

Abstract
Boswellic acids (BAs) have been shown to possess antiviral activity. Using bioinformatic methods, it was tested whether or not acetyl‐11‐keto‐β‐boswellic acid (AKBA), 11‐keto‐β‐boswellic acid (KBA), β‐boswellic acid (BBA), and the phosphorylated active metabolite of Remdesivir® (RGS‐P3) bind to functional proteins of SARS‐CoV‐2, that is, the replicase polyprotein P0DTD1, the spike glycoprotein P0DTC2, and the nucleoprotein P0DTC9. Using P0DTD1, AKBA and KBA showed micromolar binding affinity to the RNA‐dependent RNA polymerase (RdRp) and to the main proteinase complex Mpro. Phosphorylated BAs even bond in the nanomolar range. Due to their positive and negative charges, BAs and RGS‐P3 bond to corresponding negative and positive areas of the protein. BAs and RGS‐P3 docked in the tunnel‐like cavity of RdRp. BAs also docked into the elongated surface rim of viral Mpro. In both cases, binding occurred with active site amino acids in the lower micromolecular to upper nanomolar range. KBA, BBA, and RGS‐P3 also bond to P0DTC2 and P0DTC9. The binding energies for BAs were in the range of −5.8 to −6.3 kcal/mol. RGS‐P3 and BAs occluded the centrally located pore of the donut‐like protein structure of P0DTC9 and, in the case of P0DTC2, RGS‐P3 and BAs impacted the double‐wing‐like protein structure. The data of this bioinformatics study clearly show that BAs bind to three functional proteins of the SARS‐CoV‐2 virus responsible for adhesion and replication, as does RGS‐P3, a drug on the market to treat this disease. The binding effectiveness of BAs can be increased through phosphate esterification. Whether or not BAs are druggable against the SARS‐CoV‐2 disease remains to be established.
Keywords: boswellic acids, functional proteins, molecular docking, RGS‐P3, SARS‐CoV‐2
Bioinformatic methods showed that acetyl‐11‐keto‐β‐boswellic acid, 11‐keto‐β‐boswellic acid, β‐boswellic acid, and the phosphorylated active metabolite of Remdesivir® bind to functional proteins of SARS‐CoV‐2: the replicase polyprotein P0DTD1, the spike glycoprotein P0DTC2, and the nucleoprotein P0DTC9. The binding effectiveness of the boswellic acids can be increased through phosphate esterification.
Abbreviations
- 1S76
PDB code for the T7 RNA polymerase alpha‐beta methylene ATP elongation complex
- 2H2Z
PDB code for the SARS‐CoV main protease structure
- 3CL Prot
3C‐like proteinase, AKA main proteinase, or Mpro for short
- 4K4S
PDB code for the poliovirus polymerase elongation complex
- 6LU7
PDB code for the COVID‐19 main protease structure in complex with N3
- 6LVN
crystal structure of the 2019‐nCoV HR2 domain
- 6M71
PDB code for the SARS‐Cov‐2 RNA‐dependent RNA polymerase structure in complex with cofactors
- 6NUR
PDB code for the SARS‐coronavirus NSP12 structure bound to NSP7 and NSP8 co‐factors
- 6VYO
crystal structure of the RNA binding domain of the nucleocapsid phosphoprotein from SARS coronavirus 2
- ACE2
angiotensin‐converting enzyme 2
- AD4
AutoDock 4 version 4.2, software for docking
- AKBA
acetyl‐11‐keto‐β‐boswellic acid
- ATP
adenosine triphosphate
- BA
boswellic acid, general term to denominate all studied derivatives
- BBA
β‐boswellic acid CLEC4M/DC‐signal receptor
- KBA
11‐keto‐β‐boswellic acid
- Mpro
main proteinase of replicase protein 1ab
- N3
peptide‐like ligand of 6LU7
- NSP
nonstructural protein
- P0DTC1
UniProtKB code for Tt replicase polyprotein 1a (RIA SARS 2)
- P0DTC2
UniProtKB code for spike glycoprotein (SPIKE SARS 2)
- P0DTC9
UniProtKB code for nucleoprotein (NCAP SARS 2)
- P0DTD1
UniProtKB code for replicase polyprotein 1ab (R1AB_SARS2)
- P1KBA
mono‐phosphorylated derivative of KBA
- P2KBA
di‐phosphorylated derivative of KBA
- PDB
file format acronym for structure entries at the RCSB Protein Data Bank
- PDBQT
file format acronym for the Protein Data Bank with partial charges and torsions
- PKBA
phosphorylated derivative of KBA
- PL‐PRO
coronavirus papain‐like proteinase
- RdRp
SARS‐CoV‐2 RNA‐dependent RNA polymerase, AKA RNA‐directed RNA polymerase
- RGS‐P3
RGS triphosphate, active metabolite of the prodrug remdesivir (RMSDV)
- SPDBV
Swiss PDB Viewer, software for protein modeling
- Ø
symbol for an averaged value
1. INTRODUCTION
Boswellic acids (BAs)—the pharmacological ingredients of the gum resin of Boswellia species—have been shown to be active in chronic inflammatory diseases with an autoimmune character and inhibit increased expression of proinflammatory cytokines.[ 1 ] In addition, antiviral activities have been reported.[ 2 , 3 , 4 , 5 ] These observations suggest that BAs may also be active in other types of viruses including SARS‐CoV‐2.
