

Abstract
Leukemic relapse is believed to be driven by transformed hematopoietic stem cells (HSC) that harbor oncogenic mutations or have lost tumor suppressor function. Recent comprehensive sequencing studies have shown that mutations predicted to activate Ras signaling are highly prevalent in hematologic malignancies and, notably, in refractory and relapsed cases. To better understand what drives this clinical phenomenon, we expressed oncogenic NrasG12D within the hematopoietic system in mice and interrogated its effects on HSC survival. N-RasG12D conferred a survival benefit to HSCs and progenitors following metabolic and genotoxic stress. This effect was limited to HSCs and early progenitors and was independent of autophagy and cell proliferation. N-RasG12D–mediated HSC survival was not affected by inhibition of canonical Ras effectors such as MEK and PI3K. However, inhibition of the noncanonical Ras effector pathway protein kinase C (PKC) ameliorated the protective effects of N-RasG12D. Mechanistically, N-RasG12D lowered levels of reactive oxygen species (ROS), which correlated with reduced mitochondrial membrane potential and ATP levels. Inhibition of PKC restored the levels of ROS to that of control HSCs and abrogated the protective effects granted by N-RasG12D. Thus, N-RasG12D activation within HSCs promotes cell survival through the mitigation of ROS, and targeting this mechanism may represent a viable strategy to induce apoptosis during malignant transformation of HSCs.
Introduction
RAS and Ras-activating mutations have been identified as initiating genetic lesions, frequent molecular events, and prognostic markers across a range of cancers (1, 2). This includes pancreatic, lung, colon, and rhabdomyosarcoma in solid tumors, and acute myeloid and lymphoblastic leukemia and juvenile myelomonocytic leukemia in hematologic malignancies. In addition, with the wide availability of clinical sequencing, mutations predictive of hyperactive Ras signaling have also now been correlated with disease relapse and treatment refractoriness (3–9). Thus, understanding how dysregulated Ras signaling contributes to each of these outcomes is of critical importance to improve our understanding of disease development and treatment algorithms to better serve patients with cancer.
Leukemogenesis is a process during which hematopoietic stem cells and progenitors (HSC/P) are transformed into leukemic stem cells (LSC) following mutations that activate oncogenes or inactivate tumor suppressor genes (10). Importantly, LSCs have been shown to escape extensive chemo- and/or radiotherapy and persist even after patients achieve clinical remission, thus harboring a focus for disease relapse (11–13). These studies suggest that the successful treatment of leukemia depends on better defining and eliminating LSCs.
Despite accumulating evidence that RAS mutations contribute to treatment resistance in leukemia, little is known about the potential role of oncogenic Ras signaling in the survival of HSC/Ps and how it may contribute to this process. Previously, we and others reported that mice harboring an oncogenic Nras mutation, G12D, expressed from its endogenous locus develop an indolent disease with features reminiscent of chronic and juvenile myelomonocytic leukemia (CMML/JMML; refs. 14, 15). Strikingly, when additional genetic mutations were introduced in this model, oncogenic N-Ras collaborated with these lesions to reproducibly induce acute myeloid leukemia (AML; ref. 14). We subsequently reported that oncogenic N-RasG12D exerts profound effects on HSCs, resulting in their dysregulation to promote HSC self-renewal and clonal expansion (16). However, whether oncogenic N-Ras confers a survival phenotype to HSCs to promote these processes is not known.
To address this question, we utilized our previously established NrasG12D model and interrogated the effects of hyperactive N-Ras on HSC survival. We report that hyperactive N-Ras signaling confers a survival advantage to HSC/Ps. Specifically, NrasG12D HSCs and early progenitors exhibit resistance to metabolic and genotoxic stress induced by radiation and chemotherapy, and this effect was mediated by decreased reactive oxygen species (ROS) and lower mitochondrial activity. Furthermore, this effect was independent of the canonical Ras pathways MEK/ERK and PI3K/AKT/mTOR but was instead mediated via enhanced protein kinase C (PKC) activity. Ultimately, targeting noncanonical Ras effector pathways and/or destabilizing redox stress may represent novel therapeutic strategies to overcome treatment resistance in Ras-mutated hematologic malignancies.
Materials and Methods
Animals
All mice were housed in the Unit for Laboratory Animal Medicine at the University of Michigan, and protocols were approved by the University of Michigan Committee on the Use and Care of Animals (PRO00007786). Conditional NrasG12D knockin mice (14), Stat5ab knockout mice (16), and transgenic Col1A1-H2B-GFP; Rosa26-M2-rtTA mice (17) were described previously. All animals were maintained on a C57/BL6 background. Littermates or age- and gender-matched mutant and control mice (6–10 weeks of age) were used for all experiments. To activate NrasG12D, the Mx1-cre+; NrasLSL-G12D/+ mice received intraperitoneal injections of poly (I:C; GE Healthcare Life Sciences 27-4732-01) at a dose of 0.5 μg/g body mass every other day for three doses 2 weeks before analysis. Doxycycline was added to the water at a concentration of 0.2% (m/v) along with 1% sucrose. No difference of phenotype was observed between male and female mice.
Antibodies
PKC antibodies were used as follows: PKCδ (Santa Cruz Biotechnology; C-20; catalog no. SC-937) was used at dilution of 1:500, PKCδ (Cell Signaling Technology; catalog no. 2058S) was used at dilution of 1:1,000, PKCε (Santa Cruz Biotechnology; C-15; catalog no. SC-214) was used at dilution of 1:1,000, PKCθ (Santa Cruz Biotechnology, and C-18; catalog no. SC-212) was used at dilution of 1:1,000, PKCη (Santa Cruz Biotechnology; 31; catalog no. SC136036) used at dilution of 1:1,000. β-Actin (8H10D10) was purchased from Cell Signaling Technology (3700S).
Statistical analysis
Group data represent mean ± SD. N represents the number of biological replicates and is indicated in figure legends. Statistical analysis for each experiment is specifically described in the corresponding figure legend. GraphPad Prism software was used unless otherwise indicated. A minimum of three independent experiments were performed to verify reproducibility. Two-tailed Student t tests were used to assess statistical significance for two group comparisons and ANOVA tests were performed for more than two groups.
Flow cytometry and isolation of HSCs
Bone marrow cells were harvested and cell populations were isolated as described previously (16). Briefly, the bilateral hips, legs, and spine were placed in HBSS containing 3% filtered calf serum and crushed with a mortar and pestle. Staining was completed in two steps: first using HSC SLAM cocktail (all BD Biosciences), after which, c-kit enrichment was performed using MACS anti-APC or anti-biotin beads and enriched on LS magnetic column (Miltenyi). Annexin V staining was performed according to commercially provided protocols (BD Biosciences; 556547). ROS staining was performed with CellROX reagent according to manufacturer’s instructions and used at a final concentration of 5 μmol/L (Thermo Fisher Scientific; C10422). Tetra-methylrhodamine methyl ester (TMRM) was used according to manufacturer’s instructions at a final concentration of 0.5 μmol/L (Thermo Fisher Scientific; T668 and M7514). HSC sorting was completed using DIVA software on a BDAria cell sorter or Sony SA400 and analysis completed with BD LSR Fortessa using FlowJo software.
In vitro cell culture
For cytokine starvation assays, equal numbers of HSCs (typically 300–1,000) were sorted into 100 μL of SF-O3 medium (Iwai North America Inc.) supplemented with 1% bovine serum albumin, with or without cytokines [stem cell factor (SCF) and thrombopoietin (TPO); 100 ng/mL each; Peprotech 250–03 and 315–14] and cultured for 18 hours. Cleaved caspase-3/7 assays were then performed according to manufacturer’s protocol (Promega G8090).
Ex vivo analysis
ATP levels were measured immediately after sorting 400 HSCs into SF-O3 media with cytokines, according to manufacturer’s instructions using Cell-Titer Glo Assay (Promega G7570). Total and oxidized glutathione levels were measured in 1,000 sorted CD48-Lin−Sca-1+c-Kit+ (LSK) using the GSH/GSSG-Glo Assay by Promega according to instructions. To perform colony forming assays, 200 to 400 FACS-sorted HSCs were plated in duplicates into 12-well plates containing Methocult 3434 medium (Stem Cell Technologies; 3434), cultured for 7 days and counted.
mtDNA
A total of 40,000 LSKs were sorted into DirectPCR (Viagen Biotech 102-T) with proteinase K and heated overnight at 55°C. DNA was isolated following ethanol purification using standard methods. Primer sequences are as followed: ND-1: CTAGCAGAAACAAACCGGGC (forward) CCGGCTGCGTATTCTACGTT (reverse); actin: CGGCTTGCGGGTGTTAAAAG (forward) CGTGATCGTAGCGTCTGGTT (reverse).
Other
For qRT-PCR, primer sequences were as followed: PKCα: CGCTCCACACTGAATCCTC (forward) CGGCATCTTCATTAGCTCT (reverse); PKCβ: CCGCGACCTGAAACTTGACA (forward) CTCCAAATGCCCACCAGTCCA (reverse); PKCδ: TGCAAGAAGAACAACGGCAA (forward) CTGTTTAATGGCTCCACGAC (reverse); PKCε: TGACCAAGAACCCGCACAA (forward) TATTGGCTCTTCCCGCGTA (reverse); PKCθ: ATCTTTCCAGAGCCACGTTT (forward) TGTCTTCGCATCTCCTAGCAT (reverse).
