

Summary
The family of Suppressor of Cytokine Signalling (SOCS) proteins plays pivotal roles in cytokine and immune regulation. Despite their key roles, little attention has been given to the SOCS family as compared to other feedback regulators. To date, SOCS proteins have been found to be exploited by viruses such as herpes simplex virus (HSV), hepatitis B virus (HBV), hepatitis C virus (HCV), Zika virus, respiratory syncytial virus (RSV), Ebola virus, influenza A virus (IAV) and SARS‐CoV, just to name a few. The hijacking and subsequent upregulation of the SOCS proteins upon viral infection, suppress the associated JAK‐STAT signalling activities, thereby reducing the host antiviral response and promoting viral replication. Two SOCS protein family members, SOCS1 and SOCS3 are well‐studied and their roles in the JAK‐STAT signalling pathway are defined as attenuating interferon (IFN) signalling upon viral infection. The upregulation of SOCS protein by SARS‐CoV during the early stages of infection implies strong similarity with SARS‐CoV‐2, given their closely related genomic organisation. Thus, this review aims to outline the plausibility of SOCS protein inhibitors as a potential therapeutic regimen for COVID‐19 patients. We also discuss the antagonists against SOCS protein to offer an overview on the previous ‘successes’ of SOCS protein inhibition in various viral infections that may portray possible clues for COVID‐19 disease management.
Keywords: antiviral, COVID‐19, inhibition, SARS‐CoV‐2, SOCS, treatment
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- ARDS
acute respiratory distress syndrome
- CISH
cytokine‐inducible SH2‐containing protein
- COVID‐19
coronavirus disease‐2019
- CTL
cytotoxic T‐lymphocytes
- E
envelope protein
- EGF
epidermal growth factor
- ERK
extracellular signal‐regulated kinases
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HSV
herpes simplex virus
- IAV
influenza A virus
- IFN
interferon
- IFNAR
interferon α receptor
- IFNGR
interferon γ receptor
- Ig
immunoglobulin
- IGF‐1
insulin‐like growth factor I
- IL
interleukin
- IRFs
interferon‐regulatory factors
- IRS‐1
insulin receptor substrate 1
- ISGs
interferon‐stimulated genes
- JAK
Janus kinase Family
- KIR
kinase inhibitory region
- M
membrane protein
- MAPK
mitogen‐activated protein kinase
- MERS‐CoV
middle east respiratory syndrome‐coronavirus
- MHC‐I
major histocompatibility complex‐1
- N
nucleocapsid protein
- NPC
nuclear pore complex
- NSP
non‐structural proteins
- ORF
open reading frame
- PAI‐1
plasminogen activator inhibitor‐1
- PIAS3
protein inhibitor of activated STAT3
- PRRs
pattern recognition receptors
- RdRp
RNA‐dependent RNA polymerase
- RLRs
retinoic acid‐inducible gene I‐like receptors
- RSV
respiratory syncytial virus
- S
spike protein
- SARS‐CoV
severe acute respiratory syndrome‐coronavirus
- SARS‐CoV‐2
severe acute respiratory syndrome‐coronavirus‐2
- SOCS
Suppressor of Cytokine Signalling
- STAT
Signal Transduction and Activators of Transcription
- TLRs
toll‐like receptors
- TMPRSS2
transmembrane protease serine 2
- TNF
tumour necrosis factor
1. INTRODUCTION
Global public health is under enormous threat due to the ongoing COVID‐19 pandemic affecting more than 218.5 million individuals accompanied by 4.5 million related deaths worldwide. 1 The etiological agent responsible for COVID‐19 has been identified as severe acute respiratory syndrome‐coronavirus‐2 (SARS‐CoV‐2). The SARS‐CoV‐2 infection is associated with a 3.4% mortality rate, which is lower than the previous severe acute respiratory syndrome‐coronavirus (SARS‐CoV), and middle‐east respiratory syndrome‐coronavirus (MERS‐CoV) outbreaks at 9.6% and 40% mortality rate, respectively. 2 , 3 , 4 The first coronavirus outbreak caused by the SARS‐CoV was first identified in Guangdong Province, China in November 2002. 5 The animal reservoir, that is, responsible for the SARS‐CoV outbreak is suggested to be the bats. 5 Subsequently, the MERS‐CoV outbreak was reported in Jeddah, Saudi Arabia in September 2012. 6 It was postulated to be associated with the dromedary camel. 6 To date, the origin of SARS‐CoV‐2 is still being debated, with sources pointing to bats as the natural reservoir, akin to the related SARS‐CoV identified in the year 2002. 7 , 8
Coronaviruses are RNA virus that belongs to the family of Coronaviridae, which can be subdivided into four different genera: alpha (α), beta (β), gamma (γ) and delta (δ). 9 Similar to SARS‐CoV and MERS‐CoV, the SARS‐CoV‐2 is a member of the beta (β) genus, thus sharing a certain degree of similarity in general morphology and structure as its counterparts, especially concerning the SARS‐CoV with an astonishing 79% genetic identity and 94.4% amino acid similarity. 9 , 10 The SARS‐CoV‐2 infection, like its counterparts, affects mainly the respiratory system, giving symptoms such as fever, dry cough, fatigue, dyspnoea, headache, gastrointestinal discomfort, diarrhoea, conjunctivitis, skin discolouration or rashes, anosmia and dysgeusia. 8 However, the major contributor to the rapid progression of COVID‐19 lies within the large numbers of asymptomatic cases of SARS‐CoV‐2 infection in addition to the patients with the aforementioned disease symptoms. 11 , 12
Currently, the persistent symptoms of COVID‐19 pose serious health and quality of life concerns of the COVID‐19 survivors. As such, prolonged anxiety, chest pain, dizziness, hair loss, weight loss, prolonged palpitation, sleep difficulty, depression and prolonged gastrointestinal symptoms which encompasses nausea, vomiting, diarrhoea, lack of appetite, abdominal pain and dysgeusia. 13 , 14 Apart from that, other concerns such as the tempered immune system in SARS‐CoV‐2 infection may give rise to an increase of unexpected co‐infections in COVID‐19 patients. There have been many reports of coinfection in SARS‐CoV‐2. For instance, scrub typhus (caused by chiggers carrying Orientia tsutsugamushi) co‐infection in Nepal and dengue virus (transmitted by Aedes aegypti mosquitoes) co‐infection in Asia, in which, presenting similar clinical manifestations, such as fever, muscle aches, joints pain, muscle aches, fatigue and dyspnoea, just to name a few. 15 , 16 With the overlapping clinical manifestation and immunological cascades of the co‐infections, it may contribute to misdiagnosis and enhanced pathogenesis, leading to serious complications. Hence, it is vital to revisit lessons learnt from the SARS‐CoV and MERS‐CoV outbreaks, especially with respect to the regulation of cytokine signalling such as the Suppressor of Cytokine Signalling (SOCS) family, a key regulatory factor in the cytokine storm that may elicit the potentially lethal acute respiratory distress syndrome (ARDS) in COVID‐19 patients. 17 , 18
It is well‐established that inflammation is one of the major outcomes of the host‐elicited immune response, leading to a series of complicated infections. This is mediated by complex inflammatory signalling pathways, such as the Janus Kinase Family and Signal Transduction and Activators of Transcription (JAK/STAT) and NF‐κB pathways – the regulation and interaction between various cytokines, chemokines, interferons, interleukins (IL), tumour necrosis factor (TNF), endothelial cells and immune cells. 19 Cytokines are proteins that are vital for cell‐cell communication, cell differentiation, growth and defence mechanisms. 20 Cytokines also play pivotal roles in the human immune responses during infection, inflammation, cancer; and in modulating the humoral and cell‐mediated immune pathways via various cell surface receptors. 21
Upon viral infection, the phosphorylation of the N‐terminus of JAK and subsequently activation of STAT is dependent on various cytokines as well as interferons or IFN. 22 This cascade of reactions must be tightly regulated, to prevent over‐expression which leads to hyperinflammation or under‐expression which ultimately leads to poor patient outcomes. The negative regulation of the JAK/STAT pathway is mediated by the SOCS proteins. There are a total of eight known members of the SOCS family, SOCS1‐7 and CISH (Cytokine‐inducible SH2‐containing protein). 23 In this review, we discuss the roles of the SOCS family of proteins in the innate immune system and how they are modulated upon SARS‐CoV‐2 infection. Further, we review the molecular mechanisms underpinning the modes of action for the SOCS protein family that hold promise towards developing novel antiviral candidates to ameliorate the severity of COVID‐19 disease progression.
