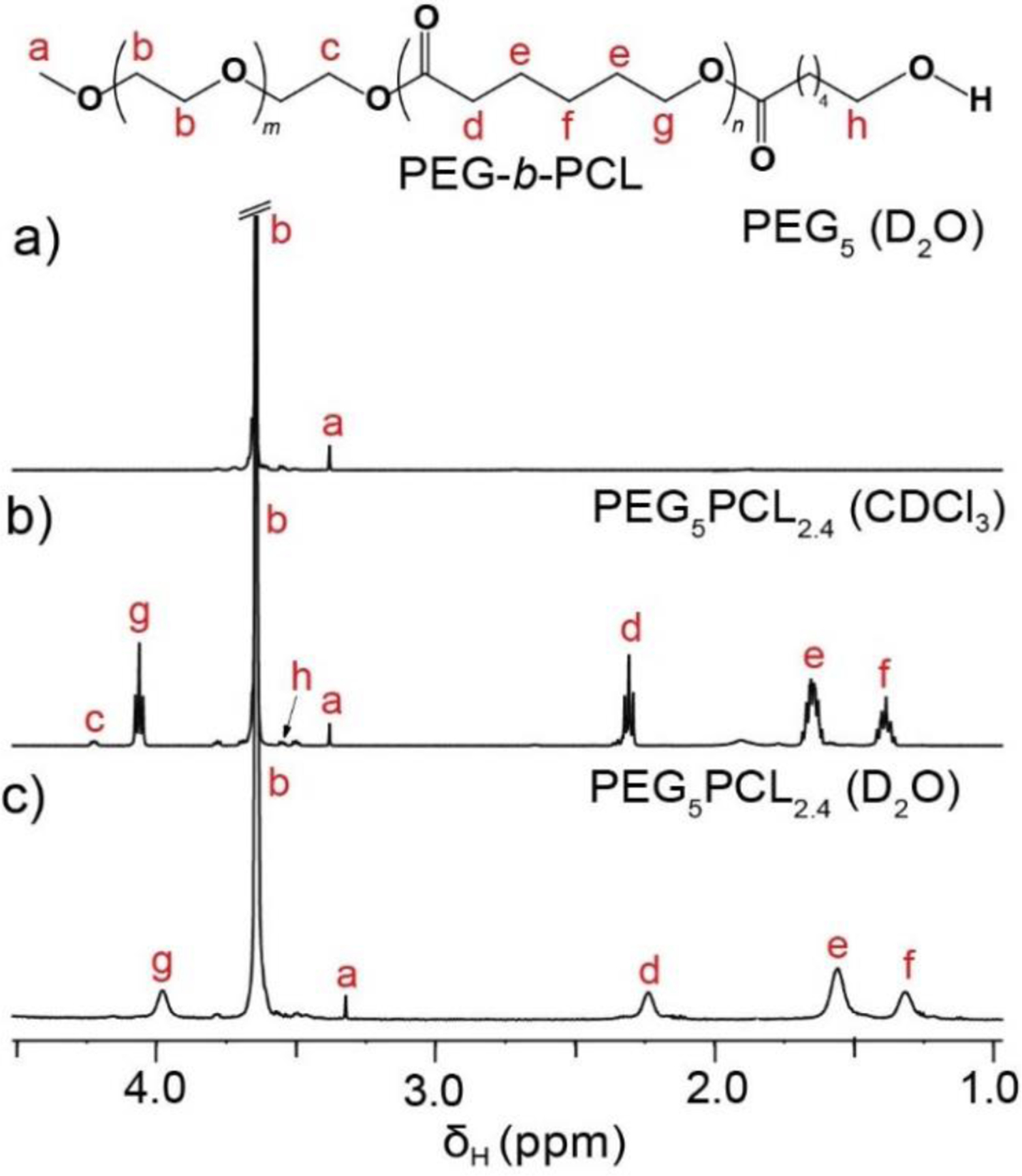
1H NMR spectra (600 MHz, 25 °C) of (a) PEG5 homopolymer in D2O, and PEG5PCL2.4 copolymer in (b) CDCl3 and (c) D2O. Aggregation of the block copolymer in D2O results in broadening of the characteristic PCL signals and a reduction in their intensity relative to the PEG signal indicative of the changing physical environment and reduced rotational mobility of protons on the hydrophobic block.
Table 1:
Composition and molecular weight characteristics of PEG macroinitiators and PEG-b-PCL block copolymers.
| Polymer | PEG Mn (kDa) |
PCL Mn (kDa)a |
Copolymer Mn (kDa)a |
Ð
Mw/Mn b |
DP PEG; PCL |
Mass fraction |
|
|---|---|---|---|---|---|---|---|
| f PEG | f PCL | ||||||
| PEG2PCL0.4 | 2.0 | 0.4 | 2.4 | 1.03 | 45; 4 | 0.83 | 0.17 |
| PEG2PCL1.1 | 2.0 | 1.1 | 3.1 | 1.02 | 45; 10 | 0.65 | 0.35 |
| PEG2PCL1.8 | 2.0 | 1.8 | 3.8 | 1.02 | 45; 16 | 0.53 | 0.47 |
| PEG2PCL4.0 | 2.0 | 4.0 | 6.0 | 1.03 | 45; 35 | 0.33 | 0.67 |
|
| |||||||
| PEG5PCL0.6 | 5.0 | 0.6 | 5.6 | 1.01 | 114; 5 | 0.89 | 0.11 |
| PEG5PCL1.3 | 5.0 | 1.3 | 6.3 | 1.02 | 114; 11 | 0.79 | 0.21 |
| PEG5PCL2.4 | 5.0 | 2.4 | 7.4 | 1.02 | 114; 21 | 0.68 | 0.32 |
| PEG5PCL4.2 | 5.0 | 4.2 | 9.2 | 1.03 | 114; 37 | 0.54 | 0.46 |
| PEG5PCL9.5 | 5.0 | 9.5 | 14.5 | 1.09 | 114; 83 | 0.34 | 0.66 |
|
| |||||||
| PEG10PCL3.2 | 10.0 | 3.2 | 13.2 | 1.02 | 227; 28 | 0.76 | 0.24 |
| PEG10PCL7.9 | 10.0 | 7.9 | 17.9 | 1.04 | 227; 69 | 0.56 | 0.44 |
| PEG10PCL10.7 | 10.0 | 10.7 | 20.7 | 1.04 | 227; 94 | 0.48 | 0.52 |
| PEG10PCL14.9 | 10.0 | 14.9 | 24.9 | 1.12 | 227; 131 | 0.40 | 0.60 |
Mn calculated from 1H NMR spectroscopy.
Mn and Ð calculated by GPC with reference to a conventional column calibration with narrow molecular weight PEG standards.
1H NMR spectra of all copolymers revealed characteristic resonances corresponding to the methylene protons of the PEG repeat unit (RU) at δH 3.65 ppm and PCL RU at δH 1.40 (-CH2CH2CH2–), 1.66 (–CH2CH2CH2–), 2.31 (–COCH2CH2–) and 4.06 (–CH2CH2O–) ppm. For lower MW polymers, resonances corresponding to the methoxy protons from the PEG end-group (δH 3.40 ppm), the methylene protons adjacent to the hydroxyl end-group of the PCL block (δH 3.56 ppm) and the methylene protons from the PEG RU adjacent to the ester group joining the PEG and PCL blocks (δH 4.23 ppm) were also clearly visible (e.g., Figure 1). Comparison of the integral ratios between the PCL and PEG RUs were used to calculate the Mn of the PCL blocks with reference to the Mn of PEG block (Table 1).
GPC DRI chromatograms of the copolymers revealed narrow Ð peaks shifting to lower retention times (i.e., higher MW) with increasing PCL DP (SI, Figure S2 and Table 1), with the exception of PEG5PCL4.2, PEG5PCL9.5 and PEG10PCL14.9 which displayed slight peak broadening most likely as a result of transesterification reactions. Nevertheless, narrow Ð values below 1.12 were obtained for the complete library, indicative of well-defined block copolymers.
3.2. Correlating PEG-b-PCL microstructure to self-assembly behaviour
The library of PEG-b-PCL copolymers prepared in this study allowed a detailed investigation of the effect of copolymer microstructure (e.g., copolymer and block Mn, and fPCL that correlates to the hydrophobic:hydrophilic balance) on their self-assembly behaviour in water and the resulting morphology of their aggregated colloids. The PEG-b-PCL copolymers were self-assembled in water using the solvent evaporation technique. Although there are various approaches to facilitate amphiphilic copolymer assembly in water (e.g., solid dispersion, dialysis, oil-in-water emulsion), the solvent evaporation technique was selected as this method enables good control over the assembly process, and is applicable to a wide variety of amphiphilic polymers and payloads.44–46 However, it is worth noting that differences in each preparation technique may modify the thermodynamic parameters of self-assembly resulting in distinct aggregates or micelles, and therefore, this study is only reflective of aggregated colloids prepared by the solvent evaporation approach.47 The aggregates were initially studied by diffusion NMR spectroscopy and DLS analysis to provide complementary information about the effect of the copolymer microstructure on the dispersity of the aggregates. Subsequently, synchrotron SAXS experiments were employed to provide details about the structure and size of the aggregates.
Diffusion NMR spectroscopy of PEG-b-PCL aggregates
The dispersity of the aggregates in D2O was analysed by interpreting the D distribution obtained from diffusion NMR experiments. The D distribution also reflects the distribution of aggregate sizes as described by the Stokes–Einstein–Sutherland equation.48,49 Analysis of the hydrophilic PEG2, PEG5, and PEG10 homopolymers (1 mg/mL) was initially conducted to establish a baseline from which to compare with the aggregates (Figure 2). The attenuation of the methylene proton signals from the PEG backbone (δH 3.65 ppm) was used to calculate D for the PEG2, PEG5, and PEG10 homopolymers (D = 14.2 × 10−11, 8.61 × 10−11 and 5.8 × 10−11 m2/s, respectively), which were similar to previously reported values.37 As expected, the D value of the homopolymers decreased with increasing MW due to an increase in the hydrodynamic size of the randomly coiled polymers in solution, and the D distributions were narrow due to the homopolymers’ narrow Ð.1H NMR spectroscopic analysis of the block copolymers in D2O revealed characteristic methylene proton resonances from both the PEG and PCL blocks, although a reduction in intensity and broadening of signals for the latter was noted (Figure 1). Previous studies have reported that the self-assembly of amphiphilic copolymers in water results in the disappearance of proton resonances of the core-forming, hydrophobic block, which has been attributed to the formation of a dehydrated ‘solid’ core.33,50 However, in this study the characteristic PCL resonances were clearly visible in the NMR spectra of the copolymers in D2O, albeit without the resolution of signal multiplicity observed in CDCl3 (Figure 1), which is attributed to a collection of species that are chemically similar yet possessing a different degree of mobility. Jette et al. also reported the presence of characteristic PCL resonances from 1H NMR analysis of PEG-b-PCL micelles and concluded that PCL resonances broaden as a result of aggregate core formation.51 The presence of the PCL resonances would indicate that the aggregates contain some protons on PCL that are rotationally mobile leading to long enough relaxation times for detection, potentially originating from a semi-hydrated core or a boundary layer between the core and corona (shell). The penetration of water into the hydrophobic core of surfactant52,53 and amphiphilic polymer54 micelles has also previously been reported.
