

Abstract:
Circular RNA (circRNA) MFACR promotes cardiomyocyte death that leads to myocardial infarction (MI). This study aimed to explore the role of MFACR in MI. T-qPCRs were performed to measure the expression levels of MFACR and miR-125b in plasma samples from both MI patients (n = 61) and healthy controls (n = 61). MFACR or miR-125b was overexpressed in AC16 cells (cardiomyocytes) to explore the interaction between them. Methylation of miR-125b gene in cells with the overexpression of MFACR was detected by methylation-specific PCR. Cell apoptosis after transfections was detected by cell apoptosis assay. MI model was constructed to further demonstrate the effect of MFACR in vivo. We found that MFACR was upregulated in MI and inversely correlated with miR-125b. In AC16 cells, hypoxia treatment increased the expression levels of MFACR and decreased the expression levels of miR-125b. In AC16 cells, overexpression of MFACR decreased the expression levels of miR-125b and increased the methylation of miR-125b gene. Under hypoxia treatment, overexpression of MFACR increased AC16 cell apoptosis, and overexpression of miR-125b decreased cell apoptosis. In addition, overexpression of miR-125b reversed the effects of overexpression of MFACR on cell apoptosis both in vivo and in vitro.
Key Words: myocardial infarction, MFACR, miR-125b, methylation, cardiomyocytes
INTRODUCTION
Myocardial infarction (MI), commonly known as heart attack, is a clinical disorder caused by reduced or ceased blood supply to the heart.1 MI can cause severe heart muscle damages, and the symptoms mainly include chest pain and discomforts in the shoulder, back, arm, neck, or jaw.2,3 It is estimated that the mortality rate of MI is about 30%, and more than half of the deaths occur before hospitalization.4,5 Even worse, about 10% of the survivors die within 1 year after discharge.4,5 MI patients are usually treated with beta blockers and aspirin, which are antiplatelet drugs.6 However, there are no available approaches to reverse the heart damages (myocardial scar).6 Therefore, novel therapeutic approaches are still needed. Besides, apoptosis of cardiomyocytes can easily lead to MI.7 Therefore, it is of great therapeutic significance to explore the molecular mechanisms of cardiomyocyte apoptosis to find effective targets for the treatment of MI.
Molecular factors play critical roles in the progression of MI.8,9 These molecular players participate in MI mainly by regulating inflammatory responses and cell apoptosis.10,11 In effect, some molecular pathways have been shown potentials in novel anti-MI therapies, such as targeted therapies that suppress MI by regulating gene expression.12 A recent study reported that KLF5 could possibly induce cell and tissue damage in MI through downregulation of miR-27a, which may provide novel insights into the treatment of MI.13 Circular RNAs (circRNAs) have no protein-coding capacity but play critical roles in human diseases by regulating gene expression.14,15 It was reported that circRNA POSTN promoted MI-induced myocardial injury and cardiac remodeling.16 Another study has also shown that mitochondrial fission and apoptosis-related circRNA (MFACR) regulated mitochondrial fission and apoptosis in the heart, which was related to MI.14 MiRNAs are small ncRNAs that are 19–22 nucleotides in length and play critical roles in various biological processes.17–20 It has been demonstrated that miRNAs are involved in the regulation of MI.7,10,21 Our microarray analysis results revealed an inverse correlation between the expression of MFACR and miR-125b, which played protective roles in cardiac dysfunction by suppressing cell apoptosis. However, the relationship between MFACR and miR-125b in MI is unknown. This study was conducted to explore the crosstalk between MFACR and miR-125b in MI.
METHODS
MI Patients and Controls
A total of 61 MI patients (acute phase) and 61 healthy controls were enrolled at the Third People's Hospital between May 2018 and June 2020. This study was approved by the ethics committee of this hospital. The patients who were diagnosed with MI underwent percutaneous coronary intervention (PCI). MI was diagnosed by ECG (pathologic Q waves and ST segment elevation) and serum biomarkers (increased troponin T or I, and CK-MB). Patients complicated with other severe clinical disorders or with initiated therapy were excluded. All healthy controls were enrolled at the physiological heath center of the aforementioned hospital. Healthy control participants with a history of MI were excluded. All participants signed the informed consent. The characteristics of MI patients were listed in Table 1.
