

Abstract
Advances in multiplexed imaging technologies have drastically improved our ability to characterize healthy and diseased tissues at the single-cell level. Co-detection by indexing (CODEX) relies on DNA-conjugated antibodies and the cyclic addition and removal of complementary fluorescently labeled DNA probes and has been used so far to simultaneously visualize up to 60 markers in situ. CODEX enables a deep view into the single-cell spatial relationships in tissues and is intended to spur discovery in developmental biology, disease and therapeutic design. Herein, we provide optimized protocols for conjugating purified antibodies to DNA oligonucleotides, validating the conjugation by CODEX staining and executing the CODEX multicycle imaging procedure for both formalin-fixed, paraffin-embedded (FFPE) and fresh-frozen tissues. In addition, we describe basic image processing and data analysis procedures. We apply this approach to an FFPE human tonsil multicycle experiment. The hands-on experimental time for antibody conjugation is ~4.5 h, validation of DNA-conjugated antibodies with CODEX staining takes ~6.5 h and preparation for a CODEX multicycle experiment takes ~8 h. The multicycle imaging and data analysis time depends on the tissue size, number of markers in the panel and computational complexity.
Introduction
Tissue architecture and cellular organization are vital for physiological processes and are determinative of pathological states such as infections, autoimmune diseases and cancer. Traditional tissue microscopy techniques, including immunohistochemistry (IHC) and immunofluorescence, have been used by pathologists and scientists for decades to study cell types, cell abundances and cell-cell interactions. Although these techniques are a mainstay of clinical diagnostics, they typically assess only one or two protein markers in a given tissue section. In the past 15 years, a number of multiplexed tissue imaging technologies have been developed that are rapidly advancing our ability to identify and spatially profile complex biological systems at the single-cell level1–19.
Co-detection by indexing (CODEX) tissue imaging with DNA-barcoded antibodies
We developed a multiplexed tissue imaging technology termed CODEX. CODEX is a fluorescence microscopy platform based on the detection of DNA-conjugated antibodies1–3 (Fig. 1a). The first iteration of CODEX rendered these antibodies visible by using fluorescent dNTP analogs and DNA polymerase primer extension1. However, the use of enzymes was costly and time consuming and required specialized buffers.
Fig. 1 |. Key components of the CODEX technology.
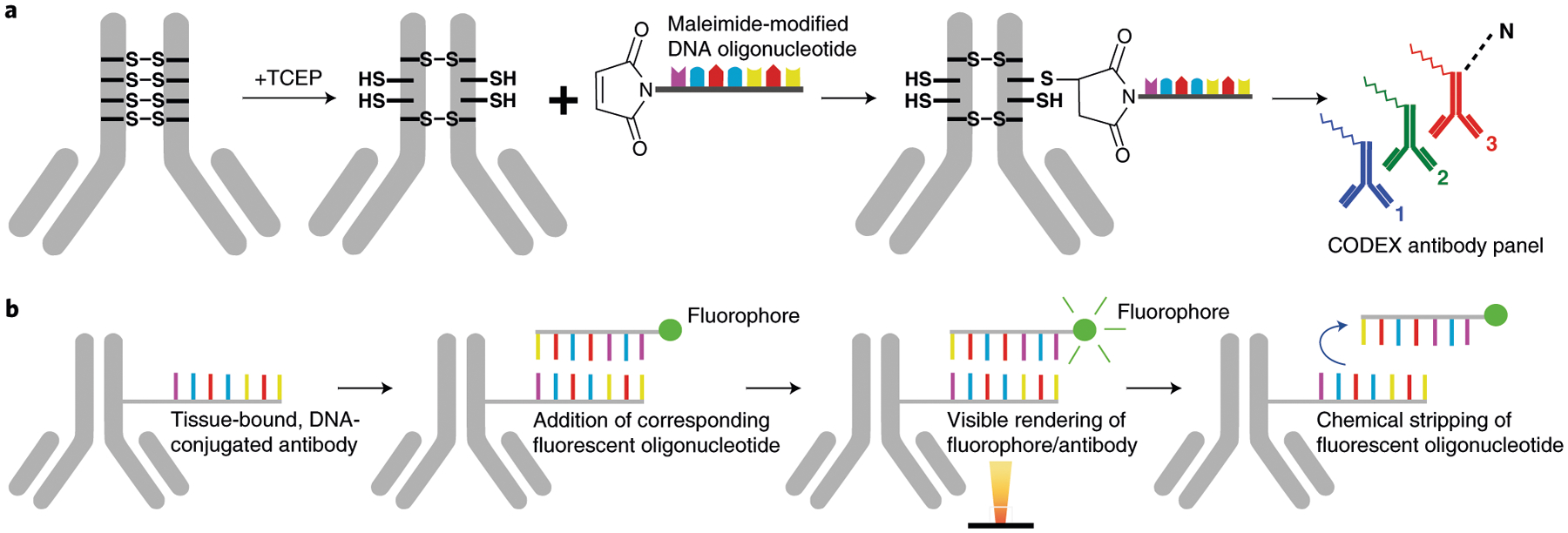
a, Antibody conjugation consists of partially reducing disulfide bonds in the IgG antibody with tris [2-carboxyethyl]phosphine (TCEP) (Steps 7–14), incubating with a unique maleimide-modified DNA oligonucleotide (Steps 16–21) and purifying the DNA-conjugated antibody (Steps 22–32). To create a CODEX antibody panel, unique DNA oligonucleotides are conjugated for up to 57 antibody targets of interest. b, Implementation of CODEX involves staining a tissue section with a unique DNA-conjugated antibody (Steps 71–107), adding the corresponding fluorescent oligonucleotide (Steps 111–115), hybridizing this fluorophore with the conjugated antibody and visualizing it with light microscopy, chemical stripping of the fluorescently tagged oligonucleotide from the tissue and iteratively repeating this process for all antibodies in the panel (Steps 133 and 134).
A newly adapted version of CODEX uses an automated microfluidics system and conventional fluorescent microscope to iteratively hybridize, image and strip fluorescently labeled DNA probes that are complementary to the tissue-bound, DNA-conjugated antibodies2,3 (Fig. 1b). This platform enables a single staining procedure, short run times, simple chemistries and simultaneous visualization, and quantification of up to 60 markers in a single tissue section has been demonstrated. This advance overcame certain limitations of spectral overlap, which can be an issue during multiplexing4, and minimizes batch effects that are observed with other multi-round immunofluorescent staining techniques5–8. The CODEX technology was licensed by Stanford University to Akoya Biosciences (https://www.akoyabio.com/), and the instrumentation and reagents are now commercially available.
Overview of the procedure
The procedure described in this protocol outlines four key sections of the CODEX method: antibody conjugation, antibody validation and titration, the multicycle reaction and subsequent data analysis. Section 1 outlines antibody conjugation, which includes partially reducing the antibody (Steps 7–15), conjugation of this partially reduced antibody with a DNA oligonucleotide (Steps 16–21) and washing and recovering the oligonucleotide-antibody conjugate (Steps 22–32). Section 2 contains the steps required for validating and titrating the antibody-oligonucleotide conjugate. This section details how to prepare the tissue with antigen retrieval (Steps 38–43), stain with the oligonucleotide-antibody conjugate (Steps 44–49), perform a post-stain fixation (Steps 50–59), hybridize with the corresponding fluorescent oligonucleotide (Steps 60–68) and image the tissue (Steps 69 and 70). Section 3 describes the procedure for setting up a CODEX multicycle reaction, including preparing the microfluidics device (Steps 108–110), mounting the coverslip onto an acrylic plate (Steps 116–132), setting the imaging parameters (Step 133) and performing H&E staining upon completion of fluorescence imaging (Steps 135–139). Section 4 details the imaging processing and data analysis procedures (Steps 140–144).
Comparison with other multiplexed phenotyping methods
Current multiplexed single-cell phenotyping assays, like flow cytometry– or mass cytometry–based methods, are able to analyze hundreds of thousands of cells with >40 antibodies simultaneously20. This has been particularly useful for deep characterization of peripheral blood mononuclear cells and tissues21. However, these assays require the dissociation of tissues into cell suspensions, resulting in loss of spatial context of the analyzed cells, as well as an underrepresentation of certain cell types, such as stromal cells22. Multiplexed tissue imaging technologies have therefore been developed to study these critical spatial features, including cell-cell interactions, environmental context and overarching tissue structure, while retaining high multiplexing capabilities and enabling the collection of single-cell protein expression information along with other cellular features.
Multiplexed imaging approaches are based on several types of detection methods. Some, like CODEX, use DNA-conjugated antibodies that are visualized by the cyclic addition and removal of fluorescently labeled DNA probes. These methods include exchange-points accumulation in nanoscale topography9, DNA exchange imaging10 and immunostaining with signal amplification by exchange reaction11. Other multiplexed imaging methods use mass spectrometry–based detection of isotope-labeled antibodies by raster laser ablation (imaging mass cytometry)12, ion beams (multiplexed ion beam imaging)13,14, amplification of endogenous nucleic acids in situ15–17 or vibrational signatures of chemical bonds to visualize molecules of interest18.
CODEX has certain advantages relative to other multiplexed imaging approaches. The commercialization of the CODEX microfluidics device minimized some technical barriers to multiplexed tissue imaging. The CODEX method works with several inverted fluorescent optical microscopes (e.g., Keyence, Zeiss and Leica). Such microscopes are often available in research laboratories, which can significantly decrease the initial infrastructure costs. A CODEX experiment is scalable, antibody panels are straightforward to customize and the system has throughput sufficient to image tissue microarrays and specimens up to 1 cm2 in size.
Currently, CODEX can simultaneously detect 57 DNA-conjugated antibodies in a single tissue section. In addition, a biotin-conjugated antibody detected by using streptavidin-phycoerythrin can be visualized in the final cycle of the multicycle reaction2. Combined with the nuclear stains Hoechst and DRAQ5, this results in 60 markers. The current limit of antibody markers is set by the number of known unique DNA oligonucleotide sequences that do not exhibit tissue, cellular DNA or oligonucleotide-oligonucleotide cross reactivity. We recently showed that the iterative washing, hybridization and stripping steps involved in a CODEX multicycle reaction do not result in decreased marker intensity or degradation of tissue morphology2. This indicates that the size of the antibody panel can be increased as new oligonucleotide sequences become available. We anticipate that routine CODEX imaging with more than a hundred markers will soon be possible, with experiment time being the largest limiting factor.
Another advantage of CODEX is that the multicycle reaction does not destroy the tissue specimen. This allows traditional downstream staining, like H&E, to be performed in tandem. This feature is critical for comparison of cell-type identifications based on fluorescence marker profiles to known morphological cellular features.
Applications of CODEX multiplexed tissue imaging
We have used CODEX to reveal single-cell biology to understand key spatial relationships in autoimmune and cancerous tissues1–3,23. CODEX is compatible with archival, formalin-fixed, paraffin-embedded (FFPE) tissue, fresh-frozen (FF) tissue, fixed-frozen tissue and spreads of single cells (Fig. 2). We have imaged human, mouse and monkey tissues and a wide range of healthy and diseased organs, including tonsil, spleen, lymph node, liver, stomach, colon, pancreas, brain, lung, kidney, breast, muscle, skin and bone marrow (Fig. 3). In one application, we stained FF sections from normal or lupus spleens of mice to demonstrate how cellular niches could be used to explore the cellular positioning of diseased spleens1. In another application, we stained FFPE tissue microarrays of samples from patients with advanced colorectal cancer to determine how spatial organization of the tumor microenvironment is linked to clinical outcome2. This work revealed a local enrichment of PD-1+CD4+ T cells that correlated with survival in high-risk patients, highlighting the importance of single-cell spatial context and cellular neighborhood analysis.
Fig. 2 |. CODEX pipeline.
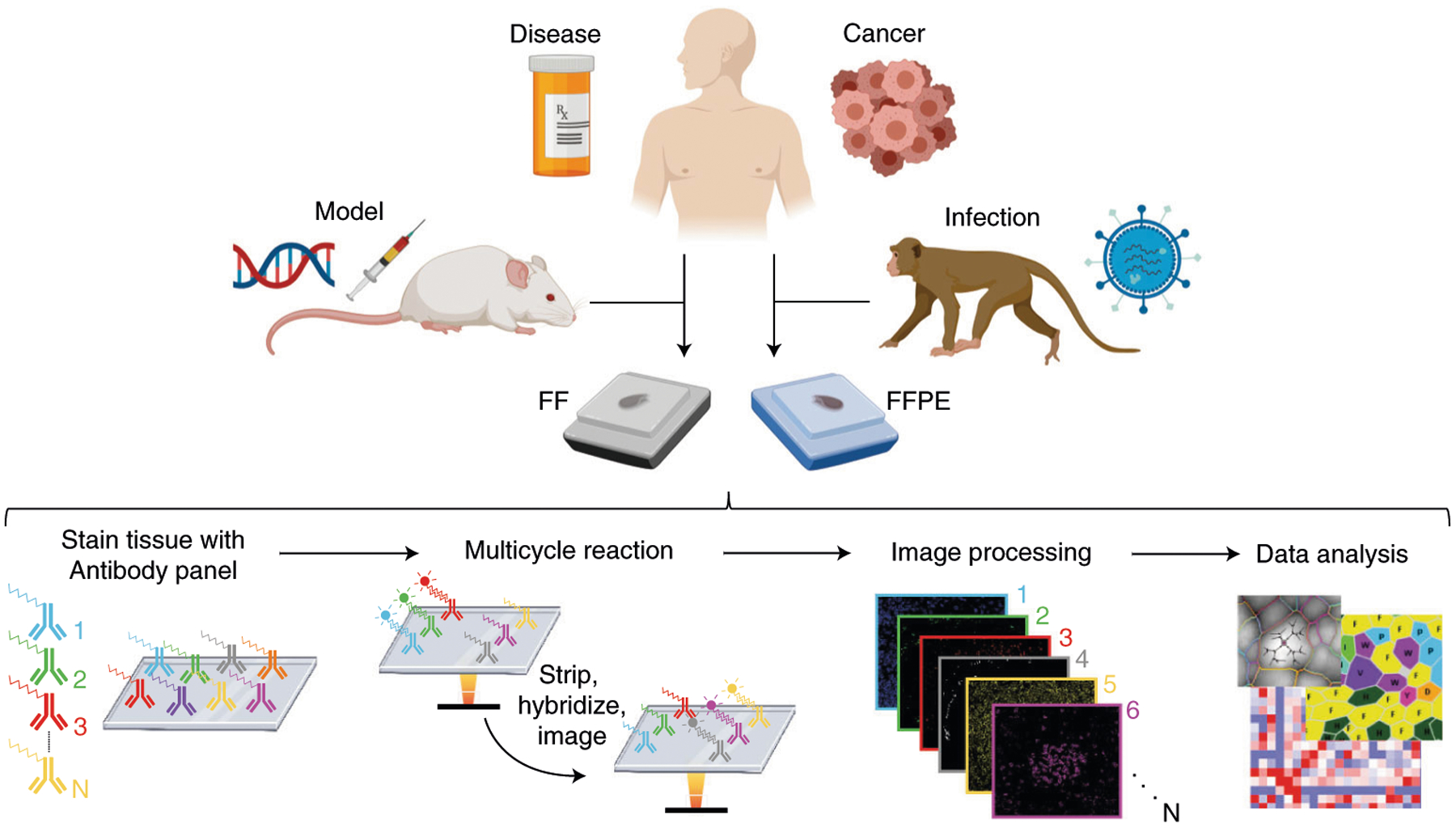
FFPE or FF tissue samples are stained with the antibody panel (Steps 71–107), a multicycle reaction is performed (i.e., iteratively imaging up to three antibodies and a nuclear stain per cycle, chemical stripping, hybridizing and re-imaging for all antibodies in the panel) (Steps 133 and 134), the raw images are computationally processed (Step 140) and data analysis is performed (Steps 141–144).
Fig. 3 |. Human FFPE tissues imaged with CODEX.
