

Abstract
The noncoding genome plays an important role in de novo gene birth and in the emergence of genetic novelty. Nevertheless, how noncoding sequences’ properties could promote the birth of novel genes and shape the evolution and the structural diversity of proteins remains unclear. Therefore, by combining different bioinformatic approaches, we characterized the fold potential diversity of the amino acid sequences encoded by all intergenic open reading frames (ORFs) of S. cerevisiae with the aim of (1) exploring whether the structural states’ diversity of proteomes is already present in noncoding sequences, and (2) estimating the potential of the noncoding genome to produce novel protein bricks that could either give rise to novel genes or be integrated into pre-existing proteins, thus participating in protein structure diversity and evolution. We showed that amino acid sequences encoded by most yeast intergenic ORFs contain the elementary building blocks of protein structures. Moreover, they encompass the large structural state diversity of canonical proteins, with the majority predicted as foldable. Then, we investigated the early stages of de novo gene birth by reconstructing the ancestral sequences of 70 yeast de novo genes and characterized the sequence and structural properties of intergenic ORFs with a strong translation signal. This enabled us to highlight sequence and structural factors determining de novo gene emergence. Finally, we showed a strong correlation between the fold potential of de novo proteins and one of their ancestral amino acid sequences, reflecting the relationship between the noncoding genome and the protein structure universe.
Comparative genomics have revealed the existence of an important amount of taxonomically restricted genes and, more specifically, of orphan genes in various eukaryotic genomes (Tautz and Domazet-Lošo 2011; Wissler et al. 2012; Van Oss and Carvunis 2019; Vakirlis et al. 2020b). These genes lack detectable homologs in outgroup species and can constitute up to 30% of a genome's genes. They can derive from clearly distinct mechanisms, including the well-known mechanisms of duplication or horizontal gene transfer followed by fast divergence (Kaessmann 2010; Tautz and Domazet-Lošo 2011; Schlötterer 2015; Van Oss and Carvunis 2019). However, de novo emergence from noncoding regions has now been proven to be an undeniable additional mechanism, and studies reporting evidence of de novo gene birth are published every year, thereby giving a new role to noncoding regions in the creation of genetic novelty (Knowles and McLysaght 2009; Tautz and Domazet-Lošo 2011; Wu et al. 2011; Murphy and McLysaght 2012; Zhao et al. 2014; Schlötterer 2015; Li et al. 2016; Vakirlis et al. 2018, 2020b; Zhang et al. 2019; Heames et al. 2020; Blevins et al. 2021). Nevertheless, how noncoding sequences can code for a functional product and consequently give rise to novel genes remains unclear. Indeed, function is intimately related to protein structure and more generally to protein structural properties. All proteomes are characterized by a large diversity of structural states. The structural properties of a protein result from its composition in hydrophobic and hydrophilic residues. Highly disordered proteins display a high hydrophilic residue content. Membrane proteins, which fold in lipidic environments but aggregate in solution, are enriched in hydrophobic residues. Finally, foldable proteins are characterized by a subtle equilibrium of hydrophobic and hydrophilic residues (Bresler and Talmud 1944). The latter are arranged together into specific patterns that dictate the formation of the secondary structures and the outcoming fold. However, contrarily to coding sequences (CDSs), the nucleotides of noncoding ones are expected to be distributed randomly along the DNA, thereby resulting in different amino acid compositions from CDSs. If and how these amino acid compositions can account for the structural states observed in proteomes are crucial questions to understand the relationship, if any, between the noncoding genome and the protein structure universe. So far, different models of de novo gene emergence have been proposed (Carvunis et al. 2012; Schlötterer 2015; Wilson et al. 2017). The “preadaptation” model proposes an “all or nothing transition to functionality” in which only sequences preadapted not to be harmful (i.e., with enough disorder not to be subjected to aggregation) will give rise to gene birth (Wilson et al. 2017). This model is supported by the observation that young genes and de novo protein domains display a higher disorder propensity than do old genes (Ekman and Elofsson 2010; Bitard-Feildel et al. 2015; Schmitz et al. 2018; Foy et al. 2019). In contrast, the proto-gene model proposes an evolutionary continuum ranging from nongenic sequences to genes (Carvunis et al. 2012). Here, genes evolve de novo through transitory proto-genes that result from pervasive expression of nongenic sequences, and proto-genes are expected to show features intermediate between nongenes and genes. In this study, the investigators reported that in yeast, young genes are less prone to disorder. Recently, Vakirlis et al. (2020a) proposed a transmembrane (TM)-first model in which the membrane environment provides a safe niche for TM-adaptive emerging peptides, which can further evolve toward more soluble peptides. These adaptive peptides have been identified with overexpression, which, according to the investigators, may not be reached outside the laboratory. Whether such peptides, although beneficial in the experimental conditions, would be produced and be beneficial in “natural” conditions deserves further investigation.
Overall, all these studies attribute to the fold potential of noncoding ORFs (including the propensities for disorder, folded state, and aggregation) an important role in the emergence of genetic novelty. However, several questions remain open. First, if the sequence and structural properties of de novo genes have been largely investigated in specific species, the raw material for de novo gene birth and the early stages preceding the fixation of the beneficial ORFs is to be further characterized (Schmitz et al. 2018). Second, if the role of the noncoding genome in de novo gene birth has been largely investigated, its role in protein evolution and structural diversity is to be further characterized as well. Indeed, de novo domains may emerge from noncoding regions through ORF extension or exonization of introns (Bornberg-Bauer and Albà 2013; Bornberg-Bauer et al. 2015). On the other hand, we can assume that protein-coding genes, whatever their evolutionary history, have had a noncoding ancestral origin (Nielly-Thibault and Landry 2019). Whether the noncoding ORFs that gave rise to novel genes can account for the structural diversity of proteomes or whether this structural diversity evolved from ancestral genes that all displayed similar structural properties (i.e., disordered, foldable, or TM-prone) is a crucial question to better understand the role, if any, of noncoding sequences in the protein structure universe.
Here, we characterized the diversity of the fold potential encoded in all intergenic ORFs (IGORFs) of Saccharomyces cerevisiae with the aim of (1) exploring whether the large diversity of structural states observed in proteomes is already present in noncoding sequences, and (2) studying the potential of the noncoding genome to produce novel protein bricks that could give birth to novel genes or be integrated into pre-existing proteins. Then, we investigated the sequence and structural factors determining de novo gene emergence by (1) characterizing the early stages of de novo gene birth through the reconstruction of 70 yeast de novo genes’ ancestral sequences and (2) characterizing the sequence and structural properties of IGORFs with a strong translation signal through ribosome profiling experiments.
Results
We extracted 105,041 IGORFs of at least 60 nucleotides (nt) in S. cerevisiae (Methods). We probed their fold potential with the hydrophobic cluster analysis (HCA) approach (Faure and Callebaut 2013a,b; Bitard-Feildel and Callebaut 2017,2018; Bitard-Feildel et al. 2018) and compared it with the one of the 6669 CDSs of S. cerevisiae. HCA highlights, from the sole information of a single amino acid sequence, the building blocks of protein folds that constitute signatures of folded domains. They consist of clusters of strong hydrophobic amino acids that have been shown to be associated with regular secondary structures (Supplemental Fig. S1; Bitard-Feildel and Callebaut 2017; Bitard-Feildel et al. 2018; Lamiable et al. 2019). These clusters are connected by linkers corresponding to loops or disordered regions. The combination of hydrophobic clusters and linkers in a sequence determines its fold potential. The latter can be appreciated in a quantitative way through the calculation of a foldability score (HCA score) that covers all the fold potential diversity of proteins.
IGORFs contain elementary building blocks of proteins
We first investigated the structural and sequence properties of proteins encoded by CDSs and IGORFs (Fig. 1; Supplemental Tables S1–S4). CDSs are longer than IGORFs and contain more HCA clusters (Mann–Whitney U test, P < 2.2 × 10−16 for both observations) (Fig. 1A,B). The HCA clusters of CDSs and IGORFs display similar sizes of about 11 residues (Mann–Whitney U test, P = 1 × 10−1) (Fig. 1C), and 96.9% of IGORFs harbor at least one HCA cluster. This result shows that the elementary building blocks of proteins are widespread in noncoding sequences. In contrast, CDSs are enriched in long linkers reflecting long flexible regions (6.3 and 11.5 residues for IGORFs and CDSs on average, respectively; Mann–Whitney U test, P = 2.6 × 10−11) (Fig. 1D). As a control, we generated scrambled intergenic sequences (Methods). The resulting random IGORFs behave similarly to real IGORFs for most properties, while being slightly shorter (Mann–Whitney U test, P = 3 × 10−3) (Supplemental Fig. S2). Whether the enrichment in long ORFs observed for real IGORFs results from high-GC-content genomic regions (STOP codons are AT-rich) is to be further investigated.
Figure 1.

Plots of the distributions of sequence and HCA-based structural properties of IGORFs and CDSs. Sequence size (A) and number of HCA clusters (B) per sequence. Size of HCA clusters (C) and size of linkers (D). The P-values were computed with the Mann–Whitney U test (one-sided for A,B,D, and two-sided for C). Asterisks denote level of significance: (***) P < 1 × 10−3; for detailed P-values, see Supplemental Tables S1–S4.
