

Abstract
Objective:
Poly ADP ribose polymerase inhibitors (PARPi) are most effective in BRCA1/2 mutated ovarian tumors. More effective treatments are needed for homologous recombination HR-proficient cancer, including CCNE1 amplified subtypes. We have shown that histone deacetylase inhibitors (HDACi) sensitize HR-proficient ovarian cancer to PARPi. This study aimed to provide complementary preclinical data for investigator-initiated phase 1/2 clinical trial of the combination of olaparib and entinostat in recurrent, HR-proficient ovarian cancer patients.
Methods:
We assessed the in vitro effects of the combination of olaparib and entinostat in SKOV-3, OVCAR-3 and primary cells derived from CCNE1 amplified high grade serous ovarian cancer (HGSOC) patients. We then tested the combination in SKOV-3 xenograft and patient-derived xenograft (PDX) model.
Results:
Entinostat potentiates the effect of olaparib in reducing cell viability and clonogenicity of HR-proficient ovarian cancer cells. The combination reduces peritoneal metastases in SKOV-3 xenograft and prolongs survival in CCNE1 amplified HR-proficient PDX model. Entinostat also enhances olaparib-induced DNA damage. Further, entinostat decreases BRCA1, a key HR repair protein, associated with decreased Ki-67, a proliferation marker, and increased cleaved PARP, a marker of apoptosis. Finally, entinostat perturbs replication fork progression, which increases genome instability.
Conclusion:
Entinostat inhibits HR repair by reducing BRCA1 and slowing replication fork progression, leading to irreparable DNA damage and ultimately cell death. This work provides preclinical support for the clinical trial of the combination of olaparib and entinostat in HR-proficient ovarian cancer and suggests potential benefit even for CCNE1 amplified subtypes.
Keywords: entinostat, olaparib HR-proficient ovarian cancer
Introduction
Poly ADP ribose polymerase inhibitors (PARPi) primarily target PARP1, which is critical for base excision repair of single-strand break DNA damage (1). PARPi also cause PARP1 trapping and PARP1-DNA adducts that indirectly lead to double-stranded breaks (DSBs). Additionally, PARP1 is involved in replication fork protection at sites of DNA repair and thus associated with stalling of replication fork progression (2). Unrepaired single-strand breaks and stalled replication forks eventually cause DSBs, fork collapse, and irreparable DNA damage that leads to cell death. The homologous recombination (HR) DNA repair pathway is an efficient method for repairing DSBs and protecting against replication stress that is regulated by BRCA1, BRCA2 and other HR repair proteins (2). PARPi induce synthetic lethality in cells with BRCA1/2 mutations because they are HR-deficient and unable to efficiently repair damage from DSBs or replication fork stress (1,3). As a result, PARPi confer substantial clinical benefit in women diagnosed with BRCA1/2 mutant ovarian cancers (4–7) but are less effective in HR-proficient ovarian cancers (8–11).
A critical clinical problem is the lack of effective treatment options for women diagnosed with poor prognosis, chemotherapy-resistant subtypes of HR-proficient ovarian cancers, such as those that harbor CCNE1 amplification (8,12–14). Tumors with CCNE1 amplification are associated with intrinsic resistance to platinum-based chemotherapy and are potential biomarkers of poor clinical outcomes (15–17). An emerging approach for treating HR-proficient ovarian cancer is to suppress HR with pharmacologic agents to create a BRCA-like, HR-deficient phenotype and thus induce contextual synthetic lethality in the presence of PARPi (18,19).
Our group has shown that histone deacetylase inhibitors (HDACi) such as suberoylanilide hydroxamic acid (SAHA), romidepsin and panobinostat are synergistic with PARPi in HR-proficient ovarian cancer cells (19–21). Histone deacetylases (HDACs) are enzymes that play a critical role in gene transcription, DNA replication and repair. They exert their action via deacetylation of histones and non-histone proteins (22–26). Specifically, treatment with HDACi that are biased towards class I HDACs (HDAC1/2/3) is associated with defective DNA repair and accumulation of DNA damage in ovarian cancer cells (27). Promising preclinical studies of PARPi-HDACi combinations from our group led to an investigator-initiated phase 1/2 clinical trial (NCT03924245). The underlying rationale for the clinical trial was originally based on our previously published results using pan-HDACi and olaparib (19–21). In designing the trial, we chose entinostat because it is a selective HDAC1/2 inhibitor and has the advantage of fewer toxicities than those associated with pan-HDACi (27,28). In addition, prior clinical trials in ovarian cancer using pan-HDACi showed minimal activity (27).
We hypothesized that entinostat with HDAC1/2 selective bias potentiates the effects of olaparib in HR-proficient ovarian cancer by increasing DSBs, by decreasing HR repair, and by stalling replication fork progression. Because there are no published preclinical data for entinostat and olaparib in ovarian cancer, we initiated parallel preclinical experiments of this drug combination. The complementary data are to refine the use of ki67, BRCA1, γ-H2AX in specimens from the clinical trial as potential biomarkers of the therapeutic efficacy of entinostat and olaparib.