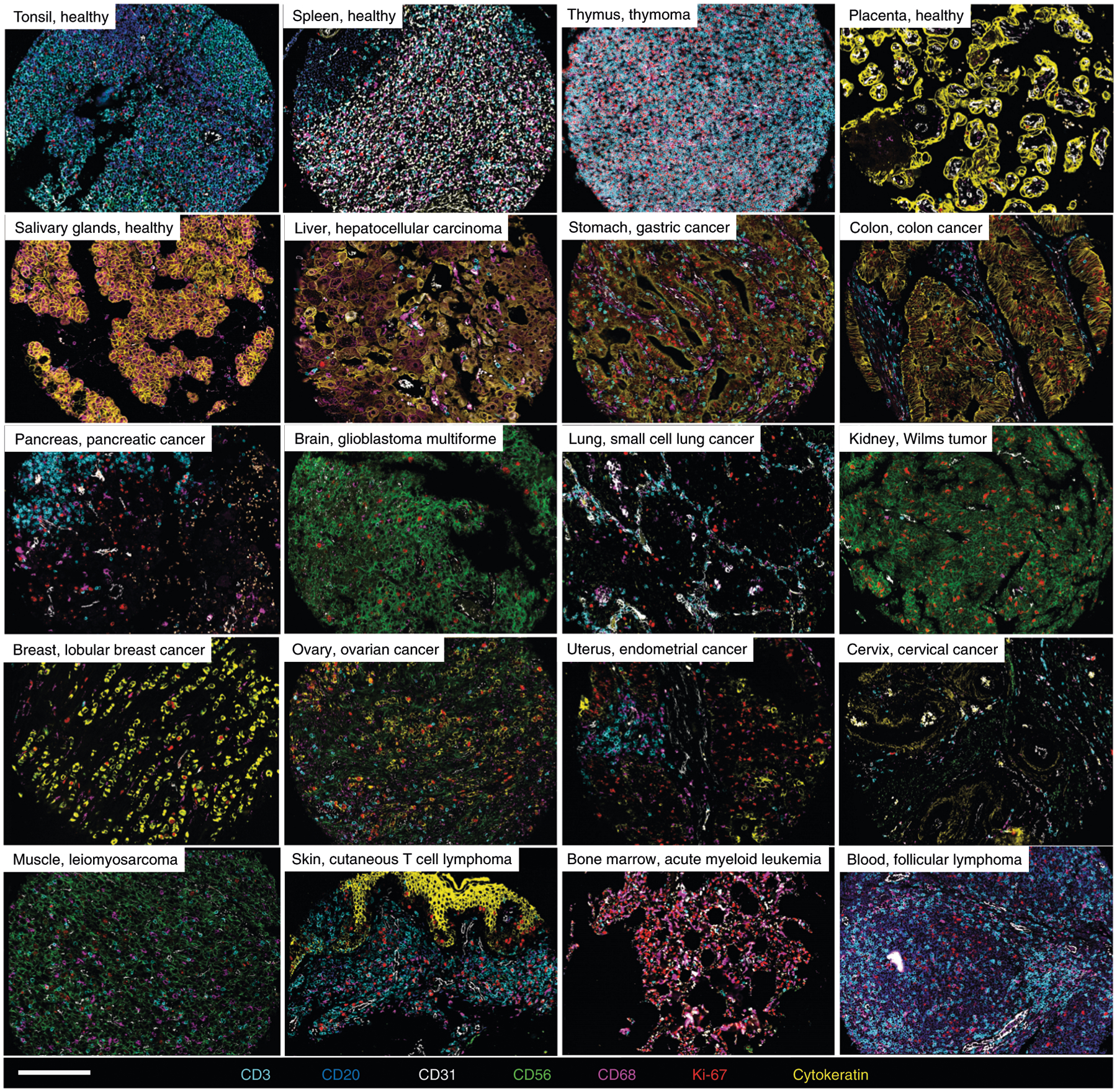
Representative images of 16 healthy and diseased tissues imaged with CODEX, highlighting seven markers—CD3, CD20, CD31, CD56, CD68, Ki-67 and cytokeratin—that are colored according to the bottom panel. Written informed consent was obtained from all patients. All samples were fully de-identified, and the study was exempt from ethics approval (no human subjects research). Scale bar, 200 μm.
Expertise needed to implement CODEX
The expertise needed to implement CODEX is similar to that needed for IHC and immunofluorescence analyses. Specifically, knowledge of specimen selection, tissue sectioning, antibody staining and microscopy are required. Additional CODEX-specific knowledge includes the (i) design of a multiparameter antibody panel, (ii) conjugation of DNA oligonucleotides to purified antibodies, (iii) hybridization reactions with complementary fluorescent oligonucleotides and (iv) multidimensional image processing and data analysis. Herein, we provide expert recommendations for each of these points.
Limitations of the CODEX technology
CODEX shares several limitations with other multiplexed imaging techniques. First, antibodies are expensive. Purified antibodies suitable for use with FFPE tissues generally cost $300–600 per 100 μg; antibodies suitable for FF tissues are often less expensive at ~$100 per 100 μg (provides enough for ~100–200 multicycle stains). Sufficient maleimide-modified DNA oligonucleotides for ~20 antibody conjugations cost ~$800 (source: TriLink BioTechnologies). Fluorescently tagged DNA oligonucleotides sufficient for ~100 reaction cycles cost ~$400 (source: Integrated DNA Technologies). In addition, each antibody requires individual conjugation and validation in a unified staining protocol. Certain antibodies or clones are not suitable for multiplexing (e.g., antibodies may have pH requirements for antigen retrieval that are incompatible with the CODEX unified staining protocol).
The CODEX platform currently lacks a signal amplification system. Despite this, we have validated >100 antibodies for FFPE tissues, including targets such as immune checkpoint markers and signaling molecules. Implementation of a signal amplification system will enable detection of low-abundance proteins that have important functions in cellular and tissue processes (e.g., transcription factors, cytokines/chemokines, etc.). Various amplification systems are currently under development; they include tetramer-based staining24, fluorescently labeling both the 3′ and 5′ ends of the fluorescently tagged oligonucleotide, branched fluorescent oligonucleotides11 and rolling circle amplification15. Signal amplification will also help overcome current issues with the baseline autofluorescence of tissues, which can be substantial in brain, lung and other tissues.
Processing and analyzing multiplexed tissue imaging data are complicated. We developed a multi-step pipeline to analyze CODEX data1–3,25, which involves image processing, single-cell segmentation, cell-type annotation and spatial analysis. Improving each of these steps is an active field of research. For instance, deep learning algorithms are being implemented for single-cell segmentation14,26,27. Cell-type clustering in a range of tissues is done by clustering algorithms such as X-shift25 and Louvain28, and clusters can be visualized by using dimensionality reduction techniques, like uniform manifold approximation and projection and t-distributed stochastic neighbor embedding29. We anticipate that these computational tools—which are generally open source and compatible across platforms—will continue to improve in accuracy, precision and scalability, while becoming more user friendly.
Future directions
To date, CODEX experiments have mainly used antibody-based detection of targets of interest. Fluorescent labeling of nucleic acid derivatives and amplification systems16,30 should extend this platform, such that DNA and RNA molecules could be investigated as well. This will allow single-cell quantitation of both genomic copy number variants and transcript enumerations within tissues. In addition, this should facilitate analyses of the spatial distribution of cytokines and chemokines within the overarching tissue context, with the potential to provide mechanistic insight into disease progression and therapeutic response31,32. Finally, microscopy techniques such as 3D tissue clearing, which enables fluorescent imaging of entire organs, and expansion microscopy for facilitating super-resolution fluorescent measurements33–35, might eventually be made compatible with CODEX.
The utility of CODEX will be further accelerated as faster imaging sources, such as high-throughput fluorescence scanners, are implemented. Use of a widefield fluorescence scanner will reduce the imaging acquisition time, making sample throughput rapid enough to meet the needs of routine pathology laboratories36. This advance will be especially useful for clinical trial–based biomarker discovery studies.
Moreover, many developments within image analysis, including the application of machine learning techniques, are transforming the accuracy of cell descriptions obtained by using microscopy26. Tools for image and feature segmentation and machine learning have facilitated automated identification of diseased tissues37. Not only could CODEX data be used within this context, but they could also be leveraged to perform automatic cell-type identification from H&E images by coupling CODEX multiplexed fluorescent marker readings with H&E data on a given tissue section. Furthermore, as segmentation algorithms improve, morphological features can be included in the cell phenotypic matrix38.
Experimental design
The CODEX method combines straightforward chemistries and tissue imaging processes. All steps can be conducted in a standard wet laboratory; access to a dark room is optional. Herein, we provide detailed descriptions for conjugating DNA oligonucleotides to purified antibodies, validating the antibody staining in FFPE and FF tissues and performing CODEX multicycle reactions. Three iterations of the CODEX microfluidics device (prototype, early-access version and commercial system) are shown in Fig. 4. We recommend using a dedicated microscope with the proper excitation/emission filters and objectives. Procedures for basic image processing and data analysis are described below. The timing for each section of the procedure is summarized in Fig. 5.
Fig. 4 |. Evolution of the CODEX technology.
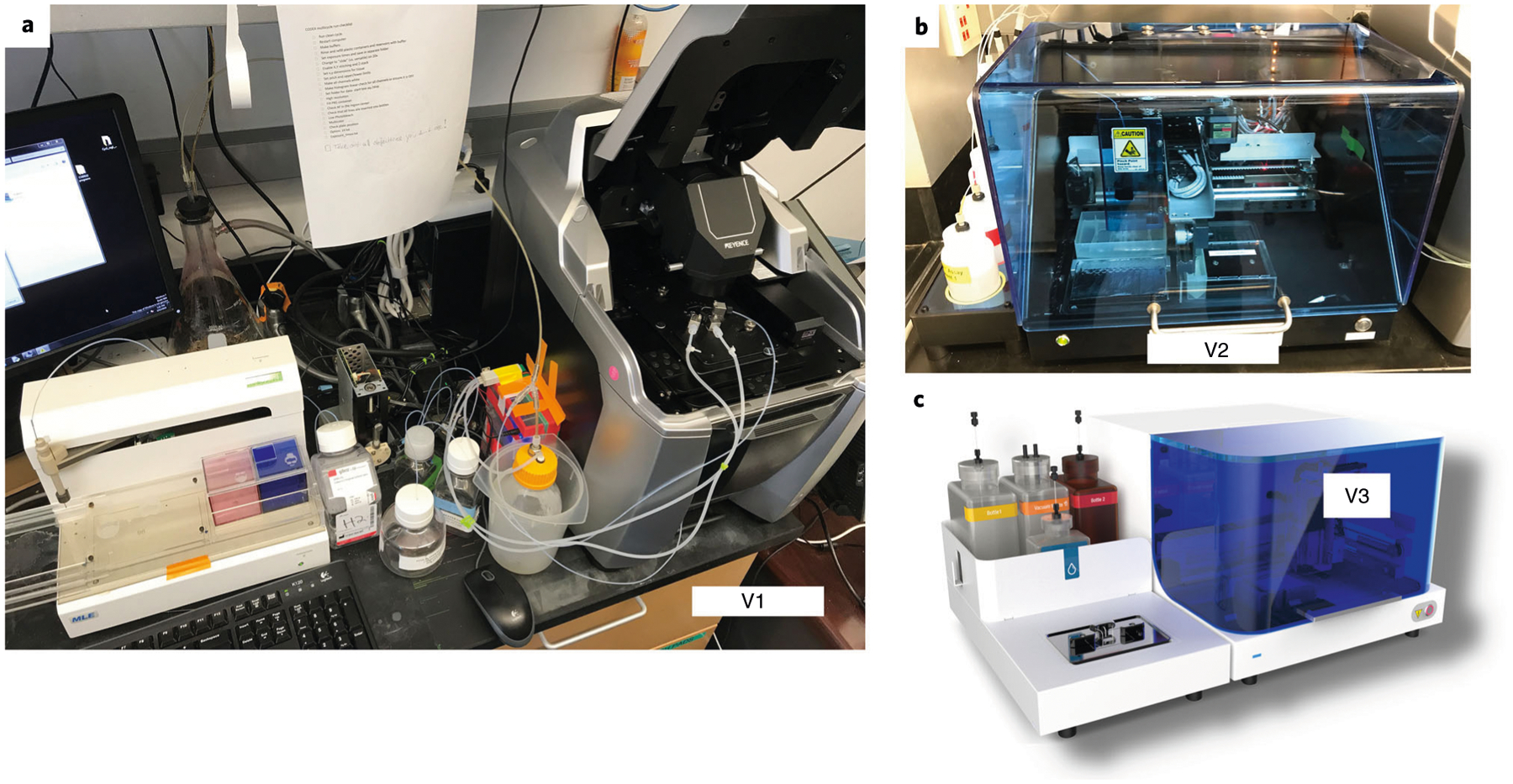
a, The prototype microfluidics device, showing tubing connections to the sample contained in a Keyence microscope. b, The early access version of the Akoya Biosciences microfluidics device. c, The commercially available Akoya Biosciences microfluidics device.
Fig. 5 |. Timing for the key elements of a CODEX experiment.
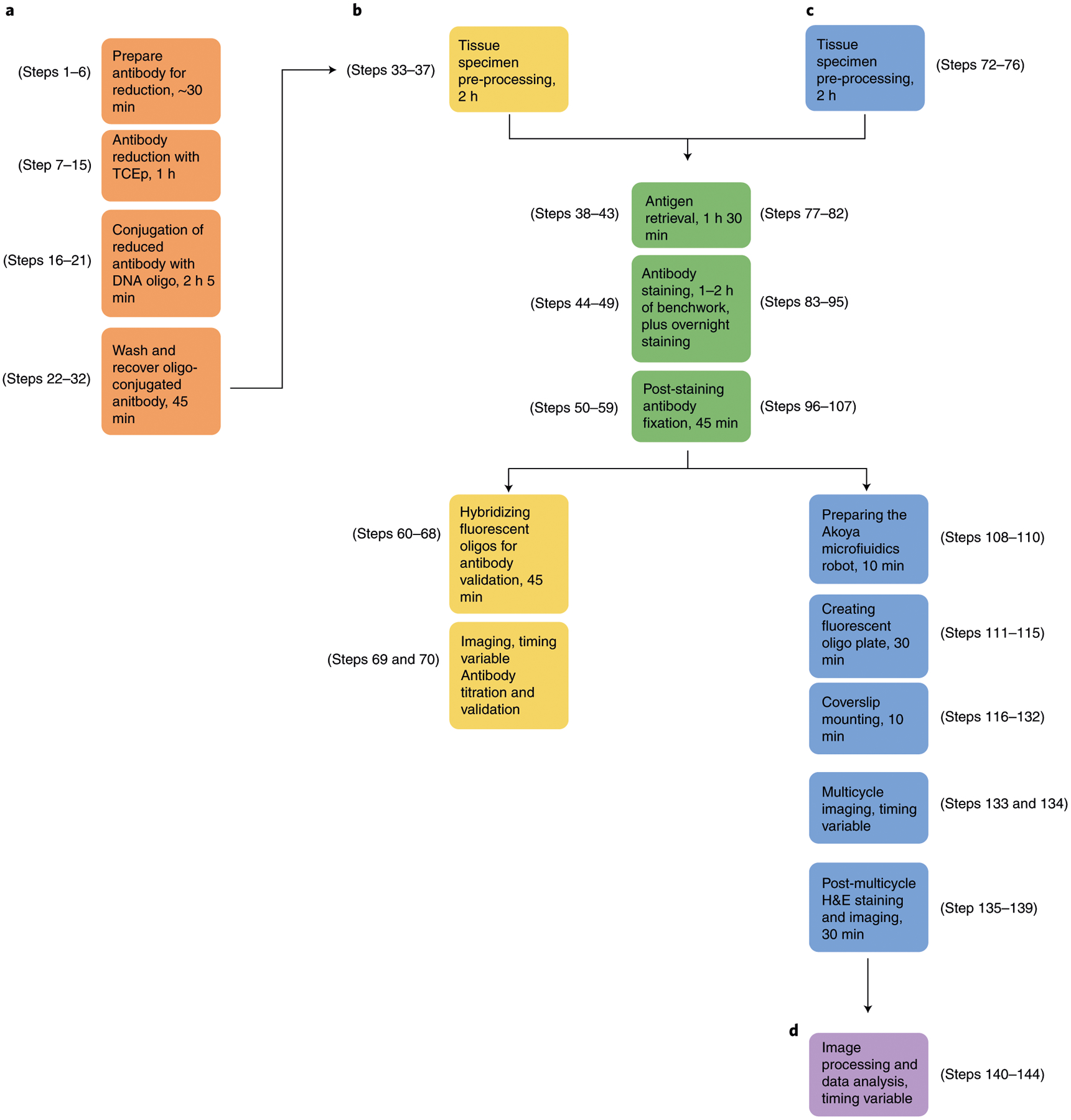
a, Steps 1–32 for conjugating DNA oligonucleotides to purified antibodies, as seen in the orange boxes. b, Steps 33–70 for validating and titrating antibodies in FFPE and FF tissue specimens, as seen in the yellow and green boxes. c, Steps 72–139 for performing a CODEX multicycle reaction, as seen in the blue and green boxes. d, Steps 140–144 for image processing and data analysis, as seen in the purple box.
Antibody conjugation and characterization
Box 1 describes the method for activating maleimide functional groups on DNA oligonucleotides. Section 1 of the protocol describes the method for conjugating these pre-activated DNA oligonucleotides to purified antibodies. Box 2 describes a method for validating the efficiency of antibody conjugation by flow cytometry. Section 2 of the protocol describes the method for validating and titrating the DNA-conjugated antibodies in FFPE tissue. Box 3 provides modifications for FF tissue.
Box 1 |. Activation of maleimide-modified DNA oligonucleotides ● Timing ~4 h 30 min, plus overnight lyophilization.
For the conjugation of DNA oligonucleotides to purified antibodies, as described in section 1 of the procedure, oligonucleotides are activated and lyophilized. This is a critical step because maleimide groups are not stable long term and are shipped in protected cycloadduct versions. To reveal ‘activate’ maleimide functional groups on the oligonucleotide, it is necessary to deprotect via a retro Diels-Alder reaction before the antibody conjugation. Generally, we will order custom maleimide-modified oligonucleotides from TriLink that are HPLC purified, 1-μmol batch size and lyophilized.
Additional reagents
-
Toluene anhydrous (Sigma, cat. no. 244511)
! CAUTION Toluene is toxic. Work in a fume hood or well-ventilated area. Proper handling and disposal procedures should be used, including disposing of toluene-contaminated pipette tips and Eppendorf tubes in designated hazardous waste containers.
-
Dry ice
! CAUTION Dry ice has a boiling temperature of −78 °C. Direct contact with the skin can cause cold burns and frostbite. Proper handling procedures should be used.
