

Abstract
Severe COVID‐19 is characterized by lung abnormalities, including the presence of syncytial pneumocytes. Syncytia form when SARS‐CoV‐2 spike protein expressed on the surface of infected cells interacts with the ACE2 receptor on neighboring cells. The syncytia forming potential of spike variant proteins remain poorly characterized. Here, we first assessed Alpha (B.1.1.7) and Beta (B.1.351) spread and fusion in cell cultures, compared with the ancestral D614G strain. Alpha and Beta replicated similarly to D614G strain in Vero, Caco‐2, Calu‐3, and primary airway cells. However, Alpha and Beta formed larger and more numerous syncytia. Variant spike proteins displayed higher ACE2 affinity compared with D614G. Alpha, Beta, and D614G fusion was similarly inhibited by interferon‐induced transmembrane proteins (IFITMs). Individual mutations present in Alpha and Beta spikes modified fusogenicity, binding to ACE2 or recognition by monoclonal antibodies. We further show that Delta spike also triggers faster fusion relative to D614G. Thus, SARS‐CoV‐2 emerging variants display enhanced syncytia formation.
Keywords: coronavirus, fusion, SARS‐CoV‐2, spike, syncytia
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Spike protein mutations expressed by emerging SARS‐CoV‐2 variants‐of‐concern differentially affect host cell‐to‐cell fusion, ACE2 receptor binding, and antibody escape.

Introduction
SARS‐CoV‐2 was initially discovered during an outbreak in Wuhan, China, before it became pandemic (Huang et al, 2020a). Since its emergence, the ancestral Wuhan strain has been supplanted by variants harboring a variety of mutations. Several of these mutations occur in the highly antigenic Spike (S) protein which endowed many of the variants with the ability to evade part of the neutralizing antibody response (Weisblum et al, 2020; Planas et al, 2021a; Liu et al, 2021b; Rees‐Spear et al, 2021; Starr et al, 2021). Individual amino acid changes in the S protein also affect viral fitness. One of the earliest identified variants contained the D614G mutation in S protein, which increased infectivity without significantly altering antibody neutralization (Yurkovetskiy et al, 2020). Several other variants have since emerged and have become globally dominant, including Alpha (B.1.1.7) first identified in the United Kingdom, Beta (B.1.351) identified in South Africa, Gamma (P.1 & P.2) identified in Brazil, and Delta (B.1.617.2) identified in India (preprint: Tegally et al, 2020; Buss et al, 2021; Frampton et al, 2021; Planas et al, 2021b; Sabino et al, 2021; preprint: Yadav et al, 2021). Some variants are more transmissible but their impact on disease severity is debated (Korber et al, 2020; Davies et al, 2021; Meng et al, 2021).
Clinically, SARS‐CoV‐2 infections range from asymptomatic or febrile respiratory disorders to severe lung injury characterized by vascular thrombosis and alveolar damage (Bussani et al, 2020). The deterioration of respiratory tissue is likely a result of both virus‐induced cytopathicity and indirect immune‐mediated damage (Buchrieser et al, 2020; Zhang et al, 2020; Zhou et al, 2020; Zhu et al, 2020). A peculiar dysmorphic cellular feature is the presence of large infected multinucleated syncytia, predominately comprised of pneumocytes (Bussani et al, 2020; Braga et al, 2021; Sanders et al, 2021). Other coronaviruses including SARS‐CoV‐1, MERS‐CoV, and HKU1 also induce syncytia formation in patient tissues and cell culture systems (Franks et al, 2003; Chan et al, 2013; Dominguez et al, 2013; Qian et al, 2013). Syncytial cells may compound SARS‐CoV‐2‐induced cytopathicity, play a role in viral persistence and dissemination, and could be a pathological substrate for respiratory tissue damage (Buchrieser et al, 2020; Braga et al, 2021; Sanders et al, 2021). Release of syncytial cells may contribute to the overall infectious dose (preprint: Beucher et al, 2021). Heterocellular syncytia containing lymphocytes have also been documented in the lungs of infected patients (Zhang et al, 2021).
The SARS‐CoV‐2 S protein is a viral fusogen. The interaction of trimeric S with the ACE2 receptor and its subsequent cleavage and priming by surface and endosomal proteases results in virus‐cell fusion (Hoffmann et al, 2020). Merging of viral and cellular membranes allows for viral contents to be deposited into the cell to begin the viral life cycle. Within the cell, newly synthesized S protein, envelope, and membrane proteins are inserted into the endoplasmic reticulum (ER) and trafficked and processed through the ER‐Golgi network (Nal et al, 2005; Duan et al, 2020; Cattin‐Ortolá et al, 2021). Virion are formed by budding into ER‐Golgi membranes and are then transported to the surface in order to be released from the cell (Klein et al, 2020). While the majority of the S protein is sequestered within the ER, motifs within its cytoplasmic tail allow for leakage from the Golgi apparatus and localization at the plasma membrane (Cattin‐Ortolá et al, 2021). The S protein at the surface of an infected cell interacts with receptors on adjacent cells, fusing the plasma membranes together and merging the cytoplasmic contents. We and others had previously shown that the S protein interacting with the ACE2 receptor induces cell–cell fusion (Buchrieser et al, 2020; Braga et al, 2021; Lin et al, 2021; Sanders et al, 2021; Zhang et al, 2021). The TMPRSS2 protease further augments cell–cell fusion (Buchrieser et al, 2020; Barrett et al, 2021; Hornich et al, 2021).
The S protein is comprised of S1 and S2 subunits. The S1 subunit includes the N‐terminal domain (NTD) and the receptor‐binding domain (RBD). The function of the NTD has yet to be fully elucidated but it may be associated with glycan binding, receptor recognition, and pre‐fusion‐to‐post‐fusion conformational changes. The NTD is also targeted by neutralizing antibodies (Krempl et al, 1997; Zhou et al, 2019; Chi et al, 2020). The RBD interacts with the ACE2 receptor and is the main target for neutralizing antibodies (Huang et al, 2020b). The S2 domain consists of the fusion peptide (FP), heptapeptide repeat sequences 1 and 2 (HR1 and HR2), the transmembrane anchor, and the C‐terminal domain. The FP inserts into the target membrane by disrupting the lipid bilayer and anchors the target membrane to the fusion machinery (Huang et al, 2020b). This exposes regions of HR1 that interact with HR2, forming a flexible loop that brings the membranes together to facilitate fusion (Huang et al, 2020b). The versatility of the S protein suggests that any mutations that may have arisen are of particular concern as they can affect fusogenicity, antibody recognition, affinity to ACE2, proteolytic processing, and incorporation into virions. There is a general paucity of information regarding how the mutations associated with variant S proteins contribute to cell–cell fusion.
S‐mediated cell–cell fusion is sensitive to innate immunity components. The interferon response to SARS‐CoV‐2 is one of the key factors down‐modulating viral entry and replication, and deficiencies in the interferon response are associated with severe or critical COVID‐19 (Arunachalam et al, 2020; Bastard et al, 2020, 2021; Hadjadj et al, 2020; van der Made et al, 2020). SARS‐CoV‐2‐induced syncytia formation by the Wuhan strain is restricted by innate immunity, in part through the action of interferon‐induced transmembrane proteins (IFITMs) (Buchrieser et al, 2020). IFITM1, 2, and 3 are restriction factors which display antiviral activity against a variety of enveloped viruses including SARS‐CoV‐2, likely by increasing membrane rigidity and hindering virus–cell fusion (Shi et al, 2021). Their effectiveness at restricting cell–cell fusion induced by the novel variants has yet to be assessed.
Here, we compared the replication and syncytia forming potential of D614G, Alpha, and Beta viruses in human cell lines and primary airway cells. We further characterized the fusogenicity of the Alpha and Beta variant S proteins and the individual contribution of each of the component mutations in syncytia formation, ACE2 binding, and evasion from a panel of antibodies. Finally, we examined the syncytia forming potential and ACE2 binding capacity of the Delta variant spike.
Results
Comparative replication kinetics of SARS‐CoV‐2 variants
We compared the replication kinetics of SARS‐CoV‐2 variants in relevant cell cultures. We first infected Caco‐2, Calu‐3, and Vero cells with Alpha, Beta, and D614G variants and generated multistep growth curves (Fig 1). Cells were infected at an equivalent, non‐saturating MOI, initially titrated in Vero cells (Appendix Fig S1A). Viral replication was assessed at 24, 48, and 72 h by flow cytometry upon staining with the pan‐SARS‐CoV‐2 anti‐S mAb102 human monoclonal antibody (Planas et al, 2021a) and then gating for S+ cells (Appendix Fig S1B). Globally, the variants replicated similarly (Fig 1). This similar replication was observed at different MOIs (Appendix Fig S1A). There were subtle differences at 24 h post‐infection, depending on the cell line and the variant. For instance, Beta replicated slightly more than D614G in Caco‐2 cells whereas Alpha replicated slight less than D614G in Vero cells (Fig 1A and C). Viral release at each time point was also assessed by extracting RNA from the supernatant and performing RT‐qPCR for the gene encoding the N protein. Viral release was again roughly similar with the different variants, especially at early time points. Alpha produced moderately more virus than D614G in all cell lines at later time points (Fig 1A–C). Beta produced more virus than D614G in Caco‐2 but less in Calu‐3 cells at later time points (Fig 1A and B).
Figure 1. Replication kinetics of D614G, Alpha, and Beta variants in cell culture.
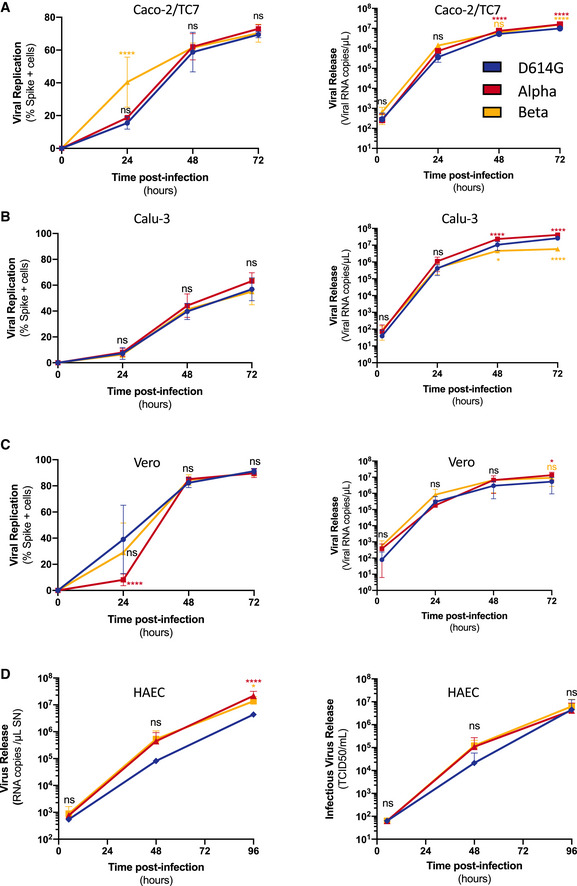
-
A–DCells were infected at the indicated MOI. Viral replication (left) and release (right) were assessed by flow cytometry and RT‐qPCR. (A) Caco2/TC7 cells (MOI 0.01), (B) Calu‐3 cells (MOI 0.001), (C) Vero cells (MOI 0.01), (D) primary human airway epithelial cells (HAEC) virus release (Right) and infectious virus release (Left) (MOI 0.01). Data are mean ± SD of at least three independent experiments. Statistical analysis: mixed‐effect analysis or two‐way ANOVA compared with D614G reference, ns: non‐significant, *P < 0. 05, ****P < 0.0001.
