

Abstract
The tumor suppressor BRCA1 accumulates at sites of DNA damage in a ubiquitin‐dependent manner. In this work, we revisit the role of RAP80 in promoting BRCA1 recruitment to damaged chromatin. We find that RAP80 acts redundantly with the BRCA1 RING domain to promote BRCA1 recruitment to DNA damage sites. We show that that RNF8 E3 ligase acts upstream of both the RAP80‐ and RING‐dependent activities, whereas RNF168 acts uniquely upstream of the RING domain. BRCA1 RING mutations that do not impact BARD1 interaction, such as the E2 binding‐deficient I26A mutation, render BRCA1 unable to accumulate at DNA damage sites in the absence of RAP80. Cells that combine BRCA1 I26A and mutations that disable the RAP80–BRCA1 interaction are hypersensitive to PARP inhibition and are unable to form RAD51 foci. Our results suggest that in the absence of RAP80, the BRCA1 E3 ligase activity is necessary for recognition of histone H2A Lys13/Lys15 ubiquitylation by BARD1, although we cannot rule out the possibility that the BRCA1 RING facilitates ubiquitylated nucleosome recognition in other ways.
Keywords: BARD1, BRCA1, DNA damage, RAP80, ubiquitin
Subject Categories: DNA Replication, Recombination & Repair; Post-translational Modifications & Proteolysis
This study reveals that the localization of BRCA1 at DNA damage sites involves two redundant ubiquitin‐dependent pathways controlled by the RNF8 and RNF168 E3 ubiquitin ligases, with a contribution from the E3 ligase activity of BRCA1.
Introduction
BRCA1 is encoded by the first familial breast and ovarian cancer tumor suppressor gene identified (Futreal et al, 1994; Miki et al, 1994). The mechanism by which BRCA1 suppresses oncogenesis is most likely linked to its function in activating DNA repair by homologous recombination (HR) (Moynahan et al, 1999; Bhattacharyya et al, 2000), although other mechanisms have also been proposed (Tarsounas & Sung, 2020). BRCA1 localizes to sites of DNA damage (Scully et al, 1997a, 1997b; Paull et al, 2000), implying that BRCA1 acts to promote DNA repair directly at DNA lesions, but this link has yet to be formally established. BRCA1 is a modular protein of 1,863 amino acid residues with a RING finger domain located at the N terminus and a coiled‐coil region as well as two tandem BRCT domains at the C terminus (Fig 1A). BRCA1 forms an obligatory heterodimer with the BRCA1‐associated RING domain protein (BARD1) through an interaction via their respective RING finger domains (Fig 1A). This interaction contributes to the stability of both proteins and confers E3 ubiquitin ligase activity to the BRCA1–BARD1 complex toward the C terminus of histone H2A, specifically the K125/K127/K129 residues (Wu et al, 1996; Kalb et al, 2014; Densham et al, 2016; Nakamura et al, 2019; Becker et al, 2021).
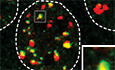
Figure 1. RAP80 is not necessary for BRCA1 recruitment to DSB sites.
-
ASchematic representation of BRCA1 and BARD1. Highlighted are RING domains (orange), PALB2‐interacting coiled‐coil region (CC; red), BRCA1 C‐terminal domains (BRCT; green), and ankyrin repeats (ANK; pink).
-
BImmunoblotting of whole‐cell extracts obtained from parental (WT) and RAP80 −/− clones in the RPE‐1 hTERT p53 −/− Cas9, RPE‐1 hTERT p53 −/− BRCA1 −/− Cas9, U2OS, U2OS 2‐6‐3 and U2OS Flp‐In/TREx cell lines with RAP80 antibodies. Tubulin is used as loading control. Representative of at least two independent immunoblots. See Fig EV1 for further details on the gene editing strategy used to knockout RAP80.
-
C, DThe indicated parental and RAP80 −/− cell lines were processed for immunofluorescence 1 h post‐irradiation (10 Gy) and stained with antibodies against ABRAXAS1 and γH2AX. Shown in (C) is the percentage of cells with > 5 ABRAXAS1 foci that colocalize with γH2AX. A minimum of 100 cells per replicate were analyzed, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs of RPE1‐hTERT p53 −/− Cas9 wild type (WT) and RAP80 −/− cells are shown in (D).
-
E, FThe indicated cell lines were processed for immunofluorescence 1 h post‐irradiation (10 Gy) and stained with antibodies against BRCA1 and γH2AX. Shown in (E) is the percentage of cells with > 5 BRCA1 foci that colocalize with γH2AX. A minimum of 100 cells per replicate were analyzed, and the bars represent mean ± SD (n = 3 biological replicates for all conditions, except n = 5 for U2OS WT, n = 2 for U2OS 2‐6‐3 cells, n = 7, 5, 4 for U2OS Flp‐In/T‐REx cells). Representative micrographs of RPE‐1 hTERT p53 −/− Cas9 wild type (WT) and RAP80 −/− are shown in (F).
-
GRPE‐1 hTERT p53 −/− Cas9 parental (WT) and RAP80 −/− cells were untreated (t = 0 h) or irradiated with a 10 Gy dose. Samples were collected at the indicated time points, processed for immunofluorescence, and stained with antibodies against BRCA1 and γH2AX. Shown is the percentage of cells with > 5 BRCA1 foci. A minimum of 100 cells per replicate were analyzed, and the datapoints represent mean ± SD (n = 3 biological replicates). All scale bars are 5 μm.
Source data are available online for this figure.
BRCA1 accumulates on the chromatin surrounding DNA damage sites in a manner that depends on histone H2AX phosphorylation and the RNF8‐ and RNF168‐catalyzed histone ubiquitylation cascade (Celeste et al, 2003; Huen et al, 2007; Kolas et al, 2007; Mailand et al, 2007; Sobhian et al, 2007; Doil et al, 2009; Stewart et al, 2009). BRCA1 does not contain any recognizable ubiquitin‐binding domain but interacts with BRCA1‐A, a large ubiquitin‐binding complex formed by ABRAXAS1, RAP80, BABAM1, BABAM2, and BRCC3 proteins (Kim et al, 2007b; Liu et al, 2007; Wang et al, 2007) (Shao et al, 2009; Kyrieleis et al, 2016; Rabl et al, 2019). BRCA1 binds to BRCA1‐A via its tandem BRCT domains that recognize phosphorylated ABRAXAS1 (also known as Abraxas or FAM175A). Within BRCA1‐A, the RAP80 subunit (also known as UIMC1) has high affinity for the Lys63‐linked ubiquitin (UbK63) chains produced by RNF8 and RNF168, thereby providing a means for BRCA1 recruitment to DNA lesions (Kim et al, 2007a; Sobhian et al, 2007; Sims & Cohen, 2009; Walters & Chen, 2009; Hu et al, 2012; Rabl et al, 2019). BRCA1‐A is one of at least three BRCA1 protein complexes mediated by the tandem BRCT domain‐dependent recognition of phosphorylated epitopes: BRCA1‐B is formed by interaction with phospho‐BACH1 and BRCA1‐C is formed by interactions with phospho‐CtIP (Wang, 2012).
Although this model of BRCA1 recruitment is attractive, loss of the BRCA1‐A complex results in increased DNA end‐resection and higher levels of HR detectable as gene conversion (Coleman & Greenberg, 2011; Dever et al, 2011; Hu et al, 2011), which is in contrast to the loss of end‐resection and HR activity seen in BRCA1‐deficient cells (Stark et al, 2004; Schlegel et al, 2006; Cruz‐Garcia et al, 2014). These observations suggest either that BRCA1 localization to DSB sites is irrelevant for its function during HR or that there are elements of BRCA1 localization to DSB sites that remain unresolved. In support of the latter possibility, RNA interference studies showed that RAP80 was dispensable for the initial BRCA1 localization at DNA damage sites but was instead proposed to be involved in the maintenance of BRCA1 on damaged DNA (Hu et al, 2011). This work implied that other mechanisms of BRCA1 recruitment to ubiquitylated chromatin must exist.
In an effort to develop a better understanding of the mechanisms of BRCA1 recruitment to DNA damage sites, we revisited the contribution of RAP80 to BRCA1 localization to DSB sites using genetic knockouts of RAP80 in multiple cell backgrounds. We found that RAP80 −/− cells have robust BRCA1 localization to DSB sites and uncovered that this was due to near‐complete redundancy with a DSB site‐targeting activity that is located in the BRCA1 RING finger domain. Mutations that alter BRCA1 RING function without impairing its BARD1 interaction (such as the E2 binding‐deficient I26A mutation) caused complete loss of BRCA1 localization to DNA damage sites and greatly impair HR in the absence of RAP80 or in the presence of mutations that disable the BRCA1/BRCA1‐A interaction. We conclude that DNA damage localization of BRCA1 is essential for its function during HR and that it is dependent on two redundant activities mediated by the BRCA1‐A complex and the BRCA1 RING domain. We finally speculate that the BRCA1 E3 ligase activity may play an important role in endowing recognition of RNF168‐catalyzed H2A Lys13/15 ubiquitylation (H2A‐K13/K15ub) by the BARD1 protein (Becker et al, 2021).