CDSs are enriched in polar and charged residues
If hydrophobic clusters of CDSs and IGORFs display similar sizes, they may not have the same amino acid composition. Therefore, for each amino acid, we calculated its propensity for being in HCA clusters of CDSs over HCA clusters of IGORFs. CDS HCA clusters are clearly enriched in polar and charged residues compared with those of IGORFs (Supplemental Fig. S3A). The same tendency is observed for CDS linkers (Supplemental Fig. S3B). Moreover, negatively charged residues are overrepresented compared with positively charged ones in both HCA clusters and linkers of CDSs. In fact, it has been shown that the charge distribution of a protein has an impact on its diffusion in the cytosol, where positively charged proteins get caught in nonspecific interactions with the abundant negatively charged ribosomes (Schavemaker et al. 2017). We show that the frequency of negatively charged residues of the yeast cytoplasmic proteins is strongly correlated with the proteins’ abundance (Spearman's correlation coefficient: Rho = 0.44, P < 2 × 10−16), suggesting that the crowded cellular environment has shaped the charge distribution of abundant proteins (Supplemental Fig. S4). This result recalls the observation made in previous studies showing that the frequency of “sticky” amino acids on the surface of globular proteins or in disordered proteins decreases as the protein cellular concentration increases (Levy et al. 2012; Macossay-Castillo et al. 2019). Finally, CDSs tend to be enriched in ancient amino acids and codons and depleted in recent ones (Supplemental Fig. S5). As observed in other studies (Trifonov 1987; Brooks and Fresco 2003), yeast CDSs are particularly enriched in GNN codons, which include those coding for negatively charged amino acids. Whether this enrichment is unrelated to codon age and simply results from amino acid content constraints, whether CDSs favor the usage of old codons for ignored reasons, or whether this observation results from a combination of both remains unclear.
IGORFs encode peptides that display a wide diversity of fold potentials, including a substantial amount of foldable peptides
We next used the HCA score in order to assess the fold potential of the peptides encoded by IGORFs. As a reference, we calculated the HCA scores for three sequence data sets consisting of 731 disordered regions, 559 globular proteins, and 1269 TM regions extracted from TM proteins, thereby expected to form aggregates in solution while being able to fold in lipidic environments (Methods) (Fig. 2A; Supplemental Fig. S6). Based on their HCA scores, we defined three categories of fold potentials (i.e., disorder prone, foldable, or aggregation-prone in solution). Here, we define as foldable, proteins that are able to fold into a compact and well-defined 3D structure or partially into an ordered structure in which the secondary structures are, however, present. Figure 2B shows that CDSs and IGORFs belonging to the low HCA score category are indeed presumed to be disordered and display low propensity for aggregation. Comparable but small proportions of CDSs and IGORFs fall into this group (4.9% and 7.7%, respectively), indicating that most coding but also noncoding sequences are not highly prone to disorder in line with the findings of Tretyachenko et al. (2017). The high HCA score category corresponds to aggregation-prone sequences with low disorder propensity. CDSs falling into this category are highly hydrophobic (Supplemental Table S5), with 81% of them annotated as uncharacterized according to UniProt (The UniProt Consortium 2019) and 60% predicted as containing at least one TM domain (Methods). Finally, the intermediate category gathers sequences that have a high potential for being completely or partially folded in solution as shown by their intermediate HCA scores comparable to those of globular proteins. Most CDSs (91.4%) and a majority of IGORFs (66.6%) fall into this category. Both are characterized by intermediate aggregation and disorder propensities, although IGORFs display a wider range of aggregation propensities (Fig. 2B). The fact that these CDSs, although predicted as foldable, show a certain propensity for aggregation, is in line with several studies that reported a high aggregation propensity of proteomes across all kingdoms of life (Greenwald and Riek 2012; Langenberg et al. 2020). This observation has been explained as the side effect of the requirement of a hydrophobic core to form globular structures (Rousseau et al. 2006b; Ganesan et al. 2016; Langenberg et al. 2020). In particular, Langenberg et al. (2020) showed a strong relationship between protein stability and aggregation propensity with aggregation-prone regions mostly buried into the protein and providing stability to the resulting fold. Like for CDSs, these regions, under the hydrophobic effect, may facilitate the stabilization of the IGORF-encoded peptide structure. Whether peptides encoded by IGORFs in the intermediate category fold into a specific 3D structure, a partially ordered structure, or a “rudimentary fold” that stabilizes itself through oligomerization, like the Bsc4 de novo protein (Bungard et al. 2017), deserves further investigation. Finally, the proportions of sequences in the different fold potential categories are different between IGORFs and CDSs, with CDSs mostly falling into the intermediate HCA score category, reflecting that being foldable is a trait that has been strongly selected by evolution. In contrast, IGORFs cover a wide range of fold potentials that is also observed in random IGORFs (4.4%, 61.7%, and 33.9% of sequences in the low, intermediate, and high HCA score categories), showing that, randomly, a wide range of fold potentials including a majority of foldable IGORFs can be expected. Overall, it is questionable whether de novo genes mainly originate from IGORFs encoding foldable peptides or from IGORFs whose corresponding peptides subsequently evolved toward foldable peptides regardless of their initial fold potential.
Figure 2.
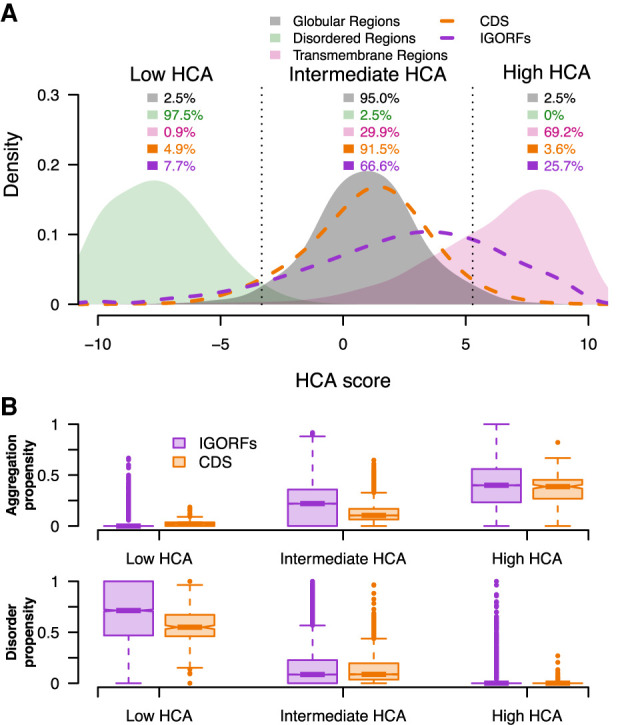
IGORFs encompass the large spectrum of fold potential of canonical proteins. (A) Distribution of the HCA scores for the three reference data sets (i.e., disordered regions, globular domains, and transmembrane regions; green, black, and pink curves, respectively) along with those for the CDSs (orange curve) and IGORFs (purple curve). There is a clear distinction between the distributions of HCA scores calculated for the three reference data sets (two-sided Kolmogorov–Smirnov test, P < 2 × 10−16 for all comparisons). Dotted black lines delineate the boundaries of the low, intermediate, and high HCA score categories, reflecting the three categories of fold potential (i.e., disorder prone, foldable, or aggregation-prone in solution). The boundaries are defined so that 95% of globular domains fall into the intermediate HCA score category, whereas the low and high HCA score categories include all sequences with HCA values that are lower or higher than those of 97.5% of globular domains, respectively. High HCA scores reflect sequences with high densities in HCA clusters that are likely to form aggregates in solution. Low HCA scores indicate sequences with high propensities for disorder, whereas intermediate scores correspond to globular proteins characterized by an equilibrium of hydrophobic and hydrophilic residues (Methods). The percentages of sequences in each category are given for all data sets. Raw data distributions are presented in Supplemental Figure S6. (B) Aggregation and disorder propensities calculated with TANGO and IUPred, respectively, are given for CDSs and IGORFs of each foldability HCA score category.
From IGORFs to de novo genes
Therefore, we traced back the evolutionary events preceding the emergence of 70 de novo genes identified in S. cerevisiae by reconstructing their ancestral IGORFs (ancIGORFs) in order to compare the foldability potential of the peptides encoded by IGORFs that gave birth to de novo genes with the one of the peptides encoded by all other IGORFs and to characterize the steps preceding the emergence of a novel gene (Methods) (Supplemental Fig. S7; Supplemental Table S6). Supplemental Figure S8 shows the example of the YOR333C de novo gene that emerged in the lineage of S. cerevisiae. The corresponding noncoding region in the ancestors preceding its emergence consists of two IGORFs separated by a STOP codon. The fusion of the two consecutive IGORFs was triggered by two 1-nt substitutions that occurred specifically in the S. cerevisiae lineage and led, respectively, to the appearance of a start codon (mutation of isoleucine into methionine through an A/G substitution) and the mutation of the STOP codon into a tyrosine through a G/C substitution. Overall, the 70 de novo genes emerged from a total of 167 ancIGORFs. A minority of de novo genes (16 cases) emerged from a single-ancIGORF that covers almost all their sequence (95% of coverage between the ancIGORF and the resulting de novo gene; i.e., single-ancIGORF de novo genes), whereas the majority (54 cases) results from the combination of multiple ancIGORFs (2.8 on average) through insertion/deletion (indel) events leading to frameshifts in the original sequence and/or STOP codon mutations as observed with the example of YOR333C (i.e., multiple-ancIGORF de novo genes). In line with the findings of Zhang et al. (2019), indels are two times more frequent than STOP codon mutations (64/33). Moreover, the multiple-ancIGORF de novo genes show sequence sizes similar to those of the single-ancIGORF ones, although the ancIGORFs they originate from are shorter than those that led to single-ancIGORF de novo genes (Supplemental Fig. S9).
