

Abstract
Background
Non‐small cell lung cancer (NSCLC) is the most common lung cancer, accounting for approximately 80% to 85% of all cases. For people with localised NSCLC (stages I to III), it has been speculated that immunotherapy may be helpful for reducing postoperative recurrence rates, or improving the clinical outcomes of current treatment for unresectable tumours. This is an update of a Cochrane Review first published in 2017 and it includes two new randomised controlled trials (RCTs).
Objectives
To assess the effectiveness and safety of immunotherapy (excluding checkpoint inhibitors) among people with localised NSCLC of stages I to III who received curative intent of radiotherapy or surgery.
Search methods
We searched the following databases (from inception to 19 May 2021): CENTRAL, MEDLINE, Embase, CINAHL, and five trial registers. We also searched conference proceedings and reference lists of included trials.
Selection criteria
We included RCTs conducted in adults (≥ 18 years) diagnosed with NSCLC stage I to III after surgical resection, and those with unresectable locally advanced stage III NSCLC receiving radiotherapy with curative intent. We included participants who underwent primary surgical treatment, postoperative radiotherapy or chemoradiotherapy if the same strategy was provided for both intervention and control groups.
Data collection and analysis
Two review authors independently selected eligible trials, assessed risk of bias, and extracted data. We used survival analysis to pool time‐to‐event data, using hazard ratios (HRs). We used risk ratios (RRs) for dichotomous data, and mean differences (MDs) for continuous data, with 95% confidence intervals (CIs). Due to clinical heterogeneity (immunotherapeutic agents with different underlying mechanisms), we combined data by applying random‐effects models.
Main results
We included 11 RCTs involving 5128 participants (this included 2 new trials with 188 participants since the last search dated 20 January 2017). Participants who underwent surgical resection or received curative radiotherapy were randomised to either an immunotherapy group or a control group. The immunological interventions were active immunotherapy Bacillus Calmette‐Guérin (BCG) adoptive cell transfer (i.e. transfer factor (TF), tumour‐infiltrating lymphocytes (TIL), dendritic cell/cytokine‐induced killer (DC/CIK), antigen‐specific cancer vaccines (melanoma‐associated antigen 3 (MAGE‐A3) and L‐BLP25), and targeted natural killer (NK) cells. Seven trials were at high risk of bias for at least one of the risk of bias domains. Three trials were at low risk of bias across all domains and one small trial was at unclear risk of bias as it provided insufficient information. We included data from nine of the 11 trials in the meta‐analyses involving 4863 participants.
There was no evidence of a difference between the immunotherapy agents and the controls on any of the following outcomes: overall survival (HR 0.94, 95% CI 0.84 to 1.05; P = 0.27; 4 trials, 3848 participants; high‐quality evidence), progression‐free survival (HR 0.94, 95% CI 0.86 to 1.03; P = 0.19; moderate‐quality evidence), adverse events (RR 1.12, 95% CI 0.97 to 1.28; P = 0.11; 4 trials, 4126 evaluated participants; low‐quality evidence), and severe adverse events (RR 1.14, 95% CI 0.92 to 1.40; 6 trials, 4546 evaluated participants; low‐quality evidence).
Survival rates at different time points showed no evidence of a difference between immunotherapy agents and the controls. Survival rate at 1‐year follow‐up (RR 1.02, 95% CI 0.96 to 1.08; I2 = 57%; 7 trials, 4420 participants; low‐quality evidence), 2‐year follow‐up (RR 1.02, 95% CI 0.93 to 1.12; 7 trials, 4420 participants; moderate‐quality evidence), 3‐year follow‐up (RR 0.99, 95% CI 0.90 to 1.09; 7 trials, 4420 participants; I2 = 22%; moderate‐quality evidence) and at 5‐year follow‐up (RR 0.98, 95% CI 0.86 to 1.12; I2 = 0%; 7 trials, 4389 participants; moderate‐quality evidence).
Only one trial reported overall response rates. Two trials provided health‐related quality of life results with contradicting results.
Authors' conclusions
Based on this updated review, the current literature does not provide evidence that suggests a survival benefit from adding immunotherapy (excluding checkpoint inhibitors) to conventional curative surgery or radiotherapy, for people with localised NSCLC (stages I to III). Several ongoing trials with immune checkpoints inhibitors (PD‐1/PD‐L1) might bring new insights into the role of immunotherapy for people with stages I to III NSCLC.
Plain language summary
Effect of immunotherapy on the prognosis for stages I to III non‐small cell lung cancer treated with surgery or radiotherapy with curative intent
Review question
Do treatments that help the body's immune system fight cancer cells (immunotherapy) make people with non‐small cell lung cancer (NSCLC) who have had surgery or radiotherapy aimed at a cure, live longer?
Background
Many people with NSCLC, who have had surgery or radiotherapy to cure their cancer, eventually die because the cancer comes back, either in the chest, or somewhere else in the body. There have been a number of trials over the years that have looked at whether immunotherapy helps people live longer. Some seemed to show a benefit, others did not.
Study characteristics
We searched four computerised databases and five trial registers to 19 May 2021. We looked for all trials that randomly allocated participants to one treatment or another (randomised controlled trials, RCTs), and included adults (aged 18 years or older) with early NSCLC (stages I to III), confirmed by laboratory testing of a sample of the tumour. We found 11 RCTs, which included over 5000 participants who had received surgery or curative radiotherapy, and were randomly allocated to receive either immunotherapy or no further treatment.
Key results
We found that giving immunotherapy, mainly vaccine‐based (aiming to activate the host immune system to induce human immune response to tumour‐specific antigens), after surgery or radiotherapy did not make people live longer. People who were given vaccine‐based immunotherapy did not seem to experience more side effects than the others. We did not find results that could tell us whether the addition of immunotherapy improved quality of life. At the moment, there is no evidence to support or refute giving immunotherapy (mainly vaccine‐based) to people with localised NSCLC (stages I to III). RCTs in progress are testing new, more promising immunotherapy drugs (e.g. checkpoint inhibitors).
Quality of the evidence
The evidence we found about overall survival and progression‐free survival was of high and moderate quality, respectively. When we looked for evidence about how many participants lived to one, two, three, or five years, it was only moderate or low quality, because the RCTs were not very well done, and their results did not agree with each other. The evidence for both any and severe adverse events was of low quality.
Summary of findings
Summary of findings 1. Immunotherapy for surgically‐treated patients with non‐small cell lung cancer (NSCLC), with or without radiotherapy with curative intent.
| Immunotherapy for surgically‐treated NSCLC patients, with or without radiotherapy with curative intent | ||||||
| Patient or population: stages I to III NSCLC patients treated with surgery or radiotherapy with curative intent Setting: hospital Intervention: immunotherapy plus surgery (adjuvant chemotherapy or chemoradiotherapy was allowed, provided it was applied to both experimental and control groups) Comparison: surgical treatment with placebo, best supportive care, no intervention | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk with surgical treatment only (control group) | Corresponding risk with immunotherapy plus surgery (experimental group) | |||||
| Overall survival (Duration of follow‐up varied between trials: median follow‐up time ranged from 37.7 months to 70 months |
The median overall survival time ranged across control groups from 22.3 to 60.2 months | The median overall survival time ranged across experimental groups from 25.6 to 62.0 months |
HR 0.94 (0.84 to 1.05) | 3848 (4 RCTs) |
⊕⊕⊕⊕
High |
|
| Progression‐free survival | The median progression‐free survival time ranged across control groups from 11.4 to 57.9 months | The median progression‐free survival time ranged across experimental groups from 14.2 to 60.5 months | HR 0.94 (0.86 to 1.03) | 3861 (5 RCTs) | ⊕⊕⊕⊝ Moderatea |
|
| Overall survival: 1‐year survival rate |
Study population | RR 1.02 (0.96 to 1.08) | 4420
(7 RCTs) |
⊕⊕⊝⊝ Lowa,b |
The presence of heterogeneity could be partly explained by the inclusion of data from trials with high risk of bias. |
|
| 824 per 1000 |
841 per 1000
(792 to 891) |
|||||
| Overall survival: 2‐year survival rate | Study population | RR 1.02 (0.93 to 1.12) | 4420 (7 RCTs) | ⊕⊕⊕⊝ Moderatea |
||
| 602 per 1000 | 605 per 1000 (551 to 664) | |||||
| Overall survival: 3‐year survival rate | Study population | RR 0.99 (0.90 to 1.09) | 4420 (7 RCTs) | ⊕⊕⊕⊝ Moderatea |
||
| 387 per 1000 | 367 per 1000 (333 to 404) | |||||
| Overall survival: 5‐year survival rate | Study population | RR 0.98
(0.86 to 1.12) |
4389
(7 RCTs) |
⊕⊕⊕⊝
Moderatea |
||
| 122 per 1000 | 81 per 1000 (71 to 92) | |||||
| Adverse events: any (Duration of follow‐up varied between trials: median follow‐up time ranged from 37.7 months to 70 months) |
Study population | RR 1.12 (0.97 to 1.28) | 4126 (4 RCTs) | ⊕⊕⊝⊝
Lowa,b
|
The presence of heterogeneity could be explained by the different agents applied in different trials. By restricting to MAGE‐A3 trials, we observed statistically significant elevation in general adverse event risk (RR 1.23, 95% CI 1.18 to 1.29; I² = 0%). | |
| 805 per 1000 | 913 per 1000 (791 to 1044) | |||||
| Adverse events: severe, grade > 2 (Duration of follow‐up varied between trials: median follow‐up time ranged from 37.7 months to 70 months) |
Study population | RR 1.14 (0.92 to 1.40) | 4546 (6 RCTs) | ⊕⊕⊝⊝ Lowa,b | The presence of heterogeneity could be explained by the involvement of low‐quality trials. | |
| 237 per 1000 | 243 per 1000 (196 to 298) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; MAGE‐A3: melanoma‐associated antigen 3: NSCLC: non‐small cell lung cancer; RCT: randomised controlled trial; RR: risk ratio. | ||||||
|
GRADE Working Group grades of evidence
High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to methodological limitations: inclusion of data from low‐quality trials. bDowngraded one level due to inconsistency: significant heterogeneity was detected during analysis.
Background
Description of the condition
Lung cancer is the most common cancer in men, and the leading cause of cancer‐related death worldwide (GLOBOCAN 2020). In 2020, there were an estimated 2.2 million new lung cancer cases (accounting for 11.4% of all new cancer cases), making up to 18% of total cancer deaths worldwide (GLOBOCAN 2020). Clinically, there are two main types of lung cancer: non‐small cell lung cancer (NSCLC), the most common type, accounting for approximately 80% to 85% of all lung cancer cases, and small cell lung cancer (SCLC; Roy 2008). This classification is important for deciding treatment and predicting prognosis (Roy 2008). Another key issue for clinical management is lung cancer staging ‐ the assessment of the extent of spread of cancer from its original source (Detterbeck 2009). For NSCLC, the best outcomes are achieved with complete surgical resection of a stage IA tumour, with up to a 70% 5‐year survival rate (Mountain 1997). However, the corresponding rates for later stages drop sharply to less than 20% for stage IIIA; the worst survival is seen in stage IV patients, with 2% living longer than five years from diagnosis.
Despite the recent advances in the treatment of NSCLC, there has been little improvement in overall survival (National Cancer Institute 2017). The current standard treatment is surgical resection (lobectomy and adequate mediastinal lymph node evaluation), with or without adjuvant chemotherapy, for those with early‐stage disease (Howington 2013). For people with unresectable, locally advanced tumours (some stage IIIA and all stage IIIB patients), curative radiotherapy combined with chemotherapy is usually offered (Ramnath 2013). Although there is a chance of being cured, overall outcomes for people with stages I to III are not very good. Even after curative treatment, many patients develop a local or distant recurrence (Lardinois 2005). We need more effective, and better tolerated therapeutic options, to prevent relapse and improve the rate of cure. Cancer immunotherapy is one possible approach.
Description of the intervention
'Cancer immunotherapy' generally refers to the use of a number of agents that activate or potentiate immune responses and increase anticancer immunity (Mellman 2011). Immunotherapy can be either active or passive. Active immunotherapy is treatment that stimulates the innate immune system to attack cancer cells (Hirschowitz 2006). Antigen‐specific cancer immunotherapeutics (ASCIs) use exogenous antigens, ideally as tumour‐specific as possible, to induce the immune system to produce an effective T‐cell response against cancer cells (Tyagi 2009). A strong adjuvant component to stimulate the immune response and a proper delivery system for promoting antigen presentation are needed for ASCIs to be effective (Mellman 2011). Passive immunotherapy provides immune effector molecules or effector cells made outside of the human body to modulate immunity. These include monoclonal antibodies and adoptive cell transfer of autologous T‐cells genetically engineered to attack tumour cells.
Currently, immunotherapy has been used successfully in people with malignant melanoma and renal cell carcinoma, because of their high immunogenicity (Drake 2014). Although there are suggestions that lung cancer is a highly immunogenic tumour, initial attempts to administer immunotherapy to people with NSCLC have failed to show clinical benefit (Dasanu 2012). However, significant improvements may come from newer agents, by identifying more relevant, targeted antigens, and developing better adjuvants and delivery systems (Finn 2008; Forde 2014). Trial reports have recorded significant objective response rates using novel agents that block immune checkpoint molecules (Topalian 2012). Also, with promising results from phase II trials, several new approaches have been tested in randomised phase III clinical trials, targeting different stages of NSCLC (Vansteenkiste 2013).