Results
To better understand the contribution of RNF168‐dependent ubiquitylation to BRCA1 accumulation to DSB sites, we generated RAP80 (UIMC1) knockouts by gene editing in multiple RPE1‐hTERT p53 −/− Cas9 (RPE1) and U‐2‐OS (U2OS) cell backgrounds (Figs 1B and EV1A). As expected, analysis of gene conversion using the traffic light reporter assay in RPE1 cells showed that loss of RAP80 causes an increase, rather than a decrease, in HR (Fig EV1B). Furthermore, RAP80 inactivation also demonstrated a complete loss of ABRAXAS1 localization to ionizing radiation (IR)‐induced foci marked by γH2AX in multiple RPE1‐ and U2OS‐derived clones (Fig 1C and D). These results indicated that these cell lines recapitulate known phenotypes associated with RAP80 inactivation.
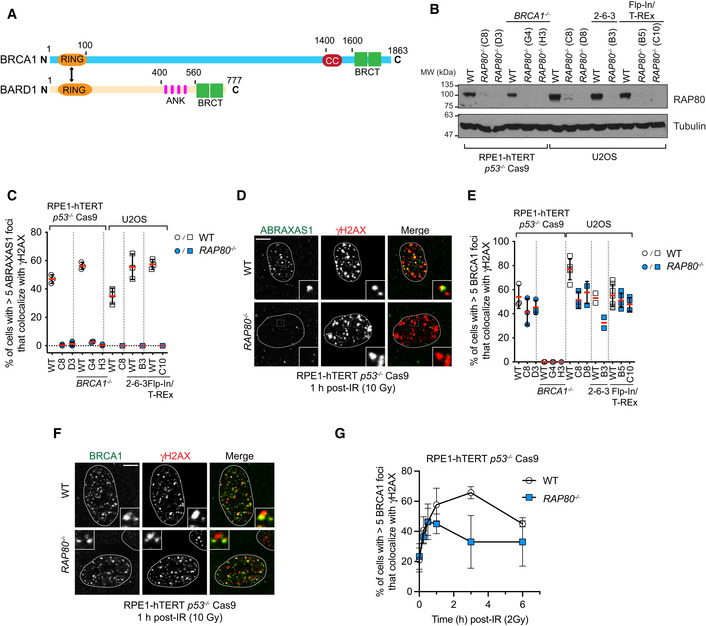
Figure EV1. Design and characterization of RAP80 −/− cell lines.
-
ATo generate RAP80 −/− cell lines, we designed single guide RNAs (sgRNA) that target the first exon of RAP80. U2OS and U2OS Flp‐In/T‐REx clones were sequence validated (Table EV1) and carry RAP80 indels leading to premature stop codons. Dark grey boxes indicate exons 1–14, red arrows indicate the location of the sgRNA, blue line indicates the epitope recognized by the RAP80 antibody used for immunoblotting.
-
BHomologous recombination was monitored by gene conversion using the traffic light reporter assay in the indicated cell lines 48 h post‐nucleofection in the absence or presence of the donor template. BFP+ mClover+ cells have undergone gene conversion. The bars represent the mean ± SD (n = 3 biological replicates).
-
C, DRPE‐1 hTERT p53 −/− Cas9 cells or their isogenic RAP80 −/−counterparts were X‐irradiated (10 Gy) and processed for BRCA1 and cyclin A immunofluorescence. Cyclin A (CycA)‐negative cells are enriched for cells in G1 whereas cyclin A‐positive cells are enriched for S/G2 cells. Shown in (C) is the quantitation and the bars represent the mean ± SD (n = 3 biological replicates). Representative micrographs are shown in (D).
-
ERepresentative micrographs of the experiment shown in Fig 1G at the 1 h timepoint. All scale bars are 5 μm.
Source data are available online for this figure.
These seven independent RAP80 −/− cell lines displayed near‐normal recruitment of BRCA1 to DSB sites as measured by IR‐induced focus formation (Fig 1E and F) and showed typical cell cycle‐restricted localization of BRCA1 in S/G2 cells (Fig EV1C and D). BRCA1 foci were completely lost in BRCA1 −/− cells (Fig 1E), indicating that the immunostaining was specific for the BRCA1 protein. We next examined BRCA1 IR‐induced focus formation and retention over time, fixing cells from 15 min to 6 h post‐irradiation. We detected a defect in the maintenance of BRCA1 foci from 1 h onward in the RAP80 −/− cell line (Figs 1G and EV1E), consistent with the phenotypes previously described using short interfering (si) RNA‐mediated depletion of RAP80 (Hu et al, 2011). We therefore conclude that RAP80, and by inference the BRCA1‐A complex, is dispensable for the initial recruitment of BRCA1 to DSB sites.
RAP80 interacts specifically with UbK63 chains. Although both RNF8 and RNF168 participate in the formation of UbK63 chains, mounting evidence suggests that RNF8 is the main source of UbK63 at DNA damage sites by ubiquitylating histone H1 (Thorslund et al, 2015). We therefore tested whether there was a differential contribution for RNF8 or RNF168 toward BRCA1 recruitment in cell lines that were proficient or deficient in RAP80. We observed that RNF8 depletion by effective siRNAs (Fradet‐Turcotte et al, 2013) led to a near‐complete loss of BRCA1 recruitment to IR‐induced foci, whereas depletion of RNF168 led, in comparison, to an incomplete decrease (Fig 2A and B). The residual BRCA1 recruitment to DSB sites observed in RNF168‐depleted cells was dependent on RAP80 as depletion of RNF168 in RAP80 −/− cells led to a complete loss of BRCA1 IR‐induced foci (Fig 2A and B). To rule out the possibility that these results were an artifact of siRNA‐mediated depletion, we examined BRCA1 recruitment in RNF8 −/− and RNF168 −/− cell lines generated in RPE1‐hTERT p53 −/− Cas9 cells (Fig EV2A). As with siRNA‐depleted cells, we observed in RNF8 −/− cells a complete loss of BRCA1 accumulation into IR‐induced foci compared with a partial reduction of BRCA1 recruitment in the RNF168 knockout cells (Fig 2C and D). Depletion of RAP80 in RNF168 −/− cells abolished the residual recruitment of BRCA1 to DNA damage sites (Fig 2C and D). Examination of localization of RAP80 to IR‐induced foci in the RNF8 −/− and RNF168 −/− cells showed that BRCA1‐A localization was completely dependent on RNF8 but only partially dependent on RNF168 (Figs EV2B and C). These results indicate that RAP80 may promote a mode of BRCA1 recruitment to DNA damage sites that is largely dependent on RNF8 and that acts in parallel to a second mode of recruitment that is dependent on RNF168‐mediated ubiquitylation of histone H2A Lys13/Lys15 residues.
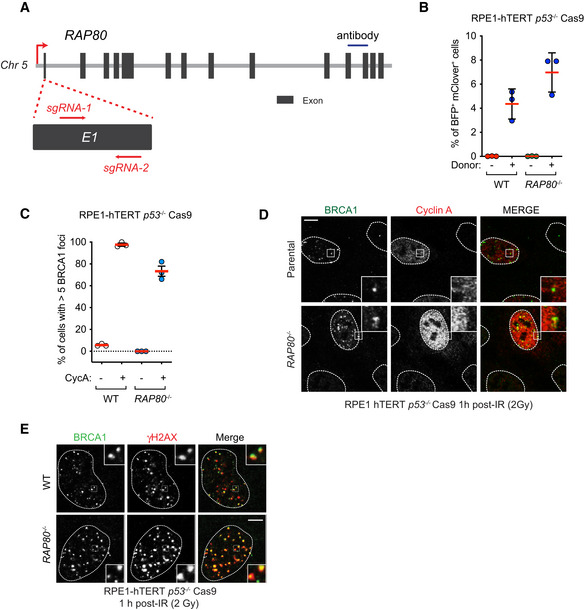
Figure 2. RAP80‐independent BRCA1 recruitment to DSB sites is dependent on RNF8 and RNF168.
-
A, BU2OS Flp‐In/T‐Rex parental (WT) and RAP80 −/− cells were transfected with siRNA pools targeting BRCA1, RNF8, RNF168, or BARD1 or with a nontargeting control siRNA (CTRL). 48 h post‐transfection cells were irradiated (2 Gy) and processed for immunofluorescence 1 h post‐IR treatment using antibodies against BRCA1 and γH2AX. Quantitation of the percentage of cells with > 5 BRCA1 foci that colocalize with γH2AX is shown in (A). A minimum of 100 cells per replicate were analyzed, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in (B).
-
C, DParental (WT) RPE1‐hTERT p53 −/− Cas9, RNF8 −/−, and RNF168 −/− cells were treated with either a nontargeting siRNA pool (CTRL) or a pool targeting RAP80. 48 h post‐transfection cells were irradiated (2 Gy) and processed for immunofluorescence 1 h post‐IR treatment using antibodies against BRCA1 and γH2AX. Quantitation of the percentage of cells with > 10 BRCA1 foci that colocalize with γH2AX is shown in (C). A minimum of 100 cells per replicate were analyzed, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in (D). All scale bars are 5 μm.
Source data are available online for this figure.
Figure EV2. Additional data supporting that loss of RNF8 or RNF168 impairs BRCA1 recruitment to DSB sites.