-
Liquid nitrogen
! CAUTION Liquid nitrogen has a boiling temperature of −196 °C. Direct contact with the skin can cause serious injury. Personal protective equipment (i.e., gloves, goggles, etc.) and proper handling procedures should be used.
-
Drierite desiccant (Thermo Fisher Scientific, cat. no. 07-578-3A)
! CAUTION Drierite is toxic. Proper handling and disposal procedures should be used.
Additional equipment
Precision balance
Digital dry bath (Genemate)
1-ml syringe and 23G (gauge) needles
PCR strip tubes, 0.2 ml (E&K Scientific, cat. no. 280008)
PCR strip caps, flat top (Sigma-Adlrich, cat. no. BR781326)
PCR strip caps, dome top (Sigma-Aldrich, cat. no. BR781327)
Long forceps (Fine Science Tools, cat. no. 11251-33)
FreeZone 4.5 plus cascade benchtop lyophilizer (Labconco, cat. no. 7386030)
Labconco Fast-Freeze flasks, complete assembly, 900 ml (Thermo Fisher Scientific, cat. no. 10-269-63)
Labconco Fast-Freeze flask adapters, steel, 3/4 inch (Thermo Fisher Scientific, cat. no. 10-269-61B)
Airtight box
Procedure
▲CRITICAL The oligonucleotide can arrive as either a white pellet or powder. It is recommended that the entire oligonucleotide pellet or powder is activated at the same time and that you do not activate more than four oligonucleotides at one time.
▲CRITICAL If activating more than one oligonucleotide at the same time, it is extremely important not to cross-contaminate the different oligonucleotides. Make sure to use a new pipette tip for each oligonucleotide to ensure that the stocks are not contaminated.
-
Make an Eppendorf tube lid label for each oligonucleotide being activated.
▲CRITICAL STEP It is important to use a label sticker on the cap of the Eppendorf tube, because toluene will wash away any labels drawn with marker.
Tare each labeled Eppendorf tube. Align the top of the tube containing the lyophilized oligonucleotide pellet or powder with the top of the tared Eppendorf tube and transfer the oligonucleotide to the Eppendorf tube by inverting and gentle tapping.
Weigh the lyophilized oligonucleotide pellet or powder in the tared Eppendorf tube.
Add 1.5 ml of anhydrous toluene with a syringe, taking care to ensure that the entire oligonucleotide is submerged. The white pellet or powder may become translucent when submerged in the toluene, but it will not dissolve. Close the lid on the Eppendorf tube and incubate for 2 h on a heat block at 90 °C in a fume hood.
-
Remove the toluene in the Eppendorf tube with a 1-ml pipette and replace with 1.5 ml of fresh toluene by using a syringe, taking care not to disturb the oligonucleotide pellet. Incubate for 2 h at 90 °C in a fume hood.
▲CRITICAL STEP If activating more than one oligonucleotide, perform this exchange one tube at a time to maintain the 90 °C temperature and to prevent the hot toluene from reaching RT.
Replace with 1.5 ml of fresh toluene with a syringe and keep at RT before the ethanol washing steps.
Carefully remove the toluene with a 1,000-μl pipette, making sure not to disturb the oligo pellet or powder.
-
Wash the oligonucleotide with 1.5 ml of 100% ethanol and slowly invert the tube two or three times. Allow the oligonucleotide pellet to settle to the bottom of the tube and carefully remove the ethanol with a 1,000-μl pipette, taking care to minimize oligonucleotide loss. Repeat four times.
▲CRITICAL STEP Make sure that the oligonucleotide pellet or powder does not get trapped in the lid during the inversions. If it does, gently invert the tube to resuspend the oligonucleotide in the ethanol.
-
After the fourth wash, completely remove the ethanol by aspirating with a 1,000-μl pipette and then a 20-μl pipette, taking care not to remove the oligonucleotide.
▲CRITICAL STEP If activating more than one oligonucleotide at a time, perform steps 10–15 one tube at a time. The additional oligonucleotides should remain suspended in ethanol until the previous oligonucleotide has undergone steps 10–15.
Dissolve the oligonucleotide pellet in buffer C (50 μl for every 1 mg of oligonucleotide based on step 3). It may require several minutes of gently mixing by pipetting up and down for the pellet to dissolve.
Measure the concentration of oligonucleotide. We measure absorbance at 260 nm by using a UV-visible spectrophotometer (e.g., Nanodrop 2000).
Add the appropriate volume of oligonucleotide for a total amount of 100 μg in each PCR tube.
Cap each PCR tube with a dome top (pre-pierced twice with a 23G needle). Vortex briefly and spin at 1,000g for 10 s at RT in a tabletop microcentrifuge.
Immediately snap-freeze the oligonucleotide mixture in liquid nitrogen by using a long forceps. Keep the PCR tube upright to ensure that the mixture remains at the bottom of the tube. Make sure to keep the PCR tube lid above the level of the liquid nitrogen so that liquid nitrogen does not enter the tube through the holes in the lid.
Immediately place the frozen PCR tube in dry ice.
Configure the lyophilizer so that the vacuum and auto-refrigeration settings are both on and fully acclimated. Lyophilize the oligonucleotide mixtures overnight. This will yield white pellets.
-
Replace the pierced dome caps with labeled flat caps. Place the PCR tubes in an airtight box containing drierite desiccant.
■PAUSE POINT The lyophilized oligonucleotides can be stored for ≤2 years at −20 °C.
Box 2 |. Validation of DNA oligonucleotide-conjugated antibodies by flow cytometry ● Timing ~1 h 30 min.
For the conjugation of DNA oligonucleotides to purified antibodies, as described in section 1 of the procedure, the conjugation can be assessed by using flow cytometry. This demonstrates whether the conjugated antibody has oligonucleotide conjugated, not the number or efficiency of oligonucleotide conjugation (this can be accomplished by running an SDS–PAGE gel). Subsequent in situ staining should be done to validate the staining efficiency.
Additional equipment
Falcon round-bottom polypropylene tubes, 5 ml (Corning, cat. no. 352053)
UltraComp eBead compensation beads (Thermo Fisher Scientific, cat. no. 01-2222-42)
Flow cytometer
Reagent setup
FACS hybridization solution. For each antibody, add 7 μl of B3 and 1 μl of the fluorescent oligonucleotide of interest to 92 μl of hybridization buffer. Mix gently in an Eppendorf tube by pipetting up and down. Prepare fresh before each experiment.
Procedure
Label a FACS tube for each oligonucleotide-conjugated antibody to be tested, including controls.
Add 200 μl of S2 to each FACS tube.
-
Add 1 drop (~50 μl) of UltraComp eBeads compensation beads. Mix vigorously by vortexing for 2 s.
▲CRITICAL STEP UltraComp eBeads bind to antibodies that were produced in mouse, rat and hamster species. If the antibody of interest was produced in another species, use a different particle system (e.g., Protein G particles).
-
Add 1 μl of the oligonucleotide-conjugated antibody (concentration: ~0.4 mg/ml) or control. Vortex briefly to mix. Incubate for 15 min at RT.
▲CRITICAL STEP For negative controls, use compensation beads with 1 μl of fluorescent oligonucleotide only (no antibody). For positive controls, use compensation beads with commercially available fluorescent FACS antibody or biotinylated antibody and fluorophore-labeled streptavidin.
Add 4 ml of S2 buffer to wash away any unbound antibody. Centrifuge at 500g for 5 min at RT. Decant the supernatant by carefully inverting the FACS tube.
Add 4 ml of hybridization buffer. Centrifuge at 500g for 5 min at RT. Decant the supernatant by carefully inverting the FACS tube.
Add 100 μl of FACS hybridization solution. Incubate for 15 min at RT in the dark.
Add 4 ml of H2 buffer to wash away unbound fluorescent oligonucleotide. Centrifuge at 500g for 5 min at RT. Decant the supernatant by carefully inverting the FACS tube.
Add 4 ml of S4 buffer. Centrifuge at 500g for 5 min at RT. Decant the supernatant by carefully inverting the FACS tubes.
Add 250 μl of S4 buffer. Vortex briefly to resuspend the compensation beads.
-
Adjust the forward and side scatter on the flow cytometer by running a small sample of compensation beads and adjusting the photomultiplier tube voltages for the channel. Make sure that the stain is within range and separated from the negative population. Record the results; an example is provided in the figure below, with positive (biotinylated antibody and fluorescent streptavidin) and negative (oligonucleotide without antibody) controls.
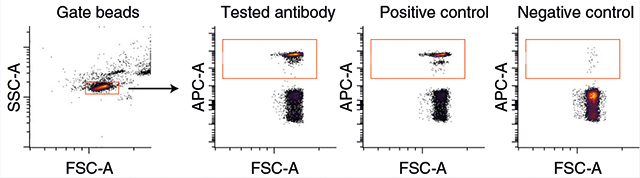
▲CRITICAL STEP Compensation beads have a negative population that enables the detection of background binding from reagents. In the figure below, this is ~45%; thus, a successfully conjugated antibody should read 55% of the beads. Definitions of abbreviations used in the figure are as follows: APC-A, allophycocyanin; FSC-A, forward scatter area; SSC-A, side scatter area.
Box 3 |. Preparing fresh-frozen tissue for CODEX ● Timing ~ 5 h of bench work, plus variable imaging time.
Methods are described for antibody validation and running a multicycle on fresh-frozen tissue. Many of the steps are the same as those used for FFPE specimens, as described in sections 2 and 3 of the procedure. Protocol differences are highlighted here. Follow procedure A for validation and titration of conjugated antibodies by CODEX in FF tissue. Follow procedure B for CODEX multicycle reaction in FF tissue.
Additional reagents
Poly-lysine (Sigma-Aldrich, cat. no. P8920-500mL)
-
Drierite desiccant (Thermo Fisher Scientific, cat. no. 07-578-3A)
! CAUTION Drierite is toxic. Proper handling and disposal procedures should be used.
Reagent setup
FF tissue fixation solution. Dilute 16% (wt/vol) PFA at a 1:10 ratio with S1 (vol/vol). Prepare fresh before each experiment and use a fresh vial of 16% (wt/vol) PFA every 2 weeks.
CODEX FF staining solution. Add 65 μl of B1, 65 μl of B2 and 43 μl of B3 to 827 μl of S2. Mix gently by pipetting up and down. Store at 4 °C for ≤2 weeks.
CODEX FF multicycle staining solution. Add 780 μl of S2, 50 μl of B1, 50 μl of B2, 50 μl of B3 and 70 μl of BC4.
Procedure A (validation and titration of conjugated antibodies by CODEX in FF tissue)
▲CRITICAL Unless otherwise stated, we perform specimen incubations in a coverslip glass jar containing 10 ml of solution.
-
Mount a 7-μm-thick FF tissue section onto a poly-lysine–treated coverslip by using standard histology procedures.
■PAUSE POINT Mounted fresh-frozen tissue sections can be stored for 1 month at −80 °C.
Place the specimen on a bed of drierite for 2 min at RT.
Place the specimen in 10 ml of 100% acetone for 10 min at RT.
Allow the specimen to dry for 2 min at RT.
To decrease the staining volume to ~100 μl, create a boundary around the specimen by using a Bondic polyacrylamide gel pen. Make sure that the area around the specimen is dry before applying the gel, so that the gel firmly adheres to the slide.
Rehydrate the specimen in 10 ml of S1 for 4 min at RT.
Add 100 μl of FF tissue fixation solution to the specimen and fix for 10 min at RT.
During step 7, prepare the conjugated antibody solution (to be used in step 11), by adding the desired antibodies to the CODEX FF staining solution to a final volume of 100 μl. Vortex briefly and spin at 1,000g for 10 s at RT in a microcentrifuge to ensure that the mixture is at the bottom of the tube.
Wash the specimen two times in 10 ml of S1, each for 2 min, at RT.
Place the specimen in 10 ml of S2 for 5 min at RT.
Add 100 μl of the conjugated antibody solution to the specimen and incubate in a humidity chamber for 3 h at RT.
Proceed with antibody fixation, fluorescent oligonucleotide hybridization and tissue imaging as described in section 2 of the main procedure (Steps 50–70).
Procedure B (CODEX multicycle reaction in FF tissue)
▲CRITICAL Unless otherwise stated, we perform specimen incubations in a coverslip glass jar containing 10 ml of solution.
-
Mount a 7-μm-thick FF tissue section onto a poly-lysine–treated coverslip by using standard histology procedures.
▲CRITICAL STEP Ensure that the tissue section is placed in the center of the coverslip, ≥4 mm from each edge.
■PAUSE POINT Mounted fresh-frozen tissue sections can be stored for 1 month at −80 °C.
Prepare the conjugated antibody cocktail as described in Steps 86–92 of the main procedure by using CODEX FF multicycle staining solution in place of CODEX FFPE blocking solution.
Proceed with steps 2–7 from procedure A of this box.
Add 100 μl of the conjugated antibody cocktail to the specimen. Incubate in a humidity chamber for 3 h at RT.
Proceed with antibody fixation, fluorescent oligonucleotide plate creation, coverslip mounting, multicycle imaging, H&E staining and data analysis as described in Steps 96–144 of the main procedure.
Execution and analysis of CODEX multicycle reactions
Section 3 of the protocol describes the method for performing a CODEX multicycle experiment on FFPE tissue. Box 3 provides modifications for FF tissue. We routinely perform CODEX experiments on FFPE and FF tissue specimens. There are numerous pros and cons for FFPE and FF samples, based on considerations of both antibody availability, compatibility and cost and tissue availability, stability, safety, morphology and applications, which are summarized in Table 1. Typically, sections are cut at a thickness of 7 μm for FF tissues and 4 μm for FFPE tissues. These thicknesses allow for adequate handling and provide one major focal plane of cells. Section 4 describes image processing and data analysis procedures; additional data processing details are provided in Supplementary Notes 1–4 and the associated Supplementary Figs. 1–3.
Table 1 |.
Comparison of CODEX tissue imaging for FFPE and FF specimens
| FFPE | FF | |
|---|---|---|
| Antibodies | ||
| Availability | Many clones are not available in purified and carrier-free format | Most flow cytometry antibodies work well |
| Utilization | Requires optimization of epitope retrieval conditions (i.e., difficult to assemble a panel of ≥50 antibodies that all stain well under the same conditions) | Little protocol optimization required |
| Cost | Expensive | Reasonable |
| Tissue specimen | ||
| Availability | Widely available (most common type of clinical specimen) | Limited availability |
| Handling | Easy to handle and store | Difficult to handle and store |
| Infectious risk | Low to none | Potentially infectious |
| Morphology | Well-preserved morphology over the course of a multicycle | Deteriorates over the course of many cycles in a multicycle |
| Applications | Retrospective clinical cohorts, with full clinical annotations; biomarker discovery of cancer or vaccine clinical trials; compatible with large-scale tissue microarrays (e.g., ≤128 samples); enabling all samples/conditions to be stained and imaged in a single multicycle | Requires prospective collection; not compatible with large-scale tissue microarrays; resulting in batch effects over numerous multicycles |
CODEX antibody panel design
Creating an antibody panel requires customization considering features of the tissues of interest (e.g., baseline auto-fluorescence and morphology) and proteins of interest. Several principles apply to every antibody panel. The Alexa488 channel tends to have more auto-fluorescence. Thus, we recommend conjugating antibodies targeting high-abundance proteins to DNA oligonucleotides for which the corresponding fluorescent oligonucleotide is labeled with Alexa488. In contrast, antibodies targeting low-abundance proteins should be conjugated to DNA oligonucleotides for which the corresponding fluorescent oligonucleotide is labeled with ATTO550 or Alexa647. We previously showed that antibody staining is remarkably stable over the course of multiple cycles2, but we did note slight signal degradation of nuclear markers in later cycles. Importantly, this does not affect strong nuclear stains such as Hoechst or tissue morphology (Extended Data Figs. 1 and 2). We therefore recommend placing antibodies that stain proteins located in the nucleus in earlier cycles. For each panel, we also recommend that the first cycle be a ‘blank’ cycle (i.e., a cycle with no added fluorescent oligonucleotides) to provide a measure of background signal, which will be subtracted during image processing.
Materials
Biological materials
Tissue from the species of interest (e.g., human, mouse or monkey) prepared according to standard pathology procedures for FFPE or FF39 ! CAUTION Working with tissue specimens requires ethical and institutional review board approval. Conforming to relevant institutional and national regulations is required. Patient consent and institutional review board review may be required for the use of archival tissue. Consult your institution for further details. In addition, fresh-frozen tissues may be infectious. Proper handling and protective procedures should be used. The tissues used in this paper were from patients who gave their written informed consent to have their tissue used for research. The samples were fully de-identified, and therefore the study was exempt from ethics approval (no human subjects research).
Reagents
Conjugation of DNA oligonucleotides to antibodies
Tween 20 (Sigma, cat. no. P1379)
Dulbecco’s PBS (DPBS), 1×, no calcium or magnesium (Thermo Fisher Scientific, cat. no. 14190–250)
Trizma HCl (Sigma, cat. no. T3253)
Trizma base (Sigma, cat. no. T1503)
Tris-(2-carboxyethyl)phosphine hydrochloride (TCEP; Thermo Fisher Scientific, cat. no. 77720; Sigma, cat. no. C4706–10G)
500 mM EDTA, pH 8.0 (Teknova, cat. no. E0308)
1 M Tris, pH 8.0 (Teknova, cat. no. T1080)
NaCl (Thermo Fisher Scientific, cat. no. S271-10)
DNA oligonucleotides (TriLink Biotechnologies; Supplementary Table 1). Follow the procedure described in Box 1 for activation of maleimide-modified DNA oligonucleotides.