2. THE SOCS FAMILY PROTEINS
Cytokines play important roles in fundamental biological processes including cytokine‐induced signal transduction pathways. Excessive or sustained release of cytokines can be detrimental, thereby emphasising the importance of negative regulators, such as the SOCS proteins, in modulating cytokine signalling (Figure 1).
FIGURE 1.
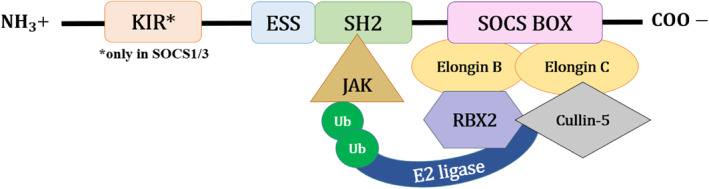
A general structural representation of the Suppressor of Cytokine Signalling (SOCS) family proteins. There are eight known members, namely SOCS1 to SOCS7 and cytokine‐inducible SH2‐containing protein. The SOCS family can be characterised by a central Src homology 2 (SH2) domain, extended SH2‐subdomain (ESS), C‐terminal SOCS box motif and N‐terminal domain with varying sequence and length. 24 Here, only the SOCS1 and SOCS3 share a distinctive kinase inhibitory region (KIR) in the N‐terminal domain, an additional direct inhibitory mechanism for JAK tyrosine kinase activity. The highly conserved SOCS box motif contains 40–60 amino acid residues and interacts with Elongin B and C, Cullin‐5 and RING‐box‐2 (RBX2), to recruit E3 ubiquitin transferase for the regulation of cytokine signalling. 25 , 26 The SH2 domain is responsible for the recognition and binding mechanism of each SOCS protein, whereas ESS contributes to the physical interaction between SOCS protein and substrate. 26
SOCS protein contributes substantially to the negative regulation of the JAK/STAT pathway. The activation of the JAK‐STAT pathway depends on the binding of the cytokines to cognate receptors which induce phosphorylation of tyrosine residues at the receptor by JAKs. Subsequently, STAT gets recruited to the site followed by dimerisation and nuclear translocation, where transcription of genes occurs. 27 Among the SOCS, SOCS1–SOCS3 and CIS primarily regulate the JAK/STAT pathway, whereas SOCS4–SOCS7 act on growth factor receptor signalling pathways. 25 Generally, SOCS protein members inhibit the JAK/STAT pathways by tagging the target proteins for ubiquitination and subsequent proteasomal degradation. This is achieved by the recruitment of the E3 ubiquitin ligase scaffold via the interaction between the SOCS box motif and Cullin‐5. 28 As the central SH2 domain determines the binding mechanism of the SOCS protein members, each SOCS protein member has different binding mechanisms.
2.1. SOCS1 and SOCS3
Compared to the other SOCS protein members, SOCS1 and SOCS3 are well‐studied in the JAK‐STAT signalling pathway, playing pivotal roles in cytokine regulation, especially in the context of virulence factors. 29 Indeed, the existence of SOCS1 and SOCS3 are mainly to ensure the viability of the organism. Studies on SOCS1 and SOCS3 knockout in mice resulted in neonatal and embryonic death, respectively. 30
SOCS1 is a 23.4 kDa protein involved in the regulation of JAK/STAT and NF‐κB pathways via ubiquitin‐mediated proteasomal degradation. In particular, SOCS1 is known to negatively regulate IFNγ‐induced signalling pathways which is evident in SOCS1 knockout mice. 31 In contrast, SOCS3 is a 24.8 kDa protein that negatively regulates the expression of pro‐inflammatory cytokines (IL‐6) and anti‐inflammatory cytokines (IL‐10) via selective binding to the gp130 receptor. 32 Despite the differences in function, SOCS1 and SOCS3 share a kinase inhibitory region (KIR) in the N‐terminal domain that functions as a pseudo‐substrate, a feature distinct from the other SOCS protein members. In addition to the generic ubiquitylation and degradation activities, SOCS1 and SOCS3 possess an additional direct inhibitory mechanism on JAK tyrosine kinase activity due to the presence of KIR, upstream of the SH2 domain. For instance, the SH2 domain in SOCS1 can directly bind to the activation loop of JAK which hinders the JAK activity and tyrosine phosphorylation of STAT1α. Moreover, SOCS1 can also bind directly to the IFNγ receptor (IFNGR) and type I IFN receptor (IFNAR) to augment the inhibitory activity on IFN signalling pathways. On the other hand, the main kinase inhibitory effect of SOCS3 is contributed by the interaction between the KIR of SOCS3 and the JAK2 kinase domain. This is due to the lower affinity of the SH2 domain in SOCS3 for the binding of the activation loop of JAK. 26 , 30 In addition, SOCS3 can interact with various cytokine receptors to downregulate JAK‐STAT signalling. For instance, the binding of SOCS3 to the Tyr401 region of the erythropoietin receptor inhibits JAK2 and STAT5 activation. 33 The binding of SOCS3 to other cytokine receptors such as Tyr757 of gp130 receptor, Tyr985 of the leptin receptor, Tyr800 of IL‐12 receptor and Tyr729 of granulocyte colony‐stimulating factor (G‐CSF) receptor, also show a similar interference to the JAK‐STAT pathway. 34
2.2. Other SOCS proteins
In contrast, the inhibitory mechanisms of other SOCS protein members are far less studied. CIS is a 28.6 kDa protein that can inhibit cytokine‐induced STAT5 signalling via substrate competition, in which the SH2 domain of CIS competitively binds to the phosphorylated tyrosine residues in activated cytokine receptors, instead of the JAKs. 32 , 35 For instance, the signal transduction of many cytokines, such as IL‐2, IL‐3, erythropoietin, growth hormone and prolactin are inhibited. 36 Similar to CIS, SOCS2 is a 22 kDa protein that also utilises competitive binding via SOCS box to hinder the growth factor‐mediated activation of STAT5b. 32 , 34 Only SOCS1 and SOCS3 are documented to interact and suppress JAK activities directly via KIR. Owing to the lack of KIR in SOCS2, the possibility of direct interaction with JAK remains minute. Interestingly, recent studies have shown otherwise, where SOCS2 can interact with JAK2 directly to negatively modulate the JAK2‐STAT5 signalling pathway, thereby diminishing the development of natural killer cells. 37 Notably, SOCS2 can antagonise other SOCS members. For example, SOCS2 antagonises SOCS1 and SOCS3 in the negative modulation of IL‐2 and IL‐3 signalling, as illustrated by the enhanced cytokine response with higher proteasomal‐dependent turnover rate of SOCS1 and SOCS3. 38