Molecular modeling and bioinformatics[ 6 , 7 ] were applied to detect and quantify any potential anti‐SARS‐CoV‐2 activities of three major Boswellia resin constituents, namely, acetyl‐11‐keto‐β‐boswellic acid (AKBA), 11‐keto‐β‐boswellic acid (KBA), and β‐boswellic acid (BBA). As biomolecular targets, three viral protein multisubunit complexes have been selected because they are involved in adhesion to the host cells and virus replication: P0DTD1, P0DTC9, and P0DTC2.
2. RESULTS
2.1. Molecular structures related to protein docking
For our screening study of boswellic acids (BAs), we initially created three three‐dimensional (3D) models of AKBA, BBA, and KBA. As their targets, we selected two crystal complexes, Mpro and RdRp (RNA‐dependent RNA polymerase). As Remdesivir® constitutes a prodrug, only its active form RGS triphosphate (RGS‐P3) was studied by AD4, not Remdesivir® itself. The ligands N3 (peptide‐like ligand of 6LU7) and RGS‐P3 were studied as reference ligands (positive control).
The favorable implications of our approach are manifold:
-
(i)
The binding sites and positions of both reference ligands (RGS‐P3 and N3) are crystallographically known (docked poses). Both were successfully “docked back” into their known poses of crystal complex as validation tests to confirm that the computer model is capable of correctly predicting the binding strengths and positions of the ligands in the crystal complex. In contrast, so‐called “blind docking” had to be carried out for BAs.
-
(ii)
Blind docking means that the final poses are determined on computational grounds without knowing their experimentally determined binding poses (“blind”). It is generally accepted that, when ligands from crystal structures can be successfully “docked back,” then also “blind docking” of hitherto unknown ligands at the same binding site can be considered as trustworthy under the same simulation conditions, that is, unchanged software settings.
-
(iii)
Moreover, advances in structure–activity relationship studies by scaffold‐hopping during the last two decades have proven not only similar chemical to similar biological activities by sharing a common molecular binding mechanism to the same target but also congeners based on completely different scaffolds, for example, in the kinome (proteome of the kinases).
For these reasons, our BA screening by AutoDock 4 was limited to the viral interactome, that is, SARS‐CoV‐2 proteins with known binding sites through experimentally determined ligand complexes (PDB codes: 6M71,[ 8 ] 6NUR,[ 9 ] 2H2Z,[ 10 ] and 6LU7[ 11 ]).
Taken together, our approach to combine so‐called “blind‐docking” of BAs (unknown outcome) with so‐called “back or self‐docking” of two reference substances (published outcome) constitutes the most valuable asset, that is, we could successfully reproduce the experimentally determined binding modes of both reference substances with their respective targets (RdRp, Mpro).
2.1.1. Virtual screening at the RGS‐P3 binding site of RdRp and the N3 binding site of Mpro
The locations of the binding sites for both target enzymes[ 9 , 11 ] were identified by superposition techniques. In our first case concerning the target protein for remdesivir, which is the prodrug of RGS‐P3, we followed the established protocol set by Shannon and coworkers earlier last year.[ 12 ] In the second case, we studied Mpro and applied the Magic Fit module of SPDBV for the 3D alignment with unliganded 2H2Z[ 10 ] and liganded 6LU7.[ 11 ] Returning to the first case, the binding site became evident only by the user‐assisted 3D alignment by Match Maker of Chimera 1.14[ 13 ] of the following four three‐dimensional models: (i) SARS‐CoV‐2 RdRp in complex with cofactors; (ii) SARS‐coronavirus NSP12 bound to NSP7 and NSP8 co‐factors; (iii) the T7 RNA polymerase alpha‐beta methylene adenosine triphosphate (ATP) elongation complex; and (iv) the poliovirus polymerase elongation complex (PDB codes: 6M71, 6NUR, 1S76,[ 14 ] and 4K4S)[ 15 ] as none of the RGS‐P3 target structures is available with a binding site‐bound drug or inhibitor. In the latter case, the bound ligand is part of the target crystal structure itself (COVID‐19 main protease in complex with an experimental inhibitor named N3 [PDB code: 6LU7]).[ 11 ]
AKBA and KBA are strong binders to both target molecules in the lower micromolecular to upper nanomolar range (see Table 1). The highest affinities coincide with the postpopulated RMSDV clusters, that is, groups of final poses in the same spatial Cartesian coordinates (x, y, z axes). This match indicates that the BAs with the stiff scaffolds do not encounter problems in occupying either the deep cleft at the active site of the RGS‐P3 binding protein or the shallow but elongated crevice (or rim) of the N3 binding site on the surface of Mpro. The individually found docking poses were merged into one 3D model to compare the results.
Table 1.
