

Abstract
Background
This analysis assessed combined safety data from 4 clinical studies of teduglutide in pediatric patients with short‐bowel syndrome–associated intestinal failure (SBS–IF).
Methods
Safety data from teduglutide‐treated patients in 4 clinical trials were pooled. The completed 12‐week and 24‐week phase 3 core studies (NCT01952080/EudraCT 2013‐004588‐30 and NCT02682381/EudraCT 2015‐002252‐27) enrolled children aged 1–17 years with SBS–IF. Patients could elect to enroll in ongoing open‐label extensions (NCT02949362/EudraCT 2016‐000863‐17 and NCT02954458/EudraCT 2016‐000849‐30). Interim data from ongoing studies were included.
Results
Safety data are reported for 89 pediatric patients treated with teduglutide for a median (range) of 51.7 (5.0–94.7) weeks. Adverse events (AEs) were reported in all patients; the most common were vomiting (51.7%), pyrexia (43.8%), upper respiratory tract infection (41.6%), and cough (33.7%). Thirty‐five patients (39.3%) had AEs considered related to teduglutide treatment; abdominal pain and vomiting were most frequent (5.6% each). Three serious AEs in 3 patients (3.4%) were considered related to teduglutide treatment: ileus, d‐lactic acidosis, and gastrointestinal obstruction due to hard stools. All 3 events resolved. One cecal polyp was detected, which was not biopsied or found on repeat colonoscopy. No cases of neoplasia occurred.
Conclusion
Based on integrated data from 4 clinical studies, including long‐term follow‐up for ≤161 weeks, teduglutide had a safety profile consistent with the individual core pediatric studies and as expected for pediatric patients with SBS–IF who never received teduglutide. The most frequent AEs reflected treatment with teduglutide, complications of the underlying disease, and typical childhood illnesses.
Keywords: gastroenterology, long‐term care, parenteral nutrition, pediatrics, short‐bowel syndrome
Clinical Relevancy Statement
Teduglutide, a glucagon‐like peptide‐2 analogue, reduces the need for parenteral support in children with short‐bowel syndrome–associated intestinal failure (SBS–IF). Safety data were pooled from 4 clinical trials of teduglutide in children with SBS–IF, including a median of 83 weeks of prospective follow‐up. Gastrointestinal events were among the most frequently reported adverse events, consistent with both the underlying disease state and the known multiple mechanisms of action of teduglutide. Teduglutide was well tolerated, and the safety profile was aligned with previous experience in adults and children with SBS–IF. No new safety concerns were identified.
Introduction
Short‐bowel syndrome (SBS) is a malabsorption disorder caused by loss of functional intestinal mass. 1 In cases of SBS‐associated intestinal failure (SBS–IF), parenteral support (PS; parenteral nutrition and/or intravenous fluids) is required to ensure sufficient fluids and nutrition to sustain adequate growth in children. 2 , 3 PS helps meet critical nutrition and fluid needs but carries risks of potentially serious complications, including liver disease, catheter‐related bloodstream infections, central venous thrombosis, and metabolic bone disease. 2
Teduglutide is an analogue of glucagon‐like peptide‐2 (GLP‐2), a key mediator of the physiologic adaptive response to intestinal resection. 4 , 5 Teduglutide is approved in North America and Europe for the treatment of patients ≥1 year of age with SBS–IF, at a dose of 0.05 mg/kg administered subcutaneously once daily 6 , 7 ; it is complementary to PS in the intestinal rehabilitation of patients with SBS–IF. The efficacy and safety of teduglutide for the treatment of pediatric patients have been evaluated in 2 phase 3 controlled studies. 8 , 9 Trends for reductions in PS volume and calories and increases in enteral nutrition volume and calories were observed in pediatric patients receiving 0.025 or 0.05 mg/kg teduglutide once daily.
Published safety data regarding the use of teduglutide in the pediatric population are limited and based on results of the 2 short‐term (≤24 weeks), phase 3 core studies. 8 , 9 To address the information gap regarding the safety of teduglutide in children with SBS–IF, including teduglutide use beyond 24 weeks, these results were pooled with interim safety data from the 2 ongoing, long‐term extension studies of teduglutide in pediatric patients to evaluate patient safety over a longer drug‐exposure period.
Methods
The pooled safety data of teduglutide‐treated pediatric patients with SBS–IF were analyzed from 4 clinical studies: 2 completed core studies and interim data from 2 ongoing extension studies. The 2 initial core studies were phase 3 clinical trials of teduglutide that enrolled children aged 1–17 years with PS‐dependent SBS (12‐week study: NCT01952080, EudraCT 2013‐004588‐30; 24‐week study: NCT02682381, EudraCT 2015‐002252‐27). The 12‐week study was an open‐label study in which patients sequentially enrolled to receive standard of care (SOC) or teduglutide (0.0125, 0.025, or 0.05 mg/kg once daily). 8 The 24‐week study included a nonblinded SOC arm and 2 randomized, double‐blind teduglutide dose groups (0.025 and 0.05 mg/kg once daily). 9 The study designs of both core studies have been previously published separately. 8 , 9
The 2 ongoing studies NCT02949362 (EudraCT 2016‐000863‐17) and NCT02954458 (EudraCT 2016‐000849‐30) are the open‐label extensions of the 12‐ and 24‐week trials, respectively. Patients were eligible for inclusion in the extension studies if they completed the 12‐week or 24‐week core study in either the teduglutide or SOC arm. The eligibility criteria for the initiation of treatment with teduglutide in the extension studies are reported in Table S1. All patients who enrolled in the extension study following the 12‐week core study experienced a gap of 2.4–3.3 years between studies due to a lag in study setup; patient safety data from this interstudy gap period were not included in this analysis. There was no gap in follow‐up between the 24‐week study and the respective extension study. The data cutoff for the interim analysis of prospective safety data included here was July 24, 2018.
