

Abstract
The nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein (NLRP) inflammasome is a key inflammatory signaling pathway activated via a two-step signaling process consisting of priming and activation steps. Several studies have shown that 1,25-dihydroxyvitamin D3 (1,25(OH)2VD3) inhibits the priming step required for NLRP3 inflammasome activation in immune cells. However, as activating the NLRP1 inflammasome in keratinocytes does not necessarily require a priming step, whether 1,25(OH)2VD3 inhibits NLRP1 activation in unprimed keratinocytes is currently unknown. In this study, we showed that 1,25(OH)2VD3 inhibits nigericin-induced NLRP1 inflammasome activation in unprimed keratinocytes. 1,25(OH)2VD3 suppressed nigericin-induced interleukin-1β (IL-1β) secretion and caspase-1 activation in human primary keratinocytes. In addition, 1,25(OH)2VD3 significantly inhibited the formation of apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) oligomers and specks, but not caspase-1 enzymatic activity, suggesting that 1,25(OH)2VD3 prevents NLRP1-ASC complex assembly in keratinocytes. Vitamin D receptor (VDR)-knockdown abolished the inhibitory effects of 1,25(OH)2VD3 on nigericin-induced ASC oligomerization and IL-1β secretion, suggesting that 1,25(OH)2VD3 suppresses inflammasome activation via VDR signaling. Furthermore, nigericin induced K+ efflux and cellular reactive oxygen species (ROS) production, and 1,25(OH)2VD3 pretreatment suppressed nigericin-induced ROS production. 1,25(OH)2VD3 increased the expression of both nuclear factor erythroid 2-related factor 2 (NRF2) and heme oxygenase-1 (HO-1), whereas HO-1 inhibition or NRF2 and HO-1 knockdown abrogated the inhibitory effects of 1,25(OH)2VD3 on IL-1β secretion. Our results indicate that 1,25(OH)2VD3 inhibits nigericin-induced activation step of NLRP1 inflammasome activation in unprimed keratinocytes. Our findings reveal the mechanism underlying the inhibitory effect of 1,25(OH)2VD3, which involves NRF2-HO-1 pathway activation through the VDR, providing further insight into the potential function of 1,25(OH)2VD3 as a therapeutic agent for inflammasome-related skin diseases.
Keywords: Vitamin D, Inflammasome, IL-1β, Reactive oxygen species, NRF2-HO-1 pathway, Normal human epidermal keratinocytes
Graphical abstract
Highlights
-
●
1,25-dihydroxyvitamin D3 inhibits nigericin-induced IL-1β secretion
-
●
VDR plays a key role for the inhibitory effect of 1,25-dihydroxyvitamin D3 on NLRP1 activation
-
●
1,25-dihydroxyvitamin D3 activates the NRF2-HO-1 pathway to suppress cellular ROS production
-
●
1,25-dihydroxyvitamin D3 inhibits the activation step of NLRP1 activation in keratinocytes
Abbreviations
- PRR
pathogen-recognition receptor
- TLR
toll-like receptor
- NLR
nucleotide-binding oligomerization domain-like receptor
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- NLRP
NLR family pyrin domain-containing proteins
- TNF
tumor necrosis factor
- IL-1
interleukin-1
- NF-κB
nuclear factor-kappa B
- NLRC4
NLR family caspase recruitment domain-containing 4
- AIM2
absent in melanoma 2
- VDR
vitamin D receptor
- NRF2
nuclear factor erythroid 2-related factor 2
- NQO-1
NAD(P)H quinone oxidoreductase 1
- HO-1
heme oxygenase-1
- KEAP1
Kelch-like ECH-associated protein 1
- ROS
reactive oxygen species
- ATP
adenosine 5′-triphosphate
- NAC
N-acetyl-l-cysteine
- ZnPP
zinc protoporphyrin
- HRP
horseradish peroxidase
- DAPI
4′-6-diamidino-2-phenylindole
- NHEKs
normal human epidermal keratinocytes
- ELISA
enzyme-linked immunosorbent assay
- qPCR
quantitative PCR
- SDS
sodium dodecyl sulfate
- TBS-T
Tris-buffered saline with 0.1% Tween-20
- PBS
phosphate-buffered saline
- siRNA
small interfering RNA
- BMDMs
bone marrow-derived macrophage cells
- NEK7
NIMA-related kinase 7
- LRRs
leucine-rich repeats
- poly(I:C)
polyinosinic-polycytidylic acid
- DPP9
dipeptidyl peptidase 9
- FIIND
function to find domain
- OSCC
oral squamous cell carcinoma
1. Introduction
The skin is constantly exposed to various environmental factors, including ultraviolet (UV) radiation and pollutants, and acts as the outermost barrier of the body [1]. Keratinocytes within the epidermis also provide innate protective immunity against bacterial pathogens through pathogen-recognition receptors (PRRs) such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs) [2]. NLRs intracellularly sense and respond to pathogen- and damage-associated molecular patterns [3], forming an inflammasome assembly with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1 [4]. Inflammasomes are multimeric protein complexes responsible for caspase-1-mediated inflammatory cytokine secretion and inflammatory cell death, termed pyroptosis. In immune cells, inflammasomes are formed and activated via a two-step process of priming and activation. In the priming step (also known as signal 1), TLR stimulation induces transcription of pro-interleukin-1β (pro-IL-1β) and inflammasome components such as NLR family pyrin domain-containing proteins (NLRPs) via nuclear factor-kappa B (NF-κB) activation [5]. In the activation step (also known as signal 2), agonists of NLR sensor proteins initiate inflammasome assembly formation, activating caspase-1 which then processes pro-IL-1β into its mature form [6]. Since basal levels of both pro-IL-1β and NLR proteins are relatively low in immunocytes, the initial priming step is indispensable for inflammasome activation. In contrast, human keratinocytes activate inflammasomes without the priming step, and this is followed by IL-1β secretion due to the constant presence of its precursor and inflammasome-forming proteins, NLR protein, ASC, and pro-caspase-1, in the cytosol [7,8]. To date, several NLRs, including NLRP1, NLRP3, and NLR family caspase recruitment domain-containing 4 (NLRC4), and absent in melanoma 2 (AIM2) have been identified as inflammasome sensors [9]. Among these, the NLRP1 inflammasome is considered a primary inflammasome in keratinocytes [10]. Although NLRP3 has also been identified as an inflammasome sensor NLRP in keratinocytes, its predominance is still debated. In addition, high NLRP1 expression levels are constantly detected in unprimed keratinocytes, whereas other inflammasome sensors remain almost undetectable [7,8]. Fenini et al. [11], used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 technology to show that UVB and the microbial toxin nigericin are sensed mainly by NLRP1, rather than NLRP3.
