

Abstract
Angiotensin converting enzyme 2 (ACE2) is a key receptor present on cell surfaces that directly interacts with the viral spike (S) protein of the severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2). It is proposed that inhibiting this interaction can be promising in treating COVID‐19. Here, the presence of ACE2 in extracellular vesicles (EVs) is reported and the EV‐ACE2 levels are determined by protein palmitoylation. The Cys141 and Cys498 residues on ACE2 are S‐palmitoylated by zinc finger DHHC‐Type Palmitoyltransferase 3 (ZDHHC3) and de‐palmitoylated by acyl protein thioesterase 1 (LYPLA1), which is critical for the membrane‐targeting of ACE2 and their EV secretion. Importantly, by fusing the S‐palmitoylation‐dependent plasma membrane (PM) targeting sequence with ACE2, EVs enriched with ACE2 on their surface (referred to as PM‐ACE2‐EVs) are engineered. It is shown that PM‐ACE2‐EVs can bind to the SARS‐CoV‐2 S‐RBD with high affinity and block its interaction with cell surface ACE2 in vitro. PM‐ACE2‐EVs show neutralization potency against pseudotyped and authentic SARS‐CoV‐2 in human ACE2 (hACE2) transgenic mice, efficiently block viral load of authentic SARS‐CoV‐2, and thus protect host against SARS‐CoV‐2‐induced lung inflammation. The study provides an efficient engineering protocol for constructing a promising, novel biomaterial for application in prophylactic and therapeutic treatments against COVID‐19.
Keywords: ACE2, extracellular vesicles, palmitoylation, SARS‐CoV‐2
Engineered EVs enriched with palmitoylated ACE2 (PM‐ACE2) efficiently capture SARS‐CoV‐2 viruses and block their interaction with the cell surface ACE2, leading to reduced infection both in vitro and in vivo. This work identifies a novel palmitoylation‐dependent mechanism whereby human cells package ACE2 into EVs, meanwhile developing PM‐ACE2‐EVs as a novel candidate for prophylactic and therapeutic treatment against COVID‐19.
1. Introduction
The expanding pandemic of coronavirus disease 2019 (COVID‐19) caused by a novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), which has resulted in over 4.3 million deaths globally, has raised significant public health concerns.[ 1 , 2 , 3 ] The recent emergence and spread of new SARS‐CoV‐2 lineage B.1.617 in India is associated with the outbreak and continuing deterioration of COVID‐19. The combination of specific mutations L452R, E484Q, and P681R in the spike protein may possibly enhance the transmissibility and immune escape. Given the scale and rapid spread of COVID‐19, there is an urgent need for effective therapeutic strategies.
SARS‐CoV‐2 is a positive‐sense RNA virus with a large single‐stranded RNA genome,[ 4 ] containing multiple open reading frames (ORFs) encoding the structural protein spike‐envelope‐membrane‐nucleocapsid and accessory proteins.[ 5 , 6 ] The spike (S) protein of SARS‐CoV‐2 plays a vital role in invading host cells, by directly interacting with the host receptor angiotensin converting enzyme 2 (ACE2),[ 7 ] which is also the host receptor of SARS‐CoV.[ 8 ] Neutralising antibodies and therapeutic strategies targeting the S or ACE2 proteins are currently under development.[ 9 , 10 , 11 , 12 ]
In addition to functioning as a coronavirus receptor, ACE2 is also considered a mono‐carboxypeptidase that removes carboxyterminal bases or hydrophobic acids from angiotensin I (Ang I), angiotensin II (Ang II), and apelin.[ 13 , 14 ] In general, ACE2 promotes the formation of Ang‐(1‐7) by cleaving Ang II.[ 15 ] However, the other biological characteristics of ACE2, including post‐translational modifications, regulation of subcellular localisation, and protein dynamics, remain elusive. In addition, whether these underlying mechanisms could be utilized to block SARS‐CoV‐2 entry remains unknown.
Extracellular vesicles (EVs), such as endosome‐origin “exosomes” and plasma membrane‐derived “ectosomes” (microparticles/microvesicles) are defined as the common but functionally specific particles delimited by a lipid bilayer and naturally released from various types of cells. “Since the consensus has not yet emerged on specific markers of EV subtypes”, studies on EVs usually divide the isolated EVs into “small EVs” (sEVs) and “medium/large EVs” (m/lEVs) based on the particle size.[ 16 , 17 , 18 ] Extracellular vesicles are capable of transporting a large range of cargoes, including proteins, lipids, and nucleic acids, to recipient cells.[ 19 , 20 , 21 ] Recently, sEVs and exomeres containing ACE2 were reported to bind SARS‐CoV‐2 through the virus spike (S) protein[ 22 ] and thus decrease SARS‐CoV‐2‐S‐pseudotyped virus infection in vitro.[ 23 ] However, the mechanism whereby human cells package ACE2 into EVs remain elusive and whether EV incorporation of ACE2 could be enhanced and employed to antagonize authentic SARS‐CoV‐2 infection in vivo needs to be urgently explored. Here, we also identified ACE2 in EVs and found that the levels of EV‐ACE2 originating from the PM are tightly controlled by S‐palmitoylation at two major residues, Cys141 and Cys498. ACE2, which is palmitoylated by zinc finger DHHC‐Type Palmitoyltransferase 3 (ZDHHC3) or de‐palmitoylated by acyl protein thioesterase 1 (LYPLA1), shows markedly enhanced or reduced location of the PM and extracellular vesicle secretion, respectively. Upon virus infection, membrane‐anchored ACE2 is secreted onto the surface of extracellular vesicles, where it can bind to the receptor binding domain (RBD) of the viral S protein, thereby blocking the association of SARS‐CoV‐2 with cell surface ACE2. This mechanism allows for the quick removal of the cell surface receptor of ACE2 for SARS‐CoV‐2, which meanwhile confers effective antiviral defence to the host. By using palmitoylation as the signal for ACE2 EV transportation, we established a standard operating procedure to generate engineered EVs with enriched ACE2 on their surface. We also demonstrated these to have high neutralization potency against authentic SARS‐CoV‐2 both in vitro and in vivo. Palmitoyl‐secretion of the EV‐ACE2 provides a new mode of anti‐viral strategy and a clear mechanistic link between palmitoylation and EV function, whereby the sensing and defensing system for virus is precisely switched.
2. Results
2.1. Extrafacial Expression of ACE2 on EVs and its Regulation by Protein Palmitoylation
We purified extracellular vesicles from Vero‐E6, MCF7, and HEK293T cells by ultracentrifugation, and subsequently performed transmission electron microscopy (TEM) and NanoSight nanoparticle tracking analysis (Figure 1a; Figure S1a,b, Supporting Information). Ultra‐performance liquid chromatography‐mass spectrometry (UPLC‐MS) and immunoblotting revealed the presence of ACE2 in EVs from Vero‐E6 and MCF7 cells, but not in HEK293T cells (Figure 1b,c). Immunogold transmission electron microscopy (IG‐TEM) also detected ACE2 exclusively in EVs from ACE2‐expressing cells and many of these ACE2 molecules were found to be anchored to the membrane (Figure 1d). Sucrose density gradient centrifugation also confirmed the presence of ACE2 in the EV fraction (Figure S1c, Supporting Information).
Figure 1.
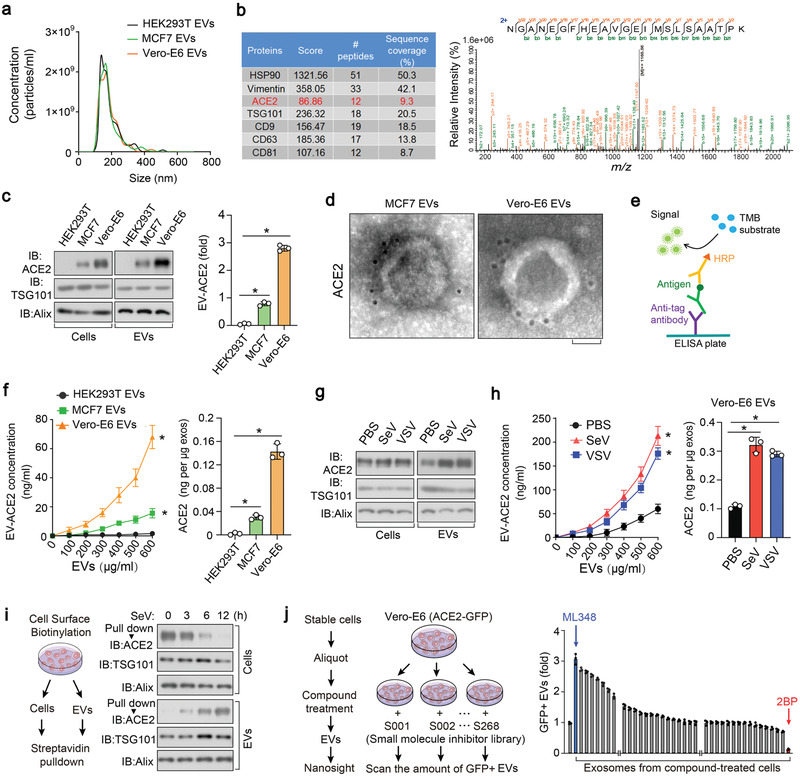
Extrafacial expression of ACE2 on EVs and its regulation by protein palmitoylation. a) Nanoparticle tracking of purified EVs from HEK293T, MCF7, and Vero‐E6 cells. b) Mass‐Spectrometry (MS) analysis of purified EVs secreted by Vero‐E6 cells, showing results for HSP90, Vimentin, ACE2, TSG101, CD9, CD63, and CD81 (left), and the peptide of ACE2 identified in MS (right). c) Left panel: immunoblot (IB) analysis of ACE2 in whole cell lysate and purified EVs derived from HEK293T, MCF7; and Vero‐E6 cell lines; right panel: quantification of the ACE2 protein levels in the EVs of three independent experiments. d) TEM (transmission electron microscope) images of EVs derived from MCF7 and Vero‐E6 cell lines incubated with anti‐ACE2 antibodies and immune‐gold labeled by gold particles. Gold particles are depicted as black dots. Scale bar, 50 nm. e) Schematic diagram of ELISA to measure ACE2 concentration on the surface of EVs. TMB, tetramethylbenzidine; HRP, horseradish peroxidase. f) ELISA of ACE2 on the surface of EVs derived from HEK293T, MCF7, and Vero‐E6 cells assayed as (e) in various concentration. Quantification of ACE2 on the surface of three types of cells were shown right. g) IB analysis of ACE2 in whole cell lysate and EVs derived from Vero‐E6 cells treated with PBS, SeV, or VSV (MOI (multiplicity of infection), 0.1 for 24h. h) ELISA (left) and normalized quantification (right) of ACE2 on the surface of EVs derived from Vero‐E6 cells treated with control PBS, SeV, or VSV (MOI = 0.1) for 24 h. i) Schematic diagram of biotin‐labeling assay (left) and IB analysis (right) to measure ACE2 levels on the cell surface and in the secreted EVs from Vero‐E6 cells treated with SeV (MOI = 0.1) for the indicated times. j) Screening procedure for cellular components or pathways critical for the secretion of ACE2 into EVs (left); nanosight analysis of GFP positive EVs derived from Vero‐E6 cells expressing ACE2‐GFP and pre‐treated with small molecule inhibitor library (right). *p < 0.05 (two‐tailed Student's t test (c, right), (f, right), (h, right), and (j, (right) or two‐way ANOVA (f, left), (h, left)). Data are analyzed of three independent experiments and shown as means ± SD (c, right), (f, h, j, right).
EVs are generated and released via a defined intracellular trafficking route.[ 24 ] We found that the knockdown of the GTPase Rab27A, a critical protein required for extracellular vesicle secretion,[ 25 ] strongly reduced the amount of ACE2‐containing EVs (Figure S1d, Supporting Information). Pre‐treatment of cells with GW4869, which inhibits the release of extracellular vesicles, also reduced the ACE2 levels in EVs (Figure S1e, Supporting Information). Moreover, we built an enzyme‐linked immunosorbent assay (ELISA) to examine the effect of ACE2 on the surface of extracellular vesicles (Figure 1e). Similar to the IG‐TEM, ELISA revealed that EV‐ACE2 exposed its extracellular domain on the surface of the EVs and had similar membrane topology as cell surface ACE2 (Figure 1d,f). Immunoblot and ELISA analysis also confirmed increased levels of ACE2 both inside the EVs and on the surface of EVs upon infection with Sendai virus (SeV) or vesicular stomatitis virus (VSV) (Figure 1g,h). To demonstrate the recycling of ACE2 from cell PMs to EVs, cell surface proteins were labeled with biotin, followed by a time‐course RNA virus infection. Biotin‐labelled ACE2 disappeared from the cell surface and appeared in the EVs after 3–12 h, confirming that the ACE2 which was enriching the EVs originated from the Vero‐E6 cell surface and could be strongly potentiated upon viral infection (Figure 1i).
To determine which proteins and pathways are critical for the secretion of EV‐ACE2, we treated Vero‐E6 cells with a library of 268 small‐molecule inhibitors that target major post‐translational modifications and cellular signaling pathways (Figure 1j). Notably, Vero‐E6 cells pre‐treated with 2‐bromopalmitate (2‐BP), a general palmitoylation inhibitor,[ 26 , 27 ] lost their ability to produce ACE2 positive EVs. Whereas cells pre‐treated with ML348, a selective inhibitor of acyl protein thioesterase 1 (APT1, [a de‐palmitoylating enzyme]),[ 28 ] strongly elevated their extracellular vesicle secretion of ACE2 (Figure 1j). These results indicate that cell surface ACE2 is released into EVs and that EV‐ACE2 levels are tightly regulated by protein palmitoylation.
2.2. S‐Palmitoylation of ACE2 is Required for its Proper Membrane Targeting and Secretion into EVs
We next sought to determine the mechanism that supports EV‐ACE2 secretion. Immunofluorescence staining revealed that a significant portion of ACE2 is located on the PM (Figure 2a). In 2‐BP‐treated cells, the PM‐localised ACE2 disappeared, leaving a large amount of ACE2 located in the cytoplasm; however, in ML348‐treated cells, the cell membrane localization of ACE2 was clearly enhanced (Figure 2a). Flow cytometry analysis also confirmed that membrane ACE2 could also be sharply reduced by 2‐BP stimulation, or increased by ML348 (Figure 2b).
Figure 2.