In the ongoing extension studies, teduglutide is provided to children with SBS–IF in treatment cycles consisting of 24 weeks of 0.05 mg/kg teduglutide once daily followed by a 4‐week follow‐up period (Figure S1). After a teduglutide treatment period, teduglutide is only reinitiated if a patient's PS plateaus or deteriorates. At the end of the 4‐week follow‐up, patients who have not reinitiated teduglutide can enter a “no‐teduglutide treatment” period of observation with safety and clinical data collection approximately every 12 weeks. Patients can be reevaluated at any time for reinitiation of teduglutide treatment. Because the clinical course determines the need for additional treatment, the length of no‐teduglutide treatment periods between treatment cycles varies.
All study protocols have been or are being conducted in accordance with the International Council for Harmonisation Good Clinical Practice guidelines and the World Medical Association Declaration of Helsinki and its amendments concerning medical research in humans. Written informed consent was provided by parents, guardians, or patients before study participation.
Study Outcomes
Outcomes assessed were study drug exposure, adverse events (AEs), vital signs, growth trajectories, laboratory findings, and occurrence of anti‐teduglutide antibodies. Compliance with teduglutide treatment was monitored by counting used/unused vials, reviewing patient diaries, and conducting interviews with caregivers and was assessed as the percentage of planned doses administered. Compliance was calculated as 100 × (doses administered/number of days receiving treatment). Patients were considered compliant with treatment if overall compliance was ≥80%. AEs reported were any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease, regardless of whether the event was considered by the clinical study investigator to be related to the study drug. The analysis includes treatment‐emergent AEs, which were defined as AEs that occurred or worsened after initiation of teduglutide.
Serious AEs (SAEs) were defined as any medical event that required inpatient hospitalization or prolongation of hospitalization, resulted in persistent or significant disability, was life‐threatening, resulted in death, or was judged by the investigator as an important medical event. Polyps of the intestine and neoplasia of any kind were considered AEs of special interest for this analysis. 6 , 7 AEs were assessed and reported by the investigator and coded using preferred terms from the Medical Dictionary for Regulatory Activities, version 19.1.
AEs, vital‐sign assessments, weight, and laboratory test results were collected at each study visit. Height was measured at baseline and at intervals of up to 12 weeks. In the core studies, colonoscopies were performed at screening for all children ≥12 years of age and for children <12 years of age if there was a positive fecal occult blood test (FOBT) with an unidentified cause. In the extension studies, patients were required to undergo colonoscopy after the equivalent of 48 weeks of teduglutide exposure. Patients received an FOBT at the end of treatment in the 12‐week core study, at weeks 12 and 24 in the 24‐week core study, at weeks 12 and 24 of each teduglutide treatment cycle in the extension studies, and annually during no‐teduglutide treatment periods of the extension studies for patients who were not teduglutide naive. Follow‐up colonoscopies were carried out for patients with positive FOBTs if clinically warranted. Serum samples were drawn and analyzed for the presence of anti‐teduglutide and neutralizing anti‐teduglutide antibodies, as described in the Supplementary Methods.
Analyses
AEs were pooled and summarized using descriptive statistics. AEs that occurred during no‐teduglutide treatment periods between treatment cycles in the extension studies were included in the analysis. For colonoscopy, growth parameter, and antibody analyses, data from the core and extension studies were pooled and analyzed according to dosing week based on the nominal study visit. In these analyses, baseline was defined as the start of teduglutide treatment.
Results
Patients
This analysis included 89 patients who were treated with teduglutide at some point in the pediatric SBS–IF clinical study program (Figure 1). Eighty‐seven patients started teduglutide treatment in the initial controlled studies; an additional 2 patients who received SOC treatment in the 24‐week core study started teduglutide treatment in the subsequent extension study. Eighteen of the 89 patients included in this analysis received teduglutide in the extension to the 12‐week core study and thus experienced a gap between studies, as described in the Methods. Patient demographics and baseline characteristics are reported in Table 1. Mean growth parameters indicated below‐average weight and height among study participants at baseline. Median (range) duration of teduglutide exposure was 51.7 (5.0–94.7) weeks (Table S2.). Sixty‐four patients (71.9%) were exposed to teduglutide for >24 weeks. Median (range) duration of patient follow‐up (including the teduglutide‐treatment and no‐teduglutide treatment period[s]) was 83.0 (8.3–161.3) weeks. Mean ± SD (range) of the “no‐teduglutide treatment period” between teduglutide treatment cycles 1 and 2 in the extension studies was 2.5 ± 5.40 (0–27.9) weeks (n = 48). All patients were treatment compliant; mean (SD) compliance was 98.8% (4.42%).
Figure 1.