Although vitamin D is a lipophilic vitamin essential for calcium and bone homeostasis, recent studies have sought to clarify its regulatory role in immunity [12]. Studies on vitamin D deficiency showed increased inflammation and sensitivity to chronic inflammatory diseases such as Crohn's disease, and vitamin D supplementation ameliorated the inflammatory phenotype [13,14]. An active metabolite of vitamin D3, 1,25-dihydroxyvitamin D3 (1,25(OH)2VD3) forms a complex with the vitamin D receptor (VDR) via ligand binding. In immune cells, the VDR-1,25(OH)2VD3 complex regulates the expression of vitamin D responsive genes and interferes with transcription factor signaling, including that mediated by NF-κB, which plays a crucial role in modulating genes involved in inflammation [15]. 1,25(OH)2VD3 also serves as an anti-inflammatory agent by reducing the release of proinflammatory cytokines such as IL-1β through inflammasome activation. In macrophages and T cells, 1,25(OH)2VD3 can inhibit the priming step of NLRP3-inflammasome activation by interfering with NF-κB signaling, leading to the release of reduced levels of IL-1β [16]. Furthermore, a recent report showed that in dsDNA-transfected keratinocytes primed by interferon gamma and IL-17A, 1,25(OH)2VD3 reduced the release of IL-1β in the NLRP1 inflammasome by suppressing the priming step [17]. However, it is still unknown whether 1,25(OH)2VD3 inhibits the activation step during inflammasome activation.
Nuclear factor erythroid 2-related factor 2 (NRF2) is a key transcription factor that regulates the expression of antioxidant enzymes such as heme oxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase 1 (NQO-1), in response to oxidative stress [18]. Under basal conditions, NRF2 is located in the cytoplasm and is susceptible to proteasome-mediated degradation following its binding to a sensor protein, Kelch-like ECH-associated protein 1 (KEAP1) [19]. Under oxidative stress conditions, a conformational change in KEAP1 caused by modification of its cysteine residues stabilizes NRF2, thus activating its transcriptional activity and inducing its nuclear translocation [20]. NRF2 signaling can negatively regulate inflammasome activation by suppressing the reactive oxygen species (ROS) generated during priming. Liu et al. [21] reported that NRF2 activation decreases mitochondrial ROS generation by lipopolysaccharide and adenosine triphosphate (ATP), resulting in the suppression of NLRP3 inflammasome activation.
Several studies have identified 1,25(OH)2VD3 as a potential inhibitor of inflammasomes, based on its inhibition of the priming step [16,17]. However, as human keratinocytes secrete proinflammatory cytokines without priming, it is unclear whether 1,25(OH)2VD3 inhibits the activation step during NLRP1 inflammasome activation in keratinocytes. In this study, we showed, for the first time, that 1,25(OH)2VD3 can inhibit the activation step of NLRP1 inflammasome activation in epidermal keratinocytes, potentially suppressing IL-1β secretion from intracellular storage. In addition, we report that the mechanism underlying the inhibitory action of 1,25(OH)2VD3 involves increasing cellular antioxidative capacity via upregulation of the VDR-NRF2-HO-1 axis.
2. Materials and methods
2.1. Reagents
We purchased 1,25(OH)2VD3 from Toronto Research Chemicals (North York, Ontario, Canada), nigericin from AdipoGen Life Sciences (San Diego, CA, USA), N-acetyl-l-cysteine (NAC) from Sigma-Aldrich (St. Louis, MO, USA), zinc protoporphyrin (ZnPP) from MedChemExpress (Monmouth junction, NJ, USA), anti-IL-1β antibody (MAB601) from R&D Systems (Minneapolis, MN, USA), anti-caspase-1 (D7F10), anti-NRF2 (D1Z9C), anti–HO–1 (D60G11), anti-Lamin A/C (2032), and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibodies from Cell Signaling Technology (Danvers, MA, USA), anti-beta actin antibody from Abcam (Cambridge, MA, USA), anti-ASC (N-15) antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-GAPDH (6C5) from Calbiochem (San Diego, CA, USA), 4′,6-diamidino-2-phenylindole (DAPI) from Dojindo Laboratories (Kumamoto, Japan), Alexa Fluor 594-conjugated anti-mouse antibodies from Thermo Fisher Scientific (Waltham, MA, USA), and CM-H2DCFDA from Invitrogen (Carlsbad, CA, USA). All other chemicals were of the highest available commercial grade.
2.2. Cell culture and treatment
Normal human epidermal keratinocytes (NHEKs, newborn/male, Thermo Fisher Scientific) were cultured in EpiLife™ medium containing 60 μM calcium supplemented with human keratinocyte growth supplement (both from Thermo Fisher Scientific) at 37 °C under a humidified atmosphere of 5% CO2. The medium was changed every other day until the culture reached approximately 50% confluence, after which it was changed every day. The cells were passaged when they reached 80%–90% confluency. NHEKs were not used beyond passage four. NHEKs were incubated with 1,25(OH)2VD3 and subsequently treated with nigericin (5 μM) to induce inflammasome activation. After 4 h of incubation, the cells and cell culture supernatants were stored at −80 °C until the indicated experimental analyses. For protein and mRNA expression analyses of NRF2 and HO-1, NHEKs were treated with 1,25(OH)2VD3. After incubation, the cells were subjected to immunoblotting or quantitative PCR (qPCR) analyses, as described below.
2.3. Enzyme-linked immunosorbent assay (ELISA)
Human IL-1β in the cell culture medium was quantified using a human IL-1 beta/IL-1F2 Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
2.4. Immunoblotting
Protein samples were prepared from culture supernatants using the protein precipitation method, as previously described [22]. In brief, three volumes of acetone were added to the culture medium and then incubated overnight at −20 °C. After centrifugation (16,100×g for 30 min at 4 °C), the supernatant was removed. Precipitated protein and cell samples were eluted using a lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 1% Triton X-100, phosphatase inhibitor cocktail (Thermo Fisher Scientific), and protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Cytoplasmic and nuclear proteins were prepared using a Minute™ Cytoplasmic and Nuclear Extraction Kit (Invent Biotechnologies, Eden Prairie, MN, USA) according to the manufacturer's instructions. The samples were subjected to sodium dodecyl sulfate (SDS) -polyacrylamide gel electrophoresis, after which proteins were transferred to polyvinylidene fluoride membranes using a semi-dry blotter (Bio-Rad Laboratories, Hercules, CA, USA). Following transfer, the membranes were incubated in a blocking solution (5% dried skimmed milk in Tris-buffered saline containing 0.1% Tween-20; TBS-T) for 1 h to reduce nonspecific binding, and then incubated with primary antibodies overnight at 4 °C. The blots were subsequently washed and incubated with HRP-conjugated secondary antibodies for 1 h. Proteins were detected using ECL Prime Western Blotting Detection Reagent (GE Healthcare Biosciences, Piscataway, NJ, USA) and ImageJ software was used for intensity analysis.