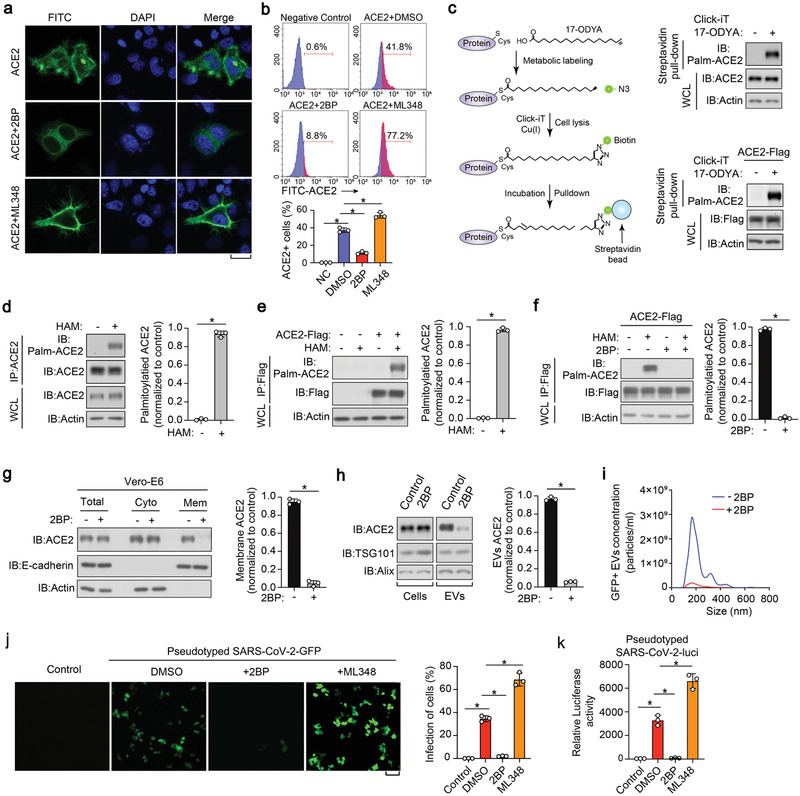
S‐palmitoylation of ACE2 is required for its proper membrane targeting and secretion into EVs. a) Immunofluorescence and DAPI (4′,6‐diamidino‐2‐phenylindole) staining of HeLa cells transfected with ACE2‐Flag and treated with control DMSO, 2BP (50 µm) or ML348 (5 µm) for 24 h. Scale bar, 20 µm. b) FACS analysis (upper panel) and quantification (lower panel) of the percentage of ACE2 positive cells from HeLa cells transfected with ACE2‐Flag and treated with control DMSO, 2BP (50 µm) or ML348 (5 µm) for 24 h. c) Left panel: schematic diagram of Click‐iT reaction used for the detection of ACE2 palmitoylation. Cells were pre‐treated with 17‐ODYA; the lysates were reacted with biotin‐azide and then analyzed with Immunoblot (IB). Right panel: profile of palmitoylated ACE2 in Vero‐E6 cells (upper panel) and ACE2‐Flag‐expressed HEK293T (lower panel) incubated with 17‐OYDA (100 µm) for 6 h. d,e) IB analysis (left panel) and quantification (right panel) of S‐palmitoylation levels of ACE2 in Vero‐E6 (d) and HEK293T (e) cells transfected with ACE2‐Flag, followed by the lysis at the presence or absence of HAM (hydroxylamine, 1 m). f) IB (left panel) and quantification (right panel) of the S‐palmitoylation levels of ACE2 in HEK293T cells transfected with ACE2‐Flag and treated with 2BP (50 µµ, 24 h), followed by the lysis at the presence or absence of HAM (1 m). g) Left panel: IB of total, cytosolic, and membrane fractions of Vero‐E6 cells treated with or without 2BP (50 µm) for 24 h; right panel: quantification of membrane ACE2 of three independent experiments as left panel. h) Left panel: IB of total cell lysates and purified EVs derived from Vero‐E6 cells treated with or without 2BP (50 µm) for 24 h. Right panel: quantification of EV‐ACE2 of three independent experiments as in left panel. i) Nanosight analysis of GFP positive EVs derived from Vero‐E6 cells expressing ACE2‐GFP and treated with or without 2BP (50 µm) for 24 h. j) Representative images of HEK293T cells stably expressing ACE2 and pretreated with 2BP (50 µm) or ML348 (5 µm) for 24 h, followed by the infection with SARS‐CoV‐2‐GFP pseudovirus (MOI, 1) for 2 days. GFP positive cells from three different views were quantified and shown right. k) SARS‐CoV‐2‐luci pseudovirus (MOI, 1) were used to infect HEK293T cells stably expressing ACE2 and pretreated with 2BP (50 µm) or ML348 (5 µm) for 24 h. Luciferase activities in cell lysates were determined at 2 days post infection. *p < 0.05 (two‐tailed Student's t test. Data are analyzed from three independent experiments and shown as mean ± SD (d–h, right, j, right, k).
Given these effects, we validated the palmitoylation of ACE2, using the Click‐iT assay (schematics in Figure 2c, left panel). Here, we incorporated azide‐containing palmitic acid into cultured cells and labeled the biotin–alkyne protein, followed by streptavidin pull‐down and immunoblot assays using ACE2 or anti‐flag antibodies. This assay confirmed the palmitoylation of ACE2 at both endogenous and exogenous levels of protein (Figure 2c, right panel). To verify this result, we performed an acyl–biotin exchange (ABE) assay, in which the free cysteine thiol groups of the proteins were immediately and irreversibly blocked by N‐ethylmaleimide (NEM), and then the palmitoylated cysteines were cleaved by hydroxylamine (HAM) and finally biotinylated for immunoblotting analysis (Figure S2a, Supporting Information). In this experiment, both endogenously or ectopically expressed ACE2 proteins were acylated (Figure 2d,e), however, their acylation levels were all diminished upon treatment with 2‐BP (Figure 2f). The increased biotin signal after HAM treatment in the ABE assay demonstrated that ACE2 incorporated palmitate through a thioester linkage (Figure 2d–f).
Next, we examined the subcellular distribution of ACE2. We found that 2‐BP could substantially and specifically reduce the content of ACE2 in the membrane fraction, however, it barely affected the overall expression of ACE2 or its cytosolic fraction (Figure 2g; Figure S2b,c, Supporting Information). In line with this, we found that 2‐BP strikingly reduced the amount of ACE2 in EVs (Figure 2h). This phenomenon was demonstrated by the NanoSight tracking system in that the number of ACE2‐green fluorescence protein (GFP) positive EVs, rather than the total number of EVs, was severely reduced by 2‐BP (Figure 2i and data not shown). These results suggest that S‐palmitoylation is required for ACE2 membrane targeting and extracellular vesicle secretion.
Previous studies revealed that viral S protein binding occurs on the outer surface of ACE2 and that SARS‐CoV‐2 uses the ACE2 receptor for cell entry.[ 29 ] Thus, we used a SARS‐CoV‐2‐GFP pseudovirus to characterize the infection efficiency in ACE2 expressing HEK293T cells. We found that the infection efficiency of the pseudotype virus was severely decreased in 2‐BP pre‐treated cells and significantly increased in ML348 pre‐treated cells (Figure 2j). Similar observations were more accurately found using the pseudovirus–luciferase reporter assay (Figure 2k). Thus, the above results indicated that palmitoylation could affect the susceptibility of cells to SARS‐CoV‐2 by altering the content of ACE2 in the membrane.
2.3. Cys141 and Cys498 Residues in ACE2 are S‐Palmitoylated
To identify the palmitoylation site of ACE2, we first employed the motif‐based predictors CSS‐palm.[ 30 ] This model predicted multiple potential ACE2 palmitoylation sites within its extracellular domain (Figure S3a, Supporting Information). To validate these predictions, flag‐tagged ACE2 was immunopurified and subjected to mass spectrometric analysis specifically for identifying palmitoylation (Figure 3a; Figure S3b, Supporting Information). This assay confirmed the palmitoylation of ACE2 residues Cys141 and Cys498 (Figure 3b–d). Sequence comparison revealed that both the Cys141 and Cys498 residues are conserved palmitoylation motifs in ACE2 orthologs (Figure 3c). Subsequently we constructed a series of corresponding ACE2 point mutants and found that simultaneous substitution of both Cys141 and Cys498 with serine (C141/498S, [2CS]), completely prevented ACE2 palmitoylation, as revealed by ABE assay and Click‐iT labelling (Figure 3e,f; and Figure S3c, Supporting Information). These results indicate that ACE2 has two major sites for palmitoylation, Cys141 and Cys498, which are likely to compensate for each other if they are blocked or removed.
Figure 3.
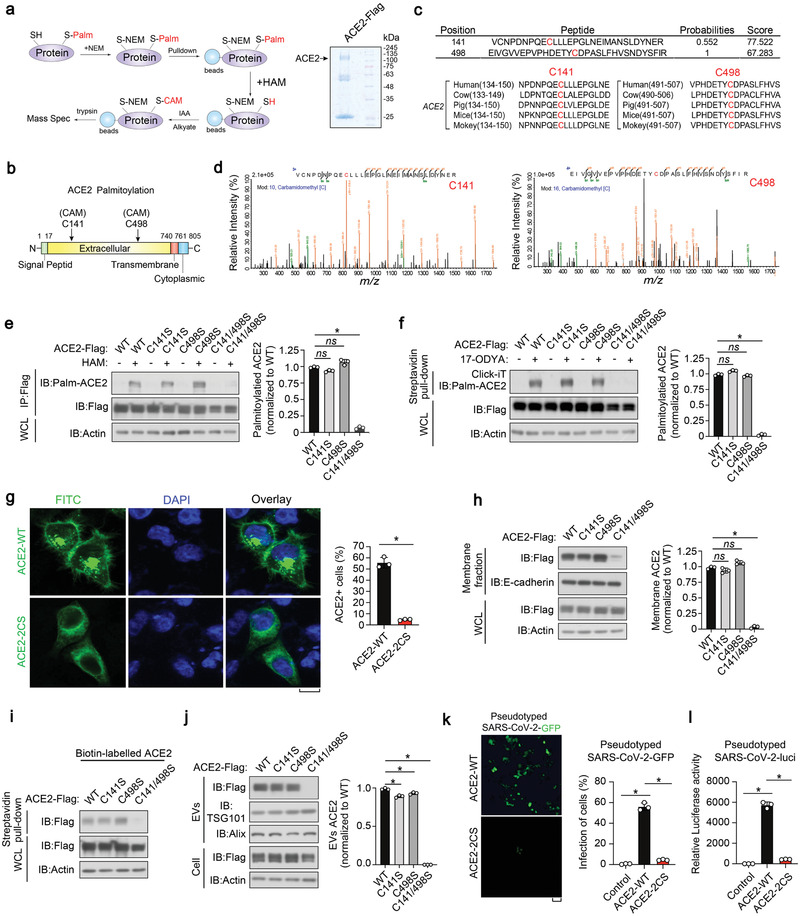
Cys141 and Cys498 residues in ACE2 are S‐palmitoylated. a) Left panel: schematic diagram of mass spectrometry (MS) analysis procedure for S‐palmitoylation modification; right panel: SDS‐PAGE of anti‐Flag (ACE2) immunoprecipitants from HEK293T cells with Coomassie brilliant blue staining. ACE2 band at the corresponding location is indicated. b) Schematic representation of S‐palmitoylation sites in the extracellular domain of ACE2. ACE2 consists of a signal peptide, an extracellular domain, a transmembrane domain and a cytoplasmic domain. c) Upper panel: peptides of S‐palmitoylated ACE2 identified in MS; lower panel: sequence alignment of identified C141 and C498 palmitoylation sites in ACE2 orthologs of different species. d) MS analysis identified ACE2 C141 site (left) and C498 site (right) that are palmitoylated. e) IB of whole cell lysate (WCL) and immunoprecipitants derived from HEK293T cells transfected with c‐terminal Flag‐tagged ACE2 WT and various CS mutants, followed by the lysis at the presence or absence of HAM (1 M). The S‐palmitoylation levels of ACE2 WT and CS mutants were quantified of three independent experiments and shown right. f) IB of WCL and streptavidin pull‐down derived from HEK293T cells transfected with c‐terminal Flag‐tagged ACE2 WT and CS mutants, followed by the Click‐iT reaction. The S‐palmitoylation of ACE2 WT and CS mutants were quantified of three independent experiments and shown right. g) Left panel: immunofluorescence and DAPI staining of HeLa cells transfected with ACE2‐WT or ‐2CS mutant. Scale bar, 20 µm; right panel: FACS analysis of the percentage of ACE2 positive cells from HeLa cells transfected with ACE2‐WT or ACE2‐2CS. n = 3 biological replicates per group. h) Left panel: IB of WCL and membrane fractions of HeLa cells stably expressing ACE2‐WT, ACE2‐CS mutants. Membrane ACE2 level was quantified of three independent experiments and shown right. i) IB analysis of WCL and streptavidin pull‐down derived from HeLa cells ectopically expressing ACE2‐WT and CS mutants. j) IB analysis (left) of total cell lysates and purified EVs derived from HEK293T cells stably expressing ACE2‐WT or CS mutants. EV‐ACE2 level was quantified of three independent experiments and shown right. k) SARS‐CoV‐2‐GFP pseudovirus were used to infect HEK293T cells stably expressing ACE2‐WT or ‐2CS mutant. Representative images (left) and quantification of GFP fluorescence intensity (right) were determined at 24 h post transduction to calculate infection (%). l) SARS‐CoV‐2‐luci pseudovirus were used to infect 293T cells stably expressing ACE2‐WT or ‐2CS mutant. Luciferase activities in cell lysates were measured at 24 h post transduction to calculate infection (%). *p < 0.05 (two‐tailed Student's t test. Data are analyzed of three independent experiments and shown as mean ± SD (e–h, right, j, right, k, right, l).
Compared with wild‐type ACE2 (ACE2‐WT), the ACE2‐2CS mutant that lost the capacity to perform palmitoylation was not able to efficiently localize to the cell surface, as revealed by immunostaining (Figure 3g, left panel) and flow cytometry (FACS) analysis (Figure 3g, right panel). In the cell fraction assay, the ACE2‐2CS mutant reduced the levels of ACE2 associated with the membrane fraction, without affecting the overall protein levels (Figure 3h). Biotin‐labeled cell surface ACE2 was also found to be severely inhibited in ACE2‐2CS transfected cells (Figure 3i). In line with this, cells expressing ACE2‐2CS barely released any EVs containing ACE2 (Figure 3j). These observations further consolidate the conclusion that site‐specific palmitoylation is required for ACE2 membrane localization and extracellular vesicle secretion. Moreover, we compared the infection of pseudotyped SARS‐CoV‐2 in control and ACE2‐2CS‐expressing cells. Fluorescence microscopy showed that the GFP‐expressing viral infection was substantially inhibited in HEK293T cells that expressed ACE2‐2CS when compared to control cells expressing ACE2‐WT (Figure 3k). In the pseudovirus–luciferase reporter assay, the value that was read in ACE2‐WT cells was more than 200 times higher than in ACE2‐2CS expressing cells (Figure 3l). Together, we identified Cys141 and Cys498 as the major palmitoylation sites of ACE2 and found that the ACE2‐2CS mutation replicated the effect of 2‐BP treatment.
2.4. ACE2 is Palmitoylated by ZDHHC3 and De‐Palmitoylated by LYPLA1
To identify the predominant palmitoyltransferase for ACE2, we established a screening approach combining ABE assay palmitoylaton detection, subcellular localization, and loss‐of‐function tests. We first performed validation using a specific small interfering RNA (siRNA) library that targeted the ZDHHC1‐24 palmitoyltransferases (Figure S4a, Supporting Information), then each palmitoyltransferase gene was silenced and screening was performed. Interestingly, knockdown of ZDHHC3, but not the other palmitoyltransferases, diminished the palmitoylation of ACE2 (Figure 4a; Figure S4a, Supporting Information). Next, we independently employed short hairpin RNA (shRNA) to knockdown ZDHHC3 and we observed a similar decrease in ACE2 palmitoylation (Figure S4b, Supporting Information). Contrastingly, ectopic expression of ZDHHC3, but not its catalytically inactive mutant C135S or other palmitoyltransferases, robustly enhanced ACE2 palmitoylation while causing an accelerated migration of ACE2 proteins during electrophoresis (Figure 4b,c). Given the reported interaction between a palmitoyltransferase and its substrate, we examined whether ZDHHC3 can bind to ACE2. As expected, co‐immunoprecipitation suggested physical interactions between ectopically expressed ACE2 and ZDHHC3 (Figure 4d; Figure S4c,d, Supporting Information) or endogenously in Vero‐E6 cells (Figure 4e; Figure S4c, Supporting Information). In agreement with this, ectopically expressed ZDHHC3 co‐localized with ACE2 in Vero‐E6 cells (Figure 4f). Furthermore, in cells depleted of ZDHHC3, ACE2 lost its membrane localization and was rarely secreted into EVs (Figure 4g,h), again replicating the effect of 2‐BP treatment. In cells overexpressing ZDHHC3, ectopically expressed ACE2 could be recruited to the cell membrane and released into EVs (Figure 4i; Figure S4e, Supporting Information). These results consistently suggest that ZDHHC3 palmitoylates, and thus, redistributes ACE2.