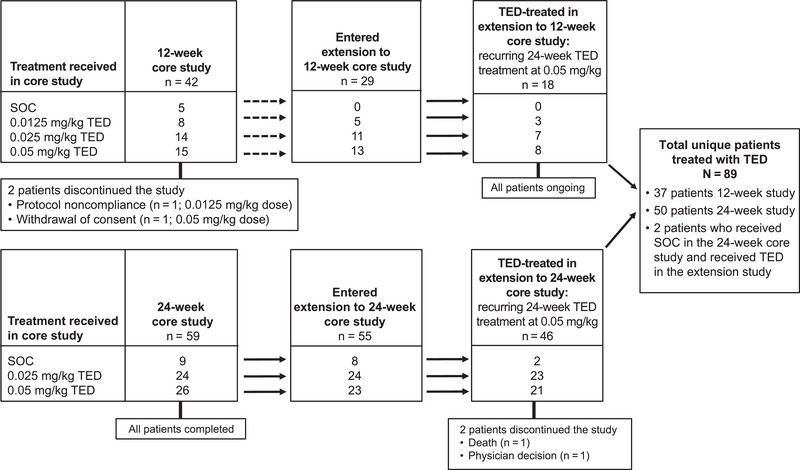
Patient disposition in the teduglutide (TED) pediatric core and extension clinical trials. TED was provided as a once‐daily subcutaneous injection. Dotted arrows represent a gap between the 12‐week core study and the subsequent extension study. SOC, standard of care.
Table 1.
Patient Demographics and Baseline Characteristics
| Parameter | N = 89 |
|---|---|
| Age, mean (SD), years | 5.6 (3.64) |
| Median (min, max), years | 5.0 (1, 15) |
| Age group, n (%), years | |
| 1 to <12 | 82 (92.1) |
| Infants, <2 | 5 (5.6) |
| Children, 2 to <12 | 77 (86.5) |
| 12 to <18 | 7 (7.9) |
| Sex, n (%) | |
| Boys | 61 (68.5) |
| Race, n (%) | |
| White | 67 (75.3) |
| Black or African American | 10 (11.2) |
| Asian | 3 (3.4) |
| Other | 3 (3.4) |
| Not available | 6 (6.7) |
| Growth parameter at baseline | |
| Weight z‐score,a mean (SD) | –0.8 (1.02) |
| Median (min, max) | –0.8 (–3.4, 1.0) |
| Height z‐score,a mean (SD) | –1.2 (1.18) |
| Median (min, max) | –1.0 (–4.3, 1.9) |
| BMI z‐score,a mean (SD) | –0.03 (1.023) |
| Median (min, max) | –0.05 (–3.6, 2.4) |
| Primary reason for SBS diagnosis, n (%) | |
| Gastroschisis | 30 (33.7) |
| Midgut volvulus | 25 (28.1) |
| Necrotizing enterocolitis | 15 (16.9) |
| Intestinal atresia | 10 (11.2) |
| Hirschsprung disease | 7 (7.9) |
| Multiple | 2 (2.2) |
| Patients with a stoma, n (%) | 17 (19.1) |
| Type of stoma | |
| Jejunostomy | 11 (12.4) |
| Ileostomy | 4 (4.5) |
| Colostomy | 2 (2.2) |
| Total remaining small‐intestine length, mean (SD),b cm | 45.9 (33.94) |
| Min, max | 0, 147.0 |
| Distal/terminal ileum present, n (%)c | 27 (31.4) |
| Ileocecal valve present, n (%)d | 22 (25.6) |
| Patients with remaining colon, n (%) | 82 (92.1) |
| Estimated percentage of colon remaining, mean (SD)e | 66.9 (32.99) |
| Colon in continuity, n (%) | 76 (85.4) |
| Colon present but not in continuity, n (%) | 6 (6.7) |
| Duration of prior PS dependence, mean (SD), years | 5.0 (3.2) |
| Baseline PS volume requirements, mean (SD),f,g mL/kg/day | 62.7 (27.77) |
| Baseline PS calories, mean (SD),f,h kcal/kg/day | 45.1 (18.41) |
| Baseline days per week of PS infusion, mean (SD)f,i | 6.7 (0.82) |
| Baseline hours per day of PS infusion, mean (SD)f,i | 12.0 (3.37) |
BMI, body mass index; max, maximum; min, minimum; PS, parenteral support; SBS, short‐bowel syndrome.
Computer programs. Centers for Disease Control and Prevention. May 16, 2014. Accessed November 20, 2020. https://www.cdc.gov/growthcharts/computer_programs.htm
n = 80.
n = 86.
Percentage based on total number of patients with data available on presence of distal/terminal ileum (n = 86).
n = 70.
Based on patients’ diary data. PS volume and PS calories were calculated on a weekly basis and divided by the number of days (ie, 7) to provide values in mL/kg/day or kcal/kg/day.
n = 82.
n = 76.
n = 85.
Adverse Events
AEs were reported in all study patients (Table 2). The most commonly reported AEs were vomiting in 51.7% of patients, pyrexia in 43.8%, upper respiratory tract infection in 41.6%, cough in 33.7%, and device‐related (central venous catheter) infection in 29.2% (Table 3). AEs considered related to teduglutide treatment by the investigator were reported in 35 patients (39.3%; Table 2); the most common were abdominal pain and vomiting (5.6% each). In addition, 27 events of injection‐site reactions and injuries in 13 patients (14.6%) were reported to be related to teduglutide treatment; all of these events were mild and nonserious. The most common related AEs associated with injection‐site administration occurring in ≥2 patients were injection‐site bruising, pain, and swelling (3 patients each) and injection‐site hemorrhage and injection‐site reaction (2 patients each). All other injection‐site–related events were reported in only 1 patient.
Table 2.
