

Abstract
Epstein‐Barr virus‐associated lymphoproliferative disease (EBV‐LPD) is frequently fatal. Innate immunity plays a key role in protecting against pathogens and cancers. The stimulator of interferon genes (STING) is regarded as a key adaptor protein allowing DNA sensors recognizing exogenous cytosolic DNA to activate the type I interferon signaling cascade. In terms of EBV tumorigenicity, the role of STING remains elusive. Here we showed that treatment with the STING inhibitor, C‐176, suppressed EBV‐induced transformation in peripheral blood mononuclear cells. In an EBV‐LPD mouse model, C‐176 treatment also inhibited tumor formation and prolonged survival. Treatment with B cells alone did not affect EBV transformation, but suppression of EBV‐induced transformation was observed in the presence of T cells. Even without direct B cell‐T cell contact in a transwell system, the inhibitor reduced the transformation activity, indicating that intercellular communication by humoral factors was critical to prevent EBV‐induced transformation. These findings suggest that inhibition of STING signaling pathway with C‐176 could be a new therapeutic target of EBV‐LPD.
Keywords: cGAS, EBV‐LPD, Epstein‐Barr virus, PAMPs, STING
Treatment with STING inhibitor, C‐176, suppressed the tumor formation in a mouse model of EBV LPDs. Although inhibitor treatment with B cells in vitro alone did not affect EBV transformation, the suppression of EBV‐induced transformation was observed in the presence of T cells

1. INTRODUCTION
Innate immunity plays an essential role in the early defense against pathogens, including viruses. Recognition of exogenous viral nucleic acid is required to induce the innate immune response, not only in antigen‐presenting cells such as macrophages and dendritic cells, but also in vascular endothelial cells, fibroblasts, and epithelial cells. 1 These cells recognize microbial pathogen‐associated molecular patterns (PAMPs) through pattern‐recognition receptors (PRRs). When PRRs recognize a PAMP, they activate the intracellular signal transduction system, which induces the innate immune response for pathogen elimination. 2 Activation of the innate immune response promotes acquired immunity, reflected in T cell activity and dendritic cell maturation. 3
The stimulator of interferon genes (STING; also known as MITA, ERIS, MPYS, and TMEM173) is an adaptor protein for PRRs recognizing viral DNAs. 4 Recently, many studies have identified various PRRs such as cyclic GMP‐AMP synthase (cGAS), interferon gamma‐inducible protein 16 (IFI16), DEAD‐box helicase 41 (DDX41), meiotic recombination 11 homolog A (MRE11), and heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1). 4 , 5 , 6 , 7 , 8 , 9 , 10 STING activation by these sensors induces the production of the humoral factors interferon (IFN)‐α/β. 11 cGAS, which plays a central role in the innate immune response, recognizes herpes virus‐derived DNA in the cytoplasm during the primary infection and viral production phases, inducing the innate immune response via STING. 8 In addition, in a methylcholanthrene (MCA)‐induced sarcoma mouse model, STING knockout mice showed a shorter survival time. 12 These data suggest that STING can promote antiviral and antitumor effects.
Epstein‐Barr virus (EBV) is a double‐stranded DNA virus that belongs to the Herpesviridae family, and commonly infects humans. EBV infects B cells and can produce EBV‐associated lymphoproliferative disorder (EBV‐LPD) in immunocompromised patients. 13 EBV‐LPD is characterized by symptoms ranging from benign polyclonal proliferation of EBV‐infected lymphocytes to malignant monoclonal proliferation. When immune suppression occurs after transplantation, malignant EBV‐LPD is challenging to treat and treatment‐related death may occur. 13 , 14
EBV shows 2 patterns of infection: latent and lytic. The transition from latent to lytic infection is known as “reactivation.” 15 In EBV‐positive lymphoma, tumor cells with both infection patterns have been observed. 16 In latently infected cells that express a limited number of virus genes, including oncogenes, the gene expression pattern is altered depending on the cell type and intracellular status. 17 Previous reports have shown that LMP2 degrades the IFN receptor, while EBNA2 and EBER induce resistance to IFN. 18 , 19 , 20 Lytic‐infected cells express approximately 80 viral genes and control IFN induction and signaling via several viral proteins. 21 , 22 Although studies have aimed to identify virus genes that negatively regulate the innate immune response, it remains unclear whether STING influences the development of EBV‐related tumors.
DNA sensor molecules, such as cGAS, STING (as adaptor proteins), and TANK binding kinase 1 (TBK1) are essential for activating IFN regulatory factor 3 (IRF3). 23 Host DHHC (Asp‐His‐His‐Cys) palmitoyl transferase palmitoylates STING, which promotes STING accumulation in the trans‐Golgi network (TGN), thereby accelerating the recruitment of downstream TBK1 and IRF3 to the TGN. 24 , 25 In recent years, STING inhibitors, including C‐176, have been identified by drug screening focused on the palmitoylation of STING. 26 These inhibitors were developed to clarify the role of STING palmitoylation in the type I IFN‐producing cascade, and in autoinflammatory disease in mice. C‐176 showed no apparent toxicity in vitro or in vivo. Moreover, C‐176 improves inflammation in the organs of three‐prime repair exonuclease 1 (TREX1)‐deficient mice, in which the overproduction of type I IFN causes Aicardi–Goutières syndrome‐like symptoms. 26
In this study, we conducted infection experiments in vitro and in vivo to clarify the role of STING in EBV tumorigenesis. In cell cultures, the STING inhibitor C‐176 suppressed the transformation of EBV. Furthermore, administering a STING inhibitor in the EBV‐LPD mouse model provided an antitumor effect and prolonged survival. Inhibitor treatment with B cells alone did not influence cell proliferation, humoral factor production, or transformation, but transformation efficiency decreased in the presence of T cells. Our findings suggest that STING‐mediated crosstalk of various inflammatory factors plays an important role in EBV‐LPD tumorigenesis.
2. MATERIALS AND METHODS
2.1. Cell culture
HEK293 cells and EBV‐positive HEK293 cells (HEK293EBV), established by the transfection of BAC DNA carrying the whole B95‐8 EBV genome (a generous gift from Dr. R. Longnecker), were grown in DMEM (Sigma‐Aldrich) supplemented with 10% FBS and hygromycin B (150 µg/ml; Takara). 27 AGS cells (a human gastric adenocarcinoma cell line) infected with a recombinant EBV 28 carrying enhanced GFP (EGFP) were cultured in Nutrient Mixture F‐12 Ham (Sigma‐Aldrich) including 10% FBS and G418 (400 µg/ml; Wako). Lymphoblast cell lines (LCLs) were established by infecting the human PBMCs from healthy donors (precision medicine, #1‐4) with a virus fluid obtained from HEK293EBV cells. LCLs and Jurkat cells were cultured in RPMI 1640 medium (Sigma‐Aldrich) supplemented with 10% FBS. All mediums contained 1% penicillin‐streptomycin (Gibco) unless otherwise specified. Primary T cells were cultured in RPMI 1640 medium containing 10% human serum, 2% l‐glutamine (Sigma‐Aldrich), 1% penicillin‐streptomycin (Sigma‐Aldrich), and recombinant human IL‐2 (PeproTech) to a final concentration of 50 IU/mL.