Figure 4.
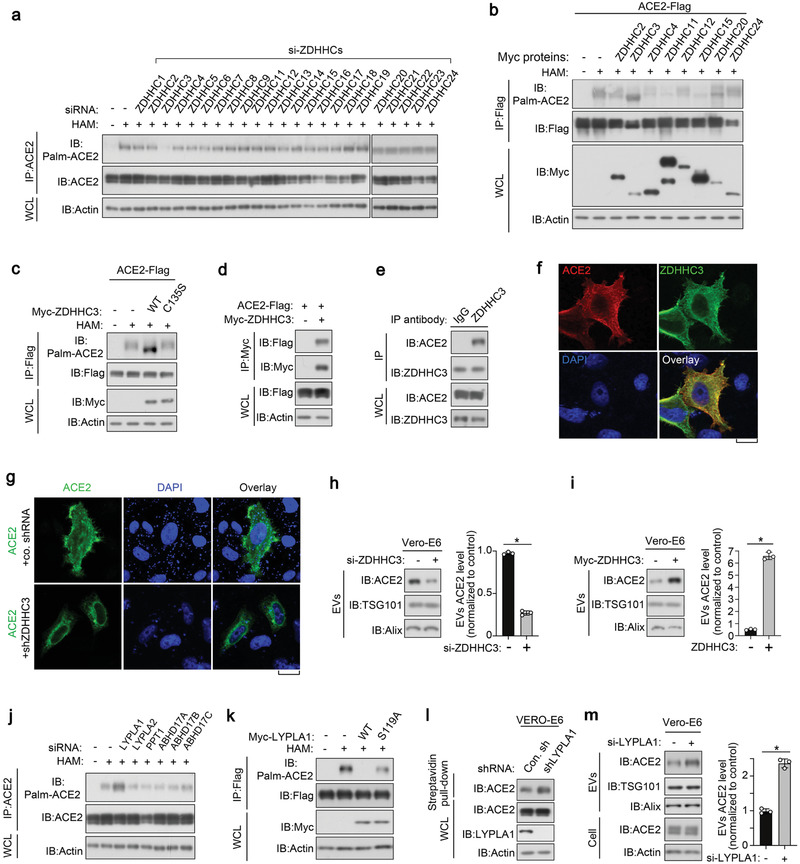
ACE2 is palmitoylated by DHHC3 and depalmitoylated by LYPLA1. a) Immunoblot (IB) analysis of whole cell lysate (WCL) and anti‐ACE2 immunoprecipitants derived from HEK293T cells stably expressing ACE2 and transfected with si‐ZDHHCs as indicated. b) IB analysis of WCL and anti‐Flag immunoprecipitants derived from HEK293T cells transfected with c‐terminal Flag tagged ACE2 and Myc‐tagged ZDHHCs as indicated. c) IB analysis of WCL and anti‐Flag immunoprecipitants derived from HEK293T cells transfected with c‐terminal Flag‐taggedACE2, Myc‐tagged ZDHHC3‐WT, and C135S mutant. d) IB analysis of whole cell lysate (WCL) and anti‐Myc immunoprecipitant from HEK293T cells transfected with ACE2‐Flag and Myc‐ZDHHC3. e) IB analysis of WCL and anti‐ZDHHC3 immunoprecipitant from Vero‐E6 cells. f) Immunofluorescence and DAPI staining of HeLa cells transfected with ACE2‐Flag and Myc‐ZDHHC. Scale bar, 20 µm. g) Immunofluorescence and DAPI staining of control and ZDHHC3 stably depleted Vero‐E6 cells transfected with ACE2. h) IB analysis of EVs from Vero‐E6 cells transfected with control siRNA or si‐ZDHHC3. ACE2 expression in EVs was quantified of three independent experiments and shown right. i) IB of EVs purified from Vero‐E6 cells transfected with or without Myc‐ZDHHC3 as indicated. EV‐ACE2 was quantified of three independent experiments and presented shown right. j) IB analysis of WCL and anti‐ACE2 immunoprecipitants derived from HEK293T cells transfected with si‐depalmitoylases as indicated. k) IB analysis of WCL and anti‐Flag immunoprecipitants derived from HEK293T cells transfected with expression plasmids encoding ACE2‐Flag, Myc‐LYPLA1‐WT, and S119A mutant. l) IB analysis of WCL and streptavidin pull‐down derived from Vero‐E6 cells infected with lentivirus encoding control shRNA (Con.sh) or LYPLA1 shRNA. m) IB analysis of ACE2 in both cells and EVs derived from Vero‐E6 cells transfected with control siRNA or si‐LYPLA1. EV‐ACE2 was quantified of three independent experiments and shown right. *p < 0.05 (two‐tailed Student's t test (h, right, i, right, m, right). Data are analyzed of three independent experiments and shown as mean ± SD (h, right, i, right, m, right).
Next, we sought to identify the S‐de‐palmitoylase for ACE2. Using siRNA for loss‐of‐function scanning, we found that the LYPLA1 knockdown, but not other de‐palmitoylating enzymes, increased ACE2 palmitoylation (Figure 4j; Figure S4f, Supporting Information). Additionally, ectopic expression of LYPLA1 almost completely removed the palmitoylation of ACE2, however its catalytically inactive mutant S119A (SA) did not have this effect (Figure 4k). In line with the function of LYPLA1 in regulating ACE2 palmitoylation, we also found that ACE2 membrane localization and extracellular vesicle secretion were upregulated upon LYPLA1 depletion (Figure 4l and data not shown). Taken together, the results show that ACE2 is palmitoylated by ZDHHC3 and de‐palmitoylated by LYPLA1.
2.5. Generation and Characterization of PM‐ACE2‐EVs; Engineered EVs Enriched with ACE2 on their Surface
EVs are nanosized vesicles that can serve as delivery systems for various therapeutics.[ 24 , 31 , 32 ] The findings reported above revealed the important role of palmitoylation in boosting ACE2 transportation into EVs. Since EV‐ACE2 shares similar membrane topology as cell surface ACE2, in that it exposes the extracellular segment to the outside of the EVs, we proposed that ACE2‐enriched EVs might be able to bind SARS‐CoV‐2 and therefore inhibit infection.
In order to maximize the delivery of ACE2 into EVs, we fused the S‐palmitoylation‐dependent PM targeting sequence of the neuronal growth cone protein GAP43 (amino acids 1 to 11, MLCCMRRTKQV)[ 33 ] to the N‐terminus of ACE2 (hereafter denoted as PM‐ACE2) (Figure 5a). Compared to the ACE2‐WT, ectopically expressed PM‐ACE2 was indeed found to be more strongly palmitoylated in the ABE assay (Figure 5b), moreover it was more concentrated on the PM in the fluorescence microscopy analysis (Figure 5c). Next, we used FACS to examine the proportion of cells with ACE2 on their surface. Notably, this proportion was significantly higher in cells transfected with PM‐ACE2 than in cells transfected with ACE2‐WT (Figure 5d). We then purified EVs from both cell types and found that PM‐ACE2 was more abundantly and efficiently released into EVs than ACE2‐WT (Figure S5a, Supporting Information). Moreover, together the ABE assay, immunofluorescence, and FACS analysis consistently revealed that fusion of the GAP43 S‐palmitoylation motif served as a very strong membrane positioning and extracellular vesicle guidance signal. It could even rescue the palmitoylation, membrane localization, and extracellular vesicle secretion capabilities of the S‐palmitoylation‐deficient ACE2‐2CS mutant (Figure S5a–d, Supporting Information).
Figure 5.
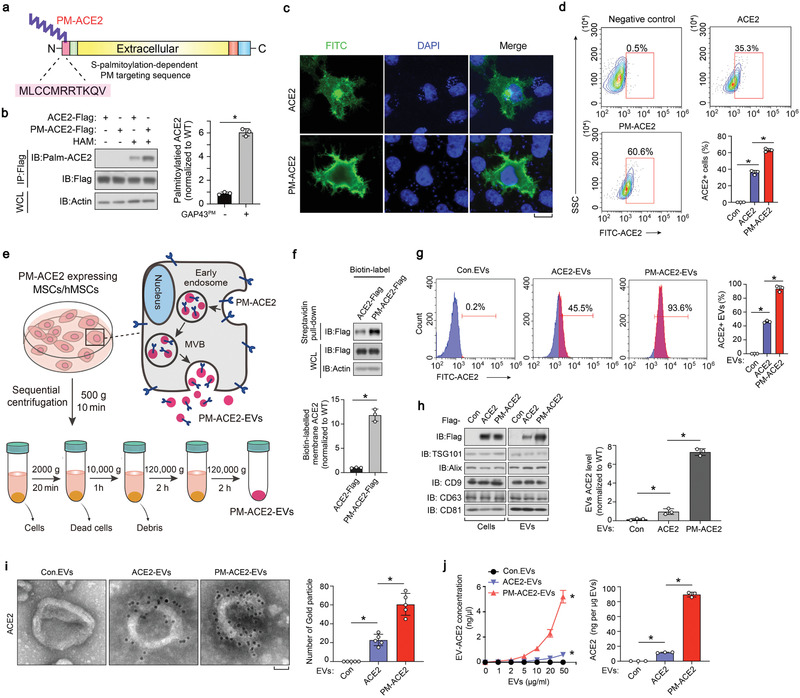
Generation and characterization of the PM‐ACE2‐EVs. a) Schematic diagram of N‐GAP43PM‐ACE2 construct. GAP43PM, amino acids 1 to 11 of GAP43. b) IB of whole cell lysate (WCL) and anti‐Flag immunoprecipitants derived from HEK293T cells transfected with c‐terminal tagged ACE2 or PM‐ACE2 plasmids and lysed at the presence or absence of HAM. Palmitoylated ACE2 was quantified of three independent experiments and shown right as mean± SD. c) Immunofluorescence and DAPI staining of HeLa cells transfected with ACE2‐WT or PM‐ACE2 (PM‐tag, GAP43 amino acids 1 to 11). Scale bar, 10 µm. d) The FACS gating strategy employed and quantification of ACE2 positive cells in HeLa cells transfected with ACE2‐WT or PM‐ACE2. e) Schematic diagram of PM‐ACE2+ EVs biogenesis and purification by differential ultracentrifugation. MVB, multiple vesicle body. f) IB analysis (top) and quantification (bottom) of WCL as well as streptavidin pull‐down derived from human mesenchymal stem cells (hMSCs) ectopically expressing ACE2‐WT and PM‐ACE2. g) FACS analysis (left) and quantification (right) of purified EVs derived from hMSCs stably expressing control vector (Con), ACE2‐WT or PM‐ACE2. h) Immunoblot (IB) analysis (left) and quantification (right) of ACE2 in whole cell lysate and EVs derived from hMSCs expressing control vector (Con), ACE2‐WT, or PM‐ACE2. i) TEM imaging (left) and gold particles quantification (right) of immune‐gold‐labeled EVs purified from hMSCs stably expressing control vector (Con), ACE2‐WT, or PM‐ACE2. Gold particles are depicted as black dots. Scale bar, 50 nm. j) ELISA (left) and normalized quantification (right) of ACE2 on the surface of EVs derived from hMSCs stably expressing control vector (Con), ACE2‐WT, or PM‐ACE2. *p < 0.05 (two‐tailed Student's t test (b, right; d, right; f, bottom; g, right; h, right; i, right; j, right or two‐way ANOVA (j, left). Data are representative of three independent experiments and shown as means ± SD (b, right; d, right; f, bottom; g, right; h, right; i, right; j).
We next tried to achieve the large scale and efficient production of clinical‐grade PM‐ACE2 enriched EVs. Considering the limited side effects of mesenchymal stem cells (MSCs) and the current employment of MSC‐EVs in various disorders,[ 34 , 35 , 36 ] we developed an approach for the production of good manufacturing practice‐grade (GMP‐grade) EVs derived from bone marrow MSCs that can express ACE2‐WT or PM‐ACE2 (Figure 5e). Cell‐surface biotinylation of these MSCs confirmed that PM‐ACE2 was more abundantly located on the cell surface than ACE2‐WT (Figure 5f). Compared to the EVs derived from control MSCs (hereafter denoted Con.EVs), TEM and NanoSight analyses showed that EVs derived from ACE2‐WT or PM‐ACE2 MSCs (hereafter denoted as ACE2‐EVs and PM‐ACE2‐EVs, respectively) showed no difference in morphology or quantity (Figure S5e,f, Supporting Information). FACS of these EVs showed that ≈46% of ACE2‐EVs were positive for ACE2, whereas 94% were positive in the PM‐ACE2‐EVs (Figure 5g). Immunoblotting showed that although the expression levels of the stably expressed PM‐ACE2 and ACE2‐WT were similar, PM‐ACE2 was more abundantly transported into EVs (Figure 5h). IG‐TEM revealed that PM‐ACE2 proteins in the EVs were more extensively anchored to the membrane than ACE2‐WT (Figure 5i). Furthermore ELISA, which precisely measures the content of ACE2 in EVs, showed that PM‐ACE2 was ≈seven times more enriched in EVs than ACE2‐WT (Figure 5j). In detection of the total number of extracellular vesicle secretion by time courses, we found the secretion rates of ACE2‐EVs and PM‐ACE2‐EV remained largely the same, but ELISA detected far more abundant PM‐ACE2‐EVs in the same experiment (Figure S5h, Supporting Information). In summary, we established a standard operating procedure to generate engineered EVs with enriched ACE2 on their surfaces.
2.6. PM‐ACE2‐EVs Show Strong Neutralization Potency Against Pseudotyped SARS‐CoV‐2
To evaluate the binding affinity between ACE2‐enriched EVs and the RBD of the SARS‐CoV‐2 S protein, we used surface plasmon resonance (SPR). It is a surface‐sensitive technique that enables the study of real‐time monitoring of biomolecular interactions. In this assay, label‐free recombinant S‐RBD protein (20 µg mL−1) was initially captured on a Series S Sensor Chip CM5 at a level of ≈3000 response units (RUs). Binding affinity analysis of the EVs‐RBD interaction was performed by flowing Con. EVs, ACE2‐EVs, and PM‐ACE2‐EVs over the S‐RBD‐captured sensor chip (Figure 6a,b). As expected, PM‐ACE2‐EVs were found to have a greater ability to bind to S‐RBD than the ACE2‐EVs; furthermore, this capability was nearly undetectable in the ACE2‐deficient Con. EVs (Figure 6c). To further characterize this association, we diluted PM‐ACE2‐EVs in a gradient, and then evaluated their binding affinity to the RBD‐sensor chip. We confirmed that PM‐ACE2‐EVs achieved specific and high‐affinity binding to the RBD, over a wide concentration range; moreover, their binding could retain linearity for a long period (Figure 6d). Furthermore, we also performed SPR assay by flowing Con. EVs, ACE2‐EVs, and PM‐ACE2‐EVs over the pseudotyped virus‐captured sensor chip (Figure S6a, Supporting Information). Consistent with the results from EVs‐RBD interaction, PM‐ACE2‐EVs were found to have a greater ability to bind with the pseudotyped virus than the ACE2‐EVs (Figure S6b, Supporting Information).
Figure 6.