Candor PBS antibody stabilizer solution (Thermo Fisher Scientific, cat. no. NC0436689)
Sodium azide (NaN3; Sigma-Aldrich, cat. no. S-8032) ! CAUTION Sodium azide is very toxic. This compound is fatal if ingested. Proper handling procedures should be used.
Purified antibodies
100 μg of purified, carrier-free antibody with specificity against the target of interest. Store according to the manufacturer’s instructions ▲CRITICAL For efficient conjugation, antibodies must be free of cysteine-containing proteins, such as BSA, which can compete for maleimide-functionalized groups. If any carrier is present, purify the antibody by using a cleanup kit (e.g., Melon gel IgG spin purification kit, Thermo Fisher Scientific, cat. no. 45206; Abcam BSA removal kit, cat. no. ab173231) before conjugating. We provide a list of the DNA-conjugated antibodies used in the presented CODEX multicycle in Supplementary Tables 2 and 3.
CODEX antibody staining
Vectabond (Vector Labs, cat. no. SP-1800)
Acetone (Thermo Fisher Scientific, cat. no. A929-4)
Xylene (Thermo Fisher Scientific, cat. no. X5-4) ! CAUTION Xylene is extremely flammable. Work in a well-ventilated area. Proper handling and disposal procedures should be used.
Ethanol, 100% (Sigma, cat. no. E7023) ! CAUTION Ethanol is flammable. Proper handling procedures should be used.
10× PBS, pH 7.4 (Thermo Fisher Scientific, cat. no. 70011069)
Dako target retrieval solution, pH 9.0 (Agilent, cat. no. S236784-2)
Tris-buffered saline (TBS) IHC wash buffer with Tween 20 (Cell Marque, cat. no. 935B-09)
Bondic polyacrylamide gel pen (Amazon, cat. no. B018IBEHQU) ! CAUTION Polyacrylamide is toxic and an irritant. Wear gloves when applying.
DPBS, 1×, no calcium or magnesium (Thermo Fisher Scientific, cat. no. 14190-250)
Mouse IgG (Sigma, cat. no. I5381-10MG)
Rat IgG (Sigma, cat. no. I4131)
Blocking component 4 (BC4; a mixture of all reporter oligonucleotides without fluorescent reporters; Supplementary Table 1)
Salmon sperm DNA, sheared, 10 mg/ml (Thermo Fisher Scientific, cat. no. AM9680)
BSA (Sigma, cat. no. A3059)
Disodium phosphate (Na2HPO4; Sigma, cat. no. S7907)
Sodium phosphate monobasic monohydrate (NaH2PO4•H2O; Sigma, cat. no. S9638)
Magnesium chloride hexahydrate (MgCl2•6 H2O; Sigma, cat. no. M2670)
NaOH (Sigma, cat. no. S8263)
Triton X-100 (Sigma, cat. no. T8787)
Paraformaldehyde ampoules, 16% (wt/vol) (Thermo Fisher Scientific, cat. no. 50-980-487)
Methanol, 100% (Thermo Fisher Scientific, cat. no. A412-4) ! CAUTION Methanol is flammable and toxic to the eyes. Proper handling and disposal procedures should be used.
Bis(sulfosuccinimidyl)suberate (BS3; Thermo Fisher Scientific, cat. no. 21580) ! CAUTION BS3 can cause skin, eye and respiratory tract irritation. Proper handling and disposal procedures should be used.
Dimethyl sulfoxide (DMSO) ampule (Sigma, cat. no. D2650)
DMSO (Sigma-Aldrich, cat. no. D2650-5X5ML) ! CAUTION DMSO readily penetrates the skin and can cause skin, eye and respiratory tract irritation. Proper handling and disposal procedures should be used.
Hoechst 33342 (Thermo Fisher Scientific, cat. no. 62249)
DRAQ5 (Cell Signaling Technology, cat. no. 4084L)
Fluorescent reporter oligonucleotides (Integrated DNA Technologies (IDT); Supplementary Table 1)
Hematoxylin, ready-to-use solution (Agilent, cat. no. S330930-2)
Eosin Y solution (Sigma, cat. no. HT110116)
Cytoseal XYL (VWR Scientific, cat. no. 48212-196)
Equipment
4 °C, −20 °C and −80 °C storage units
UV-visible spectrophotometer (Thermo Fisher Scientific, model no. NanoDrop 2000)
Tabletop microcentrifuge (Eppendorf, model no. 5424)
50-kDa Amicon Ultra 0.5-ml centrifugal filter column (EMD Millipore, cat. no. UFC505096)
Eppendorf tubes, 1.7 ml (VWR Scientific, cat. no. 87003-294)
Microcentrifuge tubes with socket screw-cap, 1.5 ml (VWR Scientific, cat. no. 89004-294)
Glass coverslips, 22 × 22 mm, 1.5-inch thickness (Electron Microscopy Sciences, cat. no. 72204-10)
Frosted glass microscope slides (Thermo Fisher Scientific, cat. no. 12-550-343)
Glass coverslip storage box (Qintay, cat. no. CS-22)
Wheaton coverslip glass jar (Thermo Fisher Scientific, cat. no. 02-912-637)
Microscope slide-staining glass jar (Ted Pella, cat. no. 432-1)
Dumont #5/45 coverslip forceps (Fine Science Tools, cat. no. 11251-33)
Six-well tissue culture plates (Thermo Fisher Scientific, cat. no. 07-20083)
Digital mini incubator (VWR Scientific, cat. no. 10055-006)
ST4020 small linear stainer (Leica, cat. no. 14050946425)
Lab Vision PT module (Thermo Fisher Scientific, cat. no. A80400012)
Slide chamber for antigen retrieval (Electron Microscopy Sciences, cat. no. 62705-01)
Humidity chamber, sealable (we use a pipette tip box with 100 ml of ddH2O on the bottom)
Adjustable speed orbital shaker (Mophorn, cat. no. B07FCY2S1P)
Heavy-duty, single-edge razor blade (Amazon, cat. no. B003O3EOFM)
Kimberly-Clark Professional Kimwipes (Thermo Fisher Scientific, cat. no. 06-666-1A)
CODEX acrylic plate (Bayview Plastic Solutions, custom; blueprints available upon request)
DMSO-resistant mounting gasket, 22 × 22 mm (Qintay, cat. no. TMG-22)
Corning black 96-well plates (Thermo Fisher Scientific, cat. no. 07-200-762)
Axygen aluminum sealing film (VWR Scientific, cat. no. 47734-817)
BZ-X710 inverted fluorescence microscope (Keyence); the currently available model for purchase is X810.
CFI Plan Apo ƛ 2×/0.10 objective (Nikon)
CFI Plan Apo ƛ 20×/0.75 objective (Nikon)
Akoya microfluidics device (Akoya Biosciences)
Keyence accessory kit: microscope objective liquid collector device and microscope liquid collector cup (Akoya Biosciences, cat. no. 9000020)
Personal computer and software requirements
Windows personal computer with a minimum of 16 GB of random access memory and a video graphic card with a minimum of 1 GB of random access memory
Storage: ≥8 terabytes; one CODEX experiment, depending on tissue size and number of markers, can be 0.5–2 terabytes in size
Optional: Nolan laboratory CODEX imaging software (https://github.com/nolanlab/)
Optional: CODEX analysis FIJI plugins (https://github.com/bmyury/CODEX-fiji-scripts)
Optional: flow cytometry software; we use CellEngine (https://www.primitybio.com/cellengine.html), but any platform can be used (e.g., Cytobank (https://cytobank.org/) or FlowJo (https://www.flowjo.com/))
Optional: Fiji/ImageJ (http://fiji.sc/Fiji) and Python (https://www.python.org/)
Reagent setup
PBS-Tween solution
Prepare a 0.1% (vol/vol) Tween solution in 1× DPBS. Store at room temperature (RT: 18–26 °C) for ≤6 months.
500 mM TCEP stock solution
Dissolve 717 mg of TCEP in 2.5 ml of ddH2O, adjust to pH 7.0 with sodium hydroxide and bring to a volume of 5 ml with ddH2O. Store at 4 °C for ≤1 year.
2.5 mM TCEP solution for antibody reduction
Add 5 μl of 500 mM TCEP and 5 μl of 500 mM EDTA (pH 8.0) to 990 μl of 1× DPBS. Mix by gently pipetting up and down. Prepare fresh before each experiment.
5 M NaCl solution
Dissolve 146.1 g of NaCl in 500 ml of ddH2O. Add 0.02 % (wt/vol) NaN3 to this solution. Store at RT for ≤1 year.
Buffer C
Add 1 ml of 1 M Tris (pH 7.0), 1 ml of 1 M Tris (pH 7.5), 30 ml of 5 M NaCl solution and 2 ml of 500 mM EDTA (pH 8.0) to 966 ml of ddH2O. Add 200 mg of NaN3 for a 0.02% (wt/vol) concentration in this solution. Store at RT for ≤1 year.
High-salt buffer C
Add 20 μl of 5 M NaCl solution to 380 μl of buffer C. Mix gently by pipetting up and down. Prepare fresh before each experiment.
High-salt PBS
Add 45 ml of 5 M NaCl solution and 25 ml of 10× DPBS to 180 ml of ddH2O. Add 50 mg of NaN3 for a 0.02% (wt/vol) concentration in this solution. Store at RT for ≤1 year.
Stock antibody stabilizer solution
Add 0.02% (wt/vol) NaN3 to Candor PBS antibody stabilizer solution. Store at 4 °C for ≤1 year.
CODEX antibody stabilizer solution
Add 1 ml of 5 M NaCl solution and 100 μl of 500 mM EDTA (pH 8.0) to 9 ml of stock antibody stabilizer solution. Vortex briefly to mix. Store at 4 °C for ≤1 year.
Antigen retrieval solution, pH 9
Dilute Dako target retrieval solution at a 1:10 ratio with ddH2O (vol/vol). Prepare fresh before each experiment.
70% ethanol solution
Dilute 100% ethanol in ddH2O to make a 70% (vol/vol) solution. Store at RT for ≤3 months.
80% Ethanol
Dilute 100% ethanol in ddH2O to make a 80% (vol/vol) solution. Store at RT for ≤3 months.
95% Ethanol
Dilute 100% ethanol in ddH2O to make a 95% (vol/vol) solution. Store at RT for ≤3 months.
1× PBS for antigen retrieval
Add 150 μl of 10× PBS to 1.35 ml of ddH2O. Store at RT for ≤6 months.
TBS IHC wash buffer with Tween 20
Dilute 20× TBS IHC wash buffer with Tween 20 at a 1:20 ratio with ddH2O (vol/vol). Store at RT for ≤1 month.
CODEX staining buffer 1 (S1)
Add 5 ml of 500 mM EDTA (pH 8.0), 50 ml of 10× DPBS, 2.5 g of BSA and 100 mg of NaN3 to 445 ml of ddH2O. Gently mix. Store at 4 °C for ≤1 year.
1 M Na2HPO4
Dissolve 70.98 g of Na2HPO4 in 500 ml of ddH2O. Add 0.02% (wt/vol) NaN3 to this solution. Store at RT for ≤1 year.
1 M NaH2PO4
Dissolve 69 g of NaH2PO4•H2O in 500 ml of ddH2O. Add 0.02% (wt/vol) NaN3 to this solution. Store at RT for ≤1 year.
CODEX staining buffer 2 (S2)
Add 250 ml of S1, 30.5 ml of 1 M Na2HPO4, 19.5 ml of 1 M NaH2PO4 and 25 ml of 5 M NaCl solution to 175 ml of ddH2O. Adjust the pH to 6.8–7.0 with sodium hydroxide. Gently mix. Store at 4 °C for ≤1 year.
Blocking reagent 1 (B1)
Dissolve 10 mg of mouse IgG in 10 ml of S2. Store in 0.5-ml aliquots at 4 °C for ≤1 year.
Blocking reagent 2 (B2)
Dissolve 10 mg of rat IgG in 10 ml of S2. Store in 0.5-ml aliquots at 4 °C for ≤1 year.
Blocking reagent 3 (B3) sheared salmon sperm DNA
This reagent comes at a 10-mg/ml concentration in ddH2O at −20 °C. Store in 0.5-ml aliquots at 4 °C for ≤1 year.
1× Tris-EDTA (TE) buffer
Add 1 ml of 1 M Tris (pH 8.0), 200 μl of 500 mM EDTA (pH 8.0) and 0.02% (wt/vol) NaN3 to 98.8 ml of ddH2O. Store at RT for ≤1 year.
BC4 solution
Prepare a mixture of the 57 nonmodified CODEX DNA oligonucleotides (Supplementary Table 1) by dissolving in TE buffer to a final concentration of 0.5 mM per oligonucleotide. Store in 0.5-ml aliquots at 4 °C for ≤1 year.
CODEX FFPE blocking solution
Add 50 μl of B1, 50 μl of B2, 50 μl of B3 and 70 μl of BC4 to 780 μl of S2. Mix gently by pipetting up and down. Store at 4 °C for ≤2 weeks.
CODEX staining buffer 4 (S4)
Add 50 ml of 5 M NaCl solution to 450 ml of S1. Gently mix. Store at 4 °C for ≤1 year.
Paraformaldehyde fixation solution
Dilute 16% (wt/vol) paraformaldehyde (PFA) at a 1:10 ratio with S4 (vol/vol). Prepare fresh before each experiment and use a fresh vial of 16% PFA every 2 weeks.
BS3 aliquots
Dissolve 50 mg of BS3 in 250 μl of DMSO (ampule). Store in 20-μl aliquots at −20 °C for ≤6 months.
Final fixative solution
Add 20 μl of BS3 (thawed to RT) to 1 ml of 1× DPBS. Mix gently by pipetting up and down. Prepare fresh before each experiment.
H2 buffer
Add 30 ml of 5 M NaCl solution, 10 ml of 1 M Tris (pH 7.5), 0.943 ml of Triton X-100, 2.03 g of MgCl2•6H2O and 0.02% (wt/vol) NaN3 to 960 ml of ddH2O. Store at RT for ≤1 year.
Hybridization buffer
Combine 100 ml of DMSO with 400 ml of H2 buffer. Stir gently to thoroughly combine. Prepare fresh before each experiment.
Stripping buffer
Add 62.5 ml of H2 buffer to 187.5 ml of DMSO. Stir gently to thoroughly combine. Prepare fresh before each experiment.
CODEX hybridization solution
For each tissue section, add 7 μl of B3, 1 μl of each complementary fluorescent oligonucleotide and the appropriate volume of H2 buffer to a final volume of 100 μl. Mix gently in an Eppendorf tube by pipetting up and down. Prepare fresh before each experiment.
Hoechst staining solution
Add 1 μl of Hoechst 33342 to 999 μl of H2 buffer. Mix gently in an Eppendorf tube by pipetting up and down. Prepare fresh before each experiment.
Plate buffer
Add 83.3 μl of Hoechst 3342 and 2.5 ml of B3 to 50 ml of H2 buffer. Store in the dark at 4 °C for ≤4 weeks.
Fluorescent oligonucleotide stock solution
Dissolve the lyophilized fluorescent oligonucleotide pellet in the appropriate volume of 1× TE buffer to a concentration of 100 μM (note: volume provided by IDT). Store in the dark at −20 °C for ≤2 years. Generally, we will order custom fluorescent reporter oligonucleotides from IDT that are HPLC purified, 1-μmol batch size and lyophilized.
Fluorescent oligonucleotide working solution
Dilute the fluorescent oligonucleotide stock solution at a 1:10 ratio with 1× TE buffer (vol/vol). Store in the dark at 4 °C for ≤1 year.
DRAQ5 staining solution
Add 2.5 μl of DRAQ5 to 247.5 μl of plate buffer. Prepare fresh when setting up the multicycle plate.
Equipment setup
▲CRITICAL Automated multicycle image acquisition and microfluidics exchange are performed by using an inverted fluorescent microscope, a microfluidics device and driver software. A detailed description of this equipment is provided below.
Microscope setup
Configure an inverted fluorescent microscope for the fluorophores to be imaged. We use a four-channel microscope (Keyence) that has appropriate excitation light sources and emission filters for DAPI, Alexa Fluor 488, Alexa Fluor 546 and Alexa Fluor 647 and works well with integrated microfluidics systems. Select the objective lens of your choice. We use a CFI Plan Apo ƛ 2×/0.10 objective (Nikon) to obtain a low-resolution overview image of the tissue. We primarily image the multicycle in high-resolution mode by using a CFI Plan Apo ƛ 20×/0.75 objective (Nikon). A 40× or 60× oil-immersion objective will provide higher resolution. Multicycle imaging is performed in low photobleach mode to limit uneven photobleaching of reporter fluorescent oligonucleotides across the tissue.
Microfluidics device and software setup
We use the commercially available microfluidics device and software from Akoya Biosciences. Restart the computer running the driver software. Open the driver software and input the multicycle parameters, including antibody names, exposure times, number of cycles and number of tissue regions. Clean the buffer containers and run a clean cycle with ddH2O to ensure that the microfluidics device is clean. After the clean cycle is complete, add the freshly prepared hybridization and stripping buffers as well as H2 buffer to the designated containers. Empty the waste containers. Secure the CODEX black plate in its designated holder. Before starting the multicycle, remove any microscope objectives that will not be used for imaging the multicycle, such as the CFI Plan Apo ƛ 2×/0.10 objective. During the multicycle reaction, hybridization of the fluorescent oligonucleotides is performed in hybridization buffer. During imaging, the tissue is kept in H2 buffer. After imaging, fluorescent oligonucleotides are removed by using stripping buffer.