2.2. STING inhibitor and agonist treatment
C‐176 and H‐151 were obtained from Sigma‐Aldrich and InvivoGen, respectively. G10 was purchased from Abcam. These reagents were dissolved in DMSO. LCLs and isolated primary T cells were treated with C‐176 or H‐151 for 24 h. Primary T cells and Jurkat cells were reacted with G10 for 2‐4 h. Reacted cells were lysed, and immunoblotting analysis for gene expression was performed.
2.3. Immunoblotting analysis
Harvested cells were lysed in loading buffer (50 mM Tris‐HCl, pH 6.8, 2% sodium dodecylsulfate (SDS), 10% glycerol, 6% 2‐mercaptoethanol, 0.0025% bromophenol blue), subjected to SDS‐PAGE (Wako), and then transferred to a PVDF membrane. The following primary antibodies were diluted in Can Get Signal Solution (TOYOBO): anti‐LMP1 antibody (S12, 1:50), anti‐GAPDH antibody (14C10; Cell Signaling Technology [CST], 1:2000), anti‐TBK1 antibody (CST), anti‐phospho‐TBK1/NAK (Ser172) antibody (D52C2, 1:1000; CST), and anti‐STING antibody (D2P2F, 1:1000; CST). As the secondary antibody, HRP‐conjugated goat anti‐mouse IgG (BioSource) or HRP‐linked anti‐rabbit IgG (CST) was used at a dilution of 2000‐5000. Forte (Merck) or Chemi‐Lumi One Ultra (Nacalai Tesque) was used as the HRP substrate, and EZ‐Analyzer (ATTO) was used to detect luminescence, which was also photographed.
2.4. RT‐PCR
LCLs were treated with DMSO or STING inhibitor (C‐176) for 24 h. Total RNAs were isolated from treated LCLs using TriPure (Sigma‐Aldrich), and RNA was then dissolved in nuclease‐free water. To detect the cellular transcripts, RT‐PCR was performed using the One‐Step TB Green PrimeScript PLUS RT‐PCR Kit (Takara) according to the manufacturer's instructions. Transcripts were detected by oligonucleotides designed to amplify gene‐specific regions. The IFN‐β primer sequence was 5′‐TGCTCTCCTGTTGTGCTTCTCC‐3′ (forward primer), 5′‐ATAGATGGTCAATGCGGCGT‐3′ (reverse primer), the IL‐6 primer sequence was 5′‐CCAGGAGCCCAGCTATGAAC‐3′ (forward primer), 5′‐CCCAGGGAGAAGGCAACTG‐3′ (reverse primer) and the CXCL10 primers were designed as 5′‐CCAGAATCGAAGGCCATCAA‐3′ (forward primer), 5′‐CATTTCCTTGCTAACTGCTTTCAG‐3′ (reverse primer). RT‐PCR was performed using the 7500 Fast Dx Real‐Time PCR instrument (Applied Biosystems), as described previously. 29
2.5. Cell proliferation assay
To measure cell proliferation, 2 × 105 LCLs were seeded onto 12‐well plates and cultured in medium containing 0.5 µM STING inhibitor (C‐176). Cells were counted for the trypan blue exclusion test every 24 h using the Countess automated cell counter (Thermo Fisher Scientific).
2.6. Cytotoxicity assay
First, 1 × 104 LCLs were seeded into a 96‐well plate and reacted for 24 h with 0, 0.25, 0.5, or 2.0 µM C‐176. To measure cell viability, CellTiter‐Glo (Promega) was added according to the manufacturer's recommendations, and the luminescence signal was detected with the GloMax Navigator Microplate Luminometer (Promega).
2.7. EBV‐LPD mouse model
We followed the relevant guidelines for Animal Experimentation of Nagoya University, Japan, and the ethical regulations. The mice were killed once they showed signs of clinical illness (20% weight loss, ruffled coat, and hunching behavior). Six‐wk‐old NOD/Shi‐scid, IL‐2RγKO (NOG) mice purchased from the Central Institute for Experimental Animals were assigned randomly to groups under specific pathogen‐free conditions, and food and water were provided. First, 1 × 107 human cord blood mononuclear cells (CBMCs), purchased from RIKEN, were co‐cultured for 2.5 h with 2 × 106 green Akata unit viral fluid derived from HEK293EBV cells and inoculated intraperitoneally (ip). Then, 150 mg/mL C‐176 dissolved in DMSO was added to 50 μL corn oil, and 562.5 nmol C‐176 was administered ip twice daily for 2 wk (total of 24 treatments). In the untreated group, an equivalent volume of DMSO was dissolved in 50 µl corn oil. Tumors that formed ip were observed under bright field and fluorescence microscopy using an M205 FA stereomicroscope (Leica).
2.8. Detection of viral DNA in mouse blood by real‐time PCR
Genomic DNA was isolated from whole blood using the DNeasy Blood & Tissue Kit (Qiagen) in accordance with the manufacturer's instructions. EBV DNA was detected by real‐time quantitative PCR, as described previously, using primer pairs and a fluorogenic DNA probe targeting the EBV BALF5 gene (sequence context: 5′‐CGGAAGCCCTCTGGACTTC‐3′ (forward) and 5′‐CCCTGTTTATCCGATGGAATG‐3′ (reverse), 5′‐PE‐TGTACACGCACGAGAAATGCGCC‐3′). 30 Amplification reactions were performed using the Fast Start Universal Probe Master (Roche) and 7500 Fast Dx RT‐PCR instrument.
2.9. Histological analysis
The mice were reflux fixed with 10% formalin neutral buffer solution (Wako), and tissues were embedded in paraffin using a Tissue‐Tek VIP 6 processor and TEC‐P‐S system (Sakura Finetek). H&E or immunohistochemical (IHC) staining was performed as described previously. 31 After deparaffinization and rehydration of the tissue sections, heat‐induced epitope retrieval was carried out in pH 9 Target Retrieval Solution (Agilent) for 30 min, after which the tissue sections were kept at room temperature for 30 min. After blocking with Protein Block (Agilent), the samples were reacted with a primary antibody, as follows: anti‐human STING (D2P2F, 1:1000; CST), anti‐LMP1 (S12, 1:50), and anti‐human CD3 (F7.2.38, 1:50; Agilent). To detect EBV‐encoded small RNA (EBER) by in situ hybridization (ISH), completely dried tissue sections were reacted with a FITC‐conjugated EBER probe Y5200 (Agilent) at 55°C for 90 min, and then with anti‐FITC antibody (M228‐3, 1:2,000; MBL). For endogenous peroxidase blocking, samples were reacted in methanol containing 0.3% hydrogen peroxide for 30 min after the primary antibody reaction. A secondary antibody reaction was carried out with EnVision Dual Link reagent (Agilent) for 30 min. For fluorescence staining, Alexa 488 anti‐rabbit IgG and Alexa 546 anti‐mouse IgG (Invitrogen) were reacted as secondary antibodies. The stained sections were mounted in Entellan New (Sigma‐Aldrich) or Prolong Gold reagent (Thermo Fisher Scientific), and whole‐slide images were acquired using a virtual slide system (VS120; Olympus). High‐resolution imaging was accomplished using confocal microscopy (LSM880; Zeiss).