Overall Summary of AEs
| N = 89 | ||
|---|---|---|
| Parameter | n (%) | Number of events |
| Any AE | 89 (100.0) | 1717 |
| Leading to treatment discontinuation | 2 (2.2) | 2 |
| Deatha | 1 (1.1) | 1 |
| AE severity b | ||
| Mild | 17 (19.1) | |
| Moderate | 36 (40.4) | |
| Severe | 36 (40.4) | |
| AE relationshipc | ||
| Not related | 89 (100.0) | 1605 |
| Related | 35 (39.3) | 112 |
| Any SAE | 69 (77.5) | 254 |
| SAE relationshipc | ||
| Not related | 69 (77.5) | 251 |
| Related | 3 (3.4) | 3 |
AE, adverse event; SAE, serious AE.
Teduglutide treatment was discontinued, and the family electively withdrew enteral and parenteral fluid and nutrition support; death was considered by the investigator to be unrelated to teduglutide treatment.
The medical assessment of severity was determined by using the following definitions. Mild: a type of AE that is usually transient and may require only minimal treatment or therapeutic intervention. The event does not generally interfere with usual activities of daily living. Moderate: a type of AE that is usually alleviated with specific therapeutic intervention. The event interferes with usual activities of daily living, causing discomfort but posing no significant or permanent risk of harm to the research participant. Severe: a type of AE that interrupts usual activities of daily living or significantly affects clinical status or may require intensive therapeutic intervention.
An individual patient may have had both an AE that was related to teduglutide and a separate AE that was not related to teduglutide.
Table 3.
AEs Occurring in ≥5.0% of Patients
| N = 89 | ||
|---|---|---|
| Parameter (preferred terms) | n (%) | Number of events |
| Any AE | 89 (100.0) | 1717 |
| Vomiting | 46 (51.7) | 145 |
| Pyrexia | 39 (43.8) | 67 |
| Upper respiratory tract infection | 37 (41.6) | 62 |
| Cough | 30 (33.7) | 50 |
| Device‐related infectiona | 26 (29.2) | 41 |
| Abdominal pain | 23 (25.8) | 65 |
| Diarrhea | 23 (25.8) | 40 |
| Headache | 18 (20.2) | 43 |
| Nasopharyngitis | 18 (20.2) | 27 |
| Viral infection | 18 (20.2) | 27 |
| Alanine aminotransferase increased | 18 (20.2) | 24 |
| Nausea | 15 (16.9) | 25 |
| Rash | 15 (16.9) | 22 |
| Influenza | 14 (15.7) | 16 |
| Dehydration | 13 (14.6) | 23 |
| C‐reactive protein increased | 13 (14.6) | 17 |
| Device breakagea | 13 (14.6) | 16 |
| Abdominal pain upper | 12 (13.5) | 21 |
| Blood bicarbonate decreased | 12 (13.5) | 15 |
| Abdominal distension | 11 (12.4) | 13 |
| Device occlusiona | 10 (11.2) | 18 |
| Fatigue | 10 (11.2) | 18 |
| Rhinorrhea | 10 (11.2) | 16 |
| Rhinitis | 9 (10.1) | 12 |
| Gastroenteritis viral | 9 (10.1) | 10 |
| Device dislocationa | 9 (10.1) | 9 |
| Aspartate aminotransferase increased | 8 (9.0) | 10 |
| Nasal congestion | 8 (9.0) | 10 |
| Anemia | 7 (7.9) | 13 |
| Oropharyngeal pain | 7 (7.9) | 8 |
| Flatulence | 7 (7.9) | 8 |
| Hematochezia | 7 (7.9) | 8 |
| Ear infection | 7 (7.9) | 8 |
| Lymphadenopathy | 7 (7.9) | 7 |
| Epistaxis | 6 (6.7) | 9 |
| γ‐Glutamyl transferase increased | 6 (6.7) | 7 |
| White blood cell–positive urine | 6 (6.7) | 7 |
| Acidosis | 6 (6.7) | 7 |
| Pain in extremity | 6 (6.7) | 7 |
| Decreased appetite | 6 (6.7) | 7 |
| Hemoglobin decreased | 6 (6.7) | 6 |
| Urinary tract infection | 5 (5.6) | 14 |
| Metabolic acidosis | 5 (5.6) | 13 |
| Gastrointestinal bacterial overgrowth | 5 (5.6) | 13 |
| Device malfunctiona | 5 (5.6) | 9 |
| Gastrostomy tube site complication | 5 (5.6) | 8 |
| Injection‐site bruising | 5 (5.6) | 8 |
| Respiratory tract infection | 5 (5.6) | 7 |
| Dizziness | 5 (5.6) | 6 |
| Ear pain | 5 (5.6) | 6 |
| Otitis media | 5 (5.6) | 6 |
| Weight decreased | 5 (5.6) | 6 |
| Constipation | 5 (5.6) | 5 |
| Fecal volume increased | 5 (5.6) | 5 |
| Device‐related sepsisa | 5 (5.6) | 5 |
| Hypoglycemia | 5 (5.6) | 5 |
AE, adverse event.
aAll device‐related events were related to central venous catheters used to administer parenteral support, not to the teduglutide injection device.
SAEs were reported in 69 of the 89 patients (77.5%; Table 4). SAEs considered related to teduglutide by the investigator occurred in 3 patients; all 3 events resolved.