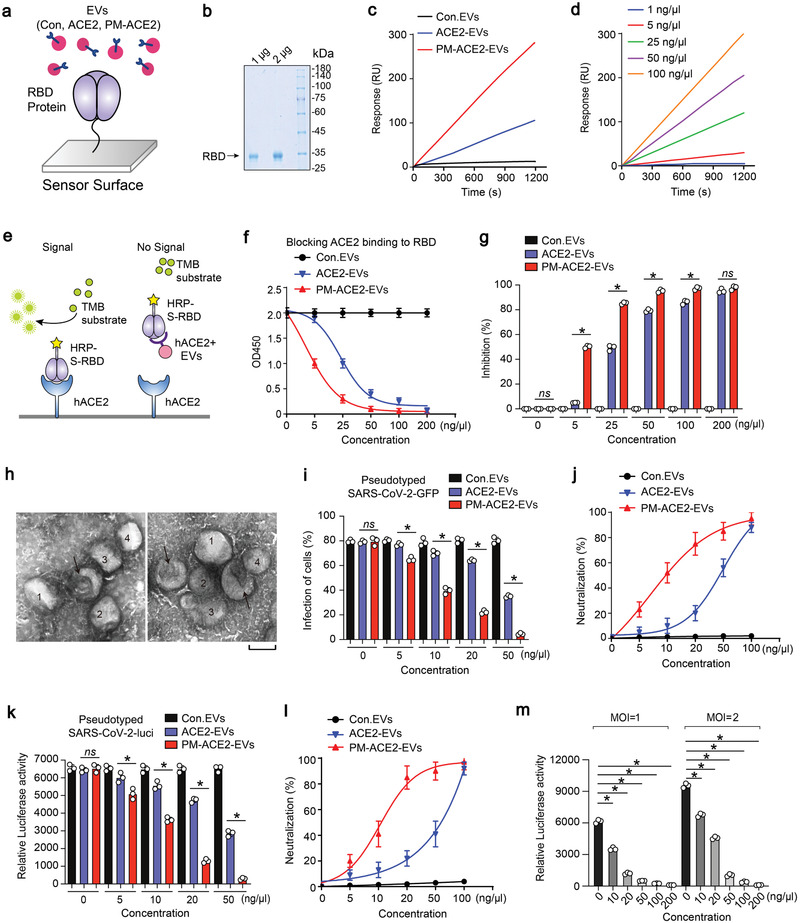
PM‐ACE2‐EVs shows neutralization potency against SARS‐CoV‐2 pseudovirus. a) Schematic diagram of functionalization of the surface‐plasmon resonance (SPR). Spike‐RBD is immobilized on chip‐gold surface at the level of 3000 RU with amine coupling standard method, and the EVs bound with receptor binding domain (RBD) protein will be captured. b) Coomassie brilliant blue staining of spike RBD protein. c) The binding affinities of control EVs (Con. EVs), ACE2‐EVs, and PM‐ACE2‐EVs with spike‐RBD protein were evaluated using SPR technique. Flow rate, 5 µL min−1; EV, 100 ng µL−1. d) SPR assay of various concentrations of PM‐ACE2‐EVs captured by RBD protein; the concentration of PM‐ACE2‐EVs were 1, 5, 25, 50, and 100 ng µL−1 as indicated. Flow rate, 5 µL min−1. e) Schematic representation of ELISA assay to measure the binding ability of hACE2 positive EVs with RBD protein. TMB, tetramethylbenzidine f) ELISA assay of the binding affinities between coupled hACE2 and spike‐RBD at the presence of Con. EVs, ACE2‐EVs, or PM‐ACE2‐EVs at the concentration of 0, 5, 25, 50, 100, and 200 ng µL−1. g) The blocking capacity of Con. EVs, ACE2‐EVs, and PM‐ACE2‐EVs toward hACE2‐RBD binding in (f) was calculated. h) The representative TEM images of PM‐ACE2‐EVs incubated with SARS‐CoV‐2 pseudovirus: the arrow indicates for EVs and the viruses are numbered. i) SARS‐CoV‐2‐GFP pseudovirus neutralization assay of ACE2‐EVs and PM‐ACE2‐EVs. Vero‐E6 cells treated with Con. EVs, ACE2‐EVs, or PM‐ACE2‐EVs (0, 5, 10, 20, 50, and 100 ng µL−1) were infected with SARS‐CoV‐2‐GFP pseudovirus (MOI, 1) for 24 h, and the GFP positive cells were determined at 48 h post infection. j) The neutralization potency of Con. EVs, ACE2‐EVs, and PM‐ACE2‐EVs was calculated based on the SARS‐CoV‐2‐GFP pseudovirus neutralization assay in (i). k) SARS‐CoV‐2‐luci pseudovirus neutralization assay of ACE2‐EVs and PM‐ACE2‐EVs. Vero‐E6 cells treated with Con.EVs, ACE2‐EVs, or PM‐ACE2‐EVs (0, 5, 10, 20, 50, 100 ng µL−1) were infected with SARS‐CoV‐2‐luci pseudovirus (MOI, 1) for 24 h, and the luciferase activity was determined at 48 h post infection. l) The neutralization potency of Con. EVs, ACE2‐EVs, or PM‐ACE2‐EVs was calculated based on the SARS‐CoV‐2‐luci pseudovirus neutralization assay in (k). m) Neutralization assay of SARS‐CoV‐2‐luci pseudovirus by different concentration of PM‐ACE2‐EVs (0, 10, 20, 50, 100, 200 ng µL−1).Vero‐E6 Cells were infected with SARS‐CoV‐2‐luci pseudovirus at MOI = 1 or 2. *p < 0.05 (two‐tailed Student's t test (g, i, k, m) or two‐way ANOVA (f, j, l). Data are representative of at least two independent experiments with triplicates and shown as means + SD (f, g, i, j, k, l, m).
Virus‐receptor binding can be mimicked in vitro via a protein–protein interaction‐based ELISA assay using purified recombinant hACE2 and the RBD of the SARS‐CoV‐2 S protein conjugated to horseradish peroxidase (HRP) (Figure 6e). We then demonstrated that the specific RBD‐hACE2 binding can be blocked or neutralized by ACE2‐enriched EVs in a dose‐dependent manner, but not by ACE2‐deficient control EVs (Figure 6f,g). PM‐ACE2‐EVs were more effective than ACE2‐EVs in this assay, particularly at lower dosages; this further confirms that the engineered PM‐ACE2‐EVs possess a higher binding inhibition capacity between RBD and hACE2 (Figure 6f,g). In the TEM, we observed an association between PM‐ACE2‐EVs and the pseudotyped SARS‐CoV‐2 (Figure 6h), suggesting that ACE2‐enriched EVs might also be effective in antagonizing SARS‐CoV‐2 infection. To verify this, we performed a SARS‐CoV‐2‐GFP pseudovirus neutralization assay, in which the ACE2 expressing HEK293T cells were incubated with serially diluted EVs and infected with the SARS‐CoV‐2‐GFP pseudovirus. Of note, PM‐ACE2‐EVs showed a significantly higher neutralisation potency (IC50 = 10 ± 0.8 ng µL−1, IC50, half‐maximal inhibitory concentration) than ACE2‐EVs (IC50 = 45 ± 1.9 ng µ−1) (Figure 6i,j; Figure S6c, Supporting Information). Similar observations were more accurately obtained using the pseudovirus‐luciferase reporter assay (Figure 6k,l; Figure S6d, Supporting Information). These data demonstrated that PM‐ACE2‐EVs inhibited the cellular attachment and invasion of SARS‐CoV‐2. Importantly, as expected from a neutralizing agent, this inhibition was dependent on the initial quantity of SARS‐CoV‐2 pseudovirus and the concentration of PM‐ACE2‐EVs (Figure 6m), thus, it is dose‐dependent. Furthermore, PM‐ACE2‐EVs still had a high neutralization efficiency (IC50 = 12 ± 0.7 ng µL−1, IC90 = 46 ± 1.3 ng µL−1) at a high multiplicity of cellular infection (MOI) (Figure 6m). We also purified free ACE2 protein as a control in the SARS‐CoV‐2 pseudovirus neutralization assay (Figure S6e, Supporting Information). Apparently, PM‐ACE2‐EVs had a stronger neutralization potency against SARS‐CoV‐2 pseudovirus than the free ACE2 protein (Figure S6e, Supporting Information). In summary, the engineered PM‐ACE2‐EVs are capable of binding to the RBD of SARS‐CoV‐2 with a high affinity, and PM‐ACE2‐EVs can inhibit the infection of pseudotyped SARS‐CoV‐2 in vitro.
2.7. PM‐ACE2‐EVs Block the Infection of Pseudotyped SARS‐CoV‐2 in hACE2 Mice In Vivo
Mice transgenic for the expression of hACE2 are vulnerable to SARS‐CoV‐2.[ 37 , 38 ] To evaluate the neutralization efficacy of engineered PM‐ACE2‐EVs against pseudotyped SARS‐CoV‐2 in vivo, specific pathogen‐free, 6‐week‐old, hACE2‐transgenic mice were injected with EVs and inoculated intranasally with pseudotyped SARS‐CoV‐2 (Figure 7a; Figure S7a, Supporting Information). Bioluminescent imaging (BLI) measurements and images showed that the mice infected with pseudotyped SARS‐CoV‐2 exhibited broad infections in multiple organs, which can be significantly and potently reduced with PM‐ACE2‐EVs treatment (Figure 7b; Figure S7b, Supporting Information). Quantified luminescence values further revealed that the neutralization efficacy of PM‐ACE2‐EVs could reach up to ≈75% (Figure 7c). Moreover, immunohistochemistry (IHC) analysis of sequential sections revealed that the SARS‐CoV‐2 S protein showed strong staining signals in alveolar and bronchial epithelial cells of mice in the control EV‐treated group. However, this was reduced by PM‐ACE2‐EVs in the therapy group (Figure 7d, left panel). Quantitative histopathology of lung sequential sections also demonstrated that the infection of pseudotyped SARS‐CoV‐2 was significantly decreased following treatment with PM‐ACE2‐EVs (Figure 7d, right panel). Furthermore, we found co‐localization of SARS‐CoV‐2‐GFP pseudovirus S protein and the GFP signals in the alveolar epithelial cells of control EV‐treated mice two days post‐infection. This phenomenon was not observed in the non‐infected group or the PM‐ACE2‐EVs treated group (Figure 7e). Similar observations of the pseudovirus S protein were obtained with IHC staining lung section bronchial epithelial cells (Figure S7c, Supporting Information). To explore the biodistribution of PM‐ACE2‐EVs in major organs of mice, especially in lung after i.v injection, we labeled PM‐ACE2‐EVs with near‐infrared (NIR) fluorescent dye of 1,1′‐dioctadecyl3,3,3′,3′‐tetramethylindotricarbocyanine iodide (DiR) and performed the in vivo analysis (Figure S7d, Supporting Information). At 24 h p.i., the animals were sacrificed, and the major organs were excised and imaged ex vivo. Strong DiR signals were observed in lung, liver, and spleen, correlating well with the in vivo imaging data but with higher fluorescent intensity (Figure S7e,f, Supporting Information). Thus, these results confirm that the engineered PM‐ACE2‐EVs can also diminish the infection of pseudotyped SARS‐CoV‐2 in vivo.
Figure 7.
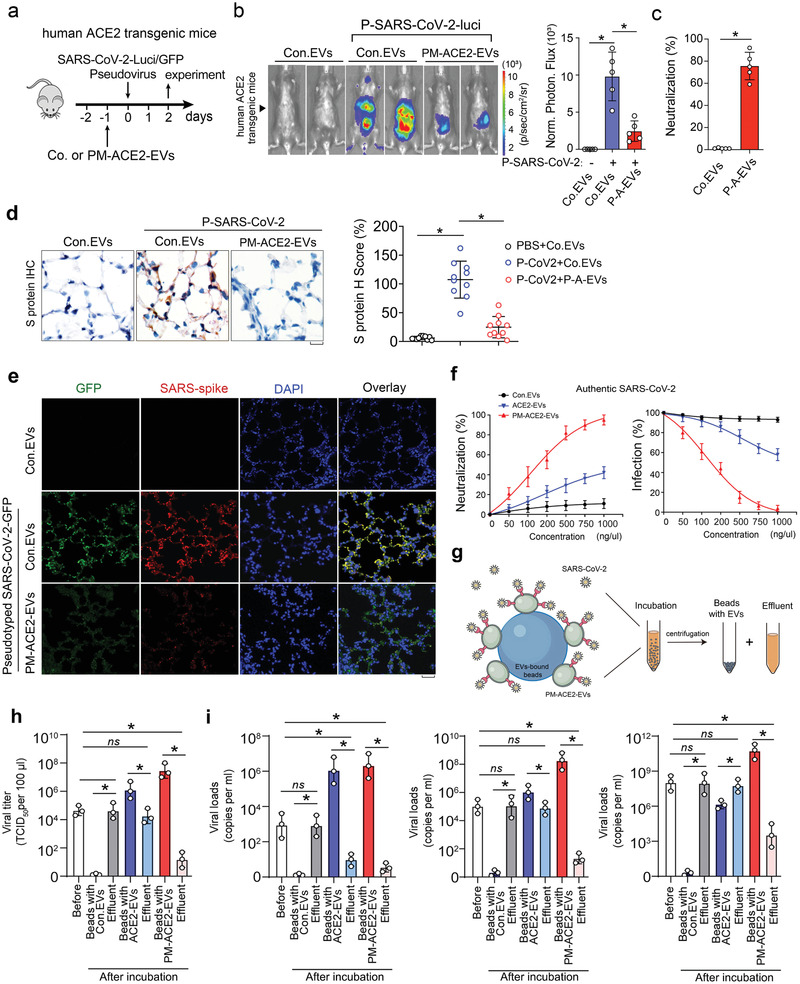
PM‐ACE2‐EVs efficiently blocks the infections of SARS‐CoV‐2 pseudovirus in vivo. a) EVs administration and experimental analysis in vivo: six‐week‐old hACE2 transgenic mice were tail vein‐injected with Con. EVs or PM‐ACE2‐EVs (100 µg per day) at day ‐1, followed by intranasal inoculation of SARS‐CoV‐2‐GFP pseudovirus or SARS‐CoV‐2‐luci pseudovirus (106 TCID50/50 µL) at the day 0. n = 5 for each group. b) Bioluminescent imaging of representative mice from each group at 2 days post infection. Normalized photon flux of all experimental mice of each group was shown right. c) The neutralization potency of PM‐ACE2‐EVs in vivo was determined based on normalized photon flux from each group. d) Representative immunohistochemistry (IHC) staining of the lung tissue from each group assayed with anti‐SARS‐CoV‐2 spike antibody. Scale bar, 20 µm. H Score of S protein from each group (2 slides per mice) was evaluated. e) Representative immunofluorescence and DAPI staining of lung tissue from each group. Scale bar, 50 µm. f) Authentic SARS‐CoV‐2 was incubated with serially diluted Con. EVs, ACE2‐EVs, or PM‐ACE2‐EVs and the mixtures were then added to Vero‐E6 cells. Left panel: neutralization against authentic SARS‐CoV‐2. Right panel: virus RNA loads were determined by RT‐qPCR. g) Schematic diagram of authentic SARS‐CoV‐2 virus enrichment by PM‐ACE2‐EVs bound beads. h) Cell culture supernatant of authentic SARS‐CoV‐2 infected Vero‐E6 cells were incubated with Con. EVs, ACE2‐EVs, or PM‐ACE2‐EVs bound aldehyde/sulphate latex beads for 1h. Virus titers were determined on Vero‐E6 cells. i) Serum samples from COVID‐19 patients with lower (left panel), intermediate (middle panel), or higher (right panel) virus titers were incubated with Con. EVs, ACE2‐EVs, or PM‐ACE2‐EVs bound aldehyde/sulphate latex beads for 1h. Viral burden was measured by RT‐qPCR assay. *p < 0.05 (two‐tailed Student's t test (b, right; c,d, right; h) or two‐way ANOVA (f). Data are representative of at least two independent experiments and shown as means ± SD (b, right; c,d, right; f, h).
