

Abstract
In breast cancer, genetic heterogeneity, the lack of actionable targets, and immune evasion all contribute to the limited clinical response rates to immune checkpoint blockade therapy. Here, we report a high-throughput screen based on the functional interaction of mouse or patient-derived breast tumour organoids and tumour-specific cytotoxic T cells for the identification of epigenetic inhibitors that promote antigen presentation and that potentiate T-cell-mediated cytotoxicity. We show that the epigenetic inhibitors GSK-LSD1, CUDC-101 and BML-210, identified by the screen, display antitumour activities in orthotopic mammary tumours in mice, that they upregulate antigen presentation mediated by the major histocompatibility complex class I on breast tumour cells, and that treatment with BML-210 substantially sensitized breast tumours to the checkpoint inhibitor programmed-death-1. Standardized measurements of tumour-cell killing activity facilitated by tumour-organoid–T cell screens may aid the identification of candidate immunotherapeutics for a range of cancers.
Breast cancer has long been considered a major clinical challenge, owing to genetic heterogeneity, the lack of sufficient targeted therapies, and the malignant nature of the disease. Over the past few years, efforts have been made to develop effective immune therapies to improve clinical outcomes in breast cancer, particularly for triple negative breast cancer (TNBC). Breast cancer, as a whole, was previously thought to be immunologically ‘cold’, as many tumours showed low lymphocyte infiltration, resulting in modest responses to immune checkpoint blockade therapy1. However, mounting evidence indicated that breast cancer is remarkably heterogeneous in terms of immune infiltration and the tumour microenvironment, and that lymphocyte infiltration into tumours is associated with good prognosis and with clinical responses to chemotherapy2–4. The first approved immunotherapy for breast cancer — the programmed-death-ligand-1 (PD-L1) inhibitor atezolizumab — is a first-line therapy for locally advanced or metastatic TNBCs that express PD-L1 in 1% or greater of tumour cells5. Promising progress has also been made in other immune checkpoint inhibitors, such as programmed death-1 (PD-1) antibodies, for treating breast cancer. Ongoing clinical trials are now focused on combinational strategies to improve the response rates of breast cancer to immune checkpoint blockade (ICB)-based therapy.
Immune profiling, integrated multi-omics analyses and tumour-microenvironment studies have revealed potential mechanisms of immune evasion in breast cancer, among which the suppression of immune recognition of tumour antigens is prevalent6, 7. In addition to loss-of-function mutations, transcriptional suppression of the genes associated with antigen processing and presentation, immune checkpoint genes, chemokines, and other immune-related genes inhibits the functional interaction of tumour cells with tumour-infiltrating lymphocytes, and thus preventing immune attack. Epigenetic drugs, such as DNA methyltransferase and histone deacetylase inhibitors, are postulated to reverse immune suppression by reprogramming the tumour cell epigenome8. Current progress on cancer cell epigenetics will advance our knowledge on immune resistance of human cancer and establish a solid foundation for preclinical development using combined epigenetic and immunotherapeutic agents as anticancer therapies9–12. Although breast cancer immunotherapy is a rapidly growing field of interest13, technical approaches and animal models for high-throughput drug discovery and validation lag behind its development. Tumour-organoid models for assessing tumour–immune recognition have a high potential to be a powerful tool for identifying drugs for immunotherapy or combination therapy. Although used for a number of cancer-drug screens, conventional 2D monolayer tumour cells and tumour spheroids14, 15 cannot well represent the tumour in vivo as the heterogeneity and diversity of cell types in the tumour niche pivotally influences therapeutic effects16, 17. Without inputs from the tumour microenvironment, it is extremely unlikely to assess potential drug effectivity in immunotherapy18. By contrast, tumour organoids preserve the complex diversity and physical architecture of the tumour tissue19, making them an ideal tool in immunotherapy drug screens. Patient-derived organoids such as colon20, breast3, prostate21, bladder22, lung23 and liver24 cancer organoids recapitulate features of their origin tissues, and model the functional interactions in the tumour microenvironment. In particular, tumour and immune-cell interactions in the tumour microenvironment are crucial for the preclinical evaluation of immunotherapy25. Co-culture of tumour organoids with autologous peripheral blood lymphocytes has been studied in breast cancer2, 26, non-small-cell lung cancer (NSCLC) and microsatellite-instable colorectal cancer27. However, a majority of CD8+ T cells in peripheral blood are non-tumour specific and thus show low antitumour killing effects in such a co-culture system28. A sufficient amount of tumour-specific T cells is essential in the tumour-organoid model for immunotherapy. Mouse OVA (chicken ovalbumin peptide, also designated as OVA257–264)-specific T cells29, human NY-ESO-1 specific CD8+ T cells30, and autologous tumour-infiltrating T cells can be used to facilitate tumour recognition and high cytotoxic activity in co-culture31. Additionally, to facilitate a high-throughput drug screen, tumour organoids used in the system need to be characterized in their structure and function. A number of factors, including organoid size, cell composition and hypoxia inside the organoid, should be taken into account.
In this study, we report a high-throughput immune-drug screening approach, in which OVA+ mouse breast tumour organoids with defined size and characterized structure were co-cultured with the CD8+ T cells that are isolated from OT-I transgenic mice29 and recognize the OVA antigen. Drug candidates that promoted antigen presentation of cancer cells and substantially enhanced the infiltration and cytotoxicity of OT-I cells were further verified in syngeneic mouse breast tumour models and in human patient-derived tumour organoids (PDOs) with autologous tumour-infiltrating T cells. Tumour organoids including PDOs generated in this study inherit molecular and cellular features of primary tumours and maintain the complex histological tumour architecture formed by epithelial, vascular endothelial and stroma cells. The cytotoxicity of autologous tumour-infiltrating CD8+ T cells provides reliable and representative evaluation for the drugs tested in the co-culture with PDOs. The standardized and unbiased protocol developed in this study can be used to generate various tumour organoid models for immune drug discovery and validation.
Results
Generation and characterization of breast tumour organoids
To facilitate our high-throughput drug screen for breast cancer immunotherapy, we established a drug screen system that is composed of OVA257–264 antigen-presented mouse breast cancer EO771 cell-derived tumour organoids and CD8+ T cells isolated from OT-I transgenic mice that recognize the OVA antigen in the context of H2Kb (Fig. 1). GFP+Luc+OVA+ EO771 cells were generated, in which green fluorescence protein (GFP) is used to label tumour cells and luciferase is expressed for measuring T cell-mediated cytotoxicity in the screen (Supplementary Fig. 1a,b). The engineered EO771 cells were orthotopically injected into the fat pad of 4th mammary gland in female C57BL/6 mice and the resulting tumours were harvested to generate tumour organoids as described in the Methods (Fig. 1). To optimize tumour organoids for a high-throughput drug screen, we studied the size, heterogeneity, hypoxia, composition and culture time of the tumour organoids. Organoid size and heterogeneity with increasing cell seeding concentrations were determined (Fig. 2a and Supplementary Fig. 1c,d). The organoid size variation was much greater as the cell seeding concentration increased, especially when higher than 2 × 105 cells/ml (2 ml per well) in the 6-well microplate. Organoid sizes were concentrated around 70 to 150 μm in diameter at the seeding concentration of 2 × 105 cells/ml after 7-day culture. We started to observe notably high cytotoxicity of CD8+ T cells from 12 h after co-culture with tumour organoids and increased up to 48 h. A moderate killing effect of T cells was desired for drug screen, as such the greater or weaker effect upon drug treatment could be detectable (Supplementary Fig. 1e,f). The proportion of dead CD8+ T cells in the tumour organoids (70–150 μm) was ~11% one day after co-culture, similar to what was observed in the 2D co-culture (Supplementary Fig. 1g,h). However, the T cell death increased considerably at two days after co-culture with the tumour organoids, suggesting that the co-culture time is optimal around one day for measuring T cell activity. Hypoxia in tumour organoids became more severe as the organoid size increased (Fig. 2b,c and Supplementary Fig. 2a-e). When the organoid size was between 100 and 150 μm in diameter, the oxygen levels at the core area of the organoids were ~ 1–5%, which is similar to the hypoxia state observed in solid tumours32, 33. Tumour cell viability in the organoids markedly declined with increasing organoid size, particularly when the organoids were larger than 150 μm in diameter (Supplementary Fig. 2f), indicating that hypoxia may cause non-specific tumour cell death if tumour organoids grow too large. Tumour organoids with modest cell death, physiologically relevant hypoxia, and relatively uniform size are optimal for a standardized high-throughput drug screen. With these measurements, we selected the tumour organoids with size of lower than 150 μm in diameter.
Fig. 1 |. A tumour-organoid-based approach for screening immunotherapy drugs.
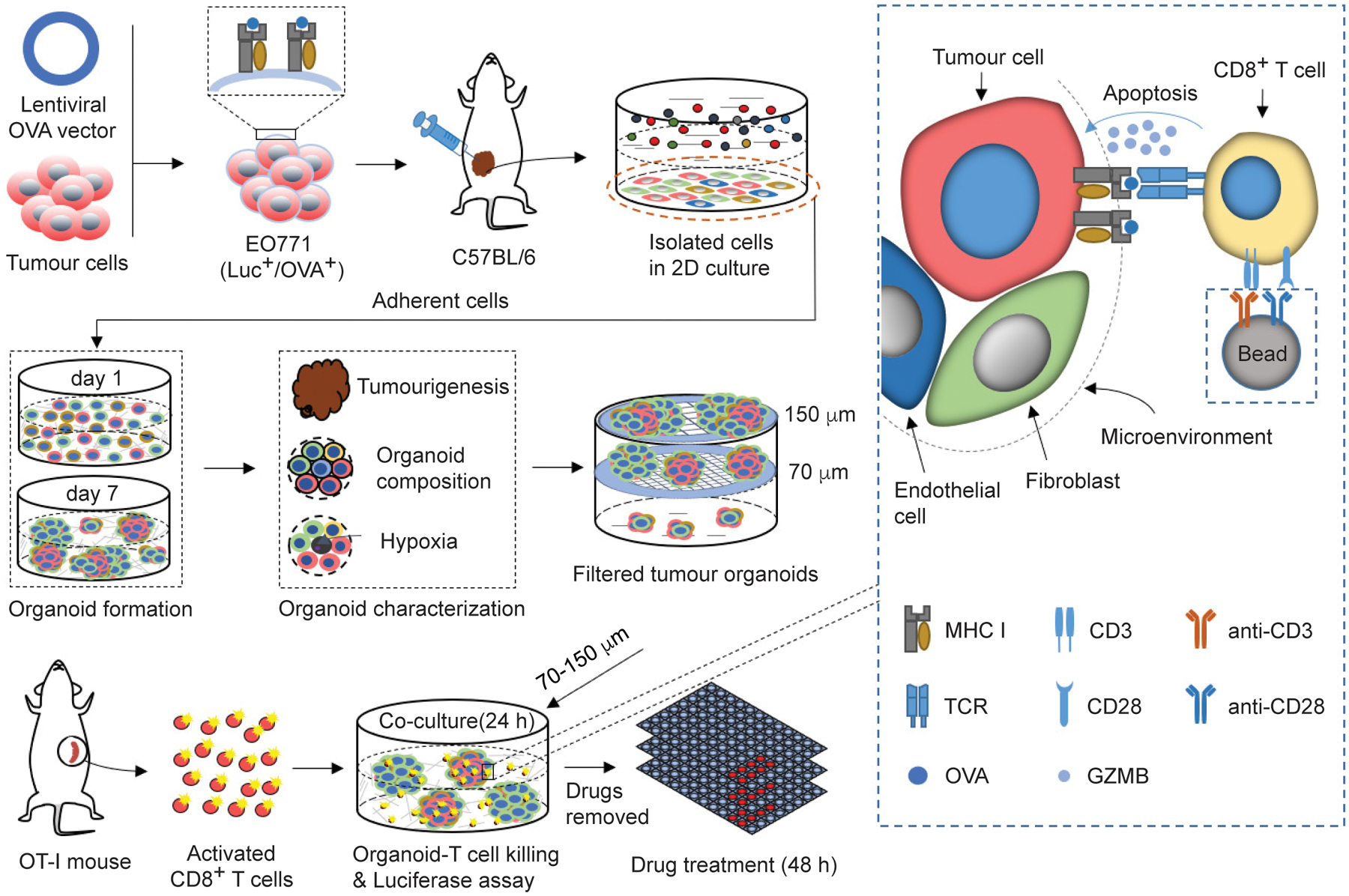
The drug screen approach is composed of breast tumour organoids and tumour-specific CD8+ T cells. A lentiviral vector expressing OVA was transduced to the Luc+ EO771 cells. The resulting OVA+ cells were orthotopically transplanted to C57BL/6 mice to generate syngeneic mammary tumours. The tumours were harvested and dissociated to single cells for 2D culture and only the adherent cells were collected to generate tumour organoids. 2 ml of tumour organoid culture medium (2 × 105 cells/ml) was seeded into 6-well culture plate with ultra-low attachment surface. After 7-day culture, tumour organoids were filtered by cell strainers with nylon mesh between 70 and 150 μm. The tumour organoids with diameter between 70 and 150 μm were treated with drugs for 48 h in the matrigel-free medium. The treated tumour organoids were then co-cultured with the pre-activated and OVA-specific CD8+ T cells from OT-I mice in the breast organoid culture medium containing 10 ng ml−1 IL2 without matrigel for 24 h. The luciferase released from the EO771 cells was measured using a Dual-luciferase report assay system (Promega) on a BioTek Cytation5 imaging reader, to assess the T cell-mediated cytotoxicity effect.
Fig. 2 |. Characterization and optimization of mouse breast tumour organoids.