SOCS4 and SOCS5 are 50.6 and 61.2 kDa proteins, respectively, that regulates epidermal growth factor (EGF) signalling. 32 , 39 It is postulated that the SH2 domain of SOCS4 and the N‐terminal region of SOCS5 interact with the EGF receptor (EGFR) via phosphorylation‐dependent and phosphorylation‐independent interactions, respectively. The docking of SOCS4 to EGFR subjects the activated EGFR to proteasomal degradation via recruitment of E3 ubiquitin ligase. Moreover, SOCS4 has the same binding affinity to STAT3 as to the EGFR, thereby suggesting that SOCS4 is capable of attenuating STAT3 activation as well. 40 , 41 On the other hand, it is proposed that SOCS5 targets the proteins (EGFR, IL‐6R, IL‐4R) for proteasomal degradation, allowing it to negatively regulate cytokine receptor signalling. As such, SOCS5 can contribute to T‐helper 1 (Th1)/Th2 differentiation via the attenuation of IL‐4 dependent STAT6 activation. 41 , 42
Akin to SOCS1–SOCS5 and CIS proteins, the inhibitory mechanism underlying SOCS6 (59.5 kDa) is likely via ubiquitination and proteasomal degradation as well, with heme‐oxidised IRP2 ubiquitin ligase‐1 as a replacement for E3 ubiquitin ligase. 43 SOCS6 is known for its role in regulating the insulin signalling pathway where it suppresses the activation of the insulin receptor substrate 1 (IRS‐1), extracellular signal‐regulated kinases (ERK1/2) and protein kinase B. 32 Similar to SOCS2, SOCS6 also possesses the ability to antagonise other SOCS protein members. Furthermore, the N‐terminal domain of SOCS6 promotes SOCS6 nuclear localisation, in which STAT3 is regulated. 44 Lastly, SOCS7 (62.9 kDa) is a well‐documented protein with its negative regulation of insulin‐like growth factor I receptor (IGF‐IR) signalling. Similarly, SOCS7 regulates the IGF‐I signalling by subjecting IRS‐1 docking on the cytoplasmic domain of activated IGF‐IR to proteasomal degradation via the SOCS box. Besides this, the interactions between the SH2 domain of SOCS7 and IRS‐2 and IRS‐4 also suppress signalling. 41 In addition, SOCS7 can modulate the JAK‐STAT signalling pathway induced by leptin and prolactin via the interaction of JAK2‐STAT3 and JAK2‐STAT5. 45
3. GENERAL FEATURES OF SARS‐CoV‐2
Similar to its beta‐coronavirus counterparts (such as SARS‐CoV and MERS‐CoV), SARS‐CoV‐2 exhibits a close resemblance in its general morphology. 46 SARS‐CoV‐2 is an enveloped non‐segmented positive‐sense RNA virus, ranging from 65 to 125 nm in diameter. 47 Its RNA genome is ∼29.9 kilobases (kb) with two‐thirds containing the main open reading frame 1a and 1b (ORF1ab) replicase from the 5′‐end – for pp1a and pp1ab polyproteins that encode non‐structural proteins (NSP) 1–11 and NSP 12–16, respectively. 48 The remaining one‐third of the genome from the 3′‐end encodes different structural proteins, such as spike (S), envelope (E), membrane (M) and nucleocapsid (N) proteins. 49 In addition, there are also variable ORFs alternating at the 3′‐end, such as the ORF3a, ORF3d, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF14 and ORF10 for the production of accessory proteins. 48 The typical organisation of the SARS‐CoV‐2 genome can be denoted as: 5′‐leader‐UTR‐replicase‐S‐E‐M‐N‐3′‐UTR‐poly(A) tail, with the aforementioned accessory genes scattered between the structural genes (S‐E‐M‐N) at the 3′ end. 50
Apart from the structural proteins, the NSPs and accessory proteins of SARS‐CoV‐2 play very crucial roles in viral replication. They are largely associated with cytokine signalling and immune evasion upon infection. NSP 1 is associated with the inhibition of IFN signalling for innate immune evasion and induces the production of chemokines, contributing to cytokine storm in COVID‐19 patients. 51 NSP3 and NSP5 (proteinase) are found to impede innate immunity, to promote cytokine expression and to cleave viral polyproteins. 52 Further, NSP7, NSP8 and NSP12 constitute the RNA‐dependent RNA polymerase (RdRp) complex that promotes viral genome replication, methylation and transcription, which may further aggravate disease progression in patients. 53 Aside from the NSPs, accessory proteins such as the ORF6 localises at the nuclear pore complex (NPC), disrupting the nuclear translocation of STAT‐1 and STAT‐2 by blocking the karyopherin/importin complex and antagonising interferon signalling – thereby suppressing the immune response. 54 ORF8 is believed to be involved in immune evasion via the downregulation of the major histocompatibility complex‐1 (MHC‐I) which functioned to present viral antigen peptides for lymphocyte recognition, thereby contributing to impaired host immune system. 55 As such, the MHC‐1 molecules present the viral peptide to cytotoxic T‐lymphocytes (CTL), subsequently the CTL releases various antiviral substances (e.g., perforins, granzyme and FasL) to induce cell death in SARS‐CoV‐2 infected cells. 55 Besides that, CTLs are also responsible for the release of various cytokines such as IFN‐γ, TNF‐α and IL‐2, an impaired CTL recognition would mean reduced expression of JAK‐STAT signalling and the following antiviral cascade response. 55 Hence, it becomes important to study the immune response with respect to these cytokine signalling pathways in the context of SARS‐CoV‐2 infection.
4. ASSOCIATED JAK‐STAT CYTOKINE SIGNALLING PATHWAYS
As the name suggests, the suppressor of cytokine signalling or SOCS family acts as a checkpoint inhibitor or negative feedback for cytokine signalling to restrict excessive inflammation. Among the SOCS family, SOCS1 and SOCS3 contain a kinase inhibitory region (KIR) that inhibits kinase activities, such as the JAK‐STAT signalling pathway, thereby playing a pivotal role in cytokine regulation. 29 Aside from JAK‐STAT, cytokine stimulation can be also induced via various other pathways, such as NF‐κB and MAPK. However, the JAK‐STAT signalling pathway represents the most classical cytokine stimulation pathway with respect to the SOCS family (Figure 2).