Results
Oncogenic N-RasG12D protects HSCs from apoptosis
We previously reported that HSCs expressing a single copy of oncogenic NrasG12D durably out-compete their wild-type counterparts through increased self-renewal and competitiveness (16). Given that tight regulation of apoptosis has been shown to provide a competitive advantage to HSCs (18), we asked whether this mechanism could be relevant after introduction of oncogenic N-RasG12D. To address this question, we generated Mx1-Cre+; NrasLSL-G12D/+ mice (referred to hereafter as NrasG12D or Nras mutant mice) using a previously described NrasG12D allele in which the G12D mutation was expressed endogenously from the Nras locus downstream of a floxed “STOP” cassette that represses transcription (14, 16). To delete the “STOP” cassette and activate the mutant G12D allele in hematopoietic tissues, we administered three doses of poly-inosine:poly-cytosine (pIpC) over a 5-day period (at a dose of 0.5 μg/g body mass) to 6- to 10-week-old NrasG12D mice. The mice were then analyzed a minimum of 2 weeks after pIpC treatment and paired with gender- and age-matched controls that lacked a Cre allele (NrasLSL-G12D/+ only mice).
To assess whether N-RasG12D protects HSCs from apoptosis, we isolated SLAM HSCs (Lineage−c-Kit+Sca-1+CD48−CD150+; Supplementary Fig. S1; ref. 19) from control and NrasG12D mice and subjected them to either cytokine starvation for 18 hours or 300 cGy of ionizing radiation (IR), which induces apoptosis via metabolic, oxidative, and genotoxic stress (20–22). Apoptotic cells were then measured using both Annexin V positivity by flow cytometry and cleaved caspase-3/7 level. HSCs cultured under growth conditions in the presence of cytokines (SCF and TPO; both at 100 ng/mL) showed no difference in apoptosis between NrasG12D and control HSCs. However, NrasG12D HSCs demonstrated reduced frequency of Annexin V-positive cells (Fig. 1A and B) and lower levels of cleaved caspase-3/7 (Fig. 1C) in the absence of cytokines. To assess if N-RasG12D afforded protection following clinically relevant therapies, we subjected NrasG12D HSCs to γ-irradiation (3 Gy; Fig. 1D), as this has been shown to induce apoptosis in HSCs (20). Similar to conditions of cytokine starvation, NrasG12D HSCs were more resistant to γ-irradiation–induced apoptosis, as demonstrated by Annexin V positivity and caspase 3/7 activation (Fig. 1E and F). Thus, N-rasG12D promoted HSC survival in vitro following metabolic and genotoxic stress.
Figure 1.
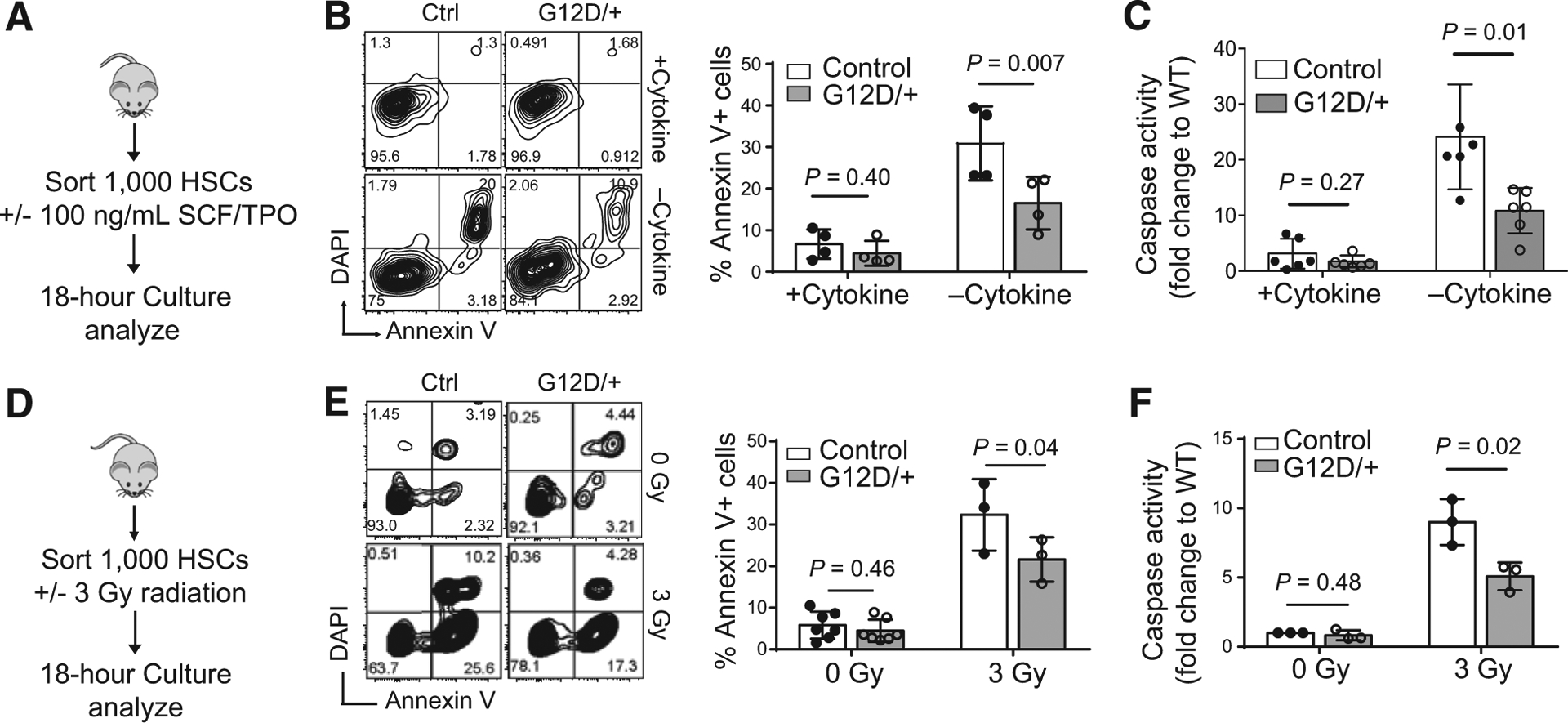
N-RasG12D/+ confers HSC survival to exogenous stress in vitro. A, Experimental design for in vitro cytokine starvation. B, Representative flow cytometry plot for Annexin V and DAPI staining with quantitative representation (n = 4). Paired Student t test was used to calculate statistical significance. C, Survival, as measured by caspase activation, of control or NrasG12D SLAM HSCs cultured in cytokine-rich (SCF+TPO; 100 ng/mL) or starved SF-03 media (n = 6). Two-tailed unpaired Student t test was used to calculate statistical significance. WT, wild-type. D, Experimental design for in vitro radiation. E, Representative flow cytometry plot for Annexin V and DAPI staining following radiation with quantitative representation (n = 7 for 0 Gy, n = 3 for 3 Gy). Paired Student t test was used to calculate statistical significance. F, Survival of FACS-sorted control or NrasG12D HSCs, as measured by caspase activation, after exposure to 300 cGy of in vitro γ-radiation (n = 3). Two-tailed unpaired Student t test was used to calculate statistical significance. Data represent mean ± SD; n, number of mice.
Given that HSCs reside in a protective bone marrow niche, we next validated the protective effects of N-RasG12D in vivo. NrasG12D mice and controls were treated with a sublethal dose (3 Gy) of γ-irradiation. Twenty-four hours following exposure, whole bone marrow (WBM) was harvested and apoptosis measured following cell surface marker staining (Fig. 2A; Supplementary Fig. S2). Both NrasG12D SLAM HSCs and LSK hematopoietic progenitor cells were protected from apoptosis as indicated by reduced frequencies of Annexin V+ cells (Fig. 2B and C). Importantly, this protective effect of N-rasG12D was restricted to hematopoietic stem and progenitor cells but not observed in differentiated lineage-committed cells (Fig. 2C). These data demonstrate that N-RasG12D exerts a prosurvival phenotype to HSCs and early progenitors in vivo and could therefore allow transformed HSC/Ps to persist following intensive therapeutic regimens. Next, to understand if HSC function is retained following in vivo radiation, 400 HSCs were FACS sorted from sublethally irradiated (3 Gy) NrasG12D and control mice and plated in methocellulose media with cytokines (SCF and TPO; both at 100 ng/mL). After 7 days of culture, Nras mutant HSCs formed significantly more colonies than control HSCs (Fig. 2D), suggesting hyperactive N-RasG12D preserves HSC function after exposure to genotoxic agents. Furthermore, to test the impact of radiation on HSC function in vivo, we administered a dose of 3 Gy γ-radiation to CD45.2 control and Nras mutant mice and 24 hours later, transplanted whole bone marrow from these mice to lethally irradiated CD45.1 recipient mice, together with wild-type CD45.1 radioprotector cells (at a ratio of 10:1; Fig. 2E). Peripheral blood chimerism was then measured up to 24 weeks posttransplantation (Fig. 2F; Supplementary Fig. S3). As reported previously (16), unirradiated Nras mutant marrow showed enhanced reconstitution relative to control (Fig. 2F). Following radiation, Nras mutant HSCs preserved the enhanced reconstitution, and showed a trend of further increase of reconstitution relative to irradiated wild-type HSCs, suggesting that despite cellular insult, N-RasG12D bestows a competitive advantage in vivo to HSCs.