2.10. Phenotyping of splenic lymphocytes in an EBV infection mouse model
Mouse splenic tissue was dissected under deep isoflurane anesthesia and homogenized with 5 mL chilled PBS. The suspension was passed through a cell strainer (100 µm; Falcon). The filtered cell suspension was centrifuged, and the cell pellet was washed twice in PBS. The final pellet was resuspended in 100 µL PBS and added to a DuraClone IM Phenotyping BASIC tube (Beckman Coulter). Mouse splenic lymphocytes were treated with PECy5 anti‐human CD45 antibody (HI30; BioLegend), PECy7 anti‐human CD3 antibody (SK7; BioSource), and FITC anti‐human CD19 antibody (J3‐119; Beckman Coulter) for 20 min on ice followed by perm/wash buffer (BioSource) for permeabilization. Cells were stained with PE anti‐human STING antibody (T3‐680; BioSource). After staining in accordance with the manufacturer's instructions, the cells were exposed to VersaLyse reagent (Beckman Coulter) for 15 min for red blood cell hemolysis, before being fixed with 4% paraformaldehyde in PBS. Lymphocyte phenotyping was performed using flow cytometry with the Gallios instrument (Beckman Coulter).
2.11. EBV transformation assay
B cells were isolated from peripheral blood or CBMCs in accordance with the manual, using the EasySep/RosetteSep Human B Cell Enrichment Cocktail (StemCell Technologies). To confirm cell isolation, cells stained in a DuraClone IM Phenotyping BASIC tube (Beckman Coulter) were analyzed by FACS analysis (Beckman Coulter; Gallios). PBMCs and B cells were seeded into a 96‐well plate and infected with a 10‐fold serial dilution of virus fluid obtained from AGS EBV cells. PBMCs were cultured in the presence of cyclosporin (0.5 µg/mL; Sigma‐Aldrich). Medium exchange was performed every 4 d with medium containing a STING inhibitor (C‐176; 0.5 µM). At 3 wk after infection, the number of wells in which transformed cells were present was counted, and the 50% transforming dose (TD50) was calculated. Fluorescence images were obtained by fluorescence microscopy (Axio Observer 7; Zeiss).
2.12. Lentiviral production
Lentivirus was produced in HEK293T cells using the lentivirus packaging plasmids pCMV‐VSV‐G (Addgene #8454) and pCMV‐R8.74 (Addgene #22036). shRNA lentiviral plasmid vectors were purchased from Vector Builder (pLV[shRNA]‐EGFP:T2A:Puro‐U6>hTMEM173[shRNA#1](VB900090‐7227ftv); pLV[shRNA]‐EGFP:T2A:Puro‐U6>hTMEM173[shRNA#3] (VB900090‐7233grx); pLV[shRNA]‐EGFP/Puro‐U6>Scramble_shRNA). HEK293T cells were transiently transfected with each lentivirus vector, together with packaging plasmids, using Lipofectamine 2000 (Thermo Fisher Scientific). Supernatants containing lentivirus were harvested at 48 and 72 h after transfection, and concentrated using Lenti‐X concentrator (Takara). Lentivirus was titrated on Jurkat cells by measuring EGFP‐positive cell rates and primary T cells were infected at an MOI of 3.
2.13. Generation of shRNA‐transduced T cells
Primary T cells were isolated with RosetteSep™ (StemCell Technologies) from healthy donor PBMCs. These cells were stimulated with anti‐CD3/CD28 beads (Invitrogen) for 3 d. At 1 d after stimulation, stimulated T cells were infected with lentivirus carrying shRNA, using a scramble sequence as a control (shScramble) or shRNAs targeting STING (ShSTING#1 and #2). On day 5 after infection, infected T cells were incubated with puromycin at a low concentration (0.5 µg/mL). Expanded T cells were restimulated with anti‐CD3/CD28 beads under puromycin at a high concentration (1.0‐2.0 µg/mL). GFP expression upon infection was determined using a Countess II counter (ThermoFisher Scientific) and shRNA knockdown efficiency was tested by immunoblotting analysis.
2.14. Transwell assay
Isolated B cells were co‐cultured with virus obtained from HEK293EBV cells for 2.5 h at room temperature. B cells were suspended in fresh RPMI medium supplemented with 10% FBS at a density of 5 × 105 cell/mL and seeded onto 24‐well plates. In total, 1 × 105 T cells were placed into the upper chamber of the transwell (0.4 µm pore size; Millipore). B cells on the lower chamber were observed by fluorescence microscopy (Axio Observer 7; Zeiss) and counted using the Countess automated cell counter (Thermo Fisher Scientific) for 7 d after infection.
2.15. Statistical analysis
Overall survival was analyzed using the Kaplan‐Meier method, and significant differences between the 2 groups were determined using the log‐rank test. The log‐rank test and Fisher exact test were performed using EZR software from Jichi Medical University Saitama Medical Center. 32 All quantitative data were analyzed using Student t test at a significance level of 1% or 5%.
3. RESULTS
3.1. STING inhibitor suppresses EBV tumor formation in the EBV‐LPD mouse model
We first assessed whether STING was required for tumorigenesis in vivo. Severely immunodeficient mice (NOG mice) transplanted with cord blood‐derived mononuclear cells co‐cultured with EBV were administered ip with C‐176 (Figure 1A). C‐176 administration prolonged survival and reduced weight loss compared with the non‐administered group (Figure 1B,C). Furthermore, C‐176 treatment resulted in a reduction in both tumor size and splenomegaly (Figure 1D). Although EBV copy levels in the peripheral blood were unaffected by C‐176, we observed reduced lymphocyte infiltration in the organs (Figure 1E,F). Therefore, these data suggested that the innate immune response mediated by STING may be involved in the development and exacerbation of EBV tumorigenesis in vivo.
FIGURE 1.