How the intervention might work
The idea of cancer immunotherapy originated with the improved understanding of immune surveillance, a process by which the immune system can recognise malignant cells as foreign, and then induce immune responses to eliminate them (Finn 2008). Physiologically, a normal cellular immune response starts with the uptake of tumour antigens by antigen‐presenting cells, such as dendritic cells or macrophages. Antigen‐presenting cells then process the antigens to T‐cells, by presenting them on their surfaces via major histocompatibility complex classes I and II. With assistance from co‐stimulatory signals, different downstream immune effectors (e.g. plasma cells, natural killer (NK) cells, and cytotoxic T‐cells) may be activated, consequently causing the apoptosis (death) of tumour cells (Figure 1). However, immune surveillance may be limited by other factors. If there is an immunosuppressive microenvironment, even malignant tumour cells that carry unusual antigens can escape an immune‐mediated attack (Drake 2006). One such resistance mechanism involves a series of immune checkpoint molecules presenting on the cell surface, including programmed death‐ligand 1 (PD‐L1) and other ligands to inhibitory T‐cell receptors, which can substantially suppress T‐cell proliferation and its killing capacity (Keir 2008).
1.
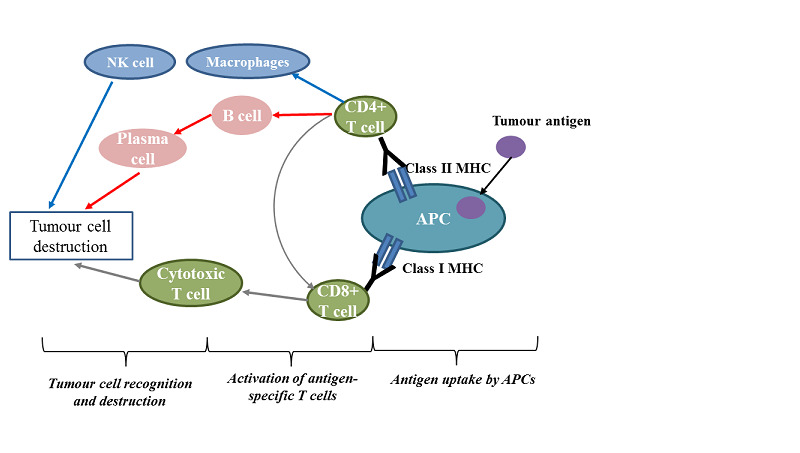
Schematic diagram of the immune components and events involved in cancer immunotherapy
APCs: antigen‐presenting cells; MHC: major histocompatibility complex; NK: natural killer
Therapeutic cancer vaccination and monoclonal antibodies that block immune checkpoints are the most widely used immunotherapies for NSCLC treatment, especially for stages I to III. Our main focus for the review was the effect of therapeutic cancer vaccines, which can be summarised by the type of antigens described below.
Cell‐based vaccines
Autologous cell vaccines, generated from lysate or whole cells from the tumours of individual patients, have the advantage of stimulating an immune response in a large variety of tumour‐specific antigens expressed by the patient's cancer cells. In contrast, allogeneic cell vaccines use mixtures of different cancer cell lines. For instance, the belagenpumatucel‐L vaccine is composed of several lung cancer cell lines (2 adenocarcinomas, 1 squamous cell carcinoma, and 1 large cell carcinoma), and two adjuvants, which form a major histocompatibility complex and antisense molecule, targeting transforming growth factor ß2 (TGF‐ß2; Giaccone 2013). Theoretically, expression of the TGF‐ß2 antisense molecule can undermine the TGF‐ß‐related immunosuppressive effect and potentiate dendritic cell activation, resulting in increased immunogenicity of gene‐modified cancer cells (Nemunaitis 2006). More recently, NK cells have been developed as another type of cell vaccine. Modulated NK cells using soluble α‐galactosylceramide (α‐GalCer) in vivo was found to be tolerated, however there was no promising clinical response due to the low levels of baseline NK cells (Hayes 2021). Furthermore, a clinical trial using ex vivo Hsp70‐derived peptide (TKDNNLLGRFELSG, TKD)/interleukin‐2 (IL‐2)‐activated autologous NK cells show it was well‐tolerated, with positive clinical responses (Multhoff 2020).
Compound‐directed vaccines
Peptide vaccines are based on amino acid sequences. However, since they can only target a few epitopes, their major shortcoming is poor immunogenicity (Kochenderfer 2007). This defect can be circumvented by incorporating an efficient delivery system or immunoadjuvants, such as the L‐BLP25 vaccine (known as stimuvax or tecemotide). The L‐BLP25 vaccine consists of a 25‐amino acid sequence from the glycoprotein mucin‐1 (MUC‐1) protein, along with an immunoadjuvant (monophosphoryl lipid A), and a liposomal delivery system (Sangha 2007). MUC‐1 is a highly glycosylated transmembrane protein found on the epithelial cell surface. In cancer cells, MUC‐1 was reported to be frequently overexpressed, with an abnormally glycosylated status (Bafna 2010). Cancer‐associated MUC‐1 can induce abnormal interactions between receptor tyrosine kinases and other cell surface receptors, which in turn lead to inappropriate activation of intracellular signalling pathways. These events then facilitate the growth, proliferation, and survival of cancer cells (Acres 2005; Bafna 2010). In preclinical trials, the L‐BLP25 vaccine induced a cellular immune response, characterised by T‐cell proliferation in response to MUC‐1.
Protein‐based vaccines can elicit an immune response targeting multiple epitopes, but efficient implementation requires that they are combined with immunoadjuvants. Melanoma‐associated antigen 3 (MAGE‐A3) is considered to be a highly exclusive tumour‐specific antigen, since normally it is only expressed in the testes and placenta, where it remains inaccessible to T‐cells because of the lack of major histocompatibility complex molecules to present the antigens (Simpson 2005). Therefore, the MAGE‐A3 vaccine is expected to be a well‐tolerated therapy with minimal side effects. In several types of cancer cells, MAGE‐A3 expression increases with tumour stage (Van den Eynde 1997). MAGE‐A3 is detected in about 35% to 50% of NSCLC tumours (Tyagi 2009).
Viral‐based vaccines
Finally, another way to produce cancer vaccines is to incorporate the target antigen into a viral backbone. One such vaccine, consisting of a modified Ankara virus, known as TG4010, has been developed to target MUC‐1 (Kochenderfer 2007).
Why it is important to do this review
Although immunotherapy for NSCLC showed disappointing results in earlier trials, more promising evidence of its efficacy has emerged in the last decade. For people with localised NSCLC (stages I to III), immunotherapy has been used to reduce the postoperative recurrence rate or negative clinical outcomes of current chemoradiotherapy for unresectable tumours. While several agents have now entered phase III clinical trials, there is a need for a systematic review to address the question of the effectiveness and safety of immunotherapy in such patients. It is also unclear what the most effective type of immunotherapeutic agents is, and which group of patients could benefit most from this treatment. Therefore, our subgroup analysis might offer supportive evidence to optimise its further development and application in the clinic.
This review was first published in 2017, and updated in May 2021.
Objectives
To assess the effectiveness and safety of immunotherapy (excluding checkpoint inhibitors) among people with localised NSCLC of stages I to III who received curative intent of radiotherapy or surgery.
Methods
Criteria for considering studies for this review
Types of studies
We only included randomised controlled trials (RCTs).
Types of participants
We included all adults (18 years or older) with histologically‐confirmed early‐stage NSCLC (stages I to III) after surgical resection (with or without chemotherapy), and those with unresectable locally advanced stage III NSCLC who had received radiotherapy, or radiotherapy combined with chemotherapy with curative intent.
Types of interventions
Surgical treatment + immunotherapy agents versus surgical treatment with placebo, best supportive care, or no intervention.
Radical radiotherapy (with or without chemotherapy) + immunotherapy agents versus radical radiotherapy (with or without chemotherapy) with placebo, best supportive care, or no intervention.
Types of outcome measures
Primary outcomes
Overall survival: defined as the interval between the date of randomisation and the date of death from any cause.
Progression‐free survival: defined as the time from randomisation to either death or disease progression, whichever occurred first. Disease progression was defined according to response evaluation criteria in solid tumours (RECIST; Therasse 2000), as at least a 20% increase in the sum of the longest diameter of target lesions, taking as reference the smallest sum of the longest diameter recorded since the treatment starts, or the appearance of one or more new lesions.
Secondary outcomes
Overall survival rates: the percentage of participants in a study who were still alive for a certain period of time.
Adverse events or side effects: graded severity with the National Cancer Institute‐Common Terminology Criteria for Adverse Events (NCI‐CTCAE 2017), including the percentage of treatment‐related deaths.
Overall response: response assessed according to RECIST guidelines (Therasse 2000), or immune‐related response criteria (Wolchok 2009).
Health‐related quality of life: measured by a validated scale.
We included all primary outcomes, as well as parts of secondary outcomes in a summary of findings table (Table 1).
Search methods for identification of studies
Electronic searches
We conducted a literature search to identify all published or unpublished RCTs. The literature search identified potential trials in all languages.
We searched the following electronic databases for potential trials.
Cochrane Central Register of Controlled Trials (CENTRAL; 2021, Issue 5) in the Cochrane Library, searched 19 May 2021 (Appendix 1).
MEDLINE PubMed (1966 to 19 May 2021; Appendix 2).
Embase (1988 to 19 May 2021; Appendix 3).
CINAHL (1982 to 19 May 2021).
The search string for MEDLINE was developed according to the Cochrane Highly Sensitive Search Strategy, sensitivity‐maximising version, as referenced in Chapter 6.4.11.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021). We adapted the terms and the search strategies for CENTRAL, MEDLINE, and Embase to search CINAHL.
Searching other resources
We searched Clinical Trial Registers (www.clinicaltrials.gov; www.controlled-trials.com), databases of the Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the World Health Organization (WHO) International Clinical Trials Registry Platform (who.int/clinical-trials-registry), to identify information about ongoing trials. We searched all databases from their inception to 19 May 2021.
We checked reference lists of all included trials and related reviews for additional references. We asked experts in the field and manufacturers of relevant drugs to provide details of outstanding clinical trials and any relevant unpublished material. We also contacted authors of identified trials and asked them to identify other published and unpublished trials.
We manually checked for potential trials in abstracts or reports from the following relevant conference proceedings (from 1990 to present): American Society of Clinical Oncology (ASCO), European Society of Medical Oncology (ESMO), European Cancer Conference Organisation (ECCO), and International Association for the Study of Lung Cancer (IASLC) World Lung Cancer Conference.
We searched for errata or retractions from eligible trials on www.ncbi.nlm.nih.gov/pubmed on 19 May 2021.
Data collection and analysis
Selection of studies
Two review authors (YY and XW for this updated review, or OT and ET for the original review) independently screened titles and abstracts of all trials that we identified as a result of the search, and labelled them as 'retrieve' (eligible, potentially eligible, or unclear) or 'do not retrieve'. For the ones coded as 'retrieve', we then referred to their full‐text study reports or publication. Two review authors (HS, RL) independently screened the full‐text reports. The procedure of study identification for inclusion and exclusion was well documented by using standard screening forms. We resolved any disagreement through discussion, or consulted a third review author (CS).
We identified and excluded duplicates and collated multiple reports of the same study, so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram (Liberati 2009), and 'Characteristics of excluded studies' table.
Data extraction and management
We used a data collection form for study characteristics and outcome data, which we tested on one study in the review. One review author (RL) extracted study characteristics from included trials. We extracted the following study characteristics.
Methods: study design (for example, parallel or cross‐over design), number of study centres and location (country), total duration (for example, date of study, follow‐up period, early stopping of trial), method of randomisation (including imbalanced randomisation ratio), methods of allocation concealment, blinding
Participants: N, age, gender, Eastern Co‐operative Oncology Group (ECOG) performance status, medical history, the severity of condition (stage), diagnostic criteria, inclusion criteria, exclusion criteria
Interventions: intervention, comparison, concomitant medications, excluded medications
Outcomes: primary and secondary outcomes specified and collected, time points reported
Notes: funding for trial, or any notable conflicts of interest of trial authors
Two review authors (YY and XW for this updated review, HS and RL for the original review) independently extracted outcome data from the included trials. We noted in the 'Characteristics of included studies' table if outcome data were reported in an unusable way. We resolved disagreements by consensus, or by involving a third review author (CS). One review author (RL) copied the data from the data collection form into the Review Manager file (Review Manager 2020). We double‐checked that the data were entered correctly by comparing the study reports with the data in the systematic review. A second review author (JZ) spot‐checked study characteristics for accuracy against the trial report.
Assessment of risk of bias in included studies
Two review authors (YY and XW for this updated review, HS and RL for the original review) independently assessed the risk of bias for each study, using RoB 1 (Higgins 2011). Any disagreements were resolved by discussion, or by involving a third assessor (JZ). We assessed the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other potential bias
We graded each potential source of bias as high, low, or unclear, and provided a quote from the study report together with a justification for our judgement in the risk of bias table. We summarised the risk of bias judgements across different trials for each of the domains listed. For overall risk of bias, we considered trials that had adequate random sequence generation, adequate allocation concealment, adequate blinding, adequate handling of incomplete outcome data, no selective outcome reporting, and were without other bias risks, as being at overall low risk of bias. We considered trials that were assessed as being at high or unclear risk of bias in the majority of domains as being at overall high risk of bias; and the remaining trials to be at low risk of bias. We considered blinding separately for different key outcomes where necessary e.g. for unblinded outcome assessment, the risk of bias for all‐cause mortality may be very different than for a participant‐reported pain scale. Where information on the risk of bias related to unpublished data or correspondence with a trialist, we noted this in the risk of bias table.
When considering treatment effects, we took into account the risk of bias for the trials that contributed to that outcome.
Measures of treatment effect
We analysed the primary outcomes based on intention‐to‐treat (ITT) analyses, where available. We measured effect estimates by hazard ratios (HRs) for time‐to‐event variables, and risk ratios (RRs) for dichotomous variables. For continuous variables, we calculated mean differences (MDs) with their corresponding 95% confidence intervals (CIs) if trials used the same measurement, and standardised mean differences (SMDs) and 95% CIs when trials use different scales. We contacted the corresponding authors for missing information about standard deviations or standard errors. For reports without available data for pooling, we tried to measure rates from figures, and calculated effect estimates from P values, t statistics, ANOVA tables, or other statistics as appropriate.