Binding energies and binding concentrations of boswellic acids and RGS‐P3 on P0DTD1 virus proteins
| Ligand | Binding energy (kcal/mol) | Binding concentration (µM) |
|---|---|---|
| Target: RNA‐dependent RNA polymerase (RdRp); UniProt code: P0DTD1; PDB code: 5M71+4K4S+1S76 | ||
| AKBA | −7.9 | 1.5 |
| KBA | −7.4 | 3.6 |
| RGS‐P3 | −10.8 | 0.01 |
| P1KBA | −9.9 | 0.06 |
| P2KBA | −10.7 | 0.01 |
| Target: Main protease of the replicase polyprotein (Mpro); UniProt code: P0DTD1; PDB code: 2H2Z/6LU7 | ||
| AKBA | −8.9 | 0.3 |
| KBA | −8.4 | 0.7 |
| N3 | −9.5 | 0.1 |
| P1KBA | −8.7 | 0.4 |
| P2KBA | −8.0* | 1.1 |
Abbreviations: AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid; N3, peptide‐like ligand of 6LU7; P1KBA, mono‐phosphorylated derivative of KBA; P2KBA, di‐phosphorylated derivative of KBA; RGS‐P3, RGS triphosphate.
2.1.2. Docking of BAs into the viral cavity of RdRp (P0DTD1)
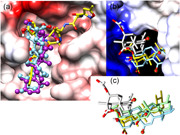
In Figure 1a, two final docked poses of KBA and AKBA occupy the elongated surface rim of viral main protease Mpro. In this figure, ball and stick models of AKBA (purple C atoms) and KBA (light blue) are shown. The position of the reference ligand N3 (yellow) was taken from crystal data (PDB code: 6LU7). The protein surface was color‐coded (from red over rosa to white and cyan to blue) to symbolize negative partial charges, electrostatic neutrality (nonpolar regions), and positive partial charges. Ligand atoms are as follows: O in red, N in blue, and H omitted for visibility. By the naked eye, the ligand–receptor complementarity can be appreciated. The oxygen‐rich head groups of BAs are linked to the receptor by a highly polar hydrogen‐bond network. The hydrophobic tail is flanked by nonpolar side chains in the binding crevice (or rim).
Figure 1.
Docking poses of KBA and AKBA on P0DTD1 virus proteins: (a) Main proteinase Mpro, (b) RNA‐dependent, and (c) superposition of KBA and AKBA. AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid
Figure 1b shows that BAs docked into the cavity of viral RdRp. The “tunnel‐like” hole that belongs to the active site of the host RNA stands for polymerase activity. The 3D model was generated based on multiple templates (PDB codes: 6M71, 6NUR, 4K4S, and 1S76). The protein surface was color‐coded (from red over pink to white, and cyan to blue) to symbolize negative partial charges, electroneutrality (nonpolar regions), and positive partial charges. Ligand atoms are as follows: O in red, N in blue, and H omitted for visibility. By the naked eye, the ligand–receptor complementarity can be appreciated. Strong ion bridges and highly polarized hydrogen bonds are established between the positively charged surface (blue, topmost) and the negatively charged ligand head groups. The aliphatic ring system is partly engulfed in a hydrophobic pocket (white surface to the foreground, bottom‐most). Two poses of ligand AKBA are shown (yellow or gray C atoms). All H atoms were omitted. The light blue stick model shows KBA; its carboxy group is hidden in the rear part. The keto groups on both BA forms are highlighted in tiny green cycles.
Computed evidence is displayed in Figure 1a, where both BAs bind to the protein surface of the replication protein of the SARS‐CoV‐2 virus via positive and negative charges of both ligands. It is noteworthy to compare the missing direct interaction with receptor side chains in stark contrast to our findings reported in the experimental and theoretical study on the modulation of glucocorticoid receptors by BAs.[ 16 ]
Figure 1c shows the superposition of two BAs (AKBA and KBA) in four final docked poses. The same orientation, as in Figure 1b, is noteworthy but without the polymerase cavity. After docking into the deep cavity at the active site, almost all final solutions lie in identical orientations and positions or close to perfect identity. For the acetylated derivative, the search algorithm found more favorable binding variations thanks to the increment of conformational freedom from the larger –O–C(═O)–CH3 side chain. This finding can be appreciated here (two models of AKBA: yellow and gray). In contrast, KBA (two stick models: green and light blue) binds with very similar poses (narrower range of possible solutions) due to its restricted conformational flexibility.
The structural similarity of BAs to steroid hormones hints at their capability to enter cell nuclei by the same mechanism as the natural glucocorticoid hormones and drug derivatives, like dexamethasone.[ 16 ] In this way, it is safe to state that BAs can enter the cell nuclei and bind to the same site of RGS‐P3 binding on the viral RdRp. The expected detrimental effect is a direct consequence of blocking the RNA reading site of the enzyme, a key step in the virus replication after cell infection.