Procedure
▲CRITICAL The procedure has four main sections. Section 1 starts at Step 1 and describes the conjugation of DNA oligonucleotides (that are prepared as detailed in Box 1) to antibodies. Box 2 provides a method for validating these conjugated antibodies by flow cytometry. Section 2 starts at Step 33 and describes the validation and titration of conjugated antibodies by staining in FFPE tissues. Box 3 provides modifications for FF tissues. Section 3 starts at Step 71 and describes the CODEX multicycle reaction. Section 4 starts at Step 140 and describes the basic procedures for data analysis, including image processing, single-cell segmentation and cell-type annotation.
Section 1: antibody conjugation with DNA oligonucleotides
▲CRITICAL Use filter pipette tips for all CODEX protocols. Keep purified antibodies at 4 °C, unless otherwise specified.
Prepare antibody for conjugation ● Timing ~30 min
-
1
Measure the concentration of the purified antibody stock. We measure absorbance at 280 nm by using a UV-visible spectrophotometer.
▲CRITICAL STEP It is important that the ratio of oligo to antibody is 2:1 by weight. Measure the antibody concentration. We recommend conjugating a minimum of 100 μg.
-
2
Add 500 μl of PBS-Tween solution to a 50-kDa–molecular weight cutoff centrifugal filter column to block nonspecific antibody binding to the column.
-
3
Centrifuge at 12,000g for 2 min at RT. The retained volume should be ~20 μl; carefully remove this liquid with a pipette, taking care not to puncture the filter. Discard the column flow-through.
▲CRITICAL STEP This step involves removing contents from the column. For all subsequent steps, the antibody will be retained within the column; be careful not to discard it.
-
4
Add 100 μg of purified antibody stock to the pre-wetted filter column from Step 3.
▲CRITICAL STEP The filter columns hold a maximum of 500 μl. If the antibody volume exceeds 400 μl, first pre-concentrate the antibody in the filter column by centrifuging at 12,000g for 8 min at RT. Discard the column flow-through and add the remainder of the antibody.
-
5
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through. Proceed with Step 6 during the centrifugation step.
-
6
During Step 5, prepare the 2.5 mM TCEP solution for antibody reduction to be used in Step 7.
Partial antibody reduction with TCEP ● Timing ~1 h
-
7
Add 360 μl of 2.5 mM TCEP solution for partial antibody reduction to the antibody in the filter column.
-
8
Vortex briefly to mix and centrifuge at 1,000g for 10 s at RT in a microcentrifuge to ensure that the mixture is at the bottom of the column.
-
9
Incubate for 30 min at RT.
▲CRITICAL STEP The antibody should not be incubated in the TCEP solution for >30 min; a longer incubation time may destabilize the antibody by reducing disulfide bonds that are critical for its structure.
-
10
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
-
11
Add 400 μl of buffer C to the partially reduced antibody in the filter column to stop the reaction.
-
12
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
-
13
Add 400 μl of buffer C to the partially reduced antibody in the filter column.
▲CRITICAL STEP The second wash step ensures that excess TCEP is removed.
-
14
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through. Proceed with Step 15 during the centrifugation step.
-
15
During Step 14, prepare the oligonucleotide solution to be used in Steps 19–21.
Conjugation of partially reduced antibody with DNA oligonucleotides ● Timing ~2 h 5 min
-
16
Dissolve 200 μg of DNA oligonucleotide (prepared as described in Box 1) in 100 μl of high-salt buffer C by pipetting up and down.
-
17
Immediately transfer the dissolved oligonucleotide to a 1.5-ml microcentrifugation tube.
-
18
Add 300 μl of high-salt buffer C to bring the final volume of the oligonucleotide solution to 400 μl.
-
19
Add 400 μl of the oligonucleotide solution to the column containing the partially reduced antibody solution.
-
20
Gently mix by pipetting up and down, taking care not to puncture the filter. Centrifuge at 1,000g for 10 s at RT in a microcentrifuge to ensure that the mixture is at the bottom of the tube.
-
21
Incubate the mixture for 2 h at RT.
Wash and recover oligonucleotide-conjugated antibody ● Timing ~45 min
-
22
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
-
23
Add 450 μl of high-salt PBS to the column (wash no. 1).
-
24
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
-
25
Add 450 μl of high-salt PBS to the column (wash no. 2).
-
26
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
-
27
Add 450 μl of high-salt PBS to the column (wash no. 3).
-
28
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
▲CRITICAL STEP Three washes in high-salt PBS ensure that any excess, unreacted maleimide-modified oligonucleotide is removed.
-
29
Resuspend the antibody mixture by adding 200 μl of CODEX antibody stabilizer solution to the column (assuming a 100-μg antibody conjugation; scale the volume accordingly); mix thoroughly by pipetting up and down and washing the sides of the filter.
▲CRITICAL STEP Be careful not to puncture the filter with the pipette tip while pipetting.
-
30
Invert the column into a new collection tube.
-
31
Centrifuge at 3,000g for 2 min at RT to collect the conjugated antibody in the bottom of the tube.
-
32
Transfer the conjugated antibody to a new screw-top tube. Label it with the name of the antibody, the oligonucleotide number and the date.
■PAUSE POINT The conjugated antibody can be stored at 4 °C for ≤2 years.
Section 2: validation and titration of DNA-conjugated antibodies by CODEX
▲CRITICAL The below protocol details how to validate and titrate DNA-conjugated antibodies by CODEX tissue imaging. Box 2 provides a complementary antibody validation method using flow cytometry.
▲CRITICAL Unless otherwise stated, we perform specimen incubations in a slide-staining glass jar containing 80 ml of solution.
Tissue specimen pre-processing ● Timing ~2 h
▲CRITICAL Prepare FFPE specimens by using Steps 33–37; for FF samples, refer to Box 3.
▲CRITICAL Select the appropriate tissue to characterize the antibody of interest. For tissue selection and antibody staining validation, we recommend using pathology expertise, previous literature, the manufacturer’s recommendations, The Human Protein Atlas (https://www.proteinatlas.org/)40 and Pathology Outlines (https://www.pathologyoutlines.com/) as guides.
-
33
Mount a 4-μm-thick FFPE tissue section onto a glass slide.
-
34
Bake the slide at 70 °C for 1 h. Proceed with Step 35 during the baking step.
-
35
During Step 34, pre-heat the PT module and antigen retrieval solution (pH 9) according to Steps 38–40.
-
36
Place the specimen in 80 ml of xylene for 30 min at RT, exchanging the solvent every 10 min.
-
37
Sequentially place the specimen in a series of solutions, each time for 3 min, at RT in the following order: 100% ethanol, 100% ethanol, 95% ethanol, 95% ethanol, 80% ethanol, 70% ethanol, ddH2O and ddH2O (vol/vol). We use an automated linear stainer for this Step. If performing manually, use 80 ml of solution for each step.
Antigen retrieval ● Timing ~ 1 h 30 min
-
38
Fill the PT module tank with 1.5 liters of 1× PBS for antigen retrieval.
-
39
Fill the slide chamber with 80 ml of antigen retrieval solution (pH 9) and place it in the PT module.
-
40
Pre-heat the PT module to 75 °C; this usually takes ~15 min.
-
41
Pause the PT module and add the specimen to the slide chamber.
-
42
Run the following program on the PT module: heat to 97 °C for 10 min and then cool to 60 °C.
▲CRITICAL STEP Using a PT module ensures that the temperature changes occur in a slow, stepwise fashion.
-
43
Remove the slide chamber from the PT module and allow it to cool to RT for 30 min.
Antibody staining ● Timing 1 h 30 min of bench work, plus overnight staining
▲CRITICAL Ensure that the entire specimen is covered with solution during all steps to prevent the tissue from drying out. To decrease the staining volume to ~100 μl, create a boundary around the specimen by using a Bondic polyacrylamide gel pen. Make sure that the area around the specimen is dry before applying the gel, so that the gel firmly adheres to the slide.
▲CRITICAL For testing and validation, we generally stain one to three antibodies per specimen, by using Alexa Fluor 488, ATTO 550 and Alexa Fluor 647 as fluorescent reporters. We recommend including positive and negative controls during this process to enable confident marker validation.
-
44
Wash the specimen in 80 ml of TBS IHC wash buffer with Tween for 10 min at RT. Place it on an orbital shaker at 100 rpm.
-
45
Remove the specimen, gently dry the slide with a Kimwipe and block the tissue with 100 μl of CODEX FFPE blocking solution for 1 h at RT in a humidity chamber.
■PAUSE POINT The specimen can be blocked for ≤3 h at RT in a humidity chamber.
-
46
Prepare the conjugated antibody solution by adding the desired antibodies to CODEX FFPE blocking solution, bringing the final volume to 100 μl. Gently mix by pipetting up and down and spin at 1,000g for 10 s at RT in a microcentrifuge to ensure that the mixture is at the bottom of the tube.
▲CRITICAL STEP We generally recommend testing antibodies at a dilution of 1:100 and subsequently titrating.
-
47
After the 1-h blocking period, discard the CODEX FFPE blocking solution by tapping the slide gently onto a Kimwipe.
-
48
Add 100 μl of the conjugated antibody solution to the specimen.
-
49
Incubate overnight at 4 °C in a sealed humidity chamber. Place on an orbital shaker at 30 rpm.
Post-staining antibody fixation ● Timing ~45 min
-
50
Wash the specimen two times in 80 ml of S2, each for 2 min, at RT. Proceed with Step 51 during the washing step.
-
51
During Step 50, prepare the PFA fixation solution to be used in Step 52.
-
52
Add 100 μl of PFA fixation solution to the specimen and incubate for 10 min at RT in a humidified chamber (fixation no. 1). PFA fixation helps fix antibodies in place before methanol treatment.
-
53
Wash the specimen in 80 ml of 1× DPBS for 1 min at RT.
-
54
Place the specimen in 80 ml of ice-cold methanol for 5 min at 4 °C (fixation no. 2). Methanol is a fixative that precipitates proteins, removes lipids from cells and improves the focus for imaging membrane-bound antibodies. This fixation helps clear and focus fluorescent reporters within cell membranes. Proceed with Step 55 during the fixation no. 2 step.
-
55
During Step 54, prepare the final fixative solution to be used in Step 57.
-
56
Wash the specimen in 80 ml of 1× DPBS for 1 min at RT.
-
57
Add 100 μl of final fixative solution to the specimen and incubate for 20 min at RT in a humidified chamber (fixation no. 3). Because aldehyde fixation is reversible, fixation with irreversible BS3 crosslinker is necessary to maintain CODEX antibodies bound to tissue for many cycles of hybridization and stripping during imaging. Proceed with Step 58 during the fixation no. 3 step.
-
58
During Step 57, prepare the hybridization and stripping buffers as well as the CODEX hybridization solution to be used in Steps 60–64.
-
59
Wash the specimen in 80 ml of 1× DPBS for 1 min at RT and then transfer the specimen to S4.
■PAUSE POINT The specimen is stable in S4 at 4 °C for ≤2 weeks.
Hybridization of fluorescent oligonucleotides for antibody validation ● Timing ~45 min
-
60
Place the specimen in 80 ml of hybridization buffer for 1 min at RT; this incubation time can be extended up to 30 min.
▲CRITICAL STEP This step ensures that the tissue equilibrates to the DMSO-based buffers that are used for hybridization.
-
61
Place the specimen in 80 ml of stripping buffer for 10 min at RT.
▲CRITICAL STEP This step removes the BC4 oligonucleotides binding to the antibody-oligos and tissue and thus increases the signal-to-noise ratio.
-
62
Place the specimen in 80 ml of hybridization buffer for 1 min at RT.
-
63
Add 95 μl of the CODEX hybridization solution to the specimen, taking care to avoid creating air bubbles when pipetting. Incubate in the dark for 10 min at RT in a humidified chamber.
-
64
Place the specimen in 80 ml of hybridization buffer for 30 s at RT.
-
65
Wash the specimen two times in 80 ml of S4, each for 1 min, at RT.
-
66
Gently remove the Bondic polyacrylamide gel with a razor blade, taking care to avoid contact with the tissue.
-
67
Dry the slide with a Kimwipe, taking care to avoid contact with the tissue.
-
68
Apply one drop of Cytoseal to the specimen and gently place a coverslip atop it. Apply light pressure to the coverslip to ensure that it is flush with the microscope slide and that excess Cytoseal is released. Allow the specimen to dry in the dark for 30 min at RT.
Imaging ● Timing variable
-
69
Rinse the slide and attached coverslip with ddH2O to remove salt residues. Dry with a Kimwipe.
-
70
Image the sample as desired to characterize the antibody staining.
▲CRITICAL STEP We recommend recording the optimal exposure time for each antibody so that this time can be used for the multicycle reaction.
? TROUBLESHOOTING
Section 3: CODEX multicycle reaction
▲CRITICAL The below procedure is for FFPE specimens. For FF samples, refer to Box 3.
▲CRITICAL Unless otherwise stated, we perform specimen incubations in a coverslip glass jar containing 10 ml of solution.
▲CRITICAL It is essential to sufficiently validate, titrate and measure the appropriate exposure times of each antibody within the multicycle panel following the protocol in Section 2 before proceeding with the multicycle reaction.
▲CRITICAL The tissue is to be mounted onto a 22 × 22 mm coverslip instead of a glass slide and must be mounted in the center of the coverslip, ≥4 mm from each edge.
Coverslip preparation ● Timing ~1 h 40 min
-
71
Treat the glass coverslips with Vectabond according to the manufacturer’s instructions.
▲CRITICAL STEP The coverslips are fragile and should always be handled with coverslip forceps to prevent cracking or breaking. It is recommended that inexperienced users stain two coverslips simultaneously in case one breaks.
■PAUSE POINT Vectabond-treated coverslips can be stored indefinitely at RT.
Tissue specimen pre-processing ● Timing ~2 h
-
72
Mount a 4-μm-thick FFPE tissue section onto a Vectabond-treated coverslip by using standard histology procedures.
▲CRITICAL STEP Confirm which side of the coverslip your tissue section is on by gently scraping the edge of the section with the coverslip forceps.
-
73
Bake the coverslip at 70 °C for 1 h. Proceed with Step 74 during the baking step.
-
74
During Step 73, pre-heat the PT module and antigen retrieval solution (pH 9) according to Steps 77–79.
-
75
Place the specimen in 10 ml of xylene for 30 min at RT, exchanging the solvent every 10 min.
-
76
Sequentially place the specimen in a series of solutions, each time for 3 min, at RT in the following order: 100% ethanol, 100% ethanol, 95% ethanol, 95% ethanol, 80% ethanol, 70% ethanol, ddH2O and ddH2O (vol/vol). We use a six-well tissue culture plate for this step, with 5 ml of volume per well.
Antigen retrieval ● Timing ~1 h 30 min
-
77
Fill the PT module tank with 1.5 liters of 1× PBS for antigen retrieval.
-
78
Fill a coverslip jar with 10 ml of antigen retrieval solution (pH 9) and place it in the PT module.
▲CRITICAL STEP To prevent the coverslip jar with the antigen retrieval solution from being submerged in the tank, we recommend using an inverted coverslip jar as a base.
-
79
Pre-heat the PT module to 75 °C; this usually takes ~15 min.
-
80
Pause the PT module and add the specimen to the coverslip glass jar.
-
81
Run the following program on the PT module: heat to 97 °C for 10 min and then cool to 60 °C.
▲CRITICAL STEP Using a PT module ensures that the temperature changes occur in a slow, stepwise fashion.
-
82
Remove the coverslip jar from the PT module and allow it to cool to RT for 30 min.
Antibody staining ● Timing 2 h of bench work, plus overnight staining
▲CRITICAL Ensure that the entire specimen is covered with solution during all steps to prevent the tissue from drying out. To decrease the staining volume to ~100 μl, create a boundary around the specimen by using a Bondic polyacrylamide gel pen. Make sure that the area around the tissue is dry before applying the gel, so that the gel firmly adheres to the slide.
-
83
Wash the specimen in 10 ml of TBS IHC wash buffer with Tween for 10 min at RT. Place it on an orbital shaker at 100 rpm.
-
84
Remove the specimen, gently dry the slide with a Kimwipe and block the tissue with 100 μl of CODEX FFPE blocking solution for 1 h at RT in a humidity chamber.
■PAUSE POINT The specimen can be blocked for ≤3 h at RT in a humidity chamber.
-
85
Prepare the conjugated antibody cocktail and resuspend within FFPE blocking solution.
-
86
Add 50 μl of CODEX FFPE blocking solution to a microcentrifuge tube. Add each antibody in the multicycle panel, at its respective dilution, for a final volume of 100 μl. Once all antibodies have been added, gently mix by pipetting up and down and spin at 1,000g for 10 s at RT in a microcentrifuge to ensure that the mixture is at the bottom of the tube.
▲CRITICAL STEP We recommend keeping the conjugated antibodies on ice while preparing the antibody cocktail.
▲CRITICAL STEP We recommend centrifuging all antibodies in the panel every month at 12,000g for 8 min at 4 °C to pellet any antibody aggregates.
-
87
Add 500 μl of PBS-Tween solution to a 50-kDa–molecular weight cutoff centrifugal filter column to block nonspecific antibody binding to the column.
-
88
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; carefully remove this liquid with a pipette, taking care not to touch the filter. Discard the column flow-through.
