

Abstract
Pancreatic cancer has an extremely poor prognosis because of its resistance to conventional therapies. Cancer stem cell (CSC)‐targeted therapy is considered a promising approach for this disease. Epithelial‐mesenchymal transition‐inducing transcription factors (EMT‐TFs) contribute to CSC properties in some solid tumors; however, this mechanism has not been fully elucidated in pancreatic cancer. Zinc finger protein, SNAIL2 (also known as SLUG), is a member of the SNAIL superfamily of EMT‐TFs and is commonly overexpressed in pancreatic cancer. Patients exhibiting high SNAIL2 expression have a poor prognosis. In this study, we showed that the suppression of SNAIL2 expression using RNA interference decreased tumorigenicity in vitro (sphere formation assay) and in vivo (xenograft assay) in 2 pancreatic cancer cell lines, KLM1 and KMP5. In addition, SNAIL2 suppression resulted in increased sensitivity to gemcitabine and reduced the expression of CD44, a pancreatic CSC marker. Moreover, experiments on tumor spheroids established from surgically resected pancreatic cancer tissues yielded similar results. A microarray analysis revealed that the mechanism was mediated by insulin‐like growth factor (IGF) binding protein 2. These results indicate that IGFBP2 regulated by SNAIL2 may represent an effective therapeutic target for pancreatic cancer.
Keywords: cancer stem cells, epithelial‐mesenchymal transition, pancreatic cancer, SNAIL2, tumor spheroid
SNAIL2 knockdown reduces tumorigenicity and resistance to gemcitabine in tumor spheroid established from resected tissue of human pancreatic cancer as well as human pancreatic cancer cell lines.

Abbreviations
- 3D
three‐dimensional
- CSC
cancer stem cell
- Dclk1(+)
doublecortin‐like kinase 1‐positive
- Dclk1
doublecortin‐like kinase 1
- EMT
epithelial‐mesenchymal transition
- EMT‐TFs
epithelial‐mesenchymal transition‐inducing transcription factors
- IGF
insulin‐like growth factor
- IGFBP
insulin‐like growth factor binding protein
- PDAC
pancreatic ductal adenocarcinoma
- PDO
patient‐derived organoid
- shSNAI2
SNAI2 gene knockdown stable clones
- Sph
spheroid
- Sph_shSNAI2
spheroid with stable SNAI2 gene knockdown
1. INTRODUCTION
Among all types of cancers, pancreatic cancer has the worst prognosis. Although its overall prognosis is improving worldwide, the 5‐y survival rate remains around 10%. 1 Poor outcomes can be attributed to the high rate of inoperability at the time of diagnosis, post‐operative recurrence, and limited efficacy of currently available conventional therapies such as irradiation and chemotherapy. The mechanisms responsible for its malignant phenotype have been examined using a variety of methods, and the CSC hypothesis is considered an important theory. CSCs, also known as cancer initiating cells, have a self‐renewal capacity similar to that of normal stem cells. In addition, they can differentiate to varying degrees into diverse cell populations, metastasize, and are invasive and resistant to anti‐cancer drugs and radiation. 2 The presence of CSCs was first reported in humans with acute myeloid leukemia. 3 In solid tumors, CSCs were initially observed in a fraction of CD44+CD24−/Low breast cancer cells. 4 Consequently, the existence of CSCs was determined based on the expression of specific surface markers and assays for evaluating tumorigenicity, which is the main indicator. Currently, CSCs have been reported in several cancer types, including brain, 5 stomach, 6 colon, 7 , 8 head and neck, 9 lung, 10 liver, 11 , 12 prostate, 13 and bladder. 14 In pancreatic cancer, a study using primary cultured human pancreatic cancer cells derived from surgical samples reported that the transplantation of CD44+/CD24+/EpCAM+ fractionated cells into immunodeficient mice resulted in tumors similar to that of the primary tumor. 15 Moreover, CD133+ cells were found to exhibit high tumorigenicity. 16 Pancreatic cancer, which is inoperable or can recur after surgery, is treated with standard chemotherapy including gemcitabine, nab‐paclitaxel, and 5‐fluorouracil. 17 Although these therapies initially suppress tumor growth, local recurrence and/or distant metastasis are often observed over time. Therefore, CSCs can contribute to these processes. An analysis of pancreatic cancer cell lines resistant to gemcitabine revealed the expression of several molecules, including CD44, CD24, and EpCAM, which are considered pancreatic CSC markers. 18 Furthermore, they are invasive and metastatic, and accumulated data strongly suggest that EMT is the main mechanism associated with these processes. EMT is regulated by EMT‐associated transcription factors (EMT‐TFs) such as the SNAIL, TWIST, and ZEB family genes. The expression of EMT‐TFs is generally increased in CSCs. 19 Therefore, EMT‐TFs contribute to the properties of CSCs in breast cancer. 20 Moreover, CSCs can be induced by EMT‐TFs in other types of cancer 21 , 22 , 23 , 24 ; however, this mechanism in pancreatic cancer has not been fully elucidated.
Recently, using genetic lineage tracing with a dual‐recombinase system and live imaging, it was shown that doublecortin‐like kinase 1‐positive [Dclk1(+)] tumor cells can continuously produce progeny cells in pancreatic intraepithelial neoplasia, primary and metastatic pancreatic ductal adenocarcinoma (PDAC), and PDAC‐derived spheroids (Sphs) in vivo and in vitro. 25 Furthermore, genes associated with CSC and EMT were enriched in mouse Dclk1(+) and human DCLK1‐high PDAC cells. 25 In the present study, we found that SNAIL2 is highly expressed among the EMT‐TFs including, SNAIL, TWIST, and ZEB family members, in human pancreatic cancer cell lines and patient‐derived Sphs. Previous reports have shown that SNAIL2 is overexpressed in pancreatic cancer and high SNAIL2 expression is associated with poor prognosis. 26 Therefore, we focused on the function of SNAIL2 and studied the molecular mechanisms of stemness regulated by SNAIL2 in pancreatic cancer.
2. MATERIALS AND METHODS
2.1. Cell culture
Pancreatic cancer cell lines including KLM‐1, KMP5, T3M‐4, PK‐8, BxPC‐3, HPAF‐II, CFPAC‐1, KMP8, Capan‐2, AsPC‐1, PK‐59, pancreatic duct epithelial cells (HPDE), and embryonic kidney 293T cells were purchased from RIKEN, Japanese Collection of Research Bioresources Cell Bank and the American Type Culture Collection. HPAF‐II, CFPAC‐1, HPDE, and 293T were cultured in Dulbecco's modified Eagle's medium (DMEM; Nacalai Tesque), whereas the other cell lines were cultured in Royal Park Memorial Institute 1640 (RPMI 1640; Nacalai Tesque). All cell culture media were supplemented with 10% fetal bovine serum (FBS; Cytiva) and 1% penicillin‐streptomycin solution (Nacalai Tesque). All cell lines were maintained at 37°C in 5% CO2 and 95% humidified air.
2.2. Human pancreatic cancer samples
Surgically resected specimens were obtained from pancreatic cancer patients at Kyoto University Hospital. The use of human subjects was approved by the Ethical Committee of Kyoto University Hospital (approved number: R1721, R1281, R2904 and G1200). All experiments were conducted in accordance with the Declaration of Helsinki as well as the guidelines and regulations of the Committee.