2.1.3. Docking of BAs into the main protease complex
Concerning the viral target Mpro, BAs are long enough to fill in a little more than half of the narrow but very extended crevice at the protease surface. As the pentacyclic triterpene scaffold is not branched and the side chains are scarce and extremely short, two adjacent pockets remain unoccupied upon binding. This provides opportunities for increment specificity and selectivity of BAs by introducing one or two larger side chains. In cycles of drug design, organic synthesis, and bioassays, novel semisynthetic derivatives could be tested. On the basis of the computed evidence, we draw the conclusion that BAs are recognized by Mpro and are amenable to derivatization for drug profiling.
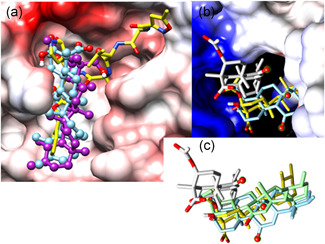
Before docking, the prodrug remdesivir was converted into its active form RGS‐P3, which is active at the site of the human RNA polymerase machinery in its triphosphate ester form (RGS‐P3). Structurally, it is similar to ATP (Figure 2).
Figure 2.
Docking poses of RGS‐P3, KBA, and AKBA on P0DTD1 virus proteins: (a) Binding site of the RdRp protein, (b) cyano group binding of RGS‐P3, and (c) superposition of RGS‐P3, KBA, and AKBA. AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid; RdRp, RNA‐dependent RNA polymerase; RGS‐P3, RGS triphosphate
Concerning the spatial subunit arrangement for the multi‐subunit RNA‐synthesis complex, some tinkering was necessary to localize the binding site of the prodrug remdesivir and its active metabolite RGS‐P3 on the target structure (PDB code: 6M71). To this end, two magnesium cations and an ATP‐like ligand (adenosyl ester of diphospho‐methyl‐phosphonic acid, APC for short) from the RNA polymerase active sites were imported upon their superposition (PDB codes: 4K4S, 1S76, and 6NUR). The affinity range of RGS‐P3 lies in the lower nanomolar range and its binding is stronger than that of BAs (see Table 1).
Figure 2a shows the binding site of RdRp with RGS‐P3 and BAs in the final docking positions (poses). Two poses of RGS‐3P (yellow stick models) span from one side to the positively charged side (blue) of the “tunnel‐like” cavity (two entries in the foreground and the white background), where its anionic tri‐phosphate rest is strongly anchored by electrostatic attraction. AKBA (light green, purple, gray) has more computed solutions due to its higher degree of conformational freedom than KBA (light blue). Intriguingly, the keto group, which triggers steroid hormone signaling,[ 16 ] remained unbounded (four small red balls). It is noteworthy that BBA (without keto group) occupies the same receptor position.
Figure 2b shows where the cyano group of RGS‐P3 binds: It is directed into a negatively charged small pocket at the entrance to the active site (fusion model of PDB codes: 6M71, 6NUR, 4K4S, 1S76). This NC‐ group docks neatly in a small pocket with favorable resonance properties with respect to the group inductive and mesomeric effects (−1, −M). The steric and electronic fitness of the small pockets is the most valuable asset to identify the binding pose of RGS‐P3 due to hitherto missing experimental evidence (no RGS‐liganded crystal structure available).
In Figure 2c, all final docked poses are in the same orientation as those in Figure 2a. All oxygen‐dotted (or polar) ligand substructures are oriented toward one side, whereas the less (or non‐) polar parts are aligned toward the more hydrophobic side of the binding cavity, evidencing the validity of established rules about electrostatic attractions and repulsions. RGS‐P3 is partly—but not fully—within the positions of BAs.
2.2. Increase in the docking affinity of BAs by phosphorylation
Administration of therapeutic doses of boswellic extract preparations to humans revealed blood concentrations of KBA in the range of 1.0 to 2.5 µM, but nearly no AKBA could be detected.[ 17 ] The binding concentration of RGS, the inactive metabolite of RMSDV, is reported here to be approx. 7.0 kcal/mol. As phosphorylated RGS (RGS‐P3) is the active form of RGS, the binding concentration of RGS, the inactive metabolite of RMSDV, is reported to be approx. 7.0 kcal/mol. As phosphorylated RGS (RGS‐P3) is the active form of RGS, the question arises if binding concentrations of BAs are in the range of available blood levels and, if not, phosphorylation of BAs would increase the binding concentration and therapeutic efficacy. AKBA was modified to elucidate the structure–activity relationship by replacing the acetyl group with phosphate ester groups.
Using the replicase polyprotein P0DTD1 as the target, the binding energy and the binding concentration of nonphosphorylated and phosphorylated BAs as well as RGS‐P3 were tested in RdRp as well as the main protease (see Table 1).
Table 1 lists the binding energies and binding concentrations of docked ligands. The values were computed by AutoDock 4.2 (AD4) as the estimated free energy of binding (E) and binding constant (K i). It is noteworthy that P1KBA is the phosphate monoester analog of AKBA, that is, a monoanionic phosphate group has replaced the acetyl side chain of AKBA. Both side chains are linked by an ester bond to PKBA or AKBA, respectively.