FIGURE 2.
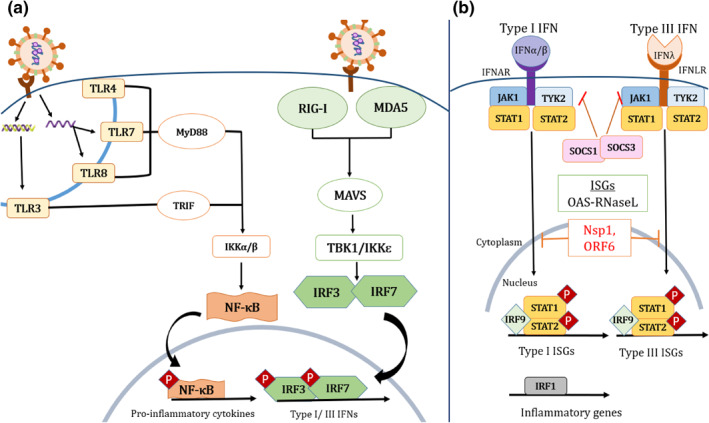
A brief schematic illustration of the Suppressor of Cytokine Signalling (SOCS)‐associated JAK‐STAT signalling pathway. (a) Pattern recognition receptors (PRRs) are expressed in various immune cells such as dendritic cells, macrophages, monocytes, neutrophils and epithelial cells. Upon infection, the immune cells recognise foreign viral antigens via PRRs such as the Toll‐like receptors (TLRs), and retinoic acid‐inducible gene I‐like receptors (RLRs), thereby stimulating the production of cytokines and interferons (IFN). (b) IFN bind to IFN receptors on the cell surface and subsequently stimulate the associated JAK‐STAT signalling pathway for the activation of antiviral interferon‐stimulated genes (ISGs). This, in turn, promotes transcription of antiviral genes such as oligoadenylate synthetase (OAS) and latent endoribonuclease (RNaseL) (shown as OAS‐RNaseL), leading to the cleavage of both host and viral RNA – thus impeding viral replication. (Figure is redrawn from Park and Iwasaki. 56 )
The JAK‐STAT pathway begins with cytokine stimulation of the cell surface receptor (such as IFN‐1 receptor). 18 These cell surface receptors are associated with JAK which subsequently phosphorylates STAT, thus triggering the downstream JAK‐STAT signalling pathway. 29 During viral infection, the cells recognise foreign viral antigens via pattern recognition receptors (PRRs), such as the intracellular nucleic acid sensors associated Toll‐like receptors (TLRs) 3, 4, 7 and 8, and retinoic acid‐inducible gene I‐like receptors (RLRs), for example, RIG‐1, MDA5. 56 , 57 Subsequently, this induces NF‐κB to produce pro‐inflammatory cytokines (such as IL‐1, IL‐6 and TNF‐α) and interferon‐regulatory factors or IRFs (such as IRF3 and IRF7). 56 The IRFs subsequently stimulate the production of type I IFNs (IFN‐α, IFN‐β, IFN‐ε, IFN‐κ and IFN‐ω) and type III IFN (IFN‐λ). 56
Following the production of IFNs, IFN‐I (IFN‐α/β) binds to the IFN‐α/β receptor‐1 (IFNAR1)/IFNAR2, that is, associated with JAK1 and TYK2 subunits which subsequently phosphorylate the cytoplasmic STAT1 and STAT2 (15X). This then stimulates the formation of STAT1‐STAT2 heterodimer transcription factor complex (STAT1/STAT2/IRF9), forming IFN‐stimulated gene factor 3 (ISGF3) for translocation to the nucleus, thereby upregulating interferon‐stimulated genes (ISGs) such as the 2′,5′–oligoadenylate synthetase (OAS), protein kinase R (PKR)—ultimately downregulating viral replication. 56 In addition, the upregulation of STAT3 also enhances the production of inflammatory cytokine IL‐6 via TLR4, which subsequently activates the NF‐κB pathway, producing more IL‐6 and other chemokines. 58
Notably, despite the aforementioned signalling cascade, other factors such as gender might come into play for the pathogenesis of COVID‐19. In which, the pattern recognition receptors (PRRs), such as the intracellular nucleic acid sensors TLRs, and RLRs are more pronounced in females than males. 59 This is shown in a study where it has suggested that the oestrogen receptor subtype, ER‐α that present only in female mice enhances TLRs and NK cells response, producing a much more robust cytokine response (TNF‐α, IL‐1 and IL‐6) compared to the male mice, causes the death of infected cells, thus, clearing the pathogen more efficiently. 60 Moreover, the predominant gender‐bias occurrence of transmembrane protease serine 2 (TMPRSS2) and ACE2 receptors in males may contribute to the high SARS‐CoV‐2 entry and replication. 60 Interestingly, the androgen receptors present only in males, are the promoters for TMPRSS2. In contrast, females can regulate the expression of ACE2 via oestrogen activities. 60 Hence, these conferred some immunoprotective measures to females compared to males.
As a critical cytokine regulator, SOCS proteins normally impede IFN signalling upon excessive STAT1 or STAT3 upregulation to limit inflammation. Unfortunately, viruses can exploit SOCS protein independently upon infection, suppressing the normal JAK‐STAT signalling pathway, reducing the antiviral ISGs (OAS, PKR)—thus subsequently downregulate the innate and adaptive immune responses to promote viral replication. 61
5. SOCS‐ASSOCIATED CYTOKINE REGULATION IN SARS‐CoV‐2 INFECTION
As mentioned, SOCS proteins regulate the JAK‐STAT pathway in a variety of ways. Hence, SOCS1 and SOCS3 can bind to the phosphotyrosine residues of JAK1, JAK2 and TYK2, inhibiting tyrosine phosphorylation of STAT1 and STAT3 via KIR, and subsequently hindering the nuclear translocation of STAT1 and STAT3. 62 This, in turn, downregulates the antiviral ISGs. While SOCS proteins can provide negative feedback to prevent excessive pro‐inflammatory cytokine production, they can be easily hijacked by the virus to promote viral replication. For instance, herpes simplex virus (HSV) induces SOCS1 and SOCS3 production, effectively inhibiting type I and type III IFN, thereby evading ISGs associated with innate immunity – thus enhancing viral replication. 29 Besides this, a similar hijack mechanism by the respiratory syncytial virus (RSV) upregulates both SOCS1 and SOCS3 via its viral NS1 and NS2 proteins, effectively inhibiting the type I IFN antiviral signalling pathway. 63 , 64 Similar SOCS1 and SOCS3 upregulation are also observed for Zika virus upon 48 h post‐infection of A549, Jar and human neural progenitor cell lines. 65 Notably, a similar phenomenon was also discovered in which SARS‐CoV can induce SOCS3 by fourfold after 48 h of infection, dampening the antiviral effects of IFNs. 66 Furthermore, the downregulation of JAK‐STAT also causes T‐cell‐dependent activation of B‐cells to fail to undergo class switch recombination to produce more robust IgA, IgG, or IgE antibodies to elicit an antiviral response against SARS‐CoV infection. 67 Given the genomic similarities between SARS‐CoV‐2 and the closely related SARS‐CoV, there exists a strong possibility for a similar phenomenon to be exhibited by SARS‐CoV‐2 on SOCS proteins.