2.3. Spheroid culture
Human pancreatic cancer spheroids were established from fresh surgical specimens obtained from patients who underwent surgical resection at Kyoto University Hospital and approved by the Ethical Committee as mentioned above (R1281). Written informed consent was obtained from each subject before inclusion in the study. The pathological characteristics of the primary tumor were as follows: mod, Pt, TS4 (8.5 × 6.5 × 3.0 cm), infiltrative type, pT3, sci, INFc, ly0, v3, ne0, mpd0, pS1, pRP1, Pv1 (Pvsp), A0, PL0, OO0, pPCM0, R0, pN2 (3/20, #16:1/5), M0. The findings were in accordance with the General Rules for the Study of Pancreatic Cancer by Japan Pancreas Society (The 7th Edition). Spheroids were processed as previously reported with some modifications. 27 Briefly, the cell aggregates were embedded in Matrigel (Corning) and covered by medium composed of 50% L‐WRN‐conditioned medium (ATCC) containing L‐Wnt3A, R‐spondin 3, and Noggin, and containing Advanced DMEM/F12 (Invitrogen), 5% FBS, 2 mmol/L L‐alanyl‐l‐glutamine (Wako), 100 units/mL penicillin, 0.1 mg/mL streptomycin (Nacalai Tesque), 2.5 µg/mL Plasmocin prophylactic (Invitrogen), 10 µM Y‐27632 (Wako), 1 µM SB431542 (Wako), 1xB27 Supplement (Thermo Fisher Scientific), 100 ng/mL Fibroblast growth factor‐Basic Recombinant Human Protein (bFGF; Thermo Fisher Scientific), and 20 ng/mL Epidermal Growth Factor Recombinant Human Protein (EGF; Thermo Fisher Scientific). Genomic DNA mutations of the spheroids were determined by next‐generation sequencing analysis using the Ion AmpliSeq 50‐gene Cancer Hotspot Panel v2 (Thermo Fisher Scientific). Sequencing, mapping alignment, and annotation was outsourced to Takara Bio.
2.4. Real‐time quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR)
Total RNA was extracted using the RNeasy mini Kit (Qiagen). Single‐strand complementary DNA (cDNA) was synthesized using the PrimeScript II 1st strand cDNA Synthesis Kit (Takara Bio), qRT‐PCR was performed using the FastStart Universal SYBR Green Master Mix (Rox) (Roche Applied Science) and the StepOnePlus real‐time system (Applied Biosystems). All reactions were performed in triplicate and mRNA expression levels were normalized to that of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). The results are presented as linearized Ct values normalized to GAPDH and the indicated reference value (2DDCt). Primers were designed using the MGH primer bank (https://pga.mgh.harvard.edu/primerbank/). All primer sequences are listed in Table S1.
2.5. Short hairpin RNA‐mediated human SNAI2 gene knockdown by lentiviral transduction and stable clone establishment
SNAI2 knockdown in pancreatic cancer cell lines and spheroids was performed by infection with a lentiviral particle that expressed human SNAI2‐targeting short hairpin RNA (shRNA). The lentivirus vector plasmid encoding human SNAI2‐targeting shRNA (Sigma‐Aldrich) was co‐transfected together with packaging plasmids consisting of pMDLg/pRRE, pRSV‐Rev, and pMD2.G (Addgene) into 293T cells using Lipofectamine 3000 (Invitrogen). After 48 h, supernatants were collected and used as lentiviral particles without enrichment. Two predesigned SNAI2‐targeting shRNAs (A: 5‐ CCGGCCCATTCTGATGTAAAGAAATCTCGAGATTTCTTTACATCAGAATGGGTTTTT‐3, B: 5‐CCGGGCGCCCTGAAGATGCATATTCCTCGAGGAATATGCATCTTCAGGGCGCTTTTTG‐3) were used and pLKO.1puro (Addgene) was used as a control. The efficiency of infection was evaluated by a green fluorescent protein‐expressing lentivirus. After 48‐72 h of viral infection, the cells were treated with medium containing puromycin (Invivogen) at 1 to 2 µg/mL for 2 wk to remove noninfected cells.
2.6. Transfection of plasmid DNA by electroporation and stable clone establishment
SNAI2 and IGFBP2 plasmid DNA was transfected into SNAI2 knockdown stable clones established from KLM1 cells to generate stable SNAIL2 and IGFBP2 overexpressing cells. The SNAIL2 and IGFBP2 overexpression vectors and control vector were purchased from Sino Biologicals. Plasmid DNA was transfected by electroporation using NEPA21 (NEPAGENE) in accordance with the manufacturer's protocol. After 48 h of transfection, the cells were treated with medium containing hygromycin B (Nacalai Tesque) at 100 µg/mL for 2 wk to remove non‐transfected cells. The transfection efficiency was verified by qRT‐PCR.
2.7. Microarray data and bioinformatic analysis
Total RNA from control vector and SNAI2 knockdown stable clones in KLM1 and human pancreatic cancer spheroids (n = 2, respectively) were analyzed using the SurePrint G3 Human Gene Expression 8x60K v3 microarrays (Agilent Technologies). Processed signal intensities were normalized by the global scaling method. A trimmed mean probe intensity was determined by removing 2% of the lower and the higher end of the probe intensities to calculate the scaling factor. Normalized signal intensities were then calculated from the target intensity on each array using this factor, so that the trimmed mean target intensity of each array was arbitrarily set to 2500. The GEO accession number for the complete microarray data in this paper is GSE155107 and GSE155108.
Gene‐Set Enrichment Analysis (GSEA) was performed using GSEA 3.0 software (http://www.broadinstitute.org/gsea/index.jsp) with 1000 gene‐set permutations using a signal‐to‐noise gene‐ranking metric with the collections c5.all.v6.1.symbols (C5). Gene annotation analysis from multiple gene lists that showed more than a 1.5‐fold change in expression was performed by Metascape (http://metascape.org/gp/index.html#/main/step1).
With respect to the following materials and methods, please refer to “Supplementary Materials and Methods” in Supporting Information.
(a) Scratch wound healing assay, (b) Sphere formation assay, (c) Chemoresistance assay using the Cell Counting Kit‐8, (d) Immunohistochemical staining, (e) Western blot analysis, (f) Flow cytometry, (g) Xenograft assay, (h) Statistics.
3. RESULTS
3.1. SNAIL2 is highly expressed in pancreatic cancer cell lines
First, we examined pancreatic cancer cell lines using a sphere formation assay, which is used to evaluate tumorigenicity in vitro. The sphere formation assay was initially utilized to identify normal neural stem cells and, subsequently, to detect CSCs. 28 , 29 Among the pancreatic cancer cell lines examined, KLM1 and KMP5 exhibited similar tumorigenic properties in terms of sphere formation (Figure 1A). Sphere formation was not observed in other cell lines. Next, we analyzed the mRNA expression of EMT‐TFs including SNAI1, SNAI2, TWIST1, TWIST2, ZEB1, and ZEB2, in KLM1 and KMP5 cells. The results indicated that the expression of the SNAI2 gene was significantly higher compared with that of the others (Figure 1B). Therefore, we focused on the function of the SNAI2 gene and evaluated the correlation between SNAIL2 and CSC properties. Notably, other pancreatic cancer cell lines expressed higher levels of the SNAI2 gene (Figure S1). There were several cell lines with higher expression of SNAI2 compared with KLM1 and KMP5; however, they did not form spheres well. We speculated that other factors in addition to EMT‐ TFs, including SNAI2, may be involved in sphere formation. To evaluate the effect of SNAI2, we generated SNAI2 gene knockdown stable clones (shSNAI2) by gene transduction of short hairpin RNAs (shRNAs) with lentiviral vectors in KLM1 and KMP5 cells. Knockdown clones were generated using 2 different types of shRNAs, each targeting a different region of the gene. SNAI2 mRNA expression was downregulated by each shSNAI2 clone compared with controls transduced with empty vector (Figure 1C). In addition, SNAI2 gene expression was downregulated at the protein level. Meanwhile, CDH1, which encodes the epithelial marker, was upregulated, whereas VIM, which encodes the mesenchymal marker, vimentin, was downregulated (Figure 1D).