Figure 2.
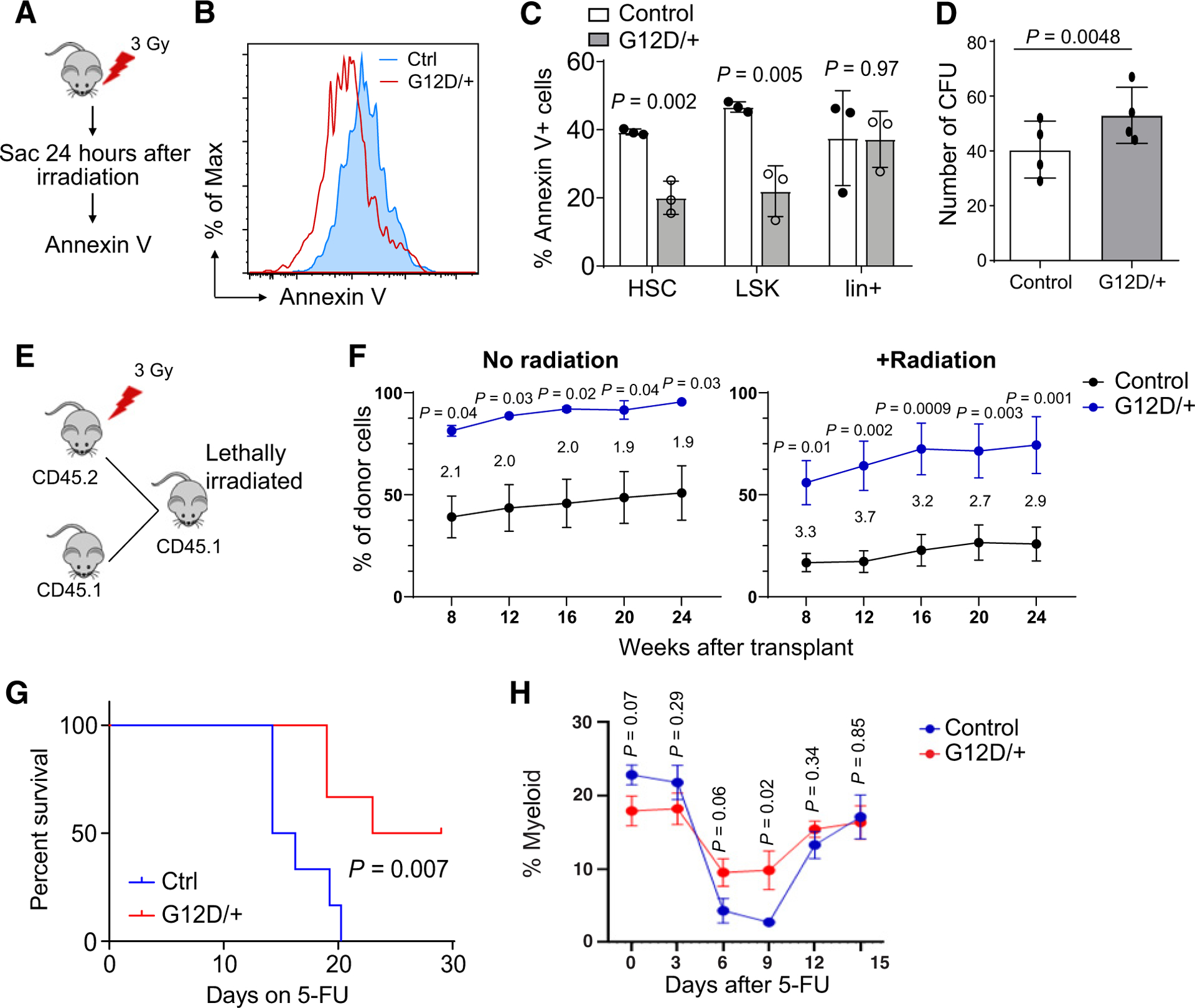
N-RasG12D protects HSCs and early progenitors from apoptosis following in vivo γ-radiation A, Experimental scheme showing exposure of control and Nras mutant mice to 300 cGy of γ-radiation in vivo. B, Representative flow cytometry histogram of Annexin V staining in control and Nras mutant mice 24 hours following in vivo radiation. C, Bone marrow from control and Nras mutant mice harvested and stained for Annexin using SLAM HSC markers (HSC: Lineage−c-kit+Sca-1+CD48−CD150+; LSK: Lineage−c-kit+Sca-1+; Lin+: all but Lineage−; n = 3). Two-tailed Student t test was used to calculate statistical significance. D, A total of 400 SLAM HSCs were FACS sorted and plated into duplicate wells containing murine Methocult media (M3434; Stem Cell Technologies) 24 hours following in vivo radiation (300 cGy) of control and Nras mutant mice. Colonies counted at day 7 of culture (n = 4). Paired t test was used for statistical analysis. CFU, colony-forming units. E, Experimental design to radiate Nras mutant and control mice prior to competitive transplant. F, Donor cell contribution to CD45+ cells in transplant recipients of Nras mutant or control donors. Wild-type competitor CD45.1 cells were mixed at a 1:1 ratio with unirradiated donor cells and at 1:10 ratio for irradiated donors (two donors were used for total of five to eight recipients). Unpaired t test was used to calculate statistical significance. Fold change for each time point is shown between the curves. G, Survival curve of control and NrasG12D mice following weekly 5-FU injections (150 mg/kg/dose; n = 6). Log-rank test was used for statistical analysis. H, The percentage of myeloid cells in peripheral blood following a single dose of 5-FU (n = 7). Two-tailed Student t test was used to calculate statistical significance. Data represent mean ± SD; n, number of mice.
We then considered additional treatment in vivo, and chose to administer four weekly doses of 5-fluorouracil (5-FU; 150 mg/kg/dose), which ablates proliferative cells within the bone marrow and relies on intact HSC function to restore hematopoiesis. Although increased HSC proliferation typically leads to increased sensitivity to 5-FU treatment, despite our previous report that N-rasG12D increases the overall proliferation of the HSC pool (16), we observed prolonged survival in NrasG12D mice after 5-FU treatment (Fig. 2G). Although all wild-type control mice became moribund or died by day 20, 50% of NrasG12D mice survived by day 30. To understand if N-RasG12D provides a survival advantage to HSCs after 5-FU, we administered a single dose of 5-FU and monitored peripheral blood cell recovery by myeloid percentage every 3 days. We observed that Nras mutant mice had a quicker recovery of peripheral blood myeloid cells (Fig. 2H), suggesting that N-RasG12D confers a survival advantage under stress.
The prosurvival phenotype of NrasG12D HSC is independent of autophagy
Evolving data have shown that autophagy is critical to HSC maintenance, self-renewal, and survival (21, 23, 24), specifically under conditions of HSC stress and during leukemogenesis (25). To test whether induction of autophagy is responsible for the survival phenotype of NrasG12D HSCs, we treated HSCs with bafilomycin A (BafA), which inhibits the late phase of autophagy by preventing fusion of autophagosomes with lysosomes, under conditions of cytokine starvation (Supplementary Fig. S4A; refs. 21, 26). Inhibition of autophagy via bafilomycin A did not diminish the resistance of NrasG12D HSCs to apoptosis as detected by caspase-3/7 activation (Supplementary Fig. S4B). To confirm these findings, we measured autophagocytic activity by selectively labeling accumulated autophagocytic vacuoles with Cyto-ID. As has been previously shown, control HSCs upregulated autophagy under conditions of cytokine starvation (21), but there was no difference in Cyto-ID levels between NrasG12D and control HSCs after cytokine starvation (Supplementary Fig. S4C). Consistent with these findings, microarray analysis of gene expression (GSE45194; ref. 16) demonstrated similar mRNA levels of key autophagy regulators in control and NrasG12D HSCs (Supplementary Fig. S4D). Taken together, these data indicate that autophagy is not a significant mediator of enhanced HSC survival induced by oncogenic N-RasG12D.
PKC signaling mediates protection from apoptosis in NrasG12D HSCs
The downstream signaling pathways following Ras activation are well-characterized to promote cell survival and proliferation in many cancers. To identify the potential signaling mediators of N-RasG12D–induced survival in HSCs, we treated HSCs with a series of compounds that inhibit the canonical downstream pathways of the Ras (2) and exposed HSCs to cytokine starvation (Fig. 3A; Supplementary Table S1). Interestingly, pharmacologic inhibition of the canonical Ras effectors, MEK (PD0325901), PI3K/AKT (GDC-0941), or mTOR (rapamycin), did not affect the survival benefit of NrasG12D HSCs under cytokine starvation (Fig. 3A). We previously found that N-RasG12D upregulates STAT5 signaling to increase HSC proliferation and self-renewal and that heterozygous knockout of Stat5ab abolished these effects (16). We therefore interrogated if the prosurvival phenotype of Nras mutant HSCs is dependent on STAT5 signaling by generating Mx1-cre+; NrasLSL-G12D/+; Stat5a/bfl/+ mice. We then injected the mice with pIpC (0.5 μg/g body mass every other day for six doses) and 2 weeks after pIpC, purified SLAM HSCs and measured apoptosis by caspase-3/7 activation after cytokine starvation. We found that heterozygous knockout of Stat5ab had minimal effects on survival in control or NrasG12D HSCs following cytokine starvation (Supplementary Fig. S4E). Together, these data suggest that STAT5 signaling is important for HSC proliferation but not survival of NrasG12D HSCs under stress conditions.