Upon viral infection, the host‐expressed IFN‐I would bind to the JAK1 and TYK2 associated IFN‐α/β receptor and trigger tyrosine phosphorylation of STAT1 or STAT3, subsequently upregulating cytokine production and antiviral ISGs (OAS, PKR), thereby halting viral replication. While the activity of the SOCS proteins can be directly hijacked to promote viral replication at an early stage, the associated JAK‐STAT signalling cascade for innate immunity can also be hijacked in response to SARS‐CoV‐2 infection at the later stage of infection, leading to enhanced cytokine production and subsequently ARDS. Further, the activity of STAT1 can be inhibited by the SARS‐CoV‐2 NSP1 and ORF6 proteins. 18 The reduction of STAT1 is compensated by the host by enhanced STAT3 activity, subsequently inducing STAT3‐ISGs. 18 Furthermore, STAT3 can be further enhanced upon acute lung injury via the upregulation of epidermal growth factor receptor (EGFR). 68 The hyperactivation of STAT3 represses miR‐34a, an inhibitor of plasminogen activator inhibitor‐1 (PAI‐1), which inadvertently enhances the level of PAI‐1, leading to the inhibition of the protein inhibitor of activated STAT3 (PIAS3) that serves as the negative feedback for STAT3. 18 Interestingly, the activation of STAT3 enhances the production of inflammatory cytokine IL‐6 via enhanced binding of PAI‐1 to TLR4, which subsequently activates the NF‐κB pathway, producing a greater amount of IL‐6 and chemokines. 69
Taken together, a SOCS antagonist may prevent further viral replication via down‐regulation of SOCS activity (hijacked by the virus upon early stage of infection), thus promoting healthy IFN signalling for JAK‐STAT and subsequent immune response via ISGs. However, the overexpression of the JAK family by the SOCS antagonist, and enhanced EGFR signalling that activates STAT3 during the late infection stage may contribute somewhat towards the cytokine storm, the main contributor to ARDS which maybe life‐threatening. Hence, the appropriate use of SOCS inhibitors at different stages of SARS‐CoV‐2 infection may be highly critical in influencing morbidity and mortality due to severe cytokine storms. In this regard, other complementary strategies should also be carefully considered and evaluated, for example, the use of convalescent serum with neutralising antibodies against SARS‐CoV‐2. 70 Convalescent plasma containing neutralising antibodies and other proteins such as anti‐inflammatory cytokines, clotting factors, defensins and natural antibodies inherited from the donor may provide passive and effective immunity to alleviate COVID‐19 pathogenesis.
6. POTENTIAL DRUGS TO REGULATE SOCS IN SARS‐CoV‐2 INFECTION
Described above are some pathways and mechanisms by which viruses, such as HSV, RSV, Zika and SARS‐CoV, can hijack the SOCS proteins to facilitate viral replication. While SOCS mediated JAK‐STAT signalling and cytokine storm are closely associated, the former often occurs during the initial stage of infection. This clearly highlights the potential of SOCS proteins as putative targets for antiviral intervention. Although there are no established treatments against SOCS proteins for SARS‐CoV‐2 infection, it may be worthwhile to revisit previous antiviral strategies. For example, the transfection of antisense oligonucleotide to HSV‐1 can effectively knock down SOCS3 in epithelial FL human amnion cells with a significant reduction in viral replication by 103 plaque‐forming units (pfu) per mL. 71 In addition, 35 μM of peptidomimetic (pJAK2 1001–1013), a modified SOCS1 antagonist, offers 40% protection against HSV‐1 infection in HEL30 keratinocytes. 72 , 73 Combination with 100 U/mL of IFN‐γ achieved 100% protection. 72 , 73 This was achieved due to the ability of pJAK2 to bind to the KIR region of SOCS1, suppressing phosphorylation of STAT1, leading to increased IFN‐γ signalling for the antiviral response. 72 , 73
Aside from that, certain miRNAs have also demonstrated the ability to suppress SOCS expression. For instance, miRNA‐19a indirectly reduces SOCS1 and SOCS3 levels via the activation of STAT3. 74 This subsequently enhances IL‐6 and IFN‐α signalling, resulting in the reduction of hepatitis C and hepatitis B virus replication. 74 Similarly, miRNA‐221 is postulated to be the mechanism underlying the antiviral effect of IFN‐α treatment in HCV patients via the downregulation of SOCS1 and SOCS3 expression, subsequently upregulating the JAK‐STAT signalling pathway for antiviral response. 75 Furthermore, the use of 100 nM of ubiquitin‐specific protease 7 (USP7) small molecules (such as P5091 and P22077) also suppress SOCS1 in HEK293T cells infected with Sendai virus and vesicular stomatitis virus, subsequently inhibiting viral replication by promoting antiviral activity mediated by IFN‐I signalling. 76 Zoledronic acid (ZA), an FDA‐approved amino‐bisphosphate drug for various diseases (such as osteoporosis, Paget's disease, postmenopausal osteoporosis, multiple myeloma and bone metastases), was shown to be able to downregulate the expression of SOCS3 effectively. 77 The study clearly shows elevated inflammatory markers such as C‐reactive protein, leptin, IL‐6 and TNF‐α, a consequence of JAK‐STAT upregulation arising from the downregulation of SOCS protein upon administration of ZA. In view of its FDA approval and with appropriate detailed clinical trials, ZA could be one of the potential drugs that could be repurposed for SARS‐CoV‐2 treatment too.
As the viral infection progresses, the hyperactivation of natural killer (NK) cells, macrophages, T cells and numerous other immune cells contribute to the production of many different chemical mediators (e.g., IP‐10, CCL2, CCL3 and CCL‐5) and proinflammatory cytokines (e.g., IFN‐γ, TNF‐α, IL‐1, IL‐6 and IL‐8) via various signalling pathway such as the IFN‐JAK‐STAT signalling pathways, leading to lung scarring and ultimately cytokine storm associated ARDS in SARS‐CoV‐2. 78 , 79 While SOCS antagonist can be useful at the early stage of infection to restore cytokine and interferon production for the antiviral response, it has diminished usage for cytokine storm inhibition at the latter stage. Hence, the immunosuppressors for JAK‐STAT signalling should also be thoroughly considered to inhibit the JAK/STAT pathway to modulate the cytokine associated hyperinflammatory response.