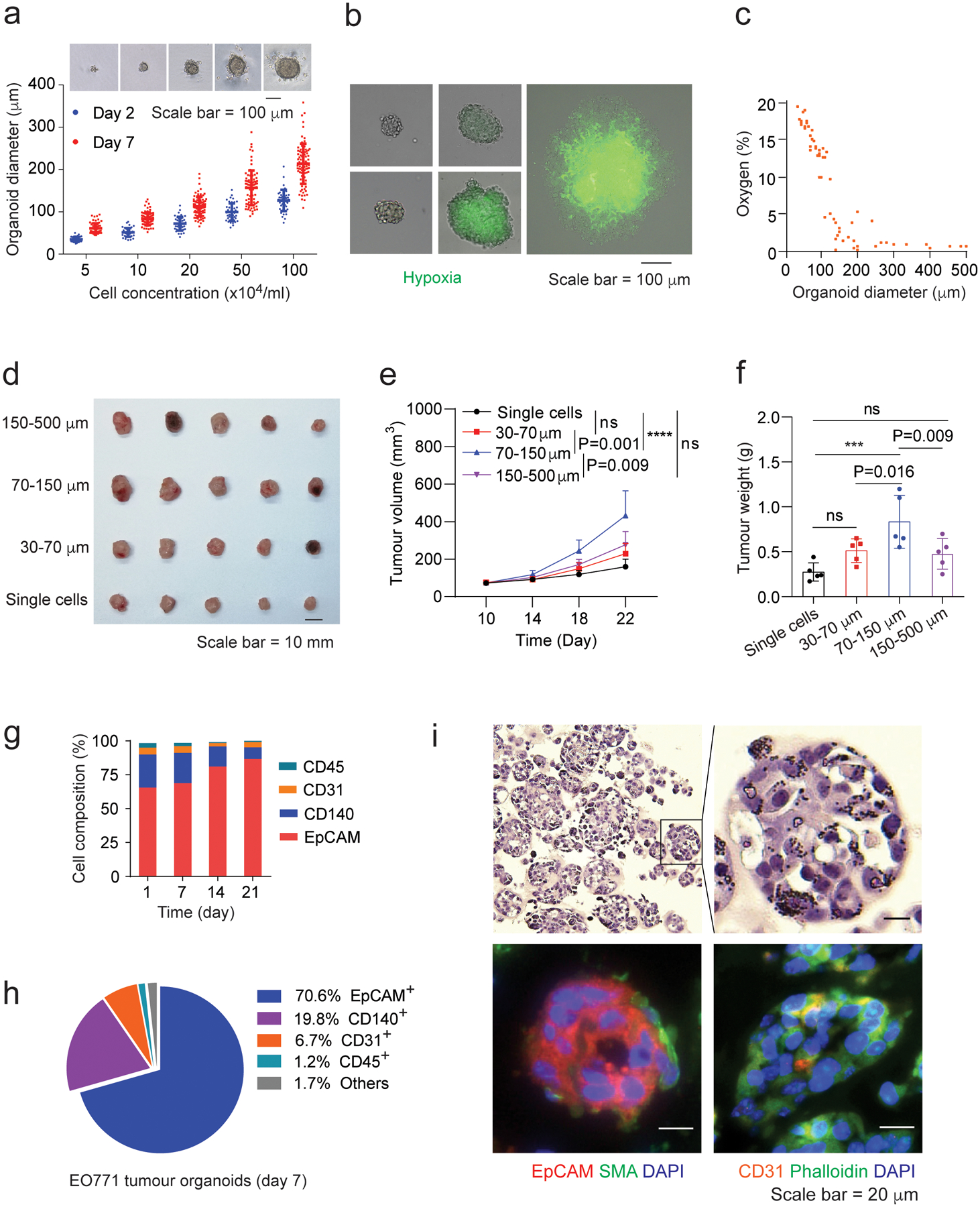
a,Organoid diameter versus cell seeding concentration at day 2 and day 7 of organoid culture. Optical images show tumour organoids at day 7. Optical images were analyzed using ImageJ to determine the organoid size. b, Hypoxia characterization of the EO771 tumour organoids under optical and fluorescence light. Image-iT Green Hypoxia reagent was used to show hypoxia in tumour organoids. The merged images with optical light and fluorescence were captured under a Leica DM4B microscope with two channels (optical light and 488 nm excitation). c, Oxygen levels in tumour organoids determined by hypoxia-caused fluorescence intensity from Supplementary Fig. 2b according to the protocol of the Image-iT Green Hypoxia reagent. d, Breast tumours derived from transplanted tumour organoids with indicated sizes. Tumour organoids containing 2 × 105 tumour cells were injected into each mouse and the resulting tumours were harvested 22 days post injection. e,f, Growth (e) and weight (f) of the tumour organoid-derived breast tumours. g, Cellular composition of the EO771 tumour organoids with indicated culture times. 20,000 events per cell sample were collected. The data were analyzed from 3 biologically parallel experiments. h, The typical cellular composition of the EO771 tumour organoids at day 7 of culture. i, H&E staining (upper panels) and immunofluorescent staining (bottom panels) of the EO771 tumour organoids. One-way ANOVA test was used for statistical analysis(e,f). Data are presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
Tumour cells and their functional interaction with associated stroma in the tumour microenvironment impacts disease initiation and progression34, 35. To further validate the optimal size of tumour organoids, we assessed the tumourigenic potential of tumour organoids with different sizes in mouse syngeneic tumour models, where the EO771-derived breast tumour organoids with same tumour cell number were transplanted into C57BL/6 mice by orthotopic injection. Tumour organoids with diameter around 70~150 μm exhibited the highest level of tumourigenesis in terms of tumour volume and weight (Fig. 2d–f), in comparison with the larger or smaller organoids. The cellular composition of tumour organoids at different time points revealed that tumour cells (positive for Epithelial Cell Adhesion Molecule, or EpCAM+) grew faster than other cell types such as tumour associated fibroblast (CD140+) and vascular endothelial cells (CD31+) (Fig. 2g). However, the cellular composition at day 7 was close to that of dissociated cells from the original tumour (day 1). In particular, the EO771 tumour organoids with the diameter of 70 to 150 μm after 7-day culture (Fig. 2h and Supplementary Fig. 2g) had 70.6% of EpCAM+ cells, 19.8% of fibroblast cells, and 6.7% endothelial cells, respectively. These cell types were also observed in immunofluorescence staining images of tumour organoids (Fig. 2i), representing the cellular composition determined by flow cytometry analysis. The results concluded that tumour organoids with size between 70~150 μm after 7-day culture well resemble their original tumours and retain strong tumourigenicity. Notably, CD45+ immune cells were excluded from the tumour organoids (Fig. 2h and Supplementary Fig. 2g). Without a variety of pre-existing immune cells in tumour organoids, we were able to measure the tumour-specific T cell-mediated cytotoxicity in the co-culture of tumour organoids with the CD8+ T cells.
Screen of epigenetic inhibitors that enhance T cell activity
Epigenetic modulation governs transcriptional program in tumour cells, which may impact their antigen presentation and response to the T cell-mediated effects. To screen small-molecule compounds that enhance the T cell-mediated cytotoxicity on tumour cells, a library of 141 compounds (Supplementary Table 1) that modulate the activity of methyltransferases, demethylases, histone acetyltransferases (HATs), histone deacetylases (HDACs), and acetylated lysine reader proteins was applied (at the concentration of 1μM) in the optimized 3D tumour organoid-T cell co-culture system. As comparison, we also used 2D tumour cell-T cell co-culture screen system in parallel. Tumour cell viability was measured as an indicator for the T cell-mediated cytotoxicity. Additionally, we also assessed the OVA antigen presentation of tumour cells in the drug-treated tumour organoids (Fig. 3a). OVA+Luc+ EO771-derived tumour organoids with diameter between 70~150 μm were generated and used in the screen (Fig. 3b). To exclude the compounds with cytotoxic activity similar to that of chemotherapeutic drugs, we first assessed their cytotoxicity on tumour organoids without T cells co-cultured. Only those compounds (121 in total) without significant cytotoxicity (P > 0.05) at the concentration of 1 μM (Fig. 3c) were selected for the immunological cytotoxicity screen in the next step, among which 8 compounds from the 3D co-culture system (Fig. 3d) and 15 compounds from the 2D co-culture system (Fig. 3e) exhibited significant activity (P < 0 .05) in enhancing the CD8+ T cell-mediated cytotoxicity. One of the primary mechanisms for immune evasion of tumour cells is reduced antigen presentation, which prevents tumour cells from the recognition and destruction of cytotoxic T cells. A total of 25 compounds markedly promoted the OVA antigen presentation on the OVA+ EO771 cells (P < 0.05, in Fig. 3f) in the 2D tumour cell culture. As expected, a reduced number of compounds were identified from the 3D tumour organoid screen than from the 2D tumour cell screen (Fig. 3g), and vice versa some compounds effective in the 3D system showed no activity in the 2D system, suggesting that tumour microenvironment and tumour stroma play an important role in modulating the immune response of tumour cells. T cell infiltration was evaluated in tumour organoids treated with the positive compounds screened from the 2D and organoid system (Extended Data Fig. 1a). The compounds screened from the organoid system induce relatively high CD8+ T cell infiltration in the tumour organoids. By contrast, only a few of the compounds from 2D system induced T cell infiltration (Extended Data Fig. 1b–d). Animal experiments confirmed that the compounds (PFI-1 and bromosporine) with poor T cell infiltration showed no anti-tumour effect (Extended Data Fig. 1e–g). The results suggested that the organoid screening system has advantage over the 2D culture approach in identifying drugs for immune drug discovery as the latter lacks the essential aspects such as T cell infiltration in tumour tissue. By combining the results from the 2D cell co-culture screen, tumour organoid co-culture screen and antigen presentation screen, three compounds (CUDC-101, GSK-LSD1 and BML-210) were identified as drug candidates that increase the antigenicity of tumour cells and enhance their response to cytotoxic T cells.
Fig. 3 |. Screening of epigenetic inhibitors that enhance T-cell-mediated tumour-cell killing.
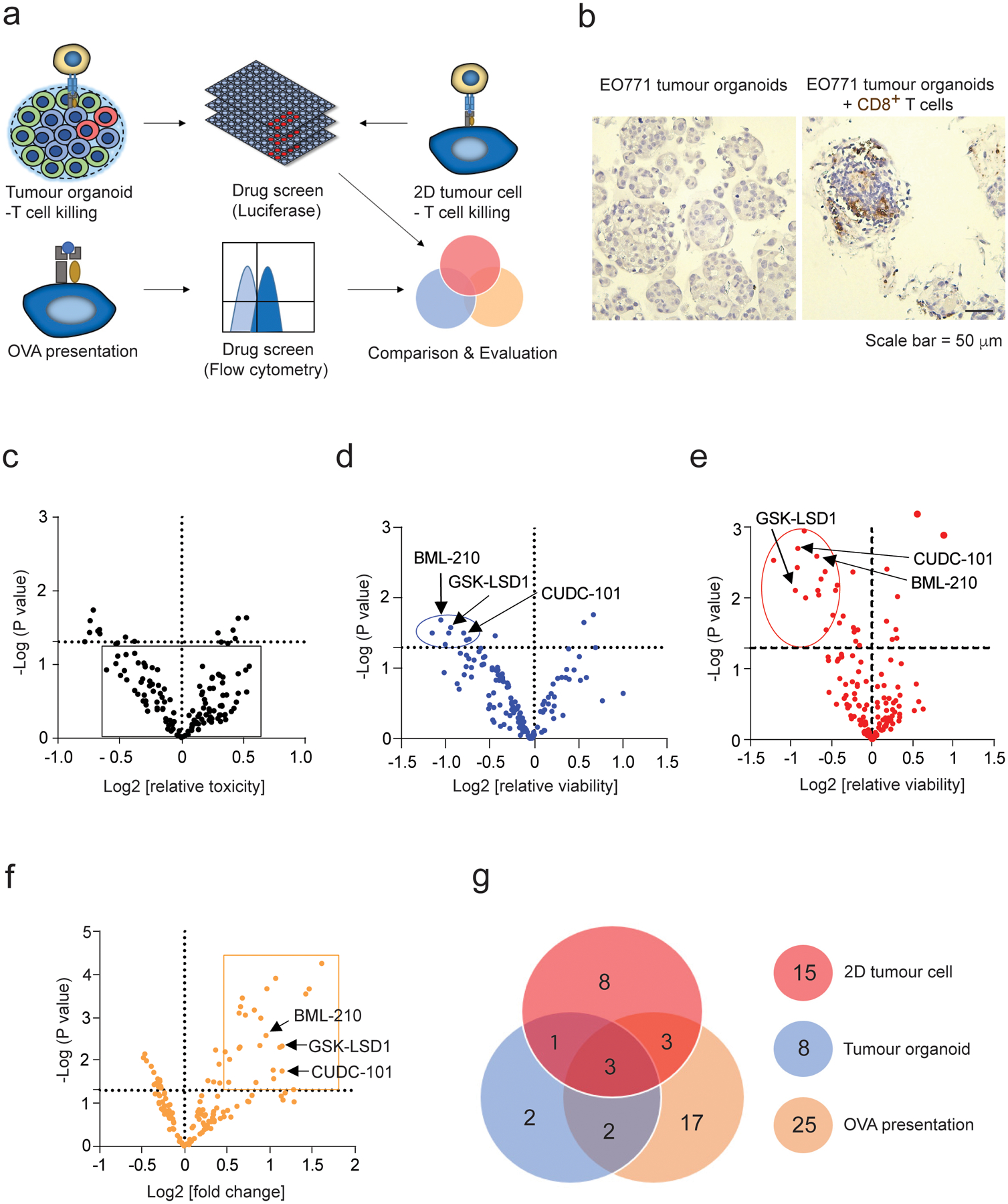
a, Schematic illustration of epigenetic drug screening based on 3D tumour organoid culture, 2D tumour cell culture, and antigen presentation on the 2D cultured tumour cells. b, Immunohistochemical (IHC) staining of CD8+ T cells in the EO771 organoids co-cultured with and without CD8+ T cells. CD8+ T cells are stained in brown colour. c, Volcano plot analysis of epigenetic inhibitors’ toxicity for the tumour cells in EO771 (Luc+OVA+) tumour organoids at the concentration of 1.0 μM. The compounds without substantial toxicity effect in the absence of CD8+ T cells were shown in the square. d, Volcano plot analysis showing the effect of epigenetic inhibitors on the CD8+ T cell-mediated cytotoxicity in the co-culture with the EO771 tumour organoids. The EO771 tumour organoids were treated with epigenetic inhibitors for 48 h and then co-cultured with CD8+ T cells for 24 h. Compounds shown in the circle significantly promoted T cell-mediated cytotoxicity (log2[relative viability] < −0.5; p < 0.05). Relative viability = (tumour cell viability of treated group)/(tumour cell viability of control group). e, Volcano plot analysis showing the effect of epigenetic inhibitors on the CD8+ T cell-mediated cytotoxicity for the EO771 (Luc+OVA+) cells in 2D culture. Compounds shown in the circle significantly promoted T cell cytotoxicity (log2[relative viability] < −0.5; p <0.05).The cytotoxicity in (d) and (e) was quantified using luciferase assay. f, Volcano plot analysis showing the effect of epigenetic inhibitors on OVA antigen presentation of the EO771 (Luc+OVA+) cells. Compounds shown in the rectangle significantly promoted OVA antigen expression (log2[OVA presentation/control] > 0.5; p < 0.05). 10,000 events per cell sample in flow cytometry were collected for data analysis. The mean fluorescence intensity (MFI) from the cells stained with APC-conjugated OVA peptide SIINFEKL antibody represents the OVA expression levels. g, Pie chart of positive compounds screened from the three screening methods. One-way ANOVA test was conducted in c-f. p value < 0.05 in the volcano plots was considered as significant difference. The data for the drug screen represent 3 biologically parallel experiments.
In the tumour organoid and T cell co-culture system, treatment with CUDC-101, GSK-LSD1 or BML-210 resulted in greater organoid dissociation (shown in smaller sizes) (Fig. 4a,b) and higher tumour cell death rates (Fig. 4c and Supplementary Fig. 3a,b), while the same treatments had no notable effect on the tumour organoids in the absence of T cells. Cytokine interferon gamma (IFNγ)36, cytolytic granule enzyme Granzyme B (GZMB)37, and tumour necrosis factor (TNFα)38 are the key indicators for cytotoxic T cell-mediated cytotoxicity. Their expression levels in the co-cultured CD8+ T cells were remarkably up-regulated when co-cultured with the drug-treated tumour organoids (Fig. 4d and Supplementary Fig. 3c). Enzyme-linked immunosorbent assay (ELISA) also showed that these CD8+ T cells secreted much higher levels of IFNγ and TNFα in comparison with those T cells co-cultured with control untreated tumour organoids(Fig. 4e). To validate the activity of these compounds in human breast tumour organoids formed by MDA-MB-468 cancer cells and human breast cancer-associated fibroblasts (CAFs), the human cancer antigen NY-ESO-1 was transduced into MDA-MB-468 cells, which can be recognized by mature human T cells specific for NY-ESO-1 bound to HLA-A2 (Extended Data Fig. 2a). Similar to the results from the mouse tumour organoid system, all the three compounds pronouncedly induced the T cell-mediated cytotoxicity (Fig. 4f, Extended Data Fig. 2b and Supplementary Fig. 3d,e). GZMB, IFNγ and TNFα expression levels in the CD8+ T cells from the treatment groups were also much higher than those in the CD8+ T cells from the control groups (Fig. 4g and Supplementary Fig. 3f). Collectively, we identified three epigenetic modulator compounds, CUDC-101, GSK-LSD1 and BML-210, that promote cytotoxic T cell-dependent anti-tumour response.
Fig. 4 |. Validation of antitumour activity of the three drug candidates in mouse and human tumour organoids.