Figure 2:
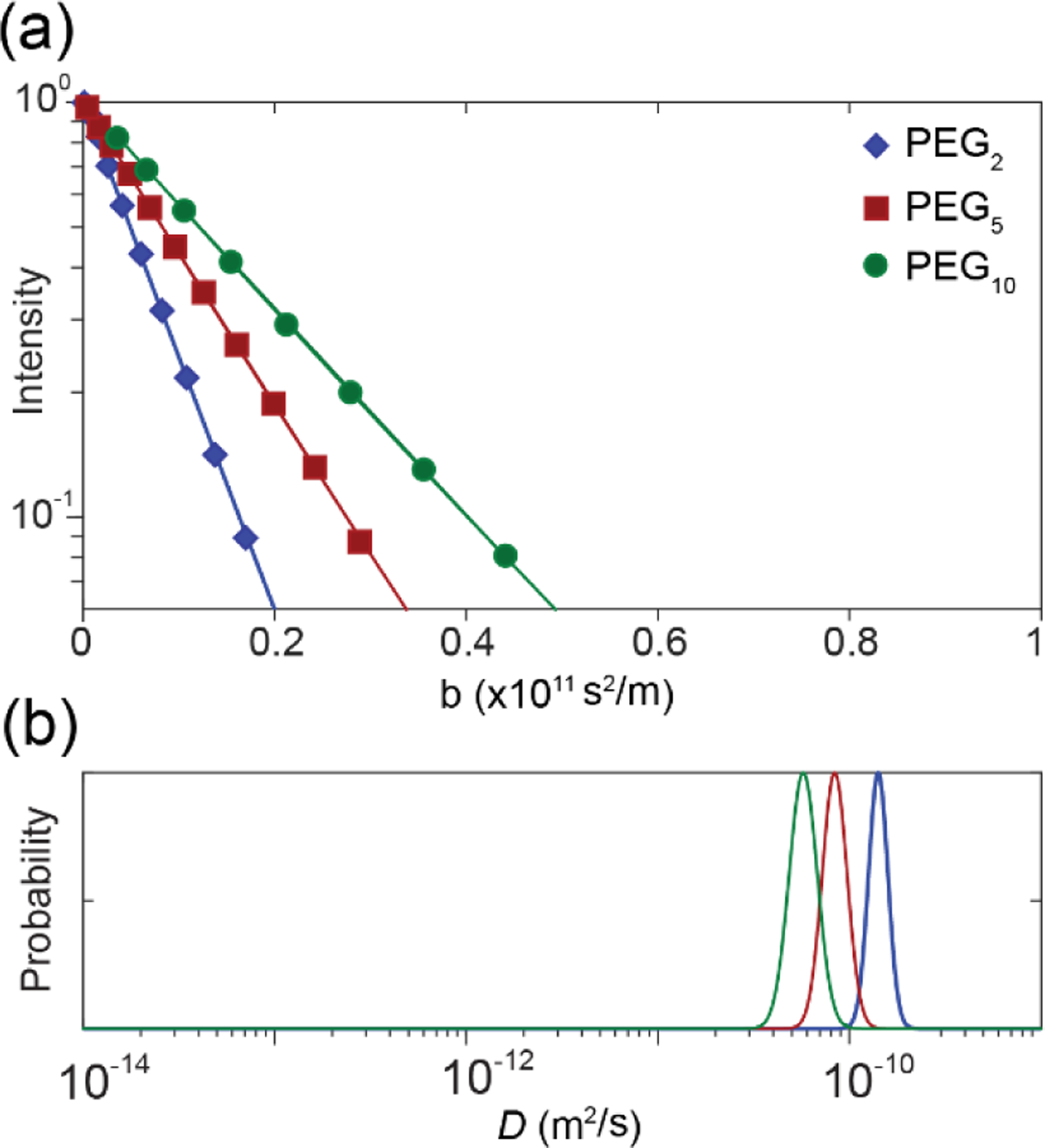
(a) PGStE NMR spectroscopy attenuation of signal intensity and (b) D distributions for the methylene protons of the PEG backbone (δH 3.65 ppm) of the PEG2 (blue), PEG5 (red) and PEG10 (green) homopolymers in D2O. The scale of the x axis was chosen for ease of comparison with subsequent figures which exhibit self-diffusion coefficients across this range.
The diffusion NMR measurement is sensitive to net displacements occurring during the observation time, . Diffusion length scales, l, are therefore well-defined by mean square displacement (MSD) through the relation (where the α is the constant depending on dimensionality and is 2 in 1D diffusion because NMR measures diffusion along the direction of the applied gradients). By this relation, aggregates with D = 10−12 m2/s (roughly the smallest D measured in this study) displace on average l = 450 nm, which is much greater than the size of the aggregated structures (at maximum a few tens of nm). Therefore, diffusion NMR measures the D of the aggregates.
In this study, the PEG and PCL resonances (δH 3.65 and 1.66 ppm, respectively) were separately analysed to produce two D distributions corresponding to the PEG and PCL blocks, respectively. As the PEG and PCL blocks are covalently tethered to one another in the copolymers one may expect that the distributions should be similar for the aggregates. However, certain weighting effects can cause deviations of the two measured D distributions, which can be indicative of underlying heterogeneity.55 In particular, differences in NMR relaxation times between blocks, a dependence of relaxation time on aggregate structure, and a broad distribution of structures can cause the measured D distributions of the two blocks to deviate from one another, an effect broadly known as relaxation weighting.55 In particular, the protons on PCL have varying degrees of mobility depending on the structure formed. This leads to the PCL signals being preferentially visible from some structures more than others. Therefore, the PCL D distribution is weighted towards a different D distribution from the PEG although the PEG and PCL are tethered. Differences between PEG and PCL D distributions indicate heterogeneity in aggregate structures.
The D distributions for the PEG2PCLy copolymer series revealed that there was a significant change in aggregate morphology as the fPCL increased (Figure 3a). The PEG and PCL D distributions for the PEG2PCL0.4 copolymer revealed similar D values to the PEG2 homopolymer distribution, although slightly broadened. The lack of a shift in D is indicative of the block copolymers existing as individually solvated molecules (unimers) or loosely associated unimers in solution, whereas the overlapping and narrow D distributions for the two blocks suggest that the blocks exist in a consistent physical environment (solvation environment) with a high degree of uniformity. These results imply that the fPCL values below 0.17 fail to promote assembly into aggregates when the PEG block Mn is 2 kDa, and that the CMC for PEG2PCL0.4 is > 1 mg/mL. As the main driving force behind aggregate formation is hydrophobic interactions between the PCL blocks in the core due to energetically unfavorable PCL-water interactions,56,57 it is evident that for the PEG2PCL0.4 copolymer the short PCL block (DP ~ 3.5) is insufficient to induce self-assembly. Thus, the enthalpic gain from aggregating a very short hydrophobic block from the solvent when attached to a relatively large hydrophilic block is not sufficient to overcome the loss in entropy associated with aggregation. Additionally, at very low PCL DPs the interaction of the hydroxyl end-group of the PCL block with water may still be significant when compared to PCL blocks with much higher DPs, which might increase their solvation and reduce hydrophobic interactions between the short PCL blocks.58 This behaviour has also been observed by Rager et al. for poly(acrylic acid-b-methyl methacrylate) (PAA-b-MMA) block copolymers with short MMA blocks, which were reported to be mainly present in aqueous solution as unassociated unimers.12
Figure 3:
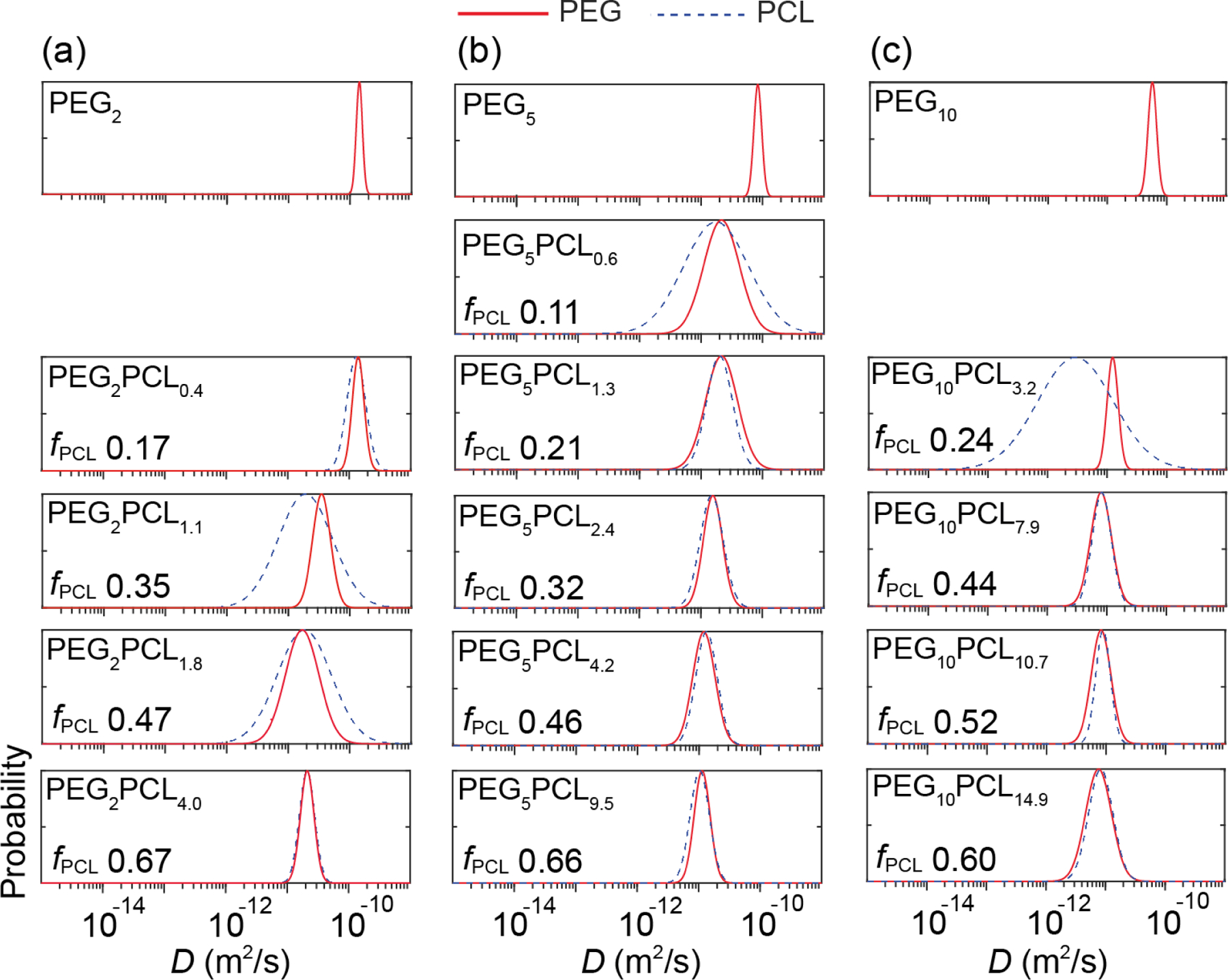
D distributions for the PEG (solid red) and PCL (dashed blue) blocks of (a) PEG2 homopolymer and the PEG2PCLy copolymer series, (b) PEG5 homopolymer and PEG5PCLy copolymer series, and (c) PEG10 homopolymer and PEG10PCLy copolymer series in D2O. Differences between PEG and PCL D distributions arise due to relaxation weighting and indicate aggregated structure heterogeneity.