As such, Baricitinib, and Ruxolitinib, a JAK1 and JAK2 inhibitor that antagonises the activity of JAK‐STAT signalling, has successfully shown clinical improvements in 12 patients, and 18 critically ill patients with COVID‐19, respectively. 80 , 81 , 82 Other potential drugs that were able to antagonise JAK‐STAT in SARS‐CoV‐2 infection are the peptidomimetic inhibitors that mimic SOCS3 binding to the JAK2 catalytic domain, namely the KIRESS and KIRCONG chimeric peptide. It has been shown to suppress STAT3 phosphorylation by up to 65% in HIV Tat protein infected mouse vascular smooth muscle cells, effectively suppressing JAK‐STAT associated cytokine production. 83 Additionally, other therapeutic options such as antibodies against inflammatory cytokines should also be considered. In which, tocilizumab, an anti‐IL6 receptor antibody, and adalimumab, an anti‐tumour necrosis factor (anti‐TNF) antibody have shown decreased pro‐inflammatory cytokines (IL‐1, IL‐6, TNF) in blood, conferring anti‐inflammatory activity, thereby reducing lung inflammation in COVID‐19 patients. 84
7. CONCLUSION
The rapid emergence of SARS‐CoV‐2 has brought into focus the reconsideration of therapeutic approaches including drug repurposing. One class of immunoregulatory proteins that plays vital roles in cytokine signalling and the immune response is the suppressor of the cytokine signalling (SOCS) protein family. While there are various therapeutic regimens including antivirals such as Remdesivir and Favipiravir, the importance of SOCS proteins in the attenuation of the IFN/JAK‐STAT pathways should not be overlooked. Among the eight SOCS protein members, SOCS1 and SOCS3 are well‐documented and studied with respect to modulating the JAK‐STAT signalling pathway. SOCS proteins are negative feedback inhibitors of JAK‐STAT activation, in the presence of IFNs. This is to ensure that there is no overexpression of IFNs that may culminate excessive JAK‐STAT signalling and subsequently exaggerated inflammatory cytokine production. However, the SOCS proteins can be easily hijacked and upregulated by various viruses such as HSV‐1, HBV, HCV, Sendai virus, vesicular stomatitis virus and importantly SARS‐CoV, thereby promoting viral replication. Given the positive results from SOCS1 and SOCS3 inhibitors in reducing viral loads, such as pJAK2 1001–1013, miRNA‐19a, ubiquitin‐specific protease 7 (USP7) and zoledronic acid (ZA), it is important to consider the potential of SOCS inhibitors in the context of SARS‐CoV‐2 infection. Nevertheless, careful considerations and investigations are warranted as SOCS antagonists might also enhance cytokine stimulation via JAK‐STAT activation, which may lead to the detrimental cytokine storm and ARDS at the late stage of viral infection.
CONFLICT OF INTEREST
All authors have read and agreed to the published version of the manuscript. All authors have declared that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
Zheng Yao Low and Ashley Jia Wen Yip contributed equally to the preparation and drafting of the manuscript. Critical revisions of the article were done by Vincent T. K. Chow and Sunil K. Lal.
ACKNOWLEDGEMENTS
This publication was supported by internal funds to Sunil K. Lal from the School of Science (SoS), Monash University Malaysia and in part by the SoS Strategic Grant 2020.
Low ZY, Wen Yip AJ, Chow VTK, Lal SK. The Suppressor of Cytokine Signalling family of proteins and their potential impact on COVID‐19 disease progression. Rev Med Virol. 2022;32(3):e2300. 10.1002/rmv.2300
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Worldometers Coronavirus Update (Live) . COVID‐19 Virus Pandemic 2021. Accessed September 1, 2021. https://www.worldometers.info/coronavirus/ [Google Scholar]
- 2. Ioannidis J. Infection fatality rate of COVID‐19 inferred from seroprevalence data. Bull World Health Organ. 2020;99:19‐33F. doi: 10.2471/blt.20.265892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zumla A, Hui D, Perlman S. Middle East respiratory syndrome. Lancet. 2015;386:995‐1007. doi: 10.1016/s0140-6736(15)60454-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peiris J, Yuen K, Osterhaus A, Stöhr K. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431‐2441. doi: 10.1056/nejmra032498 [DOI] [PubMed] [Google Scholar]
- 5. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 2016;24:490‐502. doi: 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zaki A, van Boheemen S, Bestebroer T, Osterhaus A, Fouchier R. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814‐1820. doi: 10.1056/nejmoa1211721 [DOI] [PubMed] [Google Scholar]
- 7. Andersen K, Rambaut A, Lipkin W, Holmes E, Garry R. The proximal origin of SARS‐CoV‐2. Nat Med. 2020;26:450‐452. doi: 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Low Z, Yip A, Sharma A, Lal S. SARS coronavirus outbreaks past and present—a comparative analysis of SARS‐CoV‐2 and its predecessors. Virus Gene. 2021;57:307‐317. doi: 10.1007/s11262-021-01846-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ye Z, Yuan S, Yuen K, Fung S, Chan C, Jin D. Zoonotic origins of human coronaviruses. Int J Biol Sci. 2020;16:1686‐1697. doi: 10.7150/ijbs.45472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou P, Yang X, Wang X, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. doi: 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. World Health Organization (W.H.O) . Coronavirus disease (COVID‐19). Accessed September 1, 2021. https://www.who.int/emergencies/diseases/novel‐coronavirus‐2019
- 12. Zanin L, Saraceno G, Panciani P, et al. SARS‐CoV‐2 can induce brain and spine demyelinating lesions. Acta Neurochir. 2020;162:1491‐1494. doi: 10.1007/s00701-020-04374-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fahriani M, Ilmawan M, Fajar J, et al. Persistence of long COVID symptoms in COVID‐19 survivors worldwide and its potential pathogenesis—a systematic review and meta‐analysis. Narra J. 2021;1:1‐14. doi: 10.52225/narraj.v1i2.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yusuf F, Fahriani M, Mamada S, et al. Global prevalence of prolonged gastrointestinal symptoms in COVID‐19 survivors and potential pathogenesis: a systematic review and meta‐analysis. F1000Res. 2021;10:301. doi: 10.12688/f1000research.52216.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bastola A, Sah R, Rajbhandari S, et al. SARS‐CoV‐2 and Orientia tsutsugamushi co‐infection in a young teen, Nepal: significant burden in limited‐resource countries in Asia? Narra J. 2021;1:34‐38. doi: 10.52225/narraj.v1i2.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harapan H, Ryan M, Yohan B, et al. Covid‐19 and dengue: double punches for dengue‐endemic countries in Asia. Rev Med Virol. 2020;31:e2161. doi: 10.1002/rmv.2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID‐19. J Infect. 2020;80:607‐613. doi: 10.1016/j.jinf.2020.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsuyama T, Kubli S, Yoshinaga S, Pfeffer K, Mak T. An aberrant STAT pathway is central to COVID‐19. Cell Death Differ. 2020;27:3209‐3225. doi: 10.1038/s41418-020-00633-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Annu Rev Immunol. 2000;18:143‐164. doi: 10.1146/annurev.immunol.18.1.143 [DOI] [PubMed] [Google Scholar]