Figure 3.
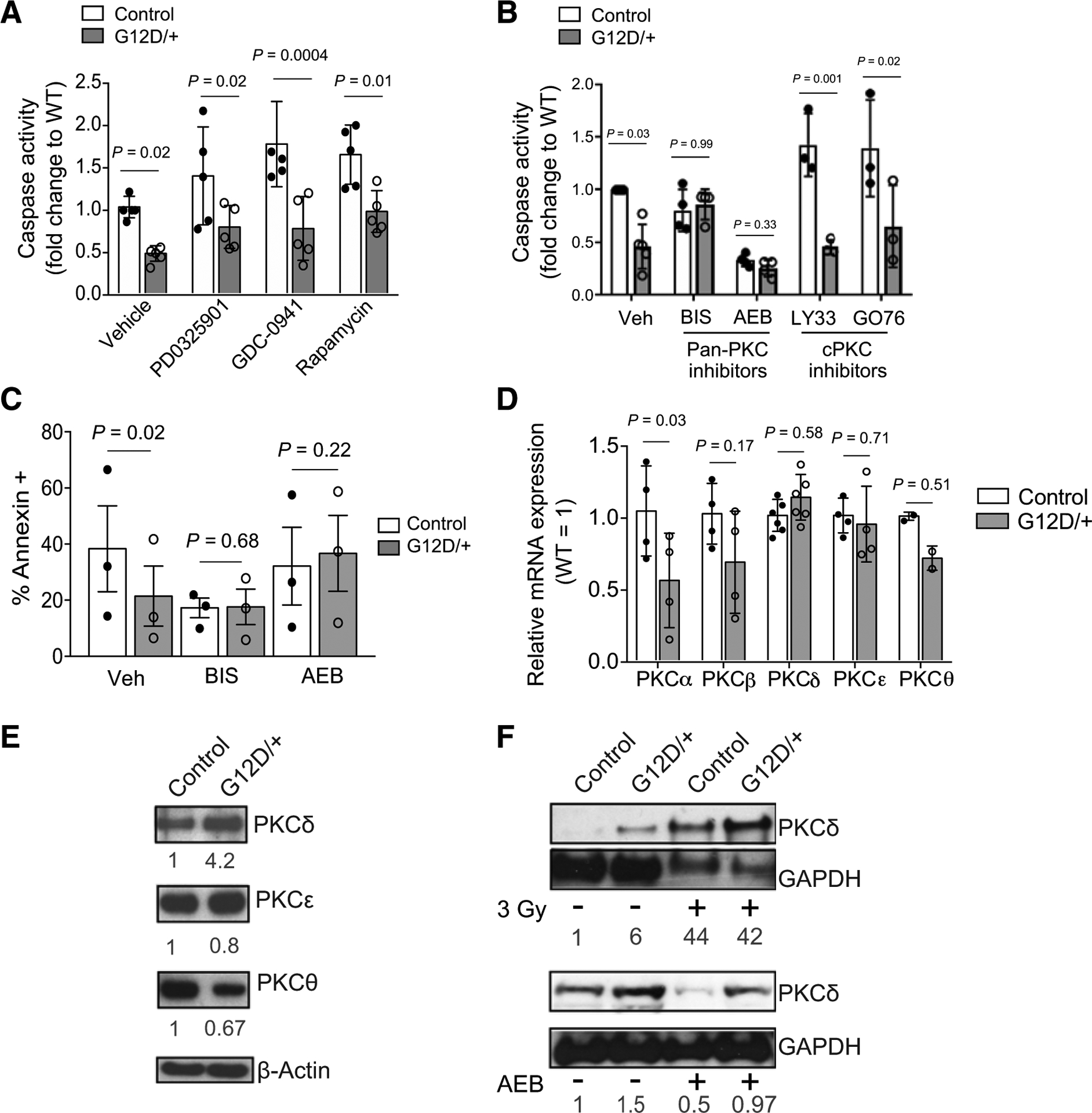
N-RasG12D–induced prosurvival phenotype of HSCs is dependent on noncanonical Ras effector PKC signaling. A, SLAM HSCs from control and Nras mutant mice were FACS sorted into SF-03 media under conditions of cytokine starvation in the presence of either vehicle control, MEK inhibitor (PD0325901; 100 nmol/L), PI3K inhibitor (GDC-0941; 399 nmol/L), or mTOR inhibitor (rapamycin; 100 nmol/L), and apoptosis was measured by caspase activation following 18 hours of culture (n = 4–6). Two-way ANOVA was used for statistical significance. WT, wild-type. B, SLAM HSCs from control and Nras mutant mice were FACS sorted into SF-03 media under conditions of cytokine starvation in the presence of either vehicle control (Veh), pan-PKC inhibitors (BIS 5 μmol/L; AEB 10 μmol/L), or inhibitors to classic PKCs (LY33 1 μmol/L; GO76 10 μmol/L). Following 18 hours of culture, apoptosis was measured by caspase activation (n = 3–5). Two-way ANOVA was used for statistical significance. C, SLAM HSCs from control and Nras mutant mice were FACS sorted into SF-03 media under conditions of cytokine starvation in the presence of either vehicle control or pan-PKC inhibitors (BIS 5 μmol/L; AEB 10 μmol/L). Following 18 hours of culture, apoptosis was measured by Annexin V staining (n = 3). Paired t test was used for statistical significance. D, mRNA levels, as measured by qRT-PCR, of PKC isoforms in SLAM HSCs isolated from control or Nras mutant mice (n = 2–6). Unpaired Student t test was used to calculate statistical significance. E, Western blots on 20,000 LSKs from control and Nras mutant mice showing the expression of novel PKC isoforms: PKCδ, PKCε, PKCθ. β-actin was used as a loading control (n = 3). Relative expression (as signal ratio of PKC isoform to β-actin) was quantified from three independent blots and is shown below each blot. F, Western blots showing the expression of PKCδ prior to and following in vivo radiation (top; LSKs) or following in vivo treatment with the PKC inhibitor AEB (bottom; cKit-enriched cells). GAPDH level was used as a loading control. Relative expression (as signal ratio of PKCδ to GAPDH) was quantified from three independent blots and is shown. Data represent mean ± SD; n, number of mice.
To further dissect downstream signaling, we considered noncanonical Ras effectors and, specifically, those that have been reported to play a role in cell survival. Of these downstream pathways, PKC isoenzymes have been reported to modulate apoptotic signaling (27). Given this, we asked if PKC signaling could contribute to the observed survival advantage in NrasG12D HSCs. There are at least 10 known PKC isoforms, which are grouped as classical PKCs (cPKC): PKCα, PKCβI, PKCβII, PKCγ; novel PKCs (nPKC): PKCδ, PKCε, PKCη, PKCθ; and atypical PKCs (aPKC): PKCζ, PKCι. PKCs (cPKC): PKCa, PKCbI, 328 PKCbII, PKCg; novel PKCs (nPKC): PKCd, PKCe, PKCh, PKCq; 329 and atypical PKCs (aPKC): PKCz, PKCi. Each group has distinct intracellular functions and inhibition of many have been interrogated as a therapeutic strategy in cancer, inflammatory diseases, and cardiac injury (27, 28). We therefore chose to administer a series of PKC inhibitors that exhibit selectivity for the various isoforms (Supplementary Table S2). We initiated these studies using two pan-PKC inhibitors, Bisindolylmaleimide I (BIS) and Sotrastaurin (AEB), and found that treatment with these inhibitors under cytokine starvation dramatically abolished the prosurvival phenotype observed in NrasG12D HSCs (Fig. 3B and C). We next chose inhibitors selective for cPKC isoforms, LY 333531 (LY33) and GO6976 (GO69), and found that they each had no effect on the survival advantage of NrasG12D HSCs, thus eliminating a significant role of the classic PKC isoforms in the antiapoptotic phenotype of NrasG12D HSCs (Fig. 3B).
To further test which PKC isoform may induce the observed findings in NrasG12D HSCs, we measured expression of several PKC isoforms in control and NrasG12D HSCs by real-time PCR. However, we did not observe increased mRNA expression of any PKC isoform in NrasG12D HSCs compared with control HSCs (Fig. 3D). Next, we questioned if PKC protein expression could be altered by N-RasG12D. Notably, both novel and classic PKC isoforms possess a common C2 domain that is activated under stress and could therefore participate in apoptosis (29, 30). Given that inhibition of classic PKC isoforms demonstrated no functional effect in NrasG12D cells, we focused on novel PKC isoforms and examined protein levels by Western blot analysis in control and NrasG12D HSC/Ps (LSK) cells. As shown, PKCδ levels were increased in NrasG12D compared with control LSKs (Fig. 3E), whereas PKCε and PKCΘ showed comparable expression levels in both. Of note, we did not detect a signal by Western blot analysis for the fourth novel PKC isoform, PKCη, in either genotype. Given that we saw differential expression of PKCδ in control and NrasG12D LSKs at baseline, we wanted to understand if PKCδ expression was increased following cellular stress and could therefore mitigate apoptosis in NrasG12D HSCs. To answer this question, we collected protein lysate from control and NrasG12D LSKs following in vivo radiation. Interestingly, PKCδ expression was increased in both control and NrasG12D LSKs after radiation (Fig. 3F). Taken together, these data suggest that PKCδ is activated in primitive NrasG12D hematopoietic cells and is further enhanced in response to radiation.