2.8. PM‐ACE2‐EVs Exhibit Ability to Neutralize Authentic SARS‐CoV‐2 In Vitro and Inhibit SARS‐CoV‐2 Infection In Vivo
We next evaluated whether infection of authentic SARS‐CoV‐2 could also be inhibited by ACE2‐enriched EVs. In Vero‐E6 cells, PM‐ACE2‐EVs were demonstrated to have much stronger effect (IC50 = 140 ± 15 ng µL) than ACE2‐EVs (IC50 = 1350 ± 110 ng µL−1) in protecting cells against authentic SARS‐CoV‐2, whereas the control EVs barely showed any effect (Figure 7f). To verify this was due to suppressed viral infection and replication in cells, we examined RNA load of SARS‐CoV‐2 and indeed found that PM‐ACE2‐EVs displayed the most robust ability to reduce SARS‐CoV‐2, as compared to that of ACE2‐EVs and control EVs (Figure 7f). These results suggested that PM‐ACE2‐EVs strongly neutralize authentic SARS‐CoV‐2.
Given the validated binding affinity between PM‐ACE2‐EVs and the RBD of SARS‐CoV‐2, we established a method to enrich SARS‐CoV2 by utilizing PM‐ACE2‐EVs bound aldehyde/sulphate latex beads, which would likely enable viral detection from extreme environments with low virus titers or elimination of virus from biological products or patient serum (Figure 7g). We first harvested supernatants from infected Vero‐E6 cells that contain authentic SARS‐CoV‐2 and performed test. As expected, beads bound with ACE2‐EVs were capable of enriching virus and thus eliminated the viral titers in the effluent; This ability was greatly enhanced by using PM‐ACE2‐EVs bound beads, in which the enrichment efficiency was approximately increased by10 times (Figure 7h). We also noticed that the beads bound with control EVs showed no enrichment at all (Figure 7h). Next, we utilized patient sera with lower, intermediate, or higher virus titers to evaluate the virus enrichment and clearance efficiency of the extracellular vesicle‐bound beads. Of note, although the beads bound with ACE2‐EVs can clear serum samples with lower virus titers, the clearance efficiency for sera samples with intermediate or higher virus titers are very low; while the beads coupled with PM‐ACE2‐EVs performed well in all sera sample tested with much better clearance efficiency which was consistent with the fact that PM‐ACE2 bound beads showed much stronger ability to enrich virus (Figure 7i).
To further evalidate PM‐ACE2‐EVs mediated protection in infection of SARS‐CoV‐2, we pre‐injected human ACE2 (hACE2) transgenic mice with PM‐ACE2‐EVs or control EVs and then inoculated via the intranasal route with 105 50% tissue culture infectious dose (TCID50) SARS‐CoV‐2. Compared to control EVs‐treated mice, viral RNA in the lung, spleen, liver, and heart were significantly reduced in PM‐ACE2‐EVs‐treated mice (Figure 8a,b; Figure S8a, Supporting Information). Consistently, lower levels of several pro‐inflammatory cytokines and chemokines, including Ifnb1, Ifng, IL1b, IL‐6, Cxcl10 and Ccl2 were detected in bulk lung homogenates from mice treated with PM‐ACE2‐EVs compared with the counterparts treated with control EVs (Figure 8c). Hematoxylin‐and‐eosin staining of lung sections indicated a severe virus‐induced lung injury, characterized by thickened alveolar septa and infiltration of inflammatory cells in the alveolar spaces in control EVs‐treated hACE2 transgenic mice, whereas only minor lung damages were observed in PM‐ACE2‐EVs pretreated mice (Figure 8d). In ashcroft score analysis of lung fibrosis, the continuous numerical scale also revealed much lower degree of fibrotic changes in PM‐ACE2‐EVs pretreated mice (Figure S8b, Supporting Information). Notably, the expression of viral S and N proteins in lung, as determined by IHC and immunofluorescence respectively, were substantially reduced in PM‐ACE2‐EVs‐treated mice than in control EVs‐treated mice (Figure 8e,f). Together, these data suggest that PM‐ACE2‐EVs can protect mice against SARS‐CoV‐2 infections.
Figure 8.
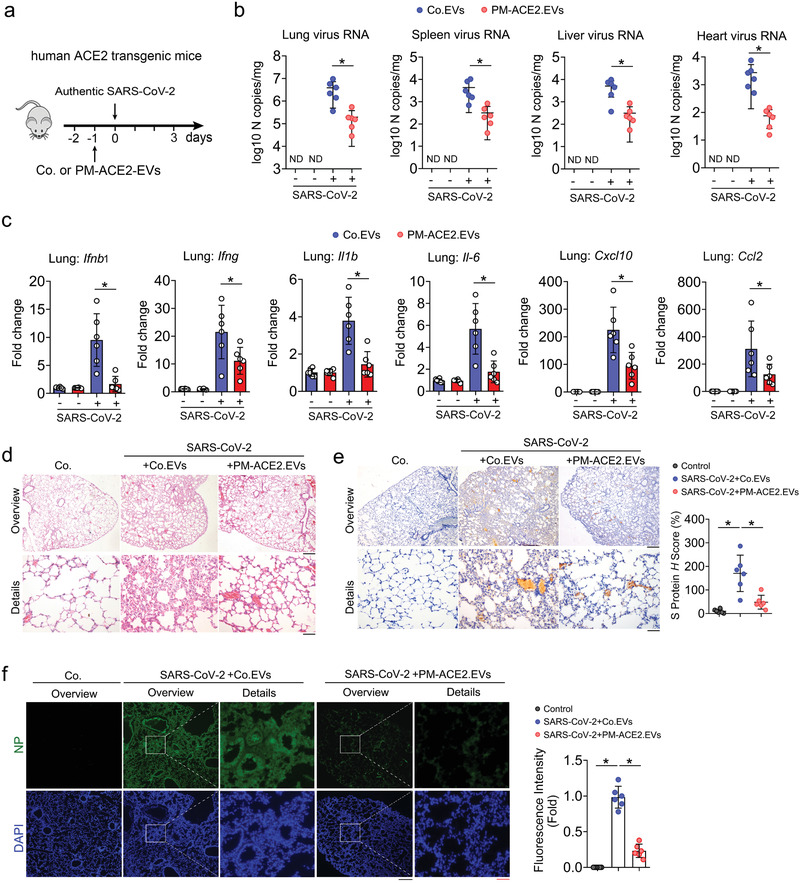
PM‐ACE2‐EVs display strong ability to enrich and neutralize authentic SARS‐CoV‐2. a) Neutralization of Con. EVs or PM‐ACE2‐EVs to authentic SARS‐CoV‐2 in hACE2 transgenic mice: six‐week‐old hACE2 transgenic mice were tail vein‐injected with Con. EVs or PM‐ACE2‐EVs (100 µg per day) at day ‐1, followed by intranasal inoculation of authentic SARS‐CoV‐2 (106 TCID50/50 µl) at the day 0. n = 6 for each group. b) Authentic SARS‐CoV‐2 burden at 3 dpi was determined in the lung, spleen, heart, and liver by RT‐qPCR. c) Relative fold changes in gene expression of cytokines and chemokines were calculated by qRT‐PCR in lung homogenates at 3 dpi from Con. EVs or PM‐ACE2‐EVs‐treated mice. n = 6 for each group. d) Hematoxylin and eosin staining analysis of tissue injury in lung sections from hACE2 transgenic mice pre‐treated with PBS, Con. EVs (100 µg per day), or PM‐ACE2‐EVs (100 µg per day) for one day and then intranasally inoculated with authentic SARS‐CoV‐2 (106 TCID50/50 µl). Scale bar, 500 µm (top) or 100 µm (bottom). e) Left panel: immunohistochemistry assay of the SARS‐CoV‐2 S protein antigen (brown) of lung sections from ACE2 transgenic mice as in (d). Scale bar, 500 µm (top) or 100 µm (bottom). Right panel: H score quantification of the S protein expressions. f) Left panel: representative immunofluorescence and DAPI staining of lung tissue from hACE2 mice as in (d). Scale bar, 500 µm (top, middle) or 100 µm (bottom). Right panel: fluorescence intensity quantification of the S protein expressions. *p < 0.05 (two‐tailed Student's t test (b,c,e, right; f, right). Data are representative of at least two independents with triplicates and shown as mean ± SD (b, c, e, right; f, right).
2.9. PM‐ACE2‐EVs Can be Used to Saturate the Viral S Protein as a Viral Trap
Above, we have identified the presence of ACE2 in EVs and reported ACE2 palmitoylation, palmitoylation‐acting enzymes, and their key roles in transporting ACE2 into EVs. By utilizing the above‐mentioned mechanisms, we have obtained engineered EVs such as PM‐ACE2‐EVs that are highly enriched with ACE2 on their surface. After verifying the efficacy of PM‐ACE2‐EVs against viral infections both in vitro and in vivo, we proposed a model by engineering PM‐ACE2‐EVs as a nano‐vesicular trap for SARS‐CoV‐2. PM‐ACE2‐EVs via their enhanced capacity to bind to the RBD of SARS‐CoV‐2's S protein can become saturated with virus, thereby suppressing viral association and attachment to cell surface ACE2 (Figure 9 ).
Figure 9.
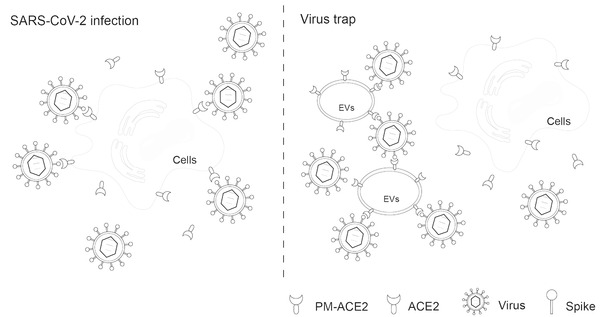
Proposed model of a trap for SARS‐CoV‐2 by engineered PM‐ACE2‐enriched EVs. Using PM‐ACE2‐EVs to saturate the viral S protein as a viral trap. Briefly, PM‐ACE2‐EVs can bind to the SARS‐CoV‐2 receptor binding domain (RBD) of the spike protein and block its interaction with cell surface ACE2, which blocks the binding of SARS‐CoV‐2‐RBD with the cell surface ACE2, inhibiting the infections of SARS‐CoV‐2. Thus, this study provides an engineering protocol of PM‐ACE2‐EVs as a novel EV‐based promising candidate for prophylactic and therapeutic treatment against COVID‐19.
3. Discussion
The S protein of SARS‐CoV‐2 has been previously reported to be vital for viral invasion of host cells by binding to ACE2 and promoting cellular entry. Considering the importance of the interaction between the S protein and ACE2, it has been proposed that preventing the S protein from recognizing ACE2 by either nanobodies or neutralizing antibodies may be an effective approach to block SARS‐CoV‐2 infection.[ 9 , 10 , 12 , 39 , 40 , 41 , 42 , 43 , 44 , 45 ] Indeed, many neutralizing antibodies against SARS‐CoV‐2 have been developed.[ 46 , 47 , 48 , 49 ]
However, RNA viruses can mutate to escape the effects of neutralizing antibodies through selection.[ 39 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 ] Therefore, effective ACE2 strategies could avoid the mutational escape of the virus and give a broad neutralizing activity. Recombinant soluble ACE2 has been shown to be sufficient for binding S protein and neutralizing SARS‐CoVs infection in cells and organoids.[ 58 ] Furthermore, a recent study has shown that an engineered trimeric ACE2 protein can bind the S protein with high affinity, to inhibit SARS‐CoV‐2.[ 59 ] However, many new drug candidates, such as proteins and nucleic acids, are highly unstable in the in vivo environment, posing a major challenge for the effectiveness of therapeutic outcomes. Therefore, ACE2 may not be an optimal inhibitor to block the early entry of SARS‐CoV‐2 infections in host cells. Due to their high stability in circulation as well as low toxicity and immunogenicity, EVs are a rapidly developing drug delivery platform for small molecules and nucleic acids in anti‐tumor treatments[ 60 , 61 ] and antiviral therapy,[ 62 , 63 , 64 ] demonstrating the safety and efficacy of extracellular vesicle delivery. Accordingly, it is conceivable that ACE2 anchored EVs interact with the RBD of SARS‐CoV‐2 S protein, thus they can potently inhibit the infection of SARS‐CoV‐2. Similar concept of research was reported using cellular nanosponges made of plasma membrane expressing ACE2 to pretreat SARS‐CoV‐2.[ 65 ] Importantly, we identified the natural existence of ACE2 in extracellular vesicle and developed a novel engineering EVs enriched with palmitoylated ACE2 for COVID‐19 therapy. Compared to soluble ACE2 protein, ACE2 anchored on the membrane of extracellular vesicles can bind the S protein with a much higher affinity. It is worth noting that ACE2 enriched EVs found in the blood, can more effectively and broadly block the interaction between S protein and host cell surface ACE2, thereby blocking the entry of SARS‐CoV‐2 into host cells.
Palmitoylation is a reversible lipid modification made with a thioester bond between an amino acid residue, typically cysteine, and a 16‐carbon fatty acid called palmitate.[ 66 , 67 , 68 ] Palmitoylation regulates the trafficking and membrane localization of proteins.[ 68 , 69 , 70 , 71 ] We have demonstrated, for the first time, that ACE2 can be palmitoylated, and that this mechanism controls ACE2 membrane localization and extracellular vesicle secretion. Screening of a small inhibitor compound library identified that 2‐bromopalmitate (2‐BP), a general palmitoylation inhibitor, reduced ACE2 levels in the EVs by disrupting the membrane localization of ACE2. Conversely, enhanced ACE2 palmitoylation by the selective inhibitor of acyl protein thioesterase 1 (APT1) increased EV‐ACE2 secretion. These findings suggest that palmitoylation regulates the extracellular vesicle secretion of ACE2. By employing both the acyl–biotin exchange (ABE) assay and the Click‐iT assay, we confirmed that ACE2 can be strongly palmitoylated at Cys141 and Cys498. Moreover, substitution of the Cys141 and Cys498 residues by serine completely abolished the palmitoylation of ACE2, resulting in a disruption of ACE2 membrane localization and diminished ACE2 secretion into the EVs. Furthermore, we identified palmitoyltransferase ZDHHC3 as the acetyltransferase responsible for ACE2 palmitoylation and showed that silencing ZDHHC3 efficiently reduced the levels of ACE2 in EVs.
Taking advantage of the essential role of palmitoylation in transporting ACE2 into EVs, we fused the S‐palmitoylation‐dependent PM targeting sequence to the N‐terminus of the ACE2 protein and constructed MSCs that expressed palmitoylated ACE2. Compared with the EVs from control cells, the forced fusion of the PM targeting sequence facilitated the accumulation of ACE2 in EVs (PM‐ACE2‐EV). Moreover, most of the ACE2 proteins were found to be located on the surface of PM‐ACE2‐EVs. Considering that EVs are approximately the same size as SARS‐CoV‐2, we tested the potential of using PM‐ACE2‐EVs as a viral trap. As expected, PM‐ACE2‐EVs can bind to the RBD of the S protein, thus blocking its interaction with cell surface ACE2. Compared to the control EVs, PM‐ACE2‐EVs exhibited a much higher neutralization potency against pseudotyped SARS‐CoV‐2. By using transgenic mice bearing human ACE2, we verified that PM‐ACE2‐EVs also significantly diminished infections of pseudotyped SARS‐CoV‐2 in vivo. These results demonstrate the versatility of engineered EVs and their outstanding physicochemical properties in neutralizing SARS‐CoV‐2. Furthermore, by employing PM‐ACE2‐EVs and beads/microspheres that enrich EVs, we proposed an innovative approach to reduce titers of SARS‐CoV‐2, which would help to reduce the virus load and thus contribute to the treatment of COVID‐19 patients.