- 20. Delgado‐Ortega M, Marc D, Dupont J, Trapp S, Berri M, Meurens F. SOCS proteins in infectious diseases of mammals. Vet Immunol Immunopathol. 2013;151:1‐19. doi: 10.1016/j.vetimm.2012.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cannon J. Inflammatory cytokines in nonpathological states. Physiology. 2000;15:298‐303. doi: 10.1152/physiologyonline.2000.15.6.298 [DOI] [PubMed] [Google Scholar]
- 22. Dalpke A, Heeg K, Bartz H, Baetz A. Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology. 2008;213:225‐235. doi: 10.1016/j.imbio.2007.10.008 [DOI] [PubMed] [Google Scholar]
- 23. Linossi E, Calleja D, Nicholson S. Understanding SOCS protein specificity. Growth Factors. 2018;36:104‐117. doi: 10.1080/08977194.2018.1518324 [DOI] [PubMed] [Google Scholar]
- 24. Wang B, Wangkahart E, Secombes C, Wang T. Insights into the evolution of the Suppressors of Cytokine Signaling (SOCS) gene family in vertebrates. Mol Biol Evol. 2018;36:393‐411. doi: 10.1093/molbev/msy230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duncan S, Baganizi D, Sahu R, Singh S, Dennis V. SOCS proteins as regulators of inflammatory responses induced by bacterial infections: a review. Front Microbiol. 2017;8:2431. doi: 10.3389/fmicb.2017.02431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454‐465. doi: 10.1038/nri2093 [DOI] [PubMed] [Google Scholar]
- 27. Thomas S, Snowden J, Zeidler M, Danson S. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015;113:365‐371. doi: 10.1038/bjc.2015.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liau N, Laktyushin A, Lucet I, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. 2018;9:1558. doi: 10.1038/s41467-018-04013-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akhtar L, Benveniste E. Viral exploitation of host SOCS protein functions. J Virol. 2010;85:1912‐1921. doi: 10.1128/jvi.01857-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H, Yoshimura A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways. Arterioscler Thromb Vasc Biol. 2011;31:980‐985. doi: 10.1161/atvbaha.110.207464 [DOI] [PubMed] [Google Scholar]
- 31. Blumer T, Coto‐Llerena M, Duong F, Heim M. SOCS1 is an inducible negative regulator of interferon λ (IFN‐λ)—induced gene expression in vivo. J Biol Chem. 2017;292:17928‐17938. doi: 10.1074/jbc.m117.788877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Delgado‐Ortega M, Melo S, Meurens F. Expression of SOCS1‐7 and CIS mRNA in porcine tissues. Vet Immunol Immunopathol. 2011;144:493‐498. doi: 10.1016/j.vetimm.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 33. Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS‐3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. 2000;275:29338‐29347. doi: 10.1074/jbc.m003456200 [DOI] [PubMed] [Google Scholar]
- 34. Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther. 2005;7:100. doi: 10.1186/ar1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liang Y, Xu W, Peng H, Pan H, Ye D. SOCS signaling in autoimmune diseases: molecular mechanisms and therapeutic implications. Eur J Immunol. 2014;44:1265‐1275. doi: 10.1002/eji.201344369 [DOI] [PubMed] [Google Scholar]
- 36. Khor C, Vannberg F, Chapman S, et al. CISH and susceptibility to infectious diseases. N. Engl J Med. 2010;362:2092‐2101. doi: 10.1056/nejmoa0905606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim W, Kim M, Kim D, et al. Suppressor of cytokine signaling 2 negatively regulates NK cell differentiation by inhibiting JAK2 activity. Sci Rep. 2017;7:46153. doi: 10.1038/srep46153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tannahill G, Elliott J, Barry A, Hibbert L, Cacalano N, Johnston J. SOCS2 can enhance interleukin‐2 (IL‐2) and IL‐3 signaling by accelerating SOCS3 degradation. Mol Cell Biol. 2005;25:9115‐9126. doi: 10.1128/mcb.25.20.9115-9126.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martens N, Uzan G, Wery M, Hooghe R, Hooghe‐Peters E, Gertler A. Suppressor of cytokine signaling 7 inhibits prolactin, growth hormone, and leptin signaling by interacting with STAT5 or STAT3 and attenuating their nuclear translocation. J Biol Chem. 2005;280:13817‐13823. doi: 10.1074/jbc.m411596200 [DOI] [PubMed] [Google Scholar]
- 40. Bullock A, Rodriguez M, Debreczeni J, Songyang Z, Knapp S. Structure of the SOCS4‐elonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS‐dependent EGFR degradation. Structure. 2007;15:1493‐1504. doi: 10.1016/j.str.2007.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sasi W, Sharma A, Mokbel K. The role of suppressors of cytokine signalling in human Neoplasms. Mol Biol Int. 2014;2014:1‐24. doi: 10.1155/2014/630797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seki Y, Hayashi K, Matsumoto A, et al. Expression of the suppressor of cytokine signaling‐5 (SOCS5) negatively regulates IL‐4‐dependent STAT6 activation and Th2 differentiation. Proc Natl Acad Sci. 2002;99:13003‐13008. doi: 10.1073/pnas.202477099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bayle J, Lopez S, Iwaï K, Dubreuil P, De Sepulveda P. The E3 ubiquitin ligase HOIL‐1 induces the polyubiquitination and degradation of SOCS6 associated proteins. FEBS Lett. 2006;580:2609‐2614. doi: 10.1016/j.febslet.2006.03.093 [DOI] [PubMed] [Google Scholar]
- 44. Hwang M, Min C, Kim H, et al. The nuclear localization of SOCS6 requires the N‐terminal region and negatively regulates Stat3 protein levels. Biochem Biophys Res Commun. 2007;360:333‐338. doi: 10.1016/j.bbrc.2007.06.062 [DOI] [PubMed] [Google Scholar]
- 45. Kario E, Marmor M, Adamsky K, et al. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. 2005;280:7038‐7048. doi: 10.1074/jbc.m408575200 [DOI] [PubMed] [Google Scholar]
- 46. Zhu Z, Lian X, Su X, Wu W, Marraro G, Zeng Y. From SARS and MERS to COVID‐19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res. 2020;21:224. doi: 10.1186/s12931-020-01479-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shereen M, Khan S, Kazmi A, Bashir N, Siddique R. COVID‐19 infection: emergence, transmission, and characteristics of human coronaviruses. J Adv Res. 2020;24:91‐98. doi: 10.1016/j.jare.2020.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yadav R, Chaudhary J, Jain N, et al. Role of structural and non‐structural proteins and therapeutic targets of SARS‐CoV‐2 for COVID‐19. Cells. 2021;10:821. doi: 10.3390/cells10040821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weiss S, Leibowitz J. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85‐164. doi: 10.1016/b978-0-12-385885-6.00009-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neuman B, Adair B, Yoshioka C, et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J Virol. 2006;80:7918‐7928. doi: 10.1128/jvi.00645-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Semper C, Watanabe N, Savchenko A. Structural characterization of nonstructural protein 1 from SARS‐CoV‐2. iScience. 2021;24:101903. doi: 10.1016/j.isci.2020.101903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khan M, Zeb M, Ahsan H, et al. SARS‐CoV‐2 nucleocapsid and Nsp3 binding: an in‐silico study. Arch Microbiol. 2020;203:59‐66. doi: 10.1007/s00203-020-01998-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hillen H, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS‐CoV‐2 polymerase. Nature. 2020;584:154‐156. doi: 10.1038/s41586-020-2368-8 [DOI] [PubMed] [Google Scholar]