2.3. Binding mode analysis of P2KBA, RGS‐P3, and N3 to RdRp and Mpro target proteins
Following the data of Table 1, it was of interest to study the binding mode of P2KBA, the most active derivative of KBA, in comparison to RGS‐P3 and N3 to P0DTD1 targets, RdRp and Mpro. The binding poses of the ligands were analyzed and the interacting amino acids at the binding sites for RdRp and Mpro were identified and documented in two‐dimensional drawings (Figure 3). To this end, the 3D space around the bound ligand was projected into the 2D drawing under the assumption that amino acids that lie in the rear (front) part of the cavity would appear above (below).
Figure 3.
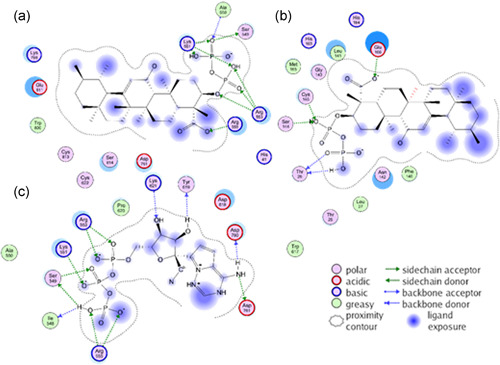
Docking poses of P2KBA and RGS‐P3 to amino acids of the RdRp and Mpro proteins of the P0DTD1 virus polyprotein: (a) P2KBA binding to RdRP, (b) P2KBA binding to Mpro, and (c) RGS‐P3 binding to RdRp. AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid; RdRp, RNA‐dependent RNA polymerase; RGS‐P3, RGS triphosphate
In Figure 3a, the KBA diphosphoric ester (P2KBA) is shown with the interacting amino acids at the binding site of RdRp. In this way, all other BA derivatives also bind KBA monophoshoric ester (P1KBA), AKBA, and KBA itself (not shown). The anionic phosphate groups and the carboxylate head group of BAs bind strongly to the cationic side chains of arginine and lysine through an elaborated concert of salt bridges and polarized hydrogen‐bonding networks. Figure 3b illustrates which residues of Mpro interact with P2KBA. The di‐phosphorylation of BAs does not enhance the binding affinity to the Mpro target in the same way as it does in the case of the RdRp target (Figure 3a). There is a trade‐off between establishing intensive hydrogen‐bond networking with the phosphate or carboxylate head group of BAs and the (water) solvent exposure of the lipophilic tail part of the pentacyclic ligand scaffold. This can be noticed in the quantitative evaluation of the docking results (see Table 1).
The BA binding site was found to be the same as that of N3 along with residues MET165 (methionine [with its ID number] 165 in 6LU7) and GLU166 (glutamate [with its ID number] 166 in 6LU7). Details about interacting amino acids with ligand N3 at the binding site of Mpro have been recently published.[ 11 ] The estimated free energy of binding of N3 is so strong that affinity can be reached in just a 10th of micromolar concentrations (see Table 1).
Figure 3c (RGS‐P3/RdRp) shows ligand RGS‐P3 with the interacting amino acids at the binding site of RdRp. The phosphate groups of RGS‐P3 bind in the same arginine and lysine‐rich region as in the BAs cases.
2.4. Binding sites and binding energy of BAs and RGS‐P3 to three functional proteins of SARS‐CoV‐2
So far, the binding and binding properties of BAs and reference substance RGS‐P3 have been studied using the replicase protein P0DTD1. In a second series of experiments, it was tested whether or not BAs also bind to the spike protein (P0DTC2) and the replicase nucleoprotein (P0DTC9). Figures 4, 5, 6 show binding of the ligands to the proteins. The binding of test substances to virus proteins was estimated again by calculation of the binding energy as kcal/mol. For every chemical structure, the top 20 bond options with the highest binding energies to the related structure of the protein have been calculated. The Ø binding energy shows how a strong chemical substance such as a ligand is bound to a protein structure.
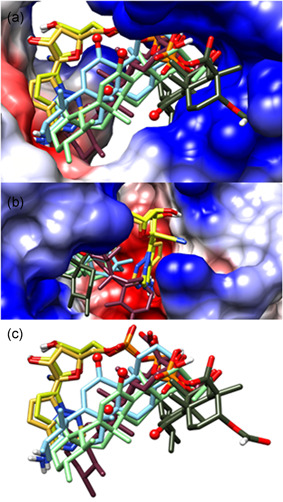
Figure 4.
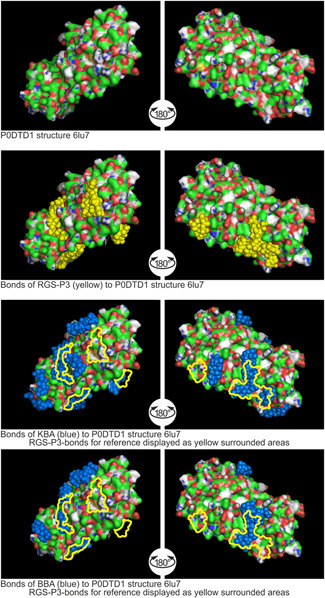
Binding sites of RGS‐P3, KBA, and BBA to replication protein P0DTD1 structure 6LU7. AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid; RGS‐P3, RGS triphosphate
Figure 5.