TABLE 1.
The Characteristics of MI Patients
| Variable | Value |
| Age (yr) | 55.9 ± 7.1 |
| Gender | |
| Male | 45.85% |
| Female | 64.15 |
| Diabetes, % | 16.21% |
| Hypertension, % | 55.35% |
| Hyperlipidemia, % | 68.03% |
| Prior myocardial infarction, % | 21.57% |
| Body mass index (kg/m2) | 29.7 ± 4.1 |
| Systolic blood pressure (mm Hg) | 124 ± 22 |
| Diastolic blood pressure (mm Hg) | 82 ± 14 |
| ST-elevation myocardial infarction, % | 47.12% |
| Serum total cholesterol, mg/dL | 120 ± 67 |
| Serum high density lipoprotein, mg/dL | 25 ± 26 |
| Serum low density lipoprotein, mg/dL | 87 ± 40 |
| Serum triglycerides, mg/dL | 110 ± 70 |
Blood Extraction and Plasma Preparations
Before therapy, blood (3 mL) was drawn from both healthy controls and MI patients. To prepare plasma samples, blood was mixed with citric acid (1:10), followed by centrifugation at 1200g for 15 minutes to collect the supernatant (plasma). Plasma samples were kept in liquid nitrogen before the subsequent assays.
Cardiomyocytes
AC16 cardiomyocyte cells (Sigma-Aldrich, St. Louis, MO) were used in this study. AC16 cells were cultivated in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen-Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 12% FBS and 1% penicillin and streptomycin (Sigma-Aldrich). AC16 cells were cultivated in an incubator at 37°C with 95% humidity and 5% CO2. Under hypoxic conditions (1% O2, 5% CO2, and 94% N2), AC16 cells were cultivated for another 96 hours. The changes of the expression levels of MFACR and miR-125b were detected at 0, 24, 48, 72, and 96 hours.
Transfections
For cell transfection, AC16 cells were precultured overnight until 50% confluence and then transfected with MFACR expression vector (Invitrogen) or mimic of miR-125b (Sigma-Aldrich) using lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Negative control (NC) experiments (NC miRNA- or empty vector-transfected cells) and Control (C) experiments (control cells with no transfections) were included in each experiment. The following experiments were performed at 48 hours post-transfection.
RNA Preparations
Total RNAs were extracted from AC16 cells and plasma samples using Ribozol reagent (VWR biotech). RNA samples were incubated with DNase I (Invitrogen) at 37°C for 85 minutes to remove genomic DNA. RNA samples were separated using 5% urea-PAGE gels to check RNA integrity. RNA purity was determined by measuring the OD 260/280 ratio of the RNA samples.
RT-qPCR
SS-IV-RT (Invitrogen) was used to reverse transcribe RNA samples with satisfactory integrity and OD values close to 2.0 into cDNA samples. With cDNA samples as the templates, qPCRs reactions were prepared using SYBR Green Real-Time PCR Master mix (Toyobo). The expression levels of MFACR were determined with 18S rRNA as the internal control. The expression of mature miR-125b was determined through the following steps: 1) poly (A) addition; 2) miRNA reverse transcriptions; 3) qPCRs. All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia) was used to complete all the steps. The 2−ΔΔCT method was used to normalize Ct values of target genes to corresponding internal controls. U6 was used as an internal control for the expression of circRNA MFACR and miR-498.
Methylation-specific PCR (MSP)
Genomic DNA was extracted from AC16 cells using conventional methods. DNA samples were converted using Methylation-Gold kit (ZYMO RESEARCH). Taq 2X master mix (NEB) was used to perform both routine PCRs and methylation-specific PCRs.