In this study, we used multiple preclinical models of HR-proficient ovarian cancer to show that entinostat enhances the anti-tumor efficacy of olaparib. We also show that entinostat potentiates olaparib-induced DNA damage by repressing HR (measured by a decrease in BRCA1 expression) and by slowing replication fork progression. The cumulative effects leading to cell death are consistent with entinostat creating a BRCA-like, HR-deficient phenotype and contextual synthetic lethality when combined with olaparib. Taken together, these results provide additional preclinical support for the clinical trial of olaparib and entinostat in HR-proficient ovarian cancer and suggest potential benefit even for poor prognosis, chemotherapy-resistant HR-proficient CCNE1 amplified subtypes.
Materials and Methods
Culture of primary and immortalized ovarian cancer cells
HR-proficient (BRCA1/2 wild-type) established epithelial ovarian cancer cell lines SKOV-3 and CCNE1 amplified OVCAR-3 (29) were obtained from American Type Culture Collection and maintained as published (19,20). The cell lines were validated by short tandem repeat analysis and tested to be free of mycoplasma contamination. Following informed patient consent, de-identified, fresh tissue was collected by the University of Kansas Medical Center (KUMC) Biospecimen Repository Core Facility (BRCF) following both institutional review board (IRB) approval and U.S. Common Rule. Short term primary cultures (KU-OC-033697 and KU-OC-031065) were derived from finely dissected tumors confirmed to be HR-proficient high-grade serous ovarian cancer and cultured in McCoy’s 5A medium (Gibco by Life technologies) with 1% Pen-Strep (Gibco by Life technologies) and 20% FBS (fetal bovine serum) (Atlanta biologicals) using standard procedures. Passage 1 and passage 2 (P1 and P2) cells were used for SRB (Sulfordhodamine B) assays. For BLI (bioluminescence imaging), SKOV-3-IP cells were initially obtained from Dr. Gordon Mills (Oregon Health & Science University, Knight Cancer Institute, Portland, Oregon) and transduced with lentiviral particles harboring the Luc2/mCherry gene (30).
Drugs and reagents
Entinostat, olaparib, rucaparib and niraparib were purchased from Selleckchem. All drugs were reconstituted in DMSO to prepare 100 mM stock, from which the desired concentrations were achieved using appropriate media. For in-vivo studies, olaparib and entinostat were dissolved in 2% DMSO combined with 30% PEG-300 in sterile saline. We have abbreviated entinostat as “Ent” and olaparib as “Ola” in figures. All other reagents were obtained from Sigma Aldrich, unless otherwise indicated. All results described are for olaparib, entinostat, or the combination, except for SRB assays using niraparib and rucaparib combinations with entinostat (Supplementary Fig. S1A).
Cell viability and clonogenic assays
Cell viability assays were performed using SRB as described (21,31). Cells were pre-treated with 0.25 μM entinostat or control (media) for 24h, followed by 72h of drug treatment with increasing concentrations of entinostat, olaparib, or the combination. For combination treatments, both drugs were administered to maintain final concentrations. Cell viability (percent compared to control) was quantified using GraphPad Prism. We used the combination index to determine interactions between the drugs, using the Chou-Talalay method (32). Clonogenic survival assays were carried out as described (19). Briefly, 500 SKOV-3 cells were seeded in 6-well plates, cultured overnight and pre-treated for 24h with media, or 0.25 μM entinostat. This was followed by 24h treatment with 0.5 μM entinostat or 10 μM olaparib or the combination of olaparib and entinostat at these concentrations. The drugs were replaced with regular media and cells were allowed to grow for 14 days. Formed colonies were stained with crystal violet and quantified using ImageJ.
Animals
NOD SCID Gamma (NSG) mice (JAX # 005557 - NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) obtained from The Jackson Laboratory were bred and maintained as a colony at the University of Kansas Medical Center. All mice were maintained and handled as per approved Institutional Animal Care and Use Committee guidelines of the University of Kansas Medical Center, Kansas City, KS (2017–2387).
SKOV-3-IP-Luc xenograft BLI:
SKOV-3-IP-Luc (2 ×106) cells in sterile PBS were injected intraperitoneally in 6–8-week-old female NSG mice. Baseline BLI was performed 10 days post injection. Mice were randomized into 4 groups carrying uniform average tumor load. Each group (n = 5 per group) were treated with vehicle (control), olaparib, entinostat or the combination. The drugs were reconstituted using vehicle described in drugs and reagents and administered via oral gavage. The entinostat and combination groups were pre-treated with 15 mg/kg entinostat, while the control and olaparib groups were administered vehicle for one week (M-F). The 4 groups were then treated with their respective drug (15 mg/kg entinostat or 100 mg/kg olaparib or the combination) for two weeks. All mice were monitored daily for adverse reactions and significant changes in body weights. BLI was performed weekly. At the end of treatment, a final BLI was performed and all mice were euthanized and necropsy performed.