-
89
Add the antibody cocktail from Step 86 to the pre-wetted filter column.
-
90
Centrifuge the column at 12,000g for 8 min at RT. The final volume should be ~20 μl; discard the column flow-through.
-
91
Resuspend the antibody cocktail in the column to a final volume of 100 μl by using CODEX FFPE blocking solution; mix thoroughly by pipetting up and down and washing the sides of the filter.
▲CRITICAL STEP Be careful not to puncture the filter with the pipette tip while pipetting.
-
92
Invert the column into a new collection tube and centrifuge at 3,000g for 2 min at RT. The 100 μl of conjugated antibody cocktail will be at the bottom of the tube.
-
93
After the 1-h blocking period, discard the CODEX FFPE blocking solution by tapping the coverslip gently onto a Kimwipe.
-
94
Add 100 μl of the conjugated antibody cocktail to the specimen.
-
95
Incubate overnight at 4 °C in a sealed humidity chamber. Place on an orbital shaker at 30 rpm.
Post-staining antibody fixation ● Timing ~45 min
-
96
Wash the specimen two times in 5 ml of S2, each for 2 min, at RT. Proceed with Step 97 during the washing step.
-
97
During Step 96, prepare the PFA fixation solution to be used in Step 98.
-
98
Add 100 μl of PFA fixation solution to the specimen and incubate for 10 min at RT in a humidified chamber (fixation no. 1).
-
99
Wash the specimen in 10 ml of 1× DPBS for 1 min at RT.
-
100
Place the specimen in 10 ml of ice-cold methanol for 5 min at 4 °C (fixation no. 2). Proceed with Step 101 during the fixation no. 2 step.
-
101
During Step 100, prepare the final fixative solution to be used in Step 103.
-
102
Wash the specimen in 10 ml of 1× DPBS for 1 min at RT.
-
103
Add 100 μl of final fixative solution to the specimen and incubate for 20 min at RT in a humidified chamber (fixation no. 3). Proceed with Step 104 during the fixation no. 3 step.
-
104
During Step 103, prepare the microfluidics device (Steps 108–110).
-
105
Wash the specimen in 10 ml of 1× DPBS for 1 min at RT.
-
106
Gently remove the Bondic polyacrylamide gel with a razor blade, taking care to avoid contact with the tissue.
-
107
Place the specimen in 10 ml of S4 until ready to mount it to the acrylic plate.
■PAUSE POINT The specimen is stable in S4 at 4 °C for ≤2 weeks.
Preparing the microfluidics device ● Timing ~10 min
-
108
Restart the computer attached to the microfluidics device.
-
109
Add the appropriate volumes of H2 buffer and hybridization and stripping buffers to the designated canisters on the microfluidics device.
-
110
Insert the desired microscope objectives and place the liquid collector cup and collector device inside the microscope; this will capture any stray liquid before it leaks into the internal instrumentation.
Creating a fluorescent oligonucleotide plate ● Timing ~30 min
▲CRITICAL Each well on the Corning black 96-well plate contains the fluorescent oligonucleotide(s) for one CODEX cycle. The total volume in each well is 250 μl, including 10 μl of each fluorescent oligonucleotide. The volume of plate buffer therefore varies according to the number of fluorescent oligonucleotides per cycle (250 μl – 10 μl × no. of fluorescent oligonucleotides):
| Cycle | DAPI | Alexa448 | ATTO55O | Alexa647 | Volume of plate buffer (μl) | Total well volume (μl) |
|---|---|---|---|---|---|---|
| 1 | Hoechst | Blank | Blank | Blank | 250 | 250 |
| 2 | Hoechst | Marker 1 | Marker 2 | Marker 3 | 220 | 250 |
| 3 | Hoechst | Blank | Marker 4 | Marker 5 | 230 | 250 |
| 4 | Hoechst | Blank | Blank | Marker 6 | 240 | 250 |
-
111
Add 250 μl of plate buffer to well A1.
▲CRITICAL STEP We recommend starting with a blank cycle, which can be used for background subtraction during imaging processing.
-
112
Add the appropriate volume of plate buffer to the remaining wells, one well for each cycle starting at well A2.
▲CRITICAL STEP If >12 wells are needed for the multicycle, continue filling wells on the next row, starting at B1.
-
113
Add 10 μl of the appropriate fluorescent oligonucleotide working solution to the designated wells (wells A2–A?) according to its cycle number to be imaged.
-
114
Add 250 μl of DRAQ5 staining solution to the final well of the multicycle.
▲CRITICAL STEP We recommend including DRAQ5 staining solution in the final cycle because this helps with downstream quantitative analysis. (i.e., clean-up gating, single-cell segmentation, etc.).
-
115
Seal the CODEX black plate with aluminum sealing film (as shown in Fig. 6a) and place it in its designated holder on the microfluidics device. The aluminum sealing film will be punctured by the Akoya microfluidics robot during the multicycle reaction. The CODEX black plate will be kept at RT during the multicycle reaction.
■PAUSE POINT The fluorescent oligonucleotide plate is stable at 4 °C for 2 weeks.
Fig. 6 |. Components required for the CODEX experiment.

a, A black plate with aluminum sealing film, containing the fluorescent oligonucleotides. b, An acrylic plate oriented with the notch at the bottom right corner (red circle). c, A DMSO-resistant mounting gasket. d, An acrylic plate with a secured coverslip. e, A mounted acrylic plate and secured coverslip within the inverted fluorescence microscope, with attached delivery and vacuum ports.
Coverslip mounting ● Timing ~10 min
▲CRITICAL The coverslip mounting protocol included herein uses a custom-made acrylic plate and acrylic plate holder (blueprints available upon request). Akoya Biosciences sells their own stage; if using their product, follow their mounting protocol.
-
116
Visually inspect the coverslip integrity.
▲CRITICAL STEP If there are any cracks, do not proceed. Never run a cracked coverslip sample on the microscope, because it can lead to spillage and irreversible microscope damage, resulting in high repair costs.
-
117
Remove the plastic film from both sides of the acrylic plate. Orient the acrylic plate with the notch at the bottom right corner (i.e., red circle), as shown in Fig. 6b.
-
118
Remove the plastic film from one side of the DMSO-resistant mounting gasket (as shown in Fig. 6c). Place it on the laboratory bench with the sticky side up.
-
119
Align and gently place the acrylic plate on top of the DMSO-resistant mounting gasket. Apply firm, outward pressure with your finger or coverslip forceps to the secured gasket to remove any trapped air. Ensure that the gasket does not extend into the well of the acrylic plate.
▲CRITICAL STEP The microscope stage is designed to fit the acrylic holder in the specific orientation shown in Fig. 6e. Specifically, the screws lock the acrylic plate into place at the notches for attachment to the microscope. Therefore, the mounting gasket and coverslip must be adhered such that the coverslip forms a well when the acrylic holder is attached to the microscope.
-
120
Flip the acrylic plate over, such that the notch is at the bottom left corner and the gasket is facing up.
-
121
Remove the second plastic film from the DMSO-resistant mounting gasket by using forceps.
-
122
Cut a small piece of cling film the size of the tissue.
-
123
Place the coverslip (from Step 107) in a six-well plate with 5 ml of H2 buffer.
-
124
Apply a small piece of cling film (pre-wet with H2 buffer) on top of the tissue to prevent tissue drying.
▲CRITICAL STEP Make sure that the cling film piece is smaller than the opening in the mounting acrylic, with ≥1 mm space on each side. This ensures that it is not stuck to the DMSO-resistant mounting gasket and that there are no gaps in the tape.
-
125
Cleanse the coverslip with a Kimwipe dipped in ddH2O to remove excess salts and then dry both sides of the coverslip with a Kimwipe, taking care to avoid contact with the tissue that is covered with the cling film.
▲CRITICAL STEP It is important that no liquid come off the side of the cling film and that the area around the cling film and the other side of the coverslip are dry. Otherwise, it will be drawn under the DMSO tape through capillary action, and the tape will not stick.
-
126
Gently invert and place the coverslip on top of the DMSO-resistant mounting gasket, such that the tissue is facing down and is inside the well of the acrylic plate. Gently apply outward pressure with your finger or forceps to the edges of the coverslip to remove any trapped air. The coverslip should now form a well with the acrylic mold as shown in Fig. 6d.
▲CRITICAL STEP If the mounting gasket gets wet while mounting the sample, the seal will be incomplete, and the sample will leak. A mounted coverslip cannot be removed from the acrylic without breaking. If a leak is suspected, a second mounting gasket can be placed on top of the coverslip to seal the leak. Because this results in increased thickness of the sample, this could lead to difficulties when imaging with a 40× oil objective if the tissue is close to the edge.
-
127
Immediately add 400 μl of H2 buffer to the acrylic plate well to ensure that the tissue stays hydrated. The small piece of cling film will float to the surface and should be removed at this time.
▲CRITICAL STEP When adding solution to or removing it from the acrylic well, take care not to eject or aspirate near the tissue or apply pressure to the coverslip, because this could dislodge the tissue or break the coverslip.
-
128
Remove the 400 μl of H2 buffer.
-
129
Add 400 μl of Hoechst staining solution to the specimen for 1 min at RT in the dark.
-
130
Wash three times by adding and aspirating 400 μl of H2 buffer, each for 10 s, at RT.
-
131
Add 400 μl of H2 buffer to the well.
-
132
Mount the acrylic plate onto a custom-designed plate holder (Fig. 4a) and secure it to the stage of the inverted fluorescence microscope. Attach the solution delivery and vacuum ports to the stage, taking care to avoid contact with the tissue as shown in Fig. 6e.
Multicycle imaging ● Timing Variable, depending on the number of cycles and size of the tissue
-
133
Image the sample as desired by setting the appropriate parameters for the microscope and microfluidics device. Typical parameters include the number of tissue regions, region size, number of Z-stacks, number of fluorescent channels and number of cycles.
▲CRITICAL STEP We highly recommend starting with a stripping cycle to remove any nonspecific oligonucleotide binding and ensure an equal background signal across cycles.
▲CRITICAL STEP If including a DRAQ5 stain in the last cycle, make sure that the microfluidics program does not perform a final washing step.
▲CRITICAL STEP We recommend using a 2× objective lens to acquire an overview image. This will facilitate finding the regions of interest for imaging during the multicycle reaction.
▲CRITICAL STEP For multicycle reactions, we generally use a 20× air objective lens, because this provides a suitable balance between image resolution and imaging time. We have also used 40× and 60× oil-immersion objectives, when higher resolution images were desired.
? TROUBLESHOOTING
-
134
Once the multicycle is complete, confirm that the images for all regions and cycles have been saved. Occasionally, the software or microfluidics hardware encounters an error during the multicycle. If this happens, you can re-image the affected regions and cycles.
Post-multicycle H&E staining and imaging ● Timing ~30 min
▲CRITICAL If using the Akoya Biosciences stage, consult them regarding the utility of post-imaging H&E staining.
-
135
Remove the microfluidic ports from the microscope stage. Do not remove the acrylic plate containing the specimen.
-
136
Remove all fluid from the acrylic well with a pipette.
-
137
Sequentially add 200 μl of the following solutions to the acrylic well, each time for 20 s unless otherwise stated, in the following order: 100% ethanol, 100% ethanol, 70% ethanol, ddH2O, hematoxylin for 60 s, warm tap water, 70% ethanol, eosin for 90 s, 70% ethanol, 95% ethanol, 100% ethanol, xylene and xylene (vol/vol).
-
138
Remove xylene from the acrylic well and add enough Cytoseal to fill the chamber (~10 drops).
-
139
Image immediately in brightfield mode, ensuring that the same regions and number of Z-stacks are captured as was done for the multicycle.
▲CRITICAL STEP Avoid contaminating the microscope or any part of the stage with eosin. Eosin is highly autofluorescent, and the slightest amount of contamination may affect subsequent experiments. After each CODEX multicycle experiment, wash the stage with 70% ethanol, soap and water.
Section 4: analysis of CODEX multiplexed imaging data
▲CRITICAL Many computational platforms exist to process and analyze multiplexed tissue imaging data. We provide the basics for using the platform developed by our laboratory to analyze CODEX data; it is freely available at https://github.com/nolanlab/. A more detailed description of these computation tools is described in Supplementary Notes 1–4 and the associated Supplementary Figs. 1–3. This software is freely accessible from github (Akoya Biosciences sells their own accompanying software).
▲CRITICAL Here, we show the computational parameters used to analyze an FFPE human tonsil; these parameters provide a good starting point for any experiment.
Image processing ● Timing Variable, depending on the amount of raw data generated (~24 h on average)
-
140
Process the raw microscope images by using the CODEX Uploader; parameters are provided in Fig. 7. This interface combines the image dimensions and drift compensation to yield the (i) concatenated individual tiles across all dimensions, (ii) best focal plane individual tiles and (iii) low-resolution montage of the stitched, best focal plane tiles for an entire region. The output files are in TIFF format. Currently supported input files are of TIFF format from Keyence and Zeiss microscope data formats and are of 16-bit type. For more detailed information about processing requirements, please see the readme file from our GitHub (https://github.com/nolanlab/CODEX#readme).
▲CRITICAL STEP We recommend using the low-resolution montage image to assess the antibody staining quality from the multicycle experiment.
Fig. 7 |. Parameters for processing raw microscope images by using the CODEX Uploader.
These include tissue size (i.e., region width and height), degree of tile overlap (0% if only imaging a single tile compared with 30% if the tissue size is greater than one tile) and the optional selection of deconvolution, background subtraction and H&E staining. These parameters will change for each experiment.
Data analysis ● Timing Variable, depending on the number of cells, variation in cell size and morphology across different cell types within the tissue
-
141
Perform single-cell segmentation on the uploaded data (i.e., TIFF files) by using the CODEX Segmenter; parameters are provided in Fig. 8. This interface is based on volumetric watershed segmentation. The output files include flow cytometry standard (FCS) and comma-separated values dataframes for each region that include channel intensities and location for each segmented cell. Additional output files include text and portable network graphics of the segmentation masks for individual tiles.
▲CRITICAL STEP We recommend performing segmentation on one to three tiles to find the optimal parameters before processing the entire dataset. See additional instructions and caveats with single-cell segmentation within Supplementary Note 3.
-
142
Perform cleanup gating on the segmented data (i.e., compensated FCS files) to remove imaging artifacts and tissue irregularities (i.e., folds), with any flow cytometry analysis software. First, gate for nucleated cells by using the Hoechst stain from channel 1 and the DRAQ5 stain. Next, gate based on the Z-position, selecting cells from the best focal planes (Fig. 9). These data are exported as FCS files.
-
143
Perform cell-type annotation on the cleaned data (i.e., FCS files) by using VorteX; parameters are provided in Fig. 10. This interface is an unsupervised, X-shift clustering program29, which processes large datasets by using the K-nearest neighbor estimation of cell event density and arranges cell types according to the antibody markers that the user selects. These data are exported as a comma-separated values file.
▲CRITICAL STEP We recommend inspecting and verifying each cell-type cluster visually within the tissue. Verifying solely on the basis of marker expression will lead to errors in cluster assignment, which will be carried forward in additional computational analysis.
? TROUBLESHOOTING
-
144
Other computation tools, which have also been developed by our laboratory, include cluster visualization within the tissue (using FIJI), minimal spanning trees and force-directed layouts (using VorteX), cellular neighborhood analysis (using Python) and complex statistical analyses (using Python). These tools are available at https://github.com/nolanlab (refs.2,29).
Fig. 8 |. Starting parameters for performing single-cell segmentation on the uploaded data by using the CODEX Segmenter.
These include cell radius, max/min/relative cutoff, cell size cutoff factor, nuclear stain channel/cycle and if desired, membrane stain channel/cycle (in general, this parameter is not used, and thus the cycle is −1). These parameters will change for each experiment.
Fig. 9 |. Cleanup gating of segmented data.