Figure 3 shows the HCA scores of the proteins encoded by the 70 de novo genes (i.e., de novo proteins) and of the peptides encoded by their corresponding ancIGORFs. The majority of de novo proteins (78%) are predicted as foldable, whereas peptides encoded by ancIGORFs display a larger range of HCA scores. However, ancIGORFs are not IGORF-like, being enriched in sequences encoding foldable peptides (75.4% and 66.6% for ancIGORFs and IGORFs respectively; one-proportion z-test, P = 9.5 × 10−3) and depleted in sequences encoding aggregation-prone ones (18.6% and 25.7% for ancIGORFs and IGORFs respectively; one-proportion z-test, P = 2.1 × 10−2).
Figure 3.

From ancIGORFs to de novo genes. Plot of the HCA score of each ancIGORF (black and white points for single and multiple ancIGORFs, respectively) along with its corresponding de novo gene (blue points). Each de novo gene is connected to its parent ancIGORF(s) with a colored line. A de novo gene is connected to several IGORFs when it results from the combination of different ancIGORFs (i.e., multiple-ancIGORF de novo genes). Green lines indicate cases in which a de novo gene is connected to a low HCA score ancIGORF, and gray and pink lines indicate connections with an intermediate and a high HCA score ancIGORFs, respectively. The HCA score densities of de novo genes and ancIGORFs are shown in gray (bottom and top of the graph, respectively).
Impact of indels and STOP codon mutations on the fold potential of a de novo protein
The overall relationship between the HCA scores of peptides encoded by ancIGORFs and their corresponding de novo proteins is characterized by a funnel shape revealing that most de novo proteins are foldable regardless of the fold potential of the peptides encoded by their IGORF parents (Fig. 3). Two hypotheses can explain this observation: (1) this funnel mostly results from the amino acid substitutions that have occurred since the fixation of the ancIGORF(s) and that led to an increase in foldability of the resulting de novo genes; (2) this funnel results from the fact that combining at least one IGORF encoding a foldable peptide with IGORFs encoding peptides with different fold potentials leads to a foldable product. Figure 4A shows that de novo genes display amino acid frequencies similar to those of ancIGORFs (Supplemental Table S5). This result shows that the mutations that occurred since the fixation of the ancIGORF did not change the overall amino acid composition of the resulting de novo genes and, thus, cannot explain the funnel shape observed in Figure 3. We then reasoned that since the divergence of the last common ancestor predating the emergence of de novo genes, single- and multiple-ancIGORF de novo genes were subjected to similar amino acid mutation rates (average sequence identity between ancIGORFs and their corresponding de novo genes: 83% and 80%, respectively), whereas the multiple-ancIGORF ones (which by definition result from the combination of several IGORFs) have also undergone indels and/or STOP codon mutations. This enabled us to quantify the impact of these different mutational events on the fold potential of the outcoming de novo proteins by calculating the correlation between the HCA score of each de novo protein and the peptides encoded by its corresponding ancIGORF(s). Figure 4B shows that single-ancIGORF de novo proteins display a clear correlation of HCA scores with those of the peptides encoded by their corresponding ancIGORFs (Spearman's correlation coefficient: Rho = 0.87, P < 1.2 × 10−5). This reveals that the amino acid mutations that occurred between the ancestor and the de novo protein did not affect the fold potential of the ancestral sequences, suggesting that the structural properties of the peptides encoded by the single-ancIGORFs were retained in the resulting de novo proteins. In contrast, the correlation is weaker for multiple-ancIGORF de novo proteins (Spearman's correlation coefficient: Rho = 0.47, P < 1.2 × 10−9). This can be attributed to the fact that 81% (44/54) of the multiple-ancIGORF de novo proteins are predicted as foldable (white dots included in the pink squares in Fig. 4B) while being associated with ancIGORFs of different foldability potentials. All foldable de novo genes include at least one foldable ancestral peptide, suggesting that, in these cases, combining disordered or aggregation-prone peptides with a foldable one has led to a foldable de novo protein as well. Supplemental Figure S7E shows the example of the de novo gene YLL020C, which results from the combination through an indel event of a long foldable ancIGORF with a short IGORF predicted as aggregation-prone. The resulting de novo gene is also predicted as foldable. Whether the foldable IGORF was the first to be selected and whether selection has only retained the combinations of IGORFs that do not affect the foldability of the pre-existing selected product deserve further investigation.
Figure 4.

Impact of amino acid substitutions and IGORF fusion on the foldability of de novo genes. (A) Radar plot reflecting the 20 amino acid frequencies of IGORFs, ancIGORFs, de novo genes, and CDSs. (B) Plot of the HCA score of each de novo gene with those of its parent ancIGORF(s). The fold potential of a single-ancIGORF de novo gene is mostly determined by the one of its parent ancIGORFs, whereas the combination of several ancIGORFs through indels and STOP codon mutations leads most of the time to a foldable product. Single- and multiple-ancIGORF de novo genes are represented by black and white points, respectively. Spearman's correlation coefficients of the relationships between single- and multiple-ancIGORF de novo genes’ HCA scores versus the score of their parent ancIGORF(s), as well as the corresponding P-values, are indicated on the plot. The contour lines mark the percentiles of the density function range in black and gray for single- and multiple-ancIGORF de novo genes, respectively. The light pink region indicates de novo genes encoding proteins predicted as foldable.
Translation of IGORFs
Next, we performed ribosome profiling experiments on S. cerevisiae (strain BY4742) and used three additional ribosome profiling data sets to define two types of translated IGORFs (Methods) (Radhakrishnan et al. 2016; Thiaville et al. 2016). The former corresponds to IGORFs that are occasionally translated with a weak translation signal (at least 10 reads in one experiment; Methods). The latter corresponds to IGORFs with a strong translation signal (more than 30 reads in at least two experiments) and with a translation that is strongly favored over the overlapping IGORFs in the other phases (i.e., highly translated IGORFs; Methods). We identified 1235 occasionally translated IGORFs and 31 highly translated ones. Figure 5 and Supplemental Figure S10 show the frequencies of the first translated codons and amino acids, respectively. For both highly and occasionally translated IGORFs, the first translated codon is enriched in AUG compared with all the other translated positions (one-proportion z-test, both P-values < 1 × 10−16). The enrichment in AUG is clearly stronger for highly translated IGORFs, whereas the first translated codons of occasionally translated IGORFs are also enriched in the NUG near-cognate codons reported as alternative start codons (one-proportion z-tests, all P-values < 2 × 10−2) (Ingolia et al. 2011; Cuevas et al. 2021). Nevertheless, because of the low read coverage of IGORFs, we cannot ensure that the first codon with a read is the first to be translated, although the enrichments in AUG or near-cognate codons support this assumption. In addition, the frequencies of the three STOP codons are comparable between all ORF categories (chi-squared tests between all pairs, P-values > 5 × 10−2) (Supplemental Table S7) with a systematic higher frequency of UAA. The S. cerevisiae genome is AT-rich, and the clear enrichment in UAA in all ORF categories, including IGORFs, is in line with previous reports conducted on different organisms showing that the frequencies of UAA and UGA STOP codons are strongly dependent on the GC content (Povolotskaya et al. 2012; Korkmaz et al. 2014; Belinky et al. 2018).
Figure 5.

Frequencies of the 61 codons at the first translated position for highly translated IGORFs (red) and occasionally translated ones (yellow). Gini indexes that reflect the statistical dispersion of the 61 codons at the first translated position are given for highly and occasionally translated IGORFs in red and yellow, respectively. Gini index values range from zero to one, and high values reflect the fact that the first translated positions are enriched in specific codons, particularly AUG and other NUG ones. Codons that are significantly observed at the first translated position compared with the other translated positions are indicated with a star (one-proportion z-test, P < 5 × 10−2). Near-cognate codons are indicated with diamonds.