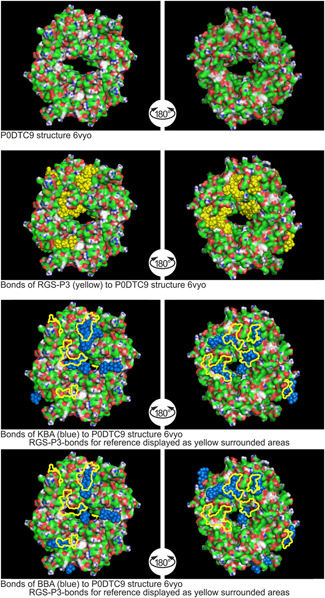
Binding sites of RGS‐P3, KBA, and BBA to nucleoprotein P0DTC9 structure 6VYO. AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid; RGS‐P3, RGS triphosphate
Figure 6.
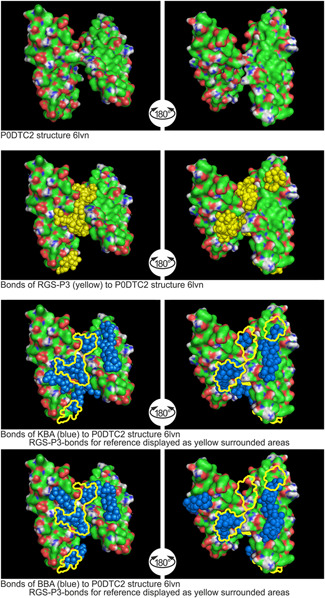
Binding sites of RGS‐P3, KBA, and BBA to spike glycoprotein P0DTC2 structure 6LVN. AKBA, acetyl‐11‐keto‐β‐boswellic acid; KBA, 11‐keto‐β‐boswellic acid; RGS‐P3, RGS triphosphate
2.4.1. Binding to P0DTD1 structure 6LU7
As shown in Figure 4, RGS‐P3, the active metabolite of remdesivir, shows Ø binding energy of −5.8 kcal/mol as confirmed status of comparison.
The KBA molecule develops more volume than RGS‐P3 due to the five rings. Its Ø binding energy is −6.1 kcal/mol, 105% force as RGS‐P3. A few binding sites of KBA are not in the region of RGS‐P3. The BBA molecule also develops more volume due to the five rings than RGS‐P3. The BBA Ø binding energy is −6.3 kcal/mol, more than those of RGS‐P3. A few binding sites of BBA are not in the region of RGS‐P3.
2.4.2. Binding to P0DTC 9‐structure 6VYO
Figure 5 shows that RGS‐P3 impacts the surface structure of the donut‐like protein structure inside the centrally located pore.
The average or mean value (symbol: Ø) reaches the value of −6.5 kcal/mol. KBA occludes the centrally located pore nearly completely. The Ø binding energy is −6.2 kcal/mol, 95% force compared to RGS‐P3. BBA occludes the centrally located pore nearly completely, too. The BBA Ø binding energy is −6.3 kcal/mol, 97% force compared to RGS‐P3.
2.4.3. Binding to P0DTC2‐structure 6LVN
The docking study with target 6LVN is shown in Figure 6.
The spike glycoprotein attaches the virion to the cell membrane by interacting with the host receptor, initiating the infection. Binding to human ACE2 and CLEC4M/DC‐SIGNR receptors and internalization of the virus into the endosomes of the host cell induce conformational changes in the spike glycoprotein.[ 18 ] RGS‐P3 impacts the surface structure at the junction of the double‐wing‐like protein structure. The Ø binding energy of RGS‐P3 was calculated to be −5.4 kcal/mol. KBA also impacts the surface structure comparably. At the binding sites of RGS‐P3, the protein structure is covered completely by KBA. The KBA Ø binding energy is −5.8 kcal/mol, which is approx. 107% of the Ø binding energy of RGS‐P3. BBA impacts the surface structure like KBA on a large scale and meets binding sites of RGS‐P3. BBA Ø binding energy shows −5.8 kcal/mol, too.
3. DISCUSSION
The computed data of this study show that BA derivatives bind to functional proteins of the SARS‐CoV‐2 virus as does the active metabolite of RMSDV (RGS‐P3). In detail: Using the replicase polyprotein P0DTD1 (Figures 1 and 2), it was found that KBA, AKBA, and BBA bind to this protein. Phosphate esters of the BAs under scrutiny are even stronger binders to the viral RdRp than native BAs. The elongated structures of the BAs occupied the active site in the elongated surface of the main protease of the virus (Mpro) again through charge complementarities. Binding of BAs was not dependent on their C‐11 keto group as is the case regarding their anti‐inflammatory activities[ 1 ] in direct interaction with the glucocorticoid receptor.[ 16 ] In this regard, RGS‐P3 spans from one site to the positively charged site of the tunnel‐like cavity of RdRp, where its anionic triphosphate rest is anchored by electrostatic attraction. Its cyano group is directed into a negatively charged small pocket at the entrance of the active site of the proteins. In contrast to BAs, RGS‐P3 did not bind to the main protease Mpro .