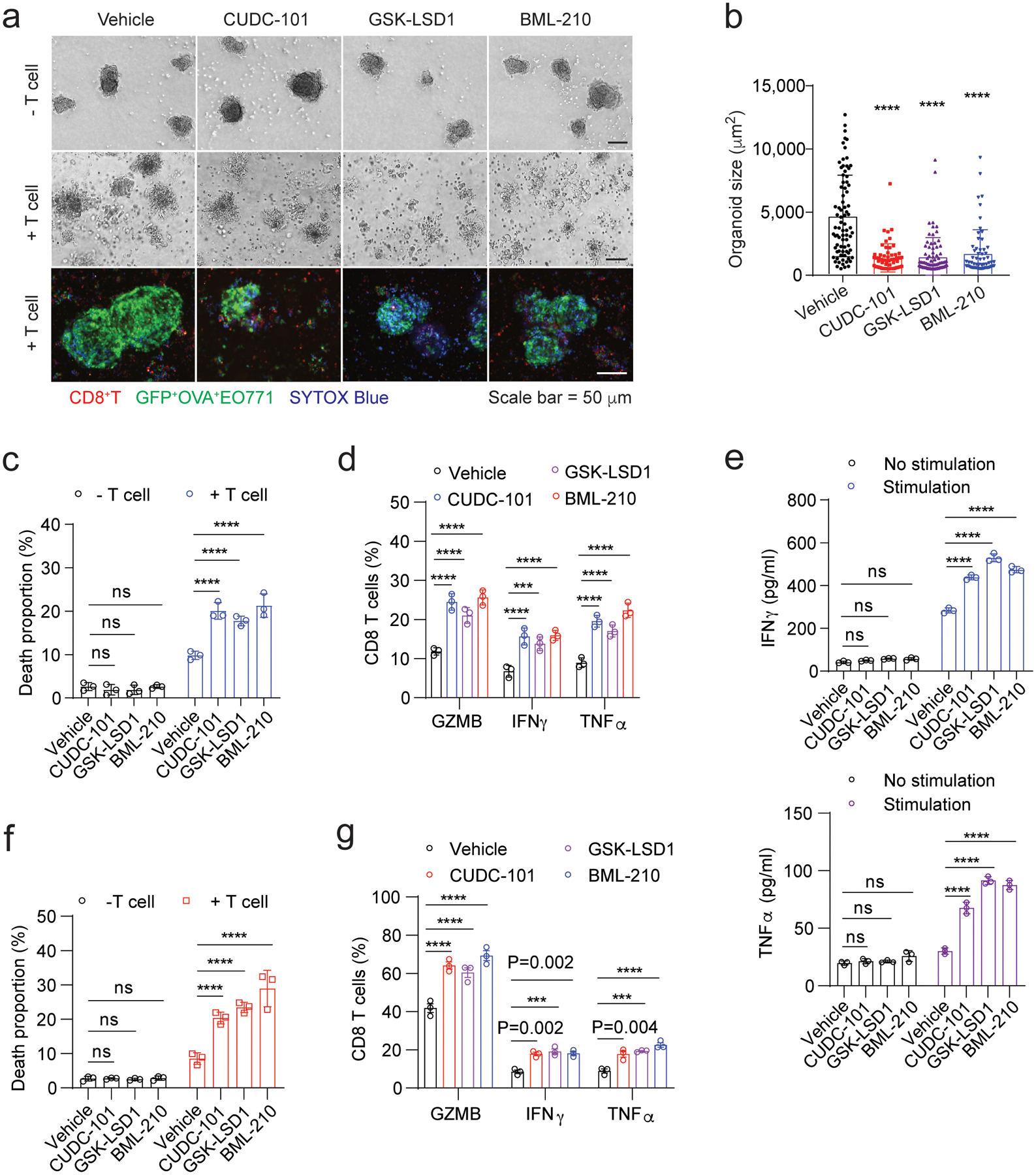
a, The cytotoxicity of OVA-specific CD8+ T cells on the OVA+ EO771 tumour organoids treated with control or drug candidates. The upper and middle panels are optical images. In the bottom confocal images, SYTOX Blue reagent staining dead cells are shown in blue colour. EO771 (GFP+Luc+OVA+) cells are shown in green colour and CD8+ T cells are shown in red colour. b, Size of OVA+ EO771 tumour organoids treated with control or drug candidates in the co-culture with OVA-specific CD8+ T cells. Optical images were analyzed using ImageJ to determine the organoid size. c, Death proportion of the OVA+ EO771 tumour cells in the tumour organoids treated with control or drug candidate, which was measured by flow cytometry. Typical dead cell gating and analysis are shown in Supplementary Fig. 3a,b. d, The percentages of OVA-specific CD8+ T cells positive for GZMB, IFNγ and TNF-α in the co-culture with tumour organoids treated with control or drug candidates. e, Levels of IFNγ and TNF-α secreted from the OVA-specific CD8+ T cells from the co-culture with the tumour organoids as indicated. The secretion of IFNγ and TNF-α was stimulated with PMA and ionomycin. f, Death proportion of NY-ESO-1+ MDA-MB-468 cells treated with control or drug candidates in the co-culture with the NY-ESO-1-specific CD8+ T cells. Typical dead cell gating and analysis are shown in Supplementary Fig. 3d,e. g, The percentages of the NY-ESO-1+ CD8+ T cells positive for GZMB, IFNγ and TNF-α in the co-culture with the MDA-MF-468 tumour organoids treated with control or drug candidates. For statistical analysis of data, one-way ANOVA test was used in (b) and two-way ANOVA test was used in (c-g). Data (c-g) are presented as mean ± SD and are representative of 3 biologically parallel experiments. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
Antitumour activity of BML-210, CUDC-101 or GSK-LSD1 in vivo
Among the three compounds identified from our screen, GSK-LSD1 was reported to inhibit the histone demethylase LSD1 and promote CD8+ T cell infiltration and anti-tumour response in a mouse melanoma model39, partly supporting the effectiveness of our tumour organoid screen system for immune drug discovery. No previous studies have been reported on the activity of BML-210 or CUDC-101 in cancer immunotherapy. We first attempted to test whether the treatment of BML-210 or CUDC-101 inhibits breast tumour growth in the context of immunocompetent animals. Mouse breast cancer EO771 cells were orthotopically implanted into the fat pad of mammary glands of female C57BL/6 mice. Once tumours were established (75–100 mm3), the mice were treated with BML-210 or CUDC-101 three times per week for two weeks (Extended Data Fig. 3a). The tumour growth and weight in the BML-210 or CUDC-101 treatment groups were notably suppressed in comparison with the control group (Fig. 5a,b and Extended Data Fig. 3b). Immune profile analysis revealed a higher tumour infiltration of total T cells (CD3+), CD4+ and CD8+ T cells in the tumours treated with each of the three drugs (Fig. 5c and Supplementary Fig. 4a). The expression of GZMB, IFNγ and TNFα in the tumour-infiltrated CD8+ T cells was also up-regulated in the drug treatment groups (Fig. 5d and Supplementary Fig. 4b), indicating enhanced cytotoxicity of the CD8+ T cells. Consequently, the tumour cell proliferation (Ki67+) from the drug treatment groups were markedly lower than the control group, while the numbers of apoptotic tumour cells (cleaved Caspase 3+) were increased in the drug treatment groups (Fig. 5e and Extended Data Fig. 3c), further suggesting the activity of the three drugs in tumour growth inhibition. Furthermore, the CD8+ T cells in the drug-treated tumours secreted more IFNγ and TNFα upon stimulation, a feature for the activity of CD8+ T cells (Fig. 5f). No visible damages were observed in the heart, kidney, spleen, liver and lung of mice treated with BML-210 or CUDC-101, suggesting the drug tolerability and low toxicity (Supplementary Fig. 4c). To prove that the tumour growth inhibition was due to anti-tumour immune response instead of direct drug treatment effect on tumour cells, similar experiments were conducted with BML-210 and CUDC-101 in the immune-deficient nude (Nu/J) mice. No significant differences were observed on tumour growth and weight between the drug treatment groups and the control group (Fig. 5g, Extended Data Fig. 3d and Supplementary Fig. 4d). Depletion of CD8+ T cells from the drug-treated mice abolished the anti-tumour activity of BML-210 or CUDC-101, while depleting CD4+ T cells did not have much effect, suggesting that BML-210 or CUDC-101 suppressed tumour growth via CD8+ T cell-mediated cytotoxicity (Fig. 5h,i and Extended Data Fig. 3e, 4).
Fig. 5 |. Antitumour activity of drug candidates in mouse breast tumour models.
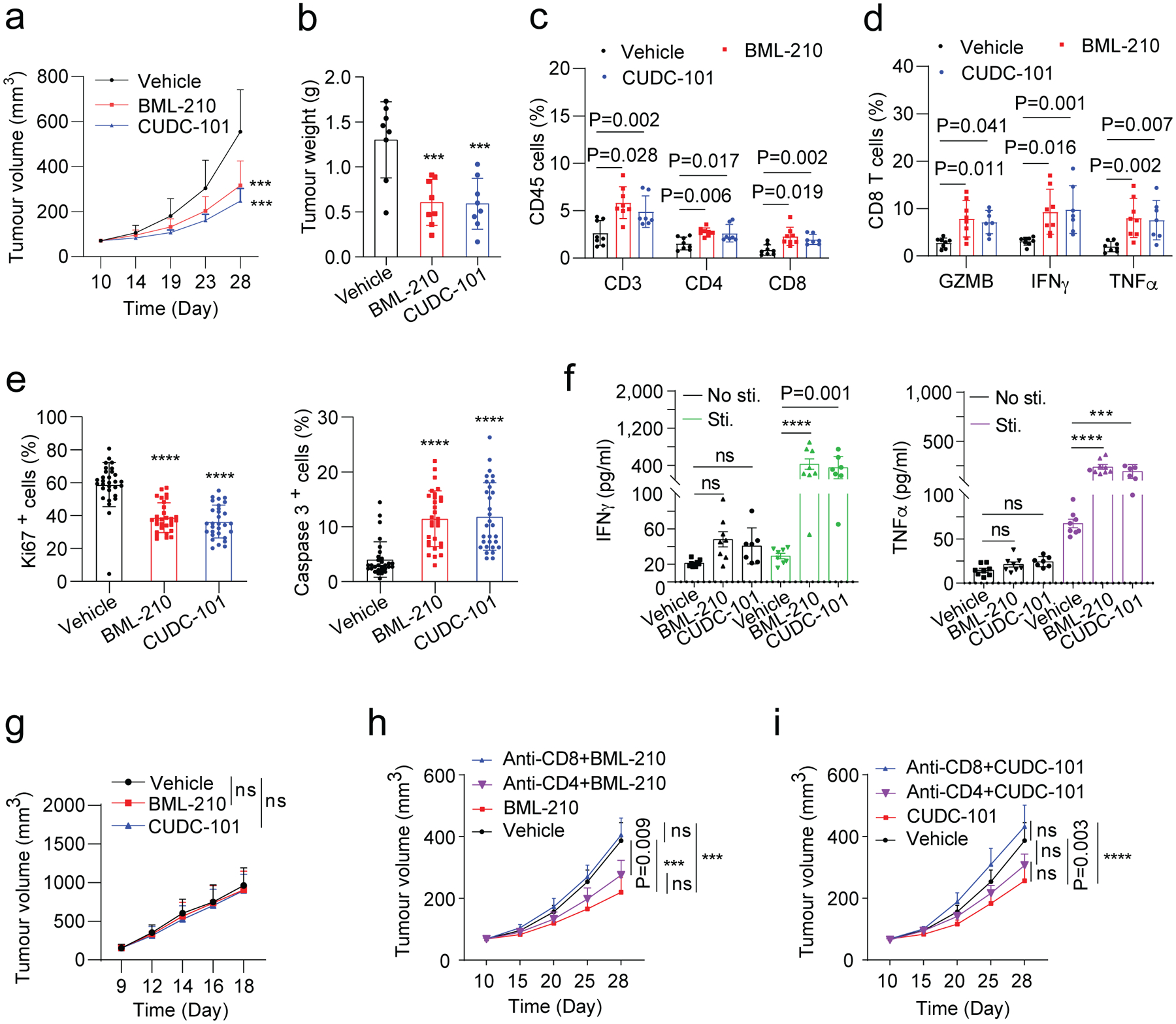
a,b, Gross dissected mammary tumour growth (a) and weight (b) of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with vehicle control, BML-210 (20 mg kg−1), or CUDC-101 (20 mg kg−1). c, Proportions of total T, CD4+ T, and CD8+ T cells in total immune (CD45+) cells in the EO771 tumours from mice treated with control, BML-210 or CUDC-101. d, Percentage of active cells in total CD8+ T cells, indicated by GZMB+, IFNγ+, TNFα+ in flow cytometry analysis. e, Quantitative results for tumour cell proliferation (Ki67+) and apoptosis (Caspase 3+) in the tumours analyzed by IHC staining images (Extended Data Fig.3c). f, Levels of secreted IFNγ and TNF-α from CD8+ T cells isolated from the tumour tissues with and without PMA and ionomycin stimulation. g, Gross dissected mammary tumour growth of the EO771 tumours from the tumour-bearing immunodeficient nude mice treated with vehicle control, BML-210 (20 mg kg−1), or CUDC-101 (20 mg kg−1). Tumours from immunodeficient mice were harvested at day 18 post injection. h,i, Gross mammary tumour growth of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with isotype control, CUDC-101 (20 mg kg−1), BML-210 (20 mg kg−1), anti-CD8 (10 mg kg−1) + CUDC-101 (20 mg kg−1), anti-CD8 (10 mg kg−1) + BML-210 (20 mg kg−1), anti-CD4 (10 mg kg−1) + CUDC-101 (20 mg kg−1) or anti-CD4 (10 mg kg−1) + BML-210 (20 mg kg−1). Tumours from tumour-bearing C57BL/6 mice were harvested at day 28 post injection. For statistical analysis of data, one-way ANOVA test was used in (a,b,e,g-i), and two-way ANOVA test was used in (c,d,f). Data are presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
Next, we wanted to confirm whether GSK-LSD1 promotes the immune response of breast tumours although its activity was reported in mouse melanoma models (Extended Data Fig. 5). In vivo studies of GSK-LSD1 were conducted in a way as described above for CUCD-101 and BML-210 (Extended Data Fig. 5a). As anticipated, the growth and weight of EO771 tumours in the GSK-LSD1 treatment group were markedly suppressed compared to the tumours in the control group (Extended Data Fig. 5b–d). Immune analyses showed a higher tumour infiltration of T cells and enhanced cytotoxicity of the CD8+ T cells in the tumours treated with GSK-LSD1 (Extended Data Fig. 5e,f). Consistently, the tumour cell proliferation from the drug treatment group were much lower and the numbers of apoptotic tumour cells were increased in the GSK-LSD1 treatment group (Extended Data Fig. 5g–i), supporting the activity of GSK-LSD1 in breast tumour growth inhibition.
Epigenetic inhibitors enhance tumour antigen presentation
As BML-210 exhibited the most potent anti-tumour activity in our in vitro and in vivo assays, we wanted to elucidate the molecular mechanism by which BML-210 enhanced the response of breast tumour cells to cytotoxic CD8+ T cells. To this end, we analyzed the genome-wide gene expression profiles to systematically identify transcriptional reprogramming in the BML-210-treated cells. Gene ontology (GO) enrichment analyses of altered gene expression profiles (up-regulated or down-regulated) showed that treatment of BML-210 led to upregulation of genes particularly enriched in lysosome, phagosome, antigen processing and presentation pathways (Fig. 6a–c). As examples, MHC-I genes (H2-d1, H2-k2, B2m), lysosome gene (Lamp1), and endosome gene (Hspa8) were induced by the BML-210 treatment (Fig. 6a). MHC-I-associated antigen processing and presentation pathway in the tumour cell is composed of a number of steps that involves protein degradation and processing, vehicle transport (endosome, lysosome and phagosome and associated cytoskeleton) and antigen loading onto MHC-I complex40, 41. Therefore, we reasoned that BML-210 treatment reprograms the gene transcription of tumour cells, leading to increased antigen processing and presentation and enhanced response to the CD8+ T cells. Gene set enrichment of the antigen processing and presentation was performed (Extended Data Fig. 6a) and the top differentially expressed genes (DEGs) were shown in the heatmap (Fig. 6c). The positively correlated DEGs were individually confirmed by quantitative PCR (qPCR), showing that most of DEGs had higher expression levels in mouse (EO771) and human (MDA-MB-468) breast cancer cells with BML-210 treatment (Fig. 6d, Extended Data Fig. 6b). H-2Kb on EO771 cells (Fig. 6e) and HLA on MDA-MB-468 cells (Extended Data Fig. 6c) were presented at much higher levels when the cells were treated with BML-210, suggesting higher levels of MHC-I-associated antigen presentation. Similarly, the increased H-2Kb or HLA associated antigen presentation was also observed in the cells treated with CUDC-101 or GSK-LSD1 (Extended data Fig. 6d–h), indicating that enhanced antigen presentation is likely a common mechanism for all the three compounds. Confocal images also confirmed that these three compounds boosted up the antigen presentation (Fig. 6f,g and Extended Data Fig. 7a–d) with no other morphological changes observed on the treated cells. Knockdown of B2M in the OVA+ EO771 cells completely abolished the effect of BML-210 on promoting the T cell-mediated cytotoxicity (Fig. 6h, Extended Data Fig. 7e,f and Supplementary Fig. 5a). As B2M is an essential component of MHC-1 molecules, this result suggests that BML-210 directly targets the MHC-I-associated antigen presentation pathway and increases the recognition of breast tumour cells by T cells.
Fig. 6 |. BML-210 treatment upregulates tumour-antigen processing and presentation.