- 54. Miorin L, Kehrer T, Sanchez‐Aparicio M, et al. SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci. 2020;117:28344‐28354. doi: 10.1073/pnas.2016650117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Y, Chen Y, Li Y, et al. The ORF8 protein of SARS‐CoV‐2 mediates immune evasion through down‐regulating MHC‐Ι. Proc Natl Acad Sci. 2021;118:e2024202118. doi: 10.1073/pnas.2024202118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Park A, Iwasaki A. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID‐19. Cell Host Microbe. 2020;27:870‐878. doi: 10.1016/j.chom.2020.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang S, Liu K, Cheng A, et al. SOCS proteins participate in the regulation of innate immune response caused by viruses. Front Immunol. 2020;11:558341. doi: 10.3389/fimmu.2020.558341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Song P, Li W, Xie J, Hou Y, You C. Cytokine storm induced by SARS‐CoV‐2. Clin Chim Acta. 2020;509:280‐287. doi: 10.1016/j.cca.2020.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Medina K, Garrett K, Thompson L, Rossi M, Payne K, Kincade P. Identification of very early lymphoid precursors in bone marrow and their regulation by estrogen. Nat Immunol. 2001;2:718‐724. doi: 10.1038/90659 [DOI] [PubMed] [Google Scholar]
- 60. Vadakedath S, Kandi V, Mohapatra R, et al. Immunological aspects and gender bias during respiratory viral infections including novel Coronavirus disease‐19 (COVID‐19): a scoping review. J Med Virol. 2021;93:5295‐5309. doi: 10.1002/jmv.27081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Okabayashi T, Kariwa H, Yokota S, et al. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J Med Virol. 2006;78:417‐424. doi: 10.1002/jmv.20556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Song M, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon‐mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273:35056‐35062. doi: 10.1074/jbc.273.52.35056 [DOI] [PubMed] [Google Scholar]
- 63. Zheng J, Yang P, Tang Y, Pan Z, Zhao D. Respiratory syncytial virus nonstructural proteins upregulate SOCS1 and SOCS3 in the different manner from endogenous IFN signaling. J Immunol Res. 2015;2015:1‐11. doi: 10.1155/2015/738547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu X, Zheng J, Zheng K, Hou Y, Zhao F, Zhao D. Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology. 2014;57:65‐73. doi: 10.1159/000357327 [DOI] [PubMed] [Google Scholar]
- 65. Seong R, Lee J, Shin O. Zika virus‐induction of the suppressor of cytokine signaling 1/3 contributes to the modulation of viral replication. Pathogens. 2020;9:163. doi: 10.3390/pathogens9030163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chiang S, Lin T, Chow K, Chiou S. SARS spike protein induces phenotypic conversion of human B cells to macrophage‐like cells. Mol Immunol. 2010;47:2575‐2586. doi: 10.1016/j.molimm.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stavnezer J, Guikema J, Schrader C. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261‐292. doi: 10.1146/annurev.immunol.26.021607.090248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Finigan J, Downey G, Kern J. Human epidermal growth factor receptor signaling in acute lung injury. Am J Respir Cell Mol Biol. 2012;47:395‐404. doi: 10.1165/rcmb.2012-0100tr [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gupta K, Xu Z, Castellino F, Ploplis V. Plasminogen activator inhibitor‐1 stimulates macrophage activation through Toll‐like Receptor‐4. Biochem Biophys Res Commun. 2016;477:503‐508. doi: 10.1016/j.bbrc.2016.06.065 [DOI] [PubMed] [Google Scholar]
- 70. Rojas M, Rodríguez Y, Monsalve D, et al. Convalescent plasma in Covid‐19: possible mechanisms of action. Autoimmun Rev. 2020;19:102554. doi: 10.1016/j.autrev.2020.102554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yokota S, Yokosawa N, Okabayashi T, Suzutani T, Fujii N. Induction of suppressor of cytokine signaling‐3 by herpes simplex virus type 1 confers efficient viral replication. Virology. 2005;338:173‐181. doi: 10.1016/j.virol.2005.04.028 [DOI] [PubMed] [Google Scholar]
- 72. Frey K, Ahmed C, Dabelic R, et al. HSV‐1‐Induced SOCS‐1 expression in keratinocytes: use of a SOCS‐1 antagonist to block a novel mechanism of viral immune evasion. J Immunol. 2009;183:1253‐1262. doi: 10.4049/jimmunol.0900570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Johnson H, Lewin A, Ahmed C. SOCS, intrinsic virulence factors, and treatment of COVID‐19. Front Immunol. 2020;11:582102. doi: 10.3389/fimmu.2020.582102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Collins A, McCoy C, Lloyd A, O’Farrelly C, Stevenson N. miR‐19a: an effective regulator of SOCS3 and enhancer of JAK‐STAT signalling. PLoS One. 2013;8:e69090. doi: 10.1371/journal.pone.0069090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu G, Yang F, Ding C, et al. MiR‐221 accentuates IFN׳s anti‐HCV effect by downregulating SOCS1 and SOCS3. Virology. 2014;462‐463:343‐350. doi: 10.1016/j.virol.2014.06.024 [DOI] [PubMed] [Google Scholar]
- 76. Yuan Y, Miao Y, Zeng C, et al. Small‐molecule inhibitors of ubiquitin‐specific protease 7 enhance type‐I interferon antiviral efficacy by destabilizing SOCS1. Immunology. 2019;159:309‐321. doi: 10.1111/imm.13147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Scheller E, Hankenson K, Reuben J, Krebsbach P. Zoledronic acid inhibits macrophage SOCS3 expression and enhances cytokine production. J Cell Biochem. 2011;112:3364‐3372. doi: 10.1002/jcb.23267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reghunathan R, Jayapal M, Hsu L, et al. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rabaan A, Al‐Ahmed S, Muhammad J, et al. Role of inflammatory cytokines in COVID‐19 patients: a review on molecular mechanisms, immune functions, immunopathology and immunomodulatory drugs to counter cytokine storm. Vaccines (Basel). 2021;9:436. doi: 10.3390/vaccines9050436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Capochiani E, Frediani B, Iervasi G, et al. Ruxolitinib rapidly reduces acute respiratory distress syndrome in COVID‐19 disease. Analysis of data collection from RESPIRE protocol. Front Med. 2020;7:466. doi: 10.3389/fmed.2020.00466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bhaskar S, Sinha A, Banach M, et al. Cytokine storm in COVID‐19—immunopathological mechanisms, clinical considerations, and therapeutic approaches: the REPROGRAM consortium position paper. Front Immunol. 2020;11:1648. doi: 10.3389/fimmu.2020.01648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D. Baricitinib therapy in COVID‐19: a pilot study on safety and clinical impact. J Infect. 2020;81:318‐356. doi: 10.1016/j.jinf.2020.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. La Manna S, Lopez‐Sanz L, Mercurio F, et al. Chimeric peptidomimetics of SOCS 3 able to interact with JAK2 as anti‐inflammatory compounds. ACS Med Chem Lett. 2020;11:615‐623. doi: 10.1021/acsmedchemlett.9b00664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Feldmann M, Maini R, Woody J, et al. Trials of anti‐tumour necrosis factor therapy for COVID‐19 are urgently needed. Lancet. 2020;395:1407‐1409. doi: 10.1016/s0140-6736(20)30858-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.