3.1. Increased affinity of KBA by derivatization through phosphorylation
Binding energies are strongly related to molar concentrations. The higher the binding energy, the lower the necessary ligand concentration and the stronger the binding. This is evident in Table 1, where RGS‐P3, the active metabolite of RMSDV (not RGS, which is the inactive metabolite of RMSDV), shows high binding energy and low molecular binding concentration, suggesting that phosphorylation of ligands of the protein might increase their binding energy and decrease the binding concentration. This also seems to hold for BAs, where, in the case of the RdRp protein of P0DTD1, diphosphorylation of KBA increased the binding energy and decreased the binding concentration into the order of RGS‐P3.
3.2. Binding sites and binding energy of BAs to the three functional proteins of SARS‐CoV‐2
Using another simulation model, it can be seen from Figures 4, 5, 6 that KBA, BBA (blue), and RGS‐P3 (yellow) bind to the P0DTD1, P0DTC9, and P0DTC2 proteins of the virus. By the naked eye, it is clear that KBA and BBA overlap each other and thus show similar binding energies. This means that BAs attack the virus at three critical stages during host infection and the viral reproduction cycle. The three stages correspond to three functional proteins that are responsible for adhesion to the host cells and viral RNA replication. In the case of P0DTC9, both BAs occlude the central pore of the donut‐like protein, and as far as P0DTC2 is concerned, they impact the surface structure at the junction of the double‐wing protein structure. RGS‐P3, the active metabolite of RMSDV, shows nearly identical binding sites.
4. CONCLUSION
BAs have been reported to possess antiviral properties. The data of this bioinformatics study suggest that they target SARS‐CoV‐2 viruses on an atomic scale on three functional proteins, which in turn are responsible for human cell adhesion or viral RNA replication. RGS‐P3, the active metabolite of Remdesivir®, served as the reference compound during our virtual screening campaign. The binding affinities of BAs can be increased through phosphorylation. However, further experimental studies are necessary to document antivirus activities in COVID‐19 disease.
5. EXPERIMENTAL
5.1. COVID‐19 virus protein targets
The following multi‐subunit and structural data were obtained from UniProtKB[ 18 ] and the European Bioinformatics Institute[ 19 ] as well as the Protein Data Bank (PDB)[ 20 ]:
P0DTD1: Viral replicase polyprotein 1ab main protease (Mpro) under PDB codes: 2H2Z and 6LU7[ 20 ] and RdRp under PDB codes: 6M71 and 6NUR.
P0DTC2: Spike glycoprotein under PDB code: 6LVN.[ 18 ]
P0DTC9: Nucleoprotein under PDB code: 6VYO.[ 18 ]
The chemical structures of the test substances used for binding studies with virus proteins are shown in Figure 7. The 3D models of the ligands were obtained from the National Library of Medicine.[ 21 ]
Figure 7.
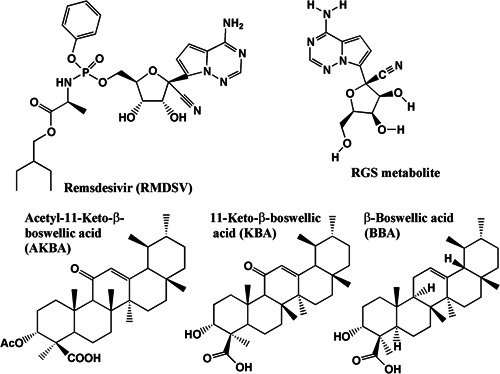
Chemical structures of RMDSV, RGS, AKBA, KBA, and BBA
5.2. Computing material
Hardware: Processor: Intel® Core™ 17‐870G5 CPU @ 3.10 GHZ 3.10 GHZ; Installed RAM: 32,0 GB, System type: 64‐Bit‐operating system, x64‐based processor.
Software: Operating system: Windows™ 10 Pro, Version: 1901.
Molecular modeling software: AutoDock Vina version 1.1.2,[ 7 ] SPDBV,[ 22 ] Vega ZZ,[ 23 ] Chimera 1.14,[ 24 ] AutoDock 4.2 (AD4),[ 25 ] AutoDock Tools version 1.5.6,[ 25 ] and PyMol version 0.99.[ 26 ]
5.3. Methods
5.3.1. Settings for the boswellic acid screening by AD4 at the RGS‐P3 binding site of RdRp and the N3 binding site of Mpro
In view of our experience with a reduced set of functional groups (one ═O; one –OH or –OR sitting on a larger stiff aliphatic ring system), we applied our published docking protocol for BAs.[ 16 ]
5.3.2. Binding sites and binding energy of test substances by AutoDock Vina
Bioinformatic calculations of molecular docking options, energies and positions of ligands to protein structures: For every combination of chemical substance and protein structure, a bioinformatical calculation has been performed using AutoDoc Vina[ 7 ] to find the top 20 docking options together with their energies and positions at the appropriate protein structure. The results have been compared to the binding situation of RGS‐P3 as the reference ligand.