Patient derived xenograft (PDX) model survival study:
De-identified, fresh tissue was obtained through the BRCF at KUMC as described for generating the primary cell lines, and PDX models were generated as previously described (33) with slight modification (Supplementary Methods 1). Tumors were immuno-stained for PAX8, p53 and WT1 expression to confirm high-grade serous histology and assayed to determine CCNE1 copy number (Supplementary Methods 2). Mice transplanted with P3 tumors for 4 weeks were randomized based on body weight into 4 treatment groups (n= 5 per group). Mice were pre-treated for one week with either vehicle or 15 mg/kg entinostat, followed by two weeks of treatment with 15 mg/kg entinostat or 100 mg/kg olaparib or the combination. After the completion of treatment, all mice were monitored for advanced disease progression (pallor, lethargy, and/or moderate-to-severe ascites accompanied by reduced grooming behavior) as a surrogate of survival. Median survival was determined using Kaplan-Meyer curves (GraphPad Prism).
Comet assay
Comet assays were performed as described in published literature (34–36). Briefly, cells were seeded in 6-well plates, followed by pre-treatment with vehicle or 0.25 μM entinostat for 24h and a 24h drug treatment 0.5 μM entinostat or 10 μM olaparib as described above. The cells were then trypsinized, washed and processed for comet assay as described (Supplementary Methods 3). For microscopic visualization, slides were stained with SYBR Gold nucleic acid stain (Invitrogen by ThermoFisher Scientific), viewed and imaged using epifluorescence microscopy at 496/522 nm excitation/emission. 40X images were used to determine the tail length using ImageJ software.
Western blot
Cells were pre-treated with vehicle or 0.25 μM entinostat for 24h followed by 72 h drug treatment 0.5 μM entinostat or 10 μM olaparib, then trypsinized, washed and the pellet was dispensed in RIPA lysis buffer (Fisher Scientific) supplemented with protease inhibitor cocktail, phosphatase inhibitor cocktail II, phosphatase inhibitor cocktail III and beta-mercaptoethanol (Bio-Rad) and sonicated followed by centrifugation at 13,000g for 45 min at 4°C. The supernatant was aspirated, and protein quantified using the Bradford assay. The protocol followed was as described previously (19,31) and immunoreactivity on the membrane was detected and analyzed using chemiluminescence and the LICOR analysis system. The primary antibody was a rabbit polyclonal against cleaved PARP that detects PARP1 fragments, but not intact PARP1 (Cell Signaling Technology; 5625; 1:100) for overnight incubation at 4 °C and a mouse monoclonal against β-actin (Life Technologies; MA1–140) was used as loading control.
Immunofluorescence
Cells were seeded on sterile coverslips in 6-well plates, treated, and processed for immunofluorescence using previously published protocols (19,37–39). Briefly, SKOV-3 cells were allowed to attach for 24h, pre-treated and then treated with the drugs for 72h as described above (Western blot). The cells were fixed in 100% methanol for 15 min, permeabilized with 0.1% Triton X-100/PBS (Bio-Rad Laboratories) for 2 min followed by blocking for 1h in 2% BSA/PBS (bovine serum albumin/phosphate buffered saline). The coverslips were incubated with primary antibodies for immunofluorescence (IF) overnight at 4°C in a humidified chamber. The first set of coverslips were stained with mouse anti-γH2Ax (Abcam, ab26350; 1:1000) followed by goat anti-mouse IgG Fab2 Alexa Fluor 555 Conjugate antibody (red; Cell Signaling: 1:500). Mouse anti-BRCA1 (EMD/Millipore; OP92; 1:100) and rabbit anti-Ki-67 (Abcam; ab92742; 1:1000) was used to double stain the second set of coverslips, detected by goat anti-mouse IgG Fab2 Alexa Flour 555 Conjugate antibody (red; Cell Signaling: 1:500) and goat anti-rabbit IgG Fab2 Alexa Fluor 488 Conjugate antibody (green, Cell Signaling: 1:500) respectively. All secondary antibodies were incubated for 1h at room temperature in a humidified chamber. The coverslips were mounted on glass slides (FisherScientific) using ProLong Gold antifade with DAPI (Cell Signaling). The slides were allowed to cure overnight in the dark and visualized using a Nikon 80i fluorescence microscope. For γH2Ax quantification, foci were counted manually to determine the average number of foci per cell. Fluorescence intensity for BRCA1 and Ki67 was determined using ImageJ.
Immunohistochemistry
Tumor tissues were fixed in 4% formalin for 24h followed by rinsing in 70% ethanol and paraffin embedding. Five micron sections were cut for immunostaining using standard methods (31,37). Antibodies used: p53 (Agilent DAKO; IR616 Clone D0–7; pre-diluted), PAX8 (Biocare medical; 901-379-070919; 1:200), WT1 (Cell Marque tissue diagnostics; 6F-H2; 1:50).