FIGURE 1.
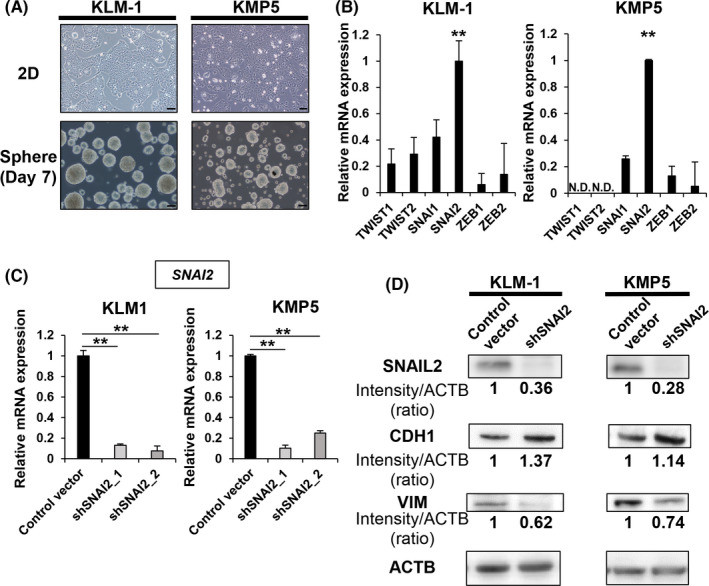
SNAIL2 is highly expressed in tumorigenic pancreatic cancer cell lines. A, Microscopy images of 2D cultures and spheres (3D culture, day 7) in KLM1 and KMP5. Scale bars: 100 µm. B, qRT‐PCR (EMT‐TF) of KLM1 and KMP5. Relative expression levels normalized by HPDE cells were compared. n = 3, each. ND, not detected. Mean + SE. **P < .01. C, qRT‐PCR (SNAI2) of control vector versus shSNAI2 from KLM1 and KMP5. n = 3, each. Mean + SE. **P < .01. D, Western blot (SNAIL2, CDH1, VIM, and ACTB) of control vector versus shSNAI2 from KLM1 and KMP5. The relative intensity (ratio) is shown below each band. shRNA was used shSNAI2_1
3.2. SNAI2 knockdown reduces tumorigenic potential
Next, a scratch wound healing assay was performed to assess changes in invasive capacity following SNAI2 gene knockdown. The results showed a decrease in the invasive capacity of all shSNAI2 clones in both cell lines (Figure S2). Therefore, SNAI2 gene knockdown changed the phenotype from a mesenchymal to an epithelial state. We determined whether SNAI2 gene knockdown affected tumorigenicity, an essential property of CSCs. Using the sphere formation assay, shSNAI2 showed a decreased number of spheres compared with controls (Figure 2A,B). In addition, the xenograft assay using a subcutaneous transplantation model in immunodeficient mice showed a difference in the number of tumors formed by the dilution of cell numbers that were transplanted (Figure 2C,D). Furthermore, the growth rate of the tumors was reduced (Figure 2E). Therefore, shSNAI2 exhibited a lower tumorigenic capacity in vitro and in vivo.
FIGURE 2.
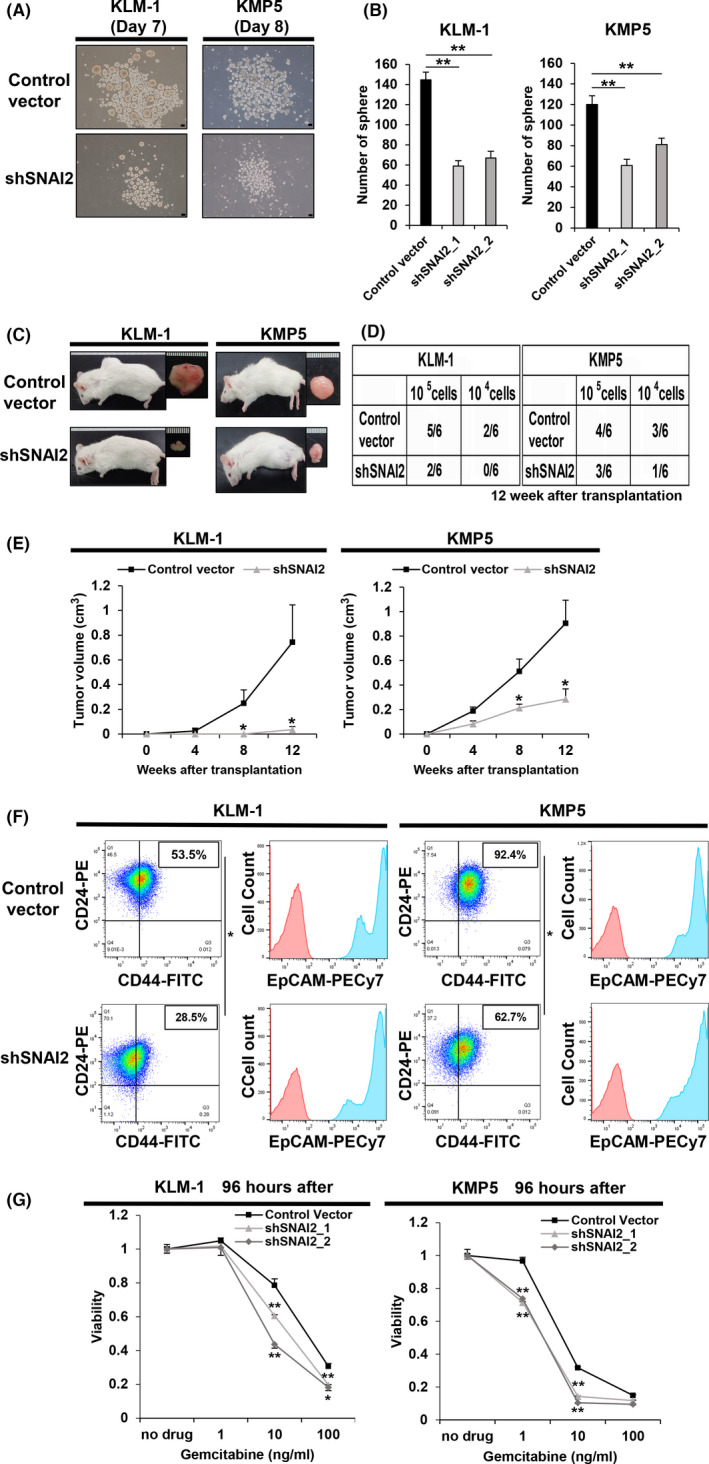
SNAI2 knockdown of tumorigenic pancreatic cancer cell lines reduces tumorigenicity, suppresses CD44 expression, and resistance to gemcitabine. A, B, Sphere formation assay of control vector versus shSNAI2 from KLM1 and KMP5 at days 7 and 8. A, Microscopy images. Scale bars: 100 µm. B, Quantification of the number of spheres. n = 3, each. Mean + SE. **P < .01. C‐E, Control vector and shSNAI2 from KLM1 and KMP5 were subcutaneously transplanted into NOD‐SCID mice (105 cells, 104 cells per site), respectively. n = 6. C, Macroscopic images of formed tumor. shRNA was used shSNAI2_1. D, Number of tumor‐forming mice per total transplanted mice at 12 wk after transplantation. E, Tumor growth curves. Mean + SE. *P < .05. F, Flow cytometric analysis of control vector and shSNAI2 from KLM1 and KMP5. shRNA was used shSNAI2_1. Two parameter plots: CD44‐FITC (X axis), CD24‐PE (Y axis), the numbers in the box represent the CD44‐positive/CD24‐positive cell ratio. Histogram: EpCAM‐PECy7 (X axis), cell count (Y axis). CD44 and CD24 double analysis was performed for the blue colored fraction of the EpCAM panel. n = 2, each. Mean + SE. *P < .05. G, The relative viability of control vector versus shSNAI2 from KLM1 and KMP5 cells 96 h after treating with gemcitabine. Viability curves at 0, 1, 10, 100 ng/mL gemcitabine. n = 3, each. Mean + SE. *P < .05, **P < .01
3.3. SNAI2 knockdown attenuates the expression of CSC markers and resistance to chemotherapy
Flow cytometry analysis of the CD44, CD24, and EpCAM fractions, which are CSC surface markers in pancreatic cancer, revealed that the CD44+/CD24+/EpCAM+ fractions, particularly CD44, decreased in shSNAI2 cells (Figure 2F). Moreover, to evaluate chemotherapy resistance, which is a characteristic of CSCs, a cell proliferation assay was performed following gemcitabine treatment. The results showed a significant reduction in the survival of shSNAI2 compared with controls, thereby indicating a reduction in gemcitabine resistance (Figure 2G). These results support the notion that SNAI2 knockdown can disrupt the properties of CSCs.