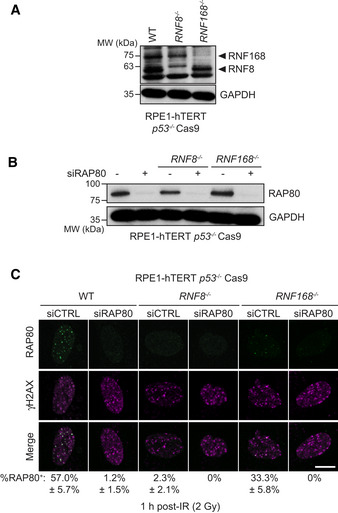
- Immunoblotting of whole‐cell extracts obtained from parental (WT), RNF8 −/−, and RNF168 −/− RPE1‐hTERT p53 −/− Cas9 cells with a mixture of RNF8 and RNF168 antibodies. GAPDH is used as a loading control.
- Immunoblotting of whole‐cell extracts from the indicated cell lines treated with either a non‐targeting siRNA control (−) or siRNA targeting RAP80 (+). GAPDH is used as a loading control. Representative of two independent immunoblots.
- The indicated parental (WT), RNF8 −/−, and RNF168 −/− cell lines were processed for immunofluorescence 1 h post‐irradiation (2 Gy) and stained with antibodies against RAP80 and γH2AX. Representative micrographs are shown with quantitation of the mean percentage of cells with > 5 RAP80 foci (± SD) that colocalize with γH2AX indicated below the micrographs (n = 3 biological replicates). A minimum of 50 cells were analyzed. The scale bar is 10 μm.
To further dissect how BRCA1 may be recruited to DNA damage via RAP80‐dependent and ‐independent pathways, we examined how a truncated protein composed of the isolated tandem BRCT domains of BRCA1 (amino acid residues 1,582–1,863; BRCA1BRCT) is recruited to DNA damage sites. We observed that contrary to the observed results for full‐length BRCA1 recruitment, localization of BRCA1BRCT into IR‐induced foci was strictly dependent on RAP80 and the ABRAXAS1‐interacting S1655 residue in the BRCT domains (Fig 3A and B). These results hinted that the putative second and RAP80‐independent mode of recruitment of BRCA1 to DNA lesions is carried out by a BRCA1 region outside the tandem BRCT domains. In order to map this additional recruitment domain, we generated stable U2OS Flp‐In/T‐Rex cell lines that express various siRNA‐resistant transgenes producing GFP‐tagged BRCA1 and variants. Consistent with the previous results, we observed that deletion of the BRCT domains or introduction of the S1655A phosphopeptide‐binding mutant in the context of full‐length BRCA1 maintains the ability of BRCA1 to form IR‐induced foci (Fig 3C and D). Furthermore, the variant BRCA1 1–1,362, containing a C‐terminal deletion of both BRCT and the PALB2‐interacting coiled‐coil regions, also formed robust IR‐induced foci in U2OS cells (Fig 3C and D). However, to our surprise, expression of a protein consisting solely of the RING finger domain (BRCA1RING, i.e., BRCA1 1–110) also localized to DNA damage sites independently of RAP80 (Fig 3C and D) with similar efficiency to the full‐length protein when focus intensity was measured (Fig EV3A). These results suggest that the RING domain may be responsible for an activity that recruits BRCA1 to DNA damage sites redundantly with RAP80.
Figure 3. RING domain participates in BRCA1 recruitment to DNA damage sites.
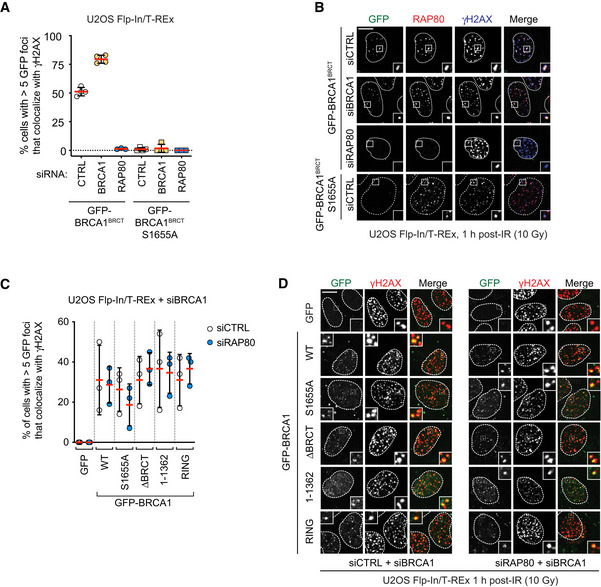
-
A, BU2OS Flp‐In/T‐REx cells with integrated transgenes encoding GFP‐BRCA1BRCT or ‐BRCA1BRCT S1655A were transfected with non‐targeting siRNA (CTRL) or siRNA targeting RAP80 or BRCA1. Following doxycycline treatment to induce transgene expression (5 μg/ml, 24 h), cells were irradiated (10 Gy) and processed for immunofluorescence 1 h post‐IR for antibodies against RAP80 and γH2AX. GFP fluorescence was used to detect BRCA1 fusions. Shown in (A) is the quantitation of a minimum of 100 cells per replicate, where the bars represent mean ± SD (n = 4). Representative micrographs are shown in (B).
-
C, DU2OS Flp‐In/T‐REx cells stably integrated with the indicated transgenes were treated with doxycycline (5 μg/ml, 36 h) to induce protein expression and transfected with an siRNA targeting BRCA1 and also either non‐targeting siRNA (CTRL) or siRNAs targeting RAP80. 1 h post‐irradiation (10 Gy), cells were processed for immunofluorescence using an antibody against γH2AX. GFP fluorescence was used to detect BRCA1 fusions. Shown in (C) is the quantitation of a minimum of 100 cells per replicate, where the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in (D). All scale bars are 5 μm.
Source data are available online for this figure.
Figure EV3. Additional data supporting that the BRCA1 RING domain acts in parallel to RAP80 for BRCA1 recruitment.
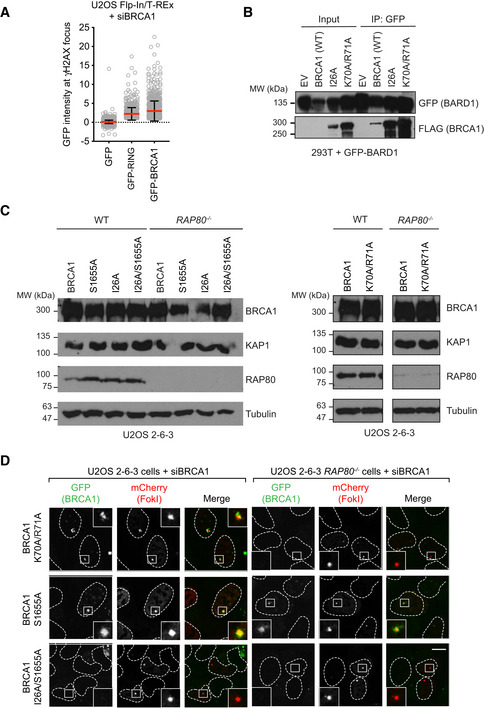
- Quantitation of GFP intensity at the γH2AX focus in U2OS Flp‐In/T‐REx cells expressing the indicated GFP fusion proteins. γH2AX IR‐induced foci were found within a nucleus and the GFP intensity under each γH2AX focal unit was measured. Intensity of GFP under each individual γH2AX focus was plotted with the background GFP intensity subtracted. Bars represent mean ± SD. n = 3,903, 676 and 871 cells analyzed.
- The BRCA1 I26A and K70A/R71A mutants interact with BARD1. Whole‐cell extracts from 293T cells expressing GFP‐BARD1 and the indicated FLAG vectors were subjected to immunoprecipitation with a GFP antibody and were then immunoblotted with FLAG or GFP antibodies. EV, empty vector. Representative of two independent experiments.
- Immunoblotting of parental U2OS 2‐6‐3 (WT) or isogenic RAP80 −/− cell lines transfected with siRNAs targeting BRCA1 and nucleofected with vectors expressing the indicated GFP fusion proteins, as described in Fig 4A and B. Tubulin and KAP1 are used as loading controls.
- Representative micrographs for the additional conditions shown in Fig 4A. The scale bars are 10 μm.
Source data are available online for this figure.
Mutations or loss of the RING domain in BRCA1 impairs its association with BARD1 and leads to BRCA1 destabilization (Hashizume et al, 2001; Joukov et al, 2001; Fabbro et al, 2002), complicating the analysis of the contribution of the RING domain to BRCA1 recruitment. We therefore explored whether we could identify mutations in the BRCA1 RING domain that impair DNA damage localization while maintaining stability. We selected two mutations: BRCA1‐I26A, which disrupts the interaction between the RING and E2 conjugating enzymes such as UbcH5c (Brzovic et al, 2003; Christensen et al, 2007), and BRCA1 K70A/R71A that disrupts the interaction between BRCA1 and the nucleosome acidic patch (McGinty et al, 2014; Witus et al, 2021). These mutations do not impair interaction with BARD1, as reported previously with recombinant proteins (Brzovic et al, 2003; McGinty et al, 2014) or in co‐immunoprecipitation studies (Fig EV3B). These two mutants, along with wild‐type BRCA1, were expressed as fusions to GFP from siRNA‐resistant transgenes in U2OS 2‐6‐3 cell lines (parental and RAP80 −/−; Fig EV3C). The U2OS 2‐6‐3 cell line contains an inducible mCherry‐LacR‐FokI fusion protein that can induce clustered DSBs at an integrated LacO array, ++which allows for facile quantitation of recruitment to DSB sites (Shanbhag et al, 2010). Upon depletion of BRCA1 by siRNA, FokI expression was induced, and GFP fusion protein recruitment to mCherry‐marked DSBs was assessed. We observed that the two BRCA1 RING mutants accumulated at DSB sites as efficiently as wild‐type BRCA1 in RAP80‐proficient cells but had greatly impaired recruitment to FokI‐induced breaks in RAP80 −/− cells, with BRCA1 I26A being the most defective (Figs 4A and B, and EV3D). These results suggested that BRCA1 recruitment to DSB sites is the result of a collaboration between the RING domain and the BRCT‐dependent interaction with BRCA1‐A. To test this idea, we also introduced the BRCA1‐S1655A mutation alone or in combination with I26A. The S1655A mutant showed reduced but RAP80‐independent recruitment to FokI‐induced DSBs that was completely abolished by the I26A mutation (Fig 4A and B).