In comparison, the D distributions for the PEG2PCL1.1 and PEG2PCL1.8 copolymers were shifted to significantly lower values, consistent with the formation of larger aggregates that diffuse more slowly through their solutions. Furthermore, the PEG and PCL distributions were broader and diverged from one another, indicating some underlying heterogeneity within the structure of the aggregates or between different aggregates. These results are consistent with TEM studies conducted by Rizis et al. on PEG2PCL2 copolymers, which were found to adopt structurally heterogeneous aggregates due to the presence of both spherical and rod-like micelles.59 For the PEG2PCL4.0 copolymer the narrow D distributions for the PEG and PCL blocks coincided and were shifted to lower D values compared to the PEG2 homopolymer, indicative of well-defined aggregates (i.e., micelles) with low size dispersity and high homogeneity.
For the PEG5PCLy copolymer series, with the exception of the PEG5PCL0.6 copolymer, the D distributions for the PEG and PCL blocks converged and were shifted to lower D values when compared to the PEG5 homopolymer (Figure 3b), indicating that they all form well-defined aggregates of increasing size. The D distributions were found to become narrower as the fPCL increased from 0.21 (PEG5PCL1.3) to 0.66 (PEG5PCL9.5), indicating the formation of lower dispersity aggregates coinciding with an increase in hydrophobic interactions. For the PEG5PCL0.6 copolymer, the shift in D values relative to the PEG5 homopolymer indicates aggregation, but the D distribution for the PCL block was significantly broader than that of the PEG block, implying some underlying heterogeneity in the aggregate structure, possibly due to the formation of loosely bound or unstable aggregates. The fact that the PEG5PCL0.6 copolymer forms aggregates when the PEG2PCL0.4 copolymer does not, suggests that either the PCL block length (DP ~ 5.3) is sufficient to induce hydrophobic interactions in the former or that the PEG block length plays a role in stabilising hydrophobic interactions.
The trends observed for the D distributions for the PEG10PCLy copolymer series (Figure 3c) somewhat resemble those for the PEG5PCLy copolymer series, with larger fPCL leading to the formation of well-defined aggregates. However, The PEG and PCL D distributions for the PEG10PCL3.2 copolymer are distinctly different, with the former being narrow and the latter very broad. This would indicate that the aggregates are structurally heterogeneous, although the reason for this is not obvious given that the fPCL (0.24) and PCL DP (~ 28) are relatively large and PEG5PCLy copolymers with similar PCL DPs formed well-defined aggregates.
The mean diffusivities and their standard deviations for the copolymers were calculated from triplicate experiments performed on three independent repeats and plotted against the fPCL to allow trends across the three copolymer series to be examined (Figure 4). Of the copolymers that formed aggregates, the standard deviations of were significant for PEG2PCL1.1 and PEG5PCL0.6, revealing the presence of metastable aggregates for which the fractions of molecules filling those states can vary from sample-to-sample. Interestingly, the standard deviations of for the PEG10PCL3.4 copolymer were significantly smaller despite distinctly different for the PEG and PCL blocks, indicating the consistent formation of heterogeneous aggregates. Overall, the were found to decrease with increasing PEG block Mn and fPCL up to 0.4, after which the remained constant for the PEG5PCLy and PEG10PCLy copolymer series.
Figure 4:
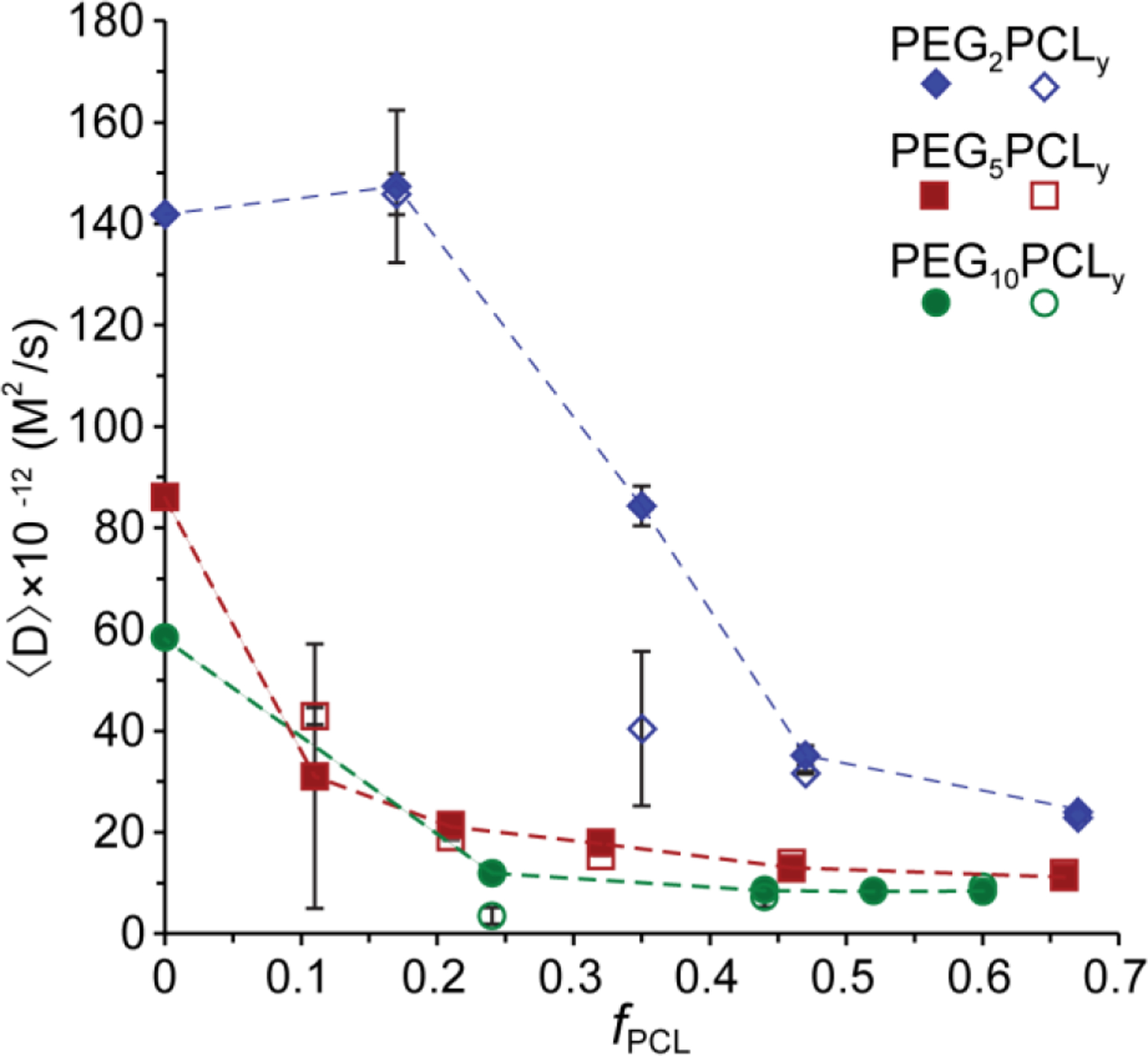
Mean (marker) and standard deviation (error bars) of measured on three separate self-assembled samples (independent experiments) for each PEGxPCLy copolymer, plotted against fPCL. In most cases, error bars are smaller than symbols. The solid and open symbols represent the of the PEG and PCL blocks, respectively, for the PEG2PCLy (blue), PEG5PCLy (red) and PEG10PCLy (green) copolymer series. Dashed lines are included as a guide to the eye and join the PEG for each series.
DLS analysis of PEG-b-PCL copolymers in water
Typically, DLS data is reported as intensity, volume or number particle size distributions (PSDs) determined mathematically from the translational diffusion coefficient, which in turn is calculated from the correlation function. The particle’s hydrodynamic diameter is calculated from the Stokes-Einstein equation for known refractive index and viscosity of samples. Importantly, this mathematical transformation, presumes that the particles being analysed are spherical in shape. However, the self-assembly of copolymers into micelles can deviate significantly from spherical structures leading to erroneous results. Furthermore, intense scattering from small populations of larger aggregates heavily influences the determination of the diffusion coefficient. In this study, measurement of the for the copolymer solutions via DLS deviated significantly from values determined by diffusion NMR spectroscopy experiments (SI, Figure S9), indicating the presence of larger (secondary) aggregates. This was further confirmed from the intensity vs Dh plots that revealed aggregates with diameters > 100 nm, particularly when the fPCL < 0.4 for the PEG2PCLy and PEG5PCLy copolymer series (SI, Figure S10). The significantly higher values determined from diffusion NMR spectroscopy experiments that are not influenced by particle size indicates that the DLS results discriminate against the smaller aggregates and the results are skewed due the presence of a small population of larger aggregates.