An 8‐year‐old girl with SBS due to necrotizing enterocolitis experienced an event of adynamic ileus of moderate severity ∼4 months after starting teduglutide. This patient had a history of intestinal dysmotility before teduglutide treatment and had initially failed screening for study entry because of an event of ileus. The patient was subsequently admitted to the study after a rescreening; no small‐bowel obstruction was noted on abdominal radiography. Teduglutide was interrupted, and oral nutrition and fluids were stopped. The event resolved 2 days later. No further events of ileus occurred during an additional 14.7 months of teduglutide treatment.
A 4‐year‐old boy with a history of small‐intestinal bacterial overgrowth had 2 events of d‐lactic acidosis assessed as severe in intensity ∼6 months apart. Both events occurred shortly after an interruption in teduglutide treatment. The investigator reported the first event as unrelated and the second event as related to teduglutide treatment. Both events were assessed by the sponsor as likely related to underlying disease and unlikely to be related to teduglutide treatment. The event resolved after interruption of teduglutide and initiation of treatment with sodium bicarbonate, antibiotics, and intravenous fluids; teduglutide treatment was subsequently reinitiated. This patient received an additional 5.7 months of teduglutide treatment after the second event.
A 6‐year‐old boy was reported to have an event of fecal impaction (fecaloma) after 4.7 months of teduglutide treatment. Teduglutide was interrupted, and the patient passed stool spontaneously without further intervention. The event was later reclassified as gastrointestinal obstruction due to hard stools. This patient received an additional 26 days of teduglutide treatment after the event.
Table 4.
SAEs Occurring in ≥5.0% of Patients
| N = 89 | ||
|---|---|---|
| Parameter (preferred terms) | n (%) | Number of events |
| Any SAE | 69 (77.5) | 254 |
| Pyrexia | 25 (28.1) | 36 |
| Device‐related infectiona | 24 (27.0) | 36 |
| Influenza | 9 (10.1) | 9 |
| Device breakagea | 8 (9.0) | 9 |
| Dehydration | 7 (7.9) | 12 |
| Upper respiratory tract infection | 6 (6.7) | 6 |
SAE, serious adverse event.
aAll device‐related events were related to central venous catheters used to administer parenteral support, not to the teduglutide injection device.
During the 4 studies, a 16‐year‐old boy with cerebral palsy and chronic pain due to hip subluxation died. In the context of severe comorbid conditions and lack of response to teduglutide, the goals of care changed for this patient. Teduglutide treatment was discontinued. The family electively withdrew enteral and parenteral fluid and nutrition support. The event was considered unrelated to teduglutide treatment by the investigator.
Two patients (2.2%) discontinued teduglutide treatment because of an AE; neither event was considered related to teduglutide. One patient with a history of cholestasis and baseline elevations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) discontinued teduglutide because of an SAE of a cholestatic liver injury pattern. The patient experienced elevated bilirubin, followed by increases in ALT and AST. The event was assessed by the investigator as related to iatrogenic iron overload; despite elevated baseline iron stores, the patient had received ∼20 months of treatment with intravenous iron sucrose infusions (15 mg/day, 6 days/week). A second patient discontinued teduglutide because of an SAE of enterocutaneous fistula. The fistula developed as a complication of an elective open gastrostomy site change.
Posttreatment colonoscopies or sigmoidoscopies of the remnant colon were performed on 34 of 82 study patients with remaining colon (41.5%). Among these 34 patients, the median cumulative treatment duration was 11.0 months (range, 0.5–16.7). One cecal polyp was identified in 1 patient, which was not biopsied; no polyps were found on a follow‐up colonoscopy performed 2 months later. No AEs of neoplasia were reported in the combined clinical trials.
Vital Signs, Growth Parameters, and Laboratory Findings
No clinically meaningful changes in vital‐sign measures or urinalysis analytes were observed during the teduglutide clinical studies. Change from baseline in growth parameters is shown in Figure S2. Weight, height, and body mass index z‐scores did not change substantially during long‐term treatment with teduglutide. Abnormal serum chemistry and hematology findings are reported in Table S3. Laboratory abnormalities that occurred in >5.0% of the patients were ALT >8 × upper limit of normal (ULN; 13.5%), lipase >3 × ULN (13.5%), phosphorus >2.254 mmol/L (6.7%), and blood urea nitrogen >12.495 mmol/L (5.6%). Except for single events of increased ALT and increased transaminases, these findings were not regarded as related to teduglutide.
Antibodies to Teduglutide
The percentage of patients with anti‐teduglutide antibodies increased from 1.1% at baseline to 33.3% at week 36 (Table 5) and plateaued thereafter. Between week 36 and week 72, the percentage of patients with anti‐teduglutide antibodies remained at or below 34.8%. Neutralizing antibodies have the theoretical potential to reduce drug activity; neutralizing antibodies to teduglutide were detected in a low number of patients overall. Between baseline and week 72, the highest percentages of patients with neutralizing antibodies were observed at weeks 60 (6.9%) and 72 (10.0%).
Table 5.