We then used the MucilAirBTM model, which consists of primary human airway epithelial cells (HAEC) grown over a porous membrane and differentiated at the air–liquid interface for over 4 weeks. This relevant model is susceptible to SARS‐CoV‐2 infection (Pizzorno et al, 2020; Robinot et al, 2021; Touret et al, 2021). The cells were infected with each variant at a similar low viral inoculum (104 TCID50). Viral RNA and infectious virus release were monitored over 96 h by RT‐qPCR and TCID50. Alpha and Beta variants produced slightly more extracellular viral RNA than D614G at later time points but not significantly higher levels of infectious particles (Fig 1D).
Taken together our data show that Alpha and Beta variants replicate similarly to the ancestral D614G strain in a panel of human cell lines and in primary cells, with some slight differences.
Syncytia formation in cells infected with SARS‐CoV‐2 variants
We next assessed the potential of SARS‐CoV‐2 variants to induce syncytia. In order to visualize cell–cell fusion, we employed our previously described S‐Fuse assay, using U2OS‐ACE2 GFP‐split cells (Buchrieser et al, 2020). In the GFP‐split complementation system, two cell lines containing half of the reporter protein are co‐cultured, producing a GFP signal only upon fusion (Fig 2A). Upon infection of S‐Fuse cells, we noticed that the Alpha and Beta variants formed larger and more numerous infected syncytia than either D614G or the ancestral Wuhan strain (Fig EV1A). We then characterized quantitatively the differences in fusogenicity by calculating the total syncytia (GFP) area and then normalizing it to nuclei number (Hoechst) (Fig EV1B). Relative to D614G, Alpha and Beta variants produced significantly more syncytia, approximately 4.5‐fold and threefold respectively, after 20 h of infection with the same MOI (Fig 2B and Appendix Fig S2A). In order to characterize syncytia formation in a cell line expressing endogenous ACE2, we generated Vero cells carrying the GFP‐split system. After 48 h of infection with the same MOI, we again found that Alpha and Beta variants produced significantly more syncytia than D614G (Fig 2C and Appendix Fig S2B) despite similar infection levels (Fig 1C). Of note, D614G produced similar levels of syncytia as the Wuhan strain in both Vero and S‐Fuse cells (Fig 2 and Appendix Fig S2).
Figure 2. SARS‐CoV‐2 variant infection increases formation of syncytia in U2OS‐ACE2 and Vero GFP‐split cells.
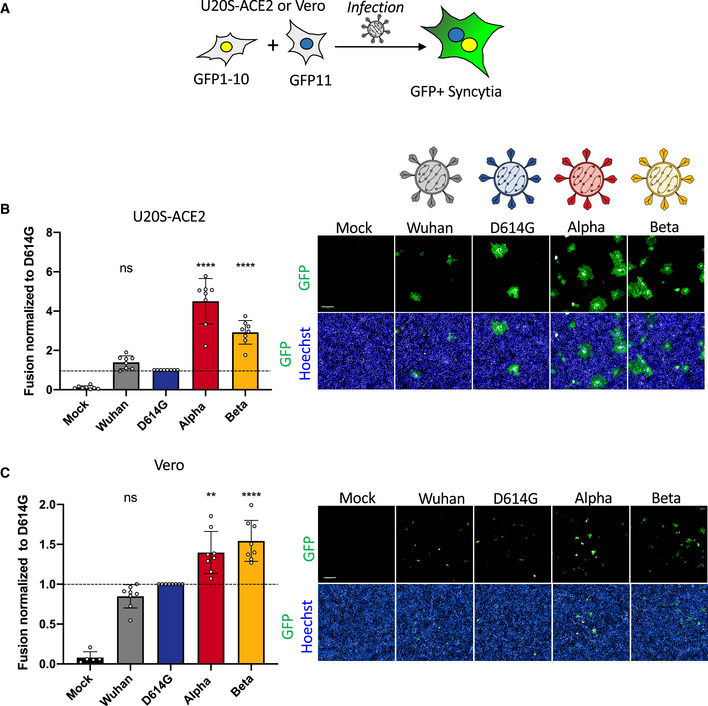
- U2OS‐ACE2 or Vero cells expressing either GFP 1–10 or GFP 11 (1:1 ratio) were infected 24 h after plating and imaged 20 h (U2OS‐ACE2) or 48 h (Vero) post‐infection.
- Left Panel: Fusion was quantified by GFP area/ number of nuclei and normalized to D614G for U2OS‐ACE2 20 h post‐infection at MOI 0.001. Right Panel: Representative images of U2OS‐ACE2 20 h post‐infection, GFP‐Split (green), and Hoechst (blue). Top and bottom are the same images with and without Hoechst channel.
- Left Panel: Quantified fusion of Vero cells infected at MOI 0.01. Right Panel: Representative images of Vero cells 48 h post‐infection, GFP‐Split (green), and Hoechst (blue).
Data information: Scale bars: 200 µm. Data are mean ± SD of eight independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference, ns: non‐significant, **P < 0.01, ****P < 0.0001.
Figure EV1. Qualitative and quantitative assessment of syncytia formation.
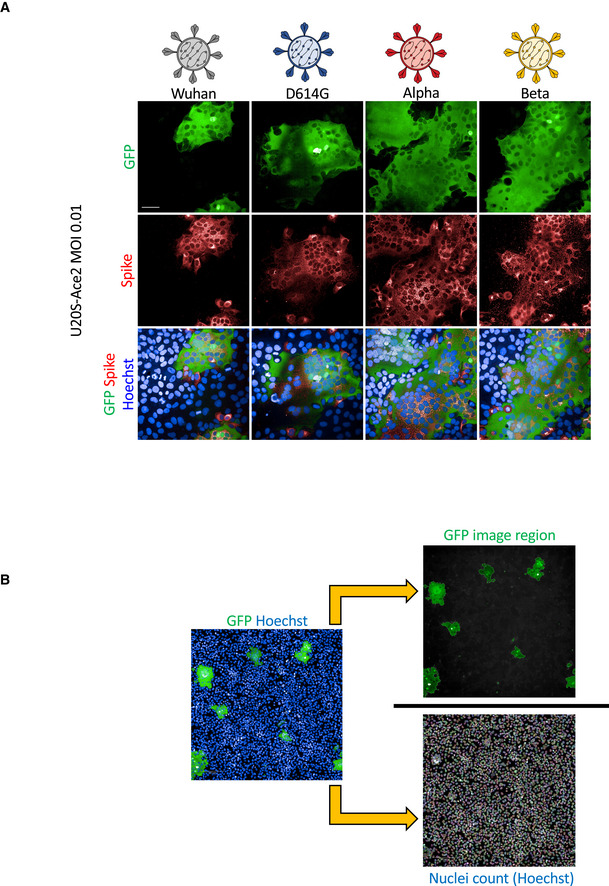
- U2OS‐ACE2 GFP‐split cells were infected at MOI 0.01 with the Wuhan, D614G, Alpha, and Beta strains for 20 h. Cells were stained for S protein with the human pan‐SARS‐CoV‐2 102 mAb and Alex647 fluorescent secondary antibody. Representative confocal images of the variant induced syncytia formation: GFP‐Split (green), Spike (red), and Hoechst (blue). Scale bars: 50 µm.
- Quantification method for syncytia formation using the Opera Phenix high content imager and harmony software: Total syncytia area (GFP area) is normalized for cell number upon quantifying the number of nuclei (Hoechst).
Therefore, Alpha and Beta variants appear more fusogenic than D614G in S‐Fuse and Vero cells.
Syncytia formation in cells expressing variant S proteins
Since syncytia formation is a consequence of the S protein expressed on the surface of an infected cell interacting with ACE2 receptors on neighboring cell, we sought to compare the fusogenic potential of the individual variant S proteins. We introduced the D614G mutation into the Wuhan protein and designed plasmids to express Alpha and Beta S proteins. We transfected the respective plasmids into Vero GFP‐split cells and quantified syncytia formation 18 h later (Fig 3A). Alpha and Beta S proteins were twofold and 1.7‐fold more fusogenic than D614G S, respectively (Fig 3B). The Wuhan S was slightly less fusogenic that the D614G S (Fig 3B). We then verified that the variation in S‐mediated fusion was not due to differential cell surface levels. Due to extensive fusion in transfected Vero cells, we were unable to compare S protein surface expression (Fig 3B and Movie EV1), as the S protein‐positive syncytia were damaged and lost during the flow cytometry procedure. As such, we transfected 293T cells, which lack ACE2 and thus will not fuse with one another upon S expression, with the different variant plasmids in order to assess S protein surface levels by flow cytometry. The variants S proteins were equally expressed after transfection (Fig EV2A–C). In order to unify the surface expression control with our fusion results, we performed an acceptor/donor experiment. 293T GFP1‐10 (donor) cells were transfected with each of the variant S plasmids. The transfected donor cells (equal number for each transfection condition) were then co‐cultured with Vero GFP11 (acceptor) cells (Fig EV2E). Some of the transfected 293T donor cells were set aside and stained for S protein to show equal surface expression between the variants (Fig EV2E). In the acceptor/donor co‐culture, we found that the 293T donor cells expressing the novel variant S proteins formed more syncytia with the Vero acceptor cells than either the D614G or Wuhan (Fig EV2E). The results of the acceptor/donor co‐culture experiment matched our fusion results in Vero cells.
Figure 3. Alpha and Beta SARS‐CoV‐2 S proteins induce more robust syncytia formation than D614G.
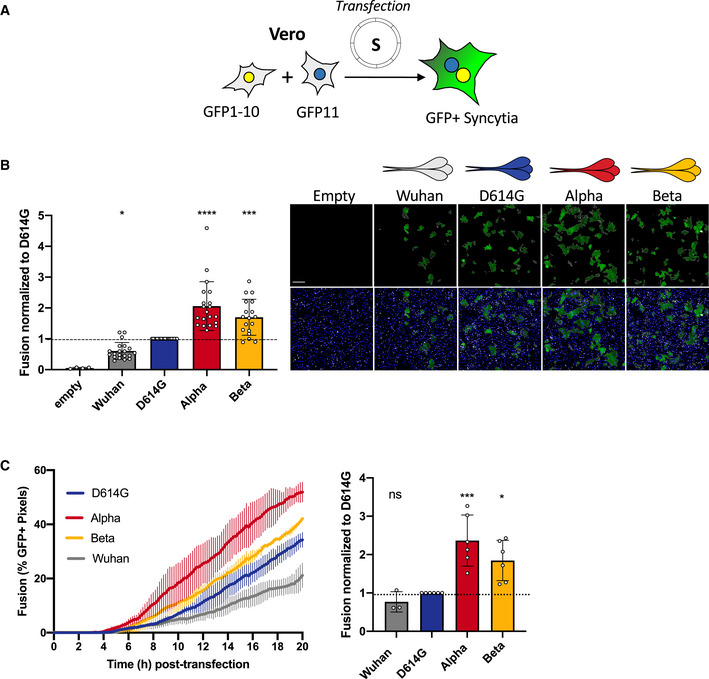
- Vero GFP‐split cells were transfected with variant S proteins and imaged 18 h post‐transfection.
- Left Panel: Fusion was quantified by GFP area/number of nuclei and normalized to D614G for each of the transfected variant S proteins. Right Panel: Representative images of Vero GFP‐split cells 18 h post‐transfection, GFP (green), and Hoechst (blue). Top and bottom are the same images with and without Hoechst channel. Scale bars: 200 µm.
- Left Panel: Quantification of variant S protein‐mediated fusion in Vero GFP‐split cells by video microscopy. Results are mean ± SD from three fields per condition from one representative experiment. Right Panel: Fusion quantification of at least three independent video microscopy experiments, 20 h post‐transfection, normalized to D614G.