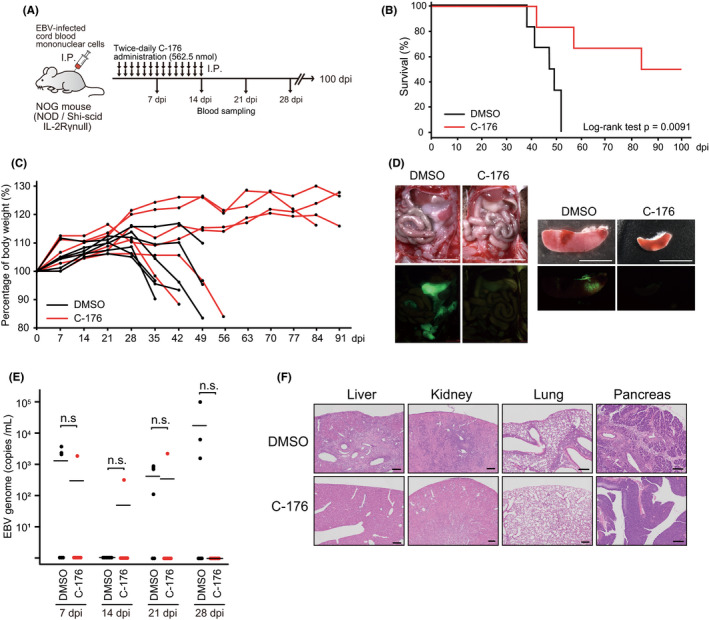
STING inhibitor suppresses EBV tumor formation in the EBV‐LPD mouse model. A, Schematic of the mouse model. A NOG mouse was inoculated ip with EBV‐infected human cord blood mononuclear cells and then administered DMSO or C‐176 (562.5 nmol) ip, twice daily for 2 wk (n = 6 mice per group). Blood sampling was carried out to identify genomic EBV DNA at the indicated time points. B, Kaplan‐Meier survival curves. C, Bodyweight changes of mice are shown. D, The ip tumor and spleen of DMSO‐treated and C‐176‐treated mice. The EBV‐encoded EGFP signal was detected under excitation light. Scale bars, 5 mm. E, The EBV genome (copies/mL) of each group is indicated. EBV DNA in blood was detected by real‐time PCR. n.s., no significant difference. F, H&E staining of DMSO‐treated and C‐176‐treated mouse organs. Scale bars, 200 μm
3.2. An inhibitor of STING suppresses EBV transformation in vitro but not STING signaling in EBV‐positive B cells
To determine whether STING is expressed in EBV‐infected cells, we next examined the expression of STING in EBV‐immortalized LCLs, which were established using PBMCs. STING was abundantly expressed in LCLs, but not in isolated primary B cells (Figure 2A), consistent with previous findings. 33
FIGURE 2.
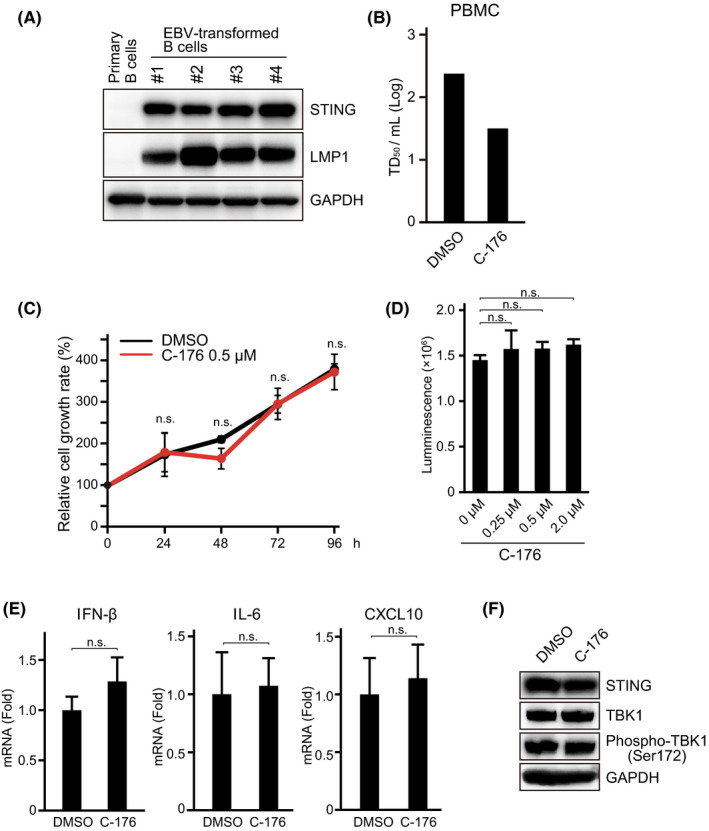
An inhibitor of STING suppresses EBV transformation in vitro but not STING signaling in EBV‐positive B cells. A, Primary B cells and EBV‐transformed B cells, known as LCLs and established from several healthy donor‐derived PBMCs, were analyzed by immunoblotting for STING. Epstein‐Barr virus LMP1 was used as an infection marker. Anti‐GAPDH antibody was used as the internal control. B, PBMCs were infected with 10‐fold serial dilutions of Akata EBV. The medium containing the C‐176 STING inhibitor (0.5 µM) was exchanged every 4 d. After 3 wk, the transformation efficiency (TD50/mL) was calculated based on the number of wells in which immortalized cells were present. C, LCLs were cultured in the presence of C‐176 and counted every 24 h for 4 d. A trypan blue exclusion test using an automated cell counter was performed. D, LCLs were treated with the indicated concentration of C‐176 for 24 h, and cell viability was quantified by a luminescent assay using CellTiter‐Glo, based on the ATP level. E, Cytokine production of EBV‐transformed B cells treated with DMSO or STING inhibitor (C‐176). LCLs treated with C‐176 for 24 h were harvested and total RNA was extracted. IFN‐β, IL‐6, and CXCL10 mRNAs were quantified by RT‐PCR. All experiments were carried out in triplicate. n.s., no significant difference, Student t test. F, Immunoblot analysis of STING, TBK1, phospho‐TBK1 (Ser 172), and GAPDH of LCLs, in the presence or absence of C‐176
To examine the effect of STING on EBV transformation activity, we infected PBMCs with Akata virus in the presence of C‐176, which has recently been identified as a STING inhibitor. At the same time, we started a cell culture in 0.5 µM C‐176 and calculated the TD50 per mL corresponding to EBV transformation activity at 3 wk after infection. 34 Figure 2B shows that the STING inhibitor C‐176 clearly decreased the transformation efficiency of PBMCs. These data indicated that STING, which plays a central role in the innate immune response, is expressed in LCLs and involved in EBV tumorigenesis in vitro.
As STING inhibitors suppressed EBV‐induced transformation, we hypothesized that STING was required for the proliferation of EBV‐immortalized cells. We counted viable cells after treatment with the C‐176 STING inhibitor. There was no significant difference in cell proliferation either with or without C‐176 (Figure 2C). A cytotoxicity assay did not show a dose‐dependent effect on cell viability (Figure 2D). In addition, STING inhibitors had no clear effect on the transcription of several humoral factors mediated by STING (Figure 2E). Furthermore, C‐176 did not affect TBK1 phosphorylation (Figure 2F). Therefore, treatment with a STING inhibitor had no significant effect on cell proliferation, at least in EBV‐immortalized B cells.