Figure 4. Nucleosome‐ and E2‐binding by the BRCA1 RING participates in promoting BRCA1 localization.
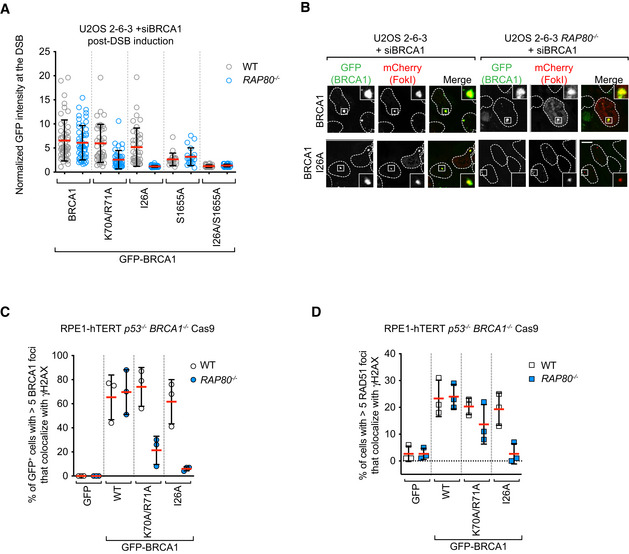
-
A, BU2OS 2‐6‐3 parental (WT) or RAP80 −/− cell lines were transfected with siRNAs targeting BRCA1, followed by nucleofection of vectors encoding the indicated GFP fusion proteins. 48 h post‐nucleofection, mCherry‐LacR‐FokI expression was induced for 5 h prior to being processed for fluorescence microscopy for GFP and mCherry. Shown in (A) is the quantitation of GFP fluorescence at the mCherry focus, where the bars represent mean ± SD (n = 50, 50, 40, 40, 40, 40, 20, 20, 30, 30 cells analyzed from 3 biological replicates). Representative micrographs for the BRCA1 and BRCA1‐I26A conditions are shown in (B). Additional micrographs for the other conditions are in Fig EV3D.
-
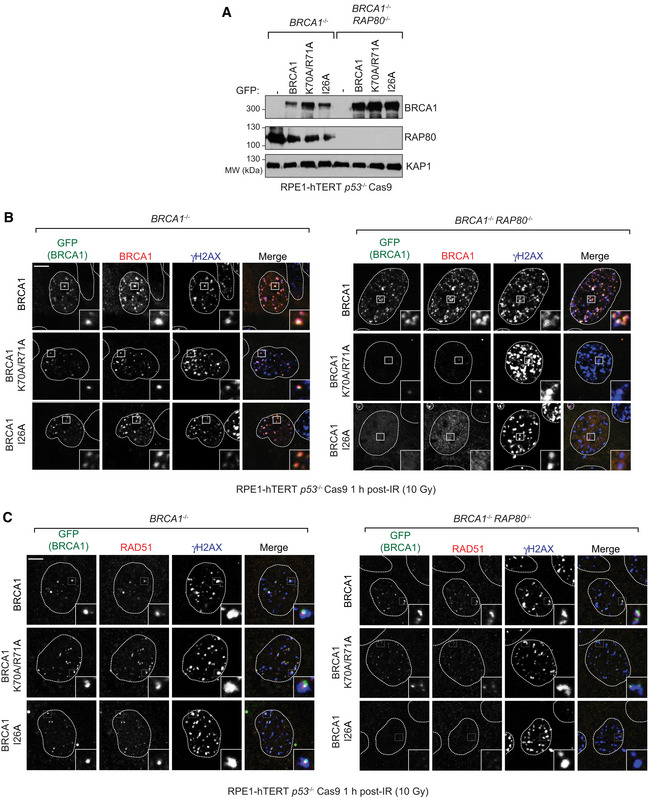
CRPE‐1 hTERT p53 −/− BRCA1 −/− Cas9 cells (WT) or their isogenic RAP80 −/− counterparts expressing the indicated GFP fusion proteins were processed 1 h post‐irradiation (10 Gy) for immunofluorescence using antibodies against BRCA1 and γH2AX. GFP fluorescence was used to detect BRCA1 fusions. A minimum of 100 cells per replicate were analyzed, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in Fig EV4B.
-
DRPE‐1 hTERT p53 −/− BRCA1 −/− Cas9 cells or their isogenic RAP80 −/− counterparts expressing the indicated GFP fusion proteins were processed 1 h post‐irradiation (10 Gy) for immunofluorescence using antibodies against RAD51 and γH2AX. GFP fluorescence was used to detect BRCA1 fusions. A minimum of 100 cells per replicate were analyzed, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in Fig EV4C. The scale bar is 5 μm in (B).
Source data are available online for this figure.
To further test the collaboration between RAP80 and the BRCA1 RING domain in an orthogonal system, we used gene editing to create a RAP80 knockout (RAP80 −/−) in an RPE1‐hTERT BRCA1 −/− p53 −/− Cas9 cell line (Fig EV4A). This cell line allowed us to assess BRCA1 IR‐induced focus formation and its dependence on RAP80 in cell lines expressing BRCA1 variants. As observed with the FokI system, we found that the BRCA1 I26A protein forms IR‐induced foci but does so in a strictly RAP80‐dependent manner (Figs 4C and EV4B). The accumulation of BRCA1 K70A/R71A at DSB sites was also largely dependent on RAP80 but to a lesser extent than on BRCA1 I26A (Figs 4C and EV4B). Together, these results indicate that both the nucleosome–RING and E2–RING interactions participate in the recruitment of BRCA1 to DSB sites in parallel to the BRCA1‐A‐dependent recognition of UbK63 chains by RAP80.
Figure EV4. Additional data supporting that the BRCA1 I26 residue collaborates with the BRCT domain in RPE1‐hTERT cells.
-
AWhole‐cell extracts from RPE‐1 hTERT p53 −/− BRCA1 −/− Cas9 cells or their isogenic RAP80 −/− counterparts expressing the indicated GFP fusion proteins were immunoblotted with antibodies against BRCA1, RAP80, and KAP1 (loading control).
-
B, CRelates to Fig 4C and D. RPE‐1 hTERT p53 −/− BRCA1 −/− Cas9 cells or their isogenic RAP80 −/− expressing the indicated GFP fusion proteins were processed 1 h post‐irradiation (10 Gy) for immunofluorescence using antibodies against γH2AX, and either BRCA1 (B) or RAD51 (C). GFP fluorescence was used also to detect BRCA1 fusions. The scale bars are 5 μm.
The identification of conditions where BRCA1 recruitment to DSB sites is severely impaired allowed us to ask whether BRCA1 accumulation into IR‐induced foci correlates with DNA repair activity by BRCA1. We first assessed RAD51 IR‐induced focus formation in the RPE1‐hTERT cell lines described above as a proxy for HR activity. Mirroring BRCA1 recruitment, we found that RAD51 focus formation was near‐normal in the RAP80‐proficient cell lines expressing the BRCA1 K70A/R71A and I26A mutants but was impaired in the RAP80 −/− cell lines, with BRCA1‐I26A showing reduction in RAD51 IR‐induced foci to the levels of the BRCA1 −/− cells expressing only GFP (Figs 4D and EV4C). These data suggest that RING mutants of BRCA1 rely on their interaction with BRCA1‐A for HR activity.