Cell Apoptosis Assay
Cells were cultivated in 6-well plates with 12,000 cells in 2 mL medium per well. Three replicate wells were set for each experiment. Under hypoxic conditions (1% O2, 5% CO2, and 94% N2), AC16 cells were cultivated for another 48 hours, followed by washing with ice-cold PBS. After that, AC16 cells were resuspended in binding buffer, followed by staining with Annexin V-FITC and propidium iodide (PI) (Dojindo, Japan). Next, flow cytometry was used to detect cell apoptosis.
MI Model
Male C57BL/6 mice (8 weeks old) were housed with free access to food and water under standard conditions with an alternated 12 h/12 hours light/dark cycle. To construct the MI model, mice were anesthetized with sodium pentobarbital (50 mg/kg) by intraperitoneal injection to receive MI introduction. MI introduction was performed by permanent ligation of the left anterior descending (LAD) artery. Mice in the control group received a sham operation with the same surgical procedures as the experimental ones, except that the LAD artery was not ligated. At indicated time points, mice were euthanized and heart tissues were collected for further experiments. The cardiac infarction was assessed by Masson staining. All animal experiments were performed in accordance with the Animal Management Rules of the Chinese Ministry of Health (document No. 55, 2001) and approved by the Animal Care Committee of the Third People's Hospital.
TUNEL Staining
To evaluate the apoptosis rate of cell apoptosis in heart tissues of MI mice, TUNEL staining was performed using the TUNEL Kit (Roche, Switzerland) following the manufacturer's instructions. The prepared paraffin sections were taken and TUNEL staining was performed according to the steps of tissue repair, TUNEL working fluid drip, DAB staining, and nuclear re-staining. The positive-staining was observed under a fluorescence microscope (Nikon, Japan) and the apoptosis rate was calculated as the following: TUNEL positive nuclei/DAPI-stained nuclei x 100%.
Western Blot Analysis
Total proteins were isolated from frozen ventricle tissues and the primary cardiomyocytes using RIPA lysis buffer (Beyotime). BCA kit (Sangon) was used to quantify protein samples. After separated by electrophoresis with 10% SDS-PAGE, proteins were transferred to PVDF membranes, which were then blocked in 5% nonfat milk at room temperature for 1 hour. After that, anti-GAPDH (ab37168, Abcam, 1:1300) and anti-cas-3 (ab184787, Abcam, 1:1000) rabbit primary antibodies were added to incubate with the membranes at 4°C for 12 hours, followed by incubation with HRP (IgG) (1:1000; ab6721; Abcam) goat secondary antibody at 24°C for 1 hour. Signals were developed using ECL (Sigma-Aldrich). Image J v1.46 software was used to analyze data.
Statistical Analyses
Data were expressed as the mean ± SD of at least 3 independent experiments. MI and the control groups were compared by unpaired t test. Shapiro Wilk normality test was performed to asses normal distribution, and ANOVA Tukey's test was used to analyze the differences among multiple transfection groups. Pearson's analysis was used for correlation analysis. GraphPad 8 software was used to analyze data. P < 0.05 was considered as statistically significant.
RESULTS
MFACR was Upregulated in MI and Inversely Correlated with miR-125b
RT-qPCRs were performed to measure the expression levels of MFACR and miR-125b. Compared with the control group, MFACR was significantly upregulated (Fig. 1A, P < 0.001), and miR-125b was significantly downregulated (Fig. 1B, P < 0.001) in the MI group. Pearson's correlation coefficient analysis showed that the expressions of MFACR and miR-125b were significantly and inversely correlated across MI (Fig. 1C) and control samples (Fig. 1D). Therefore, a crosstalk may exist between MFACR and miR-125b.
FIGURE 1.