3.3. Tumor‐infiltrating T cells express STING
To evaluate the expression of STING in vivo, we tested for its presence in tumors from EBV‐associated LPD (EBV‐LPD) mouse models. 31 , 35 , 36 Immunohistochemical analysis of the EBV‐LPD mouse model showed expression of STING in tumor tissue (Figure 3A). B cell LPD was reported in this mouse model, 35 consistent with our findings; however, CD3‐positive cells were also observed in the tumor tissue (Figure 3A). Previous reports showed that STING is expressed in human lymphocytes, including macrophages and T cells, and that the innate immune response mediated by STING is required to regulate both the innate and adaptive immune systems. 37 , 38 Moreover, it was reported that T cells provide support for the proliferation of B cells infected with EBV in vitro and in vivo. 39 Therefore, we investigated whether T cells in tumor tissues expressed STING. We observed higher levels of STING expression in LMP1‐negative cells compared with LMP1‐positive tumor cells (Figure 3B). As the tumor progressed in mice, a GFP signal indicating EBV‐positive cells was detected in the spleen, denoting the presence of a metastatic tumor (Figure 2D). Phenotypic analysis of spleen lymphocytes revealed that B and T cells, rather than natural killer (NK) cells, predominated in the spleen, suggesting that the invasive CD3‐positive cells were predominantly T cells (Figure 3C). Furthermore, multicolor‐staining analysis of the tumor revealed the expression of STING in CD3‐positive cells (Figure 3D,E). These results suggested that STING‐regulated intracellular communication between EBV‐positive tumor cells and tumor‐infiltrating T cells is important for tumorigenesis of EBV‐LPD.
FIGURE 3.
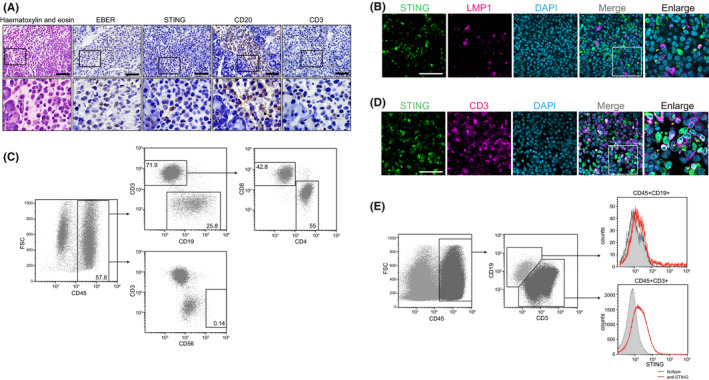
Tumor‐infiltrating T cells express STING. A, H&E and immunohistochemical staining for STING, CD20, and CD3, and in situ hybridization for EBER in tumor tissue from the EBV‐LPD model mouse. Scale bars, 50 μm. The black boxes contain magnified images. B, Co‐immunofluorescence staining for STING (green), LMP1 (magenta), and DAPI (gray) in the tumor of the EBV‐LPD mouse model. C, The relative proportions of CD45‐, CD3‐, CD8‐, CD19‐, and CD56‐positive cells in splenic lymphocytes of the EBV‐LPD mouse model were determined by flow cytometry. D, Co‐immunofluorescence staining of STING (green), CD3 (magenta), and DAPI (cyan) in the tumor of the EBV‐LPD mouse model. Scale bars, 50 μm. The white boxes contain magnified images. E, Flow cytometric analysis of STING in splenic lymphocytes in the EBV‐LPD mouse model
3.4. T cells promote EBV transformation through humoral factors
We hypothesized that the activation of STING in inflammatory T cells, rather than infected B cells, contributed to the onset of EBV‐LPD. Therefore, we compared transformation efficiency between isolated B cells alone and isolated B and T cells in the presence of C‐176. The transformation efficiency of the isolated B cells alone treated with C‐176 was equivalent to that of the untreated cells, but decreased in the presence of the isolated T cells (Figure 4A). To determine whether there was a direct interaction between the B and T cells, we performed a transformation assay using a transwell system, in which humoral factors, but not cells, could pass through the porous membrane (Figure 4B). As shown in Figure 4B, C‐176 suppressed transformation efficiency regardless of the presence or absence of a membrane. These results suggested that STING‐mediated innate immune response activity in T cells is involved in EBV tumorigenesis and regulated by humoral factors.
FIGURE 4.
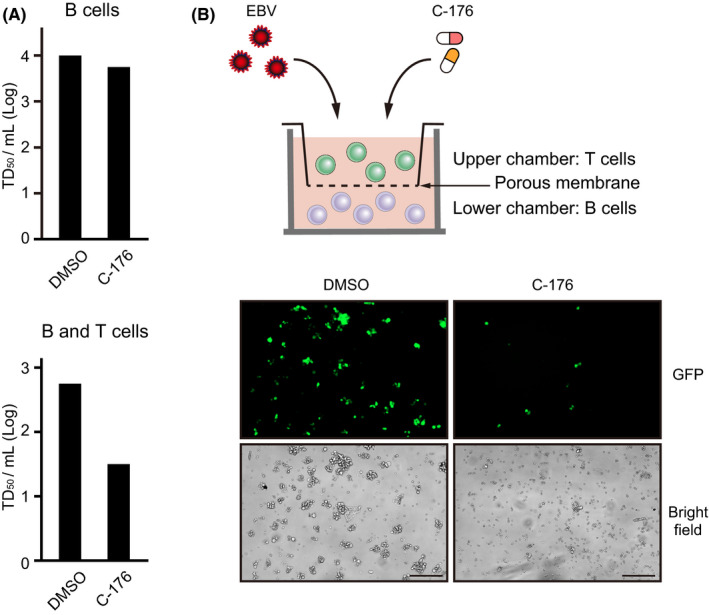
T cells promote EBV transformation through humoral factors. A, B cells isolated from PBMCs were infected with Akata EBV and cultured for 3 wk in medium containing DMSO or C‐176 (0.5 µM). Wells with transformed cells were counted within a 96‐well plate to calculate the EBV transformation efficiency (TD50/mL). This assay was also performed under B and T cell co‐culture conditions. B, Schematic of the transwell system. Isolated B cells were seeded into a 24‐well plate and cultured with medium containing C‐176 (0.5 µM), while isolated T cells were inoculated into transwell chambers with 0.4 µm polycarbonate membranes. Cells were co‐cultured for 7 d and observed by fluorescence microscopy. Scale bard, 200 μm
3.5. STING inhibitors suppressed STING signaling in T cells
Human primary T cells have been shown to express STING, and their physiological significance in terms of survival and acquired immunity have been studied. 40 Because C‐176 did not decrease the phosphorylation of TBK1 in LCLs (Figure 2F), we next evaluated the effect of STING inhibitors on STING signaling in T cells. H‐151, as well as C‐176, which inhibits the palmitoylation of STING, suppressed TBK1 phosphorylation in primary T cells (Figure 5A,B). Conversely, when primary T cells were treated with G10, a STING agonist, 41 the TBK1 phosphorylation level increased slightly (Figure 5C), which was also observed in human T cell leukemia cell line Jurkat cells (Figure 5D). These results suggested that the STING signaling pathway is commonly conserved in T cells.