Translated and ancestral IGORFs display intermediate properties between IGORFs and CDSs
Figure 6, A through D, shows the boxplot distributions of the sizes of the sequences, clusters, and linkers of all ORF categories along with their number of clusters per sequence. The HCA cluster size remains invariant for all categories except for de novo genes. In contrast, highly translated IGORFs, ancestral ones, and de novo genes overall display, for most properties, intermediate values between IGORFs and CDSs. In particular, the highly translated IGORFs and the ancIGORFs are both longer than IGORFs (Mann–Whitney U test, P = 3.4 × 10−2 and 1.3 × 10−22, respectively) and display slightly longer linkers (Mann–Whitney U test, P = 2.6 × 10−2 and 1.8 × 10−2) and higher GC contents (41.9%, 38%, and 36.1% for ancIGORFs, highly translated IGORFs, and IGORFs, respectively). To understand whether the increase in linker size could be explained by the increase in ORF length or GC content, we generated artificial IGORFs with nucleotide compositions of IGORFs and size distribution of ancIGORFs or highly translated IGORFs, respectively. Artificial IGORFs with ancIGORF lengths show linkers of similar size to those of IGORFs (Mann–Whitney U test, P = 2 × 10−1), showing that the increase in linker sizes observed for ancIGORF cannot be explained by their larger size (Supplemental Fig. S11). However, the artificial linkers are shorter than those of ancIGORFs (Mann–Whitney U test, P = 6 × 10−4), suggesting that the effect can be attributed to the nucleotide composition of ancIGORFs. Indeed, scrambling the ancIGORF nucleotides results in linker sizes similar to those of ancIGORFs, suggesting that the sole GC content of ancIGORFs is sufficient to generate long linkers. A similar trend is observed for highly translated IGORFs, although the effect is less pronounced (Supplemental Fig. S11). More generally, if for extreme hydrophobic and hydrophilic contents the sequence length has a substantial impact on cluster and linker sizes, for intermediate hydrophobic contents such as those of all ORF categories, including CDSs, the sequence length has no or small effect (Supplemental Fig. S12). As a matter of fact, artificial IGORFs with CDS sizes and IGORF nucleotide compositions are characterized by shorter linkers than those of real and scrambled CDSs (Mann–Whitney U test, P = 7.1 × 10−8 and 2 × 10−4, respectively) (Supplemental Fig. S13). All these results reveal that the size of linkers results from a subtle combination of sequence length, GC content, and, finally, the resulting amino acid composition (Supplemental Figs. S12–S14).
Figure 6.

Continuum of sequence and structural properties between the different ORF categories. Comparison of the sequence size (A), cluster number (B), cluster sizes (C), and linker sizes (D) for each ORF category (IGORFs in purple, occasionally translated IGORFs in yellow, highly translated IGORFs in red, ancIGORFs in gray, de novo genes in blue, and CDSs in orange). The P-values were computed with the Mann–Whitney U test (one-sided for A,B,D and two-sided for C). Asterisks denote level of significance: (*) P < 5 × 10−2, (**) P < 1 × 10−2, (***) P < 1 × 10−3. For each plot, the color of the asterisks indicates the ORF category used for the comparison. The exact P-values are given in Supplemental Tables S1–S4.
Discussion
In this work, we showed that the noncoding genome encodes the raw material for making proteins. In particular, we showed the widespread existence in the noncoding genome of the elementary building blocks of protein structures. Hydrophobic clusters in noncoding sequences display sizes similar to those observed in CDSs. In contrast, CDSs are enriched in longer linkers that probably contribute to optimize the local arrangements of secondary structures and provide flexibility to proteins and specificity in protein interactions. This observation is in line with several studies reporting a central role to loops in protein function and structural innovation (Blouin et al. 2004; Tendulkar et al. 2004; Espadaler et al. 2006; Papaleo et al. 2016). Like Schmitz et al. (2018), we stipulate that the increase in intrinsic structural disorder observed for old genes in work by Carvunis et al. (2012) is related to the fact that CDSs are characterized by longer linkers, thereby inducing an increase in the disorder score. As a matter of fact, most CDSs display HCA scores similar to those of globular proteins, with low disorder propensities (Fig. 2). Overall, we showed an enrichment in polar and charged residues for CDSs, which may be accompanied by an increase in specificity of protein folds and interactions through the optimization of the folding and assembly processes (Lumb and Kim 1995). De novo genes display a GC content similar to the one of CDSs, whereas their amino acid composition is rather IGORF-like. The effect is even stronger for ancIGORFs, which are characterized by the highest GC content of all ORF categories while displaying an IGORF-like amino acid composition. This suggests an important role for the GC content in de novo gene emergence, as reported by Vakirlis et al. (2018). We can hypothesize that the amino acid composition is optimized afterward while maintaining the GC content through the structure of the genetic code.
Nevertheless, how a noncoding sequence becomes coding remains unclear. In this work, we propose the IGORFs as potential elementary modules of protein birth and evolution. IGORFs could serve as starting points for de novo gene emergence or could be combined together, thus increasing protein sizes, contributing to protein modularity, and leading to more complex protein architectures. They resonate with the short protein fragments, reported so far, that result from different protein structure decompositions with the aim of partitioning protein structures into universal basic units of folding, folds, and/or function (Berezovsky et al. 2000, 2001; Lamarine et al. 2001; Papandreou et al. 2004; Alva et al. 2015; Postic et al. 2017; Nepomnyachiy et al. 2017). The sizes of these structural fragments, overall, range from 25 to 35 residues with the exception of the “themes” (average of 49 residues) (Kolodny et al. 2021) and precisely recall those of IGORFs. Additionally, we showed that IGORFs encompass all the protein fold potential diversity observed in CDSs. A majority of IGORFs encode peptides predicted as foldable, whereas an important fraction displays high HCA scores and aggregation propensities. Some of the latter, although not the majority (28%), are predicted with at least one TM domain and may “safely” locate in membranes as proposed by Vakirlis et al. (2020a). The impact of the other high HCA score IGORFs on the cell deserves further investigation. Nevertheless, we can hypothesize that if produced, most of the time their concentration will not be sufficient to be deleterious (Langenberg et al. 2020). Indeed, it seems that for CDSs, a certain degree of aggregation is tolerated at low concentration (Supplemental Fig. S15). On the other hand, although IGORFs with intermediate HCA scores may show a certain propensity for aggregation, we can hypothesize that these aggregation-prone regions, under the hydrophobic effect, may play a role in their capacity to fold, in line with the hypothesis of an amyloid origin of the globular proteins (Greenwald and Riek 2012; Langenberg et al. 2020). We hypothesize that the balanced equilibrium of hydrophobic and hydrophilic residues observed for these IGORFs (39.1% of hydrophobic residues to be compared with the 50.8% observed for high HCA score IGORFs) may render possible the burying of aggregation-prone regions and the exposure of hydrophilic residues that is accompanied by an increase in foldability. We can hypothesize that, if produced, these IGORFs could form small compact structures and/or could be stabilized through oligomerization or interactions with other proteins. Precisely, we showed that ancIGORFs predating de novo gene emergence are not IGORF-like but are rather enriched in sequences with a high propensity for foldability. Nevertheless, we can reasonably hypothesize that de novo peptides struggle to fold into a well-defined and specific 3D structure as shown with the young de novo genes BSC4 and goddard identified in the S. cerevisiae and Drosophila melanogaster lineages, respectively (Namy et al. 2003; Bungard et al. 2017; Lange et al. 2021). In particular, Bungard et al. (2017) reported that the Bsc4 protein folds partially to an ordered structure that is unlikely to be unfolded according to circular dichroism spectra and bioinformatic analyses. However, despite this “rudimentary” fold, they show through mass spectrometry and denaturation experiments that Bsc4 is able to form compact oligomers. Its hydrophobic residue content (38%) is higher than the one of CDSs (33%) and is typical of foldable IGORFs (39%). Whether this may be related to its “rudimentary” fold is questionable. We can hypothesize that the specificity of the Bsc4 structure will increase during evolution through amino acid substitutions toward hydrophilic residues.
Altogether, these results enable us to propose a model (Fig. 7) that gives a central role to IGORFs in de novo gene emergence and, to a lesser extent, in protein evolution, thus completing the large palette of protein evolution mechanisms such as duplication events, horizontal gene transfer, domain shuffling, etc. This model unifies two evolutionary processes that are usually addressed separately: the origin of novel genes and the elongation and thus evolution of pre-existing proteins, through IGORFs as elementary molecular modules widespread in noncoding regions. Once an IGORF is selected (Fig. 7A), it can be subjected to different mutational events, such as nucleotide substitutions or indels. In our model, multiple rounds of nucleotide substitutions are expected to change the amino acid landscape of the selected IGORF as shown with the enrichment of CDSs in hydrophilic residues. We can hypothesize that mutations of hydrophobic residues toward hydrophilic ones can disrupt weak clusters into linkers or can switch cluster extremities into linker extremities, thereby increasing the size of linkers (Fig. 7B). Besides, we hypothesize that the selected IGORF can elongate through indels and/or STOP codon mutations, thus incorporating a neighboring IGORF (Fig. 7C). We hypothesize that the combination of two neighboring IGORFs through indels or STOP codon mutations can lead to the creation of long linkers at the IGORFs’ junction as observed in the example of the YMR153C-A de novo gene (Supplemental Fig. S16A). Similarly, the fusion of ancIGORFs can also give rise to long clusters as observed with the YPR126C de novo gene (Supplemental Fig. S16B), although it seems that long clusters have not been retained by selection as suggested by the CDS cluster size, which is similar to the one of IGORFs. We showed, with the reconstruction of 70 yeast de novo genes and in line with work of Zhang et al. (2019), that STOP codon mutations are less frequent than indels. Bartonek et al. (2020) reported that the hydrophobicity profiles of protein sequences remain invariant after frameshift events due to the interdependence of the three reading frames. Consequently, indels or frameshift events are most of the time expected to incorporate an IGORF that encodes a peptide with a hydrophobicity profile similar to that of the pre-existing gene and may explain the fact that they are more frequent than STOP codon mutations. This suggests that the fold potential is a critical feature that needs to be conserved even in noncoding sequences, being preserved in +1, −1 phases through the structure of the genetic code. In addition, we showed that combining IGORFs encoding foldable peptides with IGORFs encoding disorder or aggregation-prone ones has a low impact on the foldability of the resulting de novo proteins of the study. We can hypothesize that the newly integrated IGORFs will benefit from the structural properties of the pre-existing IGORF network. Proteins can be seen as assemblies on an ancient protein core, whatever its evolutionary history, of either duplicated, shuffled domains, or de novo translated products encoded by neighboring IGORFs (Fig. 7D). Overall, in line with recent evolutionary fragment-based protein design developments, this model offers a rational framework for designing novel chimeric proteins by combining small elementary modules with specific structural properties (Höcker 2014; Berezovsky 2019; Bornberg-Bauer et al. 2021; Ferruz et al. 2021; Yin et al. 2021).