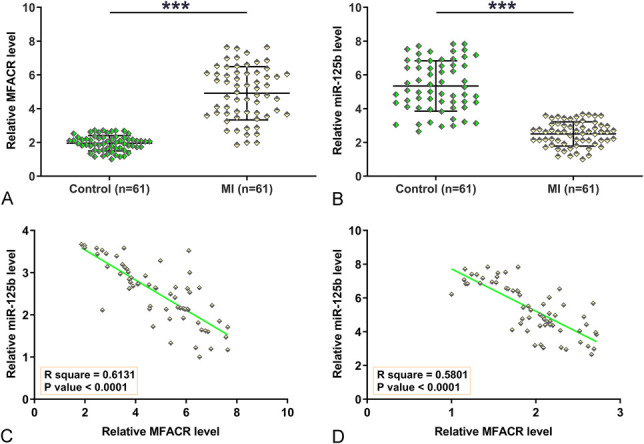
MFACR was upregulated in MI and inversely correlated with miR-125b. RNA isolations and RT-qPCRs were performed on plasma samples to analyze the expression of MFACR (A) and miR-125b (B). Pearson's correlation coefficient analysis was performed to analyze the correlations between MFACR and miR-125b across MI samples (C) and control samples (D). ***P < 0.001.
Hypoxia Treatment Altered the Expression of MFACR and miR-125b in AC16 Cells
AC16 cells were cultivated under hypoxic conditions for 24, 48, 72, and 96 hours, followed by measurement of the expression levels of MFACR and miR-125b by RT-qPCRs. It was observed that hypoxia treatment increased the expression levels of MFACR (Fig. 2A, P < 0.05) and decreased the expression levels of miR-125b (Fig. 2B, P < 0.05) in AC16 cells.
FIGURE 2.
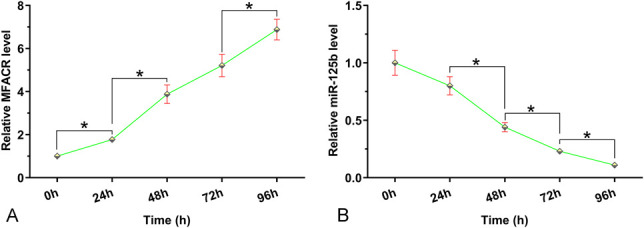
Hypoxia treatment altered the expression of MFACR and miR-125b in AC16 cells. AC16 cells were cultivated under hypoxic conditions for 24, 48, 72, and 96 hours, followed by measurement of the expression of MFACR (A) and miR-125b (B) by RT-qPCR. *P < 0.05.
Overexpression of MFACR Decreased the Expression Levels of miR-125b Through Methylation in AC16 Cells
To study the effects of crosstalk between MFACR and miR-125b, AC16 cells were transfected with MFACR expression vector or miR-125b mimic, followed by confirming the overexpression of MFACR and miR-125b every 24 hours until 96 hours. It was observed that MFACR and miR-125b were significantly overexpressed between 24 hours and 96 hours (Fig. 3A, P < 0.05). Overexpression of MFACR decreased the expression levels of miR-125b between 24 hours and 96 hours (Fig. 3B, P < 0.05), whereas overexpression of miR-125b did not affect the expression of MFACR (Fig. 3C). To study the effects of overexpression of MFACR on the methylation of miR-125b gene, MSP was performed at 96 hours post-transfection. MFACR expression vector transfection significantly increased the methylation of miR-125b (Fig. 3D). Therefore, MFACR may downregulate miR-125b through methylation.
FIGURE 3.

Overexpression of MFACR decreased the expression levels of miR-125b through methylation in AC16 cells. To study the effects of crosstalk between MFACR and miR-125b, AC16 cells were transfected with either MFACR expression vector or miR-125b mimic, followed by checking the overexpression of MFACR and miR-125b every 24 hours until 96 hours (A). The effects of overexpression of MFACR on the expression of miR-125b (B), and the effects of overexpression of miR-125b on the expression of MFACR (C) were analyzed by RT-qPCR. MSP was performed at 96 hours post-transfection to analyze the effects of the overexpression of MFACR on the methylation of miR-125b gene (D).M, methylation; U, un-methylation; *P < 0.05.