FIGURE 5.
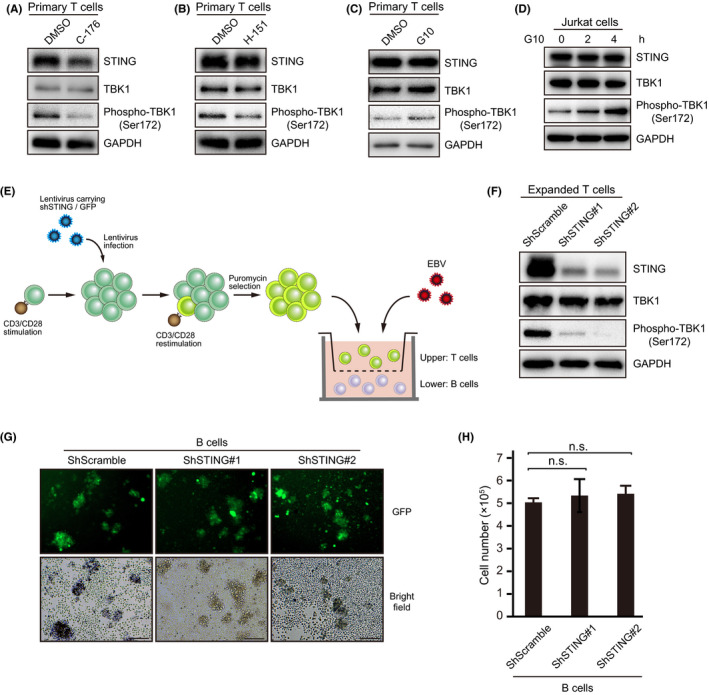
STING inhibitors suppressed STING signaling in T cells. Immunoblot analysis showing STING, TBK1, phospho‐TBK1 (Ser172), and GAPDH. A, B, Primary T cells were isolated from PBMCs and reacted with 0.5 µM C‐176 or 0.5 µM H‐151 for 24 h. C, Primary T cells were exposed to DMSO or 50 µM G10 for 2 h. D, Jurkat cells were treated with DMSO or 50 µM G10 and harvested at the indicated times. E, Human CBMC‐derived B cells were co‐cultured with EBV in vitro. On day 1 after infection, expanded T cells carrying shScramble or shSTING (#1 and #2) were seeded into the transwell chamber. F, Immunoblots for endogenous STING in shSTING‐transduced T cells. G, Fluorescence image of B cells co‐cultured with T cells. Scale bars, 200 μm. H, B cells were quantified at 7 d after infection by counting trypan blue‐stained cells
To elucidate the molecular mechanisms of T cell STING signaling in EBV tumorigenesis, we established STING‐knockdown primary T cells. Lentiviral vector targeting STING was introduced into primary T cells, and T cells were proliferated in accordance with a published method for chimeric Ag receptor T (CAR‐T) cell expansion. 42 Then, we conducted a co‐culture experiment with EBV‐infected B cells (Figure 5E). Although STING knockdown by shRNA resulted in reduced expression of STING and suppression of TBK1 phosphorylation (Figure 5F), cell growth of EBV‐infected B cells co‐cultured with STING‐knockdown T cells was comparable with that of STING‐intact T cells (Figure 5G,H). Based on these results, we were unable to determine the influence of STING on EBV‐infected B cells growth, at least in co‐culture experiments.
4. DISCUSSION
In this study, we identified the expression of STING in EBV‐transformed B cell lines and showed that administration of a STING inhibitor reduced EBV transformation activity. We also identified STING expression in tumor‐infiltrating lymphocytes, especially T cells, and in EBV‐positive B cells. Infection experiments with isolated B and T cells revealed that intercellular communication between EBV‐infected B and T cells via humoral factors, but not intercellular contact, is required for EBV transformation activity. Furthermore, administering a STING inhibitor to an EBV‐LPD mouse model suppressed EBV tumor progression, demonstrating the importance of STING for tumorigenicity.
The regulatory mechanisms by which EBV escapes the innate immune response triggered by primary infection, and the reactivation molecules, have been well characterized. 22 , 43 KSHV‐ORF52 and EBV BLRF2 homologs directly interact with cGAS to control its enzymatic activity. 44 More recently, it was found that EBV BGLF2 binds to STING and suppresses type I IFN production. 45 Thus, although EBV lytic infection‐associated genes have been well characterized, the role of STING in tumor cells, among which latently infected cells are present, remains unclear. One unexpected finding concerned the extent to which STING inhibitors significantly suppressed the transformation activity of EBV and tumorigenesis in vivo, which suggested that STING is critical for EBV tumorigenesis. Even though STING was expressed in the EBV‐transformed B cell lines, the STING inhibitor did not affect their proliferation. This suggested that EBV might utilize STING during B cell transformation and tumorigenesis, while acquiring multiple mechanisms to negatively regulate innate immunity. Notably, T cell depletion in the humanized mouse model reduced tumorigenicity efficiency, indicating that T cells support the development of EBV‐infected B cell lymphomas. 39 We found that T cells infiltrating EBV‐positive cells highly expressed STING in the tumor tissue of the EBV‐LPD mouse model (Figure 3B,C). In addition, human primary T cells expressed STING, and STING inhibitors suppressed TBK1 phosphorylation (Figure 5A,B). However, C‐176 did not affect TBK1 phosphorylation in LCLs (Figure 2F). Based on previously published literature and our results, it appears that STING expression in T cells contributes to intercellular communication between T and B cells via humoral factors. As B cell proliferation in the presence of T cells was suppressed by C‐176 at 1 wk after infection (Figure 4B), we speculate that STING in T cells is involved in the initiation but not the progression of multistage carcinogenesis. Further research is needed to identify the humoral factors produced during tumorigenesis according to the actions of STING.
Whether STING expression in tumor cells is beneficial for cell survival is controversial. In mouse primary B cells and chronic lymphocytic leukemia, stimulation with STING agonists induced cell apoptosis. 46 , 47 Moreover, proliferation and survival were intact with STING knockout in melanoma cells, and some reports indicated that the reaction of STING agonists with various carcinomas did not induce cytotoxic activity. 48 , 49 , 50 , 51 Clinically, the tumor stage has shown associations with STING expression in colorectal cancer and malignant melanoma. 52 , 53 Therefore, our findings suggest that the outcomes of STING expression in cancer cells vary depending on the cell type and tumor stage.