Summary of Anti‐Teduglutide Antibodies
| Time point | n (%) |
|---|---|
| Baseline | N = 89 |
| Negative | 88 (98.8) |
| Positive | 1 (1.1) |
| Neutralizing antibodies present | 0 |
| Week 12 | n = 37 |
| Negative | 37 (100.0) |
| Positive | 0 |
| Neutralizing antibodies present | 0 |
| Week 24 | n = 66 |
| Negative | 56 (84.8) |
| Positive | 10 (15.2) |
| Neutralizing antibodies present | 4 (6.1) |
| Week 36 | n = 57 |
| Negative | 38 (66.7) |
| Positive | 19 (33.3) |
| Neutralizing antibodies present | 0 |
| Week 48 | n = 46 |
| Negative | 30 (65.2) |
| Positive | 16 (34.8) |
| Neutralizing antibodies present | 2 (4.3) |
| Week 60 | n = 29 |
| Negative | 24 (82.8) |
| Positive | 5 (17.2) |
| Neutralizing antibodies present | 2 (6.9) |
| Week 72 | n = 10 |
| Negative | 7 (70.0) |
| Positive | 3 (30.0) |
| Neutralizing antibodies present | 1 (10.0) |
| Week 84 | n = 3 |
| Negative | 2 (66.7) |
| Positive | 1 (33.3) |
| Neutralizing antibodies present | 1 (33.3) |
Discussion
The safety data presented here are based on a median of 52 weeks of teduglutide treatment and 83 weeks of prospective follow‐up of children with SBS–IF treated with teduglutide, building on the previously published 12‐ and 24‐week pediatric core studies. No new safety risks were identified. The spectrum of gastrointestinal AEs in teduglutide‐treated patients was generally comparable to that occurring in patients in the SOC arms of the phase 3 pediatric studies. 8 , 9 The spectrum of AEs was also similar to that reported in a recent integrated analysis of safety data from the adult clinical studies of teduglutide. 10 Compared with the adult studies, upper respiratory AEs (cough, upper respiratory tract infection, nasopharyngitis, rhinorrhea, rhinitis, nasal congestion), pyrexia, vomiting, and catheter complications (device breakage, occlusion, and dislocation) were more common in the pediatric studies, as might be expected in a younger population. 11
The most commonly reported SAE in the pooled clinical studies was pyrexia (36 events, 28.1% of patients). Because of their vulnerability to potentially life‐threatening catheter‐associated bloodstream infections, children with SBS–IF who develop a fever are typically hospitalized for treatment with intravenous antibiotics while awaiting blood culture results. 12 Thus, many events of fever in these studies were SAEs. None of the events of pyrexia were deemed by the investigators or sponsor to be related to teduglutide. As expected for this patient population, catheter‐related infection was the second most frequent SAE in this analysis. Therapies that enhance intestinal adaptation and reduce PS dependence would be expected to reduce the risk of complications in these patients, particularly if they lead to earlier enteral autonomy and permit removal of the central venous catheter. 13 The sample size and duration of observation in this data set is insufficient to determine whether long‐term treatment with teduglutide reduces the rate of catheter‐related infections in children with SBS–IF.
The most commonly reported AEs assessed by the investigator as related to teduglutide treatment were vomiting and abdominal pain; none of these events resulted in discontinuation of teduglutide. Two of the 3 serious and related AEs may have been associated with effects on gastrointestinal motility. Patients with signs of dysmotility require close observation during treatment with teduglutide because of the potential for GLP‐2 analogues to increase gastrointestinal transit time. 14 The serious and related AE of gastrointestinal obstruction due to hard stool resolved spontaneously after interruption of teduglutide, and the event did not recur after resumption of teduglutide treatment. Increased fluid absorption as a result of teduglutide treatment may have contributed to hard stools in this case. The third serious and related event, d‐lactic acidosis, occurred twice in a patient with a history of gastrointestinal bacterial overgrowth, a known complication of SBS, shortly after interruptions in teduglutide treatment. 3 Patients should be closely monitored for changes in motility and intestinal absorptive function after treatment discontinuation and during treatment (re)initiation.
Elevations in aminotransferases were observed in some pediatric patients. Markedly elevated ALT (>8 × ULN) occurred in 13.5% of patients, and increased ALT, AST, and γ‐glutamyl transferase were reported as treatment‐emergent AEs for 20.2%, 9.0%, and 6.7% of patients, respectively. These rates are similar to those reported in a recent cohort of 148 children with SBS–IF who were not treated with teduglutide, of whom 19% had abnormal liver function tests. 15 Because aminotransferase levels tend to be increased in patients with SBS, patients with baseline values of AST and ALT up to 5 × ULN (12‐week core study) or 7 × ULN (24‐week core study) were permitted to enroll in the teduglutide clinical core studies. In the current analysis, all patients with abnormal liver function tests were able to remain on teduglutide treatment, except for 1 patient who discontinued teduglutide because of an SAE of a cholestatic liver injury pattern that was attributed to inadvertent iron overload unrelated to teduglutide treatment. Laboratory assessments, including bilirubin, alkaline phosphatase, lipase, and amylase to screen for biliary, gallbladder, and pancreatic disease, are recommended before initiating teduglutide and every 6 months thereafter.
With long‐term follow‐up, the prevalence of anti‐teduglutide antibodies plateaued at ∼35%, which was similar to that observed in adults. 16 In an analysis of the pediatric core studies, development of anti‐teduglutide antibodies was not associated with events of hypersensitivity (data not shown). Currently, it is unclear whether the development of anti‐drug antibodies has an impact on maintenance of efficacy, because of the limited duration of observation among the relatively small number of patients who developed anti‐drug antibodies. A very small number of patients developed neutralizing antibodies. Longer‐term treatment in larger populations will be necessary to assess the full significance of antibodies and of neutralizing antibodies in patients treated with teduglutide.