Quantitative RT-PCR
Total RNA was isolated from cells using Qiagen RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA (0.5 μg) from each sample, was transcribed to cDNA using the QuantiTect Reverse Transcription kit (Qiagen) as per the manufacturer’s instructions. Further, cDNA was diluted 1:4 with RNase-Free water and the PCR mix [TaqMan Master Mix (2x) (Thermo Fisher), 20x FAM TaqMan assay (Thermo Fisher) and 20x VIC TaqMan assay (Thermo Fisher)]. The TaqMan assays used were BRCA1 (FAM, Thermo Fisher), and the internal control 18S (VIC, Thermo Fisher). The cDNA and PCR mix were loaded in a 384-well plate and read by a ViiA 7 Real-Time PCR system (Thermo Fisher). Gene expression was determined using delta delta CT method.
DNA Fiber assay
DNA fiber assay was performed using published protocols (40–42) and as described (Supplementary Methods 4). Briefly, SKOV-3 cells were pulse-labeled with IdU followed by CldU. For treatment groups, the cells were treated with olaparib, entinostat or the combination (see Supplementary Methods). The drugs were maintained in media during the entire labeling period. The cells were harvested, washed, pelleted and 2 μL of cells were mixed with 6 μL of lysis buffer on top of a positively charged glass slide. Slides were tilted at a 20–45° angle to spread the fibers. DNA fibers were immuno-stained with rat anti-BrdU and mouse-anti-BrdU for 1.5h at RT (room temperature), washed and then incubated with anti-rat Alexa Fluor 488 and anti-mouse Alexa Fluor 546 respectively, for 1h at RT. The slides were then mounted with prolong gold antifade reagent (ThermoFisher Scientific, P36930). Images were acquired with red and green filters on a fluorescent microscope using the 63X objective (LEICA DMU 4000B; 63X/1.40–0.60 NA oil). At least 15 images were taken and at least 200 individual tracts were scored for each dataset. The length of each tract was measured manually using the segmented line tool on ImageJ software (NIH). Statistical differences in DNA fiber tract lengths were determined by Kruskal-Wallis test followed by Dunn’s multiple comparison test.
Statistical Analysis
Statistical analysis was performed using ANOVA, T-tests or other multiple comparison tests using GraphPad Prism software as described in figure legends. p<0.05 was considered statistically significant.
Results
Entinostat potentiates the effects of olaparib in reducing cell viability and clonogenic survival in HR-proficient ovarian cancer cells.
We tested the effects of olaparib and entinostat in established (SKOV-3 and OVCAR-3) and primary (KU-OC-033697 and KU-OC-031065) HR-proficient ovarian cancer cells. To mimic the clinical trial, cells were pre-treated with low-dose entinostat for 24h, followed by 72h treatment with olaparib and entinostat alone or combined. Olaparib and entinostat as single agents had limited efficacy on cell viability in the HR-proficient ovarian cancer cell lines (Fig. 1A). However, entinostat combined with olaparib significantly reduced cell viability. All combinations between olaparib and entinostat (except two lower doses in OVCAR-3) showed synergy (CI<1.0). The drug combination showed strong synergy (CI<0.3) across all cell lines, particularly SKOV-3. To further assess the effect of the combination of olaparib and entinostat in SKOV-3 cells, we performed clonogenic survival assays and found the combination significantly reduced colony formation compared to control or each drug alone (Fig. 1B).
Figure 1. Olaparib combined with entinostat decreases viability and clonogenicity in HR-proficient ovarian cancer cells.