Given the limitations of DLS to provide a true representation of the complete population of copolymer aggregates, DLS experiments were performed in triplicate on three independent repeats for all copolymer solutions to determine the PDI of the aggregates with respect to the fPCL (Figure 5). Similar to the widths of the D distributions from NMR spectroscopy experiments, the PDI acts as a measure of the variation in aggregate sizes (dispersity), with values approaching zero representative of a monodisperse system. As the fPCL increased, the mean value of PDI was found to decrease indicating the formation of well-defined and structurally similar aggregates, consistent with diffusion NMR spectroscopy experiments. For the PEG5PCL0.6 copolymer the mean value of PDI (0.46) and deviation of the PDI across multiple samples was very high, which is consistent with the presence of metastable, loosely associated unimers.
Figure 5:
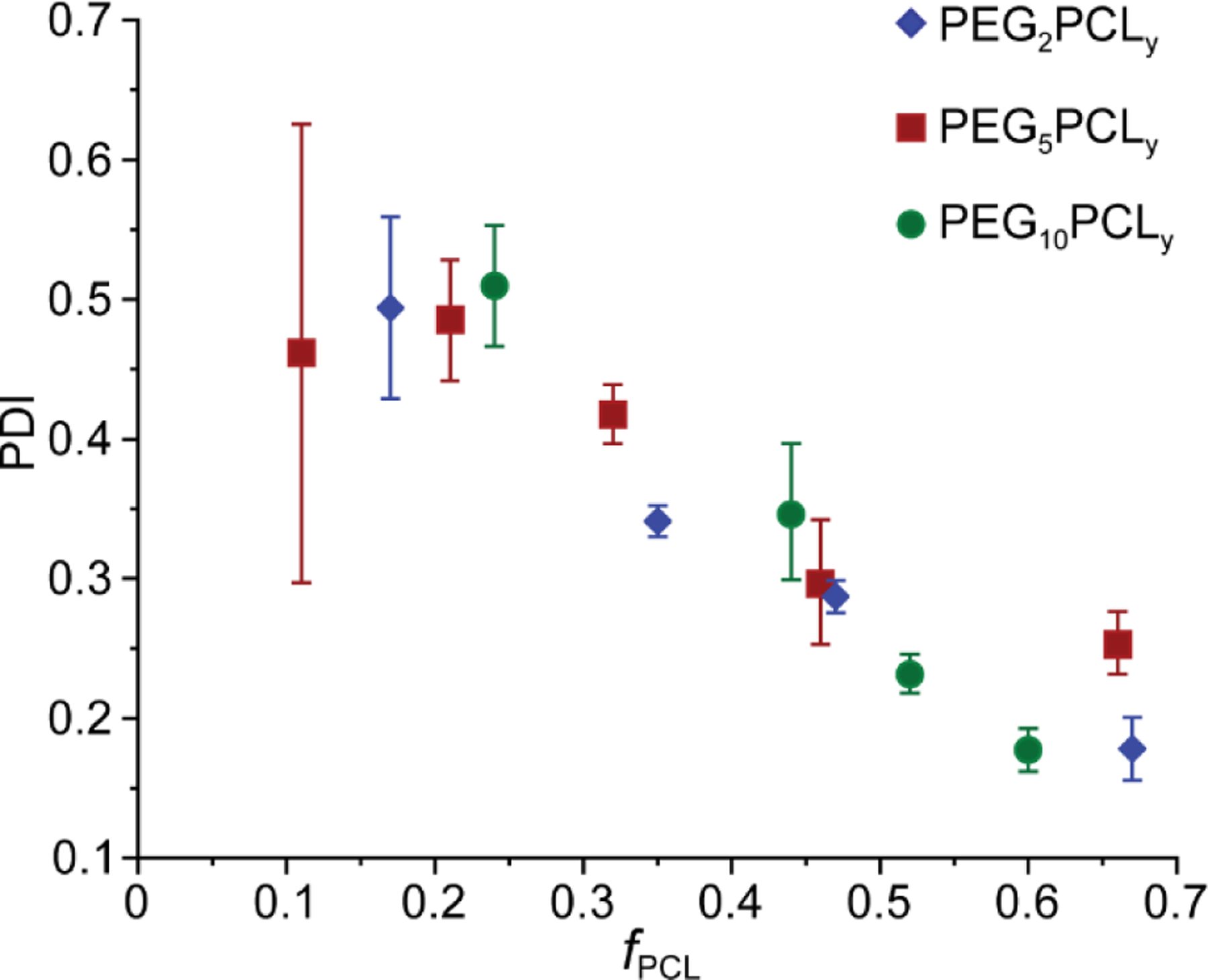
Mean (marker) and standard deviation (error bars) of PDI measured on three independent experiments of each PEGxPCLy copolymer solution, plotted against fPCL.
Synchrotron SAXS analysis of PEG-b-PCL copolymers in water
Synchrotron SAXS experiments were performed on the copolymer solutions in ultrapure water (1 mg/mL) and the scattered X-ray intensity was fitted to various form factor models to determine the shape and size of the resulting aggregates (Figure 6; refer to SI for model fitting parameters). The scattering contrast between the aqueous dispersant and hydrated PEG corona and PCL core domains was estimated form reports of the polymer electron densities and core/corona hydration in block copolymer micelles (SI, Figure S11; refer to SI for calculations).54,60 This revealed that scattering from the PCL core would dominate the scattered X-ray intensity, with the corona only scattering weakly in these micellar systems. The observed scattering patterns were therefore attributed to scattering from the hydrophobic cores of PEG-PCL micelles given the typically strong hydration of the PEG corona. Interestingly, the model fitting revealed that there is a transition in the aggregate structure across each copolymer series as the fPCL is increased, trending towards well-defined cylindrical or oblate ellipsoidal micelles.
Figure 6:
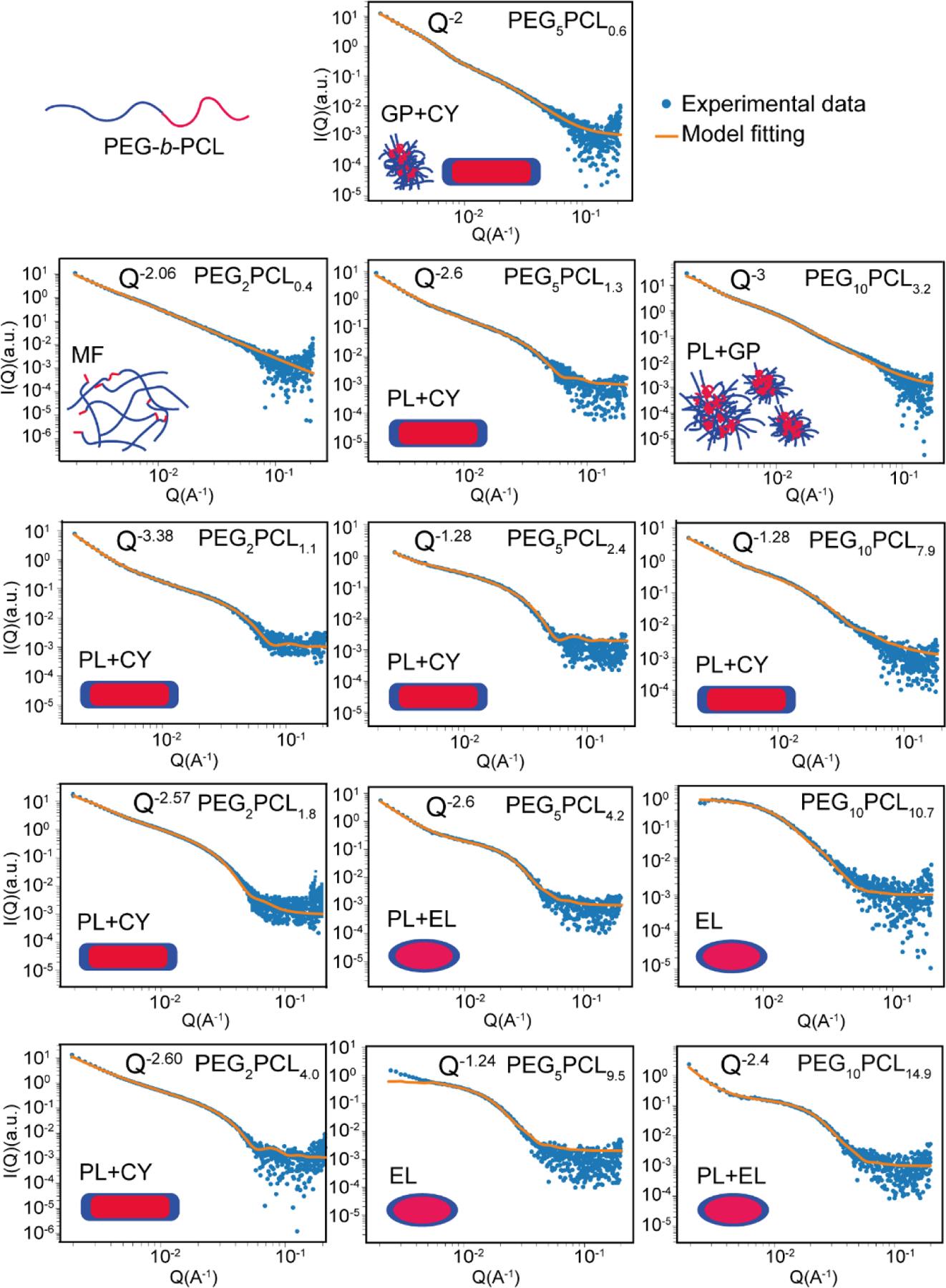
Synchrotron small-angle X-ray scattering profiles and model fitting for PEG-b-PCL block copolymers in water. Illustrations indicate the proposed structure of the aggregates, wherein the PCL cores are depicted as red, and the PEG coronas are shown in blue. The models which have been applied for fitting are denoted as follows: MF = mass fractal (random-walk polymers loosely clustered), PL = power-law scattering from secondary polymer aggregates, CY = cylindrical micelles, GP = Guinier-Porod (collapsed polymer chain), EL = ellipsoidal micelles.