Unit of analysis issues
We did not find any trials with non‐standard design (such as cluster‐randomised trials for potential 'unit of analysis error') for this updated review. But if such eligible trials emerge in future literature searches, we will carefully assess these trials (in terms of recruitment bias, baseline imbalance, loss of clusters, and comparability with individually‐randomised trials). Furthermore, we will apply proper statistical methods (such as multilevel models and generalised estimating equations) for analysis, according to the Handbook for Systematic Review of Interventions (Higgins 2021). The individual participant was the unit of analysis for this updated review.
If there had been data from a cross‐over trial with eligible intervention performances, we would have used the data from the first phase only, i.e. from randomisation to the point of cross‐over.
Had multiple trial arms been reported in a single trial, we would only have included the relevant arms. In future, if we enter two comparisons (e.g. drug A versus placebo and drug B versus placebo) from the same trial into the same meta‐analysis, we will halve the control group to avoid double counting.
Dealing with missing data
We contacted investigators or study sponsors to verify key study characteristics and obtain missing outcome data where applicable (e.g. when a study was identified as an abstract only). For full‐text reports with missing details relevant to our analysis, we also contacted the authors of the trials by email. In the case of non‐response after repeated attempts, we dropped these incomplete data from the analysis, stating this clearly in the Results section, and discussed it further under the Potential biases in the review process section of the Discussion.
Assessment of heterogeneity
We carried out tests for heterogeneity using the Chi² test, to assesses whether observed differences in results were compatible with chance alone. We used the I² statistic to quantify inconsistency across trials. The presence of heterogeneity was defined by P < 0.05 from the Chi² test, and I² > 50% (Higgins 2021). If we detected moderate or higher heterogeneity (50% to 100%), we applied a thorough exploration of possible sources of heterogeneity by means of subgroup and sensitivity analyses (as stated below). Given the limitations of the methods, the P value from the Chi² test and the value of I² were only referred to as a guide, and we exercised caution when interpreting the results.
Assessment of reporting biases
We attempted to contact study authors, asking them to provide missing outcome data. When this was not possible, and the missing data were thought to introduce serious bias, we explored the impact of including such trials in the overall assessment of results by conducting a sensitivity analysis.
We did not include sufficient trials for each outcome in the current updated review to create a funnel plot. But if we are able to pool more than 10 trials in future updates, we will do so to explore possible publication biases (intervention effect estimate versus standard error of intervention effect estimate). If we find funnel plot asymmetry, we will further investigate clinical diversity of trials as a possible explanation. If there are sufficient trials (> 10), we also will use the 'contour‐enhanced' funnel plot to differentiate asymmetry due to other factors (Peters 2008). If the supposed missing trials are in areas of higher statistical significance, the cause of the asymmetry is highly suggestive of being due to factors other than publication bias.
Data synthesis
We used Review Manager 5 for pooling data and for statistical analysis (Review Manager 2020). We used random‐effects models for primary analyses since the agents of interest had different mechanisms of action. In the future (since currently no subgroup analysis was successfully conducted), provided that the trials in some subgroups are found to be homogeneous (in terms of age, diagnostic subtype, intervention type, intervention duration), we will use both fixed‐effect and random‐effects models, and compare the results. In the absence of heterogeneity and significant reporting bias, these two models should yield the same results. In this case, we will report the results from the fixed‐effect model only. If the results are different, indicating significant heterogeneity, we will report the results from the random‐effects model only.
Subgroup analysis and investigation of heterogeneity
We had planned to perform the following exploratory subgroup analyses on the primary outcomes, using Review Manager 5 (Review Manager 2020).
Participants receiving immunotherapy who present with a different stage of NSCLC (stages I, II, or III).
Participants receiving different types of immunotherapy.
Participants with a specific biomarker: e.g. participants with a gene‐signature profile (MAGE‐A3‐positive).
In the current updated review, we did not include sufficient trials for each population (subgroup) of interest to conduct subgroup analyses. In future updates, we will perform a subgroup analysis for primary outcomes when we have at least three trials for a subgroup.
Considering the differences between subgroups, we will first examine them by visual inspection of their CIs; non‐overlapping CIs indicate a statistically significant difference in treatment effect between subgroups. Also, we will use the approach of Borenstein 2008 to formally investigate differences between two or more subgroups.
Sensitivity analysis
We performed sensitivity analyses, defined a priori, to assess the robustness of our conclusions. This was achieved by repeating the analyses to explore the influence of the following factors on effect size.
Exclusion of unpublished trials (in the current updated review, we did not include unpublished data, but will conduct this analysis if unpublished data are included in future updates).
Exclusion of lower quality trials (those at high or unclear risk of bias).
Summary of findings and assessment of the quality of the evidence
We created a summary of findings table presenting all our primary and secondary outcomes, except overall response and health‐related quality of life, using GRADEpro software (GRADEpro GDT). We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of the body of evidence as it related to the trials that contributed data to the meta‐analyses for the prespecified outcomes. We justified decisions to downgrade or upgrade the quality of the evidence using footnotes, and made comments to aid the reader's understanding of the review where necessary (Higgins 2021).
Summary of findings and assessment of the certainty of the evidence
We created a 'Summary of findings' table presenting all our primary and secondary outcomes except overall response and health‐related quality of life, using the GRADEpro software, version 3.2 (GRADEpro GDT). We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of the body of evidence as it related to the studies that contributed data to the meta‐analyses for the pre‐specified outcomes. We justified decisions to downgrade or upgrade the quality of the evidence using footnotes, and made comments to aid the reader's understanding of the review where necessary.
Results
Description of studies
Results of the search
Figure 2 shows details of the updated search results from 20/01/2017 to 19/05/2021. We identified 6218 citations in this updated version of the review (723 from CENTRAL, 3543 from MEDLINE, and 1952 from Embase). In addition, we found 3 ongoing trials in ClinicalTrials.gov (Merck 2015; Saka 2017; Wu 2011). We did not find any relevant abstract or trial from conference proceedings by handsearching.
2.
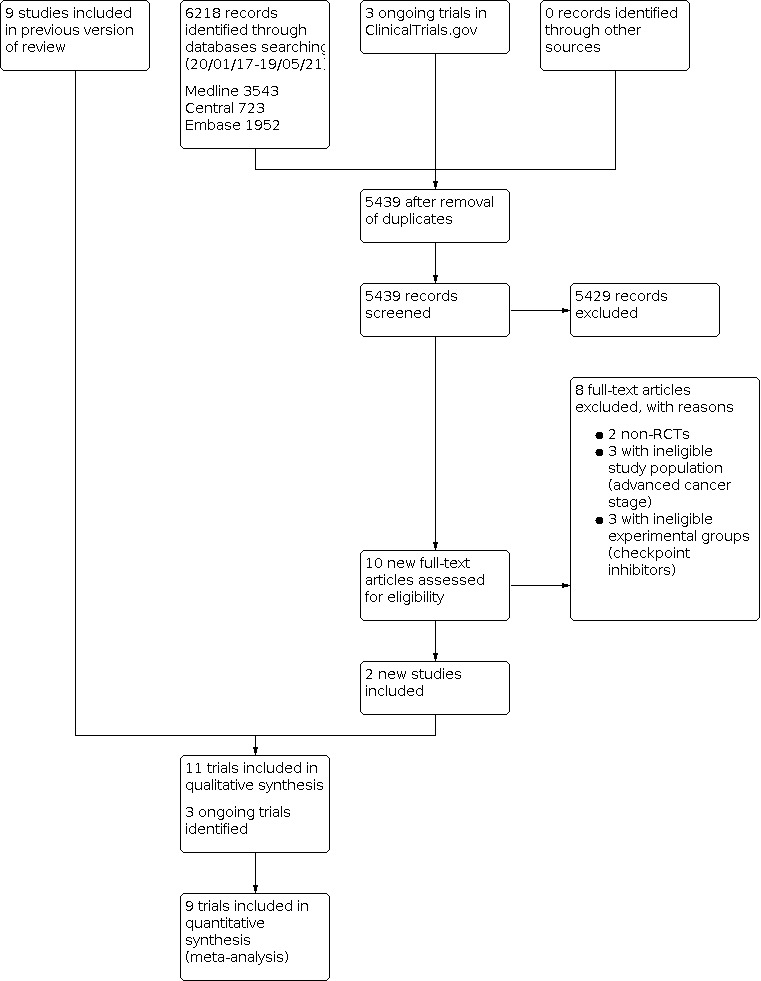
We retained 5439 references after excluding duplicates. By screening the titles and abstracts, we excluded 5429 citations, including the protocol published by Wu 2011 describing a potentially eligible trial which was terminated prematurely (16 September 2016 at ClinicalTrials.gov). We therefore had 10 new records considered to be highly relevant to our review. Among the 10 full‐text accessed, we excluded 8 trials with reasons (see Excluded studies). Two trials met our inclusion criteria (Katakami 2017; Multhoff 2020). Combined with the nine full‐text reports from the original published version of the review (Butts 2014; Fujisawa 1996; Giovanni 1996; Macchiarini 1991; Matthay 1986; Stanley 1986; Vansteenkiste 2013; Vansteenkiste 2016; Zhao 2014), our update includes 11 trials.
Included studies
Overall, we included 11 trials after carefully evaluating potentially eligible articles, corresponding to 12 individual RCTs, with a total of 5128 participants (Butts 2014; Giovanni 1996; Vansteenkiste 2013; Vansteenkiste 2016; Stanley 1986; Katakami 2017; Macchiarini 1991; Matthay 1986; Multhoff 2020; Fujisawa 1996; Zhao 2014). Two trials after 20 January 2017, were newly included in this updated review. We summarised the characteristics of the included trials in the 'Characteristics of included studies' tables.
Unfortunately, we did not manage to extract any relevant results from Matthay 1986, a small trial containing 48 Bacillus Calmette‐Guérin (BCG)‐treated and 40 control subjects. This trial reported survival time by tumour stage (stage I, stage II, and stage III), without providing any detailed data on overall survival, or adverse events, which were described in a very general way, where only fever and transient malaise were mentioned, without grading the severity. Also, because of the poor study quality of Zhao 2014, especially the conflicting data reported in the paper (survival rates in abstract, full text, and figures were different from each other), we decided not to include the results of this study in our analysis.
Study design
Four trials were double‐blind RCTs (Butts 2014; Katakami 2017; Vansteenkiste 2013; Vansteenkiste 2016). With the exception of Stanley 1986, where the details of blinding were unclear, the other six had an open‐label design (Macchiarini 1991; Matthay 1986; Multhoff 2020; Fujisawa 1996; Giovanni 1996; Zhao 2014). We included four multicentre international trials (Butts 2014; Stanley 1986; Vansteenkiste 2013; Vansteenkiste 2016), enrolling participants from the USA, Europe, and Asia. Other trials recruited participants from China (Zhao 2014), Germany (Multhoff 2020), Italy (Giovanni 1996; Macchiarini 1991), Japan (Fujisawa 1996; Katakami 2017), or the USA (Matthay 1986).
Participants
All included trials enrolled participants with histologically‐confirmed NSCLC. Two trials enrolled participants with stages I to III NSCLC (Matthay 1986; Vansteenkiste 2016). Three RCTs focused on stage I or II completely resected NSCLC (Fujisawa 1996; Stanley 1986; Vansteenkiste 2013); two trials were conducted on participants with stage II or III NSCLC (Giovanni 1996; Macchiarini 1991); and the remaining four only included participants with locally advanced NSCLC (stage III; Butts 2014; Katakami 2017; Multhoff 2020; Zhao 2014). Vansteenkiste 2013 and Vansteenkiste 2016 were separate phase II and phase III clinical trials on MAGE‐A3 immunotherapy, and they only included participants with MAGE‐A3‐positive NSCLC. Likewise, Multhoff 2020 was a phase II trial, which included participants with unresectable, membrane‐bound forms of Hsp70 (mHsp70)‐positive NSCLC. As stated above, 11 eligible trials enrolled a total of 5128 participants; after we excluded the two trials without usable information on outcomes of interest (Matthay 1986; Zhao 2014), the total number of participants that contributed to the analyses was 4863. The mean age of analysed participants was 61 years, with a range of 19 to 89 years; 75.1% of them were men (Giovanni 1996 did not report the number of men and women).
Interventions
Three trials included participants with unresectable NSCLC, in two of which, participants were receiving chemoradiotherapy and randomly assigned to receive immunotherapy (L‐BLP25) or placebo (Butts 2014; Katakami 2017). In another trial, participants were randomly assigned to natural killer (NK) cell‐based immunotherapy along with chemoradiotherapy, or chemoradiotherapy alone (Multhoff 2020). All other included RCTs included participants with surgically‐treated NSCLC. Four RCTs compared the immunotherapy group to a control group of either placebo, best supportive care, or no intervention (Fujisawa 1996; Matthay 1986; Stanley 1986; Vansteenkiste 2013); the other four trials allowed adjuvant chemotherapy (Vansteenkiste 2016; Zhao 2014), or chemoradiotherapy (Giovanni 1996; Macchiarini 1991), for both experimental and control groups. It is noteworthy that the immunotherapy agents used in these trials differed over the years. Earlier trials mainly studied active immunotherapy, such as BCG injected into the intrapleural space (Macchiarini 1991; Stanley 1986), or into the tumour (Matthay 1986). The focus of research then gradually moved to passive immunotherapy, with adoptive cell transfer (transfer factor (TF); Fujisawa 1996), tumour‐infiltrating lymphocytes (TIL; Giovanni 1996), dendritic cell/cytokine‐induced killer (DC/CIK; Zhao 2014), and natural killer (NK) cells (Multhoff 2020). Most recently, antigen‐specific cancer vaccines (MAGE‐A3; Vansteenkiste 2013; Vansteenkiste 2016) and L‐BLP25 (Butts 2014; Katakami 2017), were widely introduced and evaluated.