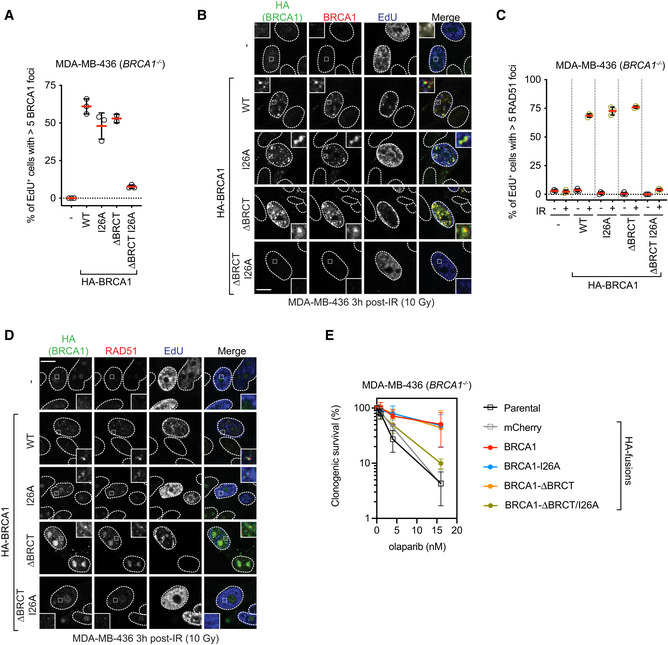
The aforementioned RPE‐hTERT BRCA1 −/− cell lines did not maintain homogenous expression of the BRCA1 variants long enough to allow an assessment of HR activity using assays such as resistance to PARP inhibition. We therefore used MDA‐MB‐436 cells, which harbor a hemizygous BRCA1 5396+1G >A mutation that causes complete loss of BRCA1 protein expression. This cell line was employed for reconstitution assays using lentivirus, as in Nacson et al (2018). We used a virus expressing BRCA1 ΔBRCT first described in Nacson et al (2018) and introduced the I26A mutation in both the ΔBRCT vector and a corresponding vector expressing an otherwise wild‐type BRCA1. We first examined BRCA1 localization to DSB sites and found that as expected from the aforementioned studies, the BRCA1 IR‐induced foci were only abrogated when we combined the BRCT domain deletion with the I26A mutation (Figs 5A and B, and EV5A). Similarly, only the BRCA1 I26A–ΔBRCT mutant showed strongly defective RAD51 focus formation in response to IR, in line with our previous results (Fig 5C and D). Using etoposide, rather than IR, treatment, we observed that although MDA‐MB‐436 cells expressing BRCA1 ΔBRCT had impaired end‐resection, as recently noted (Nacson et al, 2020), any residual end‐resection and RAD51 loading activity were abrogated by adding the I26A mutation (Fig EV5B and C). Finally, we subjected these cell lines to increasing doses of the PARP inhibitor olaparib and measured clonogenic survival. We found that the single BRCA1 I26A and ΔBRCT mutants had similar levels of PARP inhibitor resistance as wild‐type BRCA1 (Figs 5E and EV5D), whereas the BRCA1 I26A–ΔBRCT mutant showed PARP inhibitor hypersensitivity that approached that of the parental or control cell line that only expressed an HA‐mCherry fusion (Figs 5E and EV5D). These results confirm that BRCA1 recruitment to DNA damage sites and its DNA repair activity involve two redundant pathways: one that involve the interaction of the BRCT domain with BRCA1‐A and the other that involves the RING domain. This activity of the RING domain is also completely dependent on its interaction with its cognate E2.
Figure 5. Loss of BRCA1 recruitment to DSB sites causes sensitization to PARP inhibition.
-
A, BMDA‐MB‐436 cells transduced with lentivirus expressing the indicated HA fusions were irradiated (10 Gy) and 3 h later were pulsed with EdU to label S phase cells 20 min prior to fixation. Cells were processed for EdU labeling and immunofluorescence using HA and BRCA1 antibodies. Shown in (A) is the quantitation of a minimum of 100 EdU+ cells per replicate, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in (B).
-
C, DMDA‐MB‐436 cells transduced with lentivirus expressing the indicated HA fusions were irradiated (10 Gy) and 3 h later were pulsed with EdU to label S phase cells 20 min prior to fixation. Cells were processed for EdU labeling and immunofluorescence using HA and RAD51 antibodies. Shown in (C) is the quantitation of minimum of 100 EdU+ cells per replicate, and the bars represent mean ± SD (n = 3 biological replicates). Representative micrographs are shown in (D).
-
EClonogenic survival assays using the indicated dose of olaparib and MDA‐MB‐436 cells transduced with lentivirus expressing the indicated HA fusions. Data are shown as mean ± SD (n = 3 biological replicates). Representative clonogenic images are shown in Fig EV5D.
Data information: All scale bars are 5 μm.
Source data are available online for this figure.
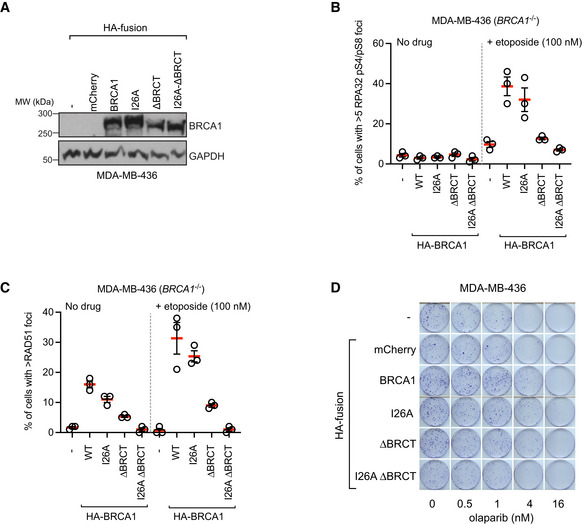
Figure EV5. Additional data supporting that the BRCA1 I26 residue collaborates with the BRCT domain in MDA‐MB‐436 cells.
-
AImmunoblotting of whole‐cell extracts obtained from MDA‐MB‐436 cells transduced with lentivirus expressing the indicated HA fusions. GAPDH was used as a loading control.
-
B, CMDA‐MB‐436 cells transduced with lentivirus expressing the indicated HA fusions were treated with etoposide for 3 h (100 nM) or left untreated. Cells were processed for immunofluorescence with RPA32 pS4/pS8 (B) or RAD51 antibodies (C). The bars represent the mean ± SD (n = 3 independent immunostainings).
-
DRepresentative images of clonogenic survival assays using the indicated dose of olaparib and MDA‐MB‐436 parental cells (‐) or cells transduced with lentivirus expressing the indicated HA‐tagged fusions.
Source data are available online for this figure.
Discussion
Our results are consistent with BRCA1 having two ubiquitin‐dependent modes of recruitment to DSB sites (Fig 6). The first recruitment pathway is dependent on the recognition of UbK63‐linked chains by the RAP80 subunit of the BRCA1‐A complex that interacts with BRCA1 via the latter's BRCT domains. The second mode of recruitment involves the BRCA1 RING domain and is critical for the recognition of H2AK13/K15ub, the mark catalyzed by RNF168. These conclusions are remarkably consistent with earlier structure–function studies published over 15 years ago by Au and Henderson (Au & Henderson, 2005) and a recently published work (Krais et al, 2021).
Figure 6. Model of ubiquitin‐dependent BRCA1 recruitment to DSB sites.
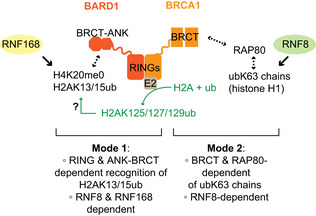
BRCA1 localizes at DSB sites via two redundant ubiquitin (ub)‐dependent processes. The first mode is mediated by the ANK‐BRCT domain of BARD1, also known as BUDR, which recognizes H4K20me0 nucleosomes modified with H2A‐K13/15ub, the product of RNF168. Our work is consistent with a role for the E3 ligase activity (green) for promoting this binding event. The second mode on BRCA1 recruitment is dependent on the BRCT domains of BRCA1 binding to BRCA1‐A whose RAP80 subunit recognizes Ly63‐linked ub chains (ubK63), most likely on histone H1.
Our results suggest the BRCA1 RING domain has two distinct molecular roles that converge on the recognition of the RNF168‐catalyzed histone marks at DNA damage sites. The first is to promote the interaction with BARD1. This is critical, given that the H2AK13/K15ub mark is itself recognized by BARD1 via the ankyrin repeat domain (ARD) in tandem with the BARD1 BRCT domain (the so‐called BUDR) (Becker et al, 2021; Dai et al, 2021; Hu et al, 2021). However, the BRCA1 I26A and K70A/R71A mutations do not perturb interaction with BARD1, and therefore, they might have a second role in H2AK13/K15ub recognition by a mechanism unrelated to the formation of the BRCA1–BARD1 heterodimer. We envisage two possibilities: first, given that the I26A mutant is greatly impaired for ubiquitylation (Brzovic et al, 2006; Kalb et al, 2014), our results support a model whereby BRCA1 catalyzes C‐terminal H2A ubiquitylation to promote its own recruitment. Although this model is attractive, it is not clear how C‐terminal H2A ubiquitylation could act to retain the BRCA1–BARD1 complex at DSB sites. An interesting idea is that the described recruitment of the SMARCAD1 chromatin remodeler by H2A K125/K127/K129 ubiquitylation displaces 53BP1 from nucleosomes (Densham et al, 2016; Densham & Morris, 2017), allowing for BRCA1–BARD1 binding to the liberated H2AK13/K15ub‐bearing nucleosomes. We also note that Densham et al (2016) observed mild sensitivity to olaparib in HeLa cells expressing BRCA1 I26A, suggesting that in some contexts, this activity is important for BRCA1 function even in RAP80‐proficient cells.
We also consider an alternative model where the integrity of the BRCA1 RING–E2 interaction is important for the correct positioning of the BARD1 BUDR for H2AK13/15ub recognition. Such a function for the BRCA1 RING in enabling ubiquitylated nucleosome binding is further supported by our observation that mutation of the acidic patch‐interacting K70/R71 residues also impairs BRCA1 recruitment to DNA lesions in the absence of RAP80. However, we note that in the recent cryo‐EM structures of the BRCA1–BARD1 complex bound to unmodified nucleosomes, the E2 is tilted away from the nucleosome and does not appear to participate in nucleosome binding (Hu et al, 2021; Witus et al, 2021). Furthermore, the phenotypic impact of the I26A mutation is much greater than that of the K70A/R71A mutation, which does not align as well with how these mutations impair interaction with nucleosomes. These two models have testable hypotheses that will be the focus of future experiments.