3.4. SNAIL2 is highly expressed in Sph tumors established from human pancreatic cancer specimens
In addition to pancreatic cancer cell lines, we established spheroid (Sph) tumors from surgically resected human pancreatic cancer specimens for the analysis of EMT‐TFs. Prior to the assay, immunohistochemical staining of human pancreatic cancer surgical specimens confirmed the expression of SNAIL2 in the nuclei of the cancer cells (Figure 3A). The method of establishing the three‐dimensional (3D) culture of Sphs was based on a similar approach as previously reported 26 with slight modification. The advantage of using Sphs in the analysis of EMT‐TFs is that the culture conditions may be more representative of the tumor microenvironment in vivo compared with the conventional serum‐dependent two‐dimensional cell culture model. The clinicopathological characteristics of patients who underwent resection for pancreatic cancer are described above. To analyze cancer genome mutations, genomic DNA was extracted from the established Sphs and analyzed with an oncogene panel test (containing 50 oncogenes/anti‐oncogenes) using next‐generation sequencing. The results showed typical mutations in the KRAS, TP53, and SMAD4 genes, all of which are common in pancreatic cancer (data not shown). 30 The data showed that the Sphs were completely derived from pancreatic cancer epithelial cells. The Sphs were spherical and partly formed glandular tubular structures similar to that of the primary tumor (Figure 3B). Next, the mRNA expression of EMT‐TFs was examined. The results indicated that SNAI2 expression was the highest among all of the EMT‐TFs (Figure 3C). Based on these findings, SNAI2 gene knockdown stable clones spheroid with stable SNAI2 gene knockdown (Sph_shSNAI2) were generated from the original Sph tumor by lentivirus‐mediated shRNA‐targeting of the SNAI2 gene. This was similar to the method used for the cell lines (Figure 3D). Sph_shSNAI2 was associated with a significant downregulation of SNAI2 mRNA expression (Figure 3E). Similar to KLM1 and KMP5, SNAIL2 was also downregulated at the protein level. In addition, E‐cadherin was upregulated and vimentin was downregulated (Figure 3F).
FIGURE 3.
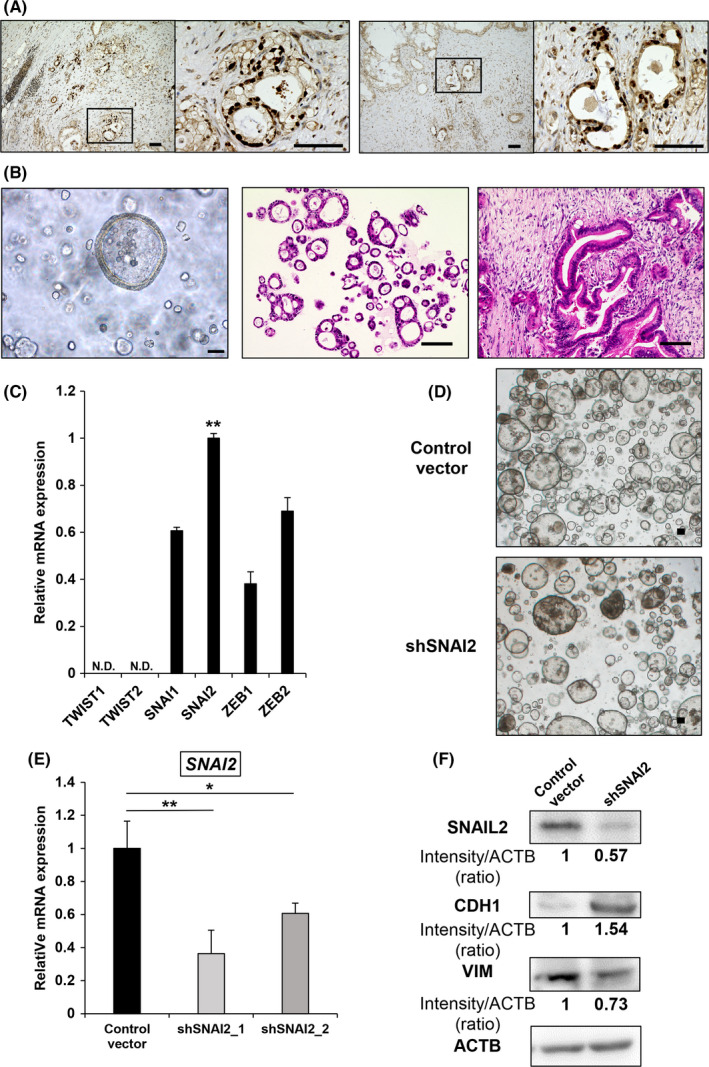
SNAI2 is highly expressed in spheroids established from surgically resected human pancreatic cancer. A, Immunohistochemical SNAIL2 staining of human pancreatic cancer (2 samples). Scale bars: 100 µm. B, Microscope image of spheroids in 3D culture (left), H&E staining of spheroids (middle), H&E staining of primary tumor (right). Scale bars: 100 µm. C, qRT‐PCR (EMT‐TF) of Sphs. Relative expression levels normalized by HPDE cells were compared. n = 3. ND, not detected. Mean + SE. **P < .01. D, Microscopic image of control vector and shSNAI2 from Sphs in 3D culture. Scale bars: 100 µm. E, qRT‐PCR (SNAI2) of control vector versus shSNAI2 from spheroids. n = 3, each. Mean + SE. *P < .05, **P < .01. F, Western blot (SNAIL2, CDH1, VIM, and ACTB) of control vector versus shSNAI2 from Sph. shRNA was used shSNAI2_1. The relative intensity (ratio) is shown below each band
3.5. SNAI2 knockdown impairs the properties of CSCs in tumor Sphs as in pancreatic cancer cell lines
Xenograft assays using Sph_shSNAI2 showed that the number of tumors formed in shSNAI2 was reduced and tumor size was decreased, consistent with the cell line data (Figure 4A,B). In addition, the tumors formed by subcutaneous transplantation into immunodeficient mice recapitulated glandular structures similar to that of primary pancreatic cancer tissue. Therefore, compared with pancreatic cancer cell lines, the Sphs exhibited very similar in vivo characteristics (Figure 4C,D). Flow cytometry revealed that the CD44+/CD24+/EpCAM+ fraction was reduced in Sph_shSNAI2 tumors, similar to cell lines, and resistance to gemcitabine was reduced based on a cell proliferation assay (Figure 4E,F). Therefore, SNAI2 knockdown impaired the properties of CSCs in patient‐derived Sphs.
FIGURE 4.