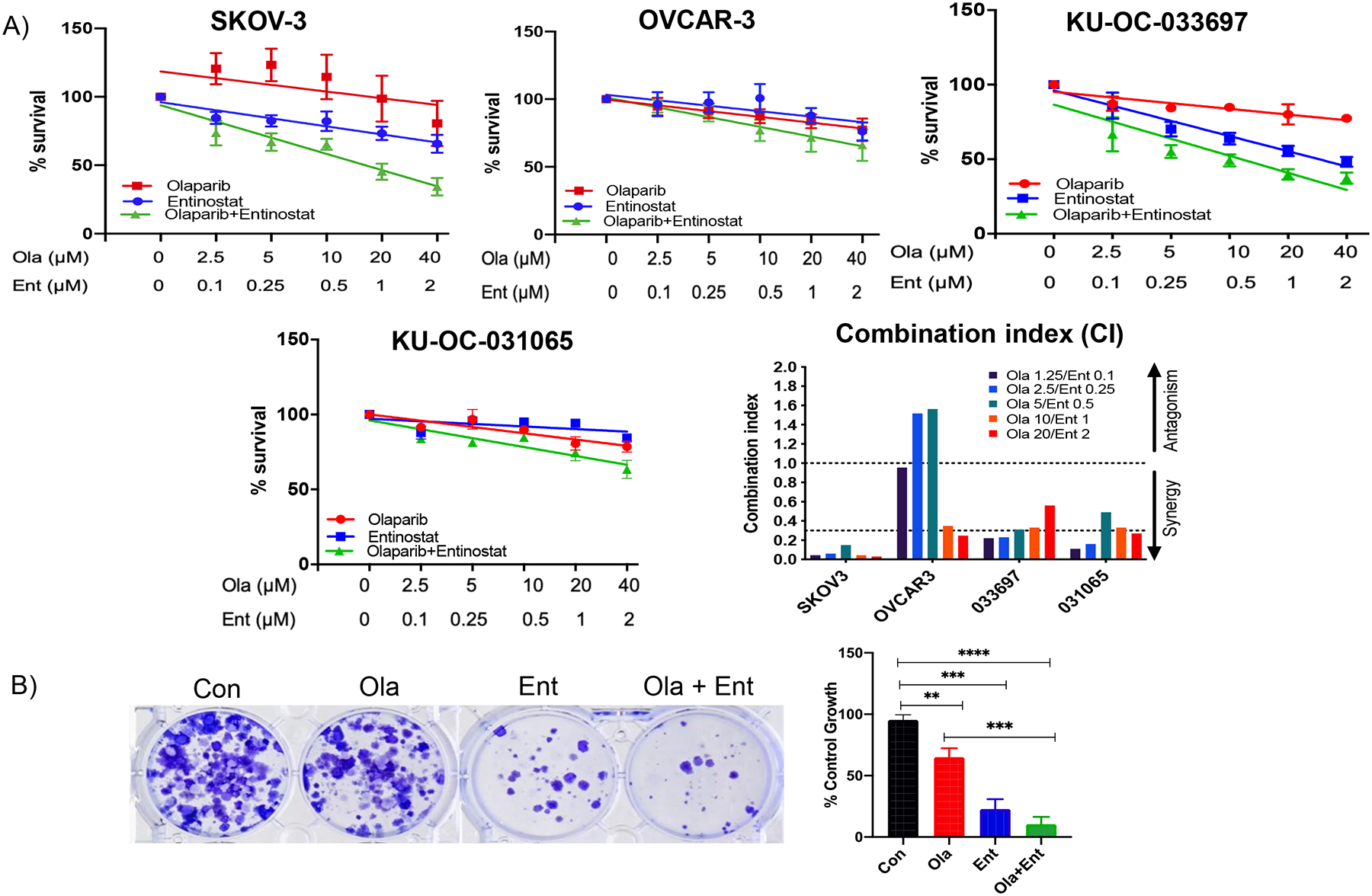
(A) Cell proliferation and combination index: Cells (SKOV-3, OVCAR-3, patient derived primary cells) were pre-treated with 0.25 μM entinostat or media for 24 h followed by 0.1, 0.25, 0.5, 1 or 2 μM Ent and/or 2.5, 5, 10, 20 or 40 μM Ola (alone or combination) for 72 h followed by SRB assay to measure cell proliferation. All cell lines and primary cells showed enhanced inhibition of cell viability in Ola+Ent treated groups compared to Ola or Ent alone. Combination index (CI) showed synergy (CI value <1) between Ent and Ola at all concentrations, in all cell lines tested, except two earlier concentrations for OVCAR-3. Strong synergy was seen at all concentrations in SKOV-3 cells and several combinations in other cells (CI value <0.3). CI was calculated by Chou-Talalay method. (B) Clonogenicity: 500 SKOV-3 cells/well were plated overnight on 6 well plates. Cells were pre-treated with 0.25 μM Ent for 24 h and subsequently treated with 0.5 μM Ent or 10 μM Ola for 24 h. Drugs were replaced by regular media, cells allowed to grow until colonies formed and stained with crystal violet using standard procedure. Quantification of clonogenic data was carried out using ImageJ and graph plotted for each treatment as percent of control. Difference between control and each treatment was significant, with larger difference between Con and Ola+Ent. **p < 0.001; ***p<0.0001; ****p<0.00001; determined by one-way ANOVA, Sidak’s multiple comparisons. [Con – Control; Ola – Olaparib; Ent – Entinostat].
Several PARPi have been approved by the FDA for the treatment of ovarian cancer but have varying efficacy (43–45). Therefore, we tested the effects of entinostat in combination with the other FDA-approved PARPi, niraparib and rucaparib, in SKOV-3 cells. Niraparib, which has stronger PARP trapping activity than both olaparib and rucaparib (46–48) showed the strongest single agent effect, which was potentiated by entinostat (Fig. S1A). The effects of rucaparib, which by itself caused negligible effect on viability, were enhanced by entinostat (Fig. S1B). This suggests that entinostat potentiates the efficacy of PARPi as a class of drugs.
The combination of olaparib and entinostat reduces peritoneal spread in an HR-proficient xenograft model and prolongs survival in a CCNE1 amplified HR-proficient PDX model.