Data information: Data are mean ± SD of at least three independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference, ns: non‐significant, *P < 0. 05, ***P < 0.001, ****P < 0.0001.
Figure EV2. SARS‐CoV‐2 variant S proteins are expressed equally at the cell surface.
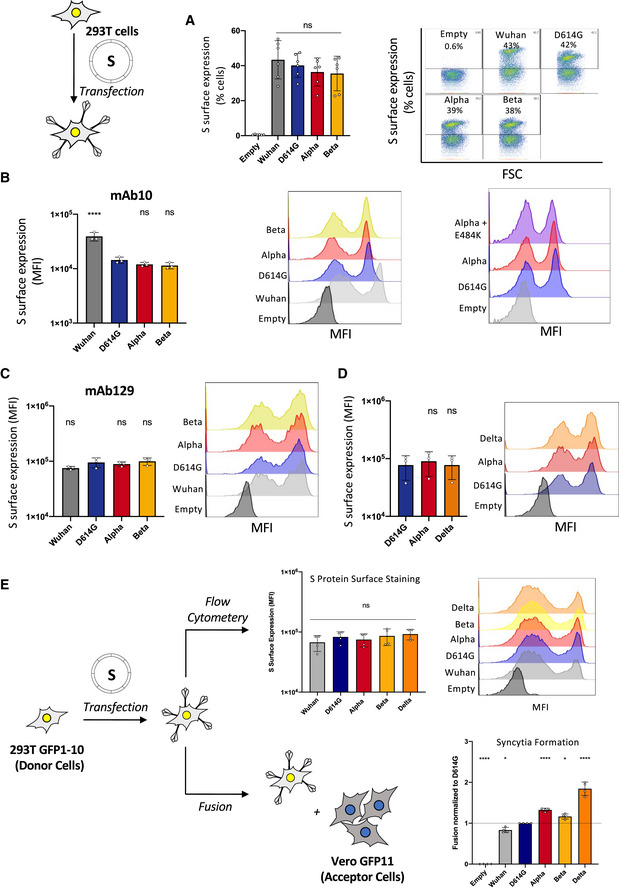
- Left Panel: Quantification of percent of cells expressing each S protein at the surface. Right Panel: Representative FACs plots.
- Quantification of median florescent intensity (MFI) of variant S protein at the cell surface and representative histograms of MFI of the Wuhan, D614G, Alpha, Beta, and Alpha + E484K variants S protein using mAb10.
- Quantification of median florescent intensity (MFI) of variant S protein at the cell surface and representative histograms of MFI of the Wuhan, D614G, Alpha, Beta, and Alpha + E484K variants S protein using mAb129.
- Quantification of median florescent intensity (MFI) of variant S protein at the cell surface and representative histograms of MFI of the Delta variant compared with the Alpha and D614G using mAb129.
- Controlled acceptor/donor experiment 293T GFP1‐10 donor cells transfected with S protein and verified to have equal S protein expression on the surface (top), were then added to Vero GFP11 acceptor cells (bottom) to assess fusion.
Data information: Flow cytometry data are mean ± SD of at least three independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference, ns: non‐significant, *P < 0. 05, ****P < 0.0001.
We then measured the kinetics of syncytia formation induced by the different S proteins in Vero GFP‐Split cells. We conducted a comparative videomicroscopy analysis where cell–cell fusion could be visualized as soon as 6 h post‐transfection. The fusion kinetics of Alpha S protein was more rapid than any of the other variants (Fig 3C and Movie EV1). Beta also induced significantly faster fusion than D614G, whereas the Wuhan S was the slowest of all the compared proteins (Fig 3C and Movie EV1).
Altogether, our data indicate that the S proteins of Alpha and Beta variants form more syncytia than the D614G or Wuhan strains.
Restriction of S‐mediated syncytia formation by IFN‐β1 and IFITMs
As the variants did not show any major difference in replication under basal conditions, we next investigated whether they were differently sensitive to the interferon response. To this aim, we pre‐treated Vero cells or U2OS‐ACE2 (S‐Fuse) cells with increasing doses of IFN‐β1 and infected them with the different variants. IFN‐β1 was equally effective at reducing viral replication of D614G, Alpha, and Beta variants in Vero cells (Fig EV3A). Preincubation of S‐Fuse cells with IFN‐β1 also abrogated infection and syncytia formation to the same extent for the different variants (Fig EV3B). Therefore, IFN‐β1 similarly inhibited viral replication and reduced syncytia formation by D614G, Alpha, and Beta variants.
Figure EV3. Impact of IFN‐β1 and IFITMs on SARS‐CoV‐2 variant replication and S protein‐mediated cell–cell fusion.
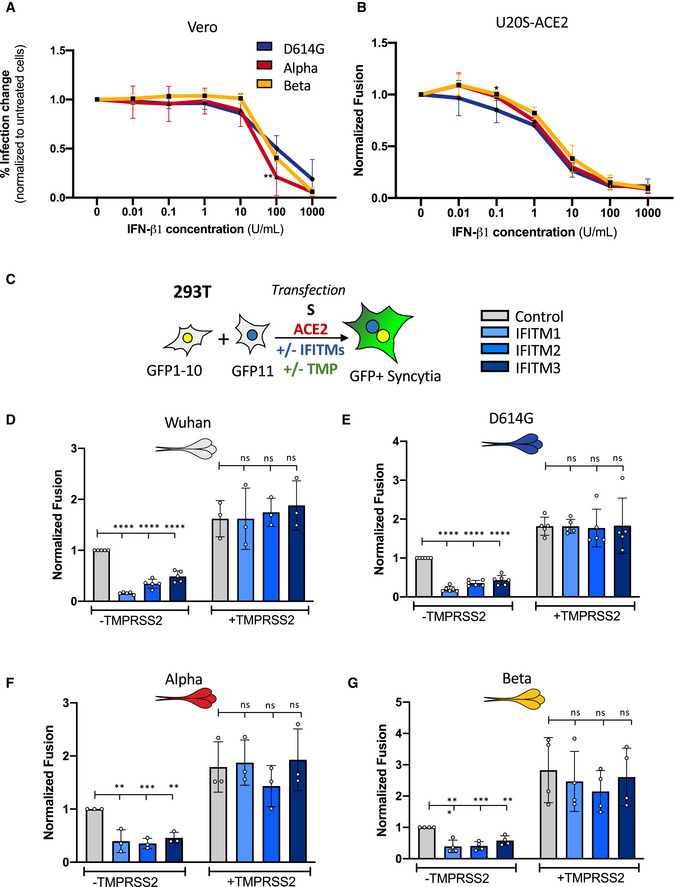
-
AVero cells were pre‐treated for 2 h with a serial dilution of IFN‐β1 prior to infection with the SARS‐CoV‐2 variants. Infected cells were maintained in media containing IFN‐β1 and analyzed by flow cytometry 48 h post‐infection to determine relative infection change.
-
BU20S‐ACE2 GFP‐split cells were pre‐treated for 2 h with a serial dilution of IFN‐β1 prior to infection with the SARS‐CoV‐2 variants. Infected cells were maintained in media containing IFN‐β1 and relative inhibition of syncytia formation 20 h post‐infection was determined via GFP signal.
-
C–GA co‐culture of 293T GFP‐Split cells were transfected with combination of S, control, ACE2, TMPRSS2, and IFITM plasmids and then imaged 18 h post‐transfection. Effect of IFITMs and TMPRSS2 on the cell–cell fusion induced by different S proteins, (D) Wuhan, (E) D614G, (F) Alpha, and (G) Beta.
Data information: Data are mean ± SD of at least three independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference or control plasmid transfection, ns: non‐significant, *P < 0. 05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
IFITMs are interferon‐stimulated transmembrane proteins that restrict early stages of the viral life cycle by inhibiting virus‐cell fusion, likely by modifying the rigidity or curvature of membranes (Compton et al, 2014; Shi et al, 2017; Zani & Yount, 2018). IFITM1 localizes at the plasma membrane while IFITM2 and 3 transit through surface and localize in endo‐lysosomal compartments (Buchrieser et al, 2020). We previously reported that IFITMs restrict Wuhan S‐mediated cell–cell fusion and that their activity was counteracted by the TMPRSS2 protease (Buchrieser et al, 2020). As infection with Alpha and Beta induce more syncytia, we further investigated if this resulted in an increased resistance to IFITM restriction. We thus characterized the impact of IFITMs on syncytia formed upon expression of D614G, Alpha, and Beta S proteins in 293T cells. The variants were all effectively restricted by IFITMs (Fig EV3C–G). The three IFITMs were expressed at similar levels in our system (not shown). The presence of TMPRSS2 increased fusion of all S proteins and reverted the restriction by IFITMs (Fig EV3C–G). Taken together, our data show that Alpha and Beta variants induce more syncytia, but their S proteins remain similarly sensitive to IFITMs.
Contribution of individual variant‐associated mutations on S protein‐mediated fusogenicity
We next sought to determine the contribution of each variant‐associated mutation to cell–cell fusion. Both Alpha and Beta S proteins contain the N501Y mutation in the RBD and the D614G mutation in the S1/S2 cleavage site (Fig 4A). Alpha S contains the ∆69/70 and ∆Y144 deletions in the N‐terminal domain (NTD), P681H and T716I mutations in the S1/S2 cleavage site, the S982A mutation in the heptad repeat 1 (HR1) site and the D1118H mutation in between HR1 and HR2. The Beta S is comprised of the L18F, D80A, D215G, and ∆242‐244 mutations in the NTD, K417N and E484K mutations in the receptor‐binding domain (RBD), and A701V in the S1/S2 cleavage site. We introduced individual mutations into the D614G background. Following reports of the emergence of the E484K mutation within the Alpha variant (Collier et al, 2021), we also generated a mutant Alpha S protein with the E484K mutation. We observed by flow cytometry that the mutant S proteins were similarly expressed at the cell surface (Fig EV2A–D). We expressed each mutant S into Vero GFP split cells and measured their potential to induce cell–cell fusion in comparison to the D614G S protein.
Figure 4. Mutations associated with Alpha and Beta S proteins differentially affect cell–cell fusion.
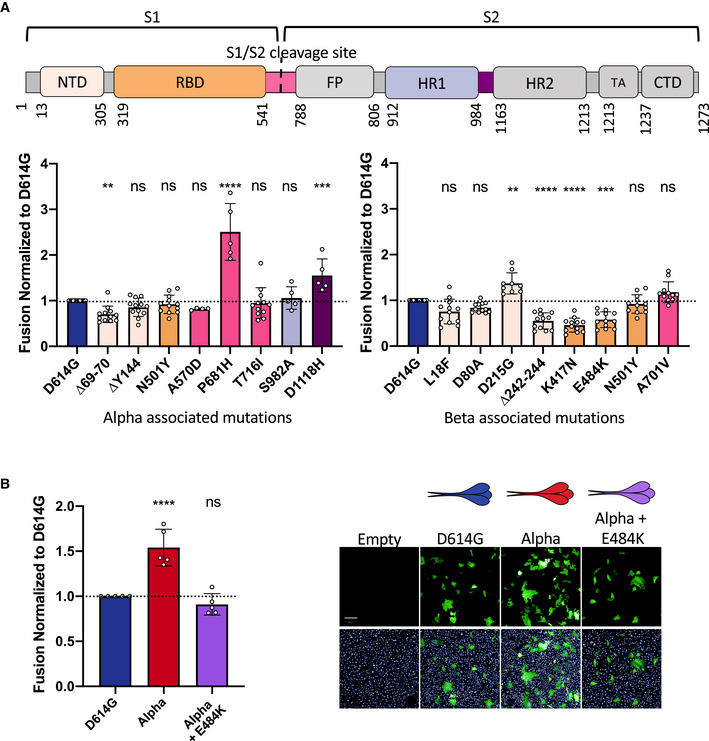
- Top Panel: Schematic representation of the S protein color‐coded for the functional regions: N‐terminal domain (NTD), receptor‐binding domain (RBD), fusion peptide (FP), heptad repeat 1,2 (HR1, HR2), transmembrane anchor (TA), C‐terminal domain (CTD). Bottom left Panel: Vero GFP‐split cells were transfected with S plasmids containing each of the individual mutations associated with Alpha variant in the D614G background. The amount of fusion was quantified at 20 h and normalized to D614G reference plasmid. Bottom right Panel: Quantified fusion for each of the individual S protein mutations associated with the Beta variant. Color code of each mutation corresponds to S protein functional regions represented in the schematic on the Top Panel. Data set for N501Y and D614G reference mutations are duplicated between bottom left and bottom right panels for presentation as these mutations are common to both variants.