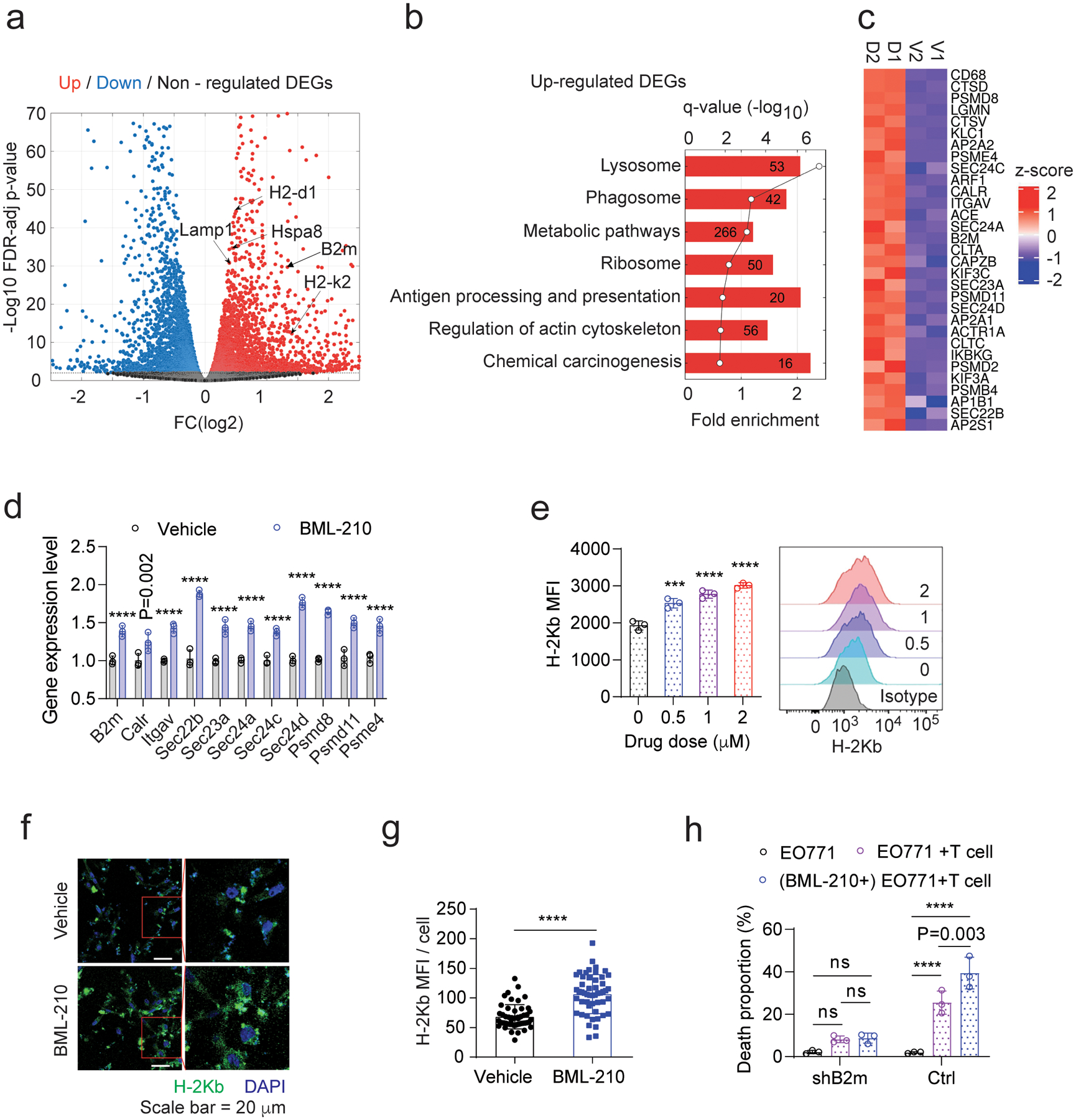
a, Volcano plot analysis of total RNA-seq data shows differentially expressed genes (DEGs) in EO771 cells treated with vehicle control or BML-210 (1.0 μM). A FDR cut-off of 0.01 was used to determine significantly differential expressed genes. b, Gene pathway analysis of up-regulated DEGs in the EO771 cells treated with BML-210. c, Heatmap shows the BML-210-induced up-regulation of DEGs enriched in antigen processing and presentation. V1 and V2 refer to the vehicle control groups (vehicle 1 and vehicle 2); D1 and D2 refer to the BML-210 drug treatment groups 1 and 2. d, Validation of up-regulated genes from (c) in mouse EO771 cells treated with control or BML-210 (1.0 μM) by quantitative RT-PCR. e, Levels of H-2Kb on the OVA+ EO771 cells treated with control or BML-210, which were determined by mean fluorescence intensity (MFI) in flow cytometry analysis. f, Confocal images showing H-2Kb (green) presented on the OVA+ EO771 cells treated with control or BML-210. Blue colour in the images refers to DAPI-stained cell nuclei. g, Quantitative analysis of images in (f) for assessing H-2Kb antigen presentation. h, Death proportions of the OVA+ EO771 tumour cells with and without B2M knockdown in the tumour organoids were measured by flow cytometry analysis. The SYTOX Blue reagent was used to determine the cell death proportion. One-way ANOVA test was conducted for statistical analysis in (e). Two-way ANOVA test was conducted for statistical analysis in (d,h). Two-sided Student’s t-test was performed for statistical analysis in (g). Data (d,e,h) from three biologically parallel experiments are presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
To examine whether these three compounds affect other cell types than tumour cells in the tumour organoid screen system, non-tumour cells (GFP− EpCAM−) from the GFP+Luc+OVA+ EO771 tumour organoids treated with each of the three compounds were gated out for analysis (Supplementary Fig. 5b). The expression levels of H-2Kb in the GFP−EpCAM− cells did not have any notable changes after drug treatments for 48 h in a dose-gradient analysis (Supplementary Fig. 5c,d). Furthermore, direct treatment of CD8+ T cells with these compounds did not affect the expression levels of GZMB, IFNγ and TNFα at different drug doses (0, 0.01, 0.1, 1 μM) (Supplementary Fig. 6). The results suggest that the compounds directly promoted the immune response of tumour cells in the tumour organoids.
BML-210 enhances the efficacy of PD-1-based ICB
To examine the BML-210-induced global immunological changes, we harvested mouse mammary tumours 26 days post orthotopic implantation of the EO771 cells for mass cytometry (CyTOF) analysis (Fig. 7a–c, Supplementary Fig. 7, 8 and Supplementary Table 2). The BML-210 treatment led to a notably enhanced infiltration of immune lymphocytes, particularly CD8+ T effector cells and natural killer (NK) cells, but decreased infiltration of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs: CD11b+Ly6G+Ly6ClowF4/80−) and increased monocytic myeloid-derived suppressor cells (Mo-MDSCs: CD11b+Ly6G−Ly6C+F4/80−)42. No significant differences were observed in tumour-associated macrophage (TAM M1: CD11b+F4/80+MHC-IIhigh and TAM M2: CD11b+F4/80+MHC-IIlow) populations. PMN-MDSCs are often immune suppressive, which restrict immune responses and suppress anti-tumour cytotoxicity of T cells43. These immunological changes are tipping the balance in the tumour microenvironment towards anti-tumour immunity. Despite the promising potential in cancer therapy, the efficacy of PD-1 blockade in metastatic TNBC is currently low, highlighting a need for strategies that render the tumour microenvironment more sensitive to PD-1 blockade. We reasoned that the enhanced antigen presentation and tumour cell recognition would improve the efficacy of the anti-PD-1-based immunotherapy. It is known that anti-PD-1 therapy modestly enhanced anti-tumour response in mouse EO771 tumour models44, 45. Thus, we assessed the therapeutic responses of breast tumours to the combinational treatment of PD-1 monoclonal antibodies (mAb) with BML-210 (20 mg kg−1) in C57BL/6 mice bearing EO771-derived tumours (Fig. 7d–f). The addition of BML-210 to the PD-1 blockade treatment resulted in much greater tumour growth control (indicated by tumour growth inhibition and reduced tumour weight) than BML-210 or PD-1 mAb single agent treatment. The results suggested that BML-210 treatment may promote global antigen presentation of tumour cells regardless of the amount of tumour neoantigens. In addition, due to the immunosuppressive tumour microenvironment, CD8+ T cells are often at inactive or exhausted states, which can be activated by the PD-1 inhibition for better cytotoxicity on the tumour cells with enhanced antigen presentation. Consistent with the results of immune profiling studies (Fig. 7a–c), much higher tumour infiltration of CD8+ T cells was observed in the combinational treatment group, compared to the control group, or the groups with single agent treatment (Fig. 7g,h). Collectively, the results of the mouse tumour models verified the effect of BML-210 in modulating the tumour immune response and sensitizing breast tumours to the ICB therapy.
Fig. 7 |. BML-210 treatment enhances antitumour responses in combination with PD-1 blockade.

a, Analysis of immune profile and microenvironment of the mouse EO771 tumours in C57BL/6 mice treated with control or BML-210 using a CyTOF panel containing 27 markers (Supplementary Table 2). tSNE representation of the immune cell subtypes and percentages of distinct immune cell populations in the tumours are shown. b,c, tSNE representation and quantitative analysis of CD8+ T cells (b) and PMN-MDSCs (CD11b+Ly6G+Ly6ClowF4/80−) (c) in the EO771 tumours with control or BML-210 treatment. The colour scale is specific for each channel. The dot plot is spectrum coloured for the indicated channel; the minimum and maximum values for the expression intensity are determined upon global expression using Cytobank. d-f, Gross dissected mammary tumour images (d), tumour growth (e) and weight (f) of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with vehicle control, BML-210 (20 mg kg−1), PD-1 antibody (200 μg per mouse), or BML-210 and PD-1 antibody combo. g,h, Histograms and quantitative results of CD8+ T cells and cleaved Caspase 3+ cells in the tumour tissues with indicated treatment. Each individual symbol in (g) and (h) represents the percentage of the positive cells from the total cells in an independent field. A total of 20 images in each group were analyzed to quantify the cell proportion. Two-sided Student’s t-test was performed for statistical analysis in (b,c). One-way ANOVA test was conducted for statistical analysis in (e,f,h). All the data are presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
Epigenetic inhibitors promote T cell activity in PDOs
There is an unmet clinical need to predict responsiveness to new cancer immunotherapy. Both syngeneic mouse models and humanized patient-derived xenograft (PDX) models are time-consuming and cost-inefficient. Here, we wanted to apply the standardized tumour organoid model to predict the therapeutic responses to BML-210, CUDC-101 or GSK-LSD1 using patient-derived organoids (PDOs). In this setting, autologous CD8+T cells were isolated from patient tumour tissues and expanded for a week. Using the protocol described in Fig. 1, PDOs were generated from 10 patients with breast cancer (Supplementary Table 3). The PDOs from each patient were characterized and compared with their originating tumours using Hematoxylin and eosin (H&E) staining and immunofluorescence imaging analysis. EpCAM and α-smooth muscle actin (α-SMA) were used as biomarkers to identify tumour epithelial cells and cancer-associated fibroblast cells, respectively, which are the main components of a breast tumour tissue. Overall, the PDOs exhibited well-organized structure similar to their original tumours (Fig. 8a). We were able to isolate reasonable numbers (> 1 × 105) of autologous T cells from 8 out of 10 breast tumour tissues. This was expected as previous treatments often result in very low level of immune infiltrate. The PDOs from each tumour were treated with BML-210, CUDC-101 or GSK-LSD1 for 48 hours in advance and then co-cultured with CD8+ T cells expanded from original autologous cells (Fig. 8b). Among the eight PDOs co-cultured with their autologous T cells, the PDOs from five tumours (PDO1, PDO3, PDO4, PDO5 and PDO8) exhibited higher T cell-mediated cytotoxicity with treatment of BML-210, CUDC-101 or GSK-LSD1. The PDO6 also exhibited higher T cell-mediated cytotoxicity when treated with BML-210 or GSK-LSD1, but showed modest effect with CUDC-101 treatment (Fig. 8c and Supplementary Fig. 9). The remaining two PDOs (PDO2 and PDO7) had only moderate increases (statistically not significant) on the T cell cytotoxicity upon the drug treatment. Thus, ~ 75% (6 out 8) of the clinical samples showed significant (P < 0.05) treatment effect of these drugs in the co-culture. Several HDAC inhibitors have been approved for the treatment of patients with hematological malignancies46. However, this class of therapeutic compounds has yet to be considered as an option for patients with breast cancer. Recent studies showed that the patients with hormone receptor-positive (HR+) breast cancer could benefit from treatment with HDAC inhibitors (tucidinostat and vorinostat)9, 47. HDACi may also upregulate PD-L1 in TNBC cells and thus potentiate immunotherapy48, but it was not tested in human breast cancer samples. LSD1 ablation by GSK-LSD1 seems to stimulate anti-tumour immunity via activation of type 1 interferon in mouse melanoma models39. Our study demonstrates that small molecule compounds for epigenetic modulation are capable to functionally promote antigen processing and presentation on breast tumour cells. Given that immune evasion of tumour cells is a major hurdle in cancer immunotherapy, the treatment of epigenetic inhibitors, such as BML-210, CUDC-101 or GSK-LSD1, can sensitize breast tumours to the immune checkpoint blockade therapy.
Fig.8 |. Treatment of BML-210, CUDC-101 or GSK-LSD1 promotes the cytotoxicity of autologous CD8+ T cells in patient-derived organoids (PDOs).
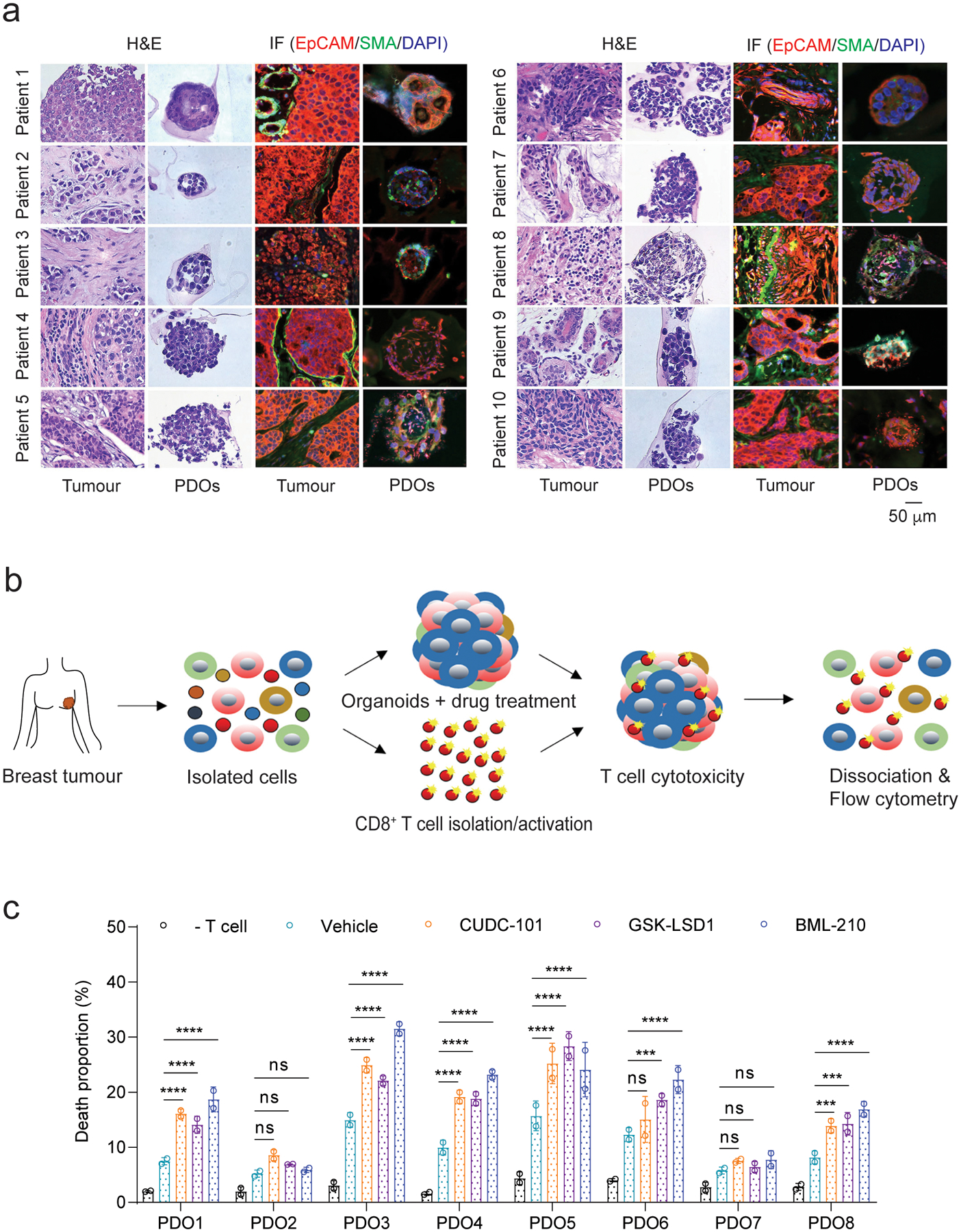
a, Characterization of PDOs from six breast cancer patients using H&E staining and immunofluorescence analysis. EpCAM+ and α-SMA+ cells represent breast cancer epithelial cells and cancer-associated fibroblast cells, respectively. b, Schematic illustration of the co-culture of PDOs and autologous tumour-infiltrating CD8+ T cells. c, Death proportions of tumour cells in the PDOs treated with control, BML-201, CUDC-210 or GSK-LSD1 after co-culture with autologous CD8+ T cells. Two-way ANOVA test was used for statistical analysis and data from two biologically parallel experiments are presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
Discussion
Personalized cancer therapy and precision medicine rely on the study of patient-derived tumour tissues and cells23, 49. In recent years, tumour organoids have emerged as an important tool for predicting clinical responses in cancer therapy50. The inclusion of 3D models to predict clinical responses to screened drugs has proven superior to traditional monolayer cultures, as they recapitulate in vivo features to a higher extend51. Human clinical samples are hardly used in high-throughput screens owing to limited tissue amounts, tissue heterogeneity, and the high costs involved52. In this study, we developed a standardized protocol to establish a tumour-organoid–T-cell system with breast tumour organoids and primary tumour-specific CD8+ T cells. This system facilitates high-throughput drug screens using mouse breast tumour organoids as well as the prediction of therapeutic responses to cancer drugs using PDOs, because 1) sufficient sources of tumour-specific CD8+ T cells are available for large-scale drug screens; 2) tumour organoids are relatively uniform in size and a pool of tumour organoids used for each drug test offsets the heterogeneity between organoids ; 3) tumour-specific T cells or autologous T cells from clinical samples preserve the original T cell receptor spectrum and 4) T-cell infiltration in organoids can be determined.