Outcome measures
Apart from Multhoff 2020, the remaining trials reported overall survival time, although measured in different ways. Matthay 1986 reported survival time by tumour stage (stage I, stage II, and stage III); but they only provided survival probability curves for stage I and stage III. Therefore, we could not extract any survival outcome data from this study. Only the four recent trials measured the difference in survival between experimental and control groups by time‐to‐event analysis and hazard ratio (HR; (Butts 2014; Katakami 2017; Vansteenkiste 2013; Vansteenkiste 2016). Fujisawa 1996 reported 5‐year and 10‐year survival rates and all other RCTs reported survival by a median or mean survival with ranges or standard deviation (SD). To enable the use of these data for meta‐analysis, we extracted data for 1‐year, 2‐year, 3‐year, and 5‐year survival rates from all included trials (where possible), from either text statements or survival curves, as secondary outcomes. Multhoff 2020 reported progression‐free survival as the primary outcome. We extracted progression‐free survival data, as HRs, from five RCTs (Butts 2014; Katakami 2017; Multhoff 2020; Vansteenkiste 2013; Vansteenkiste 2016).
The overall response was reported in only one RCT (Multhoff 2020). Adverse events were mentioned in eight trials, but we only included data from six of them in the analysis of adverse events (Butts 2014; Katakami 2017; Multhoff 2020; Stanley 1986; Vansteenkiste 2013; Vansteenkiste 2016), since Matthay 1986 and Giovanni 1996 reported adverse events in general, without grading the severity. Quality of life was reported in two RCTs (Multhoff 2020; Vansteenkiste 2016), assessed by the European Organisation for Reasearch and Treatment of Cancer (EORTC) Quality of Life Questionnaire‐Core 30 (QLQ‐C30) (Aaronson 1993) and EuroQoL‐5D (EQ‐5D)(EuroQol 1990) questionnaire, respectively.
Because of the inconsistencies in the duration of follow‐up in the different trials, these point estimates should be interpreted with caution.
Excluded studies
Please see Characteristics of excluded studies.
Risk of bias in included studies
In Figure 3 and Figure 4, we summarised the risk of bias in the included trials. Overall, we considered three trials to be well‐designed and well‐conducted, and therefore we assessed these trials at low risk of bias (Butts 2014; Vansteenkiste 2013; Vansteenkiste 2016). However, for all of them, the involvement of the sponsors during study design, analysis, and results interpretation was mentioned in their reports. One trial had unclear risks of bias for two domains of assessment, owing to limited study information for risk assessment reported in the published paper, and we considered this trial to have a unclear risk of bias (Katakami 2017). The other seven trials were at high risk of bias, mainly because of non‐blinding design (Fujisawa 1996; Giovanni 1996; Macchiarini 1991; Matthay 1986; Multhoff 2020; Stanley 1986; Zhao 2014).
3.

Risk of bias graph: review authors' judgements about each risk of bias domain, presented as percentages across all included trials.
4.

Risk of bias summary: review authors' judgements about each risk of bias domain, for each included trial.
Allocation
Four trials described how they carried out the randomisation procedure in detail (Butts 2014; Matthay 1986; Vansteenkiste 2013; Vansteenkiste 2016). The other trials did not provide details on how they randomised the participants into different treatment arms, so we considered them to be at unclear risk of selection bias (Fujisawa 1996; Giovanni 1996; Macchiarini 1991; Multhoff 2020; Stanley 1986; Zhao 2014; Katakami 2017). For Butts 2014, Katakami 2017, Vansteenkiste 2013, and Vansteenkiste 2016, randomisation was done centrally, via the Internet, minimising the risk of lack of allocation concealment. Matthay 1986 randomised with a printed table of random numbers, which was concealed from investigators with a sealed envelope.
Blinding
We judged the three trials with a double‐blind, placebo‐controlled design to be at low risk of bias for blinding of participants and researchers, as well as blinding of outcome assessors (Butts 2014; Vansteenkiste 2013; Vansteenkiste 2016). Stanley 1986 had a placebo comparator, but did not provide a detailed explanation of blinding of participants, so we evaluated it to be at unclear risk. We considered six trials to be at high risk of performance bias because they had no placebo comparator (open‐label design; (Fujisawa 1996; Giovanni 1996; Macchiarini 1991; Matthay 1986; Multhoff 2020; Zhao 2014)).
To assess the influence of detection bias, we considered the concept of each outcome; we classified overall survival, yearly survival rates, and severe adverse events as objective effects of treatment, and so they are unlikely to be affected by the non‐blinding of outcome assessment. But progression‐free survival, response, quality of life, and less severe adverse events are subjective outcomes, and could be affected by the outcome assessors’ knowledge of treatment and the participant's awareness of the assignment status. We assessed all trials to be at low risk of detection bias for objective outcomes. For subjective outcomes, the risk of detection bias was low, when data were extracted with masking for assessors (progression‐free survival and any adverse event in Butts 2014, Vansteenkiste 2013, and Vansteenkiste 2016, and quality life in Vansteenkiste 2016). And the risk of detection bias for subjective outcomes was high in the open‐label trial (progression‐free survival, overall response, and quality of life in Multhoff 2020), and unclear in the trial with blinding of the outcome assessor not specified (progression‐free survival, quality of life, and any adverse events in Katakami 2017).
Incomplete outcome data
We considered all the included trials to be at low risk of bias, either because the number of participants missing from follow‐up was very low (dropout rates below 5%), or because the primary survival analysis was done on the intention‐to‐treat population of all participants randomly allocated to treatment.
Selective reporting
The protocols of five trials were available, where all their prespecified (primary and secondary) outcomes were described in detail (Butts 2014; Katakami 2017; Multhoff 2020; Vansteenkiste 2013; Vansteenkiste 2016). Since the pre‐published protocols were consistent with their reports, we judged these five trials to be at low risk of selective outcome reporting. We considered all other trials to be at high risk for selective reporting bias, because their study protocol was unavailable and they just reported part of the primary outcomes (Fujisawa 1996; Giovanni 1996; Macchiarini 1991; Matthay 1986; Stanley 1986; Zhao 2014). Only Butts 2014, Katakami 2017, Vansteenkiste 2013, and Vansteenkiste 2016 reported overall survival, together with progression‐free survival. Others reported overall survival, but in different formats (Fujisawa 1996; Giovanni 1996; Macchiarini 1991; Matthay 1986; Stanley 1986; Zhao 2014). Multhoff 2020 reported progression‐free survival as the primary outcome and did not report overall survival.
Other potential sources of bias
Zhao 2014 reported conflicting data in their paper (survival rates in abstract, full text, and figures were different from each other), and so we considered this study to be at high risk of other potential bias. For the remaining trials, since there was no obvious potential source of bias, we classified them to be at low risk of other potential biases.
However, four trials were funded by pharmaceutical companies (Butts 2014; Katakami 2017; Vansteenkiste 2013; Vansteenkiste 2016), and the sponsors played critical roles in study design, data collection, management and statistical analysis. Therefore, we carefully compared the final reports with their previously published protocols. The consistency between these two documents mitigated this concern, by indicating that in spite of the sponsors’ involvement, these trials were conducted as planned, and with full supervision by independent administrators.
The other seven RCTs had no connection with these companies and confirmed their independence in the study implementation.
Effects of interventions
See: Table 1
Primary outcomes
Effect of immunotherapy on overall survival for participants with stages I to III NSCLC
To enable the maximum use of eligible data for the meta‐analysis of survival outcomes, we used two approaches to pool data, the first examined overall survival, and the second examined overall survival rates during different time slots. We have provided details of the second analyses under Secondary outcomes.
We extracted hazard ratios (HRs) from four trials (Butts 2014; Katakami 2017; Vansteenkiste 2013; Vansteenkiste 2016). In total, 4179 participants were involved in these trials, and 3848 of them were evaluated for overall survival (92% of all randomised participants). Using a random‐effects model, the pooled results illustrated that the study groups did not have a reduced risk of death compared to the control groups (HR 0.94, 95% confidence interval (CI) 0.84 to 1.05; high‐quality evidence; Analysis 1.1; Figure 5); we did not detect heterogeneity across the trials (I² = 0%; P = 0.56). Since only three out of the four included trials were considered to be at low risk of bias, we performed sensitivity analysis after excluding Katakami 2017. Results were consistent with the main analysis (HR 0.94, 95% CI 0.84 to 1.05) and we did not observe heterogeneity (I² = 2%; P = 0.36).
1.1. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 1: Overall survival
5.

Forest plot of comparison: Effect of immunotherapy for surgically‐treated NSCLC patients: main analyses, outcome: Overall survival.
Effect of immunotherapy on progression‐free survival for participants with stages I to III NSCLC
Five trials reported progression‐free survival (Butts 2014; Katakami 2017; Multhoff 2020; Vansteenkiste 2013; Vansteenkiste 2016). Although different agents were used in these trials, we found no heterogeneity between the results of these five trials (I² = 0%; P = 0.49); using a random‐effects model, we found that immunotherapy plus surgery showed no advantage compared to surgery alone (HR 0.94, 95% CI 0.86 to 1.03; P = 0.19; moderate‐quality evidence; Table 1; Analysis 1.2; Figure 6). We performed a further sensitivity analysis after we excluded two moderate‐/low‐quality trials (Katakami 2017; Multhoff 2020). Similar to the main analysis, we did not observe heterogeneity (I² = 39%; P = 0.19), and we did not find a difference for progression‐free survival between the two arms (HR 0.93, 95% CI 0.81 to 1.07; moderate‐quality evidence).
1.2. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 2: Progression‐free survival
6.

Forest plot of comparison: Effect of immunotherapy for surgically‐treated NSCLC patients: main analyses, outcome: Progression‐free survival.
Secondary outcomes
Effect of immunotherapy on overall survival rates for participants with stages I to III NSCLC
We added this outcome after assessing the data to capture as much information as we could on participant survival. We extracted 1‐year, 2‐year, and 3‐year survival rates from seven trials with 4420 participants (Butts 2014; Giovanni 1996; Katakami 2017; Macchiarini 1991; Stanley 1986; Vansteenkiste 2013; Vansteenkiste 2016). The results from a random‐effects model showed no clear difference in 1‐year survival probabilities for participants treated with immunotherapy compared to participants assigned to control groups (risk ratio (RR) 1.02, 95% CI 0.96 to 1.08; Analysis 1.3). The RRs were similar for 2‐year (1.02, 95% CI 0.93 to 1.12; Analysis 1.4) and 3‐year survival probability (0.99, 95% CI 0.90 to 1.09; Analysis 1.5). There was moderate heterogeneity for 1‐year (I² = 57%; P = 0.03) and 2‐year (I² = 51%; P = 0.06) survival rates; while 3‐year survival rates showed low between‐trial heterogeneity (I² = 22%; P = 0.26). Five‐year survival rates were available from seven trials that included 2834 participants from the experimental and 1555 participants from the control groups(Butts 2014; Fujisawa 1996; Katakami 2017; Macchiarini 1991; Stanley 1986; Vansteenkiste 2013; Vansteenkiste 2016). Analysis showed there was no clear benefit for 5‐year survival by adding immunotherapy (RR 0.98, 95% CI 0.86 to 1.12; Analysis 1.6), and there was no heterogeneity (I² = 0%; P = 0.75). We concluded that the quality of evidence for the 1‐year survival rate was low, primarily due to the inconsistency of effect across the trials and the involvement of trials with unclear or high risks of bias; the quality of evidence for 2‐year, 3‐year and 5‐year survival rates was moderate, because of the involvement of trials with high or unclear risk of selection bias.
1.3. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 3: Overall survival rate, 1‐year
1.4. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 4: Overall survival rate, 2‐year
1.5. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 5: Overall survival rate, 3‐year
1.6. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 6: Overall survival rate, 5‐year
Because of significant heterogeneity for the 1‐year survival rate, we repeated the analyses, restricting it to trials at low risk of bias. This sensitivity analysis suggested that the heterogeneity for 1‐year survival rates could be partly explained by the differences in study quality. And the inconsistency of effect size also was mitigated after the exclusion of low‐quality trials.
Effect of immunotherapy on adverse events for participants with stages I to III NSCLC
Four trials, with a total of 4126 evaluated participants, provided data on the proportion of participants with any adverse event, graded for severity (Butts 2014; Katakami 2017; Vansteenkiste 2013; Vansteenkiste 2016). Using a random‐effects model, our analysis showed that the addition of immunotherapy to surgery or curative radiotherapy lead to a similar risk of experiencing any adverse events among these participants (RR 1.12, 95% CI 0.97 to 1.28; P = 0.11; Analysis 1.7). The effect size varied substantially between trials (suggesting a drug‐specific effect on this outcome), which corresponded to a high level of heterogeneity between individual outcomes (I² = 95%; P < 0.00001). We also observed an increase in the general adverse event risk (RR 1.23, 95% CI 1.18 to 1.29; I² = 0%; P < 0.00001) by restricting the analysis to MAGE‐A3 trials (Vansteenkiste 2013; Vansteenkiste 2016).
1.7. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 7: Adverse events (any)
Six trials (Butts 2014; Katakami 2017; Multhoff 2020; Stanley 1986; Vansteenkiste 2013; Vansteenkiste 2016), with 2979 evaluated participants in the experimental and 1567 evaluated participants in the control groups, showed that participants receiving immunotherapy did not have different risk of severe adverse events (severity grade > 2) than their controls (RR 1.14, 95% CI 0.92 to 1.40; Analysis 1.8). We also found moderate heterogeneity among the results of these trials (I² = 56%; P = 0.04). Moreover, heterogeneity disappeared in the following sensitivity analysis where low‐quality trials were removed (RR 0.96, 95% CI 0.86 to 1.08; I² = 0%; P = 0.72), indicating that the inconsistency might have originated from the inclusion of two high risk of bias trials (Multhoff 2020; Stanley 1986), and one unclear risk of bias trial (Katakami 2017). In summary, we considered there was low‐quality evidence for both any adverse event, and severe adverse events (due to the involvement of low‐quality trials and significant heterogeneity; Table 1).