CONFLICTS OF INTEREST
Reinhard H. Caliebe is the owner of NOVOHERBS UG (haftungsbeschränkt) & Co. KG, which is involved in the business of extraction of Boswellia serrata. Hermann T. P. Ammon and Thomas Scior are researchers in the field of Boswellia pharmacology. These authors declare that there are no conflicts of interest.
ACKNOWLEDGMENTS
The authors are grateful to Professor Stefan Laufer, Tuebingen, for valuable scientific advice while preparing the manuscript. Reinhard H. Caliebe and Thomas Scior equally contributed to computer models. Open Access funding enabled and organized by Projekt DEAL.
Caliebe R. H., Scior T., Ammon H. P. T.. Binding of boswellic acids to functional proteins of the SARS‐CoV‐2 virus: Bioinformatic studies. Arch. Pharm. 2021;354:2100160. 10.1002/ardp.202100160
REFERENCES
- 1. Ammon H. P. T., Adv. Exp. Med. Biol. 2016, 928, 291. [DOI] [PubMed] [Google Scholar]
- 2. Hussein T., Miyashiro H., Nakamura N., Hatton M., Kakiuchi N., Shimotohno K., Phytother. Res. 2000, 14, 510. [DOI] [PubMed] [Google Scholar]
- 3. Mothana R. A., Mentel R., Reiss C., Lindequist U., Phytother. Res. 2006, 20, 298. [DOI] [PubMed] [Google Scholar]
- 4. von Rhein C., Weidner T., Henß L., Martin J., Weber C., Sliva K., Schnierle B. S., Antiviral Res. 2016, 125, 51. 10.1016/j.antiviral.2015.11.007 [DOI] [PubMed] [Google Scholar]
- 5. Goswami D., Mahapatra A. D., Banerjee S., Kar A., Ojha D., Mukherjee P. K., Chattopadhyay D., Phytomedicine 2018, 51, 94. 10.1016/j.phymed.2018.10.016 [DOI] [PubMed] [Google Scholar]
- 6. Liu S., Zheng Q., Wang Z., Bioinformatics 2020, 36, 3295. 10.1093/bioinformatics/btaa224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trott O., Olson A. J., J. Comput. Chem. 2010, 31, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L. W., Wang Q., Lou Z., Rao Z., Science 2020, 368, 779. 10.1126/science.abb7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirchdörfer R. N., Ward A. B., Nat. Commun. 2019, 10, 2342. 10.1038/s41467-019-10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Yin Y., Bartlam M., Rao Z., J. Mol. Biol. 2007, 366, 965. 10.1016/J.jmb.2006.11.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Nature 2020, 582, 289. https://doi.org/10,1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- 12. Shannon A., Le N. T., Selisko B., Eydoux C., Alvarez K., Guillemot J. C., Decroly E., Peersen O., Ferron F., Canard B., Antiviral Res. 2020, 178, 104793. 10.1016/j.antiviral.2020.104793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605. [DOI] [PubMed] [Google Scholar]
- 14. Yin Y. W., Steitz T. A., Cell 2004, 116, 393. 10.1016/s0092-8674(04)00120-5 [DOI] [PubMed] [Google Scholar]
- 15. Gong P., Kortus M. G., Nix J. C., Davis R. E., Peersen O. B., PLOS One 2013, 8, e60272. 10.1371/journal.pone.0060272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scior T., Verhoff M., Gutierrez‐Aztatzi I., Ammon H. P. T., Laufer S., Werz O., J. Chem. Inf. Model. 2014, 54, 978. [DOI] [PubMed] [Google Scholar]
- 17. Ammon H. P. T., Weihrauch—Anwendung in der westlichen Medizin, Springer Verlag, Berlin Heidelberg, Germany: 2018. [Google Scholar]
- 18. https://www.ebi.ac.uk/pdbe/covid-19. Accessed 17, 2021.
- 19. https://www.ebi.ac.uk/. Accessed 17, 2021.
- 20. https://www.uniport.org/uniprot. P0DTD1. Accessed 17, 2021.
- 21. https://Pubchem.ncbi.nlm.nih.gov/. Accessed 17, 2021.
- 22. Guex N., Peitsch M. C., Electrophoresis 1997, 18, 2714. [DOI] [PubMed] [Google Scholar]
- 23. Pedretti A., Mazzolari A., Gervasoni S., Fumagalli L., Vistoli G., Bioinformatics 2020, 37, 1174. 10.1093/bioinformatics/btaa774; http://www.ddl.unimit/VegaZZ [DOI] [PubMed] [Google Scholar]
- 24. http://www.cgl.ucsf.edu/chimera. Accessed 17, 2021.
- 25. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J., J. Comput. Chem. 2009, 16, 2785. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The PyMOL Molecular Graphics System, Version 1.2r3pre, New York, NY, United States: Schrödinger, LLC.