Activation of the innate immune response by STING is associated with the construction of a cancer microenvironment via crosstalk of various inflammatory factors, such as chemokines, cytokines, and growth factors, between inflammatory and cancer cells. Tumor‐infiltrating inflammatory cells can reportedly both suppress and promote cancer progression. 54 , 55 In many EBV‐positive cancers, infected cells grow in a neoplastic manner with inflammatory cell infiltration; EBV‐positive gastric cancers with infiltration of lymphocytes are known as lymphoepithelioma‐like carcinomas. 56 , 57 Moreover, infected cells are reported to release secretory inflammatory factors, such as IL‐6 and tumor necrosis factor. 58 , 59 In this case, the inflammatory cells that make up the cancer microenvironment might be closely associated with EBV tumor formation. We presume that B‐T intracellular communication in tumors will be clarified by single‐cell analysis using next‐generation sequencers.
One limitation of this study was that the experiments were performed using a STING inhibitor, and the possibility of off‐target effects cannot be ruled out. Whether C‐176, which has been shown to inhibit mouse STING more specifically compared with human STING, 26 inhibited human STING remains unclear. To rule out off‐target effects, we treated primary T cells with H‐151, which targets human STING. 26 H151 also suppressed STING signaling (Figure 5B). By contrast, no clear difference in EBV transformation was found in a co‐culture experiment with B cells and STING‐knockdown T cells. Therefore, STING in T cells alone cannot fully explain the acquisition of proliferative activity. However, the role of T cell STING in EBV tumorigenesis remains controversial, as the conditions of CD3/CD28‐stimulated and puromycin‐selected T cells are presumed to be very different from those of primary T cells. Moreover, we believe that the generation of STING‐knockdown EBV‐infected B cells may provide new insights.
In conclusion, we have identified that C‐176, a STING inhibitor, prevented EBV‐induced B cell transformation and tumorigenesis in EBV‐LPD. Taken together, these findings suggest that EBV may utilize innate immunity while avoiding antitumor immunity. Although treatment with a STING inhibitor had therapeutic effects in vivo, we were unable to identify humoral factors and T cell subsets regulating tumorigenesis via intracellular communication. Further research on the relationship between EBV and innate immunity is required to clarify the mechanisms of EBV pathogenicity.
CONFLICT OF INTEREST
H. Kimura and Y. Sato were supported by grants from Bristol Myers Squibb and Merck Sharp & Dohme, respectively. All other authors have no conflicts of interest directly relevant to the content of this article.
ACKNOWLEDGMENTS
We thank Drs. Eric Johannsen, Wolfgang Hammerschmidt, Henri‐Jacques Delecluse, and Teru Kanda for materials. We also acknowledge Yoshitaka Adachi of the Department of Hematology and Oncology, Nagoya University Graduate School of Medicine for technical support of T cell expansion; Daisuke Sugiyama of the Department of Immunology, Nagoya University Graduate School of Medicine for technical support for in vitro immunological assays. We thank all the members of our laboratory for their assistance. Histological analysis and imaging analyses were carried out in the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine. This study was supported by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JP18K15165 to TW, and 19K22560 and 20H03493 to HK), and grants from the Aichi Cancer Research Foundation; Kyousaidan; Nasu Research Foundation; Hori Sciences and Arts Foundation to TW and the Uehara Memorial Foundation; the Chemo‐Sero‐Therapeutic Research Institute to HK and the Precursory Research for Embryonic Science and Technology (PRESTO) from the Japan Agency for Medical Research and Development (AMED) (Grant No. JPMJPR19H5) to YS.
Miyagi S, Watanabe T, Hara Y, et al. A STING inhibitor suppresses EBV‐induced B cell transformation and lymphomagenesis. Cancer Sci. 2021;112:5088–5099. 10.1111/cas.15152
Shouhei Miyagi and Takahiro Watanabe contributed equally to this study.
Contributor Information
Takahiro Watanabe, Email: t.nabe.watanabe@med.nagoya-u.ac.jp.
Hiroshi Kimura, Email: hkimura@med.nagoya-u.ac.jp.
REFERENCES
- 1. Barber GN. STING‐dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88‐93. [DOI] [PubMed] [Google Scholar]
- 2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805‐820. [DOI] [PubMed] [Google Scholar]
- 3. Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM. STING‐mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. 2014;193:6124‐6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Unterholzner L, Keating SE, Baran M, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12:959‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kondo T, Kobayashi J, Saitoh T, et al. DNA damage sensor MRE11 recognizes cytosolic double‐stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A. 2013;110:2969‐2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu J, Sun L, Chen X, et al. Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L, Wen M, Cao X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science. 2019;365(6454):eaav0758. [DOI] [PubMed] [Google Scholar]
- 11. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS‐STING pathway in health and disease. Nat Rev Genet. 2019;20:657‐674. [DOI] [PubMed] [Google Scholar]
- 12. Woo SR, Fuertes MB, Corrales L, et al. STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeStefano CB, Desai SH, Shenoy AG, Catlett JP. Management of post‐transplant lymphoproliferative disorders. Br J Haematol. 2018;182:330‐343. [DOI] [PubMed] [Google Scholar]
- 14. Rezk SA, Zhao X, Weiss LM. Epstein‐Barr virus (EBV)‐associated lymphoid proliferations, a 2018 update. Hum Pathol. 2018;79:18‐41. [DOI] [PubMed] [Google Scholar]
- 15. Murata T. Regulation of Epstein‐Barr virus reactivation from latency. Microbiol Immunol. 2014;58:307‐317. [DOI] [PubMed] [Google Scholar]
- 16. Okuno Y, Murata T, Sato Y, et al. Defective Epstein‐Barr virus in chronic active infection and haematological malignancy. Nat Microbiol. 2019;4:404‐413. [DOI] [PubMed] [Google Scholar]
- 17. Münz C. Latency and lytic replication in Epstein‐Barr virus‐associated oncogenesis. Nat Rev Microbiol. 2019;17:691‐700. [DOI] [PubMed] [Google Scholar]
- 18. Shah KM, Stewart SE, Wei W, et al. The EBV‐encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene. 2009;28:3903‐3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanda K, Decker T, Aman P, Wahlström M, von Gabain A, Kallin B. The EBNA2‐related resistance towards alpha interferon (IFN‐alpha) in Burkitt's lymphoma cells effects induction of IFN‐induced genes but not the activation of transcription factor ISGF‐3. Mol Cell Biol. 1992;12:4930‐4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nanbo A, Inoue K, Adachi‐Takasawa K, Takada K. Epstein‐Barr virus RNA confers resistance to interferon‐alpha‐induced apoptosis in Burkitt's lymphoma. EMBO J. 2002;21:954‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haller O, Kochs G, Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology. 2006;344:119‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jangra S, Yuen KS, Botelho MG, Jin DY. Epstein‐Barr virus and innate immunity: friends or foes? Microorganisms. 2019;7(6):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneider WM, Chevillotte MD, Rice CM. Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu X, Li M, Wu Z, et al. Endoplasmic Reticulum Transmembrane Proteins ZDHHC1 and STING Both Act as Direct Adaptors for IRF3 Activation in Teleost. J Immunol. 2017;199:3623‐3633. [DOI] [PubMed] [Google Scholar]