In this study, cell composition, size and hypoxia of tumour organoids were characterized and optimized for immunotherapy drug screens. First, to avoid interference, immune cells from original tumours were excluded from the tumour organoids. Although it is known that immune cells are a part of tumour microenvironment18, they are not essential components of cancer organoids51, 53, 54. The complexity of culture condition in vitro and the heterogeneity of immune cells existed in different tumour organoids only lead to inaccurate measurement in drug screen. On the other hand, tumour organoids without immune cells can only be used to screen drugs that promote CD8+ T mediated cytotoxicity, yet this is the aim to be achieved in our study as the CD8+ cytotoxic T lymphocytes are on the center of current cancer immunotherapy55. Second, hypoxia signaling can trigger resistance to immune therapy56. Hypoxia in large tumour organoids results in massive cell death, suggesting that tumour organoids with size larger than 150 μm is not a good option for drug screen. Indeed, our study showed that tumour organoids with size between 70~150 μm maintain the tumour architecture formed by epithelial, vascular endothelial and stroma cells and inherit the property of tumourigenesis from the original tumour. Finally, the 7-day culture time allows tumour organoids to maintain similar cellular composition as their original tumours, an important feature for drug screen.
From the three drug candidates identified from this study, GSK-LSD1 has a confirmed activity as a lysine specific demethylase (LSD1) inhibitor. Inhibition of LSD1 enhanced tumour immunogenicity by stimulating endogenous retrovirus expression and downregulating RNA induced silencing complex in a mouse melanoma model39. Both BML-210 and CUDC-101 are Class I and Class II HDAC inhibitors (HDACis)57, 58. However, CUDC-101 is possibly an inhibitor of the receptor kinases epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2). As HDACis have been considered to leverage the efficacy of immunotherapy59, we focused on BML-210 as an HDAC inhibitor for molecular mechanism study with gene expression data. Results from immunological studies are in line with our analysis of RNA-seq data in the BML-210-treated cells, suggesting that BML-210 upregulates MHC-I-associated antigen processing and presentation. This mechanism is probably shared by GSK-LSD1 and CUDC-1 because they also promote the MHC-I antigen presentation on breast tumour cells. This study also confirmed the anti-tumour activity of BML-210 in cancer immunotherapy as single agent or in combination with PD-1 checkpoint blockade. The three drugs in this study upregulate the antigen processing and presentation machinery at least partly by transcriptional reprogramming in breast tumour cells, which would not be limited to any neoantigens. Given the low response rate in PD-1-based immunotherapy for TNBC, these drugs are potentially great candidates for the combinational therapy with PD-1 or PD-L1 blockade.
In summary, we have described a repeatable and standardized protocol for immunotherapy drug discovery, in which we characterized and optimized tumour organoids in histological feature, composition, hypoxia, tumourigenesis and culture time, facilitating a large-scale drug screen. The specific recognition between tumour organoids and CD8+ T cells makes the tumour-cell killing activity standardized and measurable in a drug screen. PDO models provided essential validation of our screening results. The treatment of BML-210 can sensitize breast tumours to the immune checkpoint blockade therapy, warranting further preclinical studies and potential clinical trials. The application of the our tumour organoid-T cell system to different types of tumours such as colorectal and lung cancers will accelerate drug identification for cancer immunotherapy.
Methods
Cell lines
Mouse breast cancer EO771 cell line and human breast cancer MDA-MB-468 cell line were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
Primary breast tumour cell culture
Mouse and human breast tumours were cut into small pieces with diameter ~2–4 mm, minced by the gentle MACS Dissociator (Miltenyi Biotec) and digested using the tumour isolation kits (Miltenyi Biotec, mouse, Cat. No. 130-096-730; human, Cat. No. 130-095-929). Detailed isolation procedure followed the manufacturer’s instructions. Single cells from mouse tumour tissue were cultured with DMEM/F12 culture medium containing 10% FBS, 2 mM Ultraglutamine I and 1% penicillin/streptomycin overnight. Single cells from patient samples were cultured in DMEM/F12 culture medium containing 10% FBS, 1% penicillin/streptomycin and 10 ng ml−1 human IL2. Next, suspended cells were removed, and the adherent cells were trypsinized and used to generate tumour organoids. Human breast cancer patient samples were provided by the Tissue Procurement and Distribution Core of Indiana University Simon Cancer Center (Supplementary Table 3).
Breast tumour organoid culture
Single cells isolated from EO771-derived tumours in tumour-bearing C57BL/6 mice were suspended into cold mouse breast cancer culture medium60 (Supplementary Table 4) containing 10% Matrigel (Corning) with a concentration of 2 × 105 cells ml−1. The cells (2 ml) were seeded into each well of pre-warmed 6-well microplate with ultra-low attachment surface (Corning) and cultured at 37°C with 5% CO2. The same volume of fresh culture medium was added every other day. After 7-day culture, tumour organoids were collected using cell strainers with pore size of 70 and 150 μm. For human tumour organoid culture, 2 ml of NY-ESO-1+ MDA-MB-468 cells with a concentration of 2 × 105 cells ml−1 were seeded into each well of the prewarmed 6-well microplate with ultra-low attachment surface. The cells were cultured for two days to generate tumour spheroids. The resulting spheroids were mixed with cancer associated fibroblasts (CAFs) with a ratio of 2:1 (tumour cells versus CAFs) and cultured in the human breast cancer organoid culture medium3 for 5 days. Finally, the MDA-MB-468 tumour organoids with diameter of 70~150 μm were used for co-culture with NY-ESO-1-specific human CD8+ T cells (ASTARTE Biologics). Patient-derived organoids (PDOs) were cultured following the same procedures as mouse EO771 tumour organoids using human breast cancer organoid culture medium3.
Hypoxia test in breast tumour organoids
The Image-iT™ Green Hypoxia reagent (Invitrogen) was used to stain tumour organoids. The protocol follows the manufacturer’s instructions. Fluorescent images were captured under a Leica DM4B microscope. The mean fluorescence intensity is the mean intensity of the projected area of hypoxia in tumour organoids, which was quantified using ImageJ software 1.50e.
Tumour cell viability assay
For 2D tumour cell culture, tumour cells were seeded into 96 well plates at 2,000 cells per well and cultured for 72 h. For tumour organoids, tumour organoids with diameter between 70 and 150 μm were seeded at 50 organoids per well into 96-well microplates with ultra-low attachment surface (Corning). Cells or tumour organoids were treated with indicated compound at the final concentration of 1μM. After 3-day culture, tumour organoids were dissociated into single cells using TrypLE Express (Corning) at 37°C with a shaking velocity of 500 rpm on a thermomixer (Eppendorf) for 15 min. The 96-well microplates were added with 10 μl/well of Premix WST-1 reagent (TaKaRa) and incubated for 1 h. The microplates were read at absorbance of 450 nm on the Epoch microreader (BioTek). The toxicity of the epigenetic drugs was quantified using the premix WST-1 reagent according to the manufacturer’s protocol.
Cytotoxicity of CD8+ T cells in the co-culture with tumour organoids
We generated mouse EO771 (Luc+OVA+) cell line that presents OVA257–264 antigen (from chicken ovalbumin) and human MDA-MB-468 (NY-ESO-1+) cell line that presents NY-ESO-1 antigen for use in the T cell-mediated cytotoxicity assay. OVA-specific CD8+ T cells were isolated from the spleen of OT-I mouse that contains transgenic inserts of Tcra-V2 and Tcrb-V5 genes. The transgenic T cell receptor can recognize the ovalbumin peptide SIINFEKL (OVA257–264). Anti-NY-ESO-1 human CD8+ T cells (ASTARTE Biologics) were used to recognize the NY-ESO-1+ MDA-MB-468 cells. Autologous human CD8+ T cells were isolated from patient tumour tissues and amplified in the DMEM/F12 medium supplemented with 10% FBS, 2 mM Ultraglutamine I, 1% penicillin/streptomycin and 10 ng ml−1 of human IL2 for 7 days. The CD8+ T cells were stimulated by the T-activator CD3/CD28 Dynabeads (Gibco, 11452D for mouse and 11131D for human) for 48 h prior to use for cytotoxicity assay. Mouse and human tumour organoids with diameter between 70 and 150 μm were collected and mixed with corresponding CD8+ T cells in DMEM/F12 medium supplemented with 10% FBS, 2 mM Ultraglutamine I, 1% penicillin/streptomycin and 10 ng ml−1 IL2 (Biolegend, mouse: 575402; human: 589102) at a ratio of organoids versus T cells at 1 : 200 in the microplates with ultra-low attachment surface. The cells were co-cultured for 24 h and subject to further analyses, such as optical and fluorescence imaging, luciferase assay or flow cytometry.
Epigenetic inhibitor screen on CD8+ T cell-mediated cytotoxicity
The epigenetic inhibitor library (Cayman chemical, Cat# 11071, Batch# 0522205, Supplementary Table 1) was used for drug screen. Three different screen methods were conducted. In tumour organoid-T cell co-culture, EO771 (Luc+OVA+) tumour organoids with diameter between 70 and 150 μm were seeded with 100 organoids/well and treated with 1 μM of drug for 48 h. The tumour organoids were centrifuged at 100 rcf for 5 min. The medium was replaced with fresh T cell culture medium containing OVA-specific T cells. A total of 20,000 T cells were added in each well. After co-culture for 24 h, cytotoxicity of tumour cells was measured using a Dual-luciferase report 1000 assay system (Promega) in which luciferase expressed exclusively in the live EO771 (Luc+OVA+) cells was detected by a BioTek Cytation5 imaging reader. Low levels of luciferase from survived tumour cells refer to high cytotoxicity in the co-culture. The luciferase intensity in each well was subtracted by the background intensity for post-data analysis. In 2D tumour cell-T cell co-culture, EO771 (Luc+OVA+) tumour cells were seeded at a density of 4,000 cell/well and treated with drug for 48 h. The medium was replaced and the tumour cells were mixed with CD8+ T cells at a ratio of 1:5 in 96-well microplate (Corning) for overnight. The same protocol as above was used to measure the cytotoxicity of tumour cells. For OVA antigen presentation assay, EO771 (GFP+Luc+OVA+) tumour cells were treated with indicated drugs for 48 h, stained with APC-conjugated OVA peptide SIINFEKL antibody for 15 min, and then were subjected to flow cytometry analysis. The mean fluorescence intensities (MFI) represent the OVA expression levels.
To evaluate intra-tumour infiltration of T cells, EO771 (Luc+OVA+) tumour organoids were treated with indicated drugs for 48 h, and then co-cultured with OT-I T cells. After co-culture for 48h, the organoids with T cells inside or attached were dissociated by TrypLE Express (Thermo Fisher) into single cells and stained with APC/Cy7-conjugated anti-mouse CD8 (Biolegend) and SYTOX Blue reagent for 15 min. The CD8+ T cell proportions from tumour organoids were analyzed by flow cytometry.
T cell effector function analysis
We measured cytokine and cytolytic granule production (IFNγ, GZMB and TNFα) of CD8+ T cells from mouse tumour samples and tumour organoid-T cell co-culture. Briefly, CD8+ T cells were placed into the 24-well plate at 1 × 106 cells/well, stimulated with 1 μM ionomycin and 50 ng ml−1 phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) for 4 h, in the presence of 5 μg ml−1 Brefeldin A (BFA), with the purpose to amplify the expression of intracellular cytokines61. The cells were stained with APC/Cy7-conjugated anti-CD8a (Biolegend) for 15 min and then fixed by 4% PFA. After washing, cells were stained with PerCP/Cy7-conjugated anti-GZMB, APC-conjugated anti-IFNγ and PE-conjugated anti-TNFα (Biolegend) for 15 min. In flow cytometric analyses, T cells stained with isotype control antibodies were used as negative controls for gating the cytokine or granule-producing cells. For T cells from tumour organoid-T cell co-culture, tumour organoids were first digested into single cells with TrypLE Express at 37°C before APC/Cy7-conjugated anti-CD8a staining. In the IFNγ and TNFα secretion assay (Biolegend), T cells were stimulated with ionomycin and PMA without the presence of BFA. The media were collected for ELISA assay.
Immunohistochemistry (IHC) and Immunofluorescence (IF) analysis of tumour organoids
Tumour organoids were gently centrifuged at 100 rcf from organoid culture medium and then embedded in 2% agar. Tumour organoids were fixed in 4% paraformaldehyde solution for overnight at room temperature. The samples were replaced with 70% ethanol and later embedded in paraffin blocks. Paraffin sections were cut at 5 μm thickness, attached on slides, heated at 60°C for 1 h and then overnight at 37°C. The slides were stained with hematoxylin and eosin (H&E). IHC staining follows the protocol described62. Mouse-specific antibodies for CD8α (Cell Signaling Technology, clone D4W2Z, 1:400 dilution), cleaved caspase3 (Cell Signaling Technology, clone D175, 1:400 dilution) and Ki67 (Cell Signaling Technology, clone D3B5, 1:400 dilution) were used for IHC staining. The DAB peroxidase substrate kit (Vector laboratories Inc.) was used to visualize the antibodies. For IF analysis, the sample slides were blocked 3% BSA for 40 min after hydration. The slides were stained with primary antibodies including EpCAM (Abcam, 1:400 dilution), FITC-conjugated anti-α-SMA (Genetex, 1:400 dilution) and AF594-conjugated anti-CD31 (Biolegend) for 1 h. For EpCAM staining, the slide was further stained with secondary antibody Alexa Fluor 647-onjugated anti-rabbit IgG (Biolegend, 1:200) for 40 min. The fluorescence images were captured under a Leica DM4B microscope.
Immunoblotting
Immunoblotting assay was performed as described in the study63. Briefly, EO771 cells expressing lentiviral control shRNA (Emptor vector pLKO.1-Puro, Sigma) or B2M shRNA was lysed in pre-cold RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor mixture (Thermo Scientific). The protein concentration from cell lysates was determined using BCA kit (Thermo Fisher Scientific). 20 μg of cell lysate from each sample was used for polyacrylamide gel electrophoresis. The polyvinylidene difluoride (PVDF) membrane with the transferred proteins was blocked with 5% non-fat milk overnight, and then incubated with primary antibody B2M (Abcam) or β-actin (Abgent) at room temperature for 2h. The membrane was washed three times and incubated with HRP-conjugated anti-mouse or anti-rabbit secondary antibody at a concentration of 1: 5,000 at room temperature for 1.5 h. The membrane was washed three times, incubated with chemiluminescent substrates (western lightning plus ECL kit, PerkinElmer), and exposed to X-ray film for imaging analysis.