- Left Panel: Quantified fusion of the Alpha + E484K variant S protein normalized to D614G S. Right Panel: Representative images of fusion at 20 h. Scale bar: 200 µm. Top and bottom are the same images with and without Hoechst channel.
Data information: Data are mean ± SD of at least four independent experiments. Top and bottom are the same images with and without Hoechst channel. Statistical analysis: statistics for both left and right panels of A were conducted together. One‐way ANOVA compared with D614G reference, ns: non‐significant, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Of the mutations that are associated with Alpha, we found that the ∆69/70 deletion in the NTD decreased cell–cell fusion whereas P681H and D1118H substitutions both increase fusion (Figs 4A and EV4E). P681H displayed the greatest fusogenicity of all investigated mutations, being almost 2.5‐fold higher than D614G S (Figs 4A and EV4E). As previously mentioned, the introduction of the D614G mutation in the S1/S2 border of the Wuhan S protein also relatively increased fusion, stressing the importance of this cleavage site in fusogenicity (Fig 3B).
Figure EV4. SARS‐CoV‐2 variant S protein‐associated mutations are expressed equally at the cell surface.
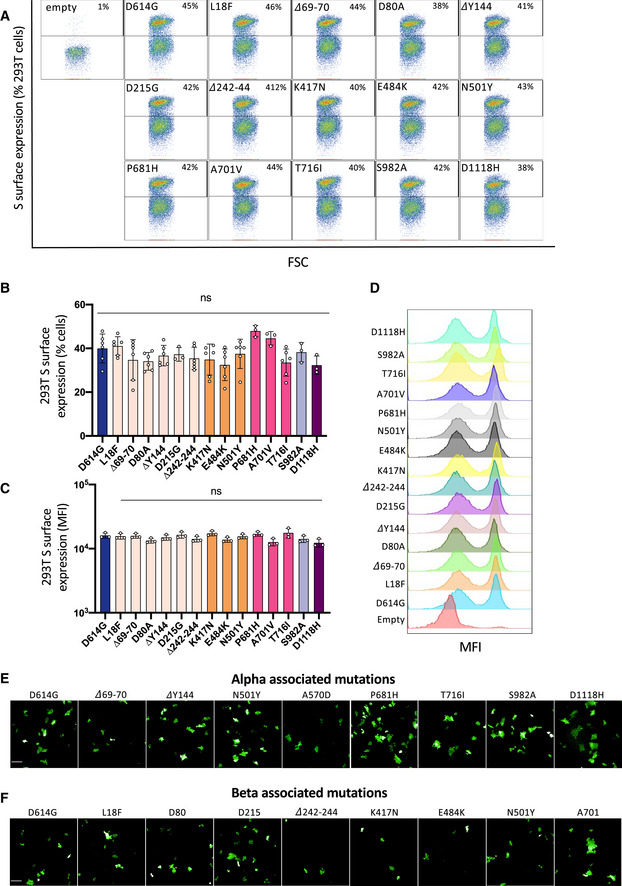
- Representative FAC plots of percent of cells expressing each mutant S protein at the surface.
- Quantification of percent of cells expressing each S protein at the surface.
- Quantification of median florescent intensity (MFI) of the mutant S protein at the cell surface.
- Representative histograms of MFI of each mutant S protein.
- Representative images of Vero GFP‐split cells 20 h after transfection with each Alpha variant‐associated mutant S protein, GFP‐Split (Green). Scale bars: 200 µm.
- Representative images of Vero GFP‐split cells 20 h after transfection with each Beta variant‐associated mutant S protein. Scale bars: 200 µm.
Data information: Data are mean ± SD of at least three independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference, ns: non‐significant.
Among the mutations associated with Beta, the ∆242‐244 deletion, as well as the K417N and E484K mutations in the RBD significantly decreased syncytia formation (Figs 4A and EV4F). Only the D251G mutation in the NTD modestly increased syncytia formation relative to D614G (Figs 4A and EV4F). The introduction of the E484K RBD mutation into the Alpha S protein significantly decreased its potential to form syncytia, despite not changing cell surface expression, further supporting the mutation’s restrictive effect on cell–cell fusion (Figs 4B and EV2B). Taken together, our data suggest that variant S proteins are comprised of mutations that play contrasting roles in cell–cell fusion. P681H, D1118H, and D215G substitutions facilitate fusion, whereas mutations ∆69/70, ∆242‐244, K417N, and E484K antagonize cell–cell fusion.
Binding of S proteins bearing individual variant‐associated mutations to ACE2
We next explored the impact of variant‐associated mutations on S binding to the ACE2 receptor. To this aim, we transiently expressed each mutant protein in 293T cells. Cells were then stained with a serial dilution of soluble biotinylated ACE2, revealed with fluorescent streptavidin, and then analyzed by flow cytometry (Fig 5A). Titration binding curves were generated, and EC50 (the amount of ACE2 needed for 50% binding) was calculated. The S protein of Alpha had the highest affinity to ACE2, confirming previous results by us and others (Planas et al, 2021; Ramanathan et al, 2021). Alpha was sequentially followed by Beta, D614G, and Wuhan S (Figs 5B and EV5A). As expected, mutations within the RBD had the most significant impact on ACE2 binding. N501Y found in both Alpha and Beta drastically increased ACE2 binding, in line with previous reports indicating that this mutation enhances affinity of the viral protein to its receptor (Ali et al, 2021; Luan et al, 2021; Tian et al, 2021). The K417N substitution present in the Beta S decreased ACE2 binding (Figs 5B and EV5C). The E484K mutant had a slightly, but not significantly, higher binding to ACE2 (Fig EV5C). This was corroborated by the observation that addition of the E484K mutation to Alpha S protein also slightly increased ACE2 binding (Figs 5B and EV5A). Mutation in the S1/S2 cleavage site, HR1/HR2 sites, or NTD did not have any significant impact on ACE2 binding (Figs 5B and EV5B–E). It is worth noting that the NTD ∆242‐244 mutant displayed a marginally lower binding to ACE2 (Figs 5B and EV5B). Therefore, the N501Y mutation is the most significant contributor to increased ACE2 binding of the variants, though it does not affect cell–cell fusion on its own. The K417N, ∆242‐244, and E484K mutations restrict fusogenicity but differently affect ACE2 binding, with the former two decreasing affinity and the latter slightly increasing.
Figure 5. ACE2 and monoclonal antibody binding to S proteins with Alpha and Beta associated mutations.
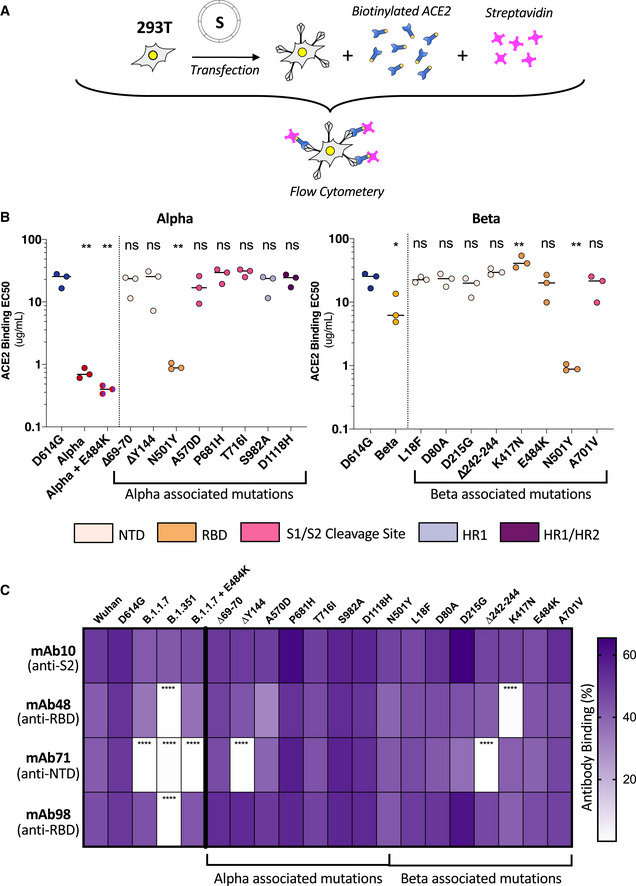
- 293T cells were transfected S proteins with each variant‐associated mutation for 24 h and stained with biotinylated ACE2 and fluorescent streptavidin before analysis by flow cytometry.
- Left Panel: EC50 values (concentration of ACE2 needed for 50% binding) for Alpha and associated mutations. Color code corresponds to location on S protein functional domains and lower EC50 values signify higher affinity to ACE2 binding. Right Panel: EC50 values for Beta and associated mutations. Data set for N501Y and D614G reference mutations are duplicated between left and right panels as mutations are common to both variants.
- S protein transfected 293T cells were stained with human monoclonal antibodies targeting the S2 (mAb10), RBD (mAb48 and mAb98), and the NTD (mAb71). Cells were analyzed by flow cytometry. The percentage of positive cells is indicated.
Data information: Data are mean of at least three independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference, ns: non‐significant, *P < 0. 05, **P < 0.01, ****P < 0.0001.
Figure EV5. ACE2 binding curves to SARS‐CoV‐2 variant S proteins and associated mutations.
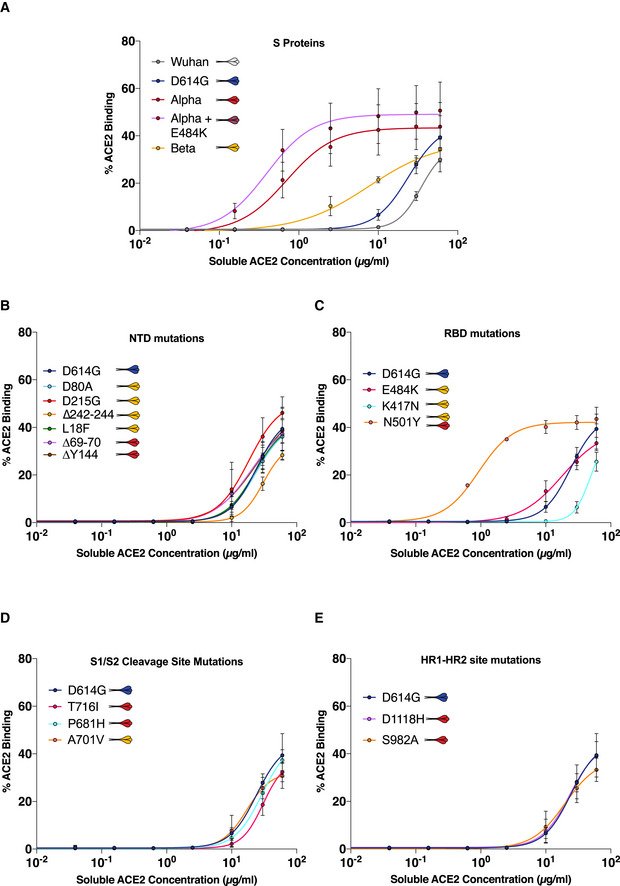
-
AACE2 binding dilution curves of each variant S protein.