Overexpression of MFACR Increased AC16 Cell Apoptosis Induced by Hypoxia through miR-125b
Cell apoptosis under hypoxic conditions was detected. Overexpression of MFACR increased AC16 cell apoptosis, and overexpression of miR-125b decreased cell apoptosis. In addition, overexpression of miR-125b reversed the effects of overexpression of MFACR on cell apoptosis (Fig. 4, P < 0.05).
FIGURE 4.
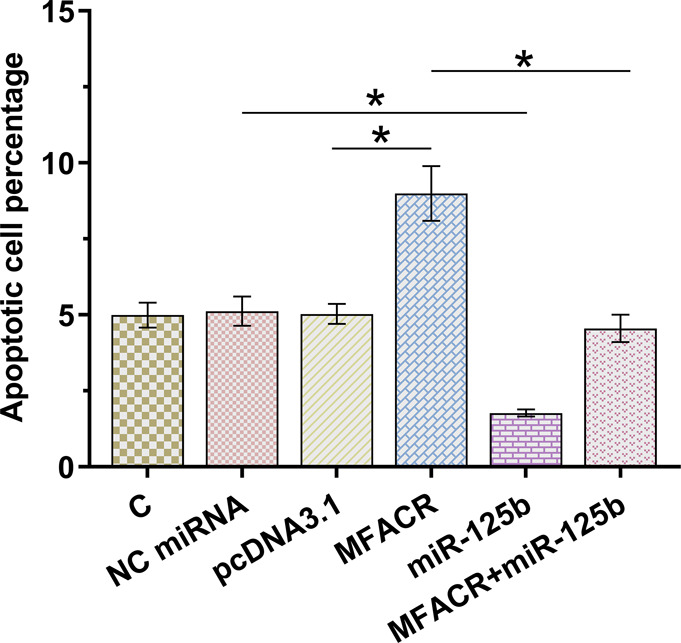
Overexpression of MFACR increased AC16 cell apoptosis induced by hypoxia through miR-125b. Under hypoxic conditions, the role of MFACR and miR-125b in regulating the apoptosis of AC16 cells was analyzed by cell apoptosis assay. *P < 0.05.
Overexpression of MFACR Intensified MI through Downregulating miR-125b
To identify the effect of MFACR and miR-125b on MI-induced myocardial infarction and cardiac remodeling in vivo, the lentiviral vectors of circPostn shRNA and miR-125b were injected in the MI mice ventricular chamber, and the effects of MFACR and miR-125b were validated (Fig. 5). Masson staining results showed that overexpression of MFACR significantly promoted MI-related myocardium injuries (Fig. 5A) and reduced the infarct size (Fig. 5B, P < 0.05).
FIGURE 5.
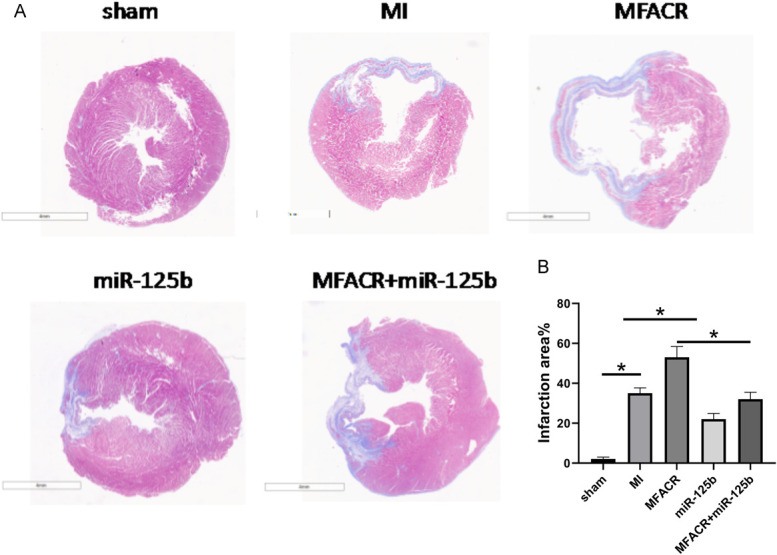
Overexpression of MFACR promoted the progression of MI through miR-125b in vivo. Masson staining was used to explore the effect of MFACR on MI-related myocardium injuries (A) and infarct size (B). *P < 0.05.