Animal studies
All animal experiments were performed in accordance with the National Institutes of Health’s Animal Use Guidelines and protocols approved by the Animal Care and Use Committee of Indiana University School of Medicine. To examine the tumourigenic potential of tumour organoids, tumour organoids with different sizes (diameter of 30–70, 70–150, and 150–500 μm) were orthotopically injected into fat pad of the 4th mammary gland in female C57BL/6 mice. Each mouse was injected with a total of 2 × 105 tumour cells. In each group, a total of 5 mice (5-week old) were used. Tumours were measured in two dimensions using a manual caliper. Tumour volume was calculated using the following formula: V = 0.5 × length × width × width. Tumour volume was measured every 3 days. Tumours were harvested 22 days after injection. The tumour growth, volume and weight were analyzed. To evaluate the anti-tumour effect of drugs in immunocompetent C57BL/6 mice and immunodeficient nude (NU/J) mice, 2 × 105 EO771 cells were orthotopically injected into fat pad of the 4th mammary gland in female mice with 5-week-old. Once tumours were established at day 10, the tumour-bearing mice were randomly divided into each treatment group (n = 8 mice/group), and were treated with vehicle control, BML-210, CUDC-101, GSK-LSD1, PFI-1 or Bromosporine (20 mg kg−1 by intraperitoneal injection) 3 times/week for 2 weeks. For CD4/CD8 depletion experiment, the tumour-bearing mice were randomly divided into 7 groups (n = 8 mice/group) including isotype control (rat IgG2a, Bioxcell), isotype control +CUDC-101, isotype control + BML-210, anti-CD8 (Clone: 53–6.72, Bioxcell) + CUDC-101, anti-CD8 + BML-210, anti-CD4 (Clone: GK1.5, Bioxcell) + CUDC-101, or anti-CD4+ BML-210. Intraperitoneal injection of inhibitors (20 mg kg−1) and anti-CD4/CD8 (10 mg kg−1) were administrated to the mice by 3 times/week for 2 weeks. The tumours were harvested at day 18 (NU/J mice) or day 28 (C57BL/6 mice) for further analyses. In immune checkpoint blockade experiment, 2 × 105 EO771 cells were orthotopically injected into fat pad of the 4th mammary gland in 5-week-old female C57BL/6 mice . From day 10, the tumour-bearing mice were randomly divided into each treatment group (n=8 mice/group), and the mice were treated with IgG2a isotype control antibody (clone 2A3, Bioxcell), BML-210 (20 mg kg−1), anti-PD-1 (200 μg/mouse, Bioxcell), or BML-210 and anti-PD-1 combo. The treatment was done 3 times/week for 2 weeks. Tumours were harvested at day 26 post injection and the tumours were analyzed by IHC staining, flow cytometry, ELISA or CyTOF assay.
Flow Cytometry
Sample acquisition was done using LSR Fortessa X-20 or LSR Fortessa (BD Biosciences) and data were analyzed using FlowJo v10.6.0 software. Live/dead cells were assessed using SYTOX Blue (Invitrogen, Dilution 1:100). For cell staining involving cell permeabilization, the BD cytofix/cytoperm fixation permeabilization kit (BD Biosciences) was used. The PBS buffer containing 0.5% BSA and 10% FBS were used to dilute antibodies. The antibodies used in flow cytometrical analyses include: AF594-conjugated anti-mouse CD31(Biolegend, Dilution 1:100), APC-conjugated anti-mouse CD140a (Biolegend, Dilution 1:400), PE-conjugated anti-mouse CD326 (Biolegend, Dilution 1:100), AF647-conjugated anti-mouse CD326(Biolegend, Dilution 1:100), BV405-conjugated anti-mouse CD45 (Biolegend, Dilution 1:100), PE/Cy7-conjugated anti-mouse CD3 (Biolegend, Dilution 1:100), AF700-conjugated anti-mouse CD4 (Biolegend, Dilution 1:100), APC/Cy7-conjugated anti-mouse CD8a (Biolegend, Dilution 1:100), APC-conjugated anti-mouse CD8 (Biolegend, Dilution 1:100), PE-conjugated anti-mouse CD11b (Biolegend, Dilution 1:200), AF647-conjugated anti-mouse CD11c (Biolegend, Dilution 1:200), PerCP/Cy5.5-conjugated anti-mouse Fn/80 (Biolegend, Dilution 1:100), BV421-conjugated anti-mouse I-A/I-E (Biolegend, Dilution 1:1000), BV650-conjugated anti-mouse CD19 (Biolegend, Dilution 1:100), APC-conjugated anti-mouse H-2Kb bound to SIINFEKL antibody (Biolegend, Dilution 1:20), PerCP/Cy5.5 anti-mouse H-2Kb Antibody (Biolegend, Dilution 1:100), PE-Anti-Human HLA-A,B,C antibody (Biolegend, Dilution 1:100), FITC-conjugated anti-mouse IgG (Biolegend, Dilution 1:100), FITC-conjugated anti-Rat IgG (Biolegend, Dilution 1:100), APC-conjugated anti-mouse IFNγ (Biolegend, Dilution 1:100), PE-conjugated anti-mouse TNFα (Biolegend, Dilution 1:100), PerCP/Cyanine5.5-conjugated anti-human/mouse Granzyme B (Biolegend, Dilution 1:100), APC-conjugated anti-human IFNγ (Biolegend, Dilution 1:100), PE-conjugated anti-human TNFα (Biolegend, Dilution 1:100). The compensation was performed using CompBeads for negative control (BD Biosciences), anti-mouse Ig,κ (BD Biosciences), anti-rat Ig,κ and anti-hamster Ig,κ (BD Biosciences). The beads were stained with the corresponding antibodies separately under the same conditions as the cells were stained in each experiment. SYTOX Blue reagent was used to stain dead cells. Half of the live cells were incubated at 60°C for 2 min to enable membrane permeable and then mixed with the other half incubated at 37°C. The mixed cells were stained with SYTOX Blue for 15 mins and used for compensation. For all the data analysis, doublet exclusion was performed and only single cells were analyzed. Dead cell populations were also excluded from analysis if the experiments were not involved in cell killing. One million events per sample were collected for immune profiling analysis in the animal experiments. For all in vitro experiments, at least ten thousand events per sample were collected for flow cytometry analysis.
Confocal Microscopy
OVA+ EO771 and NY-ESO-1+ MDA-MB-468 cells were seeded at 5 × 103 cells/well in the 8-well slide (Millicell EZ slide, R8AA09916) and treated with 1 μM drug (BML-210, CUDC-101 or GSK-LSD1) for 48 h. FITC-conjugated anti-mouse H-2Kb antibody (Biolegend, dilution 1:50) and FITC-conjugated anti-HLA-A2 (Biolegend, dilution 1:50) were used to stain OVA+ EO771 and NY-ESO-1+ MDA-MB-468 cells, respectively, for 2 h. For cell nucleus staining, cells were incubated with 0.5% Triton X-100/PBS for 10 min and stained with DAPI (Sigma-Aldrich, dilution 1:100) for 15 min. The slides were rinsed in PBS twice and mounted with ProLong Gold anti-fade mountant (Invitrogen, P36930). Fluorescence images were captured using Leica TCS SP8 (upright high-speed multiphoton) confocal imaging system. 3-D images were analyzed by the Imaris x64 8.1.2 software.
Mass Cytometry (CyTOF)
EO771 tumours in C57BL/6 mice were harvested and dissociated into single cells. 1 × 106 single cells from each sample were suspended in ice-cold buffer (PBS with 0.5% BSA and 0.02% Azide). The antibody panel (Supplementary Table 2) was used for sample staining. Samples were analyzed using the CyTOF 2 Mass Cytometer (Fluidigm). All the samples were done on bead-based normalization before analysis on Cytobank. CyTOF data were analyzed using viSNE analysis using the Barnes-Hut implementation of the t-Distributed Stochastic Neighbor embedding (tSNE) algorithm as described previously64.
Quantitative reverse transcription PCR (RT-PCR)
Mouse EO771 cells were treated with and without BML-210 drug for 48 h. Total RNA was isolated from the cells using TRIzol (Ambion, Lot: 260808) and the miRNeasy Mini kit (Qiagen, 157029493, Thermo Scientific). The quality of the RNA was quantified by a Nano Drop 2000 spectrophotometer. A total of 1μg RNA was reverse transcribed into cDNA using the qscriptXLT cDNA superMix reagent (Quantabio, 66141329). Quantitative PCR was performed using SYBR Green fastmix low Rox reagent (TaKaRa) on a Quantstudio real-time PCR system (ThermoFisher Scientific). The PCR primer sequences are listed in Supplementary Table 5.
RNA sequencing
Total RNA was extracted from EO771 cells using TRIzol (Ambion) and the miRNeasy Mini kit (Qiagen). The quantity and quality of the RNA was evaluated by an Agilent Bioanalyzer 2100. 100 ng of total RNA from each sample was used, from which ribosomal RNA was removed by the QIAseq FastSelect rRNA Removal HMR Kit (Qiagen). The cDNA library was constructed using the KAPA RNA Hyper Prep Kit (Roche Corporate) and the quality was accessed by the Qubit and Agilent Bioanalyzer. The library pool was sequenced in 100b paired-end read format on NovaSeq 6000 (Illumina) at the Center for Medical Genomics, Indiana University School of Medicine (IUSM).
RNA-seq analysis
The reads were mapped to the mouse genome mm10 using STAR (v2.5)65. RNA-seq aligner with the following parameter: “--outSAMmapqUnique 60”. Uniquely mapped sequencing reads were assigned to GENCODE M22 genes using feature Counts (v1.6.2)66 with the following parameters: “-s 2 -p -Q 10”. The count data were filtered using read count per million (CPM) > 10 in more than two of the samples, normalized using TMM (trimmed mean of M values) method and subjected to differential expression analysis using edgeR package (v3.28.1)67,68 in R software (version 3.5.3). A false discovery rate (FDR) cut-off of 0.01 was used to determine significantly differential expressed genes (DEGs). The data were visualized with volcano plot using MATLAB. The gene enrichment analysis on DEGs from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database and Gene Ontology database was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID ) functional analysis69. The analysis was visualized using MATLAB with q value (−log10) and gene number in each GO and pathway. Gene set enrichment analysis (GSEA) of the antigen processing and presentation pathway was carried out with GSEA software70. The significantly differential genes from GSEA were plotted with heatmap by using the Complex Heatmap package71 in R software (version 3.5.3).
Statistical analysis
Data were analyzed using GraphPad Prism 9. Fluorescence intensity from images was quantified using ImageJ software (Version 1.50e). Flow cytometry data were analyzed using FlowJo version 10.8.0. Group sizes and error bars are indicated as needed. Statistical analysis of two independent groups of data was performed using Two-sided Student’s t-test. Statistical analysis of data sets higher than two groups with one independent variable was performed using one-way ANOVA. Statistical analysis of multiple groups of two factors was performed using two-way ANOVA. All the data deviations are indicated as mean ± standard deviation (SD). p value indicated as *** (p < 0.001) and **** (p < 0.0001). p < 0.05 is considered significantly different.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
ED Fig. 1. Functional evaluation of the compounds screened from the 2D and tumour-organoid systems.
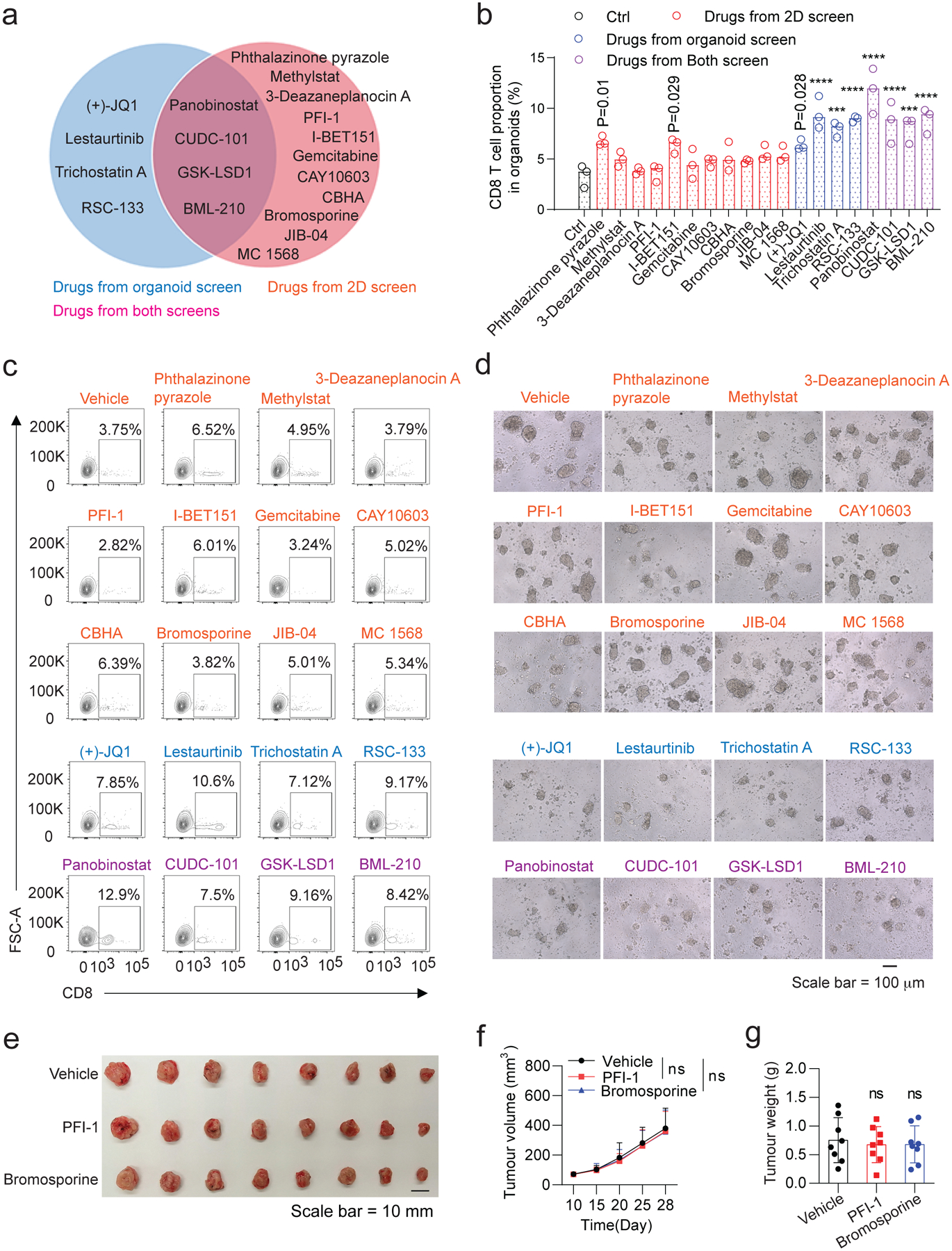
a, Pie chart of the positive compounds screened from the 2D and tumour organoid systems. b, Effect of the above compounds (a) on T cell infiltration in tumour organoids. The CD8+ T cells (from OT-I mouse) were co-cultured with the compound-treated GFP+Luc+OVA+ EO771 tumour organoids for 48h. The tumour organoids with T cells infiltrated or attached were dissociated to single cells and stained with APC/Cy7-conjugated anti-mouse CD8 and SYTOX Blue reagent for 15 min. The T cell proportions from the tumour organoids were analyzed by flow cytometry. Data from 3 biologically parallel experiments were analyzed using One-way ANOVA and presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001. c, Representative flow cytometry data showing the CD8+ T cell proportion in the GFP+Luc+OVA+ EO771 tumour organoids. A total of 20,000 events per cell sample were collected for data analysis. d, Representative optical images of CD8+ T cells co-cultured with the GFP+Luc+OVA+ EO771 tumour organoids. e-g, Gross dissected mammary tumour images (e), tumour growth (f) and weight (g) of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with vehicle control, PFI-1 (20 mg kg−1), or Bromosporine (20 mg kg−1). Tumours were harvested at day 28 post injection. For statistical analysis of data, one-way ANOVA test was used in (f,g). Data are presented as mean ± SD. ns, no significance
ED Fig. 2. Validation of antitumour activity of the three drug candidates in human tumour organoids.