-
B–EACE2 binding dilution curves of each variant‐associated mutation located in (B) S protein n‐terminal domain (NTD), (C) receptor‐binding domain (RBD), (D) S1/S2 cleavage site, and (E) heptad repeat 1–2 site (HR1‐HR2).
Data information: Data are mean ± SD of three independent experiments.
Therefore, ACE2 binding and fusogenicity are two functions of the S protein that can be partially deconvoluted through individual mutations.
Antibody binding to S proteins bearing individual variant‐associated mutations
We had previously found that certain neutralizing antibodies differentially affect SARS‐CoV‐2 D614G, Alpha, and Beta variants (Planas et al, 2021a). For instance, neutralizing monoclonal antibody 48 (mAb48) restricts D614G virus but not Alpha or Beta variants (Planas et al, 2021a). We sought to determine which mutations in variant S proteins contributed to the lack of recognition by the neutralizing antibodies. To this aim, we assessed by flow cytometry the binding of a panel of four human monoclonal antibodies (mAbs) to the different S mutants. As a control we used mAb10, a pan‐coronavirus antibody that targets an unknown but conserved epitope within the S2 region (Planchais, manuscript in preparation). mAb10 equally recognized all variants and associated individual mutations (Fig 5C). mAb48 and mAb98 target the RBD and mAb71 the NTD (Planas et al, 2021b) (Planchais, manuscript in preparation). mAb48 did not recognize the Beta variant and more specifically did not bind to the K417N mutant (Fig 5C). The mAb71 recognized neither Alpha nor Beta variants and did not bind to their respective NTD ∆Y144 and ∆242‐244 mutations. The K417N and ∆242‐244 mutations were also responsible for decreasing S‐mediated fusion, suggesting a tradeoff between antibody escape and fusion (Fig 5C). mAb98 did not recognize the Beta variant. However, none of the associated mutations were specifically responsible for the lack of binding (Fig 5C), suggesting a combined effect on the structure of the S protein that may affect antibody escape.
Therefore, several of the mutations found in the variants S proteins are advantageous in terms of antibody escape, despite slightly reducing the ability of the proteins to fuse.
S protein‐mediated syncytia formation by the Delta variant
With the emergence and rapid spread of the Delta variant, we sought to characterize its potential to form syncytia. We recently showed that the Delta variant induces large syncytia in S‐Fuse cells (Planas et al, 2021b). We thus compared the fusogenicity of the Delta S protein to that of D614G and Alpha. We transiently expressed the three S proteins in Vero GFP‐split cells. The Delta S protein triggered more cell–cell fusion than the D614G variant but was similar to the Alpha S protein (Fig 6A). The fusion kinetic of the Delta S was also similar to Alpha but more rapid than D614G (Fig 6B). We confirmed that the variant S proteins were equally expressed on the surface by transfecting them into non‐fusogenic 293T cells and performing flow cytometry upon staining with the pan‐SARS‐CoV‐2 mAb129 (Fig EV2D). We next examined the ACE2 binding potential of Delta S protein using our aforementioned soluble biotinylated ACE2. The Delta S protein has a higher binding capacity to ACE2 than the D614G S protein, but the binding was lower than the Alpha S protein (Fig 6C).
Figure 6. Delta SARS‐CoV‐2 S protein induces more syncytia formation and binds more to ACE2 than D614G.
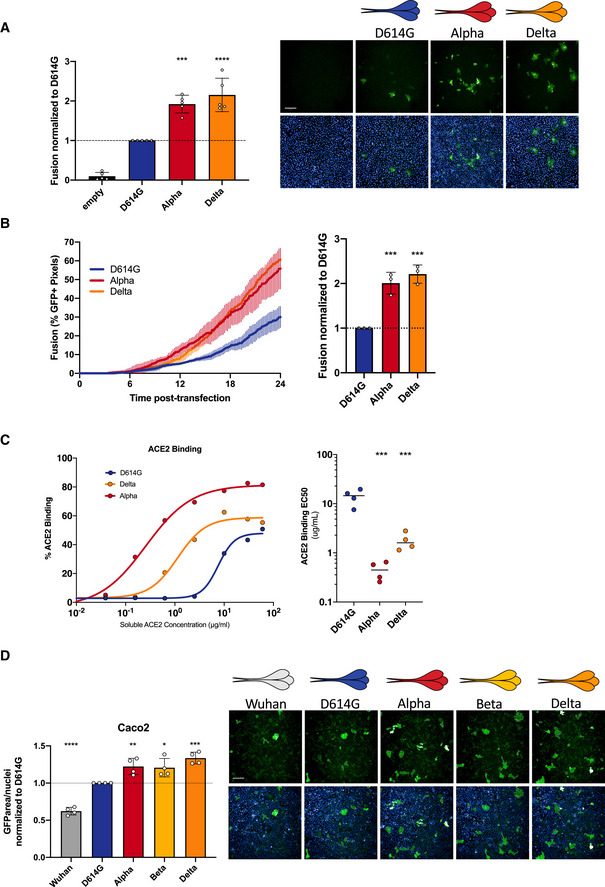
- Vero GFP‐split cells were transfected with variant S proteins and imaged 18 h post‐transfection. Left Panel: Fusion was quantified by GFP area/number of nuclei and normalized to D614G for each of the transfected variant S proteins. Right Panel: Representative images of Vero GFP‐split cells 18 h post‐transfection, GFP (green), and Hoechst (blue). Top and bottom are the same images with and without Hoechst channel. Scale bars: 200 µm.
- Left Panel: Quantification of Delta S protein‐mediated fusion in Vero GFP‐split cells by video microscopy. Results are mean ± SD from three fields per condition from one representative experiment. Right Panel: Fusion quantification of three independent video microscopy experiments, 20 h post‐transfection, normalized to D614G.
- 293T cells were transfected S proteins with each variant‐associated mutation for 24 h and stained with biotinylated ACE2 and fluorescent streptavidin before analysis by flow cytometry. Left Panel: Representative ACE2 binding dilution curves for the Delta variant in relation to Alpha and D614G. Right Panel: EC50 values (concentration of ACE2 needed for 50% binding) for Alpha for the Delta variant.
- Caco2 GFP‐split cells were transfected with variant S proteins and imaged 18 h post‐transfection. Left Panel: Fusion was quantified by GFP area/ number of nuclei and normalized to D614G for each of the transfected variant S proteins. Right Panel: Representative images of Caco2 GFP‐split cells 18 h post‐transfection, GFP (green), and Hoechst (blue). Top and bottom are the same images with and without Hoechst channel. Scale bars: 200 µm.
Data information: Data are mean ± SD of at least three independent experiments. Statistical analysis: one‐way ANOVA compared with D614G reference, *P < 0. 05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
S protein‐mediated syncytia formation and TMPRSS2
We then asked whether the TMPRSS2 protease, which cleaves S and facilitates viral fusion, may act differently on the variant S proteins. To this aim, we generated a Caco2 GFP‐split cell line and then expressed the different S proteins to examine fusogenicity. Human Caco2 cells express endogenous levels of TMPRSS2 and ACE2. In line with our results in Vero cells which lack endogenous TMPRSS2 (Fig 3B), we found that the Alpha, Beta, and Delta variants fused more than the D614G, and the Wuhan S protein fused the least (Fig 6D). Thus, the differences in variant fusogenicity can also be visualized in the presence of TMPRSS2. In order to see whether TMPRSS2 differentially processes the variants S proteins, we expressed each of them without or with TMPRSS2 in 293T cells in the absence ACE2. We examined the processing of the different S by western blot and the expression levels of S1 and S2 by flow cytometry. While all S proteins are processed, the cleavage profile induced by TMPRSS2 and the expression levels between the different variant S proteins were similar (Appendix Fig S3).
Discussion
The replication and cytopathic effects of SARS‐CoV‐2 variants are under intense scrutiny, with contrasting results in the literature (Hou et al, 2020; Frampton et al, 2021; Leung et al, 2021; preprint: Liu et al, 2021a; Touret et al, 2021). For instance, there was no major difference in the replication kinetics of Alpha and D614G strains in some reports (preprint: Thorne et al, 2021; Touret et al, 2021), whereas others suggested that Alpha may outcompete D614G in a co‐infection assay (Touret et al, 2021). Some studies proposed that the N501Y mutation may provide a replication advantage, whereas others suggested that N501Y is deleterious (Hou et al, 2020; Frampton et al, 2021; Leung et al, 2021; preprint: Liu et al, 2021a). These discrepant results may be due to the use of different experimental systems, viral strains, multiplicities of infection, and cell types.
Here, we show that Alpha and Beta variants replicate to the same extent as the early D614G strain in different human cell lines and primary airway cells. Moreover, Alpha and Beta induced more cell–cell fusion than D614G. Increased fusion was observed in U2OS‐ACE2 cells and in naturally permissive Vero cells. In agreement with infection data, transfection of Alpha and Beta S proteins in the absence of any other viral factors, produced significantly more syncytia than D614G, which in turn, fused more than the Wuhan S. Comparative video microscopy analysis revealed that Alpha S fused the most rapidly, followed by Beta, D614G, and finally Wuhan. Thus, Alpha and Beta variants display enhanced S‐mediated syncytia formation. One limitation of our study resides in the fact that were unable to look at surface expression of the variant S proteins in Vero and Caco2 without losing the large S protein‐positive syncytia. We thus used the non‐fusogenic 293T cells to control for surface expression. We further show that S‐expressing 293T cells fuse with Vero cells in donor/acceptor experiments. The experiments confirmed the enhanced fusogenicity of the variants in cells with similar levels of S protein at their surface.
We further show that Alpha and Beta remain sensitive to restriction by IFN‐β1. The fusion mediated by their respective S proteins is inhibited by IFITMs. This extends previous results by us and others demonstrating that ancestral Wuhan S is effectively inhibited by this family of restriction factors (Buchrieser et al, 2020; Shi et al, 2021). It has been recently reported in a pre‐print that Alpha may lead to lower levels of IFN‐β1 production by infected Calu‐3 cells and may be less sensitive to IFN‐β pre‐treatment, when compared to first wave viral isolates (preprint: Thorne et al, 2021). We did not detect here differences of IFN‐β1 sensitivity between the variants in Vero and U2OS‐ACE2 cells. Again, these discrepant results may reflect inherent differences between Calu‐3, Vero, and U2OS‐ACE2 cells, or the use of different viral isolates.
We then characterized the contribution of the individual mutations present in Alpha and Beta S proteins to their respective fusogenicity. The highly fusogenic Alpha S consists of more mutations that robustly increase fusion (P681H and D1118H) than mutations that decrease fusion (∆69/70). In contrast, the Beta variant is comprised of several restrictive mutations (∆242‐244, K417N, and E484K) and only one mutation that modestly increased fusion (D215G). The strongest increase in fusion was elicited by the P681H mutation at the S1/S2 border. This mutation likely facilitates proteolytic cleavage of S and thus promotes S mediate cell–cell fusion. Indeed, the analogous P681R mutation present in B.1.617.2 and B.1.617.3 variants increases S1/S2 cleavage and facilitates syncytia formation (Jiang et al, 2020; preprint: Ferreira et al, 2021). Of note, another report with indirect assessment of variant S fusogenicity suggested a mild decrease or no difference in cell–cell fusion of Alpha and Beta relative to Wuhan S (Hoffmann et al, 2021). These previous experiments were performed in 293T cells at a late time points (24 h post‐transfection), which may preclude detection of the accelerated fusion triggered by the variants.