2.5. ASC oligomer assay
ASC oligomers were detected by immunoblotting, as previously reported [7], but with some modifications. Briefly, NHEKs were lysed using 1% Triton X-100 in phosphate-buffered saline (PBS). After centrifugation at 16,100×g for 15 min at 4 °C, the pellets were chemically crosslinked with 2 mM disuccinimidyl suberate (Thermo Fisher Scientific) for 30 min at room temperature. The reaction was quenched using 20 mM Tris-HCl pH 7.5 for 15 min. After centrifugation at 16,100×g for 15 min at 4 °C, the pellets were resuspended in a lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and phosphatase inhibitor cocktail) and analyzed by western blotting.
2.6. Immunofluorescence
ASC specks were visualized using immunostaining and confocal microscopy, as previously reported [23], but with some modifications. Briefly, cells were fixed in 4% paraformaldehyde in PBS for 20 min, washed three times with PBS, and permeabilized using 0.1% Triton X-100 in PBS for 20 min. After washing three times with PBS, the cells were blocked with 5% skim milk in TBS-T, washed with TBS-T, and then incubated overnight at 4 °C with anti-ASC antibody (1:50 dilution). They were then washed three times with TBS-T and incubated with Alexa Fluor 594-labeled anti-mouse IgG antibody (1:400 dilution) for 1 h. For nuclear counterstaining, cells were treated with DAPI solution (1:400 dilution) for 20 min, washed with TBS-T, and then mounted with Fluoromount/Plus™ (Diagnostic BioSystems, Pleasanton, CA, USA). Fluorescence signals were imaged using a Leica DM IRE2 fluorescence microscope (Leica Microsystems Japan, Tokyo, Japan) and captured using a Leica MC190 HD camera (Leica Microsystems Japan). Cells with ASC specks were counted using ImageJ software, with at least four distinct fields being analyzed.
2.7. RNA silencing
For small interfering RNA (siRNA) studies, we purchased human VDR siRNA (ON-TARGETplus SMARTpool #L-003448-00), human NRF2 (NFE2L2) siRNA (ON-TARGETplus SMARTpool #L-003755-00), human HO-1 (HMOX1) siRNA (ON-TARGETplus SMARTpool #L-006372-00), and a non-targeting siRNA control pool (ON-TARGETplus SMARTpool control #D-001810-10) from Horizon Discovery (Cambridge, UK). The non-targeting siRNA was used as a negative control. NHEKs were seeded at a density of 2.0 × 105 cells/well in a 6-well plate and then transfected with 20 nM of VDR or scramble siRNA plus HiPerFect Transfection Reagent (Qiagen), according to the manufacturer's instructions. qPCR was used to assess the efficiency of siRNA-mediated suppression of target mRNA.
2.8. ROS detection
We measured cellular ROS levels using a CM-H2DCFDA probe (Invitrogen), according to the manufacturer's instructions. Briefly, cells were seeded in a 96-well plate at a density of 2 × 104 cells/well and treated with 1,25(OH)2VD3 or NAC. The cells were then incubated with 10 μM CM-H2DCFDA for 30 min. After washing twice with PBS, we measured fluorescence (Ex/Em = 492/537 nm) using an Infinite 200 Pro plate reader (Tecan, Männedorf, Switzerland) and visualized the cells under a Leica DM IRE2 fluorescence microscope (Leica Microsystems Japan). Images were captured using a Leica MC190 HD camera (Leica Microsystems Japan).
2.9. Intracellular K+ measurement
Intracellular K+ levels were measured using a protocol described by Ubels et al. [24], but with some modifications. Briefly, cells were washed twice with 280 mM sucrose in ultrapure water to remove extracellular electrolytes. The cells were lysed by adding 500 μL of ultrapure water to each well and placing the plate in a −80 °C freezer for 5 min and then rapidly thawing it in a 37 °C incubator. The lysate was centrifuged at 10,000×g for 3 min, and the protein levels in the supernatants were measured using a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). The supernatant was transferred into an Amicon Ultra-0.5 Centrifugal Filter Device and centrifuged at 16,100×g for 15 min to remove proteins bigger than 3 kDa. Intracellular K+ quantity was measured using an atomic absorption spectrophotometer (Shimadzu AA 6800, Kyoto, Japan). Ion concentrations in cell lysates were expressed as μg/mg protein.
2.10. qPCR
Total RNA was isolated from cultured cells using the RNeasy Mini Kit (Qiagen, Mississauga, Canada), according to the manufacturer's instructions. RNA was stored in RNase-free water at −80 °C until reverse transcription. Following the manufacturer's instructions, we synthesized the first-strand cDNA from 1 μg total RNA using a PrimeScript II 1st strand cDNA Synthesis Kit (Takara Bio, Shiga, Japan). Target mRNA expression levels were measured using Applied Biosystems 7500 Real Time PCR System (Applied Biosystems, Foster City, CA, USA) and the following PrimeTime® Std qPCR Assay (Integrated DNA Technologies, Coralville, Iowa, USA): GAPDH (assay ID Hs.PT.39a.22214836), NFE2L2 (assay ID Hs.PT.58.28159373), and HMOX1 (assay ID Hs.PT.58.45340055). All reactions were performed in triplicate. The housekeeping gene, GAPDH, was used for normalization. Comparative CT method was used to calculate the relative amounts of mRNA.
2.11. Statistical analyses
Data are presented as mean ± standard error of the mean of at least three independent experiments. Differences among means were analyzed by one-way analysis of variance, followed by the Tukey–Kramer test. Student's t-tests was used to test for significant differences between two groups and p < 0.05 was considered statistically significant.
3. Results
3.1. 1,25(OH)2VD3 suppresses nigericin-induced IL-1β secretion and caspase-1 activation in keratinocytes
To evaluate the effect of 1,25(OH)2VD3 on inflammasome activation in keratinocytes, we investigated whether 1,25(OH)2VD3 inhibits the IL-1β secretion and caspase-1 activation induced by the inflammasome activator nigericin. As shown in Fig. 1A, 1,25(OH)2VD3 significantly inhibited IL-1β secretion in nigericin-stimulated NHEKs, and its inhibitory effect increased as the pretreatment period increased. 100–1000 nM concentrations of 1,25(OH)2VD3 almost completely suppressed nigericin-induced IL-1β secretion (Fig. 1B). 1,25(OH)2VD3 on its own did not induce IL-1β secretion (Fig. S1A). Keratinocytes preserve pro-IL-18 as a depot precursor protein and secrete its mature form, IL-18, as well as IL-1β, through inflammasome activation [3,7]. 1,25(OH)2VD3 inhibited nigericin-induced IL-18 secretion (Fig. S1B). Immunoblotting analyses showed that 1,25(OH)2VD3 pretreatment clearly reduced IL-1β levels secreted into the culture media in NHEKs treated with nigericin (Fig. 1C and D). In addition, 1,25(OH)2VD3 inhibited nigericin-induced pro-caspase-1 cleavage into its active form in a dose-dependent manner. Furthermore, 1,25(OH)2VD3 did not directly inhibit caspase-1 enzyme activity (Fig. S1C). These results suggest that 1,25(OH)2VD3 suppresses IL-1β secretion by inhibiting inflammasome activation.