Figure 7.

Model of de novo gene emergence and protein evolution with IGORFs as elementary structural modules. (A) IGORFs encode a wide diversity of peptides from disorder-prone to aggregation-prone ones, among which, a vast amount is expected to be able to fold in solution. Upon pervasive translation, some peptides that can be deleterious or not will be degraded right away. Among the others, the blue one will confer an advantage to the organism and will be further selected, thus providing a starting point for de novo gene birth. (B) The starting point IGORF, once selected, is subjected to amino acid substitutions, thereby increasing the overall proportion of hydrophilic residues of the encoded peptide. In the present case, this induces (1) the disruption of the second cluster, resulting in the increase of the size of the central linker, and (2) the establishment of specific interactions between hydrophilic residue (red dots), which increase the specificity of the folding process and the resulting fold. (C) The STOP codon of the starting point IGORF can be mutated into an amino acid, thereby adding the yellow IGORF to the pre-existing selected IGORF and elongating its size. (D) After multiple events of amino acid substitutions and IGORF combinations through STOP codon mutations or indels, we obtain a protein that displays the canonical features of CDSs (i.e., long sequences, long linkers, enrichment in polar and charged residues), which enable the optimization of its flexibility and the increase in specificity of its folding process, 3D fold, and interactions and finally participate along with domain shuffling or duplication events in the modular architecture of genuine proteins. We note that although the figure focuses on de novo gene emergence, this model can also apply to already existing proteins.
Our model is supported by previous observations which show that (1) de novo genes are shorter than old ones (Wolf et al. 2009; Tautz and Domazet-Lošo 2011), (2) the size of de novo gene exons is similar to that of old genes (Palmieri et al. 2014; Schlötterer 2015; Neme et al. 2017), and (3) novel domains are generally observed in the C-terminal regions (Bornberg-Bauer et al. 2015; Klasberg et al. 2018). Nevertheless, a lot of questions regarding the mechanisms predating the selection of an IGORF remain open. Figure 6 displays a continuum in the presented properties between IGORFs and CDSs that recalls the proto-gene model proposed by Carvunis et al. (2012), although the continuity between the translated IGORFs and the ancestral ones is to be shown. Whether the high translation signal of highly translated IGORFs derives directly from the acquisition of a methionine or whether it derives from previously occasionally translated IGORFs that have optimized their translational activity remains unclear. Similarly, the fate of highly translated IGORFs and their relationship with ancIGORFs are to be further characterized. Indeed, among the population of highly translated ORFs, some of them may give rise to future novel genes, thereby constituting, today, the ancIGORFs of tomorrow, whereas others may be short-lived in evolutionary history. Finally, the increase in sequence and linker sizes observed between the different ORF categories opens several questions. We showed that the increase in linker size for ancIGORFs can be explained by their GC content and, finally, their amino acid composition. Precisely, ancIGORFs display a higher GC content than IGORFs (41.9% and 36.1%, respectively), suggesting a role for GC-rich genomic regions in de novo gene properties and emergence as reported in previous studies (Basile et al. 2017; Vakirlis et al. 2018). Whether this increase in GC content is accompanied by an increase in sequence length (STOP codons are AT-rich), linker size, and, finally, foldability is a very interesting question that deserves further study. Indeed, it is still unknown whether the linker size is simply the consequence of the enrichment of CDSs in hydrophilic residues and the increase in protein size or whether harboring long linkers is accompanied by an increase in foldability and is thus a selected criterion. Finally, all these results highlight an intimate relationship between sequence length, GC content, and amino acid composition, whose combination is directly related to the size of linkers and clusters and, finally, to the foldability of the resulting product. Which one or which combination has driven the evolution of CDSs? Our results cannot enable us to conclude. Nevertheless, the function of a protein derives directly from its structure and interactions and can be, more generally, related to the concepts of stability, specificity, and diversity. These concepts are in turn related to the equilibrium between hydrophobic and hydrophilic residues, protein modularity, and, finally, protein size, which may altogether shape the linker and cluster size of proteins.
In this work, we propose a model that covers the genesis of all the diversity of the structural states observed in current proteins. If IGORFs encoding foldable peptides seem to be more likely to give rise to novel genes, disordered or aggregation-prone de novo proteins may emerge occasionally (Fig. 4B). They are most of the time (79%) associated with ancIGORFs expected to encode disordered or aggregation-prone peptides as well, suggesting that the structural properties of de novo proteins are already encoded in the ancestral peptide they originate from. Whether the fold potential of a starting point IGORF conditions the structural properties of the resulting de novo protein is an exciting question that deserves further study. Indeed, we can hypothesize that once selected, an IGORF can elongate over time through the incorporation of neighboring IGORFs, provided that the latter do not affect the fold potential of the pre-existing protein. In accordance with work of Vakirlis et al. (2020a), we can reason that once a starting point IGORF is selected, it engenders novel selected effects, which, in turn, increase the constraints exerted on it and subsequently reduce the possibility of future changes. It is thus tempting to speculate that the structural properties of the peptide encoded by the starting point IGORF will be retained during evolution through the elimination of the deleterious IGORFs’ combinations. All these observations suggest that the diversity of the structural states observed in current proteins has been originally inherited from the diversity of the fold potential already encoded in the noncoding genome. If and how the noncoding genome can account for the structural diversity of proteins are other exciting questions that deserve further study.
Methods
Data sets
CDSs and IGORFs
The CDSs were extracted from the genome of S. cerevisiae S288C according to the genome annotation of the Saccharomyces Genome Database (Cherry et al. 2012). All unannotated ORFs of at least 60 nt, no matter if they start with an AUG codon, were extracted from the 16 yeast chromosomes. We only retained ORFs that are free from overlap with another gene or that partially overlap with a gene if the nonoverlapping region is >70% of the IGORFs sequence.
Data sets of reference
The disorder data set consists of 731 disordered regions extracted from intrinsically disordered proteins of the DisProt database (Hatos et al. 2020), that were used for the calibration of HCAtk (Bitard-Feildel and Callebaut 2018). The globular data set consists of 559 globular proteins extracted from the Protein Data Bank (Berman et al. 2000; Burley et al. 2021) that were used for the calibration of IUPred (Dosztányi et al. 2005; Mészáros et al. 2009; Dosztányi 2018; Mészáros et al. 2018). The TM regions data set gathers 1269 TM regions extracted from the transmembrane proteins contained in the Protein Data Bank of Transmembrane Proteins (PDBTM) (Tusnády et al. 2004, 2005; Kozma et al. 2012). We only retained TM segments longer than 20 amino acids corresponding to the minimum size of an IGORF.
Random noncoding genome
Intergenic regions were concatenated, and their nucleotides were scrambled. Then random IGORFs of at least 60 nt were extracted as explained above.
Scrambled sequences
Scrambled sequences were generated by shuffling the nucleotides of the ORFs of interest. When an in-frame STOP codon was generated, its 3 nt were randomized until they did not lead to a STOP codon.
Artificial IGORFs
We generated artificial sequences of fixed size (e.g., size of CDS) by drawing nucleotides according to the nucleotide composition of IGORFs.
Estimation of the fold potential, the aggregation, disorder, and TM propensities
The foldability potential was estimated using a score derived from the HCA approach using the HCAtk program (Bitard-Feildel and Callebaut 2018; Bitard-Feildel et al. 2018), whereas the disorder and aggregation propensities were assessed with IUPred and TANGO, respectively (Supplemental Methods; Fernandez-Escamilla et al. 2004; Linding et al. 2004; Dosztányi et al. 2005; Rousseau et al. 2006a ; Mészáros et al. 2009; Dosztányi 2018; Mészáros et al. 2018). The presence of TM domains was predicted with TMHMM (Krogh et al. 2001).