We show that the binding of variant S to soluble ACE2 paralleled their fusogenicity. Alpha bound the most efficiently to ACE2, followed by Beta, D614G, and finally Wuhan. However, the ACE2 affinity of S proteins carrying individual mutations did not exactly correlate to fusogenicity. For instance, the N501Y and D614G mutations drastically increased ACE2 affinity, but only D614G enhanced fusogenicity. The K417N substitution, and to a lesser degree ∆242–244, had a lower affinity to ACE2 and also restricted cell–cell fusion. The E484K mutation significantly restricts fusion, but mildly increases ACE2 affinity. This suggests that on the level of individual S mutations, the relationship between ACE2 affinity and increased fusogenicity is not always linear. Variant mutations may also confer advantages in an ACE2‐independent manner. Indeed, recent work has suggested that the E484 mutation may facilitate viral entry into H522 lung cells, requiring surface heparan sulfates rather than ACE2 (Puray‐Chavez et al, 2021). It would be of future interest to examine the syncytia formation potential of the variant mutations in other cell types.
We selected a panel of 4 mAbs that displayed different profiles of binding to Alpha, Beta, D614G, and Wuhan S proteins. The mAb10 targeting the S2 domain recognized all variants and was used as a positive control. Wuhan and D614G were recognized by the three other antibodies, targeting either the NTD or RBD. Alpha lost recognition by the anti‐NTD mAb71, whereas Beta was neither recognized by mab71 nor by the two anti‐RBD antibodies mAb48 and mAb 98. Upon examining the potential of S proteins carrying individual mutations to bind to human monoclonal antibodies, we found that the ones that restrict (∆242–244, K417N) or have no effect on fusogenicity (∆Y144) are also not recognized by some mAbs. This suggests that variant S proteins have undergone evolutionary trade‐off in some circumstances, selecting for mutations that provide antibody escape at the detriment of fusogenicity. In accordance with our findings, deep sequence binding analysis and in vitro evolution studies suggest the N501Y mutation increases affinity to ACE2 without disturbing antibody neutralization (preprint: Liu et al, 2021a; Starr et al, 2021; Zahradník et al, 2021). The E484K and K417N RBD mutations in the Beta variant may also increase ACE2 affinity, particularly when in conjunction with N501Y (preprint: Nelson et al, 2021; Zahradník et al, 2021). However, the resulting conformational change of the S protein RBD may also decrease sensitivity to neutralizing antibodies (preprint: Nelson et al, 2021). Future work assessing the structural and conformational changes in the S protein elicited by a combination of individual mutations or deletions may further help elucidate the increased fusogenicity and antibody escape potential of the variants.
While we had previously shown that the interaction between the S protein on the plasma membrane with the ACE2 receptor on neighboring cells is sufficient to induce syncytia formation, there is compelling evidence of the importance of the TMPRSS2 protease in S activation (Buchrieser et al, 2020; Dittmar et al, 2021; Koch et al, 2021; Ou et al, 2021). We found that the S protein of the novel variants induced more syncytia formation than the D614G and Wuhan S proteins in human Caco2 cells which express endogenous ACE2 and TMPRSS2. However, we did not detect any major differences in the processing of the variant S proteins by TMPRSS2. It will be worth further characterizing how the fusogenicity of variant‐associated mutations is influenced by other cellular proteases like furin.
The presence of infected syncytial pneumocytes was documented in the lungs of patients with severe COVID‐19 (Bussani et al, 2020; Tian et al, 2020; Xu et al, 2020). Syncytia formation may contribute to SARS‐CoV‐2 replication and spread, immune evasion, and tissue damage. A report using reconstituted bronchial epithelia found that viral infection results in the formation and release of infected syncytia that contribute to the infectious dose (preprint: Beucher et al, 2021). The neutralizing antibody response to SARS‐CoV‐2 infection has divergent effect on cell–cell fusion, with some antibodies restricting S‐mediated fusion, while other increase syncytia formation (Asarnow et al, 2021). Cell‐to‐cell spread of virus may be less sensitive to neutralization by monoclonal antibodies and convalescent plasma than cell‐free virus (preprint: Jackson et al, 2021). It is thus possible that infected syncytial cells facilitate viral spread. Within this context, it is necessary to better understand the fusogenic potential of the SARS‐CoV‐2 variants that have arisen and will continue to emerge.
We have characterized here the replication, fusogenicity, ACE2 binding, and antibody recognition of Alpha and Beta variants and the role of their S‐associated mutations. Despite the insights we provide into the S‐mediated fusogenicity of the variants, we did not address the conformational changes that the mutations individually or in combination may elicit. We further show that Alpha, Beta, and Delta S proteins more efficiently bind to ACE2 and are more fusogenic than D614G. The virological and immunological features of the Delta variant which explain its higher transmissibility rate compared to the Alpha and other variants at the population level remains an outstanding question.
Materials and Methods
Plasmids
A codon‐optimized version of the reference Wuhan SARS‐CoV‐2 Spike (GenBank: QHD43416.1) was ordered as a synthetic gene (GeneArt, Thermo Fisher Scientific) and was cloned into a phCMV backbone (GeneBank: AJ318514), by replacing the VSV‐G gene. The mutations for Alpha and Beta (Fig 4A) were added in silico to the codon‐optimized Wuhan strain and ordered as synthetic genes (GeneArt, Thermo Fisher Scientific) and cloned into the same backbone (Planas et al, 2021a). The D614G S plasmid was generated by introducing the mutation into the Wuhan reference strain via Q5 site‐directed mutagenesis (NEB). Other individual mutations were subsequently introduced into the D614G S by the same process. Plasmids were sequenced prior to use. The primers used for sequencing and the site‐directed mutagenesis are listed in the tables (Tables EV1 and EV2). pQCXIP‐Empty control plasmid, pQCXIP‐IFITM1‐N‐FLAG, pQCXIP‐IFITM2‐N‐FLAG, pQCXIP‐IFITM3‐N‐FLAG were previously described (Buchrieser et al, 2019). pQCXIP‐BSR‐GFP11 and pQCXIP‐GFP1‐10 were from Yutaka Hata ((Kodaka et al, 2015); Addgene plasmid #68716; http://n2t.net/addgene:68716; RRID: Addgene_68716 and Addgene plasmid #68715; http://n2t.net/addgene:68715; RRID: Addgene_68715). pcDNA3.1‐hACE2 was from Hyeryun Choe ((Li et al, 2003); Addgene plasmid # 1786; http://n2t.net/addgene:1786; RRID: Addgene_1786). pCSDest‐TMPRSS2 was from Roger Reeves ((Edie et al, 2018); Addgene plasmid # 53887; http://n2t.net/addgene:53887; RRID: Addgene_53887).
Cells
Vero E6, HEK293T, U2OS, Caco2/TC7, Calu3 were cultured in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS). Vero and 293T GFP‐split cells transduced cells with pQCXIP were cultured with 4 µg/ml and 1 µg/ml of puromycin (InvivoGen), respectively. U2OS GFP‐split cells transduced with pLenti6 were cultured in 1 µg/ml puromycin and 10 µg/ml blasticidin (InvivoGen). The MucilAirTM primary human bronchial epithelial model was previously described (Robinot et al, 2021). All cells lines were either purchased from ATCC or were kind donations from members of the Institut Pasteur and were routinely screened for mycoplasma.
Viruses
Experiments with SARS‐CoV‐2 isolates were performed in a BSL‐3 laboratory, following safety and security protocols approved by the risk prevention service of Institut Pasteur. The Wuhan SARS‐CoV‐2 strain (BetaCoV/France/IDF0372/2020) and the D614G strain (hCoV‐19/France/GE1973/2020) were supplied by Dr. S. van der Werf of the National Reference Centre for Respiratory Viruses (Institut Pasteur, Paris, France). The D614G viral strain was sourced through the European Virus Archive goes Global (EVAg) platform, which is funded by the European Union’s Horizon 2020 research and innovation program under grant agreement 653316. The Alpha strain was isolated in Tours, France, from an individual who returned from the United Kingdom. The Beta strain (CNRT 202100078) originated from an individual in Creteil, France. Informed consent was provided by the individuals for use of their biological materials. The viruses were isolated from nasal swabs on Vero cells and further amplified one or two passages on Vero cells. The viruses were sequenced directly from the nasal squabs and again upon passaging. Titration of viral stocks was performed by 50% tissue culture infectious dose (TCID50).
Viral release
For quantification of extracellular viral RNA, supernatants were diluted and heat‐inactivated for 20 min at 80°C. qRT‐PCR was performed from 1 µl of template RNA in a final volume of 5 μl per reaction in 384‐well plates using the Luna Universal Probe One‐Step RT‐qPCR Kit (New England Biolabs) with SARS‐CoV‐2 N‐specific primers (Table EV1) on a QuantStudio 6 Flex thermocycler (Applied Biosystems). Standard curve was performed in parallel using purified SARS‐CoV‐2 viral RNA. Infectious virus release was assessed by harvesting supernatant at each time point and preforming a TCID50 assay using Vero cells.
GFP‐split fusion assay
For cell–cell fusion assays, Vero, U2OS‐ACE2, or 293T cell lines stably expressing GFP1‐10 and GFP11 were co‐cultured at a 1:1 ratio (3 × 104, 2 × 104, and 7 × 104 cells/well total, respectively) and were transfected in suspension with a total of 100 ng of DNA with Lipofectamine 2000 (Thermo) in a 96‐well plate (uClear, #655090). 10 ng of phCMV‐SARS‐CoV2‐S and/or 25 ng of pCDNA3.1‐hACE2, 25 ng of pCSDest‐TMPRSS2, and 40 ng of pQCXIP‐IFITM were used and adjusted to 100 ng DNA with pQCXIP‐Empty (control plasmid). At 20 h post‐transfection, images covering 80–90% of the well surface, were acquired per well on an Opera Phenix High‐Content Screening System (PerkinElmer). The GFP area and the number of nuclei were quantified on Harmony High‐Content Imaging and Analysis Software (Fig EV1B). For infection, cells were plated at the aforementioned concentrations and infected the next day with a range of MOIs and fixed at 20 h (U2OS‐ACE) or 48 h (Vero) post‐infection with 4% paraformaldehyde for 30 min. For video microscopy experiments, Vero GFP‐split cells (mixed 1:1) were transfected in suspension with 50 ng of phCMV‐SARS‐CoV2‐S and 450 ng of pQCXIP‐Empty for 30 min at 37°C. Cells were washed twice and then seeded at a confluency of 2 × 105 cells per quadrant in a u‐Dish 35‐mm Quad dish (Ibidi #80416). Cells were allowed to settle, and fluorescence images were taken at 37°C every 10 min up to 24 h using a Nikon BioStation IMQ, with three fields for each condition. Fusion defined as percent of GFP pixels was calculated with ImageJ.
Flow cytometry
For ACE2 binding, 293T cells transfected with S proteins for 24 h were stained with soluble biotinylated ACE2 diluted in MACS buffer at indicated concentrations (from 60 to 0.01 µg/ml) for 30 min at 4°C. The cells were then washed twice with PBS and then incubated with Alexa Fluor 647‐conjugated streptavidin (Thermo Fisher Scientific, 1:400) for 30 min at 4°C. Finally, the cells were washed twice with PBS and then fixed with 4% paraformaldehyde. The results were acquired using an Attune Nxt Flow Cytometer (Life Technologies). Transfection efficiency was assessed by staining with pan‐SARS‐CoV‐2 human mAb129. Antibody binding to S proteins was assessed by an analogous protocol where transfected 293T cells were first stained with either human mAb10 (pan‐coronavirus anti‐S2), mAb102 and mAb129 (pan‐SARS‐CoV‐2), mAb48 and mAb98 (SARS‐CoV‐2 anti‐RBD), and mAb71 (SARS‐CoV‐2 anti‐NTD) at 1 µg/ml. The antibodies were derived from convalescent individuals by the Mouquet laboratory at the Institut Pasteur. mAb10 was generated during the early stages of the epidemic from a patient infected with the Wuhan strain and thus has a higher affinity for the Wuhan S protein (Planas et al, 2021a). For viral replication, infected cells were fixed at each time with 4% paraformaldehyde for 30 min. The cells were stained in the same manner described above with anti‐spike mAb102 and secondary Alexa Fluor 647 (1:500) in MACS buffer containing 0.05% saponin. The gating strategy to determine S protein‐positive cells is represented in the supplement (Appendix Fig S1B).