We next examined if PKCδ inhibition could, in fact, diminish the antiapoptotic effects in NrasG12D HSCs. Both genetic and chemical inhibition of PKCδ were tested to understand this. First, we used shRNA to knockdown PKCδ in both control and NrasG12D HSCs (Fig. 4A and B) and measured caspase activation following cytokine starvation, as described previously (Fig. 4C). Notably, knockdown of PKCδ abrogated the protective effects of N-RasG12D in HSCs, after cytokine starvation (Fig. 4C). Next, we wanted to understand the role of PKC signaling in vivo and irradiated (3 Gy) control and Nras mutant mice, and administered two intraperitoneal injections of either vehicle or PKC inhibitor AEB (Fig. 4D). Twenty-four hours after radiation, bone marrow was harvested, and cellular apoptosis measured by Annexin staining. Notably, administration of PKC inhibition in vivo abrogated, at least in part, the protective effects of N-RasG12D following radiation (Fig. 4E and F). Together, these data support that PKC signaling is required by N-RasG12D–induced survival of HSC/Ps under stress conditions.
Figure 4.
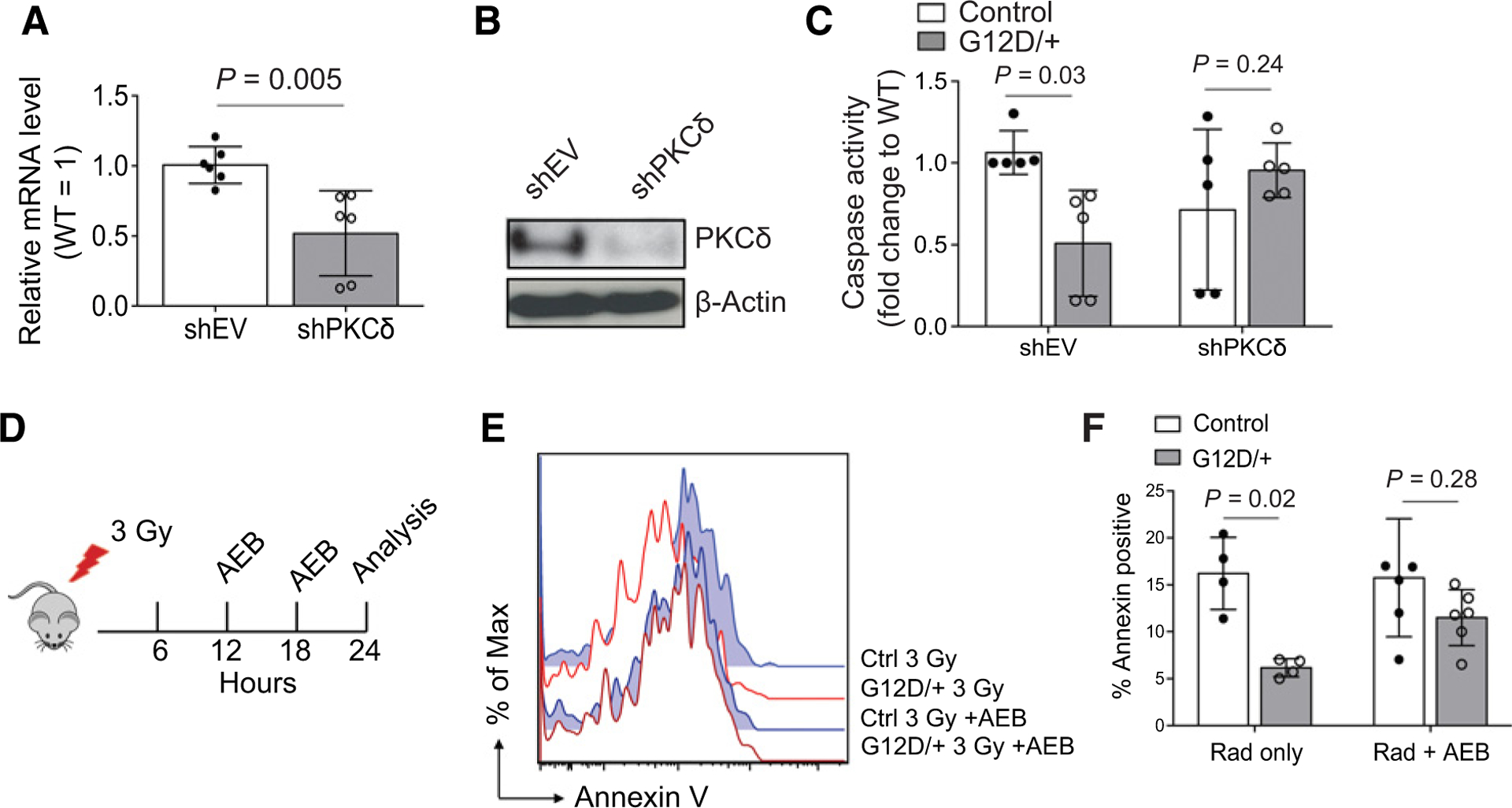
Inhibition of PKC ameliorates prosurvival benefit conferred to HSCs by N-RasG12D. A and B, c-kit–enriched cells were transfected with either empty vector control or PKCδ shRNA, and mRNA level by qRT-PCR (A) and protein level by Western blot analysis (B) was measured to verify knockdown (n = 6). Unpaired Student t test was used to calculate statistical significance. WT, wild-type. C, FACS-purified SLAM HSCs from control or Nras mutant mice were exposed to cytokine starvation following transfection with either empty vector or PKCδ shRNA, and apoptosis was measured after 18 hours of culture using caspase activation (n = 5). Two-way ANOVA was used for statistical significance. D, Experimental design using in vivo administration of the PKC inhibitor AEB (10 μg/g body mass) to sensitize HSCs to apoptosis with and without γ-radiation. E and F, Representative flow cytometry histogram (E) and quantitation (F) showing apoptosis of SLAM HSCs as measured by Annexin V staining following in vivo radiation alone or together with AEB treatment in control and Nras mutant mice (n = 4–6). Two-way ANOVA was used for statistical significance. Data represent 10 mean ± SD; n, number of mice.
N-RasG12D mitigates oxidative stress to promote survival of HSC/Ps
HSC fitness relies on the tight regulation of ROS (23, 31–35) whereas dysregulation of redox balance by γ-irradiation and chemotherapy is a well-described biochemical mechanism that induces apoptosis, specifically during Ras-mediated tumorigenesis (36–38). Thus, we asked if oncogenic N-RasG12D could alter ROS levels in HSCs and progenitors. To investigate this, we labeled freshly harvested bone marrow cells with CellRox, a fluorescent dye specific for cellular ROS, and found that NrasG12D HSCs had reduced levels of ROS relative to control HSCs (Fig. 5A). Similar to the results of apoptosis, a reduction of ROS was not present in Nras mutant lineage positive cells but did remain lower in early primitive hematopoietic cells, LSKs (Fig. 5A).
Figure 5.