1.8. Analysis.

Comparison 1: Effect of immunotherapy for surgically treated NSCLC patients: main analyses, Outcome 8: Adverse events (severe, grade > 2)
Effect of immunotherapy on overall response rate for participants with stages I to III NSCLC
The overall response was assessed only in Multhoff 2020 according to RECIST guidelines (Therasse 2000). One participant in the experimental group showed complete response, and one participant showed partial response. One participant in the control arm had a partial response. The overall response rate was 33% (2/6) in the experimental group and 14% (1/7) in the control group.
Effect of immunotherapy on health‐related quality of life for participants with stages I to III NSCLC
Vansteenkiste 2016 assessed the health‐related quality of life of their participants (1515 experimental and 757 control), using both EQ‐5D (EuroQol 1990) utility scores and visual analogue scores (VAS) (Aitken 1969). They did not find any evidence that the application of melanoma‐associated antigen 3 (MAGE‐A3) improved quality of life. Instead, the immunotherapy group reported significantly lower scores (indicating poorer life quality) on quality of life assessments on the day after the first and the third MAGE‐A3 administrations, compared to the control group.
Multhoff 2020 assessed the quality of life of participants using the QLQ‐C30 (Aaronson 1993). Differences in means of sum scores of the QLQ‐C30 varied at multiple visits, and the experimental group, in general, had a better quality of life during the follow‐up period. However, there were no differences between both studied groups across all visits.
Subgroup analysis
As described in our protocol, we had planned to perform subgroup analysis for primary outcomes according to participants at different tumour stages (I, II, or III), type of immunotherapy, and specific prognostic biomarkers that were used for participant selection. However, since we only found four trials with data for primary outcome analyses, we were not able to conduct any subgroup analyses at this stage.
Discussion
Summary of main results
This updated review, which pooled survival data from trials that focused mainly on vaccine‐based immunotherapy, showed that there was no clear evidence of additional survival benefit from immunotherapy (excluding checkpoint inhibitors) for people with stages I to III NSCLC, who had undergone surgery or received radiotherapy with curative intent. For overall survival, pooled data from four (3848 participants analysed) of the 11 included trials suggested no difference in death (hazard ratio (HR) 0.94, 95% confidence interval (CI) 0.84 to 1.05) for those receiving immunotherapy, compared to their controls. Moreover, the pooled data from five (3861 participants analysed) of the 11 included trials also indicated a similar result in risk of progression (progression‐free survival HR 0.94, 95% CI 0.86 to 1.03) for those receiving immunotherapy. Similarly, survival rate analyses of data from seven trials (4420 participants) showed no improvement of 1‐year, 2‐year, 3‐year, or 5‐year survival associated with the addition of immunotherapy agents. Sensitivity analysis showed that the exclusion of low‐quality data could not explain the observed variation in study results. Due to the small number of trials, we could not perform subgroup analyses to detect the possible differences caused by tumour stage, type of immunotherapy, or the use of a specific prognostic biomarker.
Furthermore, we observed similar risk of experiencing any adverse event (12% for MAGE‐A3 and L‐BLP25 together (P = 0.11)) for participants who were assigned to immunotherapy, compared to the control groups. The risk of having severe adverse events also showed no clear increase (RR 1.14, 95% CI 0.92 to 1.40).
Two of the 11 included trials reported controversial findings about quality of life after immunotherapy. The immunotherapy group in the MAGE‐A3 study reported significantly lower scores (indicating poorer life quality) on quality of life assessments on the day after the first and the third MAGE‐A3 administrations, however the experimental group of the NK cell study showed a better quality of life during the follow‐up period, compared to their controls. The immunotherapy group in one study reported significantly higher overall response rate, indicating better therapy response, compared with the control group.
Overall completeness and applicability of evidence
In order to reduce publication bias, our literature search sought unpublished or ongoing trials, without any limitation on publication language. However, the identified trials only partially addressed the objectives of the review, mainly because data about the effect of immunotherapy for participants with stages I to III NSCLC were insufficiently described. Survival outcomes were reported in various ways. Only five recent trials (45% of included trials) provided adequate data for overall survival and progression‐free survival (Butts 2014; Katakami 2017; Multhoff 2020; Vansteenkiste 2013; Vansteenkiste 2016). Due to the absence of a uniform approach for group comparison, data extraction and analyses from earlier trials was difficult. In addition, because of clinical (different agents used in each trial, and participants with different tumour stages) and statistical heterogeneity, the legitimacy of pooling results may be debatable. The limited number of included trials prevented us from detailed comparisons within more homogeneous subgroups. Also, some important outcomes, such as response rates and health‐related quality of life, could not be examined in this updated review, since response rates were only measured in one trial and quality of life from two trials was measured on different scales.
At the time of our literature search, new immunotherapy agents, i.e. checkpoint inhibitors (PD‐1/PD‐L1), are mainly applied to people with either advanced stage or metastatic NSCLC. Several trials that aimed to assess the efficacy and safety of checkpoint inhibitor drugs are still ongoing (NCT02273375; NCT02486718; NCT02504372). Therefore, further reviews that focus on the effectiveness of checkpoint inhibitors for people with stage I to III NSCLC will be needed when results from several trials are available.
Quality of the evidence
We identified 11 eligible RCTs. We could not extract useful data (for our outcomes of interest) from Matthay 1986, and we discarded results from Zhao 2014 because of poor study quality and contradictory reports of the primary outcomes, leaving only nine trials for further analysis. Data from trials performed before 2000 generally were of poor quality, because of the open‐label design and lack of information provided about randomisation methods (particularly allocation concealment). We considered the four newer double‐blinded RCTs to be of high or moderate quality (Butts 2014; Katakami 2017; Vansteenkiste 2013; Vansteenkiste 2016).
It should be noted that the available data for the meta‐analyses of primary outcomes was incomplete. All participants included in the final analysis were from either four or five trials, and evaluated three types of immunotherapies, although we did not downgrade the quality of the evidence because of this. Clinical and statistical heterogeneity leads to a problem of imprecision for most of our outcomes of interest. We found no suggestion of publication bias, and therefore, no serious limitation was assumed. In addition, we could not explain the inconsistency between individual trial results by our predefined subgroups.
Potential biases in the review process
For the primary analyses, we combined all intervention groups' data, regardless of the type of immunotherapeutic agents administered, and the specific biomarker used for participant selection. Consequently, our analyses were subject to high potential risk of between‐study heterogeneity, due to clinical diversity. Further, we observed significant statistical heterogeneity in the analysis of the 1‐year survival rate. But, because only a limited number of eligible trials was included in these analyses, we were not able to fully explore the reasons for the variation by subgroup or sensitivity analysis. The unexplained inconsistency between individual trial results may undermine the reliability of our effectiveness assessment.
Another issue relevant to the potential bias of our pooled results was the small proportion of trials included in the meta‐analyses of overall survival (36%) and progression‐free survival (45%), our primary outcomes (i.e. a large proportion of included trials were at high risk of selective reporting bias). Although these five trials contained more than 80% of all participants (4195/5128) from all eligible trials, such analyses were deemed to be incomplete, with over‐representation of the effects of most recent agents (Butts 2014; Katakami 2017; Multhoff 2020; Vansteenkiste 2013; Vansteenkiste 2016). This naturally limited our analysis to antigen‐specific vaccines (MAGE‐A3 and L‐BLP25) and cell‐based vaccines (NK cells). On the other hand, since other trials (with smaller numbers of participants) produced mixed results, we were unable to assess the efficacy of other types of immunotherapeutic agents in the current updated review.
Two review authors independently carried out study selection, assessment of risks of bias, and data collection, without blinding. Regarding missing data and missing information related to study quality assessment, we tried to contact authors by email to obtain these details. However, even after repeated attempts, we did not receive any response from these authors (since these trials were conducted a long time ago, the non‐response rates were expected to be high). In the end, we were unable to access sufficient information to assess the eligibility of two trials (Macchiarini 1989; Schlieben 1984). For the other trials, we deemed that there were unclear risks of certain study bias (see Figure 4), and we extracted and used available data for the meta‐analysis for each outcome. Again, this could influence the accuracy and reliability of our results because of incomplete study assessment and data pooling.
Agreements and disagreements with other studies or reviews
The survival benefit of immunotherapy for people with localised NSCLC has been discussed for decades. For early (stage I and II) or locally advanced (IIIA) NSCLC, the first attempts to use immunotherapy (mainly cellular immunity‐based therapeutic strategies) in such patients began in the 1980s, using active immunotherapy agents (e.g. Bacillus Calmette‐Guérin (BCG)) or adoptive cell transfer (e.g. transfer factor (TF)). Striking improvements in survival were observed in some clinical trials, including both RCTs (Fujisawa 1996; Macchiarini 1991), and non‐randomised clinical trials; while null results were reported in Matthay 1986. These attempts were finally thought to fail since the largest RCT (with 441 participants) found no significant difference between experimental and control groups (Stanley 1986). New efforts were launched after 2000, mainly applying vaccine‐based immunotherapy agents. Promising outcomes from small trials inspired further explorations (Vansteenkiste 2013). However, disappointing results were reported from later phase III RCTs (Butts 2014; Vansteenkiste 2016). Similar conclusions were drawn from other published reviews (Carrizosa 2015; Reckamp 2015). Nevertheless, new trials in this area have been started, with a focus on novel agents, especially immune checkpoint inhibitors, for which superior progression‐free survival has been reported in the planned interim analysis of one phase III RCT trial (Antonia 2017).
Authors' conclusions
Implications for practice.
So far, based on this updated review, there is no evidence showing better survival outcomes associated with the addition of immunotherapy (excluding checkpoint inhibitors) for people with stages I to III NSCLC. However, the probability of experiencing adverse events may be greater for people receiving immunotherapy, especially the MAGE‐A3 vaccine. A major concern for the evidence on overall survival and progression‐free survival was the small proportion of included trials (36%; 45%) that contributed usable data for group comparisons. For yearly survival rate analyses, for which more data were available, we found no clear differences between the treatment groups for 1‐year, 2‐year, 3‐year, or 5‐year survival rates, with significant heterogeneity across the trials for the 1‐year rate, which could not be fully explained by either variation in study quality or the clinical differences in the trials.
Based on these results, we consider that at present, there is insufficient evidence to support or negate the use of adjuvant immunotherapy (excluding checkpoint inhibitors) for people with stages I to III NSCLC. Several planned or ongoing trials that aim to treat people with stages IB to IIIA NSCLC with adjuvant checkpoint inhibitors (PD‐1/PD‐L1) may provide new evidence for the use of immunotherapy.
Implications for research.
We found no ongoing clinical trials on participants with stages I to III NSCLC that could provide additional evidence on the effectiveness of vaccine‐based immunotherapy from this updated review. We identified several planned or ongoing trials with a focus on adjuvant checkpoint inhibitors (PD‐1/PD‐L1). Future efforts should be put into the development of novel, effective immunotherapy, and the detection of more useful prognostic biomarkers to guide the use of these immunotherapeutic drugs.
What's new
| Date | Event | Description |
|---|---|---|
| 19 May 2021 | New search has been performed | We updated the background. Four new authors joined the team: Yuan Y, Wan X, Chen W and Yin D. Three authors left the team: Roudi R, Teghararia O, Tiselius E. |
| 19 May 2021 | New citation required but conclusions have not changed | We ran a new literature search on 19 May 2021. We identified and included two new trials (Katakami 2017; Multhoff 2020). Our conclusions remain unchanged. |
History
Protocol first published: Issue 9, 2014 Review first published: Issue 12, 2017
Acknowledgements
We thank Corynne Marchal, Managing Editor, Cochrane Lung Cancer Group for providing administrative and logistical support for the conduct of the current review. We thank Ludovic Trinquart, Noelle O’Rourke, and Sophie Paget‐Bailly for their comments on the protocol. We thank François Calais for helping us revise the search strategy and also for running the search for us. We thank Noelle O’Rourke, Fergus Macbeth, Frederic Fiteni, Bertrand Mennecier, Virginie Westeel, and Lars Lidgard for their helpful advice in this updated review.
We thank Raheleh Roudi, Eva Tiselius, and Olivia Teghararian for their help in the process of data extraction and manuscript preparation for the original review published in 2017.