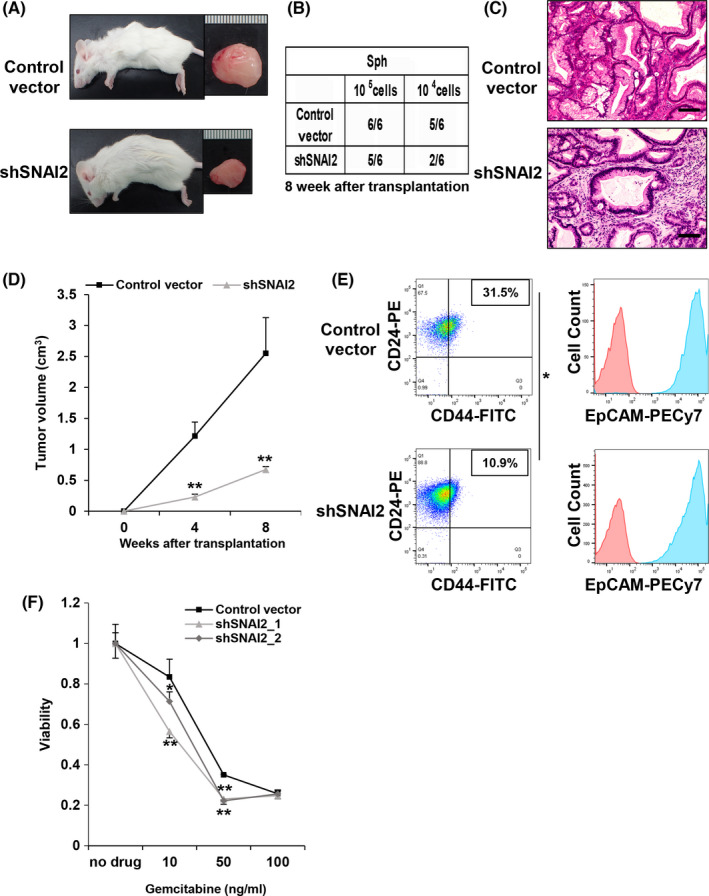
SNAI2 knockdown reduces tumorigenicity and resistance to gemcitabine in spheroids from human pancreatic cancer. A‐D, Control Vector and shSNAI2 from Sph were subcutaneously transplanted into NOD‐SCID mice (105 cells, 104 cells per site), respectively. shRNA was used shSNAI2_1. n = 6. A, Macroscopic images of formed tumor. B, Number of tumor‐forming mice per total transplanted mice at 8 wk after transplantation. C, H&E staining of formed tumors. Scale bars: 100 µm. D, Tumor growth curves. Mean + SE. **P < .01. E, Flow cytometric analysis of control vector and shSNAI2 from spheroids. shRNA was used shSNAI2_1. Two parameter plots: CD44‐FITC (X axis), CD24‐PE (Y axis), Numbers represent the CD44‐positive/CD24‐positive cell ratio. Histogram: EpCAM‐PECy7 (X axis), cell count (Y axis). CD44 and CD24 double analysis was performed for the blue colored fraction of the EpCAM panel. n = 2, each. Mean + SE. *P < .05. F, The relative viability of control vector versus shSNAI2 from spheroid 96 h after treating cells with gemcitabine. Viability curves at 0, 10, 50, 100 ng/mL gemcitabine. n = 5, each. Mean + SE. **P < .01
3.6. Microarray analysis identifies significant differentially expressed genes
To elucidate the mechanisms associated with the above results, we performed a microarray analysis comparing RNA samples between the control vector and shSNAI2 groups using KLM1 cells and Sphs. A GSEA revealed significant differences in the gene sets involved in EMT and stem cell differentiation in both the KLM1 and Sph groups (Figure 5A). Metascape was used to identify Gene Ontology (GO) terms associated with significant changes in the KLM1 and Sph groups. Among the top listed GO terms, there were EMT‐related and biological function terms as expected, however IGFBP‐related terms were also identified (Figure 5B). Therefore, we focused on the IGFBP gene family (More detailed lists are shown in Figure S3). IGFBP1 and IGFBP2, 2 major IGFBP members, are listed as hits in the IGFBP‐related results between controls and shSNAI2 in both KLM1 cells and Sphs (Table S2). We examined changes in the mRNA expression of the IGFBPs and IGFBP2 was found to be significantly downregulated in shSNAI2 compared with the controls in both KLM1 cells and Sphs (Figure 6A). The expression of IGFBP2 in human pancreatic cancer tissues was examined by immunohistochemical staining. The results indicated that many pancreatic cancer epithelial cells were positive (Figure 6B). Moreover, the expression of IGFBP2 was also downregulated at the protein level (Figure 6C). In addition, the sphere formation assay performed with the addition of IGFBP2 neutralizing antibody in KLM1 cells showed decreased sphere formation compared with the control (Figure 6D,E).
FIGURE 5.
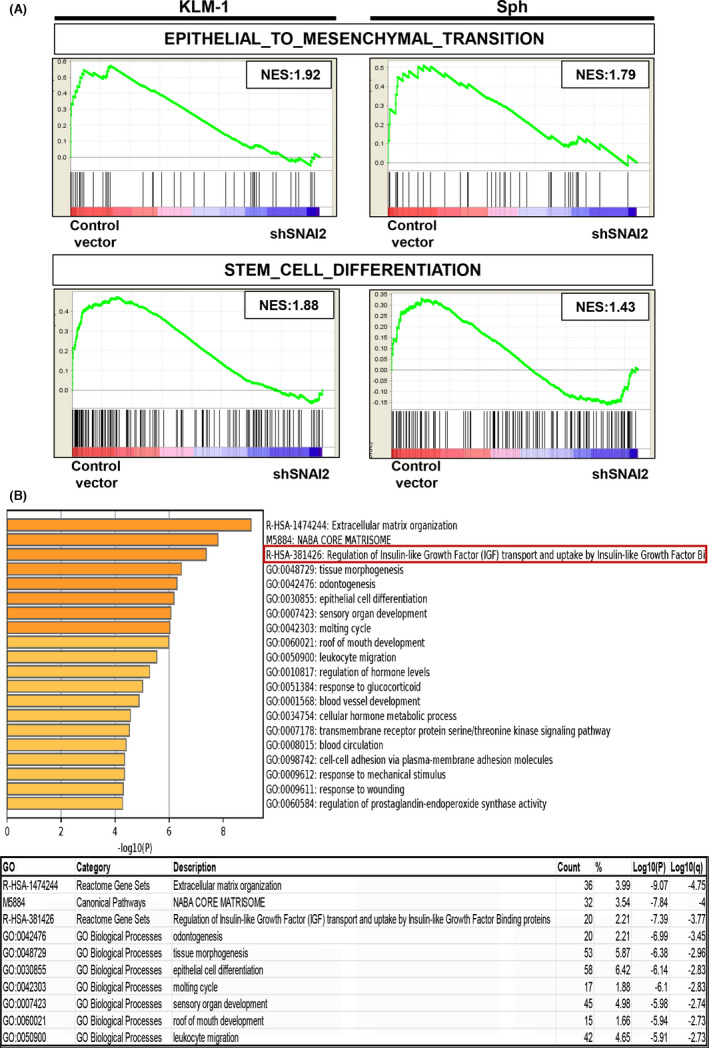
DNA microarray analysis revealed several significant gene sets. A, Gene‐set enrichment analysis plots of the enrichment of the indicated gene signatures in control vector versus shSNAI2 from KLM1 cells and Sphs using the “C5” compilation from Molecular Signature Database (MSigDB, Broad Institute). NES, normalized enrichment score. n = 2, each. B, Gene Ontology enrichment analysis of differentially expressed genes (fold change: 1.5) in control vector versus shSNAI2 common to KLM1 cells and spheroids using the Metascape online tool. Bar graph of top 20 enriched terms across input gene lists (upper), colored by P‐values. Top 10 Gene Ontology list (lower), “Count” is the number of genes in the user‐provided lists with membership in the given ontology term, “%” is the percentage of all of the user‐provided genes that are found in the given ontology term (only input genes with at least one ontology term annotation are included in the calculation), “Log10(P)” is the P‐value in log base 10, “Log10(q)” is the multi‐test adjusted P‐value in log base 10. n = 2, each
FIGURE 6.