To our knowledge, the current study reports the first EV‐based engineering technology for preventing SARS‐CoV‐2. Flexible and efficient strategies could improve the antiviral efficacy while minimizing the production cost, dose, and potential toxicity for clinical application. Our findings not only identify a novel palmitoylation‐dependent mechanism by which human cells can secrete ACE2 into EVs, but also provides an engineering protocol for PM‐ACE2‐EVs as a novel EV‐based candidate for prophylactic and therapeutic treatment against COVID‐19. We hope that this extracellular vesicle technology presented here will contribute to curbing the current COVID‐19 pandemic, as well as future disease outbreaks.
4. Experimental Section
Cell Culture
HEK293T, HeLa, MCF7, Vero‐E6, and A549 cells were purchased from ATCC and cultured in DMEM supplemented with 10% FBS (Cat No. FSP500, ExCell Bio) and 1% penicillin–streptomycin. Bone marrow‐derived human mesenchymal stem cells (hMSC) were from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Single cell suspension of mouse mesenchymal stem cells (mMSC) were obtained from bone medullary cavity of femurs and tibias with 0.5% FBS in PBS and filtered by passing through 70 µm cell strainer. The MSCs were cultured in complete growth medium with 10% FBS and 1% penicillin–streptomycin. All cell lines were free of mycoplasma and were cultured at 37 °C under 5% CO2.
Animal Studies
Mice experiments were approved by the Committee for Animal Welfare in Soochow University. For the animal experiments, specific pathogen‐free, 6‐week‐old, male and female transgenic human ACE2‐transgenic mice were purchased from the Shanghai Model Organisms Center (Shanghai, China). For the in vivo pseudovirus infection blocking experiments, six‐weeks‐old hACE2 transgenic mice were tail vein‐injected with control EVs or PM‐ACE2 EVs (100 µg per mice) at day ‐1, followed by intranasal inoculation of SARS‐CoV‐2‐GFP or SARS‐CoV‐2‐luci pseudovirus (106 TCID50/50 µL) at the day 0 (n = 5 for each group). Bioluminescent reporter imaging was used to monitor the development of infection and detect the differences of lungs among each group by IVIS spectrum in vivo imaging system. Mice were dissected at 2 days post infection to detect pseudotyped virus infection and histopathological changes by bioluminescent imaging and immunohistochemistry staining. For the in vivo authentic SARS‐CoV‐2 infection blocking experiments, six‐weeks‐old hACE2 transgenic mice were tail vein‐injected with control EVs or PM‐ACE2 EVs (100 µg per mice) at day ‐1, followed by intranasal inoculation of authentic SARS‐CoV‐2 (106 TCID50/50 µl) at the day 0 (n = 6 for each group). Mice were dissected at 3 days post infection to detect authentic virus infection and histopathological changes. All the authentic SARS‐CoV‐2‐related experiments were approved by the Ethics Committee of ZSSOM of Sun Yat‐sen University on Laboratory Animal Care and conducted in a Biosafety Level 3 (BSL‐3) facility. All animal studies were conducted in accordance with the guidelines of Soochow University or Laboratory Monitoring Committee of Guangdong Province of China.
EV Separation, Concentration, and Characterization
As previously described,[ 72 , 73 ] to obtain EVs, cells were seeded at a density of 1.2 × 107 cells/dish and cultured in DMEM supplemented with 10% EV‐depleted FBS (FBS‐derived extracellular vesicles were removed by centrifugation at 120 000 × g overnight) for 48–72 h. 200 mL cell culture supernatants were collected and subjected to sequential centrifugation steps: 500 × g for 10 min at 4 °C to remove floating and dead cells; 2000 × g at 4 °C for 20 min to remove dead cell‐derived residue; 10 000 × g at 4 °C for 1 h to remove microvesicles. The supernatants were then centrifuged at 120 000 × g at 4 °C for 2 h or overnight to concentrate EVs suspended in medium. After that, the most supernatants were discarded and the medium at the bottom of ultra tube was resuspended by PBS (pH 7.4, 0.22 µm filtered), followed by the centrifugation at 120 000 × g at 4 °C for another 2 h. Next, the whole supernatants were poured away slowly and the EVs adhered in the bottom of ultra tube were resuspended by 400 uL PBS (pH 7.4) softly. For immunoblot analysis, the EVs were lysed by 8 m urea buffer with 2.5% SDS for 0.5 h at room temperature and the concentration of total proteins was quantified by BCA Protein Assay Kit (Thermo Scientific).
The EVs purified from cell culture supernatants were analyzed by NanoSight NS500 (Malvern Instruments, Amesbury, UK), which is equipped with fast video capture and Nanoparticle tracking analysis (NTA) software.
All relevant data of the authors’ experiments were submitted to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV210209).[ 74 ]
Lentiviral Transduction and the Generation of Stable Cell Lines
Lentiviruses were prepared by co‐transfecting HEK293T cells with pLKO.1 plasmids and the three packaging plasmids pCMV‐VSVG, pMDLg‐RRE (gag/pol), and pRSV‐REV. After 48h transfection, the cell supernatants were harvested.
To obtain stable cell lines, cells were infected with supernatants with 5 ng mL−1 polybrene (Sigma). After 48 h, infected cells were cultured under selection with puromycin (2 µg mL−1) for three days. The validated lentiviral shRNAs were obtained from Sigma–Aldrich (MISSION shRNA).
Human Rab27a shRNAs: TRCN0000279985 (#1) and TRCN0000279982 (#2); human ZDHHC3 shRNAs: TRCN0000133710 (#1) and TRCN0000162097 (#2); human ZDHHC3 shRNA: TRCN0000299950.
Primers and Reagents
Sequences of siRNAs used in this study are listed in Table S1, Supporting Information. Primer sequences used for qRT–PCR are listed in Table S2, Supporting Information. ACE2 were subcloned from pcDNA3.1 recombinant plasmid (provided by Prof. Wang PH, Shandong University, China) into the pLV‐C‐Flag bc puro lentivirus vector and pLVX‐C‐AcGFP‐N1 vector. Point mutations were generated by the site‐directed mutagenesis with KOD plus (Toyobo) polymerase. All constructs were confirmed by DNA sequencing. Anti‐cancer component library were from SelleckChem. 2‐bromopalmitate (2BP) was from Sigma (#18263‐25‐7); ML348 was from MCE (HY‐100736); Hieff Trans in vitro siRNA/miRNA Transfection Reagent (Cat No. 40806; Yeasen, Shanghai, China) 17‐ODYA was from APExBIO (# 34450‐18‐5); and GW4869 was from MCE (HY‐19363).
Real‐Time RT‐PCR (qRT‐PCR)
Total RNAs were extracted from tissues homogenates of organs using the RNAiso Plus (9109, Takara), and reverse transcription was performed using the HiScript II Q RT SuperMix (R223‐01, Vazyme). Real‐time PCR was performed with ChamQ Universal SYBR qPCR Master Mix (Q711‐02, Vazyme). All target gene expression levels were normalized to GAPDH.
Primers sequences targeting N gene of SARS‐CoV‐2 were as follows:
SARS‐CoV‐2‐N‐F: 5′‐GGGGAACTTCTCCTGCTAGAAT‐3′,
SARS‐CoV‐2‐N‐R: 5′‐CAGACATTTTGCTCTCAAGCTG‐3′.
Primers sequences targeting SARS‐CoV‐2 S gene were as follows:
SARS‐CoV‐2‐S‐RBD‐qF1: 5′‐CAATGGTTTAACAGG CACAGG‐3′,
SARS‐CoV‐2‐S‐RBD‐qR1: 5′‐CTCAAGTGTCTGTGGATCACG‐3′.
Flow Cytometry Analysis
To detect the level of ACE2 on cell membrane surface, cells were prepared according to standard protocols and suspended in 2% FBS with PBS. Anti‐human ACE2 antibody (CL488‐66699, Proteintech) was used for ACE2 detection. To detect the level of ACE2+ EV, EVs were incubated with aldehyde/sulphate latex beads (4 µm, Invitrogen, no.1743119) in PBS for 1h at 4 °C. EV‐bound beads were incubated with anti‐ACE2 FITC‐conjugated antibody (CL488‐66699, Proteintech) for 30 min. The percentage of positive beads referred to as the percentage of ACE2+ EVs was calculated relative to the total number of beads analyzed per sample. All samples were analyzed with Beckman CytoFlex (Beckman) or FACSAria II (Becton Dickinson) machines. FACS data were analyzed with CytExpert software and FlowJo (TreeStar).
Immunoprecipitation and Immunoblotting
Briefly, cells were lysed with lysis buffer (20 mm Tris‐HCl pH 7.4, 2 mm EDTA, 25 mm NaCl, 10% glycerol and 1% NP40) containing protease inhibitors cocktail (MCE, No. HY‐K0010) for 15 min at 4 °C. After centrifugation at 12 000 rpm for 5 min, the protein concentrations were measured using BCA kit, and equal amounts of proteins were incubated with anti‐FLAG M2‐AGAROSE beads (Sigma, A2220) or specific antibodies and protein A/G‐Sepharose (Santa Cruz, sc‐2003) for immunoprecipitation. The precipitants were washed with same lysis buffer and eluted with sample buffer for 5 min at 100 °C. Immunoblot (IB) analysis was performed with specific primary antibodies overnight at 4 °C, followed by secondary peroxidase‐conjugated antibodies at RT for 1h.
To detect the level of cell surface ACE2, cells were biotinylated for 30 min at 4 °C and the biotinylated ACE2 on the cell surface was precipitated with streptavidin beads and analyzed by immunoblotting. For the biotinylation internalization/recycling assay, cell surface ACE2 were biotinylated for 30 min at 4 °C and then incubated at 37 °C for the indicated times. The biotinylated ACE2 on cell surface or in EVs were precipitated with streptavidin beads and subjected to immunoblotting analysis.
The following antibodies were used for immunoprecipitation, IB, and immunofluorescence: ACE2 (A4612, ABclonal, 1:2000 for IB, 1:200 for IF), ACE2 (sc‐390851, Santa Cruz, 1:2000 for IB, 1:200 for IF), ACE2 (ab‐15348, Abcam, 1:2000 for IB, 1:200 for IF), Spike (ab273433, Abcam, 1:2000 for IB, 1:200 for IF), Spike (A20022, ABclonal, 1:2000 for IB, 1:200 for IF), ZDHHC3 (ab‐31837, Abcam, 1:2000 for IB, 1:200 for IF), LYPLA1 (A4419, ABclonal, 1:2000 for IB), TSG101 (sc‐7964, Santa Cruz Biotechnology, 1:1000 for IB), Alix (sc‐53540, Santa Cruz Biotechnology, 1:1000 for IB, 1:100 for IF), Hrs (A1790, ABclonal Technology, 1:2000 for IB), Rab27a (sc‐74586, Santa Cruz Biotechnology, 1:1000 for IB), CD9 (A19027, ABclonal, 1:2000 for IB), CD63 (ab216130, Abcam, 1:2000 for IB), CD81(sc‐166029, Santa Cruz Biotechnology, 1:1000 for IB), GAPDH (G041, Applied Biological Materials, 1:5000 for IB), β‐actin (#A5441, Sigma, 1:10 000 for IB), polyclonal Myc (A‐14) (sc‐789, Santa Cruz Biotechnology, 1:1000 for WB), monoclonal Myc (9E10) (sc‐40, Santa Cruz Biotechnology, 1:1000 for WB), Flag (M2, Sigma, 1:10 000 for IB, 1:200 for IF), FITC Goat Anti‐Rabbit IgG (AS001, ABclonal, 1:200 for IF) or Rhodamine Goat Anti‐Rabbit IgG (AS040, ABclonal, 1:200 for IF).
Immunofluorescence
Briefly, cells cultured on chamber slides were washed with PBS, fixed with 4% PFA Fix Solution for 20 min, permeabilized with 0.5% Triton X‐100. Cells were then blocked with 3% bovine serum albumin (BSA) containing 0.1% Tween 20, and incubated with different specific antibodies at 4 °C overnight, followed by incubation with fluorescent‐dye‐conjugated secondary antibodies at RT for 1h. The cell nuclei were stained with DAPI (Sigma).
Immunogold Labeling and Electron Microscopy
As previously described,[ 72 , 73 ] purified EVs suspended in PBS were dropped on formvar carbon‐coated nickel grids with glow discharge treatment. Grids were stained with 2% uranyl acetate and observed using a transmission electron microscope (JEM‐1011). For immunogold labeling, the grids were blocked with 3% BSA at RT for 1h, incubated with ACE2 antibody (A4612, ABclonal, 1:200), followed by incubation with the secondary antibody conjugated with anti‐rabbit gold antibody (colloidal gold 10 nm, Sigma, G7402). The grids were washed three times with PBS after each staining step before uranyl acetate staining.
Subcellular Fractionations Assays
Subcellular fractionations assay was performed with cell fractionation kit (Cell Signaling Technology, #9038). After treating with 0.1% DMSO or 2BP (100 µm) overnight, wild type Vero‐E6 cells were harvested with 0.5 mL cold PBS, and 100 µL cell suspension was aliquoted into a 1.5 mL tube with SDS loading buffer and treated by sonicate and centrifugation. The supernatant of which was the whole cell lysate (total). The remaining 400 µL cell suspension was centrifuged and resuspended by cytoplasm isolation buffer (CIB). After incubation on ice and centrifugation, the supernatant was collected as cytoplasmic fraction and pellet was resuspended with membrane isolation buffer (MIB). After incubation on ice and higher speed centrifugation, the supernatant was collected as membrane fraction. All experiments were performed according to the manufacturer's instructions.
Acyl–Biotin Exchange Palmitoylation Assay
The Acyl‐Biotin exchange palmitoylation assay was performed using a previously reported method.[ 75 ] In brief, HEK293T cells transiently expressing Flag‐tagged ACE2 were harvested 48 h after transfection and washed with cold PBS. For the ZDHHCs‐knockdown experiments, the siRNAs were transfected 24 h prior to the transfections of the ACE2‐Flag constructs. Prior to cell lysis, N‐ethylmaleimide (NEM, E3876, Sigma‐Aldrich) was dissolved in 100% EtOH and added to the lysis buffer of PH 7.4 (50 mm Tris‐HCl pH 7.4, 1% NP‐40, 150 mm NaCl, 10% Glycerol) at the final concentration of 50 mm. Cells were then suspended in NEM containing lysis buffer for 1 h at 4 °C and the supernatants were incubated with anti‐Flag beads (Sigma, A2220) at 4 °C for 1h. After incubation, the beads were washed three times with lysis buffer of pH 7.4 and three times with lysis buffer of pH 7.2. Then, beads were incubated with a hydroxylamine (HAM) buffer at RT for 1 h, washed three times with lysis buffer of pH 7.2, and three times with lysis buffer of pH 6.2. Subsequently, beads were treated with 2 µm Biotin‐BMCC (21 900, Thermo Fisher Scientific) in lysis buffer of pH 6.2 at 4 °C for 1 h. The immunoprecipitate samples were analyzed by Western blot using anti‐Flag M2‐antibody (Sigma, 1:10 000) and streptavidin‐HRP (21 130, Thermo Fisher Scientific).
For mass spectrometry of palmitoylated ACE2, HEK293T cells transfected with hACE2‐Flag were harvested and followed with purification by immunoprecipitation with anti‐Flag beads. The free sulfhydryl were blocked by N‐ethylmaleimide, and the palmitoylation of palmitoylated cysteine residues was cut off by 1 m hydroxylamine (HAM) and alkylated with 4 mm iodoacetamide (Sigma). Then, the proteins were digested with trypsin.