The scattering profile of the PEG2PCL0.4 copolymer had a power law (Q−2.06) dependence in scattered X-ray intensity, which is consistent with random walk polymer structures (polymer in theta solvent) or disk-like aggregates (Figure 6),61 and implies that the hydrophobic PCL attached to PEG decreases its overall solubility in water. A mass fractal model was used to interpret the scattering data, and indicated the presence of polymer segments with radii of 2.5 nm in large (~87 nm) fractal aggregates with a mass fractal dimension of 2.06.62 The individual sub-units of the mass fractal were presumed to be small and mobile, corresponding to the high D observed in the diffusion NMR spectroscopy results. These small mobile units are likely well solvated PEG sections of the polymer chains, with the aggregate held together by hydrophobic interactions between the short PCL blocks in the polymer.
The scattering profiles of the PEG2PCL1.1, PEG2PCL1.8 and PEG2PCL4.0 copolymers all had power law dependences at Q ˂ 0.005 Å−1 (SI, Figure S12), and when fitted in isolation, the power law exponents were −2.63, −1.88 and −2.03, respectively. Initially, flexible/worm-like cylinder models were used to fit the data but they only give a good fit when the low Q power law exponent was close to −2.0 as the model assumes that the flexible cylinders follow a random (Gaussian) walk. However, the low Q power laws were too shallow for PEG2PCL1.1 and PEG2PCL4.0, and for PEG2PCL1.8 and the fit in the high Q region of the scattering profile was poor. The presence of the steep power law dependence in the scattering profiles at low Q was therefore attributed to surface scattering from large secondary aggregates resulting from aggregation of primary particles. Models comprising a cylindrical form factor for the smaller primary particles added to a power law representing larger aggregates (power-law+cylinder) effectively modelled the X-ray scattering data from PEG2PCL1.1, PEG2PCL1.8 and PEG2PCL4.0 copolymer solutions. The modelled power law exponents were greater than those determined from the low Q fits in isolation, suggesting that an insufficiently low Q range was measured to effectively model the aggregate fractal dimension (SI, Figure S12). The Q−1 power law at the low Q end of the cylinder form factor also makes a significant contribution to the scattering intensity at low Q in the region overlapping with the power law fit, so the power law gradient at low Q is convoluted with the cylinder form factor. Unfortunately, this meant that the cylinder lengths could not be determined as the low Q Guinier plateau was obscured by the power law scattering from the larger aggregates; however, the radii of the cylindrical aggregates were observed to increase as the fPCL was increased (Table 2). These results are consistent with previous studies by Rizis et al. on PEG-b-PCL copolymers with short PEG (2 kDa) block lengths whereby long thread-like or cylindrical aggregates were observed via TEM,59 and support the predictions of Nagarajan based upon thermodynamic considerations between the core and corona that smaller hydrophilic blocks will result in the formation of cylindrical micelles.14 A more complete discussion on molecular packing in the micelles is given at the end of this section.
Table 2:
Model fitting parameters pertinent to the shape and size of polymer aggregates observed in this study. Models are denoted as follows: MF = mass fractal, PL = power law, CY = cylinder, GP = Guinier-Porod and EL = ellipsoid.
| Polymer | Model | Low Q power law (Q−x) | Radius (nm) |
||||
|---|---|---|---|---|---|---|---|
| Particle radius | Radius of gyration (Rg) | Cylinder radius | Ellipsoid equatorial radius | Ellipsoid polar radius | |||
| PEG2PCL0.4 | MF | 2.1 | 2.6 | - | - | - | - |
| PEG2PCL1.1 | PL+CY | 3.4 | - | - | 4.6 | - | - |
| PEG2PCL1.8 | PL+CY | 2.6 | - | - | 6.6 | - | - |
| PEG2PCL4.4 | PL+CY | 2.6 | - | - | 6.4 | - | - |
|
| |||||||
| PEG5PCL0.6 | GP+CY | 2.0 | - | 13.7 | 2.1 | - | - |
| PEG5PCL1.3 | PL+CY | 2.6 | 5.6 | - | - | ||
| PEG5PCL2.4 | CY | - | - | 6.2 | - | - | |
| PEG5PCL4.2 | PL+EL | 2.6 | - | - | - | 12.1 | 7.0 |
| PEG5PCL9.5 | EL | - | - | - | 20.3 | 8.1 | |
|
| |||||||
| PEG10PCL3.2 | PL+GP | 3.0 | - | 16.3 | - | - | - |
| PEG10PCL7.9 | PL+CY | 2.6 | - | - | 9.4 | - | - |
| PEG10PCL10.7 | EL | - | - | - | 22.0 | 6.3 | |
| PEG10PCL14.9 | PL+EL | 2.4 | - | - | - | 13.5 | 6.3 |
The presence of two broad knee-like features around Q = 0.005 and 0.020 Å−1 in the scattering profile of the PEG5PCL0.6 copolymer (Figure 6 and SI, Figure S13) indicated the presence of two different aggregate populations, which would explain the broad D distribution observed in the diffusion NMR spectroscopy experiments and high PDI in the DLS results. The scattering profile was fitted with two Guinier-Porod models, one for the low Q region and another for high Q. The Guinier region at low Q describes the particle size (radius of gyration) and shape based on the low Q power law exponent and the Porod region at high Q describes the surface roughness/uniformity of the particles through a power law exponent.63 The higher Q particle population fitting indicated that the smaller particles in the system were cylindrical micelles (shape factor (s) = 1.17) (refer to SI for model fitting parameters) consistent with the PEG2PCLy polymers with higher fPCL. The whole scattering profile could be fitted effectively by adding a second Guinier-Porod model for the larger particle population at low Q with a shape factor = 2, suggesting the presence of larger aggregates with fractal dimensions equivalent to random walk polymers or disks.
Further increases in the fPCL for the PEG5PCLy copolymer series resulted in a transition from cylindrical to oblate ellipsoidal micelles. The lengths of the cylindrical micelles of PEG5PCL1.3 and PEG5PCL2.4 were indeterminate due to the low Q Guinier plateau being obscured by the dominant power law scattering from large secondary aggregates (Figure 6 and SI, Figure S13). However, their radii were determined to be on the order of 5.6–6.2 nm. The scattering from PEG5PCL4.2 and PEG5PCL9.5 at high Q was more consistent with oblate ellipsoid form factors than cylinders (Figure 6 and SI, Figure S14). The equatorial and polar radii of the ellipsoidal micelles were on the order of 12–20 nm and 7–8 nm, respectively, with the volume increasing with increasing fPCL.
In general, the form factor models used to fit the X-ray scattering profiles from the PEG5PCLy copolymers incorporated a power law (with exponents between −2.6 and −3.0) at low Q consistent with larger secondary aggregates, which were not observed in the diffusion NMR spectroscopy experiments that displayed narrow D distributions (with the exception of PEG5PCL0.6) (Figure 6). This discrepancy may result from the relative proportion of secondary aggregates being very small as the X-ray scattering from particles scales with the cubed particle volume, and therefore, the number densities of the large aggregate particles are likely to be below the threshold detection limit of the NMR spectroscopy experiments. This is also supported by the narrow PDI values obtained from DLS for the PEG5PCL4.2 and PEG5PCL9.5 copolymers, which imply that the proportion of larger secondary aggregates are small; while intensity PSDs reveal the presence of larger aggregates, volume PSDs confirm that they are very small in number (SI, Figure S10). That being said, the broad D distributions and PDI for the PEG5PCL0.6 copolymer indicate that the relative proportion of larger aggregates is higher in this system.
The scattering profile of the PEG10PCL3.2 copolymer solution was similar to that of the PEG5PCL0.6 copolymer and indicated the presence of a polydisperse system with large and small polymer aggregates (Figure 6). The low Q scattering shows a Q−3 power law dependency representative of the large aggregates; whilst scattering in the high Q region could be fitted with a Guinier-Porod model with a shape factor s = 0 and the Porod exponent of 2.5, which indicates spherical clusters comprising a mass fractal with fractal dimension consistent with partially collapsed random walk polymers. These results suggest that at lower fPCL and higher PEG block Mn (10 kDa) the copolymers are observed in a transitional state between ill-defined polymer clusters and metastable aggregates, supporting the NMR spectroscopy experiments and DLS results. Similar to the PEG5PCLy copolymer series, the PEG10PCLy series was observed to undergo a transition from metastable aggregates to cylindrical, and finally, ellipsoidal micelles as the fPCL was increased (Figure 6). PEG10PCL7.9 was observed as cylinders ~35 nm in length and 10 nm in radius. PEG10PCL10.7 and PCL10PCL14.9 were effectively modelled as oblate ellipsoids with equatorial radii of 14–22 nm and polar radii of 6.3 nm.
Overall, the synchrotron SAXS analysis reveals the emergence of defined aggregate shapes, including cylindrical and ellipsoidal micelles with increasing fPCL, regardless of the PEG Mn. Similarly, Rajagoal et al. investigated the effect of PEG and PCL MW on micelle formation for a series of PEGxPCLy copolymers using fluorescent imaging.23 Although in most cases the fPCL was significantly > 0.5, several of the copolymers (PEG2PCL2.9, PEG5PCL3.0, PEG5PCL5.9 and PEG5PCL8.8) were similar in composition to those reported in our study. Interestingly, Rajagoal et al. imaged these copolymers as spherical micelles and in no instances observed a transition from cylindrical to spherical micelles with increasing fPCL.23 These differences possibly highlight the effect of fluorescent dyes or slightly different preparation techniques on micelle formation. Furthermore, the resolution of the imaging technique may have prevented spherical micelles from being distinguished from ellipsoidal micelles.
In general, an increase in the fPCL at a constant PEG Mn led to an increase in aggregate radius (for both cylindrical and ellipsoidal micelles) (Table 2). This is further supported by a decrease in values calculated from diffusion NMR experiments (Figure 4). However, a slight decrease in the aggregate radius (equatorial radius) was observed in going from PEG10PCL10.7 to PEG10PCL14.9.
In terms of molecular packing considerations,14 increasing the length of the hydrophilic block (PEG) leads to increasing inter-headgroup repulsions in the corona. This effectively increases the surface area occupied by each polymer molecules at the core-aqueous interface (a). This lowers the critical packing parameter [CPP = V/alt, where V is the volume occupied by the hydrophobic tail block and lt is the extended length of the tail] and drives the micelle shape towards a more spherical geometry. Based on the observation that increasing the hydrophobic PCL block length for a given PEG Mn also drives the micelles towards a more spherical geometry suggests that the ratio V/ lt becomes smaller with increasing PCL block length, that is to say that the increase in the extended length of the PCL block is proportionally larger than the equivalent increase in the volume it occupies in the micelle core. This is commensurate with the increase in average particle radii measured by SAXS, with the average radii of the hydrophobic cores of the ellipsoidal particles being greater than the equivalent cylindrical particles with the same PEG Mn.