Fig. 1.

1,25(OH)2VD3inhibits nigericin-induced Interleukin (IL)-1β secretion and caspase-1 activation in normal human epidermal keratinocytes (NHEKs).
NHEKs were pretreated with 1,25(OH)2VD3 (100 nM) for the indicated times, or at the indicated concentrations for 24 h. Cells were then stimulated with nigericin (5 μM) for 4 h. (A, B) IL-1β levels in culture media were measured using ELISA. (C, D) Supernatants (SN) and cell lysates were analyzed by immunoblotting using antibodies specific for IL-1β and caspase-1. β-Actin was used as loading control. Data are presented as mean ± SEM (n = 3). **p < 0.01.
3.2. 1,25(OH)2VD3 prevents the assembly of inflammasome complexes in nigericin-stimulated keratinocytes
ASC oligomerization is an essential step in the assembly of inflammasome complexes. We hypothesized that 1,25(OH)2VD3 inhibits nigericin-inducible inflammasome activation by inhibiting ASC oligomerization in keratinocytes. To confirm this hypothesis, we performed ASC oligomer assays to assess the influence of 1,25(OH)2VD3 on ASC oligomerization. Fig. 2A shows that 1,25(OH)2VD3 pretreatment reduced ASC oligomer formation in nigericin-stimulated NHEKs in a time-dependent manner. In addition, to quantify ASC oligomers, we visualized ASC specks using immunofluorescent techniques and showed that, while >10% nigericin-treated NHEKs exhibited ASC specks, pretreatment with 1,25(OH)2VD3 significantly reduced nigericin-induced ASC specks within cells (Fig. 2B and C). These results suggest that 1,25(OH)2VD3 inhibits inflammasome complex assembly, thereby reducing inflammasome activation.
Fig. 2.

1,25(OH)2VD3prevents apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) oligomerization and ASC speck formation in nigericin-stimulated normal human epidermal keratinocytes (NHEKs).
After pretreatment with 1,25(OH)2VD3 (100 nM) for the indicated times, NHEKs were stimulated with nigericin (5 μM) for 4 h. (A) ASC oligomers were detected in whole cell lysates using disuccinimidyl suberate crosslinking by immunoblotting with antibody specific for ASC. β-Actin was used as loading control. (B, C) ASC specks were analyzed by immunostaining using antibodies specific for ASC (red) and the nuclear counterstain DAPI (blue). The white arrow represents ASC specks. Scale bar: 10 μm. Data are presented as mean ± SEM (n = 3). **p < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. 1,25(OH)2VD3 inhibits nigericin-induced inflammasome activation in keratinocytes through VDR signaling
To determine whether VDR signaling contributes to 1,25(OH)2VD3-induced downregulation of inflammasome assembly, we performed knockdown experiments using siRNA targeted at VDR. Transient transfection of NHEKs with VDR siRNA led to approximately 50% reduction in VDR protein expression (Fig. S2). VDR silencing significantly attenuated the inhibitory effect of 1,25(OH)2VD3 on nigericin-induced IL-1β secretion (Fig. 3A). In addition, Fig. 3B–D shows that siRNA-mediated knockdown of VDR abolished 1,25(OH)2VD3-induced reduction in ASC oligomer formation and specks in nigericin-stimulated NHEKs. Taken together, these results suggest that 1,25(OH)2VD3 inhibits inflammasome activation via a VDR signaling-dependent mechanism.
Fig. 3.

1,25(OH)2VD3suppresses nigericin-induced interleukin (IL)-1β release and inflammasome complex assembly via vitamin D receptor (VDR) signaling in normal human epidermal keratinocytes (NHEKs).
NHEKs were transfected with siRNA targeting VDR. NHEKs were pretreated 24 h after transfection with 1,25(OH)2VD3 (100 nM) for 24 h and then stimulated with nigericin (5 μM) for 4 h. (A) IL-1β levels in culture media were measured using ELISA. (B) Apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) oligomers were measured in whole cell lysates using disuccinimidyl suberate crosslinking by immunoblotting with antibody specific for ASC. β-Actin was used as loading control. (C, D) ASC specks were analyzed by immunostaining using antibodies specific to ASC (red) and the nuclear counterstain DAPI (blue). The white arrow represents ASC specks. Scale bar: 10 μm. Data are presented as mean ± SEM (n = 3). **p < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. 1,25(OH)2VD3 suppresses nigericin-induced ROS production, but not K+ efflux, in keratinocytes
Nigericin activates inflammasomes by promoting ROS generation and K+ efflux [25]. Therefore, to evaluate whether 1,25(OH)2VD3 inhibits nigericin-induced ROS production, we measured intracellular ROS levels using the fluorescent probe CM-H2DCFDA. Nigericin stimulation markedly increased intracellular ROS levels in NHEKs (Fig. 4A and B). Although both 1,25(OH)2VD3 and the ROS scavenger NAC significantly inhibited nigericin-induced ROS production, 1,25(OH)2VD3 required a longer pretreatment period than NAC to reduce ROS production. 1,25(OH)2VD3 did not affect intracellular ROS levels in the absence of nigericin (Fig. S3).
Fig. 4.

1,25(OH)2VD3suppresses nigericin-induced reactive oxygen species (ROS) production, but not K+efflux in normal human epidermal keratinocytes (NHEKs).
NHEKs were pretreated with 1,25(OH)2VD3 (100 nM) or the ROS scavenger N-acetyl-l-cysteine (NAC) (10 mM) for the indicated times. Following incubation, the cells were stimulated with nigericin (5 μM) for 30 min. (A) Cellular ROS levels were measured with a microplate reader using the fluorescent CM-H2DCFDA probe. (B) Intracellular ROS levels were evaluated by fluorescence microscopy using the CM-H2DCFDA probe. (C) Twenty-four h after pretreatment with 1,25(OH)2VD3 (100 nM), cells were stimulated with nigericin (5 μM) for 30 min. The intracellular potassium levels were measured using an atomic absorption spectrophotometer. Scale bar: 50 μm. Data are presented as mean ± SEM (n = 3). **p < 0.01. N.S., not significant.