- 25. Mukai K, Konno H, Akiba T, et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun. 2016;7:11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small‐molecule inhibitors. Nature. 2018;559:269‐273. [DOI] [PubMed] [Google Scholar]
- 27. Mabuchi S, Hijioka F, Watanabe T, et al. Role of Epstein‐Barr virus C promoter deletion in diffuse large B cell lymphoma. Cancers (Basel). 2021;13(3):561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maruo S, Yang L, Takada K. Roles of Epstein‐Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J Gen Virol. 2001;82:2373‐2383. [DOI] [PubMed] [Google Scholar]
- 29. Sato Y, Watanabe T, Suzuki C, et al. S‐like phase CDKs stabilize the Epstein‐Barr virus BDLF4 protein to temporally control late gene transcription. J Virol. 2019;93(8):e01707–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kimura H, Morita M, Yabuta Y, et al. Quantitative analysis of Epstein‐Barr virus load by using a real‐time PCR assay. J Clin Microbiol. 1999;37:132‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watanabe T, Sato Y, Masud HMAA, et al. Antitumor activity of cyclin‐dependent kinase inhibitor alsterpaullone in Epstein‐Barr virus‐associated lymphoproliferative disorders. Cancer Sci. 2020;111:279‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanda Y. Investigation of the freely available easy‐to‐use software 'EZR' for medical statistics. Bone Marrow Transplant. 2013;48:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gram AM, Sun C, Landman SL, et al. Human B cells fail to secrete type I interferons upon cytoplasmic DNA exposure. Mol Immunol. 2017;91:225‐237. [DOI] [PubMed] [Google Scholar]
- 34. Yanagi Y, Masud HMAA, Watanabe T, et al. Initial characterization of the Epstein⁻Barr virus BSRF1 gene product. Viruses. 2019;11(3):285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yajima M, Imadome K, Nakagawa A, et al. A new humanized mouse model of Epstein‐Barr virus infection that reproduces persistent infection, lymphoproliferative disorder, and cell‐mediated and humoral immune responses. J Infect Dis. 2008;198:673‐682. [DOI] [PubMed] [Google Scholar]
- 36. Ma SD, Xu X, Jones R, et al. PD‐1/CTLA‐4 blockade inhibits Epstein‐Barr virus‐induced lymphoma growth in a cord blood humanized‐mouse model. PLoS Pathog. 2016;12:e1005642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Imanishi T, Unno M, Kobayashi W, et al. Reciprocal regulation of STING and TCR signaling by mTORC1 for T‐cell activation and function. Life Sci Alliance. 2019;2(1):e201800282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon‐producing cells in human blood. Science. 1999;284:1835‐1837. [DOI] [PubMed] [Google Scholar]
- 39. Ma SD, Xu X, Plowshay J, et al. LMP1‐deficient Epstein‐Barr virus mutant requires T cells for lymphomagenesis. J Clin Invest. 2015;125:304‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cerboni S, Jeremiah N, Gentili M, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med. 2017;214:1769‐1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sali TM, Pryke KM, Abraham J, et al. Characterization of a Novel Human‐Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015;11:e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okuno S, Adachi Y, Terakura S, et al. Spacer Length modification facilitates discrimination between normal and neoplastic cells and provides clinically relevant CD37 CAR T cells. J Immunol. 2021;206(12):2862‐2874. [DOI] [PubMed] [Google Scholar]
- 43. Wang P, Deng Y, Guo Y, et al. Epstein‐Barr virus early protein BFRF1 suppresses IFN‐β activity by inhibiting the activation of IRF3. Front Immunol. 2020;11:513383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu JJ, Li W, Shao Y, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe. 2015;18:333‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu K, Tian H, Deng H. PPM1G restricts innate immune signaling mediated by STING and MAVS and is hijacked by KSHV for immune evasion. Sci Adv. 2020;6(47):eabd0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Walker MM, Crute BW, Cambier JC, Getahun A. B cell‐intrinsic STING signaling triggers cell activation, synergizes with B cell receptor signals, and promotes antibody responses. J Immunol. 2018;201:2641‐2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tang CH, Zundell JA, Ranatunga S, et al. Agonist‐mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016;76:2137‐2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takashima K, Takeda Y, Oshiumi H, et al. STING in tumor and host cells cooperatively work for NK cell‐mediated tumor growth retardation. Biochem Biophys Res Commun. 2016;478:1764‐1771. [DOI] [PubMed] [Google Scholar]
- 49. Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD‐1 blockade. Sci Transl Med. 2015;7:283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li T, Cheng H, Yuan H, et al. Antitumor Activity of cGAMP via Stimulation of cGAS‐cGAMP‐STING‐IRF3 Mediated Innate Immune Response. Sci Rep. 2016;6:19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liang D, Xiao‐Feng H, Guan‐Jun D, et al. Activated STING enhances Tregs infiltration in the HPV‐related carcinogenesis of tongue squamous cells via the c‐jun/CCL22 signal. Biochim Biophys Acta. 2015;1852:2494‐2503. [DOI] [PubMed] [Google Scholar]
- 52. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14:282‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76:6747‐6759. [DOI] [PubMed] [Google Scholar]
- 54. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature. 2008;454:436‐444. [DOI] [PubMed] [Google Scholar]
- 55. Wellenstein MD, de Visser KE. Cancer‐cell‐intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399‐416. [DOI] [PubMed] [Google Scholar]
- 56. Higuchi H, Yamakawa N, Imadome KI, et al. Role of exosomes as a proinflammatory mediator in the development of EBV‐associated lymphoma. Blood. 2018;131:2552‐2567. [DOI] [PubMed] [Google Scholar]
- 57. Shinozaki‐Ushiku A, Kunita A, Fukayama M. Update on Epstein‐Barr virus and gastric cancer (review). Int J Oncol. 2015;46:1421‐1434. [DOI] [PubMed] [Google Scholar]
- 58. Eliopoulos AG, Stack M, Dawson CW, et al. Epstein‐Barr virus‐encoded LMP1 and CD40 mediate IL‐6 production in epithelial cells via an NF‐kappaB pathway involving TNF receptor‐associated factors. Oncogene. 1997;14:2899‐2916. [DOI] [PubMed] [Google Scholar]
- 59. Miyauchi K, Urano E, Yoshiyama H, Komano J. Cytokine signatures of transformed B cells with distinct Epstein‐Barr virus latencies as a potential diagnostic tool for B cell lymphoma. Cancer Sci. 2011;102:1236‐1241. [DOI] [PubMed] [Google Scholar]