Correlation of NMR spectroscopy, DLS and SAXS data, and interpretation of trends in copolymer self-assembly
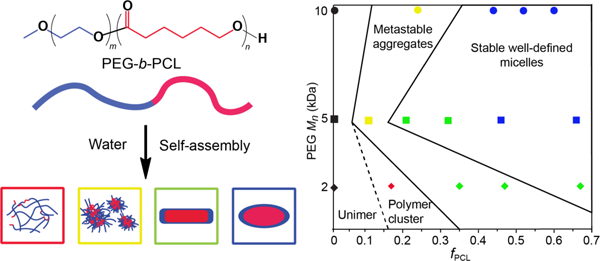
Taken together, the diffusion NMR spectroscopy, DLS and synchrotron SAXS results present a compelling picture of aggregate evolution for the copolymers investigated in this study. The diffusion NMR spectroscopy experiments revealed that the mean and spread of the D distributions can be utilised as a measure of the transition in size dispersity of the copolymer aggregates in solution with respect to changes in the PEG block Mn and fPCL; along with PDI values measured from DLS experiments. Under the conditions studied, these results indicate that there are three different aggregate states, namely loosely associated polymer clusters (mass fractal particle), metastable aggregates (very disperse systems containing different size and shape aggregates) and stable micelles (well-defined shape with a narrow dispersity) (Figure 7).
Figure 7:
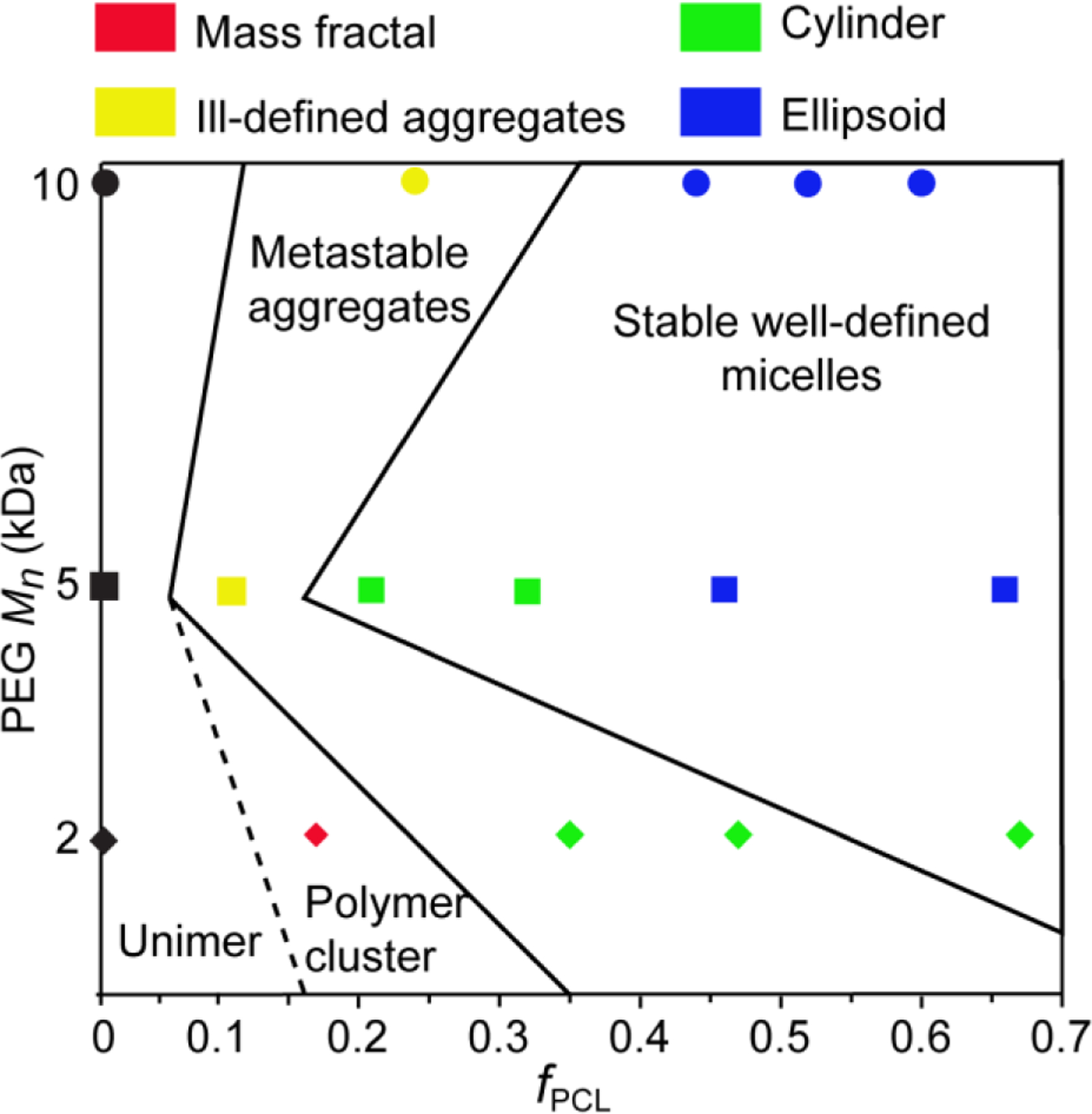
Phase diagram for aggregates formed from the PEG2PCLy (diamonds), PEG5PCLy (squares) and PEG10PCLy (circles) block copolymer series in water, indicating the aggregate structure with respect to fPCL. The diagram combines aggregation states measured by SAXS measurements (shown by colors defined in the figure legend) and by NMR and DLS (with the dashed and solid black lines dividing the samples based on aggregation stability).
For low fPCL (e.g., PEG2PCL0.4) diffusion NMR spectroscopy indicates the presence of unimers or loosely associated unimers (Figure 3) and DLS indicates the presence of very disperse aggregates (Figure 5), which implies that the system consists of loosely associated polymer clusters, which retain the mobility and diffusivity of the unimers. This is further supported by the SAXS analysis for which the scattering profile is consistent with the mass fractal model, whereby small particles in the large mass fractal particle might have diffusivities similar to unimers. For the PEG5PCL0.6 and PEG10PCL3.2 copolymers with a low fPCL, diffusion NMR spectroscopy revealed a significant decrease in the diffusivity indicative of aggregate formation. However, the large differences in the D distributions for the two blocks may be due to relaxation weighting and indicate structure size and mobility heterogeneity. The presence of aggregates with a broad distribution of sizes seen in DLS is further supported by the SAXS results that reveal the formation of a dispersed system containing large aggregates and small cylindrical micelles (metastable system). These differences in the D distributions and broad dispersity are also observed for the PEG2PCL1.1 and PEG2PCL1.8 copolymers implying that these form metastable aggregates, although SAXS would indicate that these aggregates have more defined cylindrical structures. However, the power-laws in the low Q region of the SAXS data indicates the presence of larger secondary aggregates, which might explain the differences noted in the diffusion NMR spectroscopy data for the two blocks (Figure 3 and Figure 6). With increasing fPCL, the SAXS analysis reveals that the PEG5PCL1.3, PEG5PCL2.4 and PEG10PCL7.9 copolymers form cylindrical aggregates, however, the good agreement between the D distributions for the PEG and PCL blocks in the diffusion NMR spectroscopy experiments and lower dispersity in DLS analysis implies that these are stable and relatively well-defined micelles. At larger fPCL the results from all three complementary analytical techniques indicate well-defined and stable micelles that trend towards ellipsoidal (oblate) micelles with lower aspect ratios.
Overall, several trends in the aggregation behaviour with respect to the copolymer microstructure can be concluded from this study. Whilst an increase in the fPCL results in more stable and well-defined micellar structures, the PEG Mn is also important, with lower Mn PEG (e.g., 2 kDa) failing to stabilise aggregate formation until the fPCL > 0.5. This is further supported by the fact that copolymers with similar fPCL (e.g., PEG2PCL1.8 versus PEG5PCL4.2) behave very differently, implying that the MW of the individual blocks, as well as their respective MW ratio, is critical in controlling the self-assembly process. Whilst the increase in PCL block Mn between the PEG2PCL1.8 and PEG5PCL4.2 copolymers might, in isolation, be attributable to the formation of stable aggregates, it does not explain why the PEG5PCL1.3 copolymer forms stable aggregates, and the PEG10PCL3.2 copolymer does not. Thus, it can be concluded that there is a threshold ratio between the fPCL and PEG block Mn that is responsible for controlling the formation of stable and well-defined micelles.
Furthermore, the fPCL and PEG block Mn are also responsible for controlling the aggregate structure. From a thermodynamic perspective, the self-assembly of block copolymers is governed by the interfacial energy between the core and solvent, the free energy of deformation of the polymer chains in the core, and the free energy associated with the interaction of chains in the corona.15 However, the current thermodynamic model for aggregate (micelle) formation relies on two key assumptions: (i) the aggregate core is fully segregated with no solvent, and (ii) the core and corona have a sharp interface.64 As has been shown previously, water penetration into the hydrophobic core of micelles is significant, which clearly indicates that there is a region of the core or boundary layer between the core and corona that is more hydrated.52–54 Furthermore, as we have shown in this study, the presence of characteristic PCL signals in NMR spectra of aggregates also implies the presence of a semi-hydrated core or boundary layer. If indeed there is a semi-hydrated boundary layer it would influence the interfacial energy and deformation of the blocks, which is not currently accounted for in the existing thermodynamic model of block copolymer self-assembly.
4. CONCLUSIONS
This work elucidates the key relationships between the compositional attributes of PEG-b-PCL block copolymers and their hierarchical colloidal assemblies as formed in aqueous systems. This represents the first use of a completely complementary suite of non-interfering analytical techniques for the thorough characterisation of PEG-b-PCL aggregated assemblies. Previously reported investigation of the microstructure-assembly relationships of PEG-b-PCL copolymers have been restricted by the use of a limiting range of observational techniques, and/or have been limited to smaller sample libraries.23–26 These shortfalls have been overcome in this work through a systematic evaluation using a series of complementary techniques to evaluate a larger, clearly defined library of copolymers and their assemblies. It was demonstrated that for PEG-b-PCL copolymer series investigated the fPCL confers a dominant effect on aggregate formation, stability and morphology, while the PEG block Mn plays a lesser, but still important role. At low fPCL the copolymers existed as metastable aggregates which transitioned to cylindrical and well-defined ellipsoidal micelles as the fPCL increased. This provides evidence of a critical fPCL and Mn for stable aggregate formation from amphiphilic block copolymers in water, whilst demonstrating that the size and shape of aggregates are tunable through careful selection of polymers of particular microstructural composition. This constitutes a significant advancement in knowledge within this field. As such, we anticipate that this provides a fundamental basis for understanding the parameters of aggregate formation from PEG-b-PCL block copolymers, and that this will prove valuable to the rational design of micellar drug delivery systems, where aggregate morphology may be closely correlated with their biological fate. For instance, no aggregates evaluated within this study formed perfectly spherical micelle-like aggregates, with SAXS showing that all micelle structures were elongated. However, future studies are required to determine what effects the inclusion of small hydrophobic drug molecules may have on the block copolymer self-assembly process and aggregate structure, and to determine if the trends observed for the PEG-b-PCL block copolymer system are more broadly applicable to similar amphiphilic copolymers.