Appendices
Appendix 1. CENTRAL search strategy
1. MeSH descriptor: [Lung Neoplasms] explode all trees 2. MeSH descriptor: [Carcinoma, Non‐Small‐Cell Lung] explode all trees 3. lung carcinom* 4. lung neoplasm* 5. lung cancer* 6. nsclc 7. non small cell lung 8. #1 or #2 or #3 or #4 or #5 or #6 or #7 9. MeSH descriptor: [Immunotherapy] explode all trees 10. immunother* 11. MeSH descriptor: [Cancer Vaccines] explode all trees 12. vaccin* 13. immunisation 14. immunization 15. #9 or #10 or #11 or #12 or #13 or #14 16. #8 and #15 with Publication Year from 2017 to 2021, in Trials
Appendix 2. MEDLINE PubMed search strategy
1. Carcinoma, Non‐Small‐Cell Lung[Mesh]
2. nsclc[Title/Abstract]
3. lung cancer*[Title/Abstract]
4. lung carcinoma*[Title/Abstract]
5. lung neoplasm*[Title/Abstract]
6. lung tumor*[Title/Abstract]
7. lung tumour*[Title/Abstract]
8. non‐small cell*[Title/Abstract]
9. nonsmall cell*[Title/Abstract]
10. #3 OR #4 OR #5 OR #6 OR #7
11. #8 OR #9
12. #10 AND #11
13. #1 OR #2 OR #12
14. immunotherapy[MeSH Terms]
15. immunother*[Title/Abstract]
16. Cancer vaccines[MeSH Terms]
17. vaccin*[Title/Abstract]
18. immunisation[Title/Abstract]
19. immunization[Title/Abstract]
20. #14 OR #15 OR #16 OR #17 OR #18 OR #19
21. #13 AND #20
Appendix 3. Embase search strategy
1. 'non small cell lung cancer'/exp
2. 'lung tumor'/exp
3. 'nsclc':ab,ti
4. 'lung carcinom*':ab,ti
5. 'lung cancer*':ab,ti
6. 'lung neoplasm*':ab,ti
7. 'lung tumor*':ab,ti
8. 'lung tumour*':ab,ti
9. 'nonsmall cell*':ab,ti
10. 'non‐small cell*':ab,ti
11. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10
12. 'immunotherapy'/exp
13. 'immunother*':ab,ti
14. 'cancer vaccine'/exp
15. 'vaccin*':ab,ti
16. 'immunisation':ab,ti
17. 'immunization':ab,ti
18. #12 OR #13 OR #14 OR #15 OR #16 OR #17
19. 'crossover procedure'/exp OR 'double‐blind procedure'/exp OR 'randomized controlled trial'/exp OR 'single‐blind procedure'/exp OR random* OR factorial* OR crossover* OR cross NEXT/1 over* OR placebo* OR doubl* NEAR/1 blind* OR singl* NEAR/1 blind* OR assign* OR allocat* OR volunteer*
20. #11 AND #18 AND #19
21. #11 AND #18 AND #19 NOT [19‐5‐2021]/sd
Data and analyses
Comparison 1. Effect of immunotherapy for surgically treated NSCLC patients: main analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Overall survival | 4 | Hazard Ratio (IV, Random, 95% CI) | 0.94 [0.84, 1.05] | |
| 1.2 Progression‐free survival | 5 | Hazard Ratio (IV, Random, 95% CI) | 0.94 [0.86, 1.03] | |
| 1.3 Overall survival rate, 1‐year | 7 | 4420 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.96, 1.08] |
| 1.4 Overall survival rate, 2‐year | 7 | 4420 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.93, 1.12] |
| 1.5 Overall survival rate, 3‐year | 7 | 4420 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.90, 1.09] |
| 1.6 Overall survival rate, 5‐year | 7 | 4389 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.86, 1.12] |
| 1.7 Adverse events (any) | 4 | 4126 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.97, 1.28] |
| 1.8 Adverse events (severe, grade > 2) | 6 | 4546 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.92, 1.40] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Butts 2014.
| Study characteristics | ||
| Methods | International RCT (264 centres, 33 countries), Phase III | |
| Participants |
Number of participants: 1513 (22 February 2007 to 15 November 2011, with additional observation to 22 April 2014) Number randomised
Number evaluated:
Diagnosis: patients with unresectable stage III NSCLC Inclusion
|
|
| Interventions |
Experimental group: 3 days before administration of study drug, one dose of intravenous cyclophosphamide (300 mg/m², maximum dose 600 mg) was administered to participants, initial therapy consisted of eight consecutive weekly subcutaneous injections of tecemotide (806 μg lipopeptide). Control group: 3 days before administration of study drug, a corresponding intravenous saline infusion was given to participants, initial therapy consisted of eight consecutive weekly subcutaneous injections of placebo. In the absence of progressive cancer or toxicity, maintenance tecemotide or placebo every 6 weeks was continued until disease progression. |
|
| Outcomes |
Duration of follow‐up (median): experimental group: 39.9 months (IQR 21.2 to 48.7), control group: 37.7 months (IQR 19.9 to 49.7) Duration of additional follow‐up (median): experimental group: 58.7 months, control group: 57.3 months
|
|
| Notes | Merck KGaA, the study sponsor, designed the trial in collaboration with the investigators, developed the protocol and statistical analysis plan, provided the study drug, co‐ordinated the management of study sites and the clinical data management, conducted statistical analyses, and participated in the interpretation of data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computerised interactive voice response system was used for randomisation and allocation |
| Allocation concealment (selection bias) | Low risk | A computerised interactive voice response system was used for randomisation and allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "interactive voice randomisation system staff assigned patients and were not involved in the rest of the trial. To maintain blinding, tecemotide and placebo for the primary and maintenance treatment phases were packaged in identical containers" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: 'With the exception of a designated unblinded statistician on the data monitoring board, interactive voice randomisation system staff, a designated pharmacist, and a study monitor for cyclophosphamide drug accountability records, the randomisation code was masked from the sponsor and other individuals monitoring the trial.' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1239 randomised participants were included in analysis (81.9%) |
| Selective reporting (reporting bias) | Low risk | Previous protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Fujisawa 1996.
| Study characteristics | ||
| Methods | Japanese RCT | |
| Participants |
Number of participants: 82 (between 1986 and 1990) Number randomised
Number evaluated
Diagnosis: patients with NSCLC who had undergone a complete resection and mediastinal lymph node dissection Inclusion: the eligibility criteria was:
|
|
| Interventions |
Experimental group: TF and N‐CWS: 1 vial of TF, equivalent to 5 x 10⁸ peripheral lymphocytes, was administered subcutaneously every 4 weeks, and N‐CWS (200 mg) was administered subcutaneously every 2 weeks, beginning 1 month after resections, and continuing for 1 year Control group: participants received surgery alone without any particular treatment until recurrence |
|
| Outcomes | Duration of follow‐up (median): experimental group: 99 months, control group: 83 months
|
|
| Notes | Supported in part by grants‐in‐aid for scientific research 05453481 from the Ministry of Education and Culture of Japan | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | The author reported "Patients were randomised into two groups, TF + N‐CWS or control, by closed envelope", without detailed information about how the envelope was generated and kept. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 82 randomised participants were included in analysis (100%) |
| Selective reporting (reporting bias) | High risk | Prior protocol was unavailable, and no prespecified primary outcome was reported |
| Other bias | Low risk | No obvious potential source of bias |
Giovanni 1996.
| Study characteristics | ||
| Methods | Italy; RCT | |
| Participants |
Number of participants: 113 Number randomised
Number evaluated
Diagnosis: patients with (completely or incompletely) surgically removed stage II, IIIa, or IIIb NSCLC Inclusion: eligible participants should have:
|
|
| Interventions |
Experimental group
Control group
The same radiation therapy regimen was used in both treatment arms. The radiation dose was 50 Gy/25f over a 5‐week period for completely resected participants. |
|
| Outcomes |
Duration of follow‐up: range from 6 to 40 months
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised by the method of random number (how the random number was generated was not specified in the report) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 113 randomised participants were included in analysis (100%) |
| Selective reporting (reporting bias) | High risk | Prior protocol was unavailable, and no prespecified primary outcome was reported |
| Other bias | Low risk | No obvious potential source of bias |
Katakami 2017.
| Study characteristics | ||
| Methods | Japan; multicentre (25 centres); RCT; phase I/II | |
| Participants |
Number of participants: 172 Number randomised
Number evaluated
Diagnosis: unresectable stage III NSCLC, stable disease or clinical response after primary chemoradiotherapy and performance status ≤ 1 Inclusion: eligible participants:
|
|
| Interventions | Experimental group: tecemotide (930 ug as lipopeptide) subcutaneously once weekly for 8 weeks, then every 6 weeks until disease progression or treatment withdrawal. Cyclophosphamide 300 mg/m2 (maximum dose 600 mg) was given intravenously 3 days before the first dose of tecemotide. Control group: placebo subcutaneously once weekly for 8 weeks, then every 6 weeks until disease progression or treatment withdrawal. | |
| Outcomes |
Duration of follow‐up: 34 weeks
|
|
| Notes | The work was supported by Merck Serono Co Ltd, Tokyo, Japan. The sponsor, Merck Serono Co Ltd, Tokyo, Japan, as well its parent company, Merck KGaA, Darmstadt, Germany, worked with the investigators to develop the study design, carry out the analysis and interpretation of the study data, and contributed through critical review to the writing of this report, and supported the authors in the submission. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The author reported “Central randomisation”, but the method for randomisation number generation was not described. |
| Allocation concealment (selection bias) | Low risk | Quote: “Central randomisation and treatment allocation in the phase II part was performed.” |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "This was a multicenter (25 centers), randomized, double‐blind and placebo‐controlled trial, conducted in accordance with the Declaration of Helsinki and in compliance with Good Clinical Practice." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 155 participants (90.1%) were included in the analysis. |
| Selective reporting (reporting bias) | Low risk | The prior protocol was available. No selective reporting of a particular outcome measurement or a particular outcome. Efficacy analysis was performed in three sets: ITT, mITT, and safety sets. |
| Other bias | Low risk | No obvious potential source of bias. |
Macchiarini 1991.
| Study characteristics | ||
| Methods | Italy; RCT | |
| Participants |
Number of participants: 52 (Between January 1979 and December 1980) Number randomised
Number evaluated
Diagnosis: patients stages II and III NSCLC following complete surgical resection Inclusion: eligible participants:
|
|
| Interventions |
Experimental group: chemotherapy including cyclophosphamide (1 g/m² intravenous, day 1), doxorubicin (45 mg/m² intravenous, day 1), and vincristine (1.2 mg/m² intravenous, day 1) was started within 4 weeks after operation and repeated every 3 weeks. Intrapleural BCG consisted of 5.5 per 10⁸ colony‐forming units of BCG pasteur administered into the pleural space as a single dose between postoperative days 4 and 14. 14 days after BCG administration, isionazide (INH), 300 mg/day, was administered orally for 12 weeks. Control group: chemotherapy including cyclophosphamide (1 g/m² intravenous, day 1), doxorubicin (45 mg/m² intravenous, day 1), and vincristine (1.2 mg/m² intravenous, day 1) was started within 4 weeks after operation and repeated every 3 weeks. |
|
| Outcomes |
Duration of follow‐up (median): 111 months
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 62 randomised participants were included in analysis (100%) |
| Selective reporting (reporting bias) | High risk | Previous protocol was unavailable, and no prespecified primary outcome was reported |
| Other bias | Low risk | No obvious potential source of bias |
Matthay 1986.
| Study characteristics | ||
| Methods | A USA prospective randomised trial | |
| Participants |
Number of participants: 88 Number randomised
Number evaluated
Diagnosis: patients with potentially resectable non‐small cell carcinoma of the lung Inclusion: participants suspected of having lung cancer were excluded if a definite diagnosis had not been established before thoracotomy. |
|
| Interventions |
A 12‐week course of 300 mg INH daily was given to all participants. |
|
| Outcomes |
Duration of follow‐up (median ± SD): experimental group: 986 ± 412 days; control group: 1062 ± 390 days
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified block randomisation method. Quote: "Patients were divided into 2 groups according to a table of random numbers and sealed‐envelope technique in strict numerical order of entry" |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were divided into 2 groups according to a table of random numbers and sealed‐envelope technique in strict numerical order of entry" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 85 randomised participants were included in analysis (96.6%) |
| Selective reporting (reporting bias) | High risk | No prior protocol was available, and no prespecified primary outcome was reported |
| Other bias | Low risk | No obvious potential source of bias |
Multhoff 2020.
| Study characteristics | ||
| Methods | Germany; RCT; phase II | |
| Participants |
Number of participants: 16 (between November 2014 and August 2017) Number randomised
Number evaluated
Diagnosis: histologically‐proven, inoperable, clinical stage IIIa/b squamous cell lung cancer (a subtype of NSCLC) Inclusion ‐ eligible participants
|
|
| Interventions | Experimental group: in the INT arm, patients with mHsp70‐positive tumours received TKD/IL‐2‐activated, autologous NK cells (somatic cell therapy, plasma‐derived medicinal product, IMP) subsequent to standard RCT (60 Gy to 70 Gy, platinum‐based chemotherapy). Control group: patients with mHsp70‐positive tumours were staged regularly at defined time intervals following standard RCT (60 Gy to 70 Gy, platinum‐based chemotherapy) in the control arm. | |
| Outcomes |
Duration of follow‐up: 18 months after randomisation, between November 2014 and August 2017
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label design |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 13 participants were included in the analysis (81.3%) |
| Selective reporting (reporting bias) | Low risk | The prior protocol was available. |
| Other bias | Low risk | No obvious potential source of bias |
Stanley 1986.
| Study characteristics | ||
| Methods | International randomised multicentre trial | |
| Participants |
Number of participants: 441 (February 1979 to October 1981) Number randomised
Number evaluated
Diagnosis: patients with a completely resected stage I and stage II non‐small cell bronchogenic carcinom Inclusions: this trial included only those participants with a resected non‐small cell carcinoma pT1N0M0, pT2N0M0, pT1N1M0, or pT2N1M0 (stages I and 11). Participants < 70 years of age, and those without history of previous malignancy, radiotherapy, chemotherapy, or immunosuppressive treatment. |
|
| Interventions |
Experimental group: a single dose of 1 X 10⁷ units of BCG Tice were given into the pleural space between day 6 and day 12 postoperatively. Isoniazid (INH) 300 mg/day by mouth, starting on the 14th postinjection day and continuing for 12 weeks. Control group: 35 cc of saline injected through the pleural catheter and no INH was given. |
|
| Outcomes |
Duration of follow‐up (median): 4.7 years
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Had placebo comparator without detailed explanation of blinding of participants or researchers |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 407 randomised participants were included in analysis (90.8%) |
| Selective reporting (reporting bias) | High risk | No protocol was available, and no prespecified primary outcome was reported. |
| Other bias | Low risk | No obvious potential source of bias |
Vansteenkiste 2013.