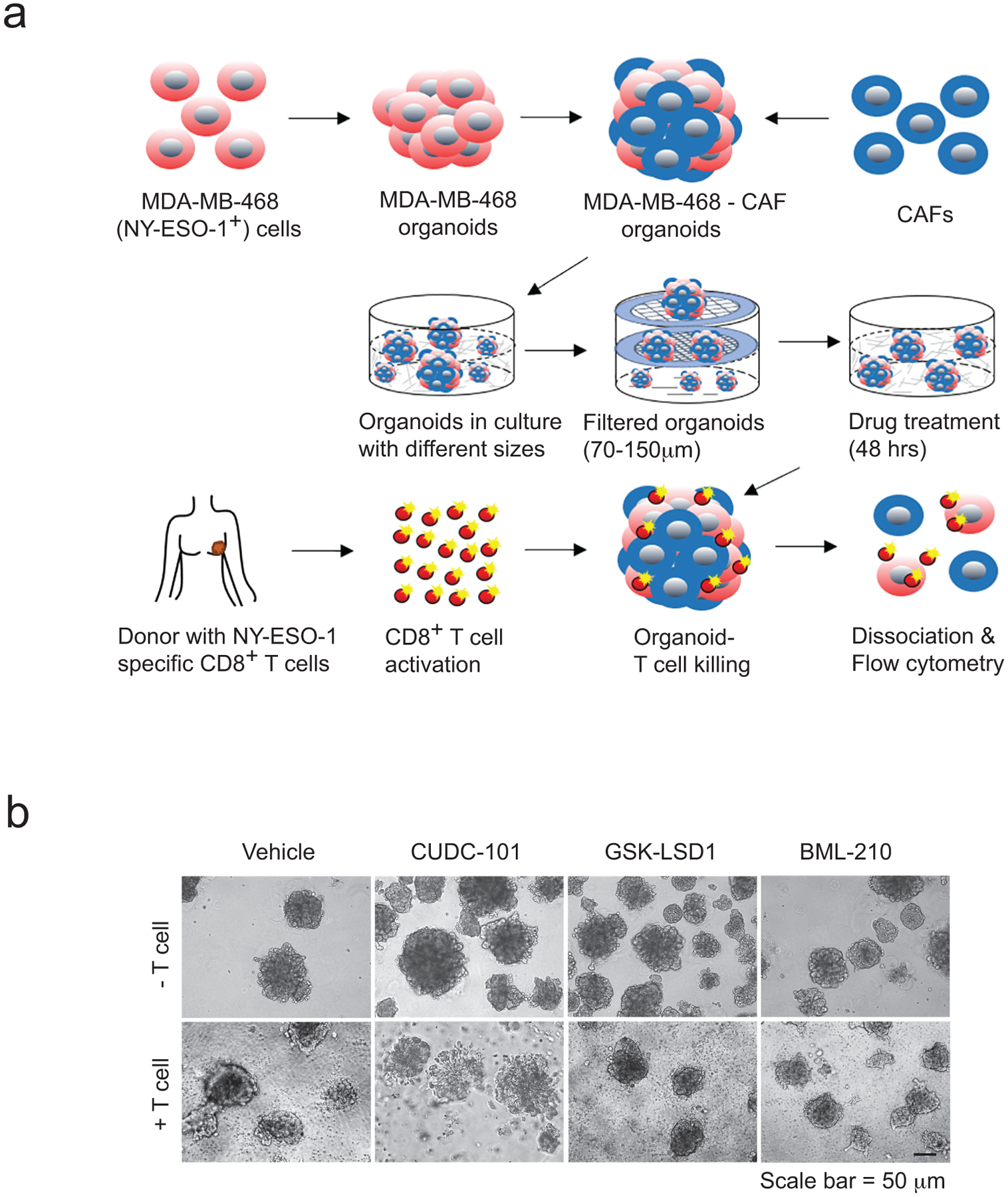
a, Schematic illustration of the drug-validation test using NY-ESO-1+ MDA-MB-468 organoids co-cultured with NY-ESO-1-specific CD8+ T cells. Human breast cancer-associated fibroblasts (CAFs) were used with MDA-MB-468 cells to generate tumour organoids. b, Optical images showing the co-culture of the CD8+ T cells and MDA-MB-468 tumour organoids treated with control or drug candidates.
ED Fig. 3. Antitumour activity of drug candidates in mouse breast tumour models.
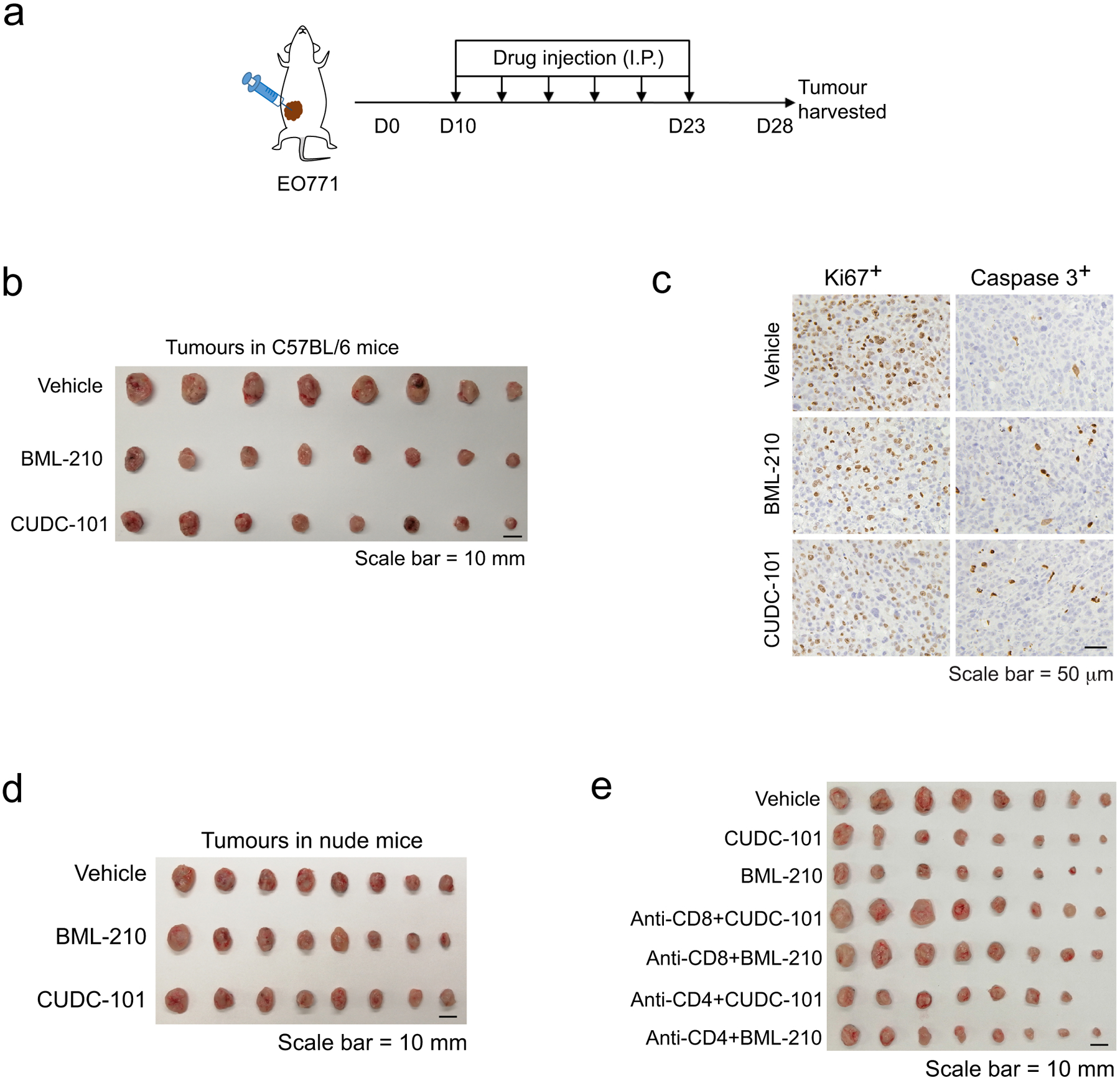
a, Drug-treatment scheme of mouse breast tumour models. b, Gross dissected mammary tumour images of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with vehicle control, BML-210 (20 mg kg−1), or CUDC-101 (20 mg kg−1). c, IHC staining images of Ki67+ and cleaved Caspase 3+ cells in the tumour tissues. d, Gross dissected mammary tumour images of the EO771 tumours from the tumour-bearing nude mice treated with vehicle control, BML-210 (20 mg kg−1), or CUDC-101 (20 mg kg−1). Tumours from nude mice were harvested at day 18 post injection. e, Gross mammary tumour images of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with isotype control, CUDC-101 (20 mg kg−1), BML-210 (20 mg kg−1), anti-CD8 (10 mg kg−1) + CUDC-101 (20 mg kg−1), anti-CD8 (10 mg kg−1) + BML-210 (20 mg kg−1), anti-CD4 (10 mg kg−1) + CUDC-101 (20 mg kg−1) or anti-CD4 (10 mg kg−1) + BML-210 (20 mg kg−1). Tumours from tumour-bearing C57BL/6 mice were harvested at day 28 post injection.
ED Fig. 4. Antitumour activity of BML-210 and CUDC-101 in mouse breast tumour models with CD4+ or CD8+ T-cell depletion.
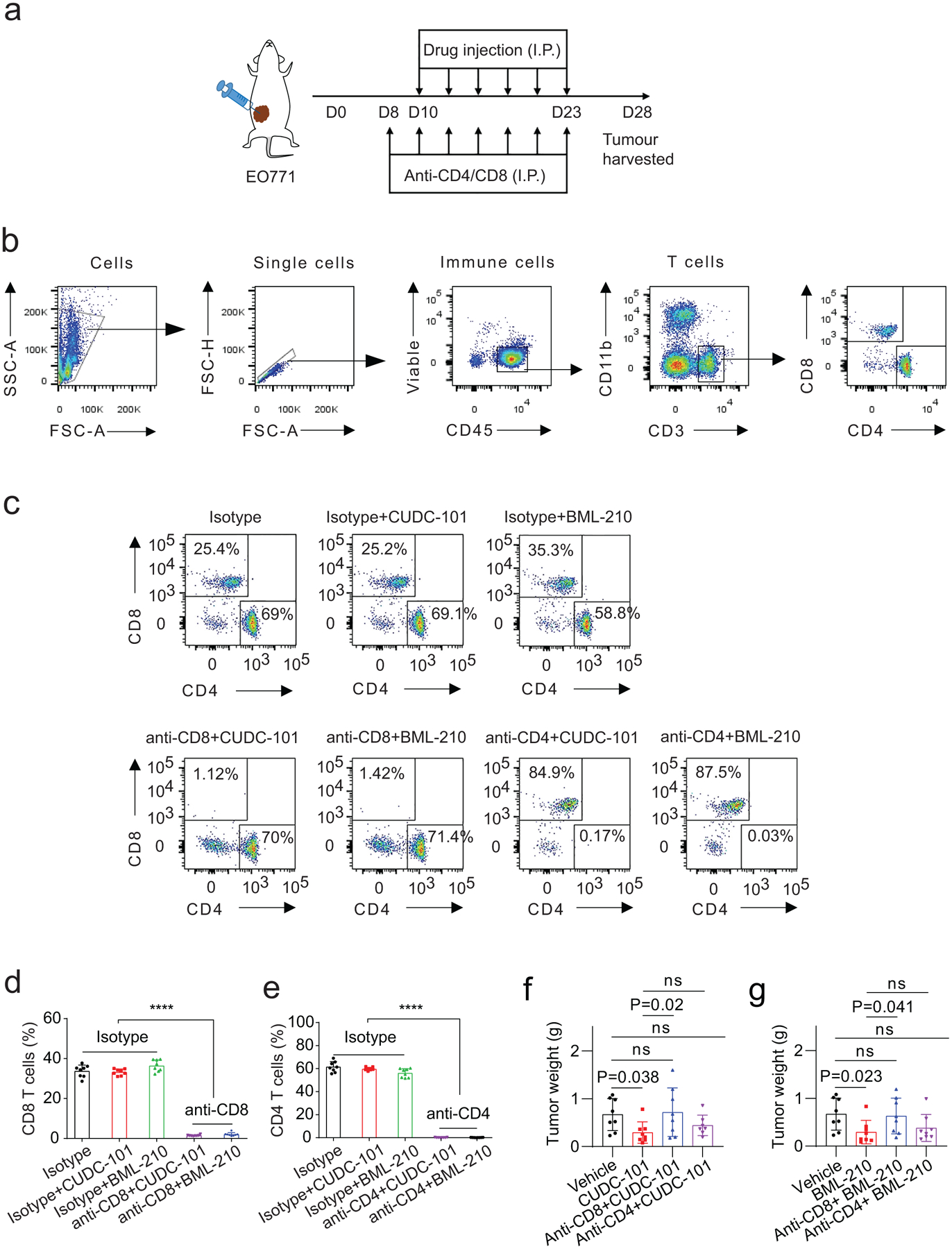
a, Drug-treatment scheme of mouse breast tumour models. b, Flow cytometry gating strategy for analysis of CD4+ and CD8+ T cells from EO771 tumours in C57BL/6 mice. c, Typical graphs showing the proportion of CD4+ and CD8+ T cells in total T cells (CD3+) from EO771 tumours treated with vehicle control or indicated compound, and with CD4 or CD8 depletion. d,e, Proportions of CD8+ (d) and CD4+ (e) T cells in total T cells from EO771 tumours with CD4 or CD8 depletion. f,g, Weight of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with control, CUDC-101 (20 mg kg−1), BML-210 (20 mg kg−1), anti-CD8 (10 mg kg−1) + CUDC-101 (20 mg kg−1), anti-CD8 (10 mg kg−1) + BML-210 (20 mg kg−1), anti-CD4 (10 mg kg−1) + CUDC-101 (20 mg kg−1) or anti-CD4 (10 mg kg−1) + BML-210 (20 mg kg−1). Tumours from tumour-bearing C57BL/6 mice were harvested at day 28 post injection. For statistical analysis of data, two-way ANOVA test was used in (d,e). One-way ANOVA test was used in (f,g). Data are presented as mean ± SD. ****, p < 0.0001; ns, no significance.
ED Fig. 5. Antitumour activity of GSK-LSD1 in mouse breast tumour models.
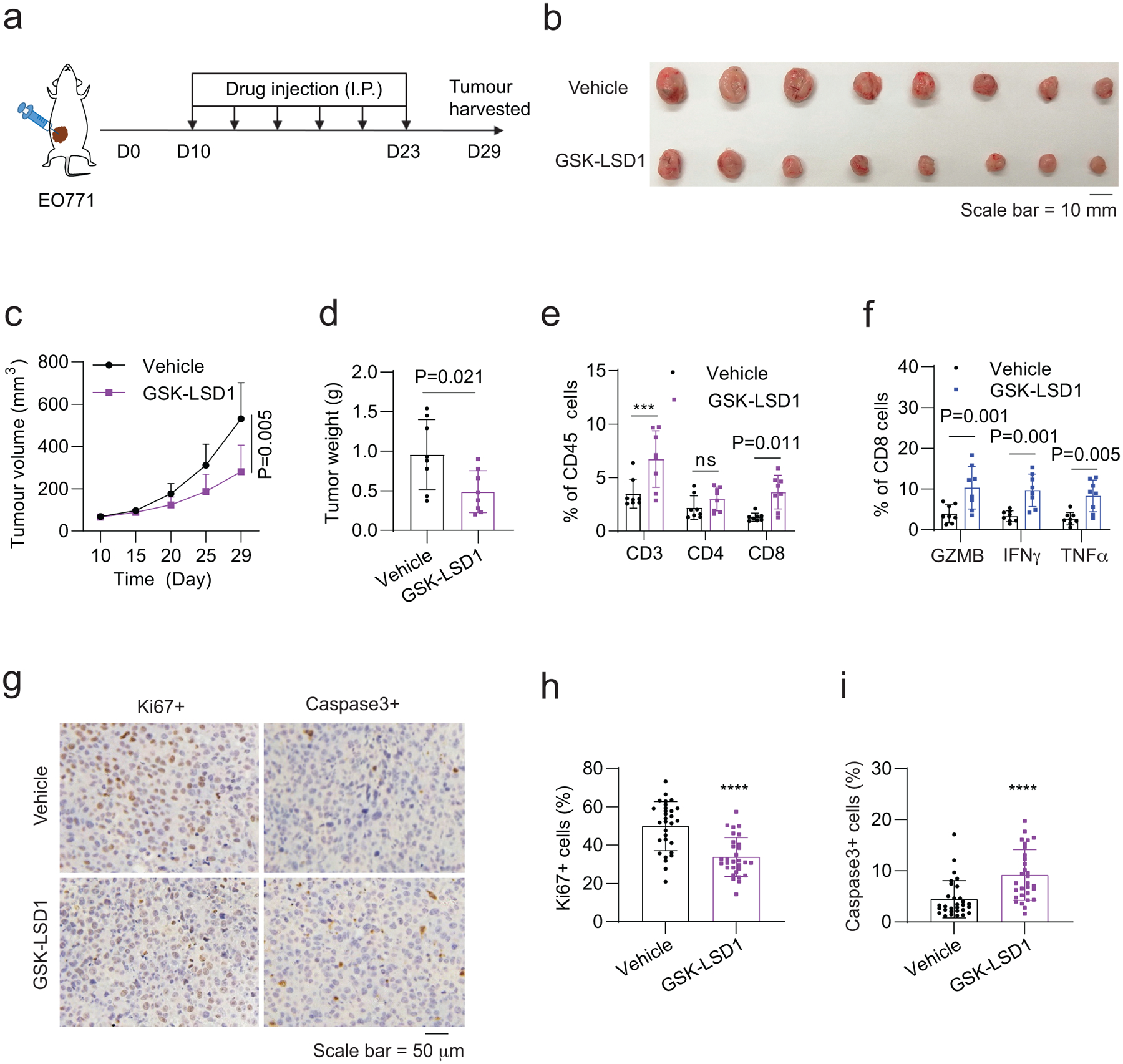
a, Drug-treatment scheme of mouse breast tumour models. b-d, Gross dissected mammary tumour images (b), tumour growth (c) and weight (d) of the EO771 tumours from the tumour-bearing C57BL/6 mice treated with vehicle control, GSK-LSD1 (20 mg kg−1). e, Proportions of total T, CD4+ T, and CD8+ T cells in total immune (CD45+) cells in the EO771 tumours treated with control, GSK-LSD1. f, Percentage of active cells in total CD8+ T cells, indicated by GZMB+, IFNγ+, TNFα+ in flow cytometry analysis. g, IHC staining images of Ki67+ and cleaved Caspase 3+ cells in the tumour tissues. h,i, Quantitative results for (g). For statistical analysis of data, Two-sided Student’s t-test was used in (c,d,h,i) and two-way ANOVA test was used in (e,f). Data are presented as mean ± SD. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
ED Fig. 6. Drug candidates promotes the expression of genes in the antigen presentation of breast tumour cells.