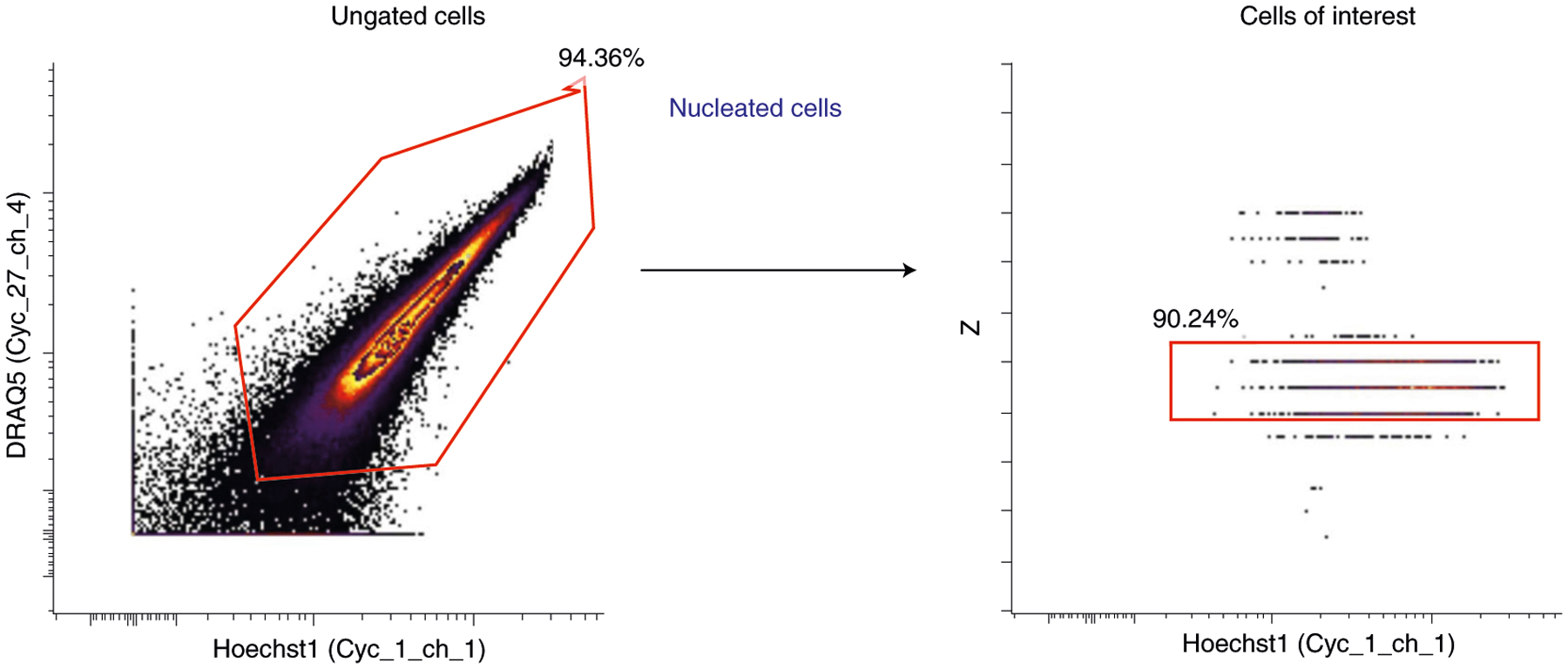
First, nucleated cells are gated by using Hoechst and DRAQ5 nuclear stains (left plot). Then, cells are gated on the basis of the Z-position of the best focal planes (right plot).
Fig. 10 |. Starting parameters for performing cell-type annotation on cleaned data by using VorteX.
These include not using numerical transformation, noise thresholding, feature re-scaling or normalization. The distance measure is angular, with density estimate N nearest neighbor spanning from 150 to 5, with 30 steps. These parameters can be adjusted as needed.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 70 | Inefficient conjugation of oligonucleotide to antibody | Improper antibody starting amount | Measure the concentration of the starting antibody stock by A280 absorbance. Ensure that the conjugation amount is 100 μg |
| Carrier proteins within antibody stocks | Check the manufacturer specifications for carrier proteins (e.g., BSA, tissue culture supernatant, ascites or other protein stabilizer). Obtain carrier-free antibodies or purify the antibody before conjugation | ||
| Over-reduction of antibody by TCEP | Ensure that antibody conjugation is performed at room temperature (i.e., avoid incubation next to heatgenerating instruments) and that reduction time does not exceed 30 min | ||
| Improper staining result after conjugation to newly activated oligonucleotide | Reduction in maleimide functional groups from oligonucleotide | Ensure the the oligonucleotide is working by conjugating it to a known working antibody (e.g., aCD3). If not, then the deprotection procedure may have gone wrong, or maleimide groups have destabilized, in which case replace it with fresh stock | |
| Unsuccessful staining (e.g., no signal or nuclear staining) | Incompatible oligonucleotide-antibody pairing | If antibody staining works with conventional immunohistochemistry, repeat conjugation with a different oligonucleotide | |
| High fluorescence background for all channels and markers | Poor tissue morphology | Select samples that were frozen/fixed immediately after removing them from the subject. Confirm proper morphology with H&E staining | |
| The coverslip was contaminated | Ensure that the coverslip was not exposed to any fluorescent material (e.g., permanent marker during ethanol/methanol steps) and that all salt precipitates are removed before mounting the coverslip to the stage. Also ensure that paraffin was completely removed during baking and xylene washing | ||
| Weak antibody signal/high background | Low-abundance target antigen | Use ATTO550 or Alexa647 channels (Alexa488 has a higher background, especially in FFPE tissue). If this is insufficient, select a corresponding fluorescent oligonucleotide that is labeled with ATTO550 or Alexa647 (lower tissue autofluorescence) on both the 5′ and 3′ ends. If that does not work, consider trying a different antibody clone | |
| The antibody has not been titrated | Consider increasing the antibody staining concentration to increase the signal | ||
| Proper antibody staining but high background (for one channel) | Antibody staining at too high a concentration | Repeat staining at a two- to fourfold lower dilution | |
| Nonspecific fluorescent globules observed | The antibody aggregates | Spin down the antibody for 8 min at 12,000g at 4 °C to pellet aggregates (we recommend doing this monthly for the entire antibody panel). Extract antibody by pipetting from the top of the supernatant | |
| 133 | High fluorescence background for all channels and markers | Nonspecific oligonucleotide fluorescent signal | Make sure to perform a stripping cycle with stripping buffer before multicycle imaging to remove excess, nonspecific oligonucleotides |
| 143 | The vortex cluster is composed of two distinct clusters | Clustering is driven by the strength of a unifying marker (e.g., GATA3 stains basal epithelium and Th2 cells) | Over cluster (i.e., beyond the elbow point) and manually merge clusters |
Timing
Section 1, antibody conjugation with DNA oligonucleotides
Steps 1–6, preparing the antibody for conjugation: ~30 min
Steps 7–15, antibody reduction with TCEP: ~1 h
Steps 16–21, conjugation of the reduced antibody with DNA oligonucleotides: ~2 h 5 min
Steps 22–32, washing and recovering the oligonucleotide-conjugated antibody: ~45 min
Section 2, validation and titration of conjugated antibodies by CODEX
Steps 33–37, tissue specimen pre-processing: ~2 h
Steps 38–43, antigen retrieval: ~1 h 30 min
Steps 44–49, antibody staining: ~1 h 30 min of bench work, plus overnight staining
Steps 50–59, post-staining antibody fixation: ~45 min
Steps 60–68, hybridizing fluorescent oligonucleotides for antibody validation: ~45 min
Steps 69 and 70, imaging: variable
Section 3, CODEX multicycle reaction
Step 71, coverslip preparation: ~1 h 40 min
Steps 72–76, tissue specimen pre-processing: ~2 h
Steps 77–82, antigen retrieval: ~1 h 30 min
Steps 83–95, antibody staining: ~2 h of bench work, plus overnight staining
Steps 96–107, post-staining antibody fixation: ~45 min
Steps 108–110, preparing the microfluidics device: ~10 min
Steps 111–115, creating a fluorescent oligonucleotide plate: ~30 min
Steps 116–132, coverslip mounting: ~10 min
Step 133 and 134, multicycle imaging: variable, depending on the number of cycles and the size of the tissue
Steps 135–139, post-multicycle H&E staining and imaging: ~30 min
Section 4, analysis of CODEX multiplexed imaging data
Step 140, image processing: variable, depending on the amount of raw data generated (~24 h on average)
Steps 141–144, data analysis: variable, depending on the variation of cell size and morphology across different cell types within the tissue
Box 1, activation of maleimide-modified DNA oligonucleotides: ~4 h 30 min, plus overnight lyophilisation
Box 2, validation of DNA oligonucleotide-conjugated antibodies by flow cytometry: ~1 h 30 min
Box 3, preparing fresh-frozen tissue for CODEX: ~5 h of bench work, plus variable imaging time
Anticipated results
Validation and titration of oligonucleotide-conjugated antibodies in FFPE and FF tissues
Purified antibodies are conjugated to unique DNA oligonucleotides as described in section 1 of the procedure. Each conjugated antibody is then stained in the appropriate FFPE or FF tissue as described in section 2 and Box 3 of the procedure. Antibody validation is critical for assessing the specificity and affinity of the conjugated antibody.
When validating antibodies, it is critical to include appropriate positive and negative controls. For instance, to validate an antibody against CD4, a positive control could be CD3, and a negative control could be CD8 or CD20. In addition, when validating antibodies to proteins with specific mutations (e.g., IDH-1 mutation in glioma), checkpoint molecules (e.g., PD-1 and LAG3) or stimulation-inducible intracellular molecules (e.g., pSTAT3), it is vital that the tissue sample being used for testing stains positive for these markers by traditional IHC. We validate all of our staining results against traditional IHC images as shown in Fig. 11a.
Fig. 11 |. Validation of CODEX antibody staining.
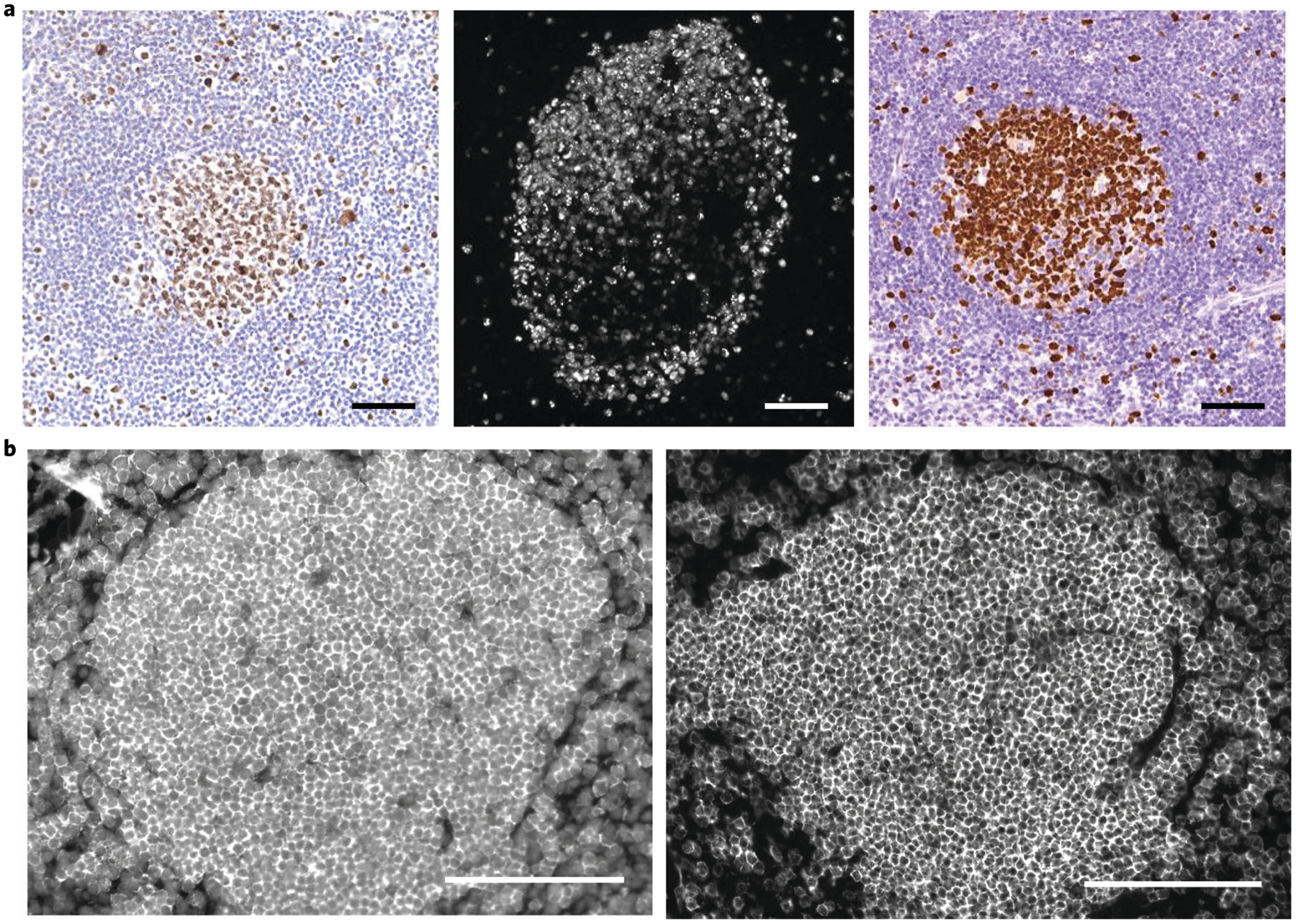
a, Successful antibody validation of Ki-67 staining of a human FFPE tonsil by manual IHC (left panel), CODEX single staining with the conjugated antibody (middle panel) and robotic IHC from The Protein Atlas (right panel). b, Comparison stainings of the mB220 conjugated-antibody with an unsuccessful (left panel; incorrect nuclear staining of many cells secondary to an adverse antibody-oligonucleotide combination) and successful (right panel; correct membrane staining of B cells) outcome. Both stainings were performed at a 1:100 dilution and 1/6-s exposure time. Written informed consent was obtained from all patients. All samples were fully de-identified, and the study was exempt from ethics approval (no human subjects research). For mouse studies, appropriate institutional regulatory board permission was obtained. Scale bars, 100 μm.
Once a conjugated antibody is validated, it must be titrated to determine the optimal concentration for use in a CODEX multicycle experiment. In the titration experiment, we generally stain the specimen with a 1:100 dilution of conjugated antibody. This dilution is then titrated depending on the signal-to-noise ratio. Most oligonucleotide-conjugated antibodies used to stain FFPE tissue are used at a dilution between 1:50 and 1:200. Most oligonucleotide-conjugated antibodies used to stain FF tissue are used at a dilution between 1:100 and 1:200. To date, we have successfully validated and titrated >200 oligonucleotide-conjugated antibodies against nuclear, cytoplasmic and membranous proteins for FFPE and FF tissues (1,2).
Certain antibody-oligonucleotide combinations are not optimal and can lead to nuclear staining and high background as seen in Fig. 11b, left panel. When this occurs, the antibody should be conjugated to a different oligonucleotide and re-tested (Fig. 11b, right panel). If a successful antibody stain cannot be obtained with a different oligonucleotide, a different antibody clone may need to be selected.
Performing and analyzing a CODEX multicycle experiment
To demonstrate the utility of CODEX, we stained and imaged an FFPE human tonsil with a panel of 57 validated antibodies including antibodies that bind to stromal, epithelial and immune markers (Supplementary Tables 2 and 3), according to section 3 of the procedure. The imaging parameters for this multicycle experiment were as follows: region size, 7 × 9 tiles (~200,000 cells); number of z-stacks, 16 (such that the best focal plane is acquired for each tile across the tissue region); objective lens, 20×; pitch, 1.5 μm; and number of cycles, 27 (such that each of the 57 antibodies is within channel 2, 3 or 4), with each cycle taking ~1 h (~30 min of imaging time and ~30 min of buffer exchanges/incubation reactions). Tile size at 20× is approximately 0.55 mm × 0.42 mm, and the total image size for this experiment was ~3.3 mm × 3.5 mm. H&E staining was performed upon completion of the multicycle reaction. The imaging dataset was then processed according to section 4 of the procedure (Supplementary Figure 1a; the user interfaces for the CODEX Uploader and Segmentation software are shown in Supplementary Figure 1b). A seven-color overlay image of the FFPE tonsil (Fig. 12a) enabled T cells, B cells, neutrophils, epithelium, macrophages and lymphatics to be distinguished at the single-cell level. Only seven markers are shown for clarity in the figure, but the 57 markers detected can be simultaneously visualized and quantified. We next performed single-cell segmentation, generating a dataframe of cell size, X/Y coordinates and quantification of each marker as well as a segmentation mask image (Supplementary Figure 1c). The data were then cleaned by manual gating (Supplementary Figure 1d).
Fig. 12 |. Results from a CODEX multicycle experiment of human FFPE tonsil.