| Study characteristics | ||
| Methods | International RCT (41 investigation sites in 12 European countries), phase II | |
| Participants |
Number of participants: 182 (September 2005 to June 2010) Number randomised
Number evaluated
Diagnosis: patients with completely resected MAGE‐A3–positive TNM stage IB or II NSCLC Inclusion
|
|
| Interventions |
Experimental group: MAGE‐A3 protein + GlaxoSmithKline’s proprietary immunostimulant AS02B was administered intramuscularly in 0.5 mL doses. First administration was 4 to 8 weeks after resection, five doses at 3‐week intervals, followed by eight doses at 3‐month intervals. Control group: placebo was sucrose in the tocopherol oil‐in‐water emulsion‐based adjuvant AS03A, was administered intramuscularly in 0.5 mL doses. First administration was 4 to 8 weeks after resection, five doses at 3‐week intervals followed by eight doses at 3‐month intervals. |
|
| Outcomes |
Duration of follow‐up (median): participants were followed for 7.3 years, with a median of 70 months after resection.
|
|
| Notes | Sponsor: Vincent G Brichard The sponsor was involved in the study design, data collection, and analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A supply randomisation list was generated at GSK Biologicals, Rixensart, using a standard SAS programme and used to identify uniquely each individual vaccine dose (= treatment box). A randomisation blocking scheme (2:1 ratio) was used to ensure that balance between treatments was maintained. |
| Allocation concealment (selection bias) | Low risk | The person in charge of the vaccination accessed the randomisation system via the Internet. The randomisation system determined the treatment randomised and provided the treatment box number to be used for the first vaccination of this patient. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind, placebo‐controlled trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "scans were evaluated by the investigator or a radiologist blinded to the treatment assignment" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 182 participants included in the analysis (100%) |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Vansteenkiste 2016.
| Study characteristics | ||
| Methods | International multicentre RCT (590 centres in 34 countries), phase III | |
| Participants |
Number of participants: 2312 (18 October 2007 to 17 July 17 2012) Number randomised:
Number evaluated:
Diagnosis: patients with completely resected stage IB, II, and IIIA MAGE‐A3‐positive NSCLC who did or did not receive adjuvant chemotherapy Inclusion: eligible participants should be:
|
|
| Interventions |
Experimental group: participants received GSK1572932A antigen‐specific up to 13 intramuscular injections during 27 months. Five doses were given every 3 weeks, followed by eight doses every 12 weeks. Control group: participants received placebo up to 13 intramuscular injections during 27 months. Five doses were given every 3 weeks, followed by eight doses every 12 weeks. Treatment started at the time of randomisation. |
|
| Outcomes |
Duration of follow‐up (median): experimental group: 38.1 months (IQR 27.9 to 48.4), control group: 39.5 months (IQR 27.9 to 50·4)
|
|
| Notes | This study was designed, funded, and interpreted by GlaxoSmithKline in co‐operation with an international steering committee. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation at the investigator site was done centrally via Internet with stratification for chemotherapy versus no chemotherapy. Within each chemotherapy stratum, a minimisation algorithm was used to allocate study group, accounting for the number of chemotherapy cycles received (1 to 2 or 3 to 4), disease stage (IB, II, or IIIA), lymph node sampling procedure (mediastinal lymph node dissection or sampling), PS score (0 to 1 or 2), and lifetime smoking status (never, past, or current)" |
| Allocation concealment (selection bias) | Low risk | The randomisation was done centrally, so that the researchers would have no way of knowing in advance the allocation of the next participant. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Individual treatment assignment was masked at all levels, masking was managed by the central randomisation system with access limited to a single sponsor database administrator who was independent from the trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Individual treatment assignment was masked at all levels, masking was managed by the central randomisation system with access limited to a single sponsor database administrator who was independent from the trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants were included in analysis (100%) |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Zhao 2014.
| Study characteristics | ||
| Methods | Chinese RCT | |
| Participants |
Number of participants: 157 (June 2010 to June 2013) Number randomised
Number evaluated
Diagnosis: stage Ⅲ NSCLC that had received complete surgery resection Inclusion:
|
|
| Interventions | Experimental group: GP chemotherapy and DC‑CIK cell immunotherapy Control group: gemcitabine (1000 mg/m²) intravenously infused at day 1 and day 8 + 30 mg/m² cisplatin intravenously injected at days 1 to 3. The treatment cycle was 21 days and each participant underwent four cycles of treatment. | |
| Outcomes |
Duration of follow‐up: the two groups both were followed up for 36 months
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 157 randomised participants were included in analysis (100%) |
| Selective reporting (reporting bias) | High risk | Prior protocol was unavailable, and no prespecified primary outcome was reported |
| Other bias | High risk | Suvival rates in abstract, full text, and figures were different from each other |
BCG: Bacillus Calmette‐Guérin CT: computed tomography DC/CIK: dendritic cell/cytokine‐induced killer ECOG: Eastern Co‐operative Oncology Group G‐GT: Gamma‐Glutamyl transferase GOT: glutamic oxaloacetic transaminase GP: gemcitabine plus platinum GPT: glutamic pyruvic transaminase HN: Hemagglutinin Neuraminidase IMP: investigational medicinal product INH: isionazide IQR: inter‐quartile range irRC: immune‐related response criteria ITT: intention‐to‐treat mITT: modified intention‐to‐treat MRI: magnetic resonance imaging N‐CWS: nocardia rubra‐cell wall skeleton NK: natural killer NSCLC: non‐small cell lung cancer PET: positron emission tomography PPD: purified protein derivative PTT: partial thromboplastin time PS: performance status RCT: randomised controlled trial rIL‐2: recombinant interleukin‐2 SD: standard deviation TF: transfer factor TIL: tumor‐infiltrating lymphocytes TKD/IL‐2: Hsp70‐derived peptide (TKDNNLLGRFELSG, TKD)/interleukin‐2 (IL‐2) TNM: cancer staging system (Tumor size, lymph Nodes, Metastasis) ULN: upper limit of normal
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bradley 2020 | Ineligible experimental groups (checkpoint inhibitors) |
| Giaccone 2020 | Ineligible experimental groups (checkpoint inhibitors) |
| Kimura 2015 | Ineligible study population (advanced cancer stage) |
| Kimura 2018 | Ineligible study population (advanced cancer stage) |
| Lee 2017 | Not a RCT |
| Mignard 2018 | Ineligible experimental groups (checkpoint inhibitors) |
| Nokihara 2017 | Ineligible study population (advanced cancer stage) |
| Rittmeyer 2017 | Ineligible study population (advanced cancer stage) |
Characteristics of ongoing studies [ordered by study ID]
Merck 2015.
| Study name | Study of tecemotide (l‐blp25) in participants with stage III unresectable non‐small cell lung cancer (NSCLC) following primary chemoradiotherapy |
| Methods | Japanese RCT, phase I/II |
| Participants |
Number of participants: 172 (December 2008 to June 2015) Number randomised
Number evaluated
Diagnosis: patients with locally advanced unresectable stage III NSCLC Inclusion ‐eligibility criteria
|
| Interventions |
Experimental group: tecemotide (L‐BLP25) + cyclophosphamide Control group: placebo + saline |
| Outcomes |
|
| Starting date | December 2008 |
| Contact information | |
| Notes | This study has been completed, but no publication was found at the time of literature searching |
Saka 2017.
| Study name | Randomized phase II study with or without adjuvant immunotherapy of alpha‐GalCer‐pulsed dendritic cells in the patients with completely resected stage II‐IIIA non‐small cell lung cancer |
| Methods | Japanese RCT, phase II |
| Participants |
Number of participants: not specified (March 2013 to March 2020) Number randomised
Number evaluated
Diagnosis: patients who had undergone a complete resection of stage II‐IIIA NSCLC followed by postoperative adjuvant therapy with cisplatin plus vinorelbine Inclusion ‐ eligibility criteria
|
| Interventions |
Experimental group: α‐GalCer‐pulsed DC immune therapy Control group: standard treatment |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | March 2013 |
| Contact information | |
| Notes | Open public recruiting. The present study began in March 2013 and will conclude in March 2020. |
Wu 2011.
| Study name | Cancer vaccine study for stage III, unresectable, non‐small cell lung cancer (NSCLC) in the Asian population (INSPIRE) |
| Methods | International RCT (45 investigation sites in 5 countries ), Phase III |
| Participants |
Number of participants: 285 (3 Dec 2009 to 10 Sep 2014) Number randomised
Number evaluated
Diagnosis: patients were locally advanced unresectable stage III NSCLC Inclusion ‐ eligibility criteria
|
| Interventions |
Experimental group: a single intravenous infusion of 300 mg/m² (to a maximum 600 mg) of low dose cyclophosphamide was given 3 days prior to first tecemotide (L‐BLP25) vaccination. After receiving single low‐dose cyclophosphamide, subjects received 8 consecutive weekly (week 1, 2, 3, 4, 5, 6, 7, and 8 primary treatment phase) subcutaneous tecemotide (L‐BLP25) vaccinations at a dose of 918 mcg and then at 6‐week intervals, beginning at week 14 (maintenance phase) until PD is documented or the subject discontinued for any other reason. The BSC was provided as per the investigator’s discretion and was not limited to palliative radiation, psychosocial support, analgesics, and nutritional support. Control group: a single intravenous infusion of 0.9% sodium chloride (saline) was administered 3 days prior to first placebo vaccination. After receiving saline solution, subjects received 8 consecutive weekly subcutaneous vaccinations with placebo at week 1, 2, 3, 4, 5, 6, 7 and 8 followed by maintenance treatment at 6‐week intervals, beginning at week 14, until PD is documented or the subject discontinued for any other reason. The BSC was provided as per the investigator’s discretion and was not limited to palliative radiation, psychosocial support, analgesics and nutritional support. |
| Outcomes | Data cut‐off date: June 2015
|
| Starting date | December 2009 |
| Contact information | Correspondence for published protocol: tony@clo.cuhk.edu.hk |
| Notes | Supported by Merck KGaA, Darmstadt, Germany This study was terminated prematurely as the sponsor decided to discontinue the programme with tecemotide in NSCLC |
BSC: Bacillus Calmette‐Guérin CT: computed tomography DC: dendritic cell ECOG: Eastern cooperative Oncology Group MRI: magnetic resonance imaging NKT: Natural killer T‐cell NSCLC: non‐small cell lung cancer PD: disease progression PET: positron emission tomography RCT: randomised controlled trial
Differences between protocol and review
The title of the published protocol was 'Immunotherapy for stage I‐III non‐small cell lung cancer treated with surgery or radiotherapy with curative intent'. However, since the available data for new immunotherapy agent was very limited, and the meta‐analysis performed in the review only pooled data for limited types of immunotherapy (mainly BCG, MAGE‐A3, L‐BLP25), we decided to narrow the title to 'immunotherapy (excluding checkpoint inhibitors)'. In the future, in order to answer specific questions about the effectiveness of specific new immunotherapy agents, such as checkpoint inhibitors, additional reviews should be planned.
Because we were not able to extract adequate data to calculate time‐to‐death hazard ratios for most of the included trials, we added a secondary outcome of 'overall survival rates: the percentage of participants in a study who were still alive for a certain period of time'. We compared overall survival in terms of yearly overall survival rates for one, two, three, and five years.
We updated the search strategies.
Four new authors (Rui Li, Raheleh Roudi, Olivia Teghararian, and Eva Tiselius) were involved during the review stage. OT and ET completed the abstract screenings and data extraction, together with RL. RL also helped with full‐text checking, data extraction, assessment of risk of bias, and manuscript drafting. RR helped with data analysis, and manuscript drafting and revision.
In the review update stage, four new authors (Yun Yuan, Wenwen Chen, Xiaoyu Wan, Dan Yin) were involved. YY, WC, XW and DY completed the abstract screenings and data extraction, together with JZ. YY helped with data analysis, and manuscript updating.
Contributions of authors
Conceiving the review: HS
Designing the review: HS, JZ, CS
Co‐ordinating the review: HS, JZ
Designing search strategies: HS, Cochrane Lung Cancer Group
Study selection, data extraction, data analysis, risk assessment: HS, JZ, YY, XW, DY, WC, CS, RL
Writing the review: JZ, YY, HS, RL
Providing general advice on the review: CS
Performing previous work that was the foundation of the current review: HS, JZ, CS
Sources of support
Internal sources
-
Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Sweden
The full texts of parts of the screened papers were retrieved from the Karolinska Institutet library.