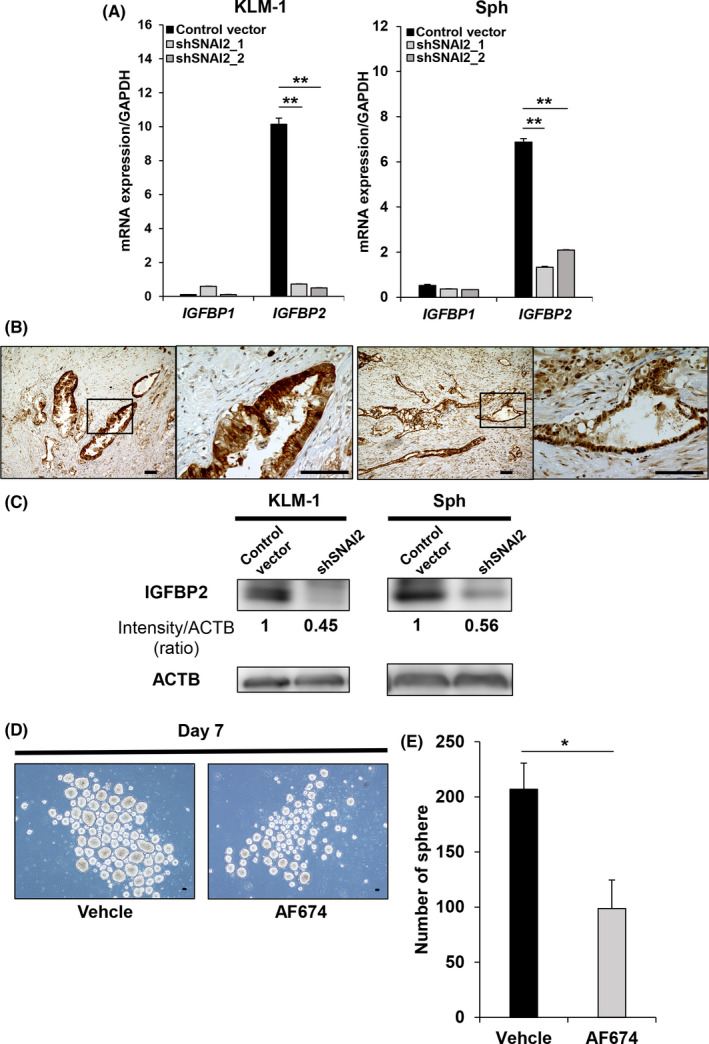
SNAI2 knockdown shows that IGFBP2 of the IGFBP family is downregulated in KLM1, KMP5, and Sphs. A, qRT‐PCR (IGFBP1 and IGFBP2) of control vector versus shSNAI2 from KLM1, KMP5, and Sphs. n = 3, each. ND, Not Detected. Mean + SE. **P < .01. B, Immunohistochemical IGFBP2 staining of human pancreatic cancer (2 samples). Scale bars: 100 µm. C, Western blot (IGFBP2 and ACTB) of control vector versus shSNAI2 from KLM1, KMP5, and Sphs. shRNA was used shSNAI2_1. The relative intensity (ratio) is shown below each band. D, E, Sphere formation assay in KLM1 after adding anti‐IGFBP2 neutralizing antibody (AF674) at day 7 compared with Vehicle. D, Microscope images. Scale bars: 100 µm. E, Quantification of number of spheres. n = 3, each. Mean + SE. *P < .05
3.7. Gene transduction of IGFBP2 restores CSC properties
The correlation between IGFBP2 and SNAIL2 through transfection of the full‐length cDNA plasmid of IGFBP2 and SNAI2 into SNAI2 knockdown clones of KLM1 cells (KLM1_shSNAI2) was assessed. We also investigated whether the properties of CSCs were restored. Successful transduction of SNAI2 or IGFBP2 into the shSNAI2 clone (sh+SNAI2, sh+IGFBP2) was confirmed at the mRNA and protein level. Also, IGFBP2 expression was slightly upregulated in sh+SNAI2. In contrast, SNAI2 expression was unchanged in the sh+IGFBP2 clone (Figure 7A,B). In the assessment of tumorigenicity, sh+IGFBP2 and sh+SNAI2 exhibited a higher number of spheres than the sh+Vector in the sphere formation assay (Figure 7C,D). In addition, a xenograft assay also showed a higher number of tumors formed with the diluted cells and a larger tumor volume in sh+IGFBP2 and sh+SNAI2 compared with the sh+Vector (Figure 7E–G). In subcutaneous tumors, the mesenchymal region was wider in sh+SNAI2 compared with sh+Vector, and appeared to be slightly wider in sh+IGFBP2 (Figure 7H). Finally, cell proliferation assays showed an almost complete restoration of resistance to gemcitabine in sh+IGFBP2 compared with sh+SNAI2 (Figure 7I). Therefore, SNAIL2 contributes to the properties of CSCs primarily through IGFBP2 in pancreatic cancer.
FIGURE 7.