Because of the intestinotrophic mechanism of action of teduglutide and reports of an increased incidence of dysplastic changes and neoplasms in rodent models exposed to exogenous GLP‐2, 17 events of intestinal polyp growth and neoplasia during teduglutide treatment are of special interest. In the pooled clinical studies, only 1 polyp, a cecal polyp, was detected, which was not biopsied or confirmed on repeat colonoscopy. No cases of cancer or neoplasia were identified. In the Study of Teduglutide Effectiveness in Parenteral Nutrition‐Dependent Short Bowel Sydrome Subjects (STEPS) phase 3 clinical study series of adults treated with teduglutide for up to 3.5 years, polyps were reported in 9 of 50 patients with colon in continuity who underwent postexposure colonoscopy. 18 Seven of these polyps had histology available; none were malignant or showed high‐grade dysplasia. By contrast, polyps were rare in this data set, even though the gut is inherently primed to grow in children. The final results from the ongoing extension studies will further characterize the long‐term safety of teduglutide treatment in pediatric patients with SBS–IF.
To facilitate the identification of colonic polyps, both the US Food and Drug Administration (FDA)– and European Medicines Agency (EMA)–approved prescriber information for teduglutide recommend FOBT in children prior to starting teduglutide, with colonoscopy/sigmoidoscopy if there is unexplained blood in the stool. 6 , 7 The guidance also recommends subsequent fecal occult blood testing annually in children and adolescents while they are receiving teduglutide and colonoscopy/sigmoidoscopy if they have new or unexplained gastrointestinal bleeding. 6 , 7 A positive FOBT can result from a variety of causes in patients with SBS, including gastritis or duodenitis, presence of stoma, friable granulation tissue at gastrostomy sites, anastomotic ulcers, diversion colitis, allergic colitis, ingestion of red meat or raw vegetables, and perianal erosions in children who wear diapers. Whereas FOBT has relatively low sensitivity and specificity for identification of polyps in children, 19 investigation of unexplained lower‐gastrointestinal bleeding via colonoscopy is SOC in children, 20 and identification of the cause of gastrointestinal bleeding is likely to impact medical care. Several patients in these studies had a positive FOBT, although none were due to polyps.
This analysis of the 4 clinical studies was limited by the long‐term, open‐label treatment period that lacks control‐group comparisons. In addition to the overall small sample size, a subset of the analysis population (n = 18) had a treatment gap of up to 3.3 years that occurred between the initial 12‐week core study and enrollment in the extension to that study. Five patients received additional teduglutide prescribed by their physicians during this retrospective observation period; data from this retrospective observation period were not included in this analysis.
In this pooled analysis of 4 clinical studies evaluating pediatric patients with SBS–IF treated with teduglutide, no new safety concerns were identified. In addition, all study patients were ≥80% treatment compliant, indicating that teduglutide was well tolerated. The extension studies are ongoing, and additional safety data over longer exposure times will be forthcoming. As with all clinical studies, patients in these trials were closely monitored, and real‐world experience with teduglutide in pediatric patients may not be identical to results reported here. Continued postmarketing safety assessments of teduglutide are underway to further understand its long‐term safety profile in the treatment of children with SBS–IF.
Statement of Authorship
A. A. Grimm and M. Yoon contributed to the conception and design of the work; S. Hill, B. A. Carter, V. Cohran, S. Horslen, S. S. Kaufman, S. A. Kocoshis, D. F. Mercer, R. J. Merritt, M. P. Pakarinen, S. Protheroe, J. F. Thompson, C. P. B. Vanderpool, R. S. Venick, and P. W. Wales contributed to the acquisition of the data; S. Hill, B. A. Carter, V. Cohran, S. Horslen, S. S. Kaufman, S. A. Kocoshis, D. F. Mercer, R. J. Merritt, M. P. Pakarinen, S. Protheroe, J. F. Thompson, C. P. B. Vanderpool, R. S. Venick, P. W. Wales, S. E. Smith, M. Yoon, and A. A. Grimm contributed to the analysis and interpretation of the data and the drafting of the manuscript. All authors critically revised the manuscript, agree to be fully accountable for ensuring the integrity and accuracy of the work, and read and approved the final manuscript.
Supporting information
Supplementary Methods, Tables S1–S3, and Figures S1–S2 are available online at http:/pen.sagepub.com
Acknowledgments
We thank the patients and their families who participated in the studies included in this analysis. We also thank the clinical study coordinators at the participating study centers.
Data availability statement: The data sets, including individual participant data behind the results reported in this article, will be available 3 months from initial request to researchers who provide a methodologically sound proposal. The data will be provided after deidentification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data requests should follow the process outlined in the Data Sharing section on http://takedaclinicaltrials.com.
Financial disclosure: Shire is a Takeda company. This analysis was funded by Shire Human Genetic Therapies, Inc, Lexington, Massachussetts, USA. The sponsor of the study participated in the design of the included studies, data collection, data analysis, data interpretation, and review and approval of the final clinical study and integrated safety reports and provided the study drug. Under the direction of the authors, editorial support was provided by Heather Heerssen, PhD, ICON plc, North Wales, Pennsylvania, USA, and was funded by Shire International GmbH, a Takeda company, Zurich, Switzerland.