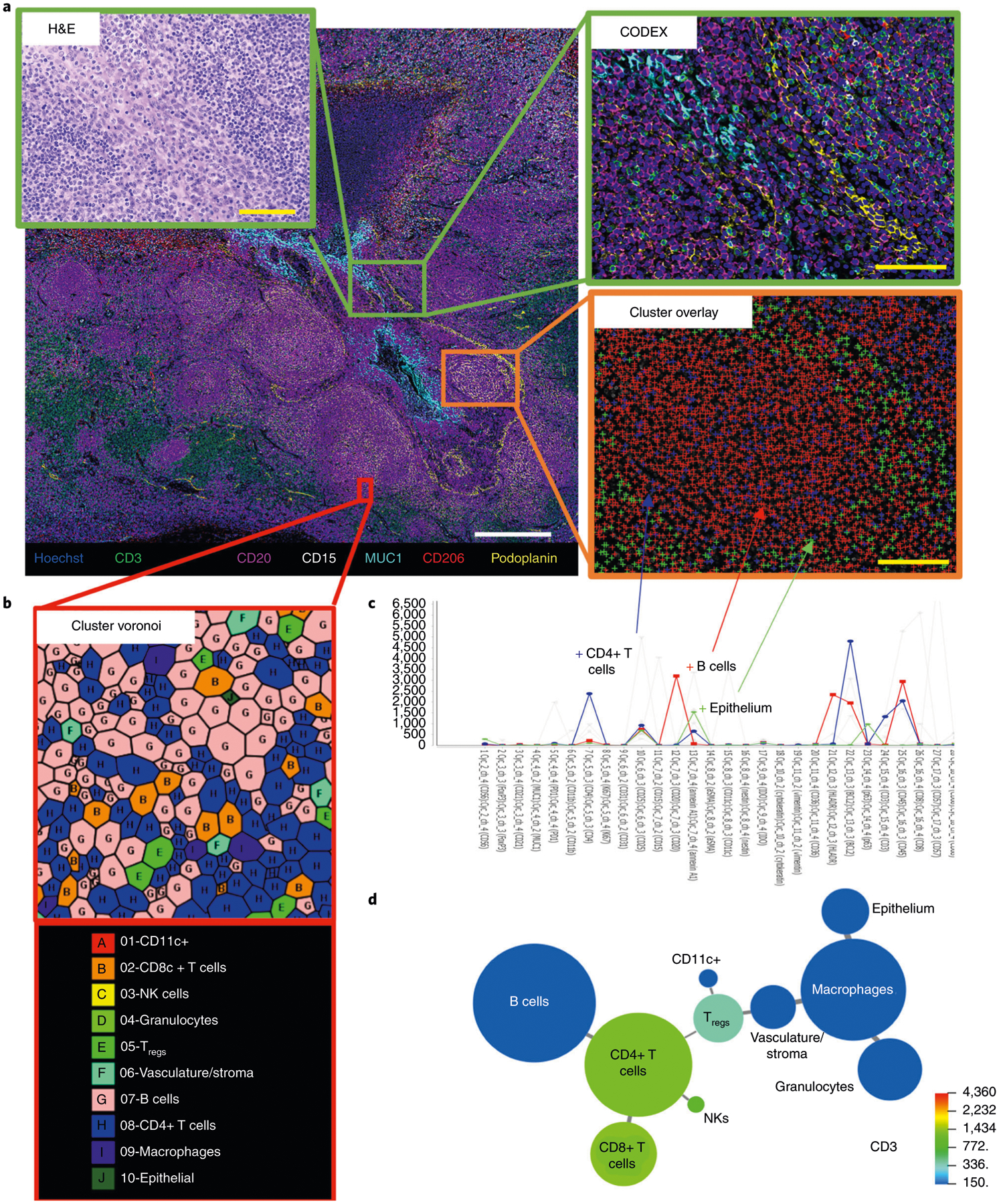
a, Seven-color fluorescent overlay image showing Hoechst, CD3, CD15, CD20, CD206, MUC-1 and podoplanin (colored according to the bottom panel) of a region of 7 × 9 tiles of tonsil; the zoomed-in green boxes show an H&E image and CODEX fluorescent image at higher resolution. b, A zoomed-in Voronoi diagram of the 10 unsupervised clusters obtained from the VorteX interface. c, The average expression profiles of CD4+ T cells, B cells and epithelial cells are shown, and a zoomed-in region (orange box) maps these clusters onto the tissue by using FIJI, with blue crosses representing CD4+ T cells, red crosses representing B cells and green crosses representing epithelial cells (gray bars in the clustering interface show other cluster profiles not highlighted here). The y axis of the graph represents the average fluorescent intensity of the cells within each cluster; the graph was cropped to zoom in on a subset of markers used in CODEX. d, A minimal spanning tree of the 10 identified clusters, with the size of each circle corresponding to the relative size of the cluster across the tissue and the color representing the average expression of CD3 in quantile scale. Written informed consent was obtained from all patients. All samples were fully de-identified, and the study was exempt from ethics approval (no human subjects research). Yellow scale bars, 100 μm. White scale bar, 500 μm. NK, natural killer; Tregs, regulatory CD4+ T cells.
Unsupervised cell-type annotation was then performed, resulting in 32 major cell-type clusters (Supplementary Figure 1e). For simplicity, we manually merged many of these clusters (e.g., plasma cells and B cells into a ‘B cell cluster’ and lymphatics, vasculature and stroma into a ‘vasculature/stroma cluster’), resulting in 10 major cell-type clusters. These final clusters include those cell types present in high abundance (B cells, CD4+ T cells and macrophages), medium abundance (epithelial, vasculature/stroma, granulocytes, CD8+ T cells and regulatory CD4+ T cells (Tregs)) and low abundance (natural killer cells and CD11c+ dendritic cells). The limited number of clusters facilitates the visual interpretation of spatial cellular relationships within the tissue by using Voronoi diagrams (Fig. 12b). It is critical to assess the accuracy of unsupervised clustering; we do this by mapping the cell-type clusters back onto the original image in ImageJ (Fig. 12c). This visual verification approach enables each cell-type cluster to be analyzed in the context of specific marker expression (i.e., by using the CODEX fluorescent images) and cell cytology (i.e., by using the H&E image).
Finally, to observe the relationships between distinct cell-type clusters, we generated a minimal spanning tree (Fig. 12d). By visualizing the minimal spanning tree in the context of CD3 expression, it is clear the T cell clusters are present at the highest frequency, confirming the accuracy of unsupervised clustering. Moreover, adaptive and immune cell types (i.e., B cells, T cells and natural killer cells) are grouped together and are separated from the grouped innate immune cell types (i.e., granulocytes and macrophages), further validating the accuracy of clustering assignments.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Tissue morphology conserved over multicycle imaging.

One tile of the CODEX multicycle for the human tonsil was segmented by using the CODEXSegm software and the nuclear stain for cycle 1 or cycle 26 (nuclear images on the top; segmentation masks on the bottom). For both instances, 2,464 cells were identified. Scale bar, 100 μm.
Extended Data Fig. 2 |. Analysis of healthy and cancerous tissue morphology during multicycle imaging.
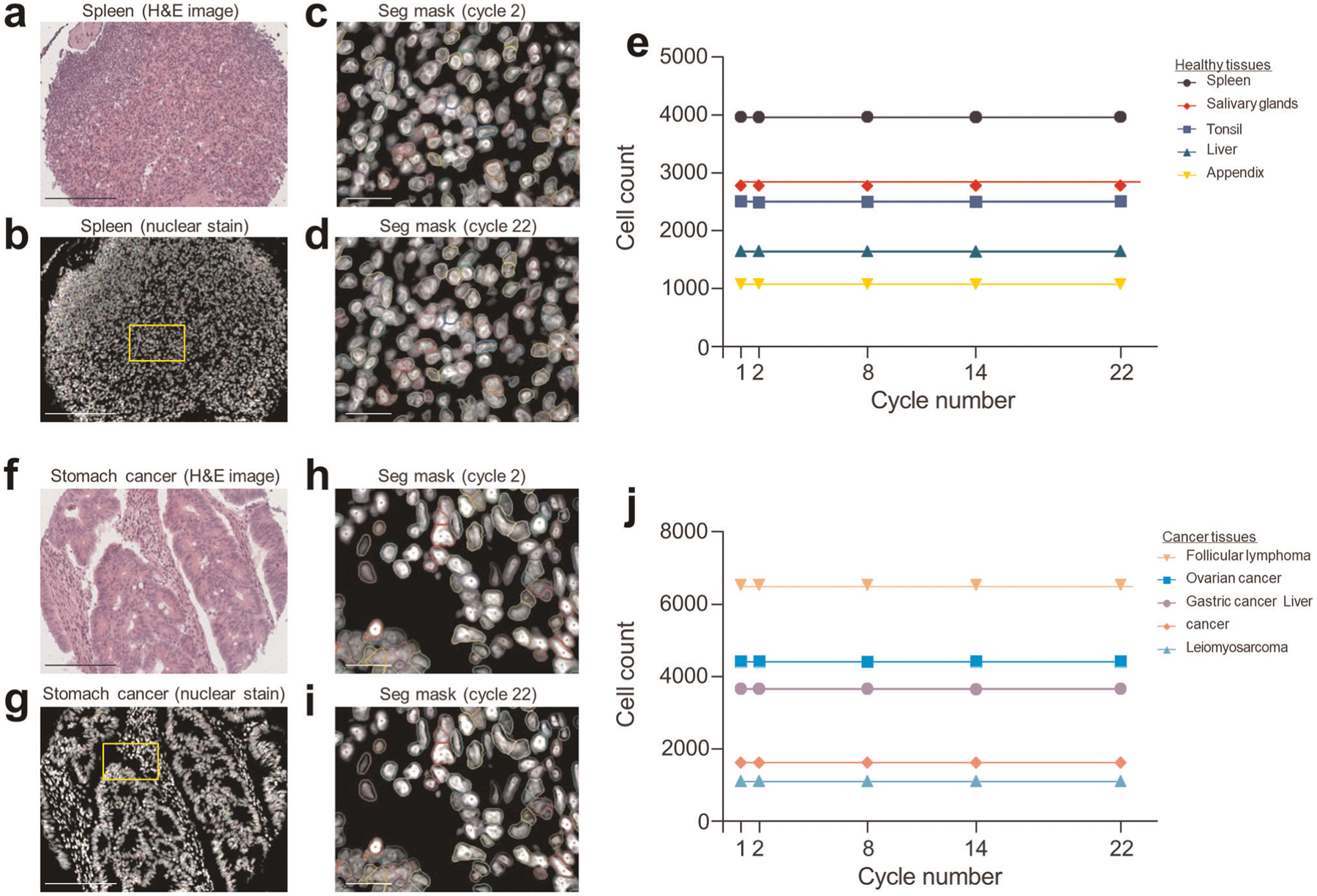
a, H&E image of a healthy spleen. Scale bar, 200 μm. b, Corresponding nuclear (Hoechst) stained image and cellular segmentation (Seg) mask. Scale bar, 200 μm. c, Zoomed-in nuclear stained image and cellular segmentation mask from cycle 2. Scale bar, 20 μm (Extended Data Applied Sciences 28 April 2020). d, Zoomed-in nuclear stained image and cellular segmentation mask from cycle 22. Scale bar, 200 μm. e, Line plot of total cell count per tissue microarray core, measured at cycles 1, 2, 8, 14 and 22, for five healthy tissues. Lines are colored according to the corresponding legend. f, H&E image of stomach cancer. Scale bar, 200 μm. g, Corresponding nuclear (Hoechst) stained image and cellular segmentation mask. Scale bar, 200 μm. h, Zoomed-in nuclear stained image and cellular segmentation mask from cycle 2. Scale bar, 20 μm. i, Zoomed-in nuclear stained image and cellular segmentation mask from cycle 22. Scale bar, 200 μm. j, Line plot of total cell count per tissue microarray core, measured at cycles 1, 2, 8, 14 and 22, for five cancer tissues. Lines are colored according to the corresponding legend.
Supplementary Material
Acknowledgements
We thank Angelica Trejo, Han Chen, Kenyi Donoso, Nilanjan Mukherjee and Gustavo Vazquez for excellent technical assistance and Dr. Xavier Rovira-Clavé for critical comments on the manuscript. This work was supported by the U.S. National Institutes of Health (2U19AI057229-16, 5P01HL10879707, 5R01GM10983604, 5R33CA18365403, 5U01AI101984-07, 5UH2AR06767604, 5R01CA19665703, 5U54CA20997103, 5F99CA212231-02, 1F32CA233203-01, 5U01AI140498-02, 1U54HG010426-01, 5U19AI100627-07, 1R01HL120724-01A1, R33CA183692, R01HL128173-04, 5P01AI131374-02, 5UG3DK114937-02, 1U19AI135976-01, IDIQ17X149, 1U2CCA233238-01 and 1U2CCA233195-01); the U.S. Department of Defense (W81XWH-14-1-0180 and W81XWH-12-1-0591); the U.S. Food and Drug Administration (HHSF223201610018C and DSTL/AGR/00980/01); Cancer Research UK (C27165/A29073); the Bill and Melinda Gates Foundation (OPP1113682); the Cancer Research Institute; the Parker Institute for Cancer Immunotherapy; the Kenneth Rainin Foundation (2018-575); the Silicon Valley Community Foundation (2017-175329 and 2017-177799-5022); the Beckman Center for Molecular and Genetic Medicine; Juno Therapeutics, Inc. (122401); Pfizer, Inc. (123214); Celgene, Inc. (133826 and 134073); Vaxart, Inc. (137364); and the Rachford & Carlotta A. Harris Endowed Chair (to G.P.N.). C.M.S. was supported by the Swiss National Science Foundation (P300PB_171189 and P400PM_183915) and the Lady Tata Memorial Trust, London, UK. D.P. was supported by an NIH T32 Fellowship (AR007422), an NIH F32 Fellowship (CA233203), a Stanford Dean’s Postdoctoral Fellowship, a Stanford Cancer Institute Fellowship and Stanford’s Dermatology Department. J.W.H. was supported by an NIH T32 Fellowship (T32CA196585) and an American Cancer Society—Roaring Fork Valley Postdoctoral Fellowship (PF-20-032-01-CSM). Parts of Fig. 2 were created with BioRender.com.
Competing interests
J.K.-D. is an employee of Akoya Biosciences, Inc. N.S. is an employee of Becton Dickinson, Inc. G.P.N. received research grants from Pfizer, Inc.; Vaxart, Inc.; Celgene, Inc.; and Juno Therapeutics, Inc. during the course of this work. N.S., Y.G. and G.P.N. are inventors on US patent 9909167, granted to Stanford University, that covers some aspects of the technology described in this paper. J.K.-D., N.S., Y.G. and G.P.N. have equity in and/or are scientific advisory board members of Akoya Biosciences, Inc. C.M.S. is a scientific advisor to Enable Medicine, Inc. The other authors declare no competing interests.
Footnotes
Code availability
The image processing and data analysis tools presented in this article are available at https://github.com/nolanlab. The code used in this protocol has been peer reviewed.
Extended data is available for this paper at https://doi.org/10.1038/s41596-021-00556-8.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-021-00556-8.
Data availability
We have uploaded both the concatenated CODEX imaging montage of the tissue (https://doi.org/10.6084/m9.figshare.12986981) and single-cell segmented data (https://doi.org/10.6084/m9.figshare.12986099) used in this paper to figshare (https://figshare.com/). The size of the raw imaging data is too large to be stored in a public repository and will therefore be stored in a private cloud-based server. Access to these data will be provided by the corresponding authors upon request.
References
- 1.Goltsev Y et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schürch CM et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy-Darling J et al. Highly multiplexed tissue imaging using repeated oligonucleotide exchange reaction. Eur. J. Immunol 51, 1262–1277 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang W, Hennrick K & Drew S A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum. Pathol 44, 29–38 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Lin JR et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, 31657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerdes MJ et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl Acad. Sci. USA 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schubert W et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol 24, 1270–1278 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Gut G, Herrmann MD & Pelkmans L Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Agasti SS et al. DNA-barcoded labeling probes for highly multiplexed Exchange-PAINT imaging. Chem. Sci 8, 3080–3091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y et al. Rapid sequential in situ multiplexing with DNA exchange imaging in neuronal cells and tissues. Nano Lett 17, 6131–6139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saka SK et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol 37, 1080–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keren L et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffitt JR et al. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl Acad. Sci. USA 113, 14456–14461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei L et al. Super-multiplex vibrational imaging. Nature 544, 465–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W, Han Y, Beliveau BJ & Gao X Combining Qdot nanotechnology and DNA nanotechnology for sensitive single-cell imaging. Adv. Mater 32, e1908410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendall SC, Nolan GP, Roederer M & Chattopadhyay PK A deep profiler’s guide to cytometry. Trends Immunol 33, 323–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartmann FJ et al. Comprehensive immune monitoring of clinical trials to advance human immunotherapy. Cell Rep 28, 819–831.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomariz A et al. Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat. Commun 9, 2532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips D et al. Highly multiplexed phenotyping of immunoregulatory proteins in the tumor microenvironment by CODEX tissue imaging. Front. Immunol 12, 687673 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newell EW et al. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat. Biotechnol 31, 623–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samusik N, Good Z, Spitzer MH, Davis KL & Nolan GP Automated mapping of phenotype space with single-cell data. Nat. Methods 13, 493–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moen E et al. Deep learning for cellular image analysis. Nat. Methods 16, 1233–1246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Blondel VD, Guillaume J-L, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008 (2008). [Google Scholar]
- 29.Wu D et al. Comparison between UMAP and t-SNE for multiplex-immunofluorescence derived single-cell data from tissue sections. Preprint at bioRxiv 10.1101/549659 (2019). [DOI] [Google Scholar]
- 30.Kishi JY et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat. Methods 16, 533–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frei AP et al. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat. Methods 13, 269–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz D et al. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst 6, 25–36.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Germain RN & Gerner MY Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc. Natl Acad. Sci. USA 114, E7321–E7330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueda HR et al. Tissue clearing and its applications in neuroscience. Nat. Rev. Neurosci 21, 61–79 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Wassie AT, Zhao Y & Boyden ES Expansion microscopy: principles and uses in biological research. Nat. Methods 16, 33–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winter PW & Shroff H Faster fluorescence microscopy: advances in high speed biological imaging. Curr. Opin. Chem. Biol 20, 46–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leng E et al. Signature maps for automatic identification of prostate cancer from colorimetric analysis of H&E- and IHC-stained histopathological specimens. Sci. Rep 9, 6992 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu PH et al. Single-cell morphology encodes metastatic potential. Sci. Adv 6, eaaw6938 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldblum JR, Lamps LW, McKenney JK & Myers JL Rosai and Ackerman’s Surgical Pathology 11th edn, Vol. 1 (Elsevier, 2018). [Google Scholar]
- 40.Uhlen M et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol 28, 1248–1250 (2010). [DOI] [PubMed] [Google Scholar]
Related links
Key reference using this protocol
- Schürch CM et al. Cell 182, 1341–1359.e19 (2020): 10.1016/j.cell.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have uploaded both the concatenated CODEX imaging montage of the tissue (https://doi.org/10.6084/m9.figshare.12986981) and single-cell segmented data (https://doi.org/10.6084/m9.figshare.12986099) used in this paper to figshare (https://figshare.com/). The size of the raw imaging data is too large to be stored in a public repository and will therefore be stored in a private cloud-based server. Access to these data will be provided by the corresponding authors upon request.