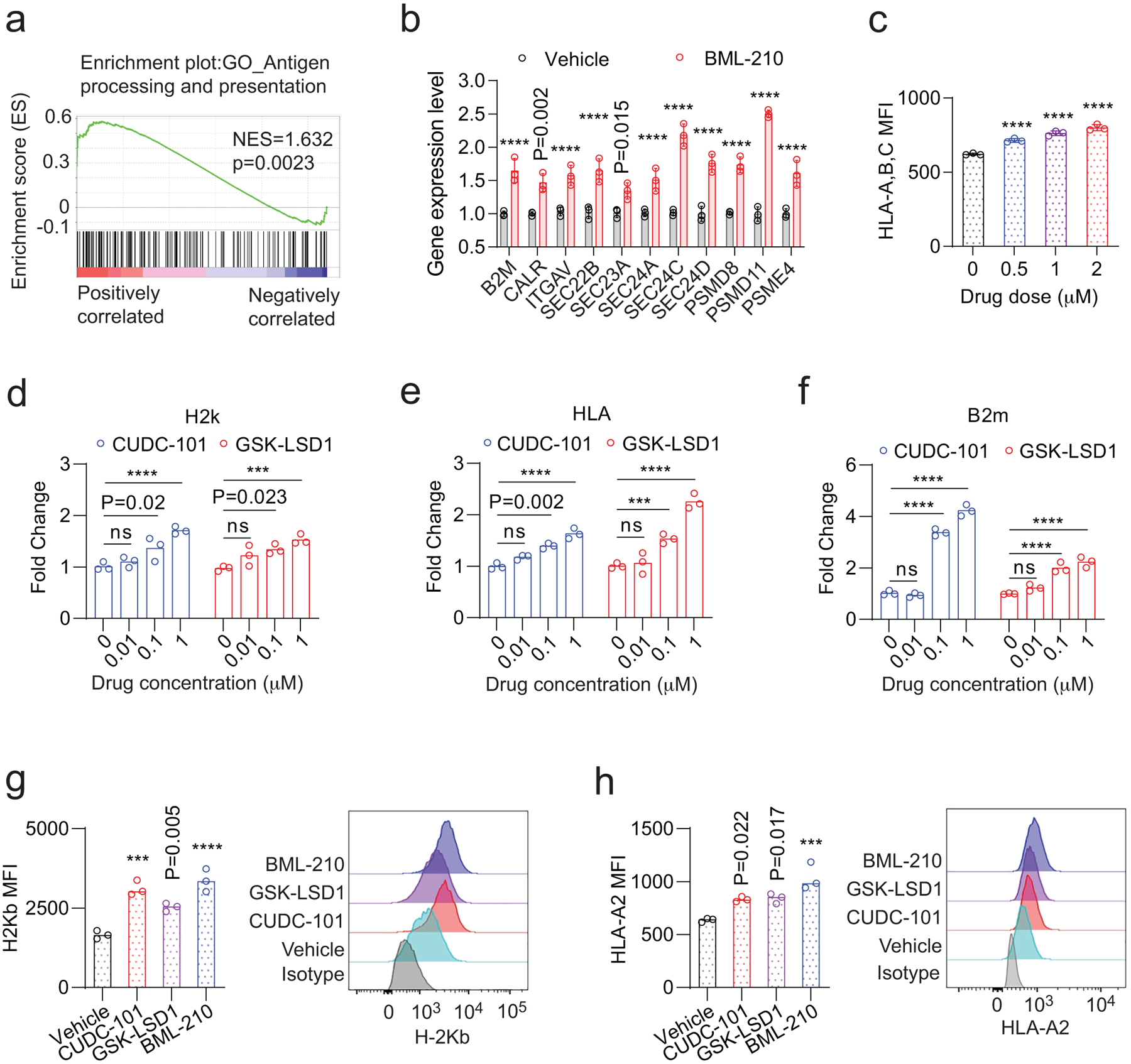
a, Gene-set enrichment plot of the antigen processing and presentation pathway in EO771 cells treated with BML-210 in comparison with the cells treated with vehicle control. b, Validation of up-regulated genes from Fig.6c in human MDA-MB-468 cells treated with vehicle control or BML-210 (1.0 μM) by quantitative RT-PCR. c, Levels of HLA-A,B,C on the NY-ESO-1+ MDA-MB-468 cells treated with control or BML-201, determined by MFI in flow cytometry analysis. d, H-2k expression levels in the OVA+ EO771 cells treated with CUDC-101 or GSK-LSD1 were determined by qRT-PCR. e, HLA gene expression levels in NY-ESO-1+ MDA-MB-468 cells treated with CUDC-101 or GSK-LSD1 were determined by qRT-PCR. f, B2M expression levels in the OVA+ EO771 cells treated with CUDC-101 or GSK-LSD1 were determined by qRT-PCR. g,h, The effect of drug treatment on the H-2Kb and HLA-A2 antigen presentation on OVA+ EO771 cells and NY-ESO-1+ MDA-MB-468 cells, respectively. Data (b,d-f) from 3 biologically parallel experiments were analyzed using Two-way ANOVA. Data (c,g,h) from 3 biologically parallel experiments were analyzed using One-way ANOVA. ***, p < 0.001; ****, p < 0.0001; ns, no significance.
ED Fig. 7. BML-210 promotes the expression of genes in the antigen presentation of breast tumour cells.
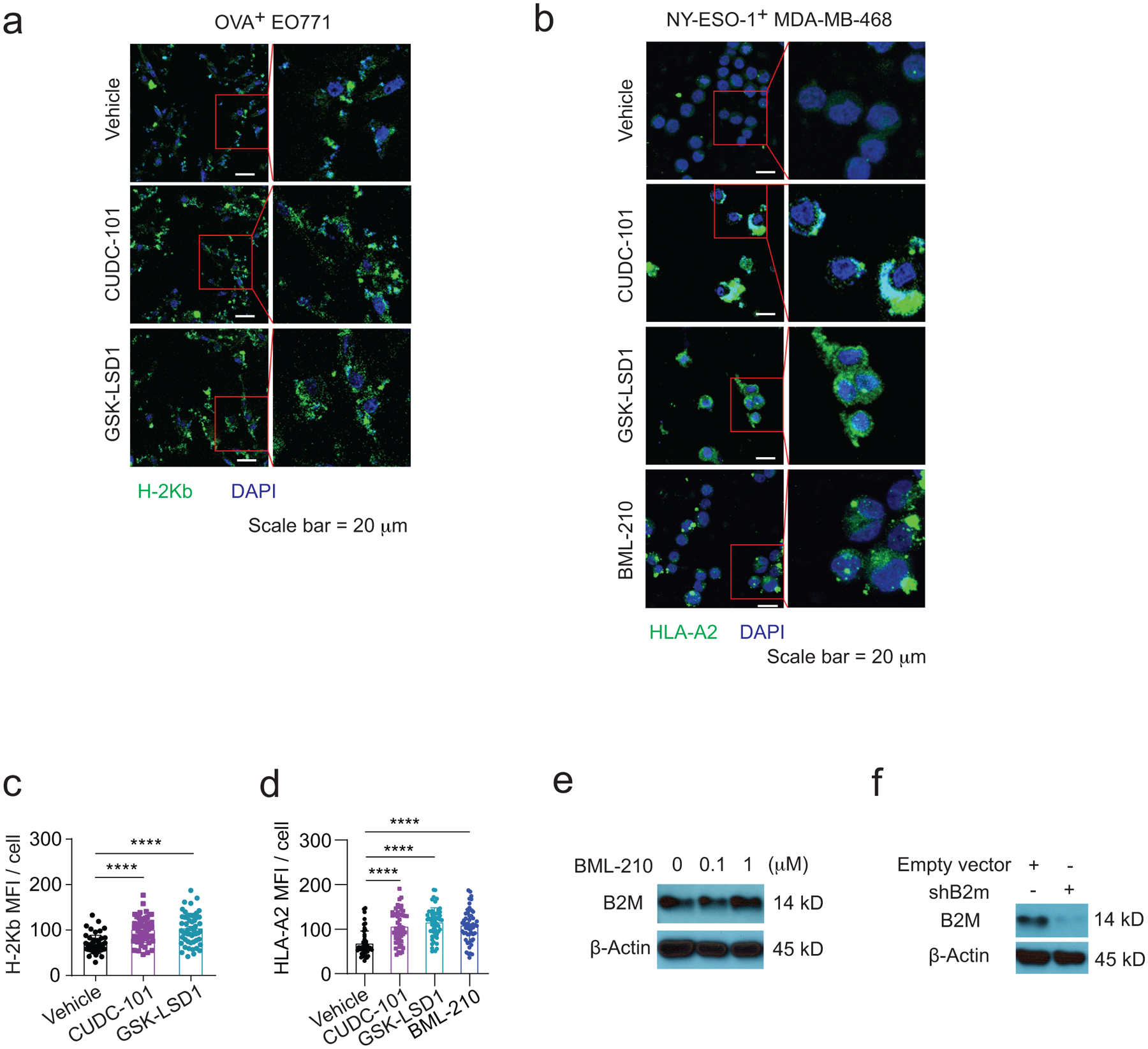
a,b, Confocal images showing H-2Kb and HLA-A2 on OVA+ EO771 cells (a) and NY-ESO-1+ MDA-MB-468 cells (b), respectively. The immunofluorescence images were analyzed by ImageJ. c, Quantitative analysis of confocal images in (a) for assessing H-2Kb antigen presentation. d, Quantitative analysis of confocal images in (b) for assessing HLA-A2 antigen presentation. e, Western blot showing B2M protein expression levels in OVA+ EO771 cells treated with BML-210 drug with 0, 0.1, 1 μM for 48h. f, Western blot showing B2M knockdown in OVA+ EO771 cells. One-way ANOVA test was conducted for statistical analysis in (c,d). Data are presented as mean ± SD. ****, p < 0.0001.
Supplementary Material
Acknowledgements
We thank the Laboratory Animal Resource Center of Indiana University for their technical support in animal studies. We are grateful to the Indiana Center for Biological Microscopy, the Flow Cytometry Resource Facility (FCRF) and the Center for Medical Genomics of Indiana University for use of instruments and technical assistance, and the Indiana University Simon Cancer Center (IUSCC) for providing human breast tissue samples. This work was supported in part by US National Institutes of Health grants R01CA203737 (X.L.), R01CA206366 (X.L. and X.H.), R01CA243023 (X.L. and X.H.), R01CA222251 (X.L.) and by Indiana University Strategic Research Initiative fund (X.L.) and Vera Bradley Foundation for Breast Cancer Research (X.L. and X.Z.).
Footnotes
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Source data for tumour burden are provided with this paper. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request. The RNA sequence data are available from the GEO database under accession number GSE182954.
Competing interests
XL and XZ are submitting a patent for the organoid-based screen method and identified drugs in this study.
References
- 1.Gatti-Mays ME et al. If we build it they will come: targeting the immune response to breast cancer. NPJ Breast Cancer 5, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dijkstra KK et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 174, 1586–1598 e1512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sachs N et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 172, 373–386 e310 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Stevanovic S et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356, 200–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmid P et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Zacharakis N et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24, 724–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim IS et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol 21, 1113–1126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villanueva L, Alvarez-Errico D & Esteller M The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol 41, 676–691 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Romero D HDAC inhibitors tested in phase III trial. Nat Rev Clin Oncol 16, 465 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Sulaiman A et al. Co-inhibition of mTORC1, HDAC and ESR1alpha retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis 9, 815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dravis C et al. Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell 34, 466–482 e466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Abreo N & Adams S Immune-checkpoint inhibition for metastatic triple-negative breast cancer: safety first? Nat Rev Clin Oncol 16, 399–400 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Wein L, Luen SJ, Savas P, Salgado R & Loi S Checkpoint blockade in the treatment of breast cancer: current status and future directions. Br J Cancer 119, 4–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feder-Mengus C, Ghosh S, Reschner A, Martin I & Spagnoli GC New dimensions in tumor immunology: what does 3D culture reveal? Trends Mol Med 14, 333–340 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Friedrich J, Seidel C, Ebner R & Kunz-Schughart LA Spheroid-based drug screen: considerations and practical approach. Nat Protoc 4, 309–324 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Junttila MR & de Sauvage FJ Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Palucka AK & Coussens LM The Basis of Oncoimmunology. Cell 164, 1233–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neal JT et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 175, 1972–1988 e1916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grassi L et al. Organoids as a new model for improving regenerative medicine and cancer personalized therapy in renal diseases. Cell Death Dis 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crespo M et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med 23, 878–884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drost J et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 11, 347–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SH et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 173, 515–528 e517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim M et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat Commun 10, 3991 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broutier L et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nature Medicine 23, 1424-+ (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minton K Studying tumour-specific T cell responses in 3D. Nat Rev Immunol 18, 602–603 (2018). [DOI] [PubMed] [Google Scholar]
- 26.de Souza N A model for tumor-immune interaction. Nat Methods 15, 762 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Cattaneo CM et al. Tumor organoid-T-cell coculture systems. Nat Protoc 15, 15–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cafri G et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun 10, 449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clarke SR et al. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol 78, 110–117 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Rapoport AP et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21, 914–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu R et al. Adoptive T-Cell Therapy Using Autologous Tumor-Infiltrating Lymphocytes for Metastatic Melanoma Current Status and Future Outlook. Cancer J 18, 160–175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banh RS et al. PTP1B controls non-mitochondrial oxygen consumption by regulating RNF213 to promote tumour survival during hypoxia. Nat Cell Biol 18, 803–813 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gogna R, Madan E, Kuppusamy P & Pati U Re-oxygenation causes hypoxic tumor regression through restoration of p53 wild-type conformation and post-translational modifications. Cell Death Dis 3, e286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quail DF & Joyce JA Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joyce JA & Pollard JW Microenvironmental regulation of metastasis. Nat Rev Cancer 9, 239–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivashkiv LB IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol 18, 545–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motyka B et al. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell 103, 491–500 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Croft M The role of TNF superfamily members in T-cell function and diseases. Nature Reviews Immunology 9, 271–285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng W et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563 e519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blum JS, Wearsch PA & Cresswell P Pathways of antigen processing. Annu Rev Immunol 31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leone P et al. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 105, 1172–1187 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Ostrand-Rosenberg S & Fenselau C Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol 200, 422–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sacchi A et al. Myeloid-Derived Suppressor Cells Specifically Suppress IFN-gamma Production and Antitumor Cytotoxic Activity of Vdelta2 T Cells. Front Immunol 9, 1271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray MJ et al. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res 18, 50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gray M, Gong J, Nguyen V, Hutchins J & Freimark B Targeting of phosphatidylserine by monoclonal antibodies augments the activity of immune checkpoint inhibitor PD-1/PD-L1 therapy in murine breast tumors. Cancer Research 76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceccacci E & Minucci S Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br J Cancer 114, 605–611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munster PN et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 104, 1828–1835 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terranova-Barberio M et al. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 8, 114156–114172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phan N et al. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun Biol 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradley CA Gastrointestinal cancer: Organoids predict clinical responses. Nat Rev Gastroenterol Hepatol 15, 189 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Clevers H Modeling Development and Disease with Organoids. Cell 165, 1586–1597 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Kondo J & Inoue M Application of Cancer Organoid Model for Drug Screening and Personalized Therapy. Cells-Basel 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sato T et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Fatehullah A, Tan SH & Barker N Organoids as an in vitro model of human development and disease. Nat Cell Biol 18, 246–254 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Appay V, Douek DC & Price DA CD8+ T cell efficacy in vaccination and disease. Nat Med 14, 623–628 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Qiu GZ et al. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol Sci 38, 669–686 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Lai CJ et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res 70, 3647–3656 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Jayathilaka N et al. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res 40, 5378–5388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banik D, Moufarrij S & Villagra A Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int J Mol Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu H et al. Organoid technology and applications in cancer research. J Hematol Oncol 11, 116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang W et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y et al. Heterozygous deletion of chromosome 17p renders prostate cancer vulnerable to inhibition of RNA polymerase II. Nat Commun 9, 4394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu YH et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520, 697–U286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van der Jeught K et al. ST2 as checkpoint target for colorectal cancer immunotherapy. Jci Insight 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCarthy DJ, Chen Y & Smyth GK Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dennis G Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003). [PubMed] [Google Scholar]
- 70.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gu Z, Eils R & Schlesner M Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.