To test the effects of olaparib and entinostat in vivo, we used the SKOV3-IP-luc model. This model of HR-proficient ovarian cancer recapitulates peritoneal metastases consistent with human ovarian cancer and allows for non-invasive visualization of peritoneal tumor progression using BLI. Similar to the clinical trial, mice were pre-treated with entinostat, followed by treatment with olaparib, entinostat, or the combination (Fig. 2A). Serial BLI measurements were obtained weekly during treatment. At the end of treatment, BLI measurements were compared to baseline for each mouse, and as treatment groups, compared to vehicle controls. Peritoneal metastases, measured by BLI, were significantly reduced in mice treated with the combination of olaparib and entinostat compared to vehicle controls (Fig. 2B). All mice were monitored on a regular basis and as tumor burden progressed, the mice were monitored daily for toxicity-induced behavioral changes such as body weight change, food and water intake, reflex, and grooming habits. During necropsy we did not detect any liver, kidney, intestine, or bowel abnormalities. Therefore, we did not perform microscopic tissue evaluation. All treatments were well tolerated, with no significant variation in behavior or body weights (Fig. S2A). These results are comparable to our previous results showing that panobinostat and olaparib did not cause any obvious organ toxicity (19)
Figure 2. Olaparib combined with entinostat decreases peritoneal tumor load in HR-proficient SKOV-3-IP-luc xenograft and increases survival in CCNE1 amplified HGSOC HR-proficient PDX mice.
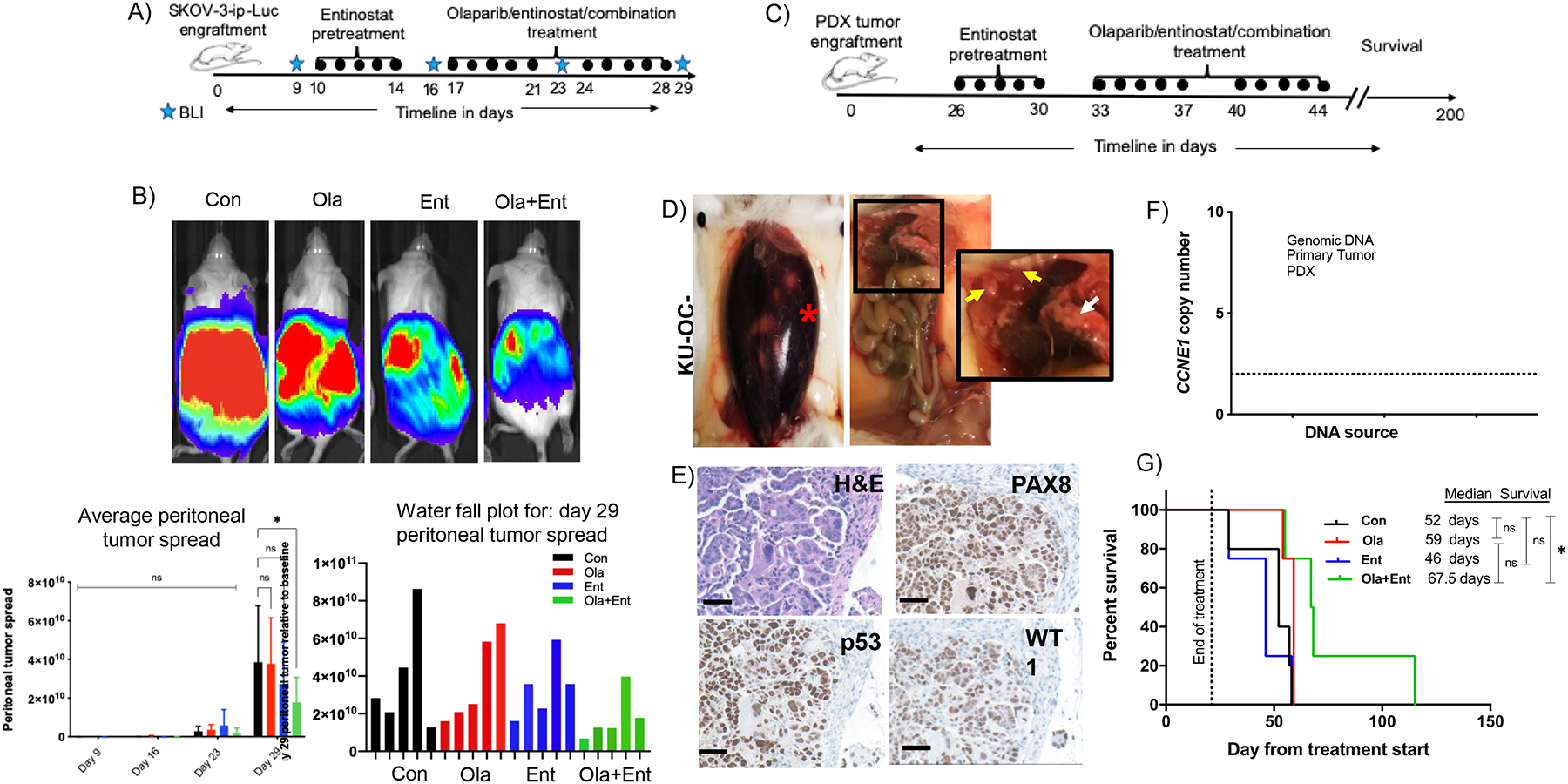
(A) SKOV-3 xenograft treatment timeline. (B) SKOV-3-xenograft in NOD-SCID mouse model: Bioluminescent imaging showing peritoneal tumor spread in four treatment groups (Control, 100 mg/kg olaparib, 15 mg/kg entinostat, combination of olaparib+entinostat) on day 29. Total flux value (indicative of peritoneal tumor spread) was plotted for each treatment group at weekly intervals and water fall plot for day 29 shows slower tumor progression in combination treatment group. Statistical significance determined using 2-way ANOVA multiple comparisons test (*p < 0.0170). (C) PDX induction and treatment timeline. (D) Large amount of ascites (red asterisk) and tumor nodules around liver yellow arrows) and omentum (white arrow) developed in NOD-SCID mice injected intraperitoneally with patient derived tumors. (E) H&E staining