Western Blot
Cells were lysed in TXNE buffer (1% Triton X‐100, 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, protease inhibitors) for 30 min on ice. Equal amounts (20–50 μg) of cell lysates were analyzed by Western blot. The following antibodies were diluted in WB buffer (PBS, 1% BSA, 0.05% Tween, 0.01% Na Azide): rabbit anti‐human TMPRSS2 (Atlas antibodies cat# HPA035787, 1:1,000), rabbit anti‐human actin (Sigma cat#A2066, 1:2,000), and human anti‐S serum derived from a convalescent individual (1:1,000). Species‐specific secondary DyLight‐coupled antibodies were used (diluted 1:10,000 in WB buffer) and proteins were revealed using a Licor Imager. Images were processed using Image Studio Lite software.
Statistical analysis
Flow cytometry data were analyzed with FlowJo v10 software (Tristar). Calculations were all performed with Microsoft Excel 365. GraphPad Prism 9 was used to generate figures and for statistical analysis. Statistical significance between different conditions was calculated using the tests indicated in the corresponding figure legends.
Author contributions
MMR, JB, MH, LG, RR, LAC, OS made experimental strategy and design. MMR, JB, MH, EB, RR, NS, LG, FG‐B, FP, RR, JD, SG, AB, DP, MZ contributed to experimentation. CP and HM contributed to vital materials and expert advice. MMR processed the data and generated the figures. MMR, JB, OS wrote and edited the manuscript. JB and OS supervised the study. All authors reviewed and approved the manuscript.
Conflict of interest
CP, HM, and OS have a pending patent application for some of the anti‐SARS‐CoV‐2 mAbs described in the present study (PCT/FR2021/070522).
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Rawdata
Acknowledgements
We thank members of the Virus and Immunity Unit for helpful discussions, Dr. Nicoletta Casartelli for her critical reading of the manuscript, and Nathalie Aulner and the UtechS Photonic BioImaging (UPBI) core facility (Institut Pasteur), a member of the France BioImaging network, for image acquisition and analysis support. Work in OS lab is funded by Institut Pasteur, Urgence COVID‐19 Fundraising Campaign of Institut Pasteur, ANRS, the Vaccine Research Institute (ANR‐10‐LABX‐77), Fondation Pour la Recherche Médicale (FRM), Labex IBEID (ANR‐10‐LABX62‐IBEID), ANR/FRM Flash Covid PROTEO‐SARS‐CoV‐2, ANR CoronaMito AAP RA‐COVID‐19 V14, and IDISCOVR. Work in UPBI is funded by grant ANR‐10‐INSB‐04‐01 and Région Ile‐de‐France program DIM1‐Health. MMR and MZ are supported by the Pasteur‐Paris University (PPU) International Doctoral Program. MMR is also supported by Institut Pasteur Department of Virology “Bourse de Soudure” fellowship. DP is supported by the Vaccine Research Institute. LG is supported by the French Ministry of Higher Education, Research and Innovation. EB is supported by the Medecine‐Sciences ENS‐PSL Program. HM laboratory is funded by the Institut Pasteur, the Milieu Intérieur Program (ANR‐10‐LABX‐69‐ 01), the INSERM, REACTing, EU (RECOVER), and Fondation de France (#00106077) grants. The funders of this study had no role in study design, data collection, analysis, interpretation, or the writing of the article.
The EMBO Journal (2021) 40: e108944.
See also: J Koch et al (December 2021)
Contributor Information
Julian Buchrieser, Email: julian.buchrieser@pasteur.fr.
Olivier Schwartz, Email: olivier.schwartz@pasteur.fr.
Data availability
This study includes no data deposited in external repositories.
References
- Ali F, Kasry A, Amin M (2021) The new SARS‐CoV‐2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med Drug Discov 10: 100086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, Sigal N, Feng Y, Bristow L, Tak‐Yin Tsang O et al (2020) Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science 369: 1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asarnow D, Wang B, Lee W‐H, Hu Y, Huang C‐W, Faust B, Ng PML, Ngoh EZX, Bohn M, Bulkley D et al (2021) Structural insight into SARS‐CoV‐2 neutralizing antibodies and modulation of syncytia. Cell 184: 3192–3204.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CT, Neal HE, Edmonds K, Moncman CL, Thompson R, Branttie JM, Boggs KB, Wu CY, Leung DW, Dutch RE (2021) Effect of clinical isolate or cleavage site mutations in the SARS‐CoV‐2 spike protein on protein stability, cleavage, and cell‐cell fusion. J Biol Chem 297: 100902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard P, Orlova E, Sozaeva L, Levy R, James A, Schmitt MM, Ochoa S, Kareva M, Rodina Y, Gervais A et al (2021) Preexisting autoantibodies to type I IFNs underlie critical COVID‐19 pneumonia in patients with APS‐1. J Exp Med 218: e20210554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, Dorgham K, Philippot Q, Rosain J, Beziat V et al (2020) Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science 370: eabd4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher G, Blondot M‐L, Celle A, Pied N, Recordon‐Pinson P, Esteves P, Faure M, Métifiot M, Lacomme S & Dacheaux D et al (2021) SARS‐CoV‐2 transmission via apical syncytia release from primary bronchial epithelia and infectivity restriction in children epithelia. bioRxiv 2021.2005.2028.446159 [PREPRINT]
- Braga L, Ali H, Secco I, Chiavacci E, Neves G, Goldhill D, Penn R, Jimenez‐Guardeño JM, Ortega‐Prieto AM, Bussani R et al (2021) Drugs that inhibit TMEM16 proteins block SARS‐CoV‐2 spike‐induced syncytia. Nature 594: 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J, Degrelle SA, Couderc T, Nevers Q, Disson O, Manet C, Donahue DA, Porrot F, Hillion KH, Perthame E et al (2019) IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise. Science 365: 176–180 [DOI] [PubMed] [Google Scholar]
- Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, Planchais C, Porrot F, Guivel‐Benhassine F, Van der Werf S et al (2020) Syncytia formation by SARS‐CoV‐2‐infected cells. EMBO J 39: e106267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss LF, Prete CA, Abrahim CMM, Mendrone A, Salomon T, de Almeida‐Neto C, França RFO, Belotti MC, Carvalho MPSS, Costa AG et al (2021) Three‐quarters attack rate of SARS‐CoV‐2 in the Brazilian Amazon during a largely unmitigated epidemic. Science 371: 288–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, Volpe MC, Colliva A, Zanconati F, Berlot G et al (2020) Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID‐19 pathology. EBioMedicine 61: 103104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattin‐Ortolá J, Welch L, Maslen SL, Skehel JM, Papa G, James LC, Munro S, Cattin‐Ortolá J, Welch LG, Maslen SL et al (2021) Sequences in the cytoplasmic tail of SARS‐CoV‐2 Spike facilitate expression at the cell surface and syncytia formation. Nat Commun 12: 5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J‐W, Chan K‐H, Choi G‐Y, To K‐W, Tse H, Cai J‐P, Yeung ML, Cheng V‐C, Chen H, Che X‐Y et al (2013) Differential cell line susceptibility to the emerging novel human betacoronavirus 2c EMC/2012: implications for disease pathogenesis and clinical manifestation. J Infect Dis 207: 1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X, Yan R, Zhang J, Zhang G, Zhang Y, Hao M, Zhang Z, Fan P, Dong Y, Yang Y et al (2020) A neutralizing human antibody binds to the N‐terminal domain of the Spike protein of SARS‐CoV‐2. Science 369: 650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier DA, De Marco A, Ferreira IATM, Meng BO, Datir RP, Walls AC, Kemp SA, Bassi J, Pinto D, Silacci‐Fregni C et al (2021) Sensitivity of SARS‐CoV‐2 B.1.1.7 to mRNA vaccine‐elicited antibodies. Nature 593: 136–141 [DOI] [PubMed] [Google Scholar]
- Compton AA, Bruel T, Porrot F, Mallet A, Sachse M, Euvrard M, Liang C, Casartelli N, Schwartz O (2014) IFITM proteins incorporated into HIV‐1 virions impair viral fusion and spread. Cell Host Microbe 16: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, Pearson CAB, Russell TW, Tully DC, Washburne AD et al (2021) Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science 372: eabg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar M, Lee JS, Whig K, Segrist E, Li M, Kamalia B, Castellana L, Ayyanathan K, Cardenas‐Diaz FL, Morrisey EE et al (2021) Drug repurposing screens reveal cell‐type‐specific entry pathways and FDA‐approved drugs active against SARS‐Cov‐2. Cell Rep 35: 108959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez SR, Travanty EA, Qian Z, Mason RJ (2013) Human coronavirus HKU1 infection of primary human type II alveolar epithelial cells: cytopathic effects and innate immune response. PLoS One 8: e70129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H (2020) The SARS‐CoV‐2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike‐based vaccine immunogens. Front Immunol 11: 576622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edie S, Zaghloul NA, Leitch CC, Klinedinst DK, Lebron J, Thole JF, McCallion AS, Katsanis N, Reeves RH (2018) Survey of human chromosome 21 gene expression effects on early development in Danio Rerio. G3 8: 2215–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira I, Datir R, Papa G, Kemp S, Meng B, Rakshit P, Singh S, Pandey R, Ponnusamy K, Radhakrishnan VS et al (2021) SARS‐CoV‐2 B.1.617 emergence and sensitivity to vaccine‐elicited antibodies. bioRxiv 10.1101/2021.05.08.443253 [PREPRINT] [DOI] [Google Scholar]
- Frampton D, Rampling T, Cross A, Bailey H, Heaney J, Byott M, Scott R, Sconza R, Price J, Margaritis M et al (2021) Genomic characteristics and clinical effect of the emergent SARS‐CoV‐2 B.1.1.7 lineage in London, UK: a whole‐genome sequencing and hospital‐based cohort study. Lancet Infect Dis 21: 1246–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, Reid AH, Selbs E, Mcevoy CPL, Hayden CDL, Fukuoka J et al (2003) Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol 34: 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Pere H, Charbit B, Bondet V, Chenevier‐Gobeaux C et al (2020) Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 369: 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Arora P, Gross R, Seidel A, Hornich BF, Hahn AS, Kruger N, Graichen L, Hofmann‐Winkler H, Kempf A et al (2021) SARS‐CoV‐2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 184: 2384–2393 e2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N‐H, Nitsche A et al (2020) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornich BF, Grosskopf AK, Schlagowski S, Tenbusch M, Kleine‐Weber H, Neipel F, Stahl‐Hennig C, Hahn AS (2021) SARS‐CoV‐2 and SARS‐CoV spike‐mediated cell‐cell fusion differ in their requirements for receptor expression and proteolytic activation. J Virol 95: e00002‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YJ, Chiba S, Halfmann P, Ehre C, Kuroda M, Dinnon KH, Leist SR, Schäfer A, Nakajima N, Takahashi K et al (2020) SARS‐CoV‐2 D614G variant exhibits efficient replication ex vivo and transmission in vivo . Science 370: 1464–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu YI, Zhang LI, Fan G, Xu J, Gu X et al (2020a) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yang C, Xu XF, Xu W, Liu SW (2020b) Structural and functional properties of SARS‐CoV‐2 spike protein: potential antivirus drug development for COVID‐19. Acta Pharmacol Sin 41: 1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L, Rodel H, Hwa S‐H, Cele S, Ganga Y, Tegally H, Bernstein M, Giandhari J, Gosnell BI, Khan K et al (2021) SARS‐CoV‐2 cell‐to‐cell spread occurs rapidly and is insensitive to antibody neutralization. bioRxiv 10.1101/2021.06.01.446516 [PREPRINT] [DOI] [Google Scholar]