Conflicts of interest: S. Hill served as a consultant/speaker for and received research support from Shire and served as an advisory board member for Baxter. B. A. Carter received research support from Shire and served as a consultant for Fresenius Kabi. V. Cohran received research support from Shire and served on the speakers bureau for Abbott Nutrition. S. Horslen, D. F. Mercer, C. P. B. Vanderpool, and R. S. Venick received research support from Shire. S. S. Kaufman received research support from and owns equity in Takeda. S. A. Kocoshis received research support from Shire and served as a speaker for Abbott Nutrition. R. J. Merritt, S. Protheroe, J. F. Thompson, and P. W. Wales served as consultants and/or speakers for and received research support from Shire. M. P. Pakarinen served as consultant and/or speaker for and received research support from Shire and served as an advisory board member for Mirum Pharmaceuticals. M. Yoon is an employee of Shire Human Genetic Therapies, Inc, a Takeda company. A. A. Grimm (current affiliation: Ultragenyx Pharmaceuticals, Inc, Novato, California, USA) and S. E. Smith (current affiliation: Kaleido Biosciences, Lexington, Massachussetts, USA) are former employees of Shire Human Genetic Therapies, Inc, a Takeda company.
This and other JPEN podcasts are available at https://nutritioncare.org/podcasts
References
- 1. Wales PW, Christison‐Lagay ER. Short bowel syndrome: epidemiology and etiology. Semin Pediatr Surg. 2010;19(1):3‐9. [DOI] [PubMed] [Google Scholar]
- 2. Mutanen A, Wales PW. Etiology and prognosis of pediatric short bowel syndrome. Semin Pediatr Surg. 2018;27(4):209‐217. [DOI] [PubMed] [Google Scholar]
- 3. Goulet O, Abi Nader E, Pigneur B, Lambe C. Short bowel syndrome as the leading cause of intestinal failure in early life: some insights into the management. Pediatr Gastroenterol Hepatol Nutr. 2019;22(4):303‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drucker DJ, Erlich P, Asa SL, Brubaker PL. Induction of intestinal epithelial proliferation by glucagon‐like peptide 2. Proc Natl Acad Sci U S A. 1996;93(15):7911‐7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Drucker DJ. Gut adaptation and the glucagon‐like peptides. Gut. 2002;50(3):428‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. GATTEX (teduglutide) . Full prescribing information. Shire‐NPS Pharmaceuticals, Inc; 2019.
- 7. Revestive (teduglutide) . Full prescribing information. Shire Pharmaceuticals Ireland Limited; 2019.
- 8. Carter BA, Cohran VC, Cole CR, et al. Outcomes from a 12‐week, open‐label, multicenter clinical trial of teduglutide in pediatric short bowel syndrome. J Pediatr. 2017;181:102‐11.e105. [DOI] [PubMed] [Google Scholar]
- 9. Kocoshis SA, Merritt RJ, Hill S, et al. Safety and efficacy of teduglutide in pediatric patients with intestinal failure due to short bowel syndrome: a 24‐week, phase III study. JPEN J Parenter Enteral Nutr. 2020;44(4):621‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pape U‐F, Iyer KR, Jeppesen PB, et al. Teduglutide for the treatment of adults with intestinal failure associated with short bowel syndrome: pooled safety data from four clinical trials. Therap Adv Gastroenterol. 2020;13:1756284820905766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hendley JO. Epidemiology, pathogenesis, and treatment of the common cold. Semin Pediatr Infect Dis. 1998;9(1):50‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hudgins JD, Goldberg V, Fell GL, Puder M, Eisenberg MA. Reducing time to antibiotics in children with intestinal failure, central venous line, and fever. Pediatrics. 2017;140(5):e20171201. [DOI] [PubMed] [Google Scholar]
- 13. Winkler MF, Smith CE. Clinical, social, and economic impacts of home parenteral nutrition dependence in short bowel syndrome. JPEN J Parenter Enteral Nutr. 2014;38(1 suppl):32S‐7S. [DOI] [PubMed] [Google Scholar]
- 14. Janssen P, Rotondo A, Mule F, Tack J. Review article: a comparison of glucagon‐like peptides 1 and 2. Aliment Pharmacol Ther. 2013;37(1):18‐36. [DOI] [PubMed] [Google Scholar]
- 15. Abi Nader E, Lambe C, Talbotec C, et al. Outcome of home parenteral nutrition in 251 children over a 14‐y period: report of a single center. Am J Clin Nutr. 2016;103(5):1327‐1336. [DOI] [PubMed] [Google Scholar]
- 16. Schwartz LK, O'Keefe SJ, Fujioka K, et al. Long‐term teduglutide for the treatment of patients with intestinal failure associated with short bowel syndrome. Clin Transl Gastroenterol. 2016;7(2):e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trivedi S, Wiber SC, El‐Zimaity HM, Brubaker PL. Glucagon‐like peptide‐2 increases dysplasia in rodent models of colon cancer. Am J Physiol Gastrointest Liver Physiol. 2012;302(8):G840‐G849. [DOI] [PubMed] [Google Scholar]
- 18. Armstrong D, Forbes A, Jeppesen PB, Lee HM, Nagy P, Seidner DL. Colon polyps in patients with short bowel syndrome before and after teduglutide: post hoc analysis of the STEPS study series. Clin Nutr. 2020;39(6):1774‐1777. [DOI] [PubMed] [Google Scholar]
- 19. Thakkar K, Alsarraj A, Fong E, Holub JL, Gilger MA, El Serag HB. Prevalence of colorectal polyps in pediatric colonoscopy. Dig Dis Sci. 2012;57(4):1050‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomson M, Tringali A, Dumonceau JM, et al. Paediatric gastrointestinal endoscopy: European Society for Paediatric Gastroenterology Hepatology and Nutrition and European Society of Gastrointestinal Endoscopy guidelines. J Pediatr Gastroenterol Nutr. 2017;64(1):133‐153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Tables S1–S3, and Figures S1–S2 are available online at http:/pen.sagepub.com