We then measured the intracellular K+ concentration to assess whether 1,25(OH)2VD3 inhibits K+ efflux upon nigericin stimulation. As shown in Fig. 4C, NHEK stimulation with nigericin markedly decreased intracellular K+ concentration, but 1,25(OH)2VD3 did not affect the K+ efflux triggered by nigericin. These results suggest that 1,25(OH)2VD3 inhibits nigericin-inducible ROS production, but not K+ efflux.
3.5. 1,25(OH)2VD3 activates the NRF2-HO-1 antioxidant axis that inhibits IL-1β secretion in nigericin-stimulated keratinocytes
To explore the effect of 1,25(OH)2VD3 on the NRF2-HO-1 axis, we first measured NRF2 mRNA expression levels using qPCR following 1,25(OH)2VD3 treatment of NHEKs. Fig. 5A shows that 1,25(OH)2VD3 significantly increased NRF2 gene expression. We also assessed the localization of NRF2. We observed NRF2 nuclear translocation as early as 1 h after treatment with 1,25(OH)2VD3 (Figs. 5B, 5C, S4A, and S4B). 1,25(OH)2VD3 clearly increased the mRNA levels of key NRF2 targets such as HO-1 and NQO-1 (Figs. 5D and S4C). Upregulated HO-1 protein expression was also confirmed in 1,25(OH)2VD3-treated NHEKs (Fig. 5E and F). To evaluate whether the NRF2-HO-1 pathway activated by 1,25(OH)2VD3 inhibits IL-1β secretion, we examined the influence of the HO-1 inhibitor, ZnPP, and siRNAs against NRF2 and HO-1 on IL-1β secretion in nigericin-stimulated NHEKs. As shown in Fig. 5G, ZnPP almost completely counteracted the inhibitory effects of 1,25(OH)2VD3 on nigericin-induced IL-1β secretion. ZnPP itself did not affect nigericin-induced secretion of IL-1β (Fig. S4D). Knocking down NRF2 and HO-1 in NHEKs reduced their expression by approximately 80% (Figs. S4E and F). Both NRF2 and HO-1 silencing significantly attenuated the inhibitory effect of 1,25(OH)2VD3 on nigericin-induced IL-1β secretion (Fig. 5H). These results suggest that 1,25(OH)2VD3 suppresses IL-1β secretion induced by nigericin by activating the NRF2-HO-1 pathway.
Fig. 5.

1,25(OH)2VD3attenuates interleukin (IL)-1β secretion via NRF2-HO-1 pathway activation in nigericin-stimulated normal human epidermal keratinocytes (NHEKs).
NHEKs were treated with 1,25(OH)2VD3 (100 nM) for the indicated times, or at the indicated concentrations for 24 h. (A) NRF2 mRNA levels were quantified by qPCR. (B, C) Cell lysates were analyzed by immunoblotting using antibody specific for NRF2. Lamin A/C and GAPDH were used as nuclear and cytoplasmic loading controls, respectively. (D) HO-1 mRNA levels were quantified by qPCR. (E, F) Cell lysates were analyzed by immunoblotting using antibody specific for HO-1. β-Actin was used as loading control. (G) NHEKs were pretreated with HO-1 inhibitor ZnPP (1 μM) for 30 min or (H) transfected with siRNAs targeting NRF2 and HO-1 for 24 h. NHEKs were cultured with 1,25(OH)2VD3 for 24 h. The cells were then stimulated with nigericin (5 μM) for 4 h. IL-1β levels in the culture media were measured using ELISA. Data are presented as mean ± SEM (n = 3). *p < 0.05 and **p < 0.01.
4. Discussion
Inflammasomes are key inflammatory signaling components. Unlike other cells, human keratinocytes can activate inflammasomes without the priming step by utilizing depot precursor proteins. Several studies have shown that 1,25(OH)2VD3 suppresses inflammasome activation [16,17]. Although its mechanism of action is assumed to involve inhibition of the priming step, no studies have focused on the inhibitory effect of 1,25(OH)2VD3 on the activation step. Here, we showed, for the first time, that 1,25(OH)2VD3 suppresses the activation step of nigericin-induced NLRP1 activation in keratinocytes by upregulating the NRF2-HO-1 pathway (Fig. 6). K+ efflux agonists such as nigericin and ATP trigger the activation step of inflammasomes in cells primed by an agonistic stimulus for TLR and TNF receptors [26]. We showed that nigericin induced IL-1β secretion in unprimed NHEKs, suggesting that, in keratinocytes, NLRP1 inflammasome activation can be initiated without the priming step. Furthermore, 1,25(OH)2VD3 significantly reduced nigericin-induced IL-1β secretion and NLRP1 inflammasome assembly in NHEKs (Fig. 1, Fig. 2). On its own, 1,25(OH)2VD3 did not affect NLRP1 expression levels, although pro-IL-1β expression levels were increased following treatment with 1,25(OH)2VD3 (Fig. S5). Verway et al. [27], reported that 1,25(OH)2VD3 activates VDR as a transcription factor that increases pro-IL-1β gene expression. Therefore, under unstimulated conditions, such as non-infection, 1,25(OH)2VD3 may potentiate epithelial immunity by increasing the pro-IL-1β pool in NHEKs. Interestingly, its inhibitory activity correlated well with the length of the pretreatment period. In addition, 1,25(OH)2VD3 did not decrease the K+ efflux levels induced by nigericin (Fig. 4C). These results suggest that 1,25(OH)2VD3 does not directly inhibit nigericin-induced inflammasome signaling.
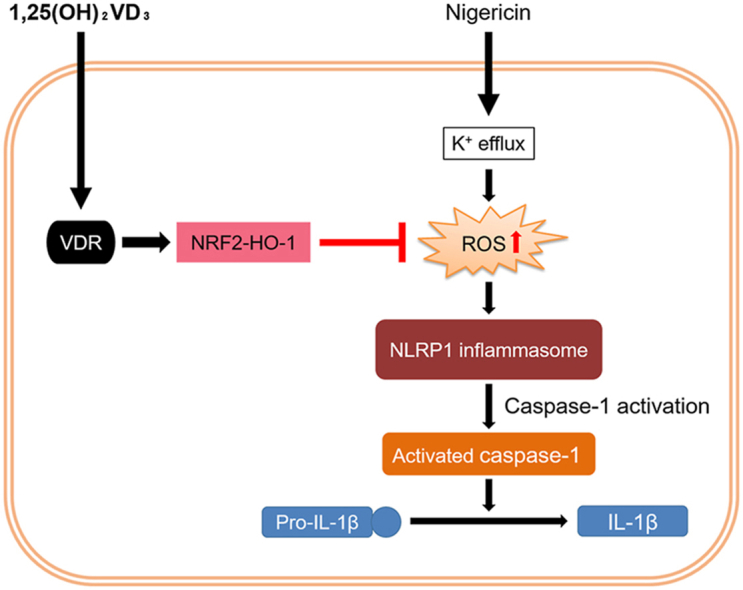
Fig. 6.