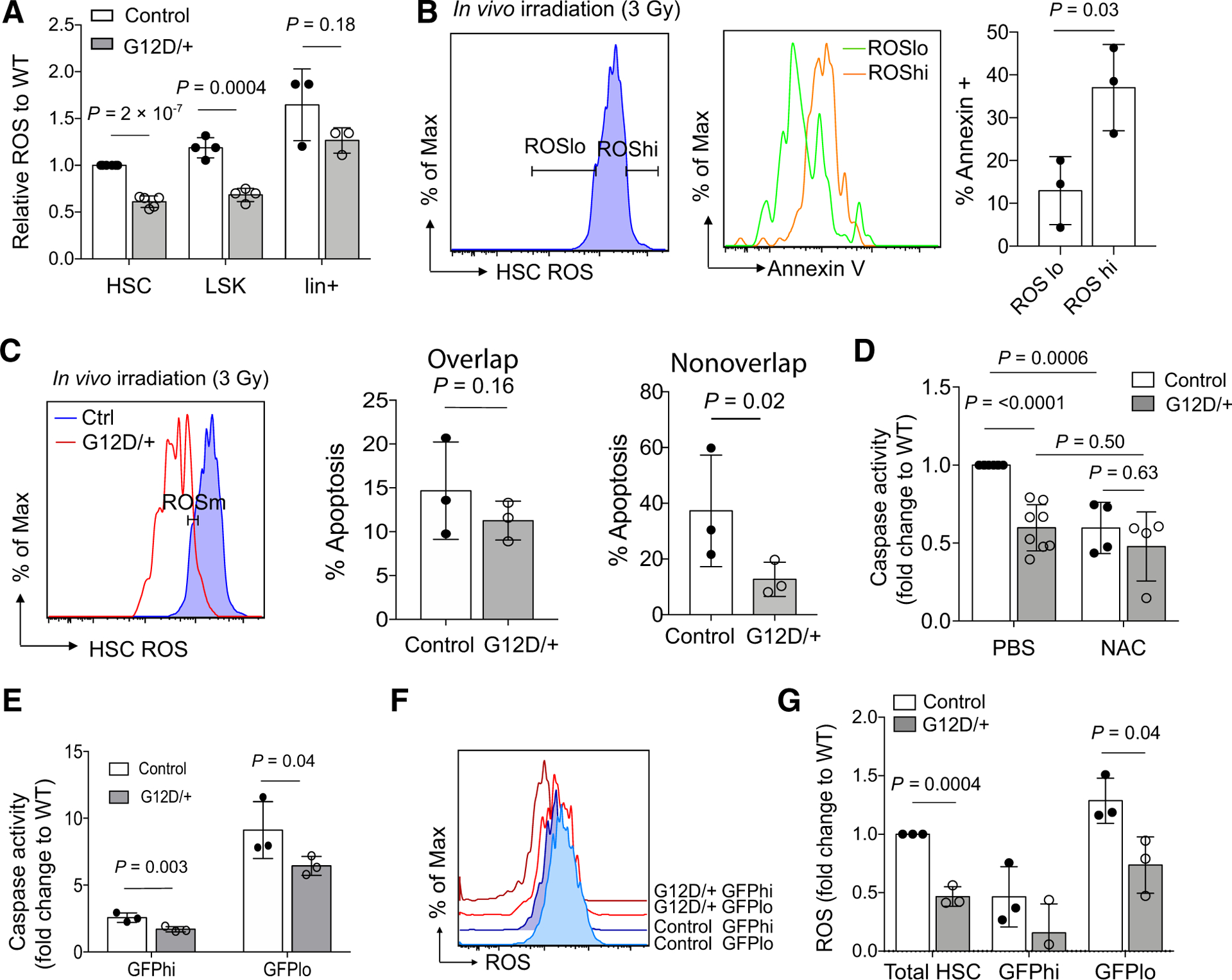
N-RasG12D lowers ROS in HSCs and early progenitors and affords a prosurvival advantage to HSCs. A, Levels of cellular ROS measured by CellRox in HSC, LSK, and lineage-positive hematopoietic compartments in control and Nras mutant mice (n = 4). Student t test was used to determine statistical significance. WT, wild-type. B, Levels of apoptosis, as measured by Annexin V staining, in ROSlo (lowest 10% of HSCs) and ROShi (highest 10% of HSCs) populations in wild-type mice following in vivo radiation (n = 3). Student t test was used to determine statistical significance. C, Levels of apoptosis, as measured by Annexin V staining, in wild-type and Nras mutant HSC with overlapping (left) or nonoverlapping (right) ROS levels (measured by CellRox staining) following in vivo radiation (n = 3). Paired Student t test was used to determine statistical significance. D, FACS-purified HSCs from control or Nras mutant mice were exposed to cytokine starvation for 18 hours in the presence or absence of N-acetyl cysteine (100 μmol/L), and apoptosis was measured by caspase activation (n = 4). Two-way ANOVA was used to determine statistical significance. E–G, Control and NrasG12D mice that carry Col1A1-H2B-GFP; Rosa26-M2-rtTA (H2B-GFP) transgenes were treated with doxycycline for 6 weeks, followed by a 16-week no doxycycline chase period. SLAM HSCs were then FACS sorted into GFPhi and GFPlo populations (Supplementary Fig. S1). Apoptosis (E) was measured by caspase activation following cytokine starvation (18 hours) in vitro (n = 3). ROS levels (F and G) were measured by CellRox (n = 3). Paired Student t test was used to determine statistical significance. Of note, no P value was calculated for GFPhi cells in G because of a negative value in the NrasG12D group. Data represent mean ± SD; n, number of mice.
Next, to determine whether levels of ROS correlate with apoptosis, we stained wild-type HSCs with both CellRox and Annexin V following in vivo radiation of control mice (3 Gy). As expected, HSCs with the highest ROS levels demonstrated the highest Annexin V positivity, whereas those with lower ROS were less apoptotic (Fig. 5B; Supplementary Fig. S5). We next examined ROS levels in control and NrasG12D mice after radiation. As hypothesized, there was no difference in Annexin V positivity between control and NrasG12D HSCs that have similar levels of ROS (Fig. 5C), whereas analysis of the wild-type and Nras mutant HSCs with nonoverlapping ROS levels confirmed that Nras mutant HSCs had lower levels of apoptosis than control HSCs (Fig. 5C). Together, these data indicate that apoptosis is diminished in NrasG12D HSCs, at least in part, due to lower levels of ROS. To understand if HSC cell death could be attenuated through reduced ROS, sorted HSCs from control and NrasG12D mice were treated with the anti-oxidant N-acetyl cysteine (NAC; 100 μmol/L) and subjected to 18 hours of cytokine starvation. Notably, NAC rescued wild-type HSCs from apoptosis compared with vehicle-treated HSCs but had minimal impact on NrasG12D HSCs (Fig. 5D), indicating that HSC survival is tightly associated with redox balance under stress and N-RasG12D–induced survival of HSCs is at least partially mediated by lowering ROS levels.
Given that redox levels are intimately coupled to cell proliferation and the metabolic demands of a cell, we used previously generated Col1A1-H2B-GFP; Rosa26-M2-rtTA (H2B-GFP) mice (17) to study survival and levels of ROS in quiescent and cycling HSCs. In these mice, HSCs can be labeled with H2B-GFP during a 6-week period of doxycycline administration and HSC proliferation can be followed during a subsequent 12- to 18-week chase period without doxycycline. We previously reported that all SLAM HSCs are GFP+ after 6 weeks of doxycycline treatment (16). After an 18-week chase period, however, HSCs can be characterized as proliferative (GFPlow, about 80% of total HSCs) or quiescent HSCs (GFPhigh, about 20% of total HSCs) and therefore readily separated by FACS (Supplementary Fig. S1). To understand if cell proliferation contributed to the survival benefit induced by N-RasG12D, quiescent (GFPhigh) and cycling (GFPlow) HSCs from control and NrasG12D mice were sorted and subjected to cytokine starvation for 18 hours. Although our previous data showed that N-RasG12D promotes proliferation of cycling (GFPlow) HSCs but reduces proliferation of quiescent (GFPhigh) HSCs (16), both quiescent and cycling NrasG12D HSC fractions were more resistant to apoptosis under conditions of cytokine starvation (Fig. 5E), indicating that the survival advantage is a fundamental effect of N-Ras signaling, rather than a consequence of changes in cell cycle. Furthermore, NrasG12D HSCs had lower levels of ROS levels in both GFPhigh and GFPlow fractions, suggesting that N-RasG12D reduces ROS irrespective of proliferative status (Fig. 5F and G). As expected, GFPlow HSCs from both genotypes had higher levels of ROS than GFPhigh HSCs, consistent with a more proliferative and metabolically active cell population (Fig. 5F and G).
To determine the mechanism by which N-RasG12D reduces ROS in HSCs, we considered processes that could either enhance ROS detoxification or decrease ROS production. First, we measured the levels of total and oxidized glutathione and did not detect any difference between the Nras mutant and control LSKs (Supplementary Fig. S6A). In addition, gene expression by microarray analysis did not reveal differences in the levels of the master antioxidant regulator, NRF2, and its targets genes, or genes with known function in HSC maintenance and redox regulation, including foxo3 and bmi1 (Supplementary Fig. S6B). To confirm these findings, we measured mRNA levels of the NRF2 target, Hmox1, Nqo1, and Gclm by qRT-PCR and did not see any significant differences between control and NrasG12D HSCs (Supplementary Fig. S6C). Taken together, enhanced antioxidant signaling is unlikely the mechanism by which N-RasG12D reduces ROS in HSCs. Next, we determined if ROS production is decreased in NrasG12D HSCs. The majority of ROS is derived from bioenergetic processes in the mitochondria (39, 40), and we thus questioned if N-RasG12D induced changes in mitochondrial metabolism in HSCs. To examine metabolic activity, we measured commonly used metabolic output parameters including mitochondrial membrane potential (via TMRM staining) and ATP (via CellTiter-Glo). We found that NrasG12D HSCs demonstrated overall lower mitochondrial membrane potential and ATP levels (Fig. 6A and B). Notably, this phenotype was again limited to HSCs and early progenitors and not seen in total bone marrow or lineage-committed cells (Fig. 6A; Supplementary Fig. S7A). Of note, we found no significant differences in mitochondrial DNA content as measured by the ratio of mitochondrial DNA to nuclear DNA by qPCR, between control and NrasG12D LSKs (Fig. 6C), suggesting N-RasG12D dysregulates mitochondrial metabolic activity rather than mitochondrial biogenesis. To validate this phenotype, we used a second mouse model, Vav1-Cre+; NrasLSL-G12D/+, which allows activation of NrasG12D within the hematopoietic compartment during embryogenesis and does not require activation by pIpC injection (41). Consistently, HSCs from Vav1-cre+; NrasLSL-G12D/+ mice showed lower levels of ROS and mitochondrial membrane potential and no differences in mitochondrial DNA content compared with wild-type control littermates (Supplementary Fig. S7B–S7D). Again, this phenotype was limited to HSCs and early progenitors and not observed in lineage-committed cells (Supplementary Fig. S7B and S7C). Taken together, these data suggest that oncogenic N-RasG12D diminishes oxidative stress specifically in the HSC and early progenitor compartments, at least in part, by remodeling metabolism.
Figure 6.