Overexpression of MFACR Increased Cardiomyocyte Apoptosis in MI Model by Downregulating miR-125b
TUNEL staining was performed to evaluate the effect of MFACR and miR-125b on cell apoptosis in the MI mice. The results showed that overexpression of MFACR increased cardiomyocyte apoptosis in MI model whereas miR-125b reversed the effect of MFACR (Fig. 6A). The date of TUNEL staining could directly show the trends (Fig. 6B, P < 0.05). To further verify that MFACR increased cardiomyocyte apoptosis, the expression of apoptosis-related protein was detected by Western blotting analysis (Fig. 7, P < 0.05). It showed that the expression levels of caspare-3 were increased in the MI group compared with that in the control group. In addition, overexpression of miR-125b alleviated cardiomyocyte apoptosis on MI model, whereas overexpression of MFACR aggravated cardiomyocyte apoptosis.
FIGURE 6.
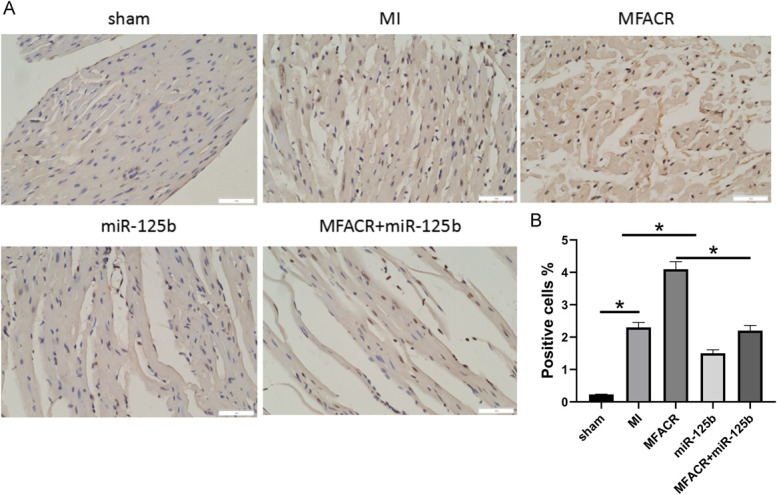
Overexpression of MFACR increased cardiomyocyte apoptosis through miR-125b in MI model. In the MI model, the role of MFACR and miR-125b in regulating the apoptosis of cardiomyocyte was analyzed by TUNEL staining assay (A); statistical analysis of TUNEL staining (B). *P < 0.05.
FIGURE 7.
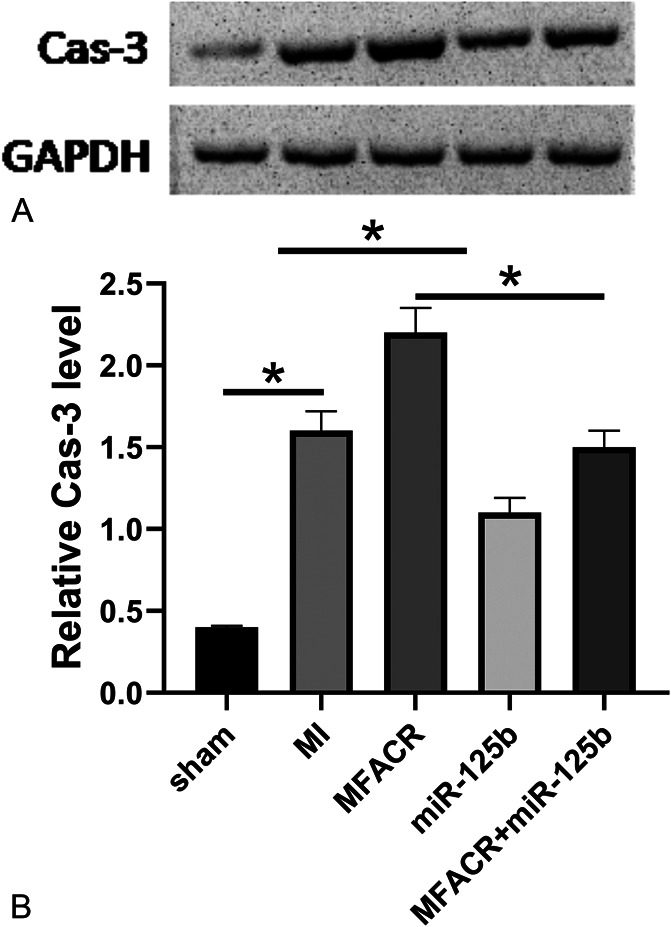
Overexpression of MFACR increased the expression levels of Cas-3 of heart tissues in the MI model through miR-125b. The expression of Cas-3 of heart tissues in MI model was detected by western blotting (A); statistical analysis of the WB (B). *P < 0.05.