Schematic illustration of the molecular mechanism through which 1,25(OH)2VD3suppresses nigericin-induced NLRP1 inflammasome activation in normal human epidermal keratinocytes (NHEKs).
Nigericin causes mitochondrial reactive oxygen species (ROS) production, which induces NLRP1 inflammasome activation and subsequent IL-1β secretion. 1,25(OH)2VD3 inhibits nigericin-induced ROS production by upregulating the VDR-NRF2-HO-1 axis and, consequently, suppresses NLRP1 inflammasome activation in nigericin-stimulated NHEKs.
We found that 1,25(OH)2VD3 reduced nigericin-induced ROS production (Fig. 4A and B). Tschopp et al. [28], raised the possibility that a decline in intracellular potassium concentration induces ROS generation. Several studies have shown that ROS production induces NLRP1 activation [29,30]. However, to the best of our knowledge, there are no reports on how ROS activates the NLRP1 inflammasome. To evaluate whether ROS production is required in the activation step of NLRP1 activation in keratinocytes, we tested the effect of NAC on nigericin-induced NLRP1 activation. NAC significantly suppressed both IL-1β secretion and inflammasome assembly induced by nigericin (Fig. S6). Our results show that, at least in keratinocytes, ROS production plays a key role in NLRP1 activation in the absence of priming stimuli. Epidermal keratinocytes are constantly exposed to UVB from sunlight. UVB induces NLRP1 activation in NHEKs through ROS production [30,31]. Therefore, 1,25(OH)2VD3 is expected to inhibit UVB-induced NLRP1 activation.
Although the mechanisms underlying NLRP inflammasome activation remain largely unclear, several proteins have recently been identified as essential activation regulators. NIMA-related kinase 7 (NEK7), a member of the NEK family, regulates NLRP3 oligomerization and activation downstream of K+ efflux [32]. NEK7 binds to the C-terminal leucine-rich repeats (LRRs) of NLRP3 in a kinase-independent manner, which is likely mediated by mitochondrial ROS [32]. Since NLRP1 contains an LRR domain similar to NLRP3, the ROS-NEK7 axis is likely involved in NLRP1 activation in keratinocytes. We found that 1,25(OH)2VD3 effectively inhibited ROS production, but differed from NAC by requiring a much longer pretreatment period. This intriguing difference suggests that 1,25(OH)2VD3 cannot scavenge ROS directly. Finally, we revealed that 1,25(OH)2VD3 inhibits IL-1β secretion in response to nigericin via prior upregulation of the NRF2-HO-1 pathway (Fig. 6). 1,25(OH)2VD3 is likely to activate the NRF2-HO-1 signaling pathway by binding to VDR. Chen et al. [33], reported that VDR binds to the NRF2 promoter region to increase NRF2 transcription. Indeed, VDR depletion using siRNA significantly abrogated the effects of 1,25(OH)2VD3 on both NLRP1 activation and IL-1β secretion (Fig. 3). Therefore, we are also interested in how 1,25(OH)2VD3 may affect the ROS-NEK7 axis during NLRP1 activation in keratinocytes.
Quite recently, NLRP1 has been reported to function as a direct sensor for double-stranded RNA [34]. According to this report, polyinosinic-polycytidylic acid (poly(I:C)), a synthetic dsRNA analog, was able to trigger IL-1β release in an NLRP1-dependent manner in several epithelial cell types such as NHEKs and immortalized human bronchial epithelial cells. Reins et al. [35] have shown that in epithelial cells, 1,25(OH)2VD3 inhibited poly(I:C)-induced IL-1β secretion. Because ROS production is involved in poly(I:C)-proinflammatory action [36], NRF2-HO-1 axis activation by 1,25(OH)2VD3 partly explain why 1,25(OH)2VD3 inhibits poly(I:C)-induced IL-1β secretion in epithelial cells.
Hereditary diseases of inflammasomes cause autoimmune and autoinflammatory disorders such as rheumatoid arthritis, systemic lupus erythematosus, cryopyrin-associated autoinflammatory disease, and pyoderma gangrenosum [37,38]. The spectrum of autoinflammatory diseases linked to the inflammasome has expanded rapidly over the last decade [39]. Some of these are categorized as inflammasomopathies, and have encouraged new approaches for diagnosis and treatment. 1,25(OH)2VD3 inhibits both the priming and activation steps in the inflammasome-IL-1 system, which is a promising treatment option for these autoinflammatory diseases. Aberrant inflammasome activation has been linked to cancer, and autoimmune and neurodegenerative diseases [40,41]. Zhong et al. [42], reported that dipeptidyl peptidase 9 (DPP9) negatively regulates NLRP1 activation, which is required for both its catalytic function and interaction with the function to find the domain (FIIND). In human cells, NLRP1 activation requires autolytic proteolysis within the FIIND domain, which is distinct from NLRP3 [30]. They also showed that talabostat, a novel drug candidate for cancer immunotherapy, initiates NLRP1 activation by inhibiting DPP9, suggesting that the NLRP1 inflammasome activated by talabostat may partially explain its anti-cancer activity [42]. In fact, 1,25(OH)2VD3 did not inhibit talabostat-induced IL-1β secretion in NHEKs, which is distinct from the secretion induced by nigericin (Fig. S7). We propose that 1,25(OH)2VD3 supplementation may help reduce the risk of unexpected pyroptosis by pathogens during talabostat-based therapy. According to their report, NLRP1 regulation by DPP9 seems to function independently of regulatory mechanisms that involve other NLRP1 domains, such as the pyrin domain and LRR in human cells [42]. Therefore, NRF2-HO-1 axis activated by 1,25(OH)2VD3 is unlikely to interfere with talabostat therapy based on NLRP1 activation due to DPP9 inhibition. Furthermore, chemotherapy in patients with oral squamous cell carcinoma (OSCC) supports the clinical validity of 1,25(OH)2VD3 supplementation [43]. Huang et al. [43], reported that vitamin D supplementation can reduce the over-expression of gasdermin family protein E in normal tissues in patients with OSCC associated with chemotherapeutic side effects primarily caused by NLRP inflammasome activation.