Protein abundances and amino acid propensities
Protein abundance data were extracted from the PaxDb database (Wang et al. 2012). To depict the impact of the avoidance of nonspecific interactions with the ribosome, we only retained cytoplasmic proteins as annotated in UniProt (The UniProt Consortium 2019). The propensity of an amino acid i to be found in a CDS cluster is defined by the log ratio of the frequencies of the amino acid i in CDS clusters versus IGORF clusters as follows:
Reconstruction of ancIGORFs
To reconstruct the ancIGORFs of S. cerevisiae, we used the genomes of the neighboring species Saccharomyces paradoxus (Durand et al. 2019), Saccharomyces arboricola (Yue et al. 2017), Saccharomyces mikatae, Saccharomyces kudriavzevii, and Saccharomyces uvarum (Scannell et al. 2011). Based on four independent studies that each listed de novo genes of the S. cerevisiae genome, we retained all de novo genes identified in at least two studies (Carvunis et al. 2012; Lu et al. 2017; Vakirlis et al. 2018; Wu and Knudson 2018). This led to a total of 171 de novo genes, among which we retained those for which we were able to identify at least two additional homologous sequences in the neighboring species, among which at least one had to be noncoding in order to reconstruct the corresponding nongenic region in the ancestor (Supplemental Table S6). Therefore, we searched for the orthologous genes of the 70 de novo genes in the neighboring species using BLASTP (e-value < 1 × 10−2) (Supplemental Fig. S7A). Then, based on the species tree and starting from the branch of S. cerevisiae, we traced back to the root and identified the first node branching with a branch for which no orthologous gene had been detected (Supplemental Fig. S7A, yellow circle). We hypothesize that the corresponding locus in the ancestor was still nongenic. We searched for the corresponding nongenic regions in the remaining species with TBLASTN (evalue < 1 × 10−2). Following the protocol described by Vakirlis and McLysaght (2019), the resulting homologous nucleotide sequences and orthologous de novo genes were subsequently aligned with MACSE v2.05 (Ranwez et al. 2011, 2018), and the corresponding phylogenetic tree was constructed with PhyML (Guindon et al. 2010). The multiple sequence alignment and its corresponding tree were given as inputs to PRANK (Löytynoja and Goldman 2010) for the reconstruction of the corresponding ancestral nongenic nucleotide sequence (Supplemental Fig. S7B,C). Finally, the ancestral nucleotide sequences were translated into the three reading frames. The resulting IGORFs were then aligned with the de novo gene of S. cerevisiae with LALIGN (Huang and Miller 1991); those sharing a homology with it were retained (Supplemental Fig. S7D).
Ribosome profiling analyses
Ribosome profiling data sets
We used five ribosome profiling data sets of wild-type S. cerevisiae, two of which were generated in the present study (NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] accession number GSE173861, samples GSM5282046 and GSM5282047) (Supplemental Methods). The three others were taken from Radhakrishnan et al. (GEO accession number GSE81269, samples GSM2147982 and GSM2147983) (Radhakrishnan et al. 2016) and Thiaville et al. (GEO accession number GSE72030, sample GSM1850252) (Thiaville et al. 2016).
Selection of ribosome protected fragments (RPFs)
Ribosome profiling reads were mapped on the genome of S. cerevisiae S288C using Bowtie (Langmead et al. 2009). For this study, we only kept the 28-mers because, on average, 90% of them were mapped on a CDS in the correct reading frame (Supplemental Fig. S17).
Periodicity
The periodicity is calculated using a metagene profile. It provides the number of footprints relative to all annotated start codons in a selected window. The metagene profile is obtained by pooling together all the annotated CDSs and counting the number of RPFs at each nucleotide position. Supplemental Figure S17 shows a clear accumulation of signal over the CDSs, and a nice periodicity over the 100 first nucleotides.
Identification of the occasionally translated IGORFs
We retained the IGORFs with at least 10 reads in at least one data set.
Identification of the highly translated IGORFs
We kept the IGORFs with at least 30 reads in at least two data sets, for which the fraction of in-frame reads was higher than 0.8.
Statistical analyses
All statistical analyses that aimed at comparing distributions were performed in R (4.0.3) (R Core Team 2020) using the Kolmogorov–Smirnov test (two-sided) when comparing whether the HCA score distributions are statistically different and using the Mann–Whitney U test for the comparison of the median cluster size, linker size, sequence size, and cluster number distributions (bilateral test for the comparison of cluster sizes and unilateral test for the other properties). We used the one-proportion z-test for the comparison of the proportion of disordered, foldable, or aggregation-prone sequences between different ORF categories. To circumvent the P-value problem inherent to large samples (Lin et al. 2013), tests were performed iteratively 1000 times on samples of 500 individuals randomly chosen from the initial sample when it was larger than 500 individuals. The averaged P-value over the 1000 iterations was subsequently calculated.
Data access
The raw ribosome profiling data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE173861. Raw and calculated data along with codes to reproduce analyses and figures are available as Supplemental Code 1, and the programs to extract the IGORFs and estimate their structural properties (ORFtrack and ORFold) are available in the ORFMine package as Supplemental Code 2 and on GitHub (https://github.com/i2bc/ORFmine).
Supplementary Material
Acknowledgments
Work by C.P. was supported by a French government fellowship.
Author contributions: C.P., M.R., and I.H. performed research. C.P., M.R., I.H., O.N., and A.L. analyzed data. C.P. and A.L. designed research. C.P., I.C., J.-C.G., O.N., O.L., and A.L. wrote the paper. A.L. conceived the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275638.121.
Competing interest statement
The authors declare no competing interests.
References
- Alva V, Söding J, Lupas AN. 2015. A vocabulary of ancient peptides at the origin of folded proteins. eLife 4: e09410. 10.7554/eLife.09410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartonek L, Braun D, Zagrovic B. 2020. Frameshifting preserves key physicochemical properties of proteins. Proc Natl Acad Sci 117: 5907–5912. 10.1073/pnas.1911203117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile W, Sachenkova O, Light S, Elofsson A. 2017. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput Biol 13: e1005375. 10.1371/journal.pcbi.1005375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinky F, Babenko VN, Rogozin IB, Koonin EV. 2018. Purifying and positive selection in the evolution of stop codons. Sci Rep 8: 9260. 10.1038/s41598-018-27570-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovsky IN. 2019. Towards descriptor of elementary functions for protein design. Curr Opin Struct Biol 58: 159–165. 10.1016/j.sbi.2019.06.010 [DOI] [PubMed] [Google Scholar]
- Berezovsky IN, Grosberg AY, Trifonov EN. 2000. Closed loops of nearly standard size: common basic element of protein structure. FEBS Lett 466: 283–286. 10.1016/S0014-5793(00)01091-7 [DOI] [PubMed] [Google Scholar]
- Berezovsky IN, Kirzhner VM, Kirzhner A, Trifonov EN. 2001. Protein folding: looping from hydrophobic nuclei. Proteins 45: 346–350. 10.1002/prot.1155 [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res 28: 235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitard-Feildel T, Callebaut I. 2017. Exploring the dark foldable proteome by considering hydrophobic amino acids topology. Sci Rep 7: 41425. 10.1038/srep41425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitard-Feildel T, Callebaut I. 2018. HCAtk and pyHCA: a toolkit and python API for the hydrophobic cluster analysis of protein sequences. bioRxiv 10.1101/249995 [DOI] [PMC free article] [PubMed]
- Bitard-Feildel T, Heberlein M, Bornberg-Bauer E, Callebaut I. 2015. Detection of orphan domains in Drosophila using “hydrophobic cluster analysis”. Biochimie 119: 244–253. 10.1016/j.biochi.2015.02.019 [DOI] [PubMed] [Google Scholar]
- Bitard-Feildel T, Lamiable A, Mornon J, Callebaut I. 2018. Order in disorder as observed by the “hydrophobic cluster analysis” of protein sequences. Proteomics 18: 1800054. 10.1002/pmic.201800054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins WR, Ruiz-Orera J, Messeguer X, Blasco-Moreno B, Villanueva-Cañas JL, Espinar L, Díez J, Carey LB, Albà MM. 2021. Uncovering de novo gene birth in yeast using deep transcriptomics. Nat Commun 12: 604. 10.1038/s41467-021-20911-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin C, Butt D, Roger AJ. 2004. Rapid evolution in conformational space: a study of loop regions in a ubiquitous GTP binding domain. Protein Sci 13: 608–616. 10.1110/ps.03299804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornberg-Bauer E, Albà MM. 2013. Dynamics and adaptive benefits of modular protein evolution. Curr Opin Struct Biol 23: 459–466. 10.1016/j.sbi.2013.02.012 [DOI] [PubMed] [Google Scholar]
- Bornberg-Bauer E, Schmitz J, Heberlein M. 2015. Emergence of de novo proteins from “dark genomic matter” by “grow slow and moult”. Biochem Soc Trans 43: 867–873. 10.1042/BST20150089 [DOI] [PubMed] [Google Scholar]