of tumor section and immunohistochemistry showing positive expression of p53, PAX8 and WT1, indicative of high-grade serous carcinoma (20X, 100micron scale bar). (F) Copy caller assay indicating the tumor was CCNE1 amplified. (G) PDX mouse model pre-treated with 15 mg/kg entinostat or vehicle for one week, followed by two-week treatment with vehicle, 100 mg/kg olaparib or 15 mg/kg entinostat or olaparib+entinostat showed that treatment with combination significantly improved the median survival (*p<0.0325, Log-rank (Mantel-Cox) test). [Con – Control; Ola – Olaparib; Ent – Entinostat].
To simulate the patient population represented in the clinical trial of entinostat and olaparib, we generated a PDX model of HR-proficient high-grade serous ovarian cancer (KU-OC-033697) (Fig. 2C–2D). High-grade serous tumors were confirmed by histological appearance and by immunostaining for PAX8, p53 and WT1 (Fig. 2E). The patient had clinically defined HR-proficient (germline BRCA1/2 wild-type) and platinum-resistant disease, and passaged tumors harbored CCNE1 amplifications (Fig. 2F). At the end of treatment, the mice were monitored closely for disease progression and survival. The combination of olaparib and entinostat significantly prolonged the median survival (67.5 days vs 52 days) compared to vehicle alone (Fig. 2G). Two mice from the combination treatment group survived greater than 100 days. There was no significant difference in median survival in mice treated with olaparib (59 days) or entinostat (46 days) compared to vehicle-treated mice. Similar to the SKOV-3-IP-luc model, treatments were well tolerated with no significant variation in body weights (Fig. S2B) and no signs of organ toxicity was noted at necropsy.
Entinostat potentiates olaparib-induced DNA damage in HR-proficient cells.
We have previously shown that HDACi upregulate the expression of γ-H2AX (a marker of DSBs), which when prolonged, is associated with DNA damage-induced cell death (19,49) Thus, γ-H2AX could serve as a potential marker of response to PARPi-HDACi combinations and is being tested as a marker in the clinical trial of olaparib and entinostat. In this study, we observed a robust increase in the number of γ-H2AX foci in SKOV-3 ovarian cancer cells treated with the combination of olaparib and entinostat compared to cells treated with each drug alone (Fig. 3A).
Figure 3. Olaparib combined with entinostat causes significant DNA damage in HR-proficient ovarian cancer cells.