DISCUSSION
In this study, we investigated the involvement of MFACR and miR-125b in MI. We found that MFACR was upregulated, whereas miR-125 was downregulated in MI. Interestingly, MFACR may downregulate miR-125b through methylation to promote the apoptosis of AC16 cells induced by hypoxia.
A recent study reported that knockdown of MFACR reduced the apoptosis of cardiomyocytes and attenuated mitochondrial fission by increasing the expression levels of miR-652–3p.22 However, the expression pattern and function of MFACR in MI are unknown. In this study we showed that MFACR was upregulated in MI. Interestingly, hypoxia treatment significantly increased the expression levels of MFACR in AC16 cells. Therefore, the upregulation of MFACR in MI patients is likely induced by the hypoxic conditions in the heart. Moreover, cell apoptosis analysis showed that overexpression of MANCR increased the apoptosis of AC16 cells induced by hypoxia. Therefore, hypoxia-inducible MFACR may promote MI progression by promoting cell apoptosis.
MiR-125b can target TRAF6 to regulate p53-mediated apoptotic signaling and the activity of nuclear factor κB to suppress the apoptosis of cardiomyocytes, thereby suppressing the development of cardiac dysfunction.23 It was reported that miR-652–3p, which was directly targeted and downregulated by MFACR, directly downregulated MTP18 and attenuated mitochondrial fission, cardiomyocyte apoptosis, and MI in vitro and in vivo.24 Consistently, our study observed the downregulation of miR-125b in MI. Interestingly, hypoxia treatment decreased the expression levels of miR-125b in AC16 cells, and overexpression of miR-125b reduced cell apoptosis under hypoxic conditions. We also observed that overexpression of miR-125b reversed the promoting effect of MFACR on MI in vivo. Therefore, overexpression of miR-125b may serve as a potential therapeutic target for MI. However, clinical trials are needed to verify our hypothesis.
In this study, we showed that overexpression of MFACR decreased the expression levels of miR-125b by increasing the methylation of the miR-125 gene. In addition, we constructed an MI animal model to further verify our results. Our study was in the context of the pathophysiology of MI and the underlying mechanisms still need to be further explored for potential therapeutic options.
CONCLUSION
In conclusion, MFACR is upregulated in MI and miR-125b is downregulated in MI. MFACR may downregulate miR-125b through methylation to promote the apoptosis of cardiomyocytes to promote the progression of MI.
Footnotes
The authors report no conflicts of interest.
Contributor Information
Shujuan Wang, Email: ij1037@163.com.
Long Li, Email: kb8292@163.com.
Weijie Deng, Email: iz7550@163.com.