The observation that multiple established and likely pathogenic mutations in BRCA1 affect the RING domain (Findlay et al, 2018; Bouwman et al, 2020) and that RING‐less BRCA1 protein isoforms can promote resistance to DNA damage (Drost et al, 2016; Wang et al, 2016) suggests a possible avenue for the development of agents that overcome resistance caused by RING‐less BRCA1 proteins. Indeed, targeting members of the BRCA1‐A complex in ways that would impair UbK63 recognition by RAP80 or that would target the binding of phosphorylated ABRAXAS1 by the BRCT domains should render RING‐mutated BRCA1 variants hypersensitive to PARP inhibitors or cisplatin, although being relatively innocuous to cells with wild‐type BRCA1. Although this may be difficult to accomplish through a conventional inhibitor strategy, we note that approaches such as targeted protein degradation (Neklesa et al, 2017) could provide a route for the development of agents that disrupt the DSB recruitment and HR activity of RING‐less BRCA1 variants.
Materials and Methods
Cell lines and culture conditions
RPE1‐hTERT, HEK293, and MDA‐MB‐436 cells were obtained from ATCC and cultured in DMEM (Life Technologies). U2OS cells were obtained from ATCC and were cultured in McCoy's 5A (Life Technologies). U2OS Flp‐In/T‐REx cells were a kind gift from Brad Wouters (OICR) and were cultured in McCoy's 5A medium and supplemented with 15.5 μg/ml zeocin (Invitrogen) and 5 μg/ml blasticidin (Invitrogen). U2OS 2‐6‐3 cells (U2OS ER‐mCherry‐LacI‐FokI‐DD cells) were a kind gift from R. Greenberg (University of Pennsylvania, Philadelphia, PA, USA) and were cultured and transfected as previously described (Shanbhag et al, 2010). All culture media were supplemented with 10% fetal bovine serum (FBS, Wisent). Cell lines were grown in an environmental incubator at 37°C and 5% CO2, except for BRCA1‐deficient cells which were cultured in low oxygen (3% O2, 5% CO2) at 37°C. All cell lines were routinely authenticated by STR analysis and tested to ensure the absence of mycoplasma. Cells were grown on plastic dishes and flasks using standard tissue culture practice. Cells were frozen in 10% DMSO in medium using Mr. Frosty containers (Nalgene) according to manufacturer's instructions. For long‐term storage, cells were moved to liquid nitrogen. All culture media were supplemented with 10% fetal bovine serum (FBS; Wisent).
All inducible BRCA1 cell lines were generated using the Flp‐In/T‐Rex system, as described by the manufacturer (Invitrogen). In brief, pDEST‐pcDNA5‐FRT/TO‐GFP‐derived plasmids were co‐transfected with the pOG44 vector (encoding Flp recombinase) into U2OS Flp‐In/T‐REx cells. Recombination events were selected with 200 μg/ml hygromycin B (Roche). Expression of BRCA1 variants was induced by the addition of 5 μg/ml doxycycline (Inalco) for 24 h.
RNA interference
U2OS, U2OS Flp‐In/T‐REx, U2OS 2‐6‐3, RPE1‐hTERT TP53 −/− RNF8 −/− Cas9, or RPE1‐hTERT TP53 −/− RNF168 −/− Cas9 cells were transfected with siRNA using either a forward or a reverse transfection mode. In both cases, complexes were formed in serum‐free media (Opti‐MEM; Life Technologies) by adding siRNA (dissolved in 1× RNA buffer; Thermo Fisher Scientific) and the lipid‐based transfection reagent RNAiMAX (Invitrogen) according to the manufacturer's instructions. The final concentration of siRNA complexes was 10 nM. The following OnTarget Plus siRNA reagents were purchased from Horizon Discovery/Dharmacon: BRCA1 (D‐003461‐05), RAP80 pool (L‐006995‐00‐0020), BARD1 pool (L‐003873‐00‐0020), RNF8 pool (L‐006900‐00‐0020), and RNF168 pool (L‐007152‐00‐0020).
Gene‐edited cell lines
Cell lines were generating using U2OS, U2OS Flp‐In/T‐REx, U2OS 2‐6‐3, or RPE1‐hTERT TP53 −/− Cas9 as parental cell lines. The RAP80 −/− knockout cell lines were generated by electroporation (Lonza Amaxa II Nucleofector) of plasmids expressing sgRNAs (5′‐ATTGTGATATCCGATAGTGAT‐3′ and 5′‐GTTCTGTCAGTGTGAAGAGG‐3′) and Cas9, followed by the 2A‐puromycin cassette (pX459, Addgene #62988). Twenty‐four h after transfection, cells were selected with puromycin for 24–48 h (1 μg/ml for U2OS, U2OS Flp‐In, and U2OS 2‐6‐3 cell lines, and 10–15 μg/ml for RPE1‐hTERT TP53 −/− Cas9, and RPE1‐hTERT TP53 −/− BRCA1 −/− Cas9 cell lines), followed by single clone isolation. Clones were screened by immunoblotting and immunofluorescence to verify the loss of RAP80 expression and subsequently characterized by PCR and sequencing. The genomic region targeted by the CRISPR–Cas9 was amplified by PCR using Turbo Pfu polymerase (Agilent), and the PCR product was cloned into the pCR2.1 TOPO vector (Invitrogen) before sequencing. The RPE1‐hTERT TP53 −/− BRCA1 −/− Cas9 cell lines were described previously (Noordermeer et al, 2018). The RPE1‐hTERT TP53 −/− RNF8 −/− Cas9 cell line was generated by electroporation of phU6‐gRNA (Addgene #53188) plasmids expressing sgRNAs (5′‐CCCAGAGTCTAAATGGTGTT‐3′ and 5′‐GGAAGAGGAACAGCATCTTC‐3′). Cells were isolated from single clones. The RPE1‐hTERT TP53 −/− RNF168 −/− Cas9 cell line was generated by electroporation of pX459 plasmids expressing sgRNAs (5′‐CGCTCTAAGCTTGCACTCCC‐3′ and 5′‐GCCGGGTATCGTCGTGGACT‐3′). Cells were selected with 15 μg/ml of puromycin, followed by single clone isolation. Both RPE1‐hTERT TP53 −/− RNF8 −/− Cas9 and RPE1‐hTERT TP53 −/− RNF168 −/− Cas9 clones were screened by immunofluorescence of BRCA1 and immunoblotting of RNF8 and RNF168 to verify the loss of RNF8 or RNF168 expression.
Plasmids
To generate the BRCA1 expression vectors used in this study, we generated Gateway‐compatible entry clones in pDONR221 (Invitrogen) by amplifying BRCA1 coding regions from GFP‐BRCA1 expression vectors (gift from J. Lukas). These entry clones were used to generate destination vectors in pDEST‐pcDNA5‐GFP‐FRT/TO (kind gift of A‐C Gingras). The deletion mutants of BRCA1 were created by deletion PCR. To generate the BRCA1 GFP‐BRCA1BRCT expression vector, we PCR‐amplified the region encoding amino acid residues 1,582–1,863 from a GFP‐BRCA1 expression vector and ligated the product in the BamHI and EcoRI restriction sites of pcDNA5‐GFP‐FRT/TO. The BRCA1‐I26A, BRCA1‐K70A/R71A, BRCA1‐S1655A, BRCA1‐I26A/S1655A, and GFP‐BRCT‐S1655A plasmids were generated by site‐directed mutagenesis PCR (QuikChange; Agilent), and all plasmids were sequence‐verified. To generate BRCA1 constructs resistant to BRCA1 siRNA #5 (Dharmacon, D‐003461‐05), we introduced the following underlined silent mutations in BRCA1: 5′AGTATAATC 3′. For CRISPR‐Cas9 genome editing, DNA corresponding to sgRNAs was cloned into pX459 (Addgene #62988). The pDEST‐IRES‐GFP vectors containing HA‐BRCA1 and HA‐BRCA1‐ΔBRCT were constructed and provided by N. Johnson (Nacson et al, 2018). The BRCA1‐I26A mutation was generated by site‐directed mutagenesis (Agilent) using the BRCA1 and BRCA1‐ΔBRCT in the pENTR1A Gateway entry vector (Invitrogen) and shuttled into a pDEST‐IRES‐GFP destination vector (Life Technologies) using Gateway LR Clonase II Enzyme Mix (Invitrogen).