- Jiang X, Zhang Z, Wang C, Ren H, Gao L, Peng H, Niu Z, Ren H, Huang H, Sun Q (2020) Bimodular effects of D614G mutation on the spike glycoprotein of SARS‐CoV‐2 enhance protein processing, membrane fusion, and viral infectivity. Signal Transduct Target Ther 5: 268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Cortese M, Winter SL, Wachsmuth‐Melm M, Neufeldt CJ, Cerikan B, Stanifer ML, Boulant S, Bartenschlager R, Chlanda P (2020) SARS‐CoV‐2 structure and replication characterized by in situ cryo‐electron tomography. Nat Commun 11: 5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J, Uckeley ZM, Doldan P, Stanifer M, Boulant S, Lozach PY (2021) TMPRSS2 expression dictates the entry route used by SARS‐CoV‐2 to infect host cells. EMBO J 40: e107821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaka M, Yang Z, Nakagawa K, Maruyama J, Xu X, Sarkar A, Ichimura A, Nasu Y, Ozawa T, Iwasa H et al (2015) A new cell‐based assay to evaluate myogenesis in mouse myoblast C2C12 cells. Exp Cell Res 336: 171–181 [DOI] [PubMed] [Google Scholar]
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B et al (2020) Tracking changes in SARS‐CoV‐2 spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell 182: 812–827.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempl C, Schultze B, Laude H, Herrler G (1997) Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J Virol 71: 3285–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K, Shum MH, Leung GM, Lam TT, Wu JT (2021) Early transmissibility assessment of the N501Y mutant strains of SARS‐CoV‐2 in the United Kingdom, October to November 2020. Euro Surveill 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC et al (2003) Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Li Q, Wang Y, Shi Y (2021) Syncytia formation during SARS‐CoV‐2 lung infection: a disastrous unity to eliminate lymphocytes. Cell Death Differ 28: 2019–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu J, Plante KS, Plante JA, Xie X, Zhang X, Ku Z, An Z, Scharton D, Schindewolf C et al (2021a) The N501Y spike substitution enhances SARS‐CoV‐2 transmission. bioRxiv 10.1101/2021.03.08.434499 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, VanBlargan LA, Bloyet L‐M, Rothlauf PW, Chen RE, Stumpf S, Zhao H, Errico JM, Theel ES, Liebeskind MJ et al (2021b) Identification of SARS‐CoV‐2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 29: 477–488.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan B, Wang H, Huynh T (2021) Enhanced binding of the N501Y‐mutated SARS‐CoV‐2 spike protein to the human ACE2 receptor: insights from molecular dynamics simulations. FEBS Lett 595: 1454–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B, Kemp SA, Papa G, Datir R, Ferreira IA, Marelli S, Harvey WT, Lytras S, Mohamed A, Gallo G et al (2021) Recurrent emergence of SARS‐CoV‐2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep 35: 109292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nal B, Chan C, Kien F, Siu L, Tse J, Chu K, Kam J, Staropoli I, Crescenzo‐Chaigne B, Escriou N et al (2005) Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E. J Gen Virol 86: 1423–1434 [DOI] [PubMed] [Google Scholar]
- Nelson G, Buzko O, Spilman P, Niazi K, Rabizadeh S, Soon‐Shiong P (2021) Molecular dynamic simulation reveals E484K mutation enhances spike RBD‐ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 10.1101/2021.01.13.426558 [PREPRINT] [DOI] [Google Scholar]
- Ou T, Mou H, Zhang L, Ojha A, Choe H, Farzan M (2021) Hydroxychloroquine‐mediated inhibition of SARS‐CoV‐2 entry is attenuated by TMPRSS2. PLoS Pathog 17: e1009212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorno A, Padey B, Julien T, Trouillet‐Assant S, Traversier A, Errazuriz‐Cerda E, Fouret J, Dubois J, Gaymard A, Lescure F‐X et al (2020) Characterization and treatment of SARS‐CoV‐2 in nasal and bronchial human airway epithelia. Cell Rep Med 1: 100059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D, Bruel T, Grzelak L, Guivel‐Benhassine F, Staropoli I, Porrot F, Planchais C, Buchrieser J, Rajah MM, Bishop E et al (2021a) Sensitivity of infectious SARS‐CoV‐2 B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat Med 27: 917–924 [DOI] [PubMed] [Google Scholar]
- Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel‐Benhassine F, Rajah MM, Planchais C, Porrot F, Robillard N, Puech J et al (2021b) Reduced sensitivity of SARS‐CoV‐2 variant Delta to antibody neutralization. Nature 596: 276–280 [DOI] [PubMed] [Google Scholar]
- Puray‐Chavez M, LaPak KM, Schrank TP, Elliott JL, Bhatt DP, Agajanian MJ, Jasuja R, Lawson DQ, Davis K, Rothlauf PW et al (2021) Systematic analysis of SARS‐CoV‐2 infection of an ACE2‐negative human airway cell. Cell Rep 36: 109364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Dominguez SR, Holmes KV (2013) Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS‐CoV) in virus entry and syncytia formation. PLoS One 8: e76469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan M, Ferguson ID, Miao W, Khavari PA (2021) SARS‐CoV‐2 B.1.1.7 and B.1.351 spike variants bind human ACE2 with increased affinity. Lancet Infect Dis 21: 1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees‐Spear C, Muir L, Griffith SA, Heaney J, Aldon Y, Snitselaar JL, Thomas P, Graham C, Seow J, Lee N et al (2021) The effect of spike mutations on SARS‐CoV‐2 neutralization. Cell Rep 34: 108890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinot R, Hubert M, de Melo GD, Lazarini F, Bruel T, Smith N, Levallois S, Larrous F, Fernandes J, Gellenoncourt S et al (2021) SARS‐CoV‐2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat Commun 12: 4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino EC, Buss LF, Carvalho MPS, Prete CA, Crispim MAE, Fraiji NA, Pereira RHM, Parag KV, da Silva Peixoto P, Kraemer MUG et al (2021) Resurgence of COVID‐19 in Manaus, Brazil, despite high seroprevalence. Lancet 397: 452–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Jumper CC, Ackerman PJ, Bracha D, Donlic A, Kim H, Kenney D, Castello‐Serrano I, Suzuki S, Tamura T et al (2021) SARS‐CoV‐2 requires cholesterol for viral entry and pathological syncytia formation. Elife 10: e65962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Kenney AD, Kudryashova E, Zani A, Zhang L, Lai KK, Hall‐Stoodley L, Robinson RT, Kudryashov DS, Compton AA et al (2021) Opposing activities of IFITM proteins in SARS‐CoV‐2 infection. EMBO J 40: e106501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Schwartz O, Compton AA (2017) More than meets the I: the diverse antiviral and cellular functions of interferon‐induced transmembrane proteins. Retrovirology 14: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, Li JZ, Bloom JD (2021) Prospective mapping of viral mutations that escape antibodies used to treat COVID‐19. Science 371: 850–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, Doolabh D, Pillay S, San EJ, Msomi N et al (2020) Emergence and rapid spread of a new severe acute respiratory syndrome‐related coronavirus 2 (SARS‐CoV‐2) lineage with multiple spike mutations in South Africa. medRxiv 10.1101/2020.12.21.20248640 [PREPRINT] [DOI] [Google Scholar]
- Thorne LG, Bouhaddou M, Reuschl A‐K, Zuliani‐Alvarez L, Polacco B, Pelin A, Batra J, Whelan MVX, Ummadi M, Rojc A et al (2021) Evolution of enhanced innate immune evasion by the SARS‐CoV‐2 B.1.1.7 UK variant. bioRxiv 10.1101/2021.06.06.446826 [PREPRINT] [DOI] [Google Scholar]
- Tian F, Tong B, Sun L, Shi S, Zheng B, Wang Z, Dong X, Zheng P (2021) N501Y mutation of spike protein in SARS‐CoV‐2 strengthens its binding to receptor ACE2. eLife 10: e69091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY (2020) Pulmonary pathology of early‐phase 2019 novel Coronavirus (COVID‐19) pneumonia in two patients with lung cancer. J Thorac Oncol 15: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touret F, Luciani L, Baronti C, Cochin M, Driouich J‐S, Gilles M, Thirion L, Nougairède A, de Lamballerie X (2021) Replicative fitness of a SARS‐CoV‐2 20I/501Y.V1 variant from lineage B.1.1.7 in human reconstituted bronchial epithelium. mBio 12: e00850‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Made CI, Simons A, Schuurs‐Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, van Deuren RC, Steehouwer M, van Reijmersdal SV, Jaeger M et al (2020) Presence of genetic variants among young men with severe COVID‐19. JAMA 324: 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JCC, Muecksch F, Rutkowska M, Hoffmann HH et al (2020) Escape from neutralizing antibodies by SARS‐CoV‐2 spike protein variants. eLife 9: e61312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu LI et al (2020) Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med 8: 420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav PD, Sapkal GN, Abraham P, Ella R, Deshpande G, Patil DY, Nyayanit DA, Gupta N, Sahay RR, Shete AM et al (2021) Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccinees. bioRxiv 10.1101/2021.04.23.441101 [PREPRINT] [DOI] [PubMed] [Google Scholar]
- Yurkovetskiy L, Wang X, Pascal KE, Tomkins‐Tinch C, Nyalile TP, Wang Y, Baum A, Diehl WE, Dauphin A, Carbone C et al (2020) Structural and functional analysis of the D614G SARS‐CoV‐2 spike protein variant. Cell 183: 739–751 e738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradník J, Marciano S, Shemesh M, Zoler E, Chiaravalli J, Meyer B, Rudich Y, Dym O, Elad N, Schreiber G et al (2021) SARS‐CoV‐2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol 6: 1188–1198 [DOI] [PubMed] [Google Scholar]
- Zani A, Yount JS (2018) Antiviral protection by IFITM3 in vivo . Curr Clin Microbiol Rep 5: 229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, Jia X, Wu M, Shi B, Xu S et al (2020) Viral and host factors related to the clinical outcome of COVID‐19. Nature 583: 437–440 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zheng Y, Niu Z, Zhang BO, Wang C, Yao X, Peng H, Franca DN, Wang Y, Zhu Y et al (2021) SARS‐CoV‐2 spike protein dictates syncytium‐mediated lymphocyte elimination. Cell Death Differ 28: 2765–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Chen Y, Zhang S, Niu P, Qin K, Jia W, Huang B, Zhang S, Lan J, Zhang L et al (2019) Structural definition of a neutralization epitope on the N‐terminal domain of MERS‐CoV spike glycoprotein. Nat Commun 10: 3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Ren L, Zhang LI, Zhong J, Xiao Y, Jia Z, Guo LI, Yang J, Wang C, Jiang S et al (2020) Heightened innate immune responses in the respiratory tract of COVID‐19 patients. Cell Host Microbe 27: 883–890.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu NA, Wang W, Liu Z, Liang C, Wang W, Ye F, Huang B, Zhao LI, Wang H, Zhou W et al (2020) Morphogenesis and cytopathic effect of SARS‐CoV‐2 infection in human airway epithelial cells. Nat Commun 11: 3910 [DOI] [PMC free article] [PubMed] [Google Scholar]