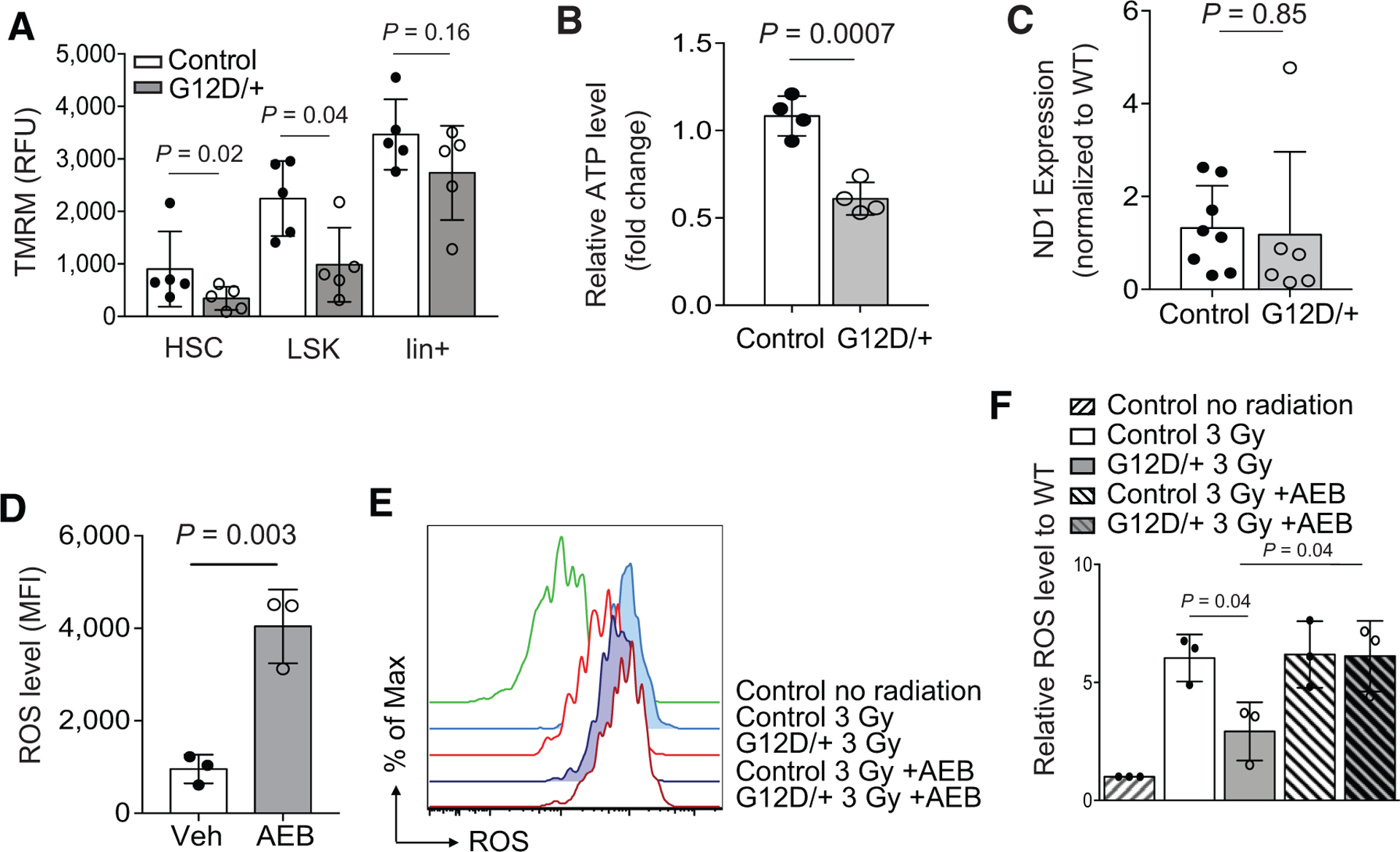
N-RasG12D reduces mitochondrial activity and ATP contents in HSCs. A, Mitochondrial membrane potential as measured by TMRM in hematopoietic populations from control or Nras mutant (G12D/+) mice (n = 5). Paired Student t test was used to determine statistical significance. RFU, relative fluorescent units. B, ATP levels of HSCs (400 SLAM HSCs) from control and Nras mutant mice (n = 4). Unpaired Student t test was used to determine statistical significance. C, Mitochondrial content, as measured by the ratio of mitochondrial (copies of ND1 gene) to nuclear (copies of β-actin gene) DNA. DNA was extracted from FACS-sorted 40,000 control and NrasG12D LSKs by DirectTail lysis reagent with proteinase K. ND1 (mitochondrial-encoded gene) copies were quantified by qPCR and normalized to β-actin (n = 8). Unpaired Student t test was used to determine statistical significance. D, Wild-type SLAM HSCs were treated in vitro with the pan-PKC inhibitor AEB, and levels of ROS were measured by flow cytometry using CellRox (n = 3). Unpaired Student t test was used to calculate statistical significance. MFI, mean fluorescence intensity. E and F, Control or Nras mutant mice were irradiated (300 cGy) and injected with two doses (6 and 12 hours after radiation) of AEB (10 μg/g body mass), and ROS levels were measured 24 hours after radiation (n = 3). ANOVA was used to calculate statistical significance. Data represent mean ± SD; n, number of mice.
Given the role of PKC signaling in apoptosis and cellular metabolism, we next determined if PKC mediates survival of Nras mutant HSCs by modulating ROS metabolism. A recent report identified PKCδ as an important modulator of HSCs, which directly linked metabolic activity with HSC function and survival (42). We observed that PKC inhibition at least partially blocked the survival of Nras mutant HSCs (Fig. 4E and F). We next determined if PKC inhibition would raise levels of ROS in HSCs. First, we exposed 3,000 FACS-sorted wild-type CD48−LSKs to PKC inhibitor, AEB, and cultured for 12 hours. Following this incubation, we observed significantly higher levels of ROS (Fig. 6D) in the AEB-treated group. To test the effect of PKC inhibition on ROS levels in vivo, we sublethally irradiated (3 Gy) control and NrasG12D mice and then administered two doses of AEB via intraperitoneal injection at 12 and 18 hours after radiation (Fig. 4D). We found that in vivo treatment with AEB significantly decreased PKCδ protein level (Fig. 3F), and abrogated N-RasG12D–mediated reductions in ROS (Fig. 6E and F). Together with the findings that PKC inhibition abolished N-RasG12D–induced survival benefit after stress, these data support the hypothesis that N-RasG12D utilizes PKC signaling, specifically via activation of PKCδ, to mitigate oxidative stress and promote survival in HSCs and that this effect may be mediated, at least in part, through reprogrammed metabolism.
Discussion
The pathologic role of oncogenic RAS mutations has been well-established across a spectrum of solid tumors through elegant work in pancreatic, lung, and colon cancers. In hematologic malignancies, although mutations in RAS and Ras-activating genes are frequently detected (2), the pathologic role of oncogenic Ras mutations, particularly in adult leukemia, are still being debated. As sequencing sensitivity improves and data from larger clinical studies become available, mutations in RAS and Ras pathways are being identified as frequent molecular lesions in pediatric cancers and are enriched when evaluating high-risk, refractory and relapsed cancers across a spectrum of disease subtypes. These studies include pediatric embryonal rhabdomyosarcoma (43), neuroblastoma (4, 44), AML (3), and relapsed acute lymphoblastic leukemia, myelodysplastic syndrome, and neuroblastoma (4, 5, 45, 46). In addition, mutations in RAS and Ras-activating genes in hematologic malignancies are associated with treatment resistance across a range of therapies, including IDH, FLT3, and JAK inhibitors and steroids (3, 7, 9, 47). Here, we show that oncogenic Ras signaling supports HSC/P survival through reduced ROS after exposure to metabolic or genotoxic stress, therefore providing mechanistic evidence of how oncogenic Ras maintains transformed HSC/P pools after treatment that promotes disease relapse.
Uncontrolled tumor growth requires massive cell and substrate turnover and, thus, many cancers have altered metabolism and dysregulated levels of ROS (48). Previous studies have linked the activation of several oncogenes, including RAS, BCR-ABL, and FLT3-ITD, with increased levels of ROS during malignant transformation (49–51). In contrast, during early tumorigenesis, oncogenic K-Ras was shown to induce the redox regulator, NRF2, thus lowering ROS and enhancing early pancreatic tumor formation (52). We show that, analogous to this effect, endogenously expressed oncogenic N-Ras confers a similar “redox low” phenotype in HSCs, although likely through a different mechanism. NrasG12D HSCs display similar levels of redox gene expression (Supplementary Fig. S6B and S6C) and comparable levels of total and oxidized glutathione (Supplementary Fig. S6A), but rather exhibit lower mitochondrial activity (Fig. 6A–C). This subsequently leads to a reduction of ROS and ultimately confers a survival benefit (Fig. 5). These results are in line with other studies describing how cancer “stem-like” populations drive treatment resistance via redox programs (36, 53, 54). Unique to HSCs, however, is that dissipation of redox stress may also provide a functional advantage, as oxidative stress is intimately linked to their differentiation, proliferation, and, ultimately, depletion (32, 55–58). Our data also show that the reduced ROS (Fig. 5A) and metabolic phenotype (Fig. 6A–C; Supplementary Fig. S7A–S7D) was limited to the HSC and early progenitor compartment and not lineage-committed cells. Thus, defining the altered biochemical dependencies and pathways in transformed HSC/Ps may provide fundamental insights into both normal and malignant hematopoiesis and highlights how studying these primitive cells may identify discrete targets distinct from the leukemic bulk.