Supplementary Material
Acknowledgments
SK acknowledges Clinical and Health Sciences, University of South Australia for Scholarship support. SAXS experiments were conducted on the SAXS/WAXS beamline of the Australian Synchrotron (ANSTO), and we acknowledge the support of ANSTO in providing facilities, expertise and funding this research (Grant M15933). N.H.W. and S.J.M. gratefully acknowledge support from the South Australian Department of State Development grant (Grant Nr: IRGP 22). This work was partly funded by the Australian Research Council with A.J.C. being the recipient of a Discovery Early Career Research Award (DE190100531). This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union’s Horizon 2020 Research and Innovation Programme under the SINE2020 project, grant agreement No 654000.
Footnotes
CRediT authorship contribution statement
Khandokar Sadique Faisal: Methodology, Investigation, Formal analysis, Validation, Writing – original draft. Andrew J. Clulow: Methodology, Investigation, Formal analysis, Validation, Writing – review and editing. Marta Krasowska: Methodology, Investigation, Writing – review and editing. Todd Gillam: Supervision, Writing – original draft, Writing – review and editing. Stanley J. Miklavcic: Validation, Writing – review and editing. Nathan H. Williamson: Validation, Supervision, Writing – review and editing. Anton Blencowe: Conceptualization, Validation, Supervision, Project administration, Funding acquisition, Writing – review and editing.
References
- (1).Gong J; Chen M; Zheng Y; Wang S; Wang Y, Polymeric micelles drug delivery system in oncology. J. Control. Release 2012, 159 (3), 312–323. doi: 10.1016/j.jconrel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- (2).Gaucher G; Dufresne MH; Sant VP; Kang N; Maysinger D; Leroux JC, Block copolymer micelles: Preparation, characterisation and application in drug delivery. J. Control. Release 2005, 109 (1–3), 169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- (3).Kim T-Y; Kim D-W; Chung J-Y; Shin SG; Kim S-C; Heo DS; Kim NK; Bang Y-J, Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin. Cancer Res 2004, 10 (11), 3708. doi: 10.1158/1078-0432.CCR-03-0655. [DOI] [PubMed] [Google Scholar]
- (4).AIHW Cancer in Australia 2010: an overview http://www.aihw.gov.au/publication-detail/?id=6442472459 (accessed 25/05/2015).
- (5).Hamaguchi T; Kato K; Yasui H; Morizane C; Ikeda M; Ueno H; Muro K; Yamada Y; Okusaka T; Shirao K; Shimada Y; Nakahama H; Matsumura Y, A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br. J. Cancer 2007, 97 (2), 170–6, doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hamaguchi T; Tsuji A; Yamaguchi K; Takeda K; Uetake H; Esaki T; Amagai K; Sakai D; Baba H; Kimura M; Matsumura Y; Tsukamoto T, A phase II study of NK012, a polymeric micelle formulation of SN-38, in unresectable, metastatic or recurrent colorectal cancer patients. Cancer Chemother. Pharmacol 2018, 82 (6), 1021–1029. doi: 10.1007/s00280-018-3693-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mai Y; Eisenberg A, Self-assembly of block copolymers. Chem. Soc. Rev 2012, 41 (18), 5969–5985. doi: 10.1039/C2CS35115C. [DOI] [PubMed] [Google Scholar]
- (8).Holder SJ; Sommerdijk NAJM, New micellar morphologies from amphiphilic block copolymers: disks, toroids and bicontinuous micelles. Polym. Chem 2011, 2 (5), 1018–1028. doi: 10.1039/C0PY00379D. [DOI] [Google Scholar]
- (9).Allen C; Maysinger D; Eisenberg A, Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf. B. Biointerfaces 1999, 16 (1), 3–27. doi: 10.1016/S0927-7765(99)00058-2. [DOI] [Google Scholar]
- (10).Tanford C, Thermodynamics of micelle formation: prediction of micelle size and size distribution. Proc. Natl. Acad Sci. U.S.A 1974, 71 (5), 1811–1815. doi: 10.1073/pnas.71.5.1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nicholas CV; Luo Y-Z; Deng N-J; Attwood D; Collett JH; Price C; Booth C, Effect of chain length on the micellisation and gelation of block copoly(oxyethylene/oxybutylene/oxyethylene) EmBnEm. Polymer 1993, 34 (1), 138–144. doi: 10.1016/0032-3861(93)90296-M. [DOI] [Google Scholar]
- (12).Rager T; Meyer WH; Wegner G; Winnik MA, Influence of chain length and salt concentration on block copolymer micellisation. Macromolecules 1997, 30 (17). 4911–4919. doi: 10.1021/ma961866l. [DOI] [Google Scholar]
- (13).Israelachvili JN; Mitchell DJ; Ninham BW, Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc., Faraday Trans. 2 1976, 72 (0), 1525–1568. doi: 10.1039/F29767201525. [DOI] [Google Scholar]
- (14).Nagarajan R, Amphiphilic surfactants and amphiphilic polymers: principles of molecular assembly. In Amphiphiles: Molecular Assembly and Applications, American Chemical Society: 2011; Vol. 1070, pp 1–22. [Google Scholar]
- (15).Nagarajan R; Ganesh K, Block copolymer self‐assembly in selective solvents: Spherical micelles with segregated cores. J. Chem. Phys 1989, 90 (10), 5843–5856. doi: 10.1063/1.456390. [DOI] [Google Scholar]
- (16).Nagarajan R; Ruckenstein E, Theory of surfactant self-assembly: a predictive molecular thermodynamic approach. Langmuir 1991, 7 (12), 2934–2969. doi: 10.1021/la00060a012. [DOI] [Google Scholar]
- (17).Lund R; Pipich V; Willner L; Radulescu A; Colmenero J; Richter D, Structural and thermodynamic aspects of the cylinder-to-sphere transition in amphiphilic diblock copolymer micelles. Soft Matter 2011, 7 (4), 1491–1500. doi: 10.1039/C0SM00894J. [DOI] [Google Scholar]
- (18).Riley T; Stolnik S; Heald CR; Xiong CD; Garnett MC; Illum L; Davis SS; Purkiss SC; Barlow RJ; Gellert PR, Physicochemical evaluation of nanoparticles assembled from poly(lactic acid)-poly(ethylene glycol) (PLA-PEG) block copolymers as drug delivery vehicles. Langmuir 2001, 17 (11), 3168–3174. doi: 10.1021/la001226i. [DOI] [Google Scholar]
- (19).Zhang L; Eisenberg A, multiple morphologies and characteristics of “crew-cut” micelle-like aggregates of polystyrene-b-poly(acrylic acid) diblock copolymers in aqueous solutions. J. Am. Chem. Soc 1996, 118 (13), 3168–3181. doi: 10.1021/ja953709s. [DOI] [Google Scholar]
- (20).Research, F. D. A. Food Additive Status List FDA: 2019. https://www.fda.gov/food/food-additives-petitions/food-additive-status-list. (accesed 20–7–2019). [Google Scholar]
- (21).Ibrahim MS; El-Wassefy NA; Farahat DS, 8 - Biocompatibility of dental biomaterials, In Biomaterials for Oral and Dental Tissue Engineering, Tayebi L; Moharamzadeh K, Eds. Woodhead Publishing: 2017; pp 117–140. [Google Scholar]
- (22).Grossen P; Witzigmann D; Sieber S; Huwyler J, PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J. Control. Release 2017, 260, 46–60. doi: 10.1016/j.jconrel.2017.05.028. [DOI] [PubMed] [Google Scholar]
- (23).Rajagopal K; Mahmud A; Christian DA; Pajerowski JD; Brown AEX; Loverde SM; Discher DE, curvature-coupled hydration of semicrystalline polymer amphiphiles yields flexible worm micelles but favors rigid vesicles: polycaprolactone-based block copolymers. Macromolecules 2010, 43 (23), 9736–9746. doi: 10.1021/ma101316w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sun X; Liu X; Li C; Wang Y; Liu L; Su F; Li S, Self-assembled micelles prepared from poly(ɛ-caprolactone)–poly(ethylene glycol) and poly(ɛ-caprolactone/glycolide)–poly(ethylene glycol) block copolymers for sustained drug delivery. J. Appl. Polym. Sci 2018, 135 (9), 45732. doi: 10.1002/app.45732. [DOI] [Google Scholar]
- (25).Qi W; Ghoroghchian PP; Li G; Hammer DA; Therien MJ, Aqueous self-assembly of poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) copolymers: disparate diblock copolymer compositions give rise to nano- and meso-scale bilayered vesicles. Nanoscale 2013, 5 (22), 10908–10915. doi: 10.1039/C3NR03250G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li X; Cooksey TJ; Kidd BE; Robertson ML; Madsen LA, Mapping coexistence phase diagrams of block copolymer micelles and free unimer chains. Macromolecules 2018, 51 (20), 8127–8135. doi: 10.1021/acs.macromol.8b01220. [DOI] [Google Scholar]
- (27).Yoon K; Kang HC; Li L; Cho H; Park M-K; Lee E; Bae YH; Huh KM, Amphiphilic poly(ethylene glycol)-poly(ε-caprolactone) AB2 miktoarm copolymers for self-assembled nanocarrier systems: synthesis, characterisation, and effects of morphology on antitumor activity. Polym. Chem 2015, 6 (4), 531–542. doi: 10.1039/C4PY01380H. [DOI] [Google Scholar]
- (28).Heald CR; Stolnik S; Kujawinski KS; De Matteis C; Garnett MC; Illum L; Davis SS; Purkiss SC; Barlow RJ; Gellert PR, Poly(lactic acid)-poly(ethylene oxide) (PLA-PEG) nanoparticles: NMR studies of the central solidlike PLA core and the liquid PEG corona. Langmuir 2002, 18 (9), 3669–3675. doi: 10.1021/la011393y. [DOI] [Google Scholar]