Click‐iT Identification of ACE2 Palmitoylation
48 h after the transfection of ACE2‐WT or ACE2 mutants into HEK293T cells, 100 µm 17‐ODYA (17‐Octadecynoic acid, B6373, APExBIO) was added to the cell medium for 12 h or overnight. Cells were lysed with lysis buffer (1% NP‐40, 50 mm Tris‐HCl pH 7.5, 150 mm NaCl, 10% Glycerol) containing protease and phosphatase inhibitors (HY‐K0010, HY‐K0023, MCE). The lysate was sonicated to disperse the DNA and centrifuged afterwards. Then, the supernatant was transferred to a clean 1.5 mL microcentrifuge tube. The following experiments were performed according to the manual using Click‐iT protein reaction buffer kit (C10276, Invitrogen). Subsequently, the complex of biotin alkyne–azide–plamitic‐protein was immunoprecipitated by streptavidin beads. The immunoprecipitate samples were analyzed by western blot.
Mass Spectrometry
SDS‐PAGE gels were minimally stained with Coomassie brilliant blue, cut into one range that contains ACE2 according to the protein marker, and digested with trypsin. Eluted peptides from the SDS‐PAGE gel were identified on a Thermo Fisher QE‐HFX mass spectrometer. Spectral data were then analyzed by Proteome Discovery software and searched from the human protein RefSeq database in Uniprot.
Surface–Plasmon Resonance (SPR)
The interaction between EVs and COVID‐19 Spike‐RBD protein or SARS‐CoV‐2 pseudovirus was detected by the Biacore T200 System (GE Healthcare). COVID‐19 Spike‐RBD protein (40592‐VNAH, Sino Biological. molecular weight: 25.1 kDa, theoretical pI: 8.89) was dissolved at a concentration of 0.25 mg mL−1 in pH 7.4 PBS (0.22 µm filtered). The concentration of RBD protein was diluted to 20 µg mL−1, respectively with 10 mm sodium acetate at different pH (pH 6.0, pH 5.0, pH 4.0) to conduct the pre‐concentration of CM5 sensor chip. The CM5 sensor chip surface was activated by using sulpho‐NHS/EDC chemistry and prepared to a target immobilization level of 3000 RU with 20 ng mL−1 COVID‐19 Spike‐RBD protein in 10 mm sodium acetate (pH 5), and blocked the CM5 chip surface by 1 m ethanolamine (pH 8.0). The Con. EVs, ACE2‐EVs, and PM‐ACE2‐EVs in the same particle concentration were flowed at the rate of 10 µL min−1 for 1200 s to allow for the association. The capture surface was regenerated by 50 mm NaOH. SARS‐CoV‐2 pseudovirus (GENEWIZ, China) was diluted at a concentration of 106 particles per mL with 10 mm sodium acetate at different pH (pH 6.5, pH 6.0, pH 5.5) to conduct the pre‐concentration of CM5 sensor chip. The CM5 sensor chip surface was activated by using sulpho‐NHS/EDC chemistry and prepared to a target immobilization level of 3000 RU with SARS‐CoV‐2 pseudovirus in 10 mm sodium acetate (pH 6.5), and M5 chip surface blocked by 1 m ethanolamine (pH 8.0). The ACE2‐EVs and PM‐ACE2‐EVs in the same particle concentration were flowed at the rate of 5 µL min−1 for 1200 s to allow for the association. The capture surface was regenerated by regeneration buffer (10% isopropanol, 5 mm NaOH in pH 7.5 PBS).
Production of SARS‐CoV‐2 Pseudovirus
To generate SARS‐CoV‐2‐GFP pseudovirus (GENEWIZ), HEK293T cells were cotransfected with a plasmid encoding codon‐optimized SARS‐CoV‐2 S protein (1‐3819) (pMD2.S), a green fluorescent protein (GFP) vector (pLenti‐CMV‐coGFP), and psPAX by using polyethyleneimine. To generate SARS‐CoV‐2‐luci pseudovirus (Genscript), HEK293T cells were cotransfected with pMD2.S encoding SARS‐CoV‐2 S protein, luciferase vector (pLVx‐luci), gap/pol, and pRSV‐REV by using polyethyleneimine. After transfection for 8 h, the medium was replaced with new complete culture medium. 48 h after transfection, the supernatant containing SARS‐CoV‐2‐GFP pseudovirus or SARS‐CoV‐2‐luci pseudovirus were collected, clarified by 0.45‐µm pore‐size filter, concentrated by centrifugation for 2 h at 30 000 g and stored in 200 uL at aliquots at −80 −°C until use. The pseudovirus titer was tested with Lenti‐X p24 Rapid Titer Kit (Clontech, No.632200).
SARS‐CoV‐2 Pseudovirus Neutralization Assay
For SARS‐CoV‐2 pseudovirus neutralization assay, serially diluted EVs and SARS‐CoV‐2‐GFP pseudovirus (GENEWIZ, China) or SARS‐CoV‐2‐luci pseudovirus (Genscript, China) were added to 48‐well culture plates containing 5 × 104 cells. The cells were then maintained at 37 °C with 5% CO2 for 48 h. For SARS‐CoV‐2‐luci pseudovirus neutralization assay, the obtained relative fluorescent were normalized to the negative control wells and used to calculate the percentage of neutralization of each concentration, and the luciferase activity of each group was measured with a PerkinElmer luminometer. For SARS‐CoV‐2‐GFP pseudovirus neutralization assay, GFP‐positive cells were detected by fluorescent inverted microscope (CKX53, Olympus). Cells without virus infection and EVs treatment were used as blank controls, and cells with virus infection but without EVs treatment were used as virus controls. Percent neutralization was calculated as (Sample signals – Blank control signals) / (Virus control signals – Blank control signals) × 100%. Data were fitted using a three‐parameter logistic model in GraphPad Prism 7.
Authentic SARS‐CoV‐2 Virus Inhibition or Authentic SARS‐CoV‐2 Neutralization
The authentic SARS‐CoV‐2 virus inhibition assay was performed according to a method previously reported.[ 76 ] Briefly, Vero‐E6 cells were passaged at a cell number of 3 × 104 per well in 96‐well plate. After incubating the cells for 18 h, 100 TCID50 of SARS‐CoV‐2 was incubated with serially diluted EVs at 37 °C for 1 h. Then, cells were infected with the EV‐virus mixtures and cultured for another 72 h. Cells were stained with crystal violet and absorbance at 570 nm/630nm was measured. Supernatants at 3 days post infection were collected to detect viral RNA titer.
Enzyme‐Linked Immunosorbent Assay (ELISA)
The analysis of ACE2 concentration in culture supernatants was performed according to the manual of ELISA Kits (ab235649, abcam). Briefly, EVs were added to the plate followed by the addition of anti‐hACE2 antibody cocktails which contained capture antibody and detector antibody. The plate was incubated for 1 h at RT on a plate shaker at 450 rpm. After washing steps, TMB substrate was added and incubated for 15 min. The chromogenic reaction was quantified following the addition of stop solution. The absorbance of the samples was measured at 450 nm.
The binding capacity of EV‐ACE2 against spike glycoprotein (RBD) was measured by SARS‐CoV‐2 surrogate virus neutralization test kit (GenScript, China).[ 77 ] Briefly, EVs were pre‐incubated with the HRP‐RBD to allow the binding of EVs to HRP‐RBD. Then the mixture was added to the plate pre‐coated with human ACE2 protein. After washing steps, TMB substrate (Invitrogen) was added to each well and the plate was incubated in the dark at ≈20–25 °C for 15 min. The chromogenic reaction was quantified following the addition of stop solution (KPL SeraCare). The absorbance of the samples was measured at 450 nm. All experiments were performed according to the manufacturer's instructions.
Histological Analysis
For routine histology, the tissue paraffin sections were stained with H&E. For IHC, lung tissues sections were incubated with the primary antibodies to Spike (ab273433, Abcam, 1:1000), Spike (A20022, ABclonal, 1:1000).[ 78 ] The quantification of ACE2 protein expression was obtained by measuring the H score. The H‐score was calculated as: the formula 3 × the percentage of strongly staining cells + 2 × the percentage of moderately staining cells + the percentage of weakly staining cells, yielding an H‐score range of 0 to 300. For IF, after re‐hydration and antigen retrieval, tissue sections were blocked by 3% BSA for 1 h, and then stained with specific antibodies, followed by incubation with secondary antibodies. Nuclei were stained with DAPI.
Quantitative Histologic Analysis
To detect the quantitative histologic evaluation of virus‐induced fibrotic changes, a numerical fibrotic scale (Ashcroft scale[ 79 ]) was measured as follows. Briefly, the grade of lung fibrosis was scored on a scale of 0 (normal) to 8 (total fibrosis) using a predetermined scale of severity (numerical fibrotic scale). After observing 25 fields in each lung section at a magnification of 100×, the mean score of all the fields was obtained as the fibrosis score in each sample. The histologic analysis strictly followed the blind principle.
Statistical Analyses
Statistical analyses between or among groups were performed with a two‐tailed Student's t test or two‐way ANOVA using Office Excel (Microsoft) and Prism 7.0 software (GraphPad). p value < 0.05 is was considered statistically significant in the Figures (*p < 0.05, n.s., not significant). Data are analyzed of three independent experiments.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
F.X., P.S., and T.P. contributed equally to this work. F.X. and F.Z. designed the experiments and analyzed the data; F.X., S.P., T.P., H.L., X.Z., and H.H. performed the experiments; A.W., F.W., J.H., H.Y., and L.Z. provided valuable discussion; F.X., L.Z., and F.Z. wrote the manuscript.
Supporting information
Supporting Information
Supporting Table 1
Supporting Table 2
Acknowledgements
The authors thank Zhengli Shi (CAS Key Laboratory of Special Pathogens and Biosafety, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China), Pei‐Hui Wang (Jinan Infectious Diseases Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China) for reagents. The current work was supported by the Chinese National Natural Science Funds (U20A201376, 82041009, 3171232, 31701234, 31871405, 91753139, 31925013, 31671457, 31571460, 31870902, 32070907 and 31871405), Jiangsu National Science Foundation (BK20180043), the Science and Technology Plan Project of Suzhou (SYS2019020), National Postdoctoral Program for innovative Talents (BX201700165), A project Funded by the Priority Acadamic Program Development of Jiangsu Higher Education Institutions.
Xie F., Su P., Pan T., Zhou X., Li H., Huang H., Wang A., Wang F., Huang J., Yan H., Zeng L., Zhang L., Zhou F., Engineering Extracellular Vesicles Enriched with Palmitoylated ACE2 as COVID‐19 Therapy. Adv. Mater. 2021, 33, 2103471. 10.1002/adma.202103471
Contributor Information
Long Zhang, Email: L_Zhang@zju.edu.cn.
Fangfang Zhou, Email: zhoufangfang@suda.edu.cn.
Data Availability Statement
Research data are not shared.
References
- 1. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature 2020, 579, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med. 2020, 382, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coronaviridae V., Nat. Microbiol. 2020, 5, 536.32123347 [Google Scholar]
- 4. Fung T. S., Liu D. X., Annu. Rev. Microbiol. 2019, 73, 529. [DOI] [PubMed] [Google Scholar]
- 5. Masters P. S., Adv. Virus Res. 2006, 66, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu D. X., Fung T. S., Chong K. K., Shukla A., Hilgenfeld R., Antiviral Res. 2014, 109, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmann M., Kleine‐Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A., Muller M. A., Drosten C., Pohlmann S., Cell 2020, 181, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li W., Moore M. J., Vasilieva N., Sui J., Wong S. K., Berne M. A., Somasundaran M., Sullivan J. L., Luzuriaga K., Greenough T. C., Choe H., Farzan M., Nature 2003, 426, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang C., Li W., Drabek D., Okba N. M. A., van Haperen R., Osterhaus A., van Kuppeveld F. J. M., Haagmans B. L., Grosveld F., Bosch B. J., Nat. Commun. 2020, 11, 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X., Li R., Pan Z., Qian C., Yang Y., You R., Zhao J., Liu P., Gao L., Li Z., Huang Q., Xu L., Tang J., Tian Q., Yao W., Hu L., Yan X., Zhou X., Wu Y., Deng K., Zhang Z., Qian Z., Chen Y., Ye L., Cell Mol. Immunol. 2020, 17, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang S., Hillyer C., Du L., Trends Immunol. 2020, 41, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiang Y., Nambulli S., Xiao Z., Liu H., Sang Z., Duprex W. P., Schneidman‐Duhovny D., Zhang C., Shi Y., Science 2020, 370, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R. E., Acton S., Circ. Res. 2000, 87, E1. [DOI] [PubMed] [Google Scholar]
- 14. Turner A. J., Hooper N. M., Trends Pharmacol. Sci. 2002, 23, 177. [DOI] [PubMed] [Google Scholar]
- 15. Wysocki J., Ye M., Rodriguez E., Gonzalez‐Pacheco F. R., Barrios C., Evora K., Schuster M., Loibner H., Brosnihan K. B., Ferrario C. M., Penninger J. M., Batlle D., Hypertension 2010, 55, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stein J. M., Luzio J. P., Biochem. J. 1991, 274, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cocucci E., Meldolesi J., Trends Cell Biol. 2015, 25, 364. [DOI] [PubMed] [Google Scholar]
- 18. Thery C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin‐Smith G. K., Ayre D. C., Bach J. M., Bachurski D., Baharvand H., Balaj L., Baldacchino S., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Bedina Zavec A., Benmoussa A., Berardi A. C., Bergese P., Bielska E., Blenkiron C., Bobis‐Wozowicz S., Boilard E., Boireau W., Bongiovanni A., et al., J. Extracell. Vesicles 2018, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thery C., Zitvogel L., Amigorena S., Nat. Rev. Immunol. 2002, 2, 569. [DOI] [PubMed] [Google Scholar]
- 20. Valadi H., Ekstrom K., Bossios A., Sjostrand M., Lee J. J., Lotvall J. O., Nat. Cell Biol. 2007, 9, 654. [DOI] [PubMed] [Google Scholar]
- 21. Thakur B. K., Zhang H., Becker A., Matei I., Huang Y., Costa‐Silva B., Zheng Y., Hoshino A., Brazier H., Xiang J., Williams C., Rodriguez‐Barrueco R., Silva J. M., Zhang W., Hearn S., Elemento O., Paknejad N., Manova‐Todorova K., Welte K., Bromberg J., Peinado H., Lyden D., Cell Res. 2014, 24, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Q., Jeppesen D. K., Higginbotham J. N., Franklin J. L., Crowe J. E. Jr., Coffey R. J., Gastroenterology 2021, 160, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cocozza F., Nevo N., Piovesana E., Lahaye X., Buchrieser J., Schwartz O., Manel N., Tkach M., Thery C., Martin‐Jaular L., J. Extracell. Vesicles 2020, 10, e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El‐Andaloussi S., Lee Y., Lakhal‐Littleton S., Li J., Seow Y., Gardiner C., Alvarez‐Erviti L., Sargent I. L., Wood M. J., Nat. Protoc. 2012, 7, 2112. [DOI] [PubMed] [Google Scholar]