Vitamin D analogs are beneficial for the treatment of psoriasis [44]. Topical vitamin D analogs are widely used as a first-line therapeutic option for psoriasis, either alone or in combination with corticosteroids. In terms of long-term unfavorable effects such as tachyphylaxis, which is seen with topical steroid treatment, vitamin D analogs can be used as long-term treatments, with no undesirable side effects. Since psoriasis was until recently considered a T-cell-dependent disease involving immunity processes, numerous studies have focused on immune cells and pathways amenable to therapeutic targeting [45]. It is now recognized that keratinocytes are also involved in innate immune responses and pathological processes. For example, releasing proinflammatory cytokines from keratinocytes as early upstream events can increase IL-1β and IL-18 levels in psoriasis lesional skin [46]. In addition, single nucleotide polymorphisms in the NLRP1 gene are associated with increased risk of psoriasis [47]. Therefore, the activation step of inflammasomes in keratinocytes may be a reasonable target for preventing potential crosstalk between immune cells and keratinocytes. Vitamin D deficiency is correlated with psoriasis-associated pathogenesis [48]. This explains why topical administration of vitamin D or its analogs is one of the most logical choices for treating this disease.
To our knowledge, this is the first report describing the inhibitory effect of 1,25(OH)2VD3 on the activation step in the NLRP1 inflammasome-mediated IL-1β secretion process, which is based on the upregulation of the NRF2-HO-1 pathway in keratinocytes. This study suggests that the activation step of the NLRP1 inflammasome in epidermal keratinocytes may be targeted by 1,25(OH)2VD3, and offers a new approach for treating inflammasome-related skin diseases.
Funding
This work was supported by a research grant from the DHC Corporation.
Declaration of competing interest
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102203.
Contributor Information
Takahisa Nakajo, Email: tnakajo@dhc.co.jp.
Takeshi Katayoshi, Email: t-katayoshi@dhc.co.jp.
Natsuko Kitajima, Email: nkitajima@dhc.co.jp.
Kentaro Tsuji-Naito, Email: knaito@dhc.co.jp.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Denda M. Skin barrier function as a self-organizing system. Forma. 2000;15:227–232. [Google Scholar]
- 2.Pivarcsi A., Nagy I., Kemeny L. Innate immunity in the skin: how keratinocytes fight against pathogens. Curr. Immunol. Rev. 2005;1:29–42. doi: 10.2174/1573395052952941. [DOI] [Google Scholar]
- 3.Jacobs S.R., Damania B. NLRs, inflammasomes, and viral infection. J. Leukoc. Biol. 2012;92:469–477. doi: 10.1189/jlb.0312132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 5.Bauernfeind F.G., Horvath G., Stutz A., Alnemri E.S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B.G., Fitzgerald K.A., Hornung V., Latz E. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pétrilli V., Papin S., Dostert C., Mayor A., Martinon F., Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 7.Zhong F.L., Mamaï O., Sborgi L., Boussofara L., Hopkins R., Robinson K., Szeverényi I., Takeichi T., Balaji R., Lau A., Tye H., Roy K., Bonnard C., Ahl P.J., Jones L.A., Baker P.J., Lacina L., Otsuka A., Fournie P.R., Malecaze F., Lane E.B., Akiyama M., Kabashima K., Connolly J.E., Masters S.L., Soler V.J., Omar S.S., McGrath J.A., Nedelcu R., Gribaa M., Denguezli M., Saad A., Hiller S., Reversade B. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. 2016;167:187–202. doi: 10.1016/j.cell.2016.09.001. e17. [DOI] [PubMed] [Google Scholar]
- 8.Feldmeyer L., Keller M., Niklaus G., Hohl D., Werner S., Beer H.D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr. Biol. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 9.Martinon F., Gaide O., Pétrilli V., Mayor A., Tschopp J. NALP inflammasomes; A central role in innate immunity. Semin. Immunopathol. 2007;29:213–229. doi: 10.1007/s00281-007-0079-y. [DOI] [PubMed] [Google Scholar]
- 10.Burian M., Yazdi A.S. NLRP1 is the key inflammasome in primary human keratinocytes. J. Invest. Dermatol. 2018;138:2507–2510. doi: 10.1016/j.jid.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Fenini G., Grossi S., Contassot E., Biedermann T., Reichmann E., French L.E., Beer H.D. Genome editing of human primary keratinocytes by CRISPR/Cas9 reveals an essential role of the NLRP1 inflammasome in UVB sensing. J. Invest. Dermatol. 2018;138:2644–2652. doi: 10.1016/j.jid.2018.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Berridge M.J. Vitamin D and depression: cellular and regulatory mechanisms. Pharmacol. Rev. 2017;69:80–92. doi: 10.1124/pr.116.013227. [DOI] [PubMed] [Google Scholar]
- 13.Bouillon R., Carmeliet G., Verlinden L., Van Etten E., Verstuyf A., Luderer H.F., Lieben L., Mathieu C., Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr. Rev. 2008;29:726–776. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X.-X., Liu Y., Luo J., Huang Z.-D., Zhang C., Fu Y. Vitamin D deficiency associated with Crohn's disease and ulcerative colitis: a meta-analysis of 55 observational studies. J. Transl. Med. 2019;17:323. doi: 10.1186/s12967-019-2070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D.-H., Meza C.A., Clarke H., Kim J.-S., Hickner R.C. Vitamin D and endothelial function. Nutrients. 2020;12:1–17. doi: 10.3390/nu12020575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wöbke T.K., Sorg B.L., Steinhilber D. Vitamin D in inflammatory diseases. Front. Physiol. 2014;5:244. doi: 10.3389/fphys.2014.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zwicker S., Hattinger E., Bureik D., Batycka-Baran A., Schmidt A., Gerber P.-A., Rothenfusser S., Gilliet M., Ruzicka T., Wolf R. Th17 micro-milieu regulates NLRP1-dependent caspase-5 activity in skin autoinflammation. PLoS One. 2017;12 doi: 10.1371/journal.pone.0175153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li N., Alam J., Venkatesan M.I., Eiguren-Fernandez A., Schmitz D., Di Stefano E., Slaughter N., Killeen E., Wang X., Huang A., Wang M., Miguel A.H., Cho A., Sioutas C., Nel A.E. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004;173:3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 19.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J.D., Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loboda A., Damulewicz M., Pyza E., Jozkowicz A., Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016;73:3221–3247. doi: 10.1007/s00018-016-2223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Zhou W., Zhang X., Lu P., Du Q., Tao L., Ding Y., Wang Y., Hu R. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem. Pharmacol. 2016;112:37–49. doi: 10.1016/j.bcp.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Fenini G., Grossi S., Gehrke S., Beer H.-D., Satoh T.K., Contassot E., French L.E. The p38 mitogen-activated protein kinase critically regulates human keratinocyte inflammasome activation. J. Invest. Dermatol. 2018;138:1380–1390. doi: 10.1016/j.jid.2017.10.037. [DOI] [PubMed] [Google Scholar]