External sources
No sources of support provided
Declarations of interest
JZ: declares no conflict of interest
YY: declares no conflict of interest
XW: declares no conflict of interest
DY: declares no conflict of interest
RL: declares no conflict of interest
WC: declares no conflict of interest
CS: declares no conflict of interest
HS: declares no conflict of interest
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Butts 2014 {published data only}
- Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer(START): a randomised, double-blind, phase 3 trial. Lancet Oncolology 2014;15(1):59-68. [DOI] [PubMed] [Google Scholar]
Fujisawa 1996 {published data only}
- Fujisawa T, Yamaguchi Y. Postoperative immunostimulation after complete resection improves survival of patients with stage I non-small cell lung carcinoma. Cancer 1996;78(9):1892-8. [PubMed] [Google Scholar]
Giovanni 1996 {published data only}
- Giovanni BR, Paolo Z, Sandro M, Paolo M, Riccardo A, Giovanni F, et al. A randomized trial of adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 versus standard therapy in the postoperative treatment of resected non-small cell lung carcinoma. Cancer 1996;78(2):244-51. [DOI] [PubMed] [Google Scholar]
Katakami 2017 {published data only}
- Katakami N, Hida T, Nokihara H, Imamura F, Sakai H, Atagi S, et al. Phase I/II study of tecemotide as immunotherapy in Japanese patients with unresectable stage III non-small cell lung cancer. Lung Cancer 2017;105:23-30. [DOI] [PubMed] [Google Scholar]
Macchiarini 1991 {published data only}
- Macchiarini P, Hardin M, Angeletti CA. Long-term evaluation of intrapleural Bacillus Calmette-Guérin with or without adjuvant chemotherapy in completely resected stages II and III non-small-cell lung cancer. American Journal of Clinical Oncology 1991;14(4):291-7. [DOI] [PubMed] [Google Scholar]
Matthay 1986 {published data only}
- Matthay RA, Mahler DA, Beck GJ, Loke J, Baue AE, Carter DC, et al. Intratumoral Bacillus Calmette-Guérin immunotherapy prior to surgery for carcinoma of the lung: results of a prospective randomized trial. Cancer Research 1986;46(11):5963-8. [PubMed] [Google Scholar]
Multhoff 2020 {published data only}EudraCT2008‐002130‐30
- Multhoff G, Seier S, Stangl S, Sievert W, Shevtsov M, Werner C, et al. Targeted natural killer cell-based adoptive immunotherapy for the treatment of patients with NSCLC after radiochemotherapy: a randomized phase II clinical trial. Clinical Trials: Immunotherapy 2020;26:5368-79. [DOI] [PubMed] [Google Scholar]
Stanley 1986 {published data only}
- Stanley K, Ludwig Lung Cancer Study Group (LLCSG). Immunostimulation with intrapleural BCG as adjuvant therapy in resected non-small cell lung cancer. Cancer 1986;58(11):2411-6. [DOI] [PubMed] [Google Scholar]
Vansteenkiste 2013 {published data only}
- Vansteenkiste J, Zielinski M, Linder A, Dahabreh J, Gonzalez EE, Malinowski W, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. Journal of Clinical Oncology 2013;31(19):2396-403. [DOI] [PubMed] [Google Scholar]
Vansteenkiste 2016 {published data only}
- Vansteenkiste JF, Cho BC, Vanakesa T, Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncology 2016;17(6):822-35. [DOI] [PubMed] [Google Scholar]
Zhao 2014 {published data only}
- Zhao M, Li H, Li L, Zhang Y. Effects of a gemcitabine plus platinum regimen combined with a dendritic cell-cytokine induced killer immunotherapy on recurrence and survival rate of non-small cell lung cancer patients. Experimental and Therapeutic Medicine 2014;7(5):1403-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Bradley 2020 {published data only}
- Bradley JD, Hu C, Komaki RR, Masters GA, Blumenschein GR, Schild SE et al. Long-term results of NRG oncology RTOG 0617: standard versus high-dose chemoradiotherapy with or without cetuximab for unresectable Stage III non-small-cell lung cancer. Journal of Clinical Oncology 2020 Mar 1;38(7):706-14. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Giaccone 2020 {published data only}
- Giaccone G, Felip E, Cobo M, Campelo RG, Denis F, Quoix E et al. Activity of OSE-2101 in HLA-A2+ non-small cell lung cancer (NSCLC) patients after failure to immune checkpoint inhibitors (ICI): step 1 results of phase III ATALANTE-1 randomised trial. Annals of Oncology 2020;31(4):S814-S815. [DOI: 10.1016/j.annonc.2020.08.1574] [DOI] [Google Scholar]
Kimura 2015 {published data only}
- Kimura H, Matsui Y, Ishikawa A, Nakajima T, Yoshino M, Sakairi Y. Randomized controlled phase III trial of adjuvant chemo-immunotherapy with activated killer T cells and dendritic cells in patients with resected primary lung cancer. Cancer Immunology, Immunotherapy 2015 Jan;64(1):51-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kimura 2018 {published data only}
- Kimura H, Matsui Y, Ishikawa A, Nakajima T, Lizasa T. Randomized controlled phase III trial of adjuvant chemo-immunotherapy with activated cytotoxic T cells and dendritic cells from regional lymph nodes of patients with lung cancer. Cancer Immunology, Immunotherapy 2018 Aug;67(8):1231-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2017 {published data only}
- Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8(+) T-cell infiltration. Clinical Cancer Research 2017 Aug 15;23(16):4556-68. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mignard 2018 {published data only}
- Mignard X, Antoine M, Moro-Sibilot D, Dayen C, Mennecier B, Gervais R et al. IoNESCO trial: Immune neoajuvant therapy in early stage non-small cell lung cancer. Revue des Maladies Respiratoires 2018 Nov;35(9):983-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Nokihara 2017 {published data only}
- Nokihara H, Lu S, Mok TSK, Nakagawa K, Yamamoto N, Shi Y et al. Randomized controlled trial of S-1 versus docetaxel in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy (East Asia S-1 Trial in Lung Cancer). Annals of Oncology 2017 Nov 1;28(11):2698-2706. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rittmeyer 2017 {published data only}
- Rittmeyer, A Barlesi, F Waterkamp, D Park, K Ciardiello, F von Pawel, J et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017 Jan 21;389(10066):255-65. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
Merck 2015 {unpublished data only}
- NCT00960115. Study of Tecemotide (L-BLP25) in Participants With Stage III Unresectable Non-small Cell Lung Cancer (NSCLC) Following Primary Chemoradiotherapy. www.clinicaltrials.gov/ct2/show/NCT00960115 (first received 17 August 2009).
Saka 2017 {published data only}
- Saka H, Kitagawa C, Ichinose Y, Takenoyama M, Ibata H, Kato T, et al. A randomized phase II study to assess the effect of adjuvant immunotherapy using α-GalCer-pulsed dendritic cells in the patients with completely resected stage II–IIIA non-small cell lung cancer: study protocol for a randomized controlled trial. Trials 2017;18:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2011 {unpublished data only}
- Wu YL, Park K, Soo RA, Sun Y, Tyroller K, Wages D, et al. INSPIRE: A phase III study of the BLP25 liposome vaccine (L-BLP25) in Asian patients with unresectable stage III non-small cell lung cancer. BMC Cancer 2011;11:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Aaronson 1993
- Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. Journal of the National Cancer Institute 1993;85(5):365-76. [PMID: ] [DOI] [PubMed] [Google Scholar]
Acres 2005
- Acres B, Limacher JM. MUC1 as a target antigen for cancer immunotherapy. Expert Review of Vaccines 2005;4(4):493-502. [DOI] [PubMed] [Google Scholar]
Aitken 1969
- Aitken RC. Measurement of feelings using visual analogue scales. Proceedings of the Royal Society of Medicine 1969;62(10):989–93. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Antonia 2017
- Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. New England Journal of Medicine 2017;377(20):1919-29. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bafna 2010
- Bafna S, Kaur S, Batra SK. Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010;29(20):2893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Borenstein 2008
- Borenstein M, Hedges LV, Higgins JP, Rothstein HR. Introduction to meta-analysis (Statistics in Practice). Chichester: John Wiley & Sons, 2008. [Google Scholar]
Carrizosa 2015
- Carrizosa DR, Gold KA. New strategies in immunotherapy for non-small cell lung cancer. Translational Lung Cancer Research 2015;4(5):553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dasanu 2012
- Dasanu CA, Sethi N, Ahmed N. Immune alterations and emerging immunotherapeutic approaches in lung cancer. Expert Opinion on Biological Therapy 2012;12(7):923-37. [DOI] [PubMed] [Google Scholar]
Detterbeck 2009
- Detterbeck FC, Boffa DJ, Tanoue LT. The new lung cancer staging system. Chest 2009;136(1):260-71. [DOI] [PubMed] [Google Scholar]
Drake 2006
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Advances in Immunology 2006;90:51-81. [DOI] [PubMed] [Google Scholar]
Drake 2014
- Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nature Reviews. Clinical Oncology 2014;11(1):24-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
EuroQol 1990
- EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy 1990 Dec;16(3):199-208. [PMID: ] [DOI] [PubMed] [Google Scholar]
Finn 2008
- Finn OJ. Cancer immunology. New England Journal of Medicine 2008;358(25):2704-15. [DOI] [PubMed] [Google Scholar]
Forde 2014
- Forde PM, Kelly RJ, Brahmer JR. New strategies in lung cancer: translating immunotherapy into clinical practice. Clinical Cancer Research 2014;20(5):1067-73. [DOI] [PubMed] [Google Scholar]
Giaccone 2013
- Giaccone G, Bazhenova L, Nemunaitis J, Juhasz E, Ramlau R, Van Den Heuval MM, et al. A phase III study of belagenpumatucel-L therapeutic tumor cell vaccine for non-small cell lung cancer (NSCLC). In: European Cancer Congress 2013. Amsterdam, 2013:Abstract n2.
GLOBOCAN 2020
- International Agency for Research on Cancer (IARC) . Cancer incidence and mortality worldwide. https://gco.iarc.fr/databases.php (accessed 8 November 2021).
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro. Version accessed prior to 1 November 2021. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015. Available from gradepro.org.
Hayes 2021
- Conall Hayes. Cellular immunotherapies for cancer. Irish Journal of Medical Science 2021;190:41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Higgins 2021
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook. [DOI] [PMC free article] [PubMed]
Hirschowitz 2006
- Hirschowitz EA, Hiestand DM, Yannelli JR. Vaccines for lung cancer. Journal of Thoracic Oncology 2006;1(1):93-104. [PubMed] [Google Scholar]
Howington 2013
- Howington JA, Blum MG, Chang AC, Balekian AA, Murthy SC. Treatment of stage I and II non-small cell lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143(5 Suppl):e278S-e313S. [DOI] [PubMed] [Google Scholar]
Keir 2008
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annual Review of Immunology 2008;26:677-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kochenderfer 2007
- Kochenderfer JN, Gress RE. A comparison and critical analysis of preclinical anticancer vaccination strategies. Experimental Biology and Medicine 2007;232(9):1130-41. [DOI] [PubMed] [Google Scholar]
Lardinois 2005
- Lardinois D, Suter H, Hakki H, Rousson V, Betticher D, Ris HB. Morbidity, survival, and site of recurrence after mediastinal lymph-node dissection versus systematic sampling after complete resection for non-small cell lung cancer. Annals of Thoracic Surgery 2005;80(1):268-74. [DOI] [PubMed] [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Medicine 2009;6(7):e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mellman 2011
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480(7378):480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mountain 1997
- Mountain CF. Revisions in the international system for staging lung cancer. Chest 1997;111:1710–17. [DOI] [PubMed] [Google Scholar]
National Cancer Institute 2017
- National Cancer Institute. Cancer Stat Facts: Lung and Bronchus Cancer. www.seer.cancer.gov/statfacts/html/lungb.html (accessed 14 November 2017).
NCI‐CTCAE 2017
- National Cancer Institute (NCI). Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf November 27, 2017.
NCT02273375
- NCT02273375. A phase III prospective double-blind placebo controlled randomized study of adjuvant MEDI4736 in completely resected non-small cell lung cancer. https://clinicaltrials.gov/ct2/show/NCT02273375.
NCT02486718
- NCT02486718. Study to assess safety and efficacy of atezolizumab (MPDL3280A) compared to best supportive care following chemotherapy in patients with lung cancer [IMpower010]. https://clinicaltrials.gov/ct2/show/NCT02486718.
NCT02504372
- NCT02504372. Study of pembrolizumab (MK-3475) vs placebo for participants with non-small cell lung cancer after resection with or without standard adjuvant therapy (MK-3475-091/KEYNOTE-091) (PEARLS). https://clinicaltrials.gov/ct2/show/NCT02504372.
Nemunaitis 2006
- Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. Journal of Clinical Oncology 2006;24(29):4721-30. [DOI] [PubMed] [Google Scholar]
Peters 2008
- Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Contour-enhanced meta-analysis funnel plots help distinguish publication bias from other causes of asymmetry. Journal of Clinical Epidemiology 2008;61(10):991–6. [DOI] [PubMed] [Google Scholar]
Ramnath 2013
- Ramnath N, Dilling TJ, Harris LJ, Kim AW, Michaud GC, Balekian AA, et al. Treatment of stage III non-small cell lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143(5 Suppl):e314S-e40S. [DOI] [PubMed] [Google Scholar]
Reckamp 2015
- Reckamp KL. Advances in immunotherapy for non-small cell lung cancer. Clinical Advances in Hematology & Oncology 2015;13(12):847-53. [PubMed] [Google Scholar]
Review Manager 2020 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.4. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2020.
Roy 2008
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. New England Journal of Medicine 2008;359(13):1367-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sangha 2007
- Sangha R, Butts C. L-BLP25: a peptide vaccine strategy in non small cell lung cancer. Clinical Cancer Research 2007;13(15 Pt 2):s4652-4. [DOI] [PubMed] [Google Scholar]
Simpson 2005
- Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nature Reviews 2005;5(8):615-25. [DOI] [PubMed] [Google Scholar]
Therasse 2000
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of Canada, National Cancer Institute of the United States. Journal of the National Cancer Institute 2000;92(3):205-16. [DOI] [PubMed] [Google Scholar]
Topalian 2012
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. New England Journal of Medicine 2012;366(26):2443-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tyagi 2009
- Tyagi P, Mirakhur B. MAGRIT: the largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clinical Lung Cancer 2009;10(5):371-4. [DOI] [PubMed] [Google Scholar]
Van den Eynde 1997
- Van den Eynde BJ, Bruggen P. T cell defined tumor antigens. Current Opinion in Immunology 1997;9(5):684-93. [DOI] [PubMed] [Google Scholar]
Wolchok 2009
- Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clinical Cancer Research 2009;15(23):7412-20. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Song 2014
- Song H, Zhu J, Suo C, Lu D. Immunotherapy for stage I‐III non‐small cell lung cancer treated with surgery or radiotherapy with curative intent. Cochrane Database of Systematic Reviews 2014, Issue 9. Art. No: CD011300. [DOI: 10.1002/14651858.CD011300] [DOI] [Google Scholar]
Zhu 2017
- Zhu J, Li R, Tiselius E, Roudi R, Teghararian O, Suo C et al. Immunotherapy (excluding checkpoint inhibitors) for stage I to III non-small cell lung cancer treated with surgery or radiotherapy with curative intent. Cochrane Database of Systematic Reviews 2017 Dec 16;12(12):CD011300. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]