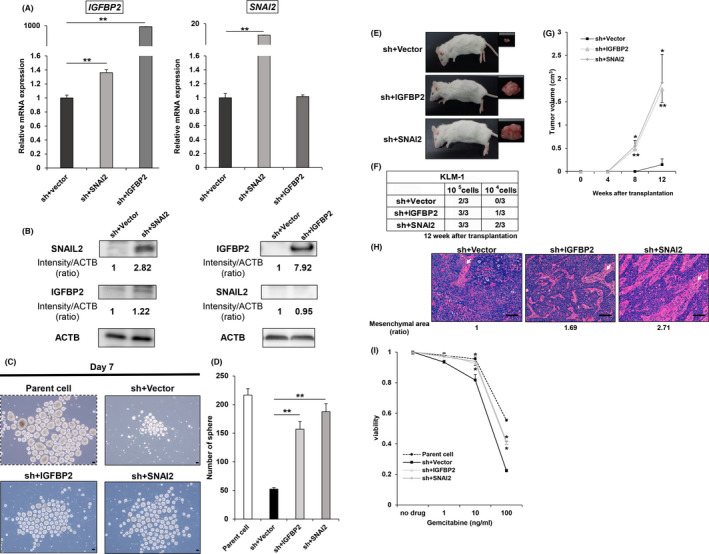
Gene transduction of IGFBP2 and SNAI2 into shSNAI2 restores mRNA and protein expression and improves tumorigenicity and resistance to gemcitabine. A, qRT‐PCR (SNAI2 and IGFBP2) in sh+Vector, sh+IGFBP2 and sh+SNAI2 from KLM1. n = 3, each. Mean + SE. **P < .01. B, Western blot (SNAIL2, IGFBP2, and ACTB) of sh+Vector versus sh+IGFBP2 and sh+SNAI2 from KLM1 cells. The relative intensity (ratio) is shown below each band. C, D, Sphere formation assay of parental cells, sh+Vector, sh+IGFBP2 and sh+SNAI2 from KLM1 cell at day 7. C, Microscope images. Scale bars: 100 µm. D, Quantification of the number of spheres. n = 3, each. Mean + SE. **P < .01. E‐H, sh+Vector, sh+IGFBP2, and sh+SNAI2 cells from KLM1 cells were subcutaneously transplanted into NOD‐SCID mice (105 cells, 104 cells per site), respectively. n = 3. E, Macroscope images of the formed tumor. F, Number of tumor‐forming mice per total transplanted mice at 12 wk after transplantation. G, Tumor growth curves. Mean + SE. *P < .05, **P < .01. H, H&E staining of the formed tumor. The mesenchymal area (indicated by arrows) ratio is shown below each picture. Scale bars: 100 µm. I, The relative viability of parent cells, sh+Vector versus sh+IGFBP2, and sh+SNAI2 from KLM1 cells 96 h after treatment with gemcitabine. Viability curves at 0, 1, 10, 100 ng/mL gemcitabine. n = 4, each. Mean + SE. **P < .01
4. DISCUSSION
EMT is a key cellular event in embryonic development and mesoderm formation. Moreover, it is required for pathological processes such as wound healing, tissue regeneration, organ fibrosis, and tumor progression. Moreover, recent studies have shown that CSCs may be established through the EMT in several types of solid tumors including pancreatic cancer. 31 Among the EMT‐TFs, SNAIL1 and TWIST1 have been examined with respect to their association with cancer stemness. For example, SNAIL1 was recently found to regulate gastric carcinogenesis through the 2 downstream genes, CCN3 and NEFL. 32 Zheng and colleagues reported that the deletion of SNAIL or TWIST enhanced sensitivity to gemcitabine and increased overall survival in mice with pancreatic cancer. However, there were no changes in invasiveness, systemic dissemination, and metastasis in vivo. 33 In humans, Hotz and co‐workers found that, based on immunohistochemical staining of 36 surgical samples with anti‐EMT‐TF antibodies, SNAIL1 was expressed in 78% of human pancreatic cancer tissues and 50% of the patients presented with positive expression of SNAIL2. In contrast, TWIST1 expression was weak or absent. 34
The data obtained in our study were quite different from that of previous reports. As shown in Figures 1B and 3C, the expression of SNAI2 was the highest among all EMT‐TFs (eg, SNAI, TWIST, and ZEB family genes) in pancreatic cancer cell lines as well as pancreatic tumor Sphs. These results are consistent with data obtained from TCGA database (Figure S4). However, TCGA data are obtained from whole samples and are, therefore, limited by the influence of stromal components.
SNAIL2 (also known as SLUG) is encoded by the human SNAI2 gene, a member of the SNAIL superfamily of C2H2‐type zinc finger transcription factors. It is a direct repressor of E‐cadherin, which binds to the E‐box motif. 35 SNAIL2 has tumor‐related functions in several cancer types, although there are no data regarding its role in CSCs in pancreatic cancer. As shown in Figures 2, 3 and 4, SNAI2 knockdown significantly reduced the CSC population, which resulted in lower tumorigenic potential and chemoresistance in both pancreatic cancer cell lines and tumor Sphs. Therefore, other EMT‐TFs cannot compensate for the function of SNAIL2 in terms of homeostasis in cancer stemness.
In the present study, we used not only human pancreatic cancer cell lines, but also tumor Sphs cultured in 3D Matrigel‐based constructs, with component cells derived from surgical tissues collected from pancreatic cancer patients. 36 The technology of 3D cultured Sphs was first established in normal mouse and human intestines. Subsequently, it expanded to other healthy organs and cancer tissues. Therefore, it is currently referred to as a PDO or tumor spheroid. 37 This innovative method has been applied to other digestive organs involved in pancreatic cancer. Boj and colleagues reported that pancreatic tumor organoids can be rapidly generated from resected tissues and biopsies, and orthotopically transplanted neoplastic organoids recapitulate the full spectrum of tumor development by forming early‐grade neoplasms that progress to locally invasive and metastatic carcinomas. 38 Based on previous reports, we successfully established tumor Sphs from patients with pancreatic cancer.
Other important findings in the current study are shown in Figures 6 and 7. A bioinformatic search of microarray data revealed that SNAIL2‐regulated cancer stemness is mediated by IGFBP2. The IGFBP superfamily is a group of secreted proteins that are structurally, functionally, and evolutionarily related, and includes 6 IGFBPs and 10 IGFBP‐related proteins. The IGFBPs exhibit high affinity for IGFs and thereby modulate the mitogenic, anti‐apoptotic, and metabolic actions of IGFs. 39 Among the IGFBPs, IGFBP2 has been correlated with the regulation of IGF activity in the nervous system, peripheral tissues, and organs. In addition to binding to circulating IGFs, the IGF‐regulatory activities of IGFBP2 involve interactions with components including the extracellular matrix, cell surface proteoglycans, and integrin receptors. Furthermore, cytoplasmic IGFBP2, most likely in the absence of the IGFs, interacts with regulatory proteins, such as transcription factors and cytoplasm‐nuclear transporters, and its activities are mediated through specific functional domains. These IGFBP2 activities, intrinsic or dependent on IGFs, contribute to its functional roles in growth, development, metabolism, and malignancy. 40 Previous studies have reported that the blockade of IGFBP2 may be an effective approach to inhibiting tumor growth and metastasis in various solid tumors including glioma, 41 ovarian cancer, 42 and breast cancer. 43 There have been a few reports regarding the regulation of IGFBP2 and we hypothesize that SNAI2 may be involved in mechanisms that regulate IGFBP2 transcription. Several studies have reported that IGFBP2 is associated with disease progression and may be a useful biomarker or therapeutic target in pancreatic cancer. Liu and colleagues reported that knockdown of IGFBP2 in pancreatic cancer cell lines Bxpc‐3 and CFPAC attenuated EMT via the hedgehog pathway. It inhibited proliferation and increased sensitivity to gemcitabine treatment in vitro and in vivo. 44 Furthermore, Gao and co‐workers showed that IGFBP2 overexpression in other pancreatic cancer cell lines (AsPc‐1 and PANC‐1) induced EMT through the nuclear factor kappa B and PI3K/AKT pathways and promoted cell invasion in vitro and metastasis in vivo. 45 These reports suggested that IGFBP2 is involved in the tumorigenesis of pancreatic cancer. However, in our report, the microarray analysis showed no significant changes in signaling pathways such as hedgehog, NF‐kB, or PI3K/AKT. In contrast, several signaling pathways including Wnt, MAPK, and PPAR were significantly associated with the changes (Figure S3). These have also been previously reported as pathways related to IGFBP2 activity. 46
We searched the literature to investigate the mechanism of IGFBP2 gene regulation by SNAIL2, but we did not find any reports of SNAI2 binding to the promoter of IGFBP2 in previous studies. Therefore, we performed the analysis of the promoter sequence of human IGFBP2 gene using JASPAR, an online tool of the functional analysis of various transcription factors created by Scandinavian researchers (URL: http://jaspar.genereg.net/). First, we have obtained 696 bp of genomic sequence located upstream of ATG start codon of human IGFBP2 gene (NCBI Gene ID: 3485). Next, we have analyzed the possible binding sites of human SNAIL2 in the above genomic sequence by JASPAR. Then, we have found 2 predicted binding sites, the first one is located at 115‐127 upstream of the ATG codon and the second one is located at 635‐647 (Figure S5). This result suggested that human SNAIL2 binds to the promoter region of the human IGFBP2 gene and directly regulates the expression of this gene. Although further studies are needed to elucidate the underlying mechanism, our findings indicate that IGFBP2 may represent a novel therapeutic target.
In conclusion, SNAIL2 is a critical factor for tumorigenicity as well as chemoresistance in human pancreatic cancer, and IGFBP2 is the main downstream target regulated by SNAIL2.