- (29).Nilsson M; Håkansson B; Söderman O; Topgaard D, Influence of polydispersity on the micellisation of triblock copolymers investigated by pulsed field gradient nuclear magnetic resonance. Macromolecules 2007, 40 (23), 8250–8258. doi: 10.1021/ma071302p. [DOI] [Google Scholar]
- (30).Li T; Senesi AJ; Lee B, Small angle x-ray scattering for nanoparticle research. Chem. Rev 2016, 116 (18), 11128–11180. doi: 10.1021/acs.chemrev.5b00690. [DOI] [PubMed] [Google Scholar]
- (31).Friedrich H; Frederik PM; de With G; Sommerdijk NAJM, Imaging of Self-Assembled Structures: Interpretation of TEM and Cryo-TEM Images. Angew. Chem. Int. Ed 2010, 49 (43), 7850–7858. doi: 10.1002/anie.201001493. [DOI] [PubMed] [Google Scholar]
- (32).Moore J; Cerasoli E, Particle light scattering methods and applications. In Encyclopedia of Spectroscopy and Spectrometry (Third Edition), Lindon JC; Tranter GE; Koppenaal DW, Eds. Academic Press: Oxford, 2017; pp 543–553. [Google Scholar]
- (33).Glavas L; Olsén P; Odelius K; Albertsson A-C, Achieving micelle control through core crystallinity. Biomacromolecules 2013, 14 (11), 4150–4156. doi: 10.1021/bm401312j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Szymusiak M; Kalkowski J; Luo H; Donovan AJ; Zhang P; Liu C; Shang W; Irving T; Herrera-Alonso M; Liu Y, Core–shell structure and aggregation number of micelles composed of amphiphilic block copolymers and amphiphilic heterografted polymer brushes determined by small-angle x-ray scattering. ACS Macro Lett 2017, 6 (9), 1005–1012. doi: 10.1021/acsmacrolett.7b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ungaro F; Conte C; Ostacolo L; Maglio G; Barbieri A; Arra C; Misso G; Abbruzzese A; Caraglia M; Quaglia F, Core-shell biodegradable nanoassemblies for the passive targeting of docetaxel: features, antiproliferative activity and in vivo toxicity. Nanomed. Nanotechnol. Biol. Med 2012, 8 (5), 637–646. doi: 10.1016/j.nano.2011.08.012. [DOI] [PubMed] [Google Scholar]
- (36).Tanner JE, Use of the Stimulated Echo in NMR Diffusion Studies. J. Chem. Phys 1970, 52 (5), 2523–2526. doi: 10.1063/1.1673336. [DOI] [Google Scholar]
- (37).Håkansson B; Nydén M; Söderman O, The influence of polymer molecular-weight distributions on pulsed field gradient nuclear magnetic resonance self-diffusion experiments. Colloid Polym. Sci 2000, 278 (5), 399–405. doi: 10.1007/s003960050532. [DOI] [Google Scholar]
- (38).Williamson NH; Nyden M; Roding M, The lognormal and gamma distribution models for estimating molecular weight distributions of polymers using PGSE NMR. J Magn Reson 2016, 267, 54–62. doi: 10.1016/j.jmr.2016.04.007. [DOI] [PubMed] [Google Scholar]
- (39).Kirby NM; Mudie ST; Hawley AM; Cookson DJ; Mertens HDT; Cowieson N; Samardzic-Boban V, A low-background-intensity focusing small-angle X-ray scattering undulator beamline. J. Appl. Crystallogr 2013, 46 (6), 1670–1680. doi: 10.1107/S002188981302774X. [DOI] [Google Scholar]
- (40).Ilavsky J; Jemian PR, Irena: tool suite for modeling and analysis of small-angle scattering. J. Appl. Crystallogr 2009, 42 (2), 347–353. doi: 10.1107/S0021889809002222. [DOI] [Google Scholar]
- (41).Doucet, et al. SasView Version 5.0.2 10.5281/zenodo.3752443 [DOI]
- (42).Mohanty AK; Jana U; Manna PK; Mohanta GP, Synthesis and evaluation of MePEG-PCL diblock copolymers: surface properties and controlled release behavior. Prog. Biomater 2015, 4 (2–4), 89–100. doi: 10.1007/s40204-015-0040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Labet M; Thielemans W, Synthesis of polycaprolactone: a review. Chem. Soc. Rev 2009, 38 (12), 3484–3504. doi: 10.1039/B820162P. [DOI] [PubMed] [Google Scholar]
- (44).Fairley N; Hoang B; Allen C, Morphological control of poly(ethylene glycol)-block-poly(ε-caprolactone) copolymer aggregates in aqueous solution. Biomacromolecules 2008, 9 (9), 2283–2291. doi: 10.1021/bm800572p. [DOI] [PubMed] [Google Scholar]
- (45).Jain S; Bates FS, On the origins of morphological complexity in block copolymer surfactants. Science 2003, 300 (5618), 460–464. doi: 10.1126/science.1082193. [DOI] [PubMed] [Google Scholar]
- (46).Liu PDG, Block Copolymer Nanofibers and Nanotubes. In Block Copolymers in Nanoscience, 2006; pp 233–255.
- (47).Hussein YHA; Youssry M, Polymeric micelles of biodegradable diblock copolymers: enhanced encapsulation of hydrophobic drugs. Materials (Basel, Switzerland) 2018, 11 (5), 688. doi: 10.3390/ma11050688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Edward JT, Molecular volumes and the Stokes-Einstein equation. J. Chem. Educ 1970, 47 (4), 261. doi: 10.1021/ed047p261. [DOI] [Google Scholar]
- (49).Einstein A, Zur Elektrodynamik bewegter Körper. Ann. Phys 1905, 322 (10), 891–921. doi: 10.1002/andp.19053221004. [DOI] [Google Scholar]
- (50).Hrkach JS; Peracchia MT; Bomb A; Lotan n.; Langer R, Nanotechnology for biomaterials engineering: structural characterisation of amphiphilic polymeric nanoparticles by 1H NMR spectroscopy. Biomaterials 1997, 18 (1), 27–30. 10.1016/S0142-9612(96)00077-4. [DOI] [PubMed] [Google Scholar]
- (51).Jette KK; Law D; Schmitt EA; Kwon GS, Preparation and drug loading of poly(ethylene glycol)-block-poly(ε-caprolactone) micelles through the evaporation of a cosolvent azeotrope. Pharm. Res 2004, 21 (7), 1184–1191. doi: 10.1023/B:PHAM.0000033005.25698.9c. [DOI] [PubMed] [Google Scholar]
- (52).Long JA; Rankin BM; Ben-Amotz D, Micelle structure and hydrophobic hydration. J. Am. Chem. Soc 2015, 137 (33), 10809–10815. doi: 10.1021/jacs.5b06655. [DOI] [PubMed] [Google Scholar]
- (53).Wadsäter M; Barauskas J; Rogers S; Skoda MWA; Thomas RK; Tiberg F; Nylander T, Structural effects of the dispersing agent polysorbate 80 on liquid crystalline nanoparticles of soy phosphatidylcholine and glycerol dioleate. Soft Matter 2015, 11, 1140–1150. doi: 10.1039/C4SM02296C [DOI] [PubMed] [Google Scholar]
- (54).Manet S; Lecchi A; Impéror-Clerc M; Zholobenko V; Durand D; Oliveira CLP; Pedersen JS; Grillo I; Meneau F; Rochas C, Structure of micelles of a nonionic block copolymer determined by SANS and SAXS. J. Phys. Chem. B 2011, 115, 11318–11329. doi: 10.1021/jp200212g [DOI] [PubMed] [Google Scholar]
- (55).Fleischer G, The effect of polydispersity on measuring polymer self-diffusion with the n.m.r. pulsed field gradient technique. Polymer 1985, 26 (11), 1677–1682. doi: 10.1016/0032-3861(85)90285-X. [DOI] [Google Scholar]
- (56).Kidd BE; Li X; Piemonte RC; Cooksey TJ; Singh A; Robertson ML; Madsen LA, Tuning biocompatible block copolymer micelles by varying solvent composition: dynamics and populations of micelles and unimers. Macromolecules 2017, 50 (11), 4335–4343. doi: 10.1021/acs.macromol.6b02579. [DOI] [Google Scholar]
- (57).Blandamer MJ; Cullis PM; Soldi LG; Engberts JBFN; Kacperska A; Van Os NM; Subha MCS, Thermodynamics of micellar systems: comparison of mass action and phase equilibrium models for the calculation of standard Gibbs energies of micelle formation. Adv. Colloid Interface Sci 1995, 58 (2), 171–209. doi. 10.1016/0001-8686(95)00252-L. [DOI] [PubMed] [Google Scholar]
- (58).Fujiwara S; Itoh T; Hashimoto M; Tamura Y, Molecular dynamics simulation of micelle formation in amphiphilic solution. Mol. Simul 2007, 33 (1–2), 115–119. doi: 10.1080/08927020601052948. [DOI] [Google Scholar]
- (59).Rizis G; van de Ven TG; Eisenberg A, Crystallinity-driven morphological ripening processes for poly(ethylene oxide)-block-polycaprolactone micelles in water. Soft Matter 2014, 10 (16), 2825–35. doi: 10.1039/c3sm52950a. [DOI] [PubMed] [Google Scholar]
- (60).Sun Y; Wang Q; Zhang S; Li H; Zhang J; Li D; Li W Synthesis of aromatic-doped polycaprolactone with tunable degradation behavior. Polym. Chem 2018, 9, (28), 3931–3943. doi: 10.1039/C8PY00374B. [DOI] [Google Scholar]
- (61).Beaucage G, Small-angle scattering from polymeric mass fractals of arbitrary mass-fractal dimension. J. Appl. Crystallogr 1996, 29 (2), 134–146. doi: 10.1107/S0021889895011605. [DOI] [Google Scholar]
- (62).Mildner DFR; Hall PL, Small-angle scattering from porous solids with fractal geometry. J. Phys. D: Appl. Phys 1986, 19 (8), 1535–1545. doi: 10.1088/0022-3727/19/8/021. [DOI] [Google Scholar]
- (63).Hammouda B, A new Guinier-Porod model. J. Appl. Crystallogr 2010, 43 (4), 716–719. doi: 10.1107/S0021889810015773. [DOI] [Google Scholar]
- (64).Nagarajan R; Ganesh K, Solubilisation in spherical block copolymer micelles: Scaling analysis based on star model. J. Chem. Phys 1993, 98 (9), 7440–7450. doi: 10.1063/1.464708. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.