Immunofluorescence microscopy
Cells were grown on glass coverslips, and ionizing radiation was delivered with a Faxitron X‐ray cabinet (#43855D) at 120 kV output voltage, 3 mA continuous current, and 3.07 Gy/min. Unless otherwise stated, 1 h post‐IR cells were fixed with 4% (w/v) paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.25% (v/v) Triton X‐100 + 2.5% BSA PBS for 10 min at room temperature, and blocked with PBS + 0.1% Tween‐20 + 5% BSA PBS for 30 min at room temperature. Alternatively, cells were pre‐extracted 10 min on ice with NuEx buffer (20 mM HEPES, pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP‐40, 1 mM DTT and a cocktail of protease inhibitors [cOmplete, EDTA‐free; Roche]), followed by 10‐min 4% PFA fixation. Cells were then incubated with the primary antibody diluted in PBS + 0.1% Tween‐20 + 5% BSA for 1–2 h at room temperature. Cells were next washed with PBS and then incubated with secondary antibodies diluted in PBS + 5% BSA supplemented with 0.8 μg/ml of DAPI to stain DNA for 1 h at room temperature. Secondary antibodies were purchased from Molecular Probes (Alexa Fluor 488 goat anti‐mouse IgG, Alexa Fluor 488 goat anti‐rabbit IgG, Alexa Fluor 488 donkey anti‐mouse IgG, Alexa Fluor 555 goat anti‐mouse IgG, Alexa Fluor 555 goat anti‐rabbit IgG, Alexa Fluor 647 goat anti‐mouse IgG, Alexa Fluor 647 goat anti‐rabbit IgG). The coverslips were mounted onto glass slides with ProLong Gold mounting agent (Invitrogen). Confocal images were taken using a Zeiss LSM780 laser scanning microscope. For examination of S phase, cells were pre‐incubated with 20 μM of EdU (Life Technologies; A10044) for 30 min after irradiation and 30 min prior to fixation and processed as follows: Cells were incubated with primary antibody diluted in PBS + 0.1% Tween‐20 + 5% BSA for 1 h at room temperature. Cells were next washed with PBS and then incubated with secondary antibodies diluted in PBS + 5% BSA supplemented with 0.8 μg/ml of DAPI to stain DNA for 1 h at room temperature. Cells were then fixed for 5 min in 4% PFA, washed, and stained with EdU staining buffer (100 mM Tris–HCl pH 8.5, 10 μM Alexa Fluor 647‐azide, 1 mM CuSO4, 100 mM ascorbic acid). Unless otherwise stated, analysis was performed on 100 cells per condition, where cells with greater than 10 (GFP, BRCA1) or 5 (RAD51) irradiation‐induced foci that co‐localize with γH2AX were considered positive. Experiments were performed in triplicate. For experiments in U2OS 2‐6‐3 cells, 25–30 images per condition were acquired, and GFP intensity at the mCherry‐FokI focus was measured relative to the background intensity of the nucleus. To differentiate the true signal present in BRCA1 (WT) and RING (BRCA1 1–110) samples from noise, a clustering mixture model was used, assuming the GFP control would follow a Gaussian distribution and the true signal would follow a gamma distribution. First, the Gaussian parameters were estimated by the method of moments on the GFP control data alone; second, for the other samples, these parameters were held static, and the gamma parameters were also estimated using the method of moments and optimized using the expectation maximization algorithm. The data given in Fig EV3A show the GFP control signal and the true signal estimated in the GFP‐BRCA1 and ‐RING samples (i.e., with noise subtracted).
Lentiviral production and cell line generation
HEK293T cells were transfected with pDEST‐IRES‐GFP HA‐BRCA1 variant plasmids along with psPAX2 packaging and VSV‐G/pMD2.G envelope plasmids, as described previously (Nacson et al, 2018). MDA‐MB‐436 cells were infected with the resulting lentivirus, as described previously (Nacson et al, 2018), and infected cells were selected based on their GFP expression, where cells were double‐sorted for populations expressing GFP‐HA‐mCherry, ‐HA‐BRCA1, ‐HA‐BRCA1‐I26A, ‐HA‐BRCA1‐ΔBRCT, and ‐HA‐BRCA‐ΔBRCT‐I26A. Cells were tested for protein expression by immunoblotting.
Clonogenic survival assays
Cells were seeded in 10‐cm dishes (500–1,000 cells per 10‐cm plate, depending on the cell line and genotype). For drug sensitivity assays, cells were seeded into media containing a range of olaparib (SelleckChem S1060) concentrations. Plates were cultured in low‐oxygen conditions (3% O2, 5% CO2 at 37°C). Medium was refreshed after 7 days in all cases. At the end of the experiment, medium was removed, cells were rinsed with PBS, and stained with 0.4% (w/v) crystal violet (Sigma; C0775) in 20% (v/v) methanol for 30 min. The stain was aspirated, and plates were rinsed twice in deionized H2O and air‐dried. Colonies were counted using a GelCount (Oxford Optronix), and data were plotted as surviving fractions relative to untreated cells.
Antibodies
We employed the following antibodies for immunofluorescence: mouse anti‐γH2AX (clone JBW301, Millipore, 1:1,000), mouse anti‐BRCA1 (clones MS110 and MS13 Calbiochem, 1:100), rabbit anti‐BRCA1 (#07‐434, Millipore 1:1,000), rabbit anti‐RAD51 serum (70‐001; lot 1; BioAcademia, 1:150,000), rabbit anti‐RAP80 rabbit (A300‐763A, Bethyl Laboratories Inc., 1:200), anti‐RAP80 rabbit (NBP1‐87156, Novus Biologicals, 1:500), rabbit anti‐ABRAXAS (A302‐180A, Bethyl Laboratories Inc., 1:500), rabbit anti‐cyclin A (sc‐751, Santa Cruz; 1:200), mouse anti‐HA.11 (MMS‐101R, Covance; 1:1,000), and rabbit anti‐pS4/S8 RPA32 (A300‐245A, Bethyl Laboratories Inc., 1:1,000) We employed the following antibodies for immunoblotting: rabbit anti‐BRCA1 (homemade, 1:1,000) (Noordermeer et al, 2018), rabbit anti‐RNF8 (kind gift from J. Chen, 1:2,500), rabbit anti‐RNF168 (homemade, 1:2,500) (Stewart et al, 2009), rabbit anti‐RAP80 (A300‐763A, Bethyl Laboratories Inc., 1:5,000), mouse anti‐tubulin (clone DM1A, Calbiochem, 1:1,000), rabbit anti‐KAP1 (Bethyl, 1:10,000), rabbit anti‐GAPDH (G9545, Sigma Aldrich, 1:5,000), mouse anti‐FLAG M2‐Peroxidase (HRP) (A8592, Sigma Aldrich, 1:1,000), and rabbit anti‐GFP (ab290, Abcam; 1:10,000).
Traffic light reporter assay
RPE1‐hTERT TP53 −/− Cas9 and RPE1‐hTERT TP53 −/− RAP80 −/− Cas9 cells were transduced with pCVL.TrafficLightReporter.Ef1a.Puro (Noordermeer et al, 2018) lentivirus at a low MOI (0.3) and selected with puromycin (15 μg/ml). Cells (7 × 105) were nucleofected with 5 μg of pCVL.SFFV.d14mClover.Ef1a.HA.NLS.Sce(opt).T2A. TagBFP (Noordermeer et al, 2018) plasmid DNA in 100 μL electroporation buffer (25 mM Na2HPO4 pH 7.75, 2.5 mM KCl, 11 mM MgCl2), using program T23 on a Nucleofector 2b (Lonza). After 48–72 h, mClover fluorescence was assessed in BFP‐positive cells using a Fortessa X‐20 (BD Biosciences, San Jose) flow cytometer.
Author contributions
AS conceptualization, investigation, writing—original draft, writing—review and editing, and visualization. NC investigation and resources. SA investigation and visualization. AMH investigation. SMN resources and review and editing. AFT resources and review and editing. DD conceptualization, writing—original draft, writing—review and editing, visualization, supervision, and funding acquisition.
Conflict of interest
DD is a shareholder and advisor for Repare Therapeutics.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Expanded View
Acknowledgements
We thank C. Escribano‐Diaz for her initial observations on RAP80 and BRCA1 that helped spur this project, R. Szilard for critical reading of the manuscript, and members of the Durocher Lab for helpful discussions. We also thank N. Johnson, G. Mer, and R. Chapman for sharing and discussing results prior to publication. We are indebted to R. Greenberg for the U2OS 2‐6‐3 cell line, N. Johnson for sharing lentiviral expression vectors for BRCA1, A. Bang for expert assistance with flow cytometry, and M. Bashkurov for help with image analysis. AS was supported for most of this study by a CIHR doctoral award. Work in the DD lab was supported by grants from CIHR (FDN143343) with additional support from the Krembil Foundation.
EMBO reports (2021) 22: e53679.
See also: A Panagopoulos & M Altmeyer (December 2021)
Data availability
The data in this manuscript did not require deposition in a public repository.