DISCLOSURE
Shigeo Takaishi has research funding from Sumitomo Dainippon Pharma Inc. The other authors declare no competing financial interests in this study.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Table S1
Supplementary Material
Table S2
Supplementary Material
ACKNOWLEDGEMENTS
We thank S. Yokoyama, previously Medical Innovation Center DSK Project, for mouse breeding and technical support, M. Nishikawa, Kyoto Institute of Nutrition and Pathology Inc, for immunohistochemical staining, and S. Watanabe, Sumitomo Dainippon Pharma Inc, for genomic DNA mutation analysis of pancreatic tumor spheroids, S. Ogawa, Department of Gastroenterology and Hepatology, Graduate School of Medicine, Kyoto University, for preparation and providing complementary DNA from HPDE cells.
Masuo K, Chen R, Yogo A, et al. SNAIL2 contributes to tumorigenicity and chemotherapy resistance in pancreatic cancer by regulating IGFBP2. Cancer Sci. 2021;112:4987–4999. 10.1111/cas.15162
Funding information
This work was supported by a Grants‐in‐Aid KAKENHI (17K09460), a research program from the Ministry of Education, Culture, Sports, Science, and Technology, and research funding for the DSK Project was provided by Sumitomo Dainippon Pharma, Inc
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7‐33. [DOI] [PubMed] [Google Scholar]
- 2. Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66(19):9339‐9344. 10.1158/0008-5472.can-06-3126 [DOI] [PubMed] [Google Scholar]
- 3. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 4. Al‐Haji M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983‐3988. https://www.pnas.org/content/100/7/3983.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821‐5828. [PubMed] [Google Scholar]
- 6. Takaishi S, Okumura T, Tu S, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27(5):1006‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106‐110. [DOI] [PubMed] [Google Scholar]
- 8. Ricci‐Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon‐cancer‐initiating cells. Nature. 2007;445(7123):111‐115. [DOI] [PubMed] [Google Scholar]
- 9. Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104(3):973‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504‐514. [DOI] [PubMed] [Google Scholar]
- 11. Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132(7):2542‐2556. [DOI] [PubMed] [Google Scholar]
- 12. Yang ZF, Ho DW, Ng MN, et al . Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13(2):153‐166. 10.1016/j.ccr.2008.01.013 [DOI] [PubMed] [Google Scholar]
- 13. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946‐10951. 10.1158/0008-5472.can-05-2018 [DOI] [PubMed] [Google Scholar]
- 14. Chan KS, Espinosa I, Chao M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor‐initiating cells. Proc Natl Acad Sci USA. 2009;106(33):14016‐14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030‐1037. [DOI] [PubMed] [Google Scholar]
- 16. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313‐323. [DOI] [PubMed] [Google Scholar]
- 17. Okusaka T, Furuse J. Recent advances in chemotherapy for pancreatic cancer: evidence from Japan and recommendations in guidelines. J Gastroenterol. 2020;55(4):369‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine‐resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14:3629‐3637. [DOI] [PubMed] [Google Scholar]
- 19. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell‐like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009;26:611‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mani SA, Guo W, Liao MJ, et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hwang WL, Yang MH, Tsai ML, et al. SNAIL regulates interleukin‐8 expression, stem cell‐like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279‐291. [DOI] [PubMed] [Google Scholar]
- 22. Liu CW, Li CH, Peng YJ, et al. Snail regulates Nanog status during the epithelial‐mesenchymal transition via the Smad1/Akt/GSK3β signaling pathway in non‐small‐cell lung cancer. Oncotarget. 2014;5(11):3880‐3894. 10.18632/oncotarget.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun Y, Song GD, Sun N, Chen JQ, Yang SS. Slug overexpression induces stemness and promotes hepatocellular carcinoma cell invasion and metastasis. Oncol Lett. 2014;7(6):1936‐1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen C, Wei Y, Hummel M, et al. Evidence for epithelial‐mesenchymal transition in cancer stem cells of head and neck squamous cell carcinoma. PLoS ONE. 2011;6(1):e16466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maruno T, Fukuda A, Goto N, et al. Visualization of stem cell activity in pancreatic cancer expansion by direct lineage tracing with live imaging. Elife. 2021;10:e55117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Recouvreux MMR, Galenkamp KMO, et al. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J Exp Med. 2020;217(9):e20200388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells as organoids. Nat Protoc. 2013;8:2471‐2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/ progenitor cells. Genes Dev. 2003;17(10):1253‐1270. http://genesdev.cshlp.org/content/17/10/1253.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396‐401. [DOI] [PubMed] [Google Scholar]
- 30. Tsuda M, Fukuda A, Kawai M, Araki O, Seno H. The role of the SWI/SNF chromatin remodeling complex in pancreatic ductal adenocarcinoma. Cancer Sci. 2021;112(2):490‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez‐Aznar E, Wiesmuller L, Sainz B Jr, Hermann PC. EMT and stemness‐key players in pancreatic cancer stem cells. Cancers (Basel). 2019;11(8):1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen R, Masuo K, Yogo A, et al. SNAIL regulates gastric carcinogenesis through CCN3 and NEFL. Carcinogenesis. 2021;42(2):190‐201. [DOI] [PubMed] [Google Scholar]
- 33. Zheng X, Carstens JL, Kim J, et al. Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13(16):4769‐4776. [DOI] [PubMed] [Google Scholar]
- 35. Nieto MA. The snail superfamily of zinc‐finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155‐166. [DOI] [PubMed] [Google Scholar]
- 36. Seino T, Kawasaki S, Shimokawa M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22(3):454‐467. [DOI] [PubMed] [Google Scholar]
- 37. Date S, Sato T. Mini‐gut organoids: reconstitution of the stem cell niche. Annu Rev Cell Dev Biol. 2015;31:269‐289. [DOI] [PubMed] [Google Scholar]
- 38. Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hwa V, Oh Y, Rosenfeld RG. The insulin‐like growth factor‐binding protein (IGFBP) superfamily. Endocr Rev. 1999;20(6):761‐787. [DOI] [PubMed] [Google Scholar]
- 40. Russo VC, Azar WJ, Yau SW, Sabin MA, Werther GA. IGFBP‐2: The dark horse in metabolism and cancer. Cytokine Growth Factor Rev. 2015;26(3):329‐346. [DOI] [PubMed] [Google Scholar]
- 41. Fukushima T, Tezuka T, Shimomura T, Nakano S, Kataoka H. Silencing of insulin‐like growth factor‐binding protein‐2 in human glioblastoma cells reduces both invasiveness and expression of progression‐associated gene CD24. J Biol Chem. 2007;282(25):18634‐18644. [DOI] [PubMed] [Google Scholar]
- 42. Lee EJ, Mircean C, Shmulevich I, et al. Insulin‐like growth factor binding protein 2 promotes ovarian cancer cell invasion. Mol Cancer. 2005;4(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. So AI, Levitt RJ, Eigl B, et al. Insulin‐like growth factor binding protein‐2 is a novel therapeutic target associated with breast cancer. Clin Cancer Res. 2008;14(21):6944‐6954. [DOI] [PubMed] [Google Scholar]
- 44. Liu H, Li L, Chen H, et al. Silencing IGFBP‐2 decreases pancreatic cancer metastasis and enhances chemotherapeutic sensitivity. Oncotarget. 2017;8(37):61674‐61686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao S, Sun Y, Zhang X, et al. IGFBP2 activates the NF‐kappaB pathway to drive epithelial‐mesenchymal transition and invasive character in pancreatic ductal adenocarcinoma. Cancer Res. 2016;76(22):6543‐6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li T, Forbes ME, Fuller GN, et al. IGFBP2: integrative hub of developmental and oncogenic signaling network. Oncogene. 2020;39(11):2243‐2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Table S1
Supplementary Material
Table S2
Supplementary Material