- 25. Ostrowski M., Carmo N. B., Krumeich S., Fanget I., Raposo G., Savina A., Moita C. F., Schauer K., Hume A. N., Freitas R. P., Goud B., Benaroch P., Hacohen N., Fukuda M., Desnos C., Seabra M. C., Darchen F., Amigorena S., Moita L. F., Thery C., Nat. Cell Biol. 2010, 12, 19. [DOI] [PubMed] [Google Scholar]
- 26. Rana M. S., Kumar P., Lee C. J., Verardi R., Rajashankar K. R., Banerjee A., Science 2018, 359, eaao6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Resh M. D., Methods 2006, 40, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adibekian A., Martin B. R., Chang J. W., Hsu K. L., Tsuboi K., Bachovchin D. A., Speers A. E., Brown S. J., Spicer T., Fernandez‐Vega V., Ferguson J., Hodder P. S., Rosen H., Cravatt B. F., J. Am. Chem. Soc. 2012, 134, 10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q., Science 2020, 367, 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren J., Wen L., Gao X., Jin C., Xue Y., Yao X., Protein Eng., Des. Sel. 2008, 21, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. E. L. A. S., Mager I., Breakefield X. O., Wood M. J., Nat. Rev. Drug Discovery 2013, 12, 347. [DOI] [PubMed] [Google Scholar]
- 32. Kamerkar S., LeBleu V. S., Sugimoto H., Yang S., Ruivo C. F., Melo S. A., Lee J. J., Kalluri R., Nature 2017, 546, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fukata Y., Dimitrov A., Boncompain G., Vielemeyer O., Perez F., Fukata M., J. Cell Biol. 2013, 202, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gimona M., Pachler K., Laner‐Plamberger S., Schallmoser K., Rohde E., Int. J. Mol. Sci. 2017, 18, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pachler K., Lener T., Streif D., Dunai Z. A., Desgeorges A., Feichtner M., Oller M., Schallmoser K., Rohde E., Gimona M., Cytotherapy 2017, 19, 458. [DOI] [PubMed] [Google Scholar]
- 36. Mendt M., Kamerkar S., Sugimoto H., McAndrews K. M., Wu C. C., Gagea M., Yang S., Blanko E. V. R., Peng Q., Ma X., Marszalek J. R., Maitra A., Yee C., Rezvani K., Shpall E., LeBleu V. S., Kalluri R., JCI Insight. 2018, 3, e99263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bao L., Deng W., Huang B., Gao H., Liu J., Ren L., Wei Q., Yu P., Xu Y., Qi F., Qu Y., Li F., Lv Q., Wang W., Xue J., Gong S., Liu M., Wang G., Wang S., Song Z., Zhao L., Liu P., Zhao L., Ye F., Wang H., Zhou W., Zhu N., Zhen W., Yu H., Zhang X., et al., Nature 2020, 583, 830. [DOI] [PubMed] [Google Scholar]
- 38. Hassan A. O., Case J. B., Winkler E. S., Thackray L. B., Kafai N. M., Bailey A. L., McCune B. T., Fox J. M., Chen R. E., Alsoussi W. B., Turner J. S., Schmitz A. J., Lei T., Shrihari S., Keeler S. P., Fremont D. H., Greco S., McCray P. B. Jr., Perlman S., Holtzman M. J., Ellebedy A. H., Diamond M. S., Cell 2020, 182, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Monteil V., Kwon H., Prado P., Hagelkruys A., Wimmer R. A., Stahl M., Leopoldi A., Garreta E., Hurtado Del Pozo C., Prosper F., Romero J. P., Wirnsberger G., Zhang H., Slutsky A. S., Conder R., Montserrat N., Mirazimi A., Penninger J. M., Cell 2020, 181, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L., Cell Res. 2020, 30, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kreye J., Reincke S. M., Kornau H. C., Sanchez‐Sendin E., Corman V. M., Liu H., Yuan M., Wu N. C., Zhu X., Lee C. D., Trimpert J., Holtje M., Dietert K., Stoffler L., von Wardenburg N., van Hoof S., Homeyer M. A., Hoffmann J., Abdelgawad A., Gruber A. D., Bertzbach L. D., Vladimirova D., Li L. Y., Barthel P. C., Skriner K., Hocke A. C., Hippenstiel S., Witzenrath M., Suttorp N., Kurth F., et al., Cell 2020, 183, 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kreer C., Zehner M., Weber T., Ercanoglu M. S., Gieselmann L., Rohde C., Halwe S., Korenkov M., Schommers P., Vanshylla K., Di Cristanziano V., Janicki H., Brinker R., Ashurov A., Krahling V., Kupke A., Cohen‐Dvashi H., Koch M., Eckert J. M., Lederer S., Pfeifer N., Wolf T., Vehreschild M., Wendtner C., Diskin R., Gruell H., Becker S., Klein F., Cell 2020, 182, 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones B. E., Brown‐Augsburger P. L., Corbett K. S., Westendorf K., Davies J., Cujec T. P., Wiethoff C. M., Blackbourne J. L., Heinz B. A., Foster D., Higgs R. E., Balasubramaniam D., Wang L., Zhang Y., Yang E. S., Bidshahri R., Kraft L., Hwang Y., Zentelis S., Jepson K. R., Goya R., Smith M. A., Collins D. W., Hinshaw S. J., Tycho S. A., Pellacani D., Xiang P., Muthuraman K., Sobhanifar S., Piper M. H., et al., Sci. Transl. Med. 2021, 13, eabf1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim C., Ryu D. K., Lee J., Kim Y. I., Seo J. M., Kim Y. G., Jeong J. H., Kim M., Kim J. I., Kim P., Bae J. S., Shim E. Y., Lee M. S., Kim M. S., Noh H., Park G. S., Park J. S., Son D., An Y., Lee J. N., Kwon K. S., Lee J. Y., Lee H., Yang J. S., Kim K. C., Kim S. S., Woo H. M., Kim J. W., Park M. S., Yu K. M., et al., Nat. Commun. 2021, 12, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Divine R., Dang H. V., Ueda G., Fallas J. A., Vulovic I., Sheffler W., Saini S., Zhao Y. T., Raj I. X., Morawski P. A., Jennewein M. F., Homad L. J., Wan Y. H., Tooley M. R., Seeger F., Etemadi A., Fahning M. L., Lazarovits J., Roederer A., Walls A. C., Stewart L., Mazloomi M., King N. P., Campbell D. J., McGuire A. T., Stamatatos L., Ruohola‐Baker H., Mathieu J., Veesler D., Baker D., Science 2021, 372, eabd9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pinto D., Park Y. J., Beltramello M., Walls A. C., Tortorici M. A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., Peter A., Guarino B., Spreafico R., Cameroni E., Case J. B., Chen R. E., Havenar‐Daughton C., Snell G., Telenti A., Virgin H. W., Lanzavecchia A., Diamond M. S., Fink K., Veesler D., Corti D., Nature 2020, 583, 290. [DOI] [PubMed] [Google Scholar]
- 47. Brouwer P. J. M., Caniels T. G., van der Straten K., Snitselaar J. L., Aldon Y., Bangaru S., Torres J. L., Okba N. M. A., Claireaux M., Kerster G., Bentlage A. E. H., van Haaren M. M., Guerra D., Burger J. A., Schermer E. E., Verheul K. D., van der Velde N., van der Kooi A., van Schooten J., van Breemen M. J., Bijl T. P. L., Sliepen K., Aartse A., Derking R., Bontjer I., Kootstra N. A., Wiersinga W. J., Vidarsson G., Haagmans B. L., Ward A. B., et al., Science 2020, 369, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., Gong Y., Xiao H., Fan Z., Tan S., Wu G., Tan W., Lu X., Fan C., Wang Q., Liu Y., Zhang C., Qi J., Gao G. F., Gao F., Liu L., Science 2020, 368, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rogers T. F., Zhao F., Huang D., Beutler N., Burns A., He W. T., Limbo O., Smith C., Song G., Woehl J., Yang L., Abbott R. K., Callaghan S., Garcia E., Hurtado J., Parren M., Peng L., Ramirez S., Ricketts J., Ricciardi M. J., Rawlings S. A., Wu N. C., Yuan M., Smith D. M., Nemazee D., Teijaro J. R., Voss J. E., Wilson I. A., Andrabi R., Briney B., et al., Science 2020, 369, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baum A., Fulton B. O., Wloga E., Copin R., Pascal K. E., Russo V., Giordano S., Lanza K., Negron N., Ni M., Wei Y., Atwal G. S., Murphy A. J., Stahl N., Yancopoulos G. D., Kyratsous C. A., Science 2020, 369, 1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xie X., Liu Y., Liu J., Zhang X., Zou J., Fontes‐Garfias C. R., Xia H., Swanson K. A., Cutler M., Cooper D., Menachery V. D., Weaver S. C., Dormitzer P. R., Shi P. Y., Nat. Med. 2021, 27, 620. [DOI] [PubMed] [Google Scholar]
- 52. Wibmer C. K., Ayres F., Hermanus T., Madzivhandila M., Kgagudi P., Oosthuysen B., Lambson B. E., de Oliveira T., Vermeulen M., van der Berg K., Rossouw T., Boswell M., Ueckermann V., Meiring S., von Gottberg A., Cohen C., Morris L., Bhiman J. N., Moore P. L., Nat. Med. 2021, 27, 622. [DOI] [PubMed] [Google Scholar]
- 53. Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J. C., Muecksch F., Rutkowska M., Hoffmann H. H., Michailidis E., Gaebler C., Agudelo M., Cho A., Wang Z., Gazumyan A., Cipolla M., Luchsinger L., Hillyer C. D., Caskey M., Robbiani D. F., Rice C. M., Nussenzweig M. C., Hatziioannou T., Bieniasz P. D., Elife 2020, 9, e61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C. O., Finkin S., Schaefer‐Babajew D., Cipolla M., Gaebler C., Lieberman J. A., Oliveira T. Y., Yang Z., Abernathy M. E., Huey‐Tubman K. E., Hurley A., Turroja M., West K. A., Gordon K., Millard K. G., Ramos V., Da Silva J., Xu J., Colbert R. A., Patel R., Dizon J., Unson‐O'Brien C., Shimeliovich I., Gazumyan A., Caskey M., Bjorkman P. J., et al., Nature 2021, 592, 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Greaney A. J., Starr T. N., Gilchuk P., Zost S. J., Binshtein E., Loes A. N., Hilton S. K., Huddleston J., Eguia R., Crawford K. H. D., Dingens A. S., Nargi R. S., Sutton R. E., Suryadevara N., Rothlauf P. W., Liu Z., Whelan S. P. J., Carnahan R. H., Crowe J. E. Jr., Bloom J. D., Cell Host Microbe 2021, 29, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choi B., Choudhary M. C., Regan J., Sparks J. A., Padera R. F., Qiu X., Solomon I. H., Kuo H. H., Boucau J., Bowman K., Adhikari U. D., Winkler M. L., Mueller A. A., Hsu T. Y., Desjardins M., Baden L. R., Chan B. T., Walker B. D., Lichterfeld M., Brigl M., Kwon D. S., Kanjilal S., Richardson E. T., Jonsson A. H., Alter G., Barczak A. K., Hanage W. P., Yu X. G., Gaiha G. D., Seaman M. S., et al., N. Engl. J. Med. 2020, 383, 2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Avanzato V. A., Matson M. J., Seifert S. N., Pryce R., Williamson B. N., Anzick S. L., Barbian K., Judson S. D., Fischer E. R., Martens C., Bowden T. A., de Wit E., Riedo F. X., Munster V. J., Cell 2020, 183, 1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Monteil V., Kwon H., Prado P., Hagelkrüys A., Wimmer R. A., Stahl M., Leopoldi A., Garreta E., Hurtado del Pozo C., Prosper F., Romero J. P., Wirnsberger G., Zhang H., Slutsky A. S., Conder R., Montserrat N., Mirazimi A., Penninger J. M., Cell 2020, 181, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guo L., Bi W., Wang X., Xu W., Yan R., Zhang Y., Zhao K., Li Y., Zhang M., Cai X., Jiang S., Xie Y., Zhou Q., Lu L., Dang B., Cell Res. 2021, 31, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alvarez‐Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J., Nat. Biotechnol. 2011, 29, 341. [DOI] [PubMed] [Google Scholar]
- 61. Wang J., Zheng Y., Zhao M., Front. Pharmacol. 2016, 7, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhu X., He Z., Yuan J., Wen W., Huang X., Hu Y., Lin C., Pan J., Li R., Deng H., Liao S., Zhou R., Wu J., Li J., Li M., Cell. Microbiol. 2015, 17, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qiu Y., Ma J., Zeng Y., Biomed. Environ. Sci. 2018, 31, 215. [DOI] [PubMed] [Google Scholar]
- 64. Madison M. N., Roller R. J., Okeoma C. M., Retrovirology 2014, 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Q., Honko A., Zhou J., Gong H., Downs S. N., Vasquez J. H., Fang R. H., Gao W., Griffiths A., Zhang L., Nano Lett. 2020, 20, 5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Linder M. E., Deschenes R. J., Nat. Rev. Mol. Cell Biol. 2007, 8, 74. [DOI] [PubMed] [Google Scholar]
- 67. Jiang H., Zhang X., Chen X., Aramsangtienchai P., Tong Z., Lin H., Chem. Rev. 2018, 118, 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aicart‐Ramos C., Valero R. A., Rodriguez‐Crespo I., Biochim. Biophys. Acta 2011, 1808, 2981. [DOI] [PubMed] [Google Scholar]
- 69. Smotrys J. E., Linder M. E., Annu. Rev. Biochem. 2004, 73, 559. [DOI] [PubMed] [Google Scholar]
- 70. Runkle K. B., Kharbanda A., Stypulkowski E., Cao X. J., Wang W., Garcia B. A., Witze E. S., Mol. Cell 2016, 62, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lu Y., Zheng Y., Coyaud E., Zhang C., Selvabaskaran A., Yu Y., Xu Z., Weng X., Chen J. S., Meng Y., Warner N., Cheng X., Liu Y., Yao B., Hu H., Xia Z., Muise A. M., Klip A., Brumell J. H., Girardin S. E., Ying S., Fairn G. D., Raught B., Sun Q., Neculai D., Science 2019, 366, 460. [DOI] [PubMed] [Google Scholar]
- 72. Gao L., Wang L., Dai T., Jin K., Zhang Z., Wang S., Xie F., Fang P., Yang B., Huang H., van Dam H., Zhou F., Zhang L., Nat. Immunol. 2018, 19, 233. [DOI] [PubMed] [Google Scholar]
- 73. Xie F., Zhou X., Fang M., Li H., Su P., Tu Y., Zhang L., Zhou F., Adv. Sci. 2019, 6, 1901779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Consortium E.‐T., Van Deun J., Mestdagh P., Agostinis P., Akay O., Anand S., Anckaert J., Martinez Z. A., Baetens T., Beghein E., Bertier L., Berx G., Boere J., Boukouris S., Bremer M., Buschmann D., Byrd J. B., Casert C., Cheng L., Cmoch A., Daveloose D., De Smedt E., Demirsoy S., Depoorter V., Dhondt B., Driedonks T. A., Dudek A., Elsharawy A., Floris I., Foers A. D., et al., Nat. Methods 2017, 14, 228. [DOI] [PubMed] [Google Scholar]
- 75. Wan J., Roth A. F., Bailey A. O., Davis N. G., Nat. Protoc. 2007, 2, 1573. [DOI] [PubMed] [Google Scholar]
- 76. Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong‐Poi H., Crackower M. A., Fukamizu A., Hui C. C., Hein L., Uhlig S., Slutsky A. S., Jiang C., Penninger J. M., Nature 2005, 436, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tan C. W., Chia W. N., Qin X., Liu P., Chen M. I., Tiu C., Hu Z., Chen V. C., Young B. E., Sia W. R., Tan Y. J., Foo R., Yi Y., Lye D. C., Anderson D. E., Wang L. F., Nat. Biotechnol. 2020, 38, 1073. [DOI] [PubMed] [Google Scholar]
- 78. Zhou F., Drabsch Y., Dekker T. J., de Vinuesa A. G., Li Y., Hawinkels L. J., Sheppard K. A., Goumans M. J., Luwor R. B., de Vries C. J., Mesker W. E., Tollenaar R. A., Devilee P., Lu C. X., Zhu H., Zhang L., Dijke P. T., Nat. Commun. 2014, 5, 3388. [DOI] [PubMed] [Google Scholar]
- 79. Ashcroft T., Simpson J. M., Timbrell V., J. Clin. Pathol. 1988, 41, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Table 1
Supporting Table 2
Data Availability Statement
Research data are not shared.