REFERENCES
- 1.Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). Eur Heart J. 2019;40:237–269. [DOI] [PubMed] [Google Scholar]
- 2.Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet 2017;389:197–210. [DOI] [PubMed] [Google Scholar]
- 3.Anderson JL, Morrow DA. Acute myocardial infarction. N Engl J Med. 2017;376:2053–2064. [DOI] [PubMed] [Google Scholar]
- 4.Asaria P, Elliott P, Douglass M, et al. Acute myocardial infarction hospital admissions and deaths in England: a national follow-back and follow-forward record-linkage study. Lancet Public Health 2017;2:e191–e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castro-Dominguez Y, Dharmarajan K, McNamara RL. Predicting death after acute myocardial infarction. Trends Cardiovasc Med. 2018;28:102–109. [DOI] [PubMed] [Google Scholar]
- 6.Luan Y, Li Y, Zhao L, et al. Primary prevention of myocardial infarction: aspirin is not as useful as it seems. Ann Transl Med. 2020;8:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng Q, Li L, Bi X. Long non-coding RNA SNHG7 knockdown protects mouse cardiac fibroblasts against myocardial infarction by regulating miR-455-3p/PTAFR axis. J Cardiovasc Pharmacol. 2021;77:796–804. [DOI] [PubMed] [Google Scholar]
- 8.Maemura K, Layne MD, Watanabe M, et al. Molecular mechanisms of morning onset of myocardial infarction. Ann N Y Acad Sci. 2001;947:398–402. [DOI] [PubMed] [Google Scholar]
- 9.Vilahur G, Juan-Babot O, Peña E, et al. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J Mol Cell Cardiol 2011;50:522–533. [DOI] [PubMed] [Google Scholar]
- 10.Saxena A, Russo I, Frangogiannis NG. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res. 2016;167:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol. 2012;8:863–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013;43:986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian Z, Zhang Y, Lyu X. Promoting roles of KLF5 in myocardial infarction in mice involving microRNA-27a suppression and the following GFPT2/TGF-β/Smad2/3 axis activation. Cell Cycle (Georgetown, TX) 2021:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han B, Chao J, Yao H. Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol Ther. 2018;187:31–44. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Li C, Tan C, et al. Circular RNAs: a new frontier in the study of human diseases. J Med Genet. 2016;53:359–365. [DOI] [PubMed] [Google Scholar]
- 16.Cheng N, Wang M, Wu Y, et al. Circular RNA POSTN promotes myocardial infarction-induced myocardial injury and cardiac remodeling by regulating miR-96-5p/BNIP3 Axis. Front Cell Dev Biol. 2020;8:618574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tu W, Gong J, Song J, et al. miR-20a/TCF4 axis-mediated inhibition of hepatocytes proliferation impairs liver regeneration in mice PHx model by regulating CDC2 and CDC6. J Cell Mol Med. 2021;25:5220–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Chen S, Lu J, et al. MicroRNA-363-3p promote the development of acute myeloid leukemia with RUNX1 mutation by targeting SPRYD4 and FNDC3B. Medicine 2021;100:e25807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng L, Han C, Nie C, et al. Identification of potential serum exosomal microRNAs involved in acinar-ductal metaplasia that is a precursor of pancreatic cancer associated with chronic pancreatitis. Medicine 2021;100:e25753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Zhu Y, Wu C, et al. Adipose-derived mesenchymal stem cells-derived exosomes carry MicroRNA-671 to alleviate myocardial infarction through inactivating the TGFBR2/smad2 Axis. Inflammation 2021;44:1815–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Yu R, Wen S, et al. Overexpressing microRNA-203 alleviates myocardial infarction via interacting with long non-coding RNA MIAT and mitochondrial coupling factor 6. Arch Pharm Res. 2021;44:525–535. [DOI] [PubMed] [Google Scholar]
- 22.Wang K, Gan TY, Li N, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ 2017;24:1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma H, Wang X, Ha T, et al. MicroRNA-125b prevents cardiac dysfunction in polymicrobial sepsis by targeting TRAF6-mediated nuclear factor κB activation and p53-mediated apoptotic signaling. J Infect Dis. 2016;214:1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Gan T, Li N, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cel Death Differ. 2017;24:1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]