- Bornberg-Bauer E, Hlouchova K, Lange A. 2021. Structure and function of naturally evolved de novo proteins. Curr Opin Struct Biol 68: 175–183. 10.1016/j.sbi.2020.11.010 [DOI] [PubMed] [Google Scholar]
- Bresler SE, Talmud D. 1944. On the nature of globular proteins. CR Acad Sci USSR 43: 310–314. [Google Scholar]
- Brooks DJ, Fresco JR. 2003. Greater GNN pattern bias in sequence elements encoding conserved residues of ancient proteins may be an indicator of amino acid composition of early proteins. Gene 303: 177–185. 10.1016/S0378-1119(02)01176-9 [DOI] [PubMed] [Google Scholar]
- Bungard D, Copple JS, Yan J, Chhun JJ, Kumirov VK, Foy SG, Masel J, Wysocki VH, Cordes MH. 2017. Foldability of a natural de novo evolved protein. Structure 25: 1687–1696.e4. 10.1016/j.str.2017.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, Christie CH, Dalenberg K, Di Costanzo L, Duarte JM, et al. 2021. RCSB protein data bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 49: D437–D451. 10.1093/nar/gkaa1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvunis A-R, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, Charloteaux B, Hidalgo CA, Barbette J, Santhanam B, et al. 2012. Proto-genes and de novo gene birth. Nature 487: 370–374. 10.1038/nature11184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. 2012. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40: D700–D705. 10.1093/nar/gkr1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas MVR, Hardy M-P, Hollý J, Bonneil É, Durette C, Courcelles M, Lanoix J, Côté C, Staudt LM, Lemieux S, et al. 2021. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep 34: 108815. 10.1016/j.celrep.2021.108815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztányi Z. 2018. Prediction of protein disorder based on IUPred. Protein Sci 27: 331–340. 10.1002/pro.3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztányi Z, Csizmók V, Tompa P, Simon I. 2005. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol 347: 827–839. 10.1016/j.jmb.2005.01.071 [DOI] [PubMed] [Google Scholar]
- Durand É, Gagnon-Arsenault I, Hallin J, Hatin I, Dubé AK, Nielly-Thibault L, Namy O, Landry CR. 2019. Turnover of ribosome-associated transcripts from de novo ORFs produces gene-like characteristics available for de novo gene emergence in wild yeast populations. Genome Res 29: 932–943. 10.1101/gr.239822.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekman D, Elofsson A. 2010. Identifying and quantifying orphan protein sequences in fungi. J Mol Biol 396: 396–405. 10.1016/j.jmb.2009.11.053 [DOI] [PubMed] [Google Scholar]
- Espadaler J, Querol E, Aviles FX, Oliva B. 2006. Identification of function-associated loop motifs and application to protein function prediction. Bioinformatics 22: 2237–2243. 10.1093/bioinformatics/btl382 [DOI] [PubMed] [Google Scholar]
- Faure G, Callebaut I. 2013a. Comprehensive repertoire of foldable regions within whole genomes. PLoS Comput Biol 9: e1003280. 10.1371/journal.pcbi.1003280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G, Callebaut I. 2013b. Identification of hidden relationships from the coupling of hydrophobic cluster analysis and domain architecture information. Bioinformatics 29: 1726–1733. 10.1093/bioinformatics/btt271 [DOI] [PubMed] [Google Scholar]
- Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L. 2004. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22: 1302–1306. 10.1038/nbt1012 [DOI] [PubMed] [Google Scholar]
- Ferruz N, Noske J, Höcker B. 2021. Protlego: a Python package for the analysis and design of chimeric proteins. Bioinformatics 37: 3182–3189. 10.1093/bioinformatics/btab253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy SG, Wilson BA, Bertram J, Cordes MH, Masel J. 2019. A shift in aggregation avoidance strategy marks a long-term direction to protein evolution. Genetics 211: 1345–1355. 10.1534/genetics.118.301719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A, Siekierska A, Beerten J, Brams M, Van Durme J, De Baets G, Van der Kant R, Gallardo R, Ramakers M, Langenberg T, et al. 2016. Structural hot spots for the solubility of globular proteins. Nat Commun 7: 10816. 10.1038/ncomms10816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald J, Riek R. 2012. On the possible amyloid origin of protein folds. J Mol Biol 421: 417–426. 10.1016/j.jmb.2012.04.015 [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hatos A, Hajdu-Soltész B, Monzon AM, Palopoli N, Álvarez L, Aykac-Fas B, Bassot C, Benítez GI, Bevilacqua M, Chasapi A, et al. 2020. DisProt: intrinsic protein disorder annotation in 2020. Nucleic Acids Res 48: D269–D276. 10.1093/nar/gkz975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heames B, Schmitz J, Bornberg-Bauer E. 2020. A continuum of evolving de novo genes drives protein-coding novelty in drosophila. J Mol Evol 88: 382–398. 10.1007/s00239-020-09939-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höcker B. 2014. Design of proteins from smaller fragments: learning from evolution. Curr Opin Struct Biol 27: 56–62. 10.1016/j.sbi.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Huang X, Miller W. 1991. A time-efficient, linear-space local similarity algorithm. Adv Appl Math 12: 337–357. 10.1016/0196-8858(91)90017-D [DOI] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. 10.1016/j.cell.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H. 2010. Origins, evolution, and phenotypic impact of new genes. Genome Res 20: 1313–1326. 10.1101/gr.101386.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasberg S, Bitard-Feildel T, Callebaut I, Bornberg-Bauer E. 2018. Origins and structural properties of novel and de novo protein domains during insect evolution. FEBS J 285: 2605–2625. 10.1111/febs.14504 [DOI] [PubMed] [Google Scholar]
- Knowles DG, McLysaght A. 2009. Recent de novo origin of human protein-coding genes. Genome Res 19: 1752–1759. 10.1101/gr.095026.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodny R, Nepomnyachiy S, Tawfik DS, Ben-Tal N. 2021. Bridging themes: short protein segments found in different architectures. Mol Biol Evol 38: 2191–2208. 10.1093/molbev/msab017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz G, Holm M, Wiens T, Sanyal S. 2014. Comprehensive analysis of stop codon usage in bacteria and its correlation with release factor abundance. J Biol Chem 289: 30334–30342. 10.1074/jbc.M114.606632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma D, Simon I, Tusnády GE. 2012. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res 41: D524–D529. 10.1093/nar/gks1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305: 567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- Lamarine M, Mornon J-P, Berezovsky IN, Chomilier J. 2001. Distribution of tightened end fragments of globular proteins statistically matches that of topohydrophobic positions: towards an efficient punctuation of protein folding? Cell Mol Life Sci 58: 492–498. 10.1007/PL00000873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamiable A, Bitard-Feildel T, Rebehmed J, Quintus F, Schoentgen F, Mornon J-P, Callebaut I. 2019. A topology-based investigation of protein interaction sites using hydrophobic cluster analysis. Biochimie 167: 68–80. 10.1016/j.biochi.2019.09.009 [DOI] [PubMed] [Google Scholar]
- Lange A, Patel PH, Heames B, Damry AM, Saenger T, Jackson CJ, Findlay GD, Bornberg-Bauer E. 2021. Structural and functional characterization of a putative de novo gene in Drosophila. Nat Commun 12: 1667. 10.1038/s41467-021-21667-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberg T, Gallardo R, van der Kant R, Louros N, Michiels E, Duran-Romaña R, Houben B, Cassio R, Wilkinson H, Garcia T, et al. 2020. Thermodynamic and evolutionary coupling between the native and amyloid state of globular proteins. Cell Rep 31: 107512. 10.1016/j.celrep.2020.03.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ED, De S, Teichmann SA. 2012. Cellular crowding imposes global constraints on the chemistry and evolution of proteomes. Proc Natl Acad Sci 109: 20461–20466. 10.1073/pnas.1209312109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z-W, Chen X, Wu Q, Hagmann J, Han T-S, Zou Y-P, Ge S, Guo Y-L. 2016. On the origin of de novo genes in Arabidopsis thaliana populations. Genome Biol Evol 8: 2190–2202. 10.1093/gbe/evw164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Lucas HC Jr, Shmueli G. 2013. Too big to fail: large samples and the p-value problem. Inf Syst Res 24: 906–917. 10.1287/isre.2013.0480 [DOI] [Google Scholar]
- Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L. 2004. A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J Mol Biol 342: 345–353. 10.1016/j.jmb.2004.06.088 [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. 2010. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics 11: 579. 10.1186/1471-2105-11-579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T-C, Leu J-Y, Lin W-C. 2017. A comprehensive analysis of transcript-supported de novo genes in Saccharomyces sensu stricto yeasts. Mol Biol Evol 34: 2823–2838. 10.1093/molbev/msx210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb KJ, Kim PS. 1995. A buried polar interaction imparts structural uniqueness in a designed heterodimeric coiled coil. Biochemistry 34: 8642–8648. 10.1021/bi00027a013 [DOI] [PubMed] [Google Scholar]