Once activated, Ras proteins trigger extensive downstream signaling cascades that often promote uncontrolled cell proliferation (1) and, illustratively, tumor growth. Despite extensive clinical efforts to target oncogenic Ras signaling through MEK, AKT, and mTOR pathways, the efficacy of inhibiting these pathways is often modest and short-lived, suggesting a refractory population persists throughout treatment. Here, we show that the biochemical effects of oncogenic Ras signaling within the HSC/P compartment confer a survival advantage (Figs. 1 and 2) and therefore may form the basis of relapse. Importantly, these effects are not mediated by the canonical Ras effector pathways, but depends on a noncanonical Ras effector, PKCδ (Figs. 3 and 4). Although there are likely isoform, tissue and context-specific effects of PKC signaling and apoptosis, PKCδ has been identified to regulate HSC maintenance and metabolism (42). Our studies here indicate that PKCδ is activated by oncogenic N-Ras and mediates the prosurvival phenotype and metabolic remodeling of Nras mutant HSCs. Interestingly, although the study from Rao and colleagues (42) showed that PKCδ knockout increased ROS and mitochondrial activity likely through increased mitochondrial mass, our results here revealed that PKC activation in NrasG12D HSCs led to increased ROS and mitochondrial activity without affecting mitochondrial mass. Therefore, the mechanism through which PKC regulates mitochondria function seems to be complex and context dependent. Finally, PKCδ has been shown to promote tumorigenesis in oncogenic K-Ras–mediated lung cancer and additional isoforms have been associated with inferior survival in lung and breast cancers (59, 60). Furthermore, inhibition of PKC has been reported to mitigate growth of lymphoma (61) and pancreatic cancer (62), and has been identified as one of the kinases targeted by dasatinib (63) and midostaurin (64). Combined, these studies highlight how PKC may contribute to tumorigenesis and serve as a therapeutic target, and thus, warrants further investigation.
Supplementary Material
Significance:
Targeting oncogenic N-Ras–mediated reduction of ROS in hematopoietic stem cells through inhibition of the noncanonical Ras effector PKC may serve as a novel strategy for treatment of leukemia and other Ras-mutated cancers.
Acknowledgments
The authors thank the University of Michigan Flow Cytometry Shared Resource Laboratory (supported by the NCI of the NIH under award number P30CA046592) and the University of Michigan Unit for Laboratory Animal Medicine for technical assistance and mouse colony maintenance, respectively. They also thank R. Coolon for genotyping and mouse colony management. This work was supported by NIH/NHLBI T32 (5-T32-HL007622), NIH K12 (5-K12-HD028820-25), Hyundai Hope on Wheels Young Investigator Award, and a St. Baldrick’s Fellow Award with Tough Like Ike Support (to G.M. Ney), and NIH/NHLBI R01HL132392 and 1R01HL150707, American Cancer Society (125080-RSG-13-253-01-LIB), American Society of Hematology, the V Foundation for Cancer Research, Gabrielle’s Angel Foundation, and University of Michigan Rogel Cancer Center research grants (to Q. Li).
Footnotes
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors’ Disclosures
G.M. Ney reports grants from NIH, St. Baldrick’s Foundation, and Hyundai Hope on Wheels during the conduct of the study, as well as patents 8,324,258 and 8,497,307 issued, licensed, and with royalties paid from Lycera Inc. No disclosures were reported by the other authors.
References
- 1.Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell 2017;170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012;120:3397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017;130:732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 2015;47:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 2014;124:3420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jerchel IS, Hoogkamer AQ, Aries IM, Steeghs EMP, Boer JM, Besselink NJM, et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018;32:931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov 2019;9:1050–63. [DOI] [PubMed] [Google Scholar]
- 8.Santos FPS, Getta B, Masarova L, Famulare C, Schulman J, Datoguia TS, et al. Prognostic impact of RAS-pathway mutations in patients with myelofibrosis. Leukemia 2020;34:799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A, et al. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal 2014;7:ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell 2008;132:681–96. [DOI] [PubMed] [Google Scholar]
- 11.Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med 2010;2:17ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 2014;111: 2548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood 2011;117:2022–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Liu Y, Li Z, Wang Z, Tan LX, Ryu MJ, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 2011;118:368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Q, Bohin N, Wen T, Ng V, Magee J, Chen SC, et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature 2013;504:143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol 2009;27:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domen J The role of apoptosis in regulating hematopoietic stem cell numbers. Apoptosis 2001;6:239–52. [DOI] [PubMed] [Google Scholar]
- 19.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005;121:1109–21. [DOI] [PubMed] [Google Scholar]
- 20.Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010;7:174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013;494:323–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams GT, Smith CA, Spooncer E, Dexter TM, Taylor DR. Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature 1990;343:76–9. [DOI] [PubMed] [Google Scholar]
- 23.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017;543:205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011;208:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumitomo Y, Koya J, Nakazaki K, Kataoka K, Tsuruta-Kishino T, Morita K, et al. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016;128:1614–24. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature 2016;529:37–42. [DOI] [PubMed] [Google Scholar]
- 27.Whelan RD, Parker PJ. Loss of protein kinase C function induces an apoptotic response. Oncogene 1998;16:1939–44. [DOI] [PubMed] [Google Scholar]
- 28.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov 2012;11:937–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuang S, Demirs JT, Kochevar IE. Protein kinase C inhibits singlet oxygen-induced apoptosis by decreasing caspase-8 activation. Oncogene 2001;20: 6764–76. [DOI] [PubMed] [Google Scholar]
- 30.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev 2008;88:1341–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006;12:446–51. [DOI] [PubMed] [Google Scholar]
- 32.Mantel CR, O’Leary HA, Chitteti BR, Huang X, Cooper S, Hangoc G, et al. Enhancing hematopoietic stem cell transplantation efficacy by mitigating oxygen shock. Cell 2015;161:1553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007;1:101–12. [DOI] [PubMed] [Google Scholar]
- 34.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007;128:325–39. [DOI] [PubMed] [Google Scholar]
- 35.Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011;118:2941–50. [DOI] [PubMed] [Google Scholar]
- 36.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009;458:780–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013; 12:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A 2011;108:8773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005; 120:483–95. [DOI] [PubMed] [Google Scholar]
- 40.Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 2006;27:639–45. [DOI] [PubMed] [Google Scholar]
- 41.Georgiades P, Ogilvy S, Duval H, Licence DR, Charnock-Jones DS, Smith SK, et al. VavCre transgenic mice: a tool for mutagenesis in hematopoietic and endothelial lineages. Genesis 2002;34:251–6. [DOI] [PubMed] [Google Scholar]
- 42.Rao TN, Gupta MK, Softic S, Wang LD, Jang YC, Thomou T, et al. Attenuation of PKCdelta enhances metabolic activity and promotes expansion of blood progenitors. EMBO J 2018;37:e100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood 2013;121: 2186–98. [DOI] [PubMed] [Google Scholar]
- 44.Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018; 362:1165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med 2017;376:536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2016;113:11306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aries IM, van den Dungen RE, Koudijs MJ, Cuppen E, Voest E, Molenaar JJ, et al. Towards personalized therapy in pediatric acute lymphoblastic leukemia: RAS mutations and prednisolone resistance. Haematologica 2015;100:e132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell 2017;168:657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997; 275:1649–52. [DOI] [PubMed] [Google Scholar]
- 50.Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem 2000;275:24273–8. [DOI] [PubMed] [Google Scholar]
- 51.Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood 2008; 111:3173–82. [DOI] [PubMed] [Google Scholar]
- 52.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475:106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017;549:476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017;170:1079–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009;461:537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003;423:302–5. [DOI] [PubMed] [Google Scholar]
- 57.Yu WM, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013;12:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, et al. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun 2016;7:13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Symonds JM, Ohm AM, Carter CJ, Heasley LE, Boyle TA, Franklin WA, et al. Protein kinase C delta is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res 2011;71:2087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zurgil U, Ben-Ari A, Rotem-Dai N, Karp G, Krasnitsky E, Frost SA, et al. PKCeta is an anti-apoptotic kinase that predicts poor prognosis in breast and lung cancer. Biochem Soc Trans 2014;42:1519–23. [DOI] [PubMed] [Google Scholar]
- 61.Chang G, Zheng J, Xiao W, Chang S, Wei Q, Wu H, et al. PKC inhibition of sotrastaurin has antitumor activity in diffuse large B-cell lymphoma via regulating the expression of MCT-1. Acta Biochim Biophys Sin 2018;50: 399–407. [DOI] [PubMed] [Google Scholar]
- 62.Cheng J, Tian L, Ma J, Gong Y, Zhang Z, Chen Z, et al. Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase Cdelta activation in pancreatic ductal adenocarcinoma. Mol Oncol 2015;9:105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tobio A, Alfonso A, Botana LM. C-kit mutations determine dasatinib mechanism of action in HMC-1 neoplastic mast cells: dasatinib differently regulates PKCdelta translocation in HMC-1(560) and HMC-1(560,816) cell lines. Immunopharmacol Immunotoxicol 2015;37:380–7. [DOI] [PubMed] [Google Scholar]
- 64.Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv 2018;2:444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.