References
- Au WW, Henderson BR (2005) The BRCA1 RING and BRCT domains cooperate in targeting BRCA1 to ionizing radiation‐induced nuclear foci. J Biol Chem 280: 6993–7001 [DOI] [PubMed] [Google Scholar]
- Becker JR, Clifford G, Bonnet C, Groth A, Wilson MD, Chapman JR (2021) BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination. Nature 596: 433–437 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK (2000) The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross‐linking agent cisplatin. J Biol Chem 275: 23899–23903 [DOI] [PubMed] [Google Scholar]
- Bouwman P, van der Heijden I, van der Gulden H, de Bruijn R, Braspenning ME, Moghadasi S, Wessels LFA, Dutch‐Belgian VUS workgroup , Vreeswijk MPG, Jonkers J (2020) Functional categorization of BRCA1 variants of uncertain clinical significance in homologous recombination repair complementation assays. Clin Cancer Res 26: 4559–4568 [DOI] [PubMed] [Google Scholar]
- Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D 3rd, Fukuda M, Ohta T, Klevit R (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin‐ligase complex. Proc Natl Acad Sci USA 100: 5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE (2006) A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1‐directed ubiquitination. Mol Cell 21: 873–880 [DOI] [PubMed] [Google Scholar]
- Celeste A, Fernandez‐Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675–679 [DOI] [PubMed] [Google Scholar]
- Christensen DE, Brzovic PS, Klevit RE (2007) E2‐BRCA1 RING interactions dictate synthesis of mono‐ or specific polyubiquitin chain linkages. Nat Struct Mol Biol 14: 941–948 [DOI] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA (2011) The BRCA1‐RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem 286: 13669–13680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Garcia A, Lopez‐Saavedra A, Huertas P (2014) BRCA1 accelerates CtIP‐mediated DNA‐end resection. Cell Rep 9: 451–459 [DOI] [PubMed] [Google Scholar]
- Dai L, Dai Y, Han J, Huang Y, Wang L, Huang J, Zhou Z (2021) Structural insight into BRCA1‐BARD1 complex recruitment to damaged chromatin. Mol Cell 81: 2765–2777e6 [DOI] [PubMed] [Google Scholar]
- Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza‐Martin M, Fletcher A, Blair‐Reid S, Beesley J, Johal B et al (2016) Human BRCA1‐BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 23: 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Densham RM, Morris JR (2017) The BRCA1 Ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus 8: 116–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, Valerie N, Xu B, Povirk LF, Valerie K (2011) Mutations in the BRCT binding site of BRCA1 result in hyper‐recombination. Aging (Albany NY) 3: 515–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doil C, Mailand N, Bekker‐Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J et al (2009) RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136: 435–446 [DOI] [PubMed] [Google Scholar]
- Drost R, Dhillon KK, van der Gulden H, van der Heijden I Brandsma I, Cruz C, Chondronasiou D, Castroviejo‐Bermejo M, Boon U, Schut E et al (2016) BRCA1185delAG tumors may acquire therapy resistance through expression of RING‐less BRCA1. J Clin Invest 126: 2903–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro M, Rodriguez JA, Baer R, Henderson BR (2002) BARD1 induces BRCA1 intranuclear foci formation by increasing RING‐dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem 277: 21315–21324 [DOI] [PubMed] [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, Shendure J (2018) Accurate classification of BRCA1 variants with saturation genome editing. Nature 562: 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet‐Turcotte A, Canny MD, Escribano‐Díaz C, Orthwein A, Leung CCY, Huang H, Landry M‐C, Kitevski‐LeBlanc J, Noordermeer SM, Sicheri F et al (2013) 53BP1 is a reader of the DNA‐damage‐induced H2A Lys 15 ubiquitin mark. Nature 499: 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal P, Liu Q, Shattuck‐Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett L, Haugen‐Strano A, Swensen J, Miki Y et al (1994) BRCA1 mutations in primary breast and ovarian carcinomas. Science 266: 120–122 [DOI] [PubMed] [Google Scholar]
- Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T (2001) The RING heterodimer BRCA1‐BARD1 is a ubiquitin ligase inactivated by a breast cancer‐derived mutation. J Biol Chem 276: 14537–14540 [DOI] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM (2011) RAP80‐directed tuning of BRCA1 homologous recombination function at ionizing radiation‐induced nuclear foci. Genes Dev 25: 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Paul A, Wang B (2012) Rap80 protein recruitment to DNA double‐strand breaks requires binding to both small ubiquitin‐like modifier (SUMO) and ubiquitin conjugates. J Biol Chem 287: 25510–25519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Botuyan MV, Zhao D, Cui G, Mer E, Mer G (2021) Mechanisms of BRCA1‐BARD1 nucleosome recognition and ubiquitylation. Nature 596: 438–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J (2007) RNF8 transduces the DNA‐damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131: 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V, Chen J, Fox EA, Green JB, Livingston DM (2001) Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc Natl Acad Sci USA 98: 12078–12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R, Mallery DL, Larkin C, Huang JT, Hiom K (2014) BRCA1 is a histone‐H2A‐specific ubiquitin ligase. Cell Rep 8: 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X (2007a) Ubiquitin‐binding protein RAP80 mediates BRCA1‐dependent DNA damage response. Science 316: 1202–1205 [DOI] [PubMed] [Google Scholar]
- Kim H, Huang J, Chen J (2007b) CCDC98 is a BRCA1‐BRCT domain‐binding protein involved in the DNA damage response. Nat Struct Mol Biol 14: 710–715 [DOI] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM et al (2007) Orchestration of the DNA‐damage response by the RNF8 ubiquitin ligase. Science 318: 1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krais JJ, Wang Y, Patel P, Basu J, Bernhardy AJ, Johnson N (2021) RNF168‐mediated localization of BARD1 recruits the BRCA1‐PALB2 complex to DNA damage. Nat Commun 12: 5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrieleis OJP, McIntosh PB, Webb SR, Calder LJ, Lloyd J, Patel NA, Martin SR, Robinson CV, Rosenthal PB, Smerdon SJ (2016) Three‐dimensional architecture of the human BRCA1‐A histone deubiquitinase core complex. Cell Rep 17: 3099–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wu J, Yu X (2007) CCDC98 targets BRCA1 to DNA damage sites. Nat Struct Mol Biol 14: 716–720 [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker‐Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (2007) RNF8 ubiquitylates histones at DNA double‐strand breaks and promotes assembly of repair proteins. Cell 131: 887–900 [DOI] [PubMed] [Google Scholar]
- McGinty RK, Henrici RC, Tan S (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514: 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck‐Eidens D, Futreal P, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett L, Ding W et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266: 66–71 [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M (1999) Brca1 controls homology‐directed DNA repair. Mol Cell 4: 511–518 [DOI] [PubMed] [Google Scholar]
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X et al (2018) BRCA1 Mutation‐specific responses to 53BP1 loss‐induced homologous recombination and PARP inhibitor resistance. Cell Rep 24, 3513–3527.e3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacson J, Di Marcantonio D, Wang Y, Bernhardy AJ, Clausen E, Hua X, Cai KQ, Martinez E, Feng W, Callen E et al (2020) BRCA1 mutational complementation induces synthetic viability. Mol Cell 78, 951–959.e956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Saredi G, Becker JR, Foster BM, Nguyen NV, Beyer TE, Cesa LC, Faull PA, Lukauskas S, Frimurer T et al (2019) H4K20me0 recognition by BRCA1‐BARD1 directs homologous recombination to sister chromatids. Nat Cell Biol 21: 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Winkler JD, Crews CM (2017) Targeted protein degradation by PROTACs. Pharmacol Ther 174: 138–144 [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez‐Quilón A, Moatti N, Zimmermann M et al (2018) The shieldin complex mediates 53BP1‐dependent DNA repair. Nature 560: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886–895 [DOI] [PubMed] [Google Scholar]
- Rabl J, Bunker RD, Schenk AD, Cavadini S, Gill ME, Abdulrahman W, Andrés‐Pons A, Luijsterburg MS, Ibrahim AFM, Branigan E et al (2019) Structural basis of BRCC36 function in DNA repair and immune regulation. Mol Cell 75, 483–497.e489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel BP, Jodelka FM, Nunez R (2006) BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res 66: 5181–5189 [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM (1997a) Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90: 425–435 [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM (1997b) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265–275 [DOI] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska‐Metcalf IU, Balane‐Bolivar C, Janicki SM, Greenberg RA (2010) ATM‐dependent chromatin changes silence transcription in cis to DNA double‐strand breaks. Cell 141: 970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao G, Patterson‐Fortin J, Messick TE, Feng D, Shanbhag N, Wang Y, Greenberg RA (2009) MERIT40 controls BRCA1‐Rap80 complex integrity and recruitment to DNA double‐strand breaks. Genes Dev 23: 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JJ, Cohen RE (2009) Linkage‐specific avidity defines the lysine 63‐linked polyubiquitin‐binding preference of rap80. Mol Cell 33: 775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA (2007) RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316: 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al‐Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M et al (2009) The RIDDLE syndrome protein mediates a ubiquitin‐dependent signaling cascade at sites of DNA damage. Cell 136: 420–434 [DOI] [PubMed] [Google Scholar]
- Tarsounas M, Sung P (2020) The antitumorigenic roles of BRCA1‐BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol 21: 284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker‐Jensen S et al (2015) Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527: 389–393 [DOI] [PubMed] [Google Scholar]
- Walters KJ, Chen X (2009) Measuring ubiquitin chain linkage: Rap80 uses a molecular ruler mechanism for ubiquitin linkage specificity. EMBO J 28: 2307–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ (2007) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B (2012) BRCA1 tumor suppressor network: focusing on its tail. Cell Biosci 2: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, Kim HH, George E, Swisher EM, Simpkins F et al (2016) RING domain‐deficient BRCA1 promotes PARP inhibitor and platinum resistance. J Clin Invest 126: 3145–3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witus SR, Burrell AL, Farrell DP, Kang J, Wang M, Hansen JM, Pravat A, Tuttle LM, Stewart MD, Brzovic PS et al (2021) BRCA1/BARD1 site‐specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nat Struct Mol Biol 28: 268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R (1996) Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet 14: 430–440 [DOI] [PubMed] [Google Scholar]