- Macossay-Castillo M, Marvelli G, Guharoy M, Jain A, Kihara D, Tompa P, Wodak SJ. 2019. The balancing act of intrinsically disordered proteins: enabling functional diversity while minimizing promiscuity. J Mol Biol 431: 1650–1670. 10.1016/j.jmb.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B, Simon I, Dosztányi Z. 2009. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 5: e1000376. 10.1371/journal.pcbi.1000376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B, Erdős G, Dosztányi Z. 2018. IUPred2a: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46: W329–W337. 10.1093/nar/gky384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DN, McLysaght A. 2012. De novo origin of protein-coding genes in murine rodents. PLoS One 7: e48650. 10.1371/journal.pone.0048650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Duchateau-Nguyen G, Hatin I, Hermann-Le Denmat S, Termier M, Rousset J. 2003. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res 31: 2289–2296. 10.1093/nar/gkg330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neme R, Amador C, Yildirim B, McConnell E, Tautz D. 2017. Random sequences are an abundant source of bioactive RNAs or peptides. Nat Ecol Evol 1: 0127. 10.1038/s41559-017-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepomnyachiy S, Ben-Tal N, Kolodny R. 2017. Complex evolutionary footprints revealed in an analysis of reused protein segments of diverse lengths. Proc Natl Acad Sci 114: 11703–11708. 10.1073/pnas.1707642114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielly-Thibault L, Landry CR. 2019. Differences between the raw material and the products of de novo gene birth can result from mutational biases. Genetics 212: 1353–1366. 10.1534/genetics.119.302187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri N, Kosiol C, Schlötterer C. 2014. The life cycle of Drosophila orphan genes. eLife 3: e01311. 10.7554/eLife.01311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R. 2016. The role of protein loops and linkers in conformational dynamics and allostery. Chem Rev 116: 6391–6423. 10.1021/acs.chemrev.5b00623 [DOI] [PubMed] [Google Scholar]
- Papandreou N, Berezovsky IN, Lopes A, Eliopoulos E, Chomilier J. 2004. Universal positions in globular proteins: from observation to simulation. Eur J Biochem 271: 4762–4768. 10.1111/j.1432-1033.2004.04440.x [DOI] [PubMed] [Google Scholar]
- Postic G, Ghouzam Y, Chebrek R, Gelly J-C. 2017. An ambiguity principle for assigning protein structural domains. Sci Adv 3: e1600552. 10.1126/sciadv.1600552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povolotskaya IS, Kondrashov FA, Ledda A, Vlasov PK. 2012. Stop codons in bacteria are not selectively equivalent. Biol Direct 7: 30. 10.1186/1745-6150-7-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Chen Y-H, Martin S, Alhusaini N, Green R, Coller J. 2016. The DEAD-box protein Dhh1p couples mRNA decay and translation by monitoring codon optimality. Cell 167: 122–132.e9. 10.1016/j.cell.2016.08.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery EJ. 2011. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS One 6: e22594. 10.1371/journal.pone.0022594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez V, Douzery EJ, Cambon C, Chantret N, Delsuc F. 2018. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol Biol Evol 35: 2582–2584. 10.1093/molbev/msy159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Rousseau F, Schymkowitz J, Serrano L. 2006a. Protein aggregation and amyloidosis: confusion of the kinds? Curr Opin Struct Biol 16: 118–126. 10.1016/j.sbi.2006.01.011 [DOI] [PubMed] [Google Scholar]
- Rousseau F, Serrano L, Schymkowitz JW. 2006b. How evolutionary pressure against protein aggregation shaped chaperone specificity. J Mol Biol 355: 1037–1047. 10.1016/j.jmb.2005.11.035 [DOI] [PubMed] [Google Scholar]
- Scannell D, Zill O, Rokas A, Payen C, Dunham M, Eisen M, Rine J, Johnston M, Hittinger C. 2011. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3 (Bethesda) 1: 11–25. 10.1534/g3.111.000273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schavemaker PE, Śmigiel WM, Poolman B. 2017. Ribosome surface properties may impose limits on the nature of the cytoplasmic proteome. eLife 6: e30084. 10.7554/eLife.30084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C. 2015. Genes from scratch—the evolutionary fate of de novo genes. Trends Genet 31: 215–219. 10.1016/j.tig.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JF, Ullrich KK, Bornberg-Bauer E. 2018. Incipient de novo genes can evolve from frozen accidents that escaped rapid transcript turnover. Nat Ecol Evol 2: 1626–1632. 10.1038/s41559-018-0639-7 [DOI] [PubMed] [Google Scholar]
- Tautz D, Domazet-Lošo T. 2011. The evolutionary origin of orphan genes. Nat Rev Genet 12: 692–702. 10.1038/nrg3053 [DOI] [PubMed] [Google Scholar]
- Tendulkar AV, Joshi AA, Sohoni MA, Wangikar PP. 2004. Clustering of protein structural fragments reveals modular building block approach of nature. J Mol Biol 338: 611–629. 10.1016/j.jmb.2004.02.047 [DOI] [PubMed] [Google Scholar]
- Thiaville PC, Legendre R, Rojas-Benítez D, Baudin-Baillieu A, Hatin I, Chalancon G, Glavic A, Namy O, de Crécy-Lagard V. 2016. Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb Cell 3: 29–45. 10.15698/mic2016.01.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretyachenko V, Vymětal J, Bednárová L, Kopecký V, Hofbauerová K, Jindrová H, Hubálek M, Souček R, Konvalinka J, Vondrášek J, et al. 2017. Random protein sequences can form defined secondary structures and are well-tolerated in vivo. Sci Rep 7: 15449. 10.1038/s41598-017-15635-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonov E. 1987. Translation framing code and frame-monitoring mechanism as suggested by the analysis of mRNA and 16 S rRNA nucleotide sequences. J Mol Biol 194: 643–652. 10.1016/0022-2836(87)90241-5 [DOI] [PubMed] [Google Scholar]
- Tusnády GE, Dosztányi Z, Simon I. 2004. Transmembrane proteins in the Protein Data Bank: identification and classification. Bioinformatics 20: 2964–2972. 10.1093/bioinformatics/bth340 [DOI] [PubMed] [Google Scholar]
- Tusnády GE, Dosztányi Z, Simon I. 2005. PDB_TM: selection and membrane localization of transmembrane proteins in the Protein Data Bank. Nucleic Acids Res 33: D275–D278. 10.1093/nar/gki002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. 2019. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis N, McLysaght A. 2019. Computational prediction of de novo emerged protein-coding genes. In Computational methods in protein evolution (ed. Sikosek T), pp. 63–81. Springer, New York. [DOI] [PubMed] [Google Scholar]
- Vakirlis N, Hebert AS, Opulente DA, Achaz G, Hittinger CT, Fischer G, Coon JJ, Lafontaine I. 2018. A molecular portrait of de novo genes in yeasts. Mol Biol Evol 35: 631–645. 10.1093/molbev/msx315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis N, Acar O, Hsu B, Coelho NC, Van Oss SB, Wacholder A, Medetgul-Ernar K, Bowman RW, Hines CP, Iannotta J, et al. 2020a. De novo emergence of adaptive membrane proteins from thymine-rich genomic sequences. Nat Commun 11: 781. 10.1038/s41467-020-14500-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis N, Carvunis A-R, McLysaght A. 2020b. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. eLife 9: e53500. 10.7554/eLife.53500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oss SB, Carvunis A-R. 2019. De novo gene birth. PLoS Genet 15: e1008160. 10.1371/journal.pgen.1008160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C. 2012. PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics 11: 492–500. 10.1074/mcp.O111.014704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Foy SG, Neme R, Masel J. 2017. Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nat Ecol Evol 1: 0146. 10.1038/s41559-017-0146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissler L, Godmann L, Bornberg-Bauer E. 2012. Evolutionary dynamics of simple sequence repeats across long evolutionary time scale in genus Drosophila. Trends Evol Biol 4: e7. 10.4081/eb.2012.e7 [DOI] [Google Scholar]
- Wolf YI, Novichkov PS, Karev GP, Koonin EV, Lipman DJ. 2009. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc Natl Acad Sci 106: 7273–7280. 10.1073/pnas.0901808106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Knudson A. 2018. Tracing the de novo origin of protein-coding genes in yeast. mBio 9: e01024-18. 10.1128/mBio.01024-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D-D, Irwin DM, Zhang Y-P. 2011. De novo origin of human protein-coding genes. PLoS Genet 7: e1002379. 10.1371/journal.pgen.1002379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Goncearenco A, Berezovsky IN. 2021. Deriving and using descriptors of elementary functions in rational protein design. Front Bioinform 1: 8. 10.3389/fbinf.2021.657529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J-X, Li J, Aigrain L, Hallin J, Persson K, Oliver K, Bergström A, Coupland P, Warringer J, Lagomarsino MC, et al. 2017. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat Genet 49: 913–924. 10.1038/ng.3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ren Y, Yang T, Li G, Chen J, Gschwend AR, Yu Y, Hou G, Zi J, Zhou R, et al. 2019. Rapid evolution of protein diversity by de novo origination in Oryza. Nat Ecol Evol 3: 679–690. 10.1038/s41559-019-0822-5 [DOI] [PubMed] [Google Scholar]
- Zhao L, Saelao P, Jones CD, Begun DJ. 2014. Origin and spread of de novo genes in Drosophila melanogaster populations. Science 343: 769–772. 10.1126/science.1248286 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.