- 23.Domiciano T.P., Wakita D., Jones H.D., Crother T.R., Verri W.A., Arditi M., Shimada K. Quercetin inhibits inflammasome activation by interfering with ASC oligomerization and prevents interleukin-1 mediated mouse vasculitis. Sci. Rep. 2017;7:41539. doi: 10.1038/srep41539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ubels J.L., Van Dyken R.E., Louters J.R., Schotanus M.P., Haarsma L.D. Potassium ion fluxes in corneal epithelial cells exposed to UVB. Exp. Eye Res. 2011;9:425–431. doi: 10.1016/j.exer.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong Z., Zhou J., Li H., Gao Y., Xu C., Zhao S., Chen Y., Cai W., Wu J. Curcumin suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Mol. Nutr. Food Res. 2015;59:2132–2142. doi: 10.1002/mnfr.201500316. [DOI] [PubMed] [Google Scholar]
- 26.Katsnelson M.A., Rucker L.G., Russo H.M., Dubyak G.R. K+ Efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015;194:3937–3952. doi: 10.4049/jimmunol.1402658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verway M., Bouttier M., Wang T.-T., Carrier M., Calderon M., An B.-S., Devemy E., McIntosh F., Divangahi M., Behr M.A., White J.H. Vitamin D induces interleukin-1β expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog. 2013;6 doi: 10.1371/journal.ppat.1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tschopp J., Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010;3:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 29.Xu T., Sun L., Shen X., Chen Y., Yin Y., Zhang J., Huang D., Li W., Li W. NADPH oxidase 2-mediated NLRP1 inflammasome activation involves in neuronal senescence in hippocampal neurons in vitro. Int. Immunopharm. 2019;69:60–70. doi: 10.1016/j.intimp.2019.01.025. [DOI] [PubMed] [Google Scholar]
- 30.Fenini G., Karakaya T., Hennig P., Di Filippo M., Beer H.D. The NLRP1 inflammasome in human skin and beyond. Int. J. Mol. Sci. 2020;21:1–16. doi: 10.3390/ijms21134788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park K., Park D., Kim Y., Nam T., Jang K., Chung H., Kim U. The essential role of Ca 2+ signals in UVB-induced IL-1β secretion in keratinocytes. J. Invest. Dermatol. 2019;139:1362–1372. doi: 10.1016/j.jid.2018.12.005. https://doi:10.1016/j.jid.2018.12.005 [DOI] [PubMed] [Google Scholar]
- 32.Liu G., Chen X., Wang Q., Yuan L. NEK7: a potential therapy target for NLRP3-related diseases. BioSci. Trends. 2020;14:74–82. doi: 10.5582/bst.2020.01029. [DOI] [PubMed] [Google Scholar]
- 33.Chen L., Yang R., Qiao W., Zhang W., Chen J., Mao L., Goltzman D., Miao D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell. 2019;18 doi: 10.1111/acel.12951. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Bauernfried S., Scherr M.J., Pichlmair A., Duderstadt K.E., Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science. 2021;371 doi: 10.1126/science.abd0811. https://doi:10.1126/science.abd0811 [DOI] [PubMed] [Google Scholar]
- 35.Reins R.Y., Baidouri H., McDermot A.M. Vitamin D activation and function in human corneal epithelial cells during TLR-induced inflammation. Invest. Ophthalmol. Vis. Sci. 2015;13:7715–7727. doi: 10.1167/iovs.15-17768. https://doi:10.1167/iovs.15-17768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H., Zhang J., Dai Y., Xu J. Nerve growth factor inhibits TLR3-induced inflammatory cascades in human corneal epithelial cells. J. Inflamm. 2019;16:27. doi: 10.1186/s12950-019-0232-0. https://doi:10.1186/s12950-019-0232-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moghaddas F., Masters S.L. Monogenic autoinflammatory diseases: Cytokinopathies. Cytokine. 2015;74:237–246. doi: 10.1016/j.cyto.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Yang C.-A., Chiang B.-L. Inflammasomes and human autoimmunity: a comprehensive review. J. Autoimmun. 2015;61:1–8. doi: 10.1016/j.jaut.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Kastner D.L., Aksentijevich I., Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis B.K., Wen H., Ting J.P.-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venegas C., Kumar S., Franklin B.S., Dierkes T., Brinkschulte R., Tejera D., Vieira-Saecker A., Schwartz S., Santarelli F., Kummer M.P., Griep A., Gelpi E., Beilharz M., Riedel D., Golenbock D.T., Geyer M., Walter J., Latz E., Heneka M.T. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. 2017;552:355–361. doi: 10.1038/nature25158. [DOI] [PubMed] [Google Scholar]
- 42.Zhong F.L., Robinson K., Teo D.E.T., Tan K.-Y., Lim C., Harapas C.R., Yu C.-H., Xie W.H., Sobota R.M., Au V.B., Hopkins R., D'Osualdo A., Reed J.C., Connolly J.E., Masters S.L., Reversade B. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem. 2018;293:18864–18878. doi: 10.1074/jbc.RA118.004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Z., Zhang Q., Wang Y., Chen R., Wang Y., Huang Z., Zhou G., Li H., Rui X., Jin T., Li S., Zhang Y., Huang Z. Inhibition of caspase-3-mediated GSDME-derived pyroptosis aids in noncancerous tissue protection of squamous cell carcinoma patients during cisplatin-based chemotherapy. Am. J. Canc. Res. 2020;10:4287–4307. [PMC free article] [PubMed] [Google Scholar]
- 44.Giustina A., Bouillon R., Binkley N., Sempos C., Adler R.A., Bollerslev J., Dawson-Hughes B., Ebeling P.R., Feldman D., Heijboer A., Jones G., Kovacs C.S., Lazaretti-Castro M., Lips P., Marcocci C., Minisola S., Napoli N., Rizzoli R., Scragg R., White J.H., Formenti A.M., Bilezikian J.P. Controversies in vitamin D: a statement from the third international conference. JBMR Plus. 2020;4 doi: 10.1002/jbm4.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albanesi C., Madonna S., Gisondi P., Girolomoni G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front. Immunol. 2018;9:1549. doi: 10.3389/fimmu.2018.01549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albanesi C., Pastore S. Pathobiology of chronic inflammatory skin diseases: interplay between keratinocytes and immune cells as a target for anti-inflammatory drugs. Curr. Drug Metabol. 2010;11:210–227. doi: 10.2174/138920010791196328. [DOI] [PubMed] [Google Scholar]
- 47.Ekman A.-K., Verma D., Fredrikson M., Bivik C., Enerbäck C. Genetic variations of NLRP1: susceptibility in psoriasis. Br. J. Dermatol. 2014;6:1517–1520. doi: 10.1111/bjd.13178. [DOI] [PubMed] [Google Scholar]
- 48.Mostafa W.Z., Hegazy R.A. Vitamin D and the skin: focus on a complex relationship: a review. J. Adv. Res. 2015;6:793–804. doi: 10.1016/j.jare.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.