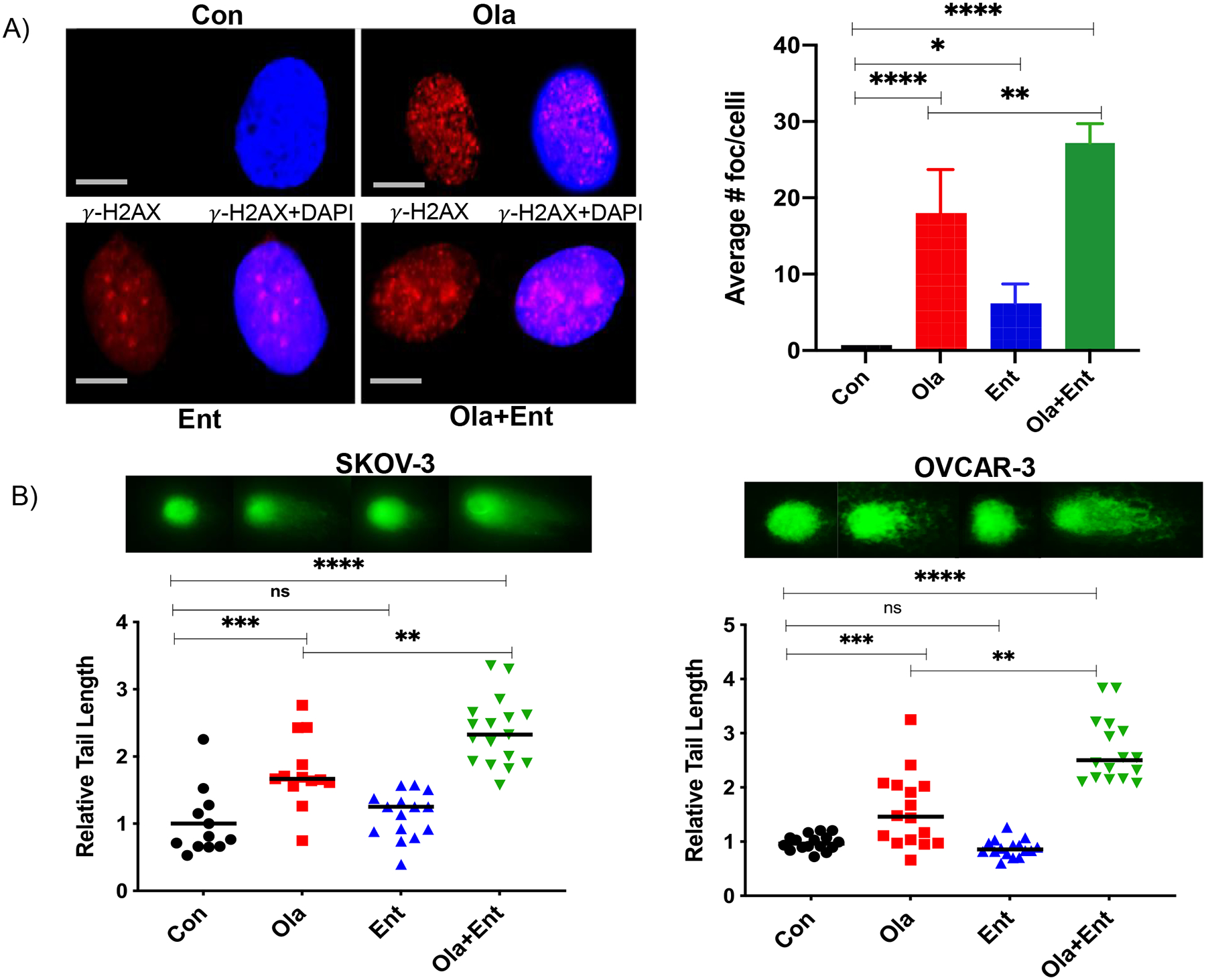
(A) γ-H2AX foci localization in SKOV-3 cells showed high number of foci in Ola treated cells, whereas merged foci were seen in Ola+Ent treated cells, indicative of extensive DNA damage. Statistical analysis showed enhanced foci in Ola+Ent (60X, 50-micron scale bar; *p<0.01; **p< 0.001; ***p,0.001; ****p<0.0001; one-way ANOVA; Sidak’s multiple comparison tests). (B) HR-proficient ovarian cancer cells pre-treated with 0.25 μM Ent for 24 h followed by 0.5 μM Ent or 10 μM Ola alone or Ola+Ent for 24 h were processed for comet assay to determine DNA damage. Both SKOV-3 and OVCAR-3 showed significant DNA damage with Ola and even greater extent of DNA damage with Ola+Ent (*p<0.01; **p< 0.001; ***p,0.001; ****p<0.0001; one-way ANOVA; Sidak’s multiple comparison tests). Fluorescent images show the classic comet tails indicative of DNA damage and the graphs show the relative length of comet tails for individual cells. [Con – Control; Ola – Olaparib; Ent – Entinostat].
To measure DNA damage, we performed a comet assay in SKOV-3 and OVCAR-3 ovarian cancer cells using published protocols (34–36). Relative tail length (a measurement of total DNA damage per cell) was significantly increased in cells treated with the combination of olaparib and entinostat compared to controls and to each drug alone (Fig. 3B). This confirmed that the combination of olaparib and entinostat caused significantly more DNA damage than olaparib alone. Notably, entinostat caused no DNA damage by itself, but potentiated olaparib’s DNA damaging efficacy significantly.
Entinostat suppresses BRCA1 expression and slows replication fork progression in HR-proficient cells.
Our group and others have demonstrated that HDACi reduce HR repair through suppression of HR genes, which in turn contributes to the accumulation of DSBs, irreparable DNA damage, and ultimately, cell death (19,20,50–52). Therefore, we measured the effects of entinostat on BRCA1 levels, which is a critical protein and surrogate marker for HR repair. As expected, immunofluorescence (IF) analysis following treatment with entinostat (alone and combined with olaparib) significantly reduced BRCA1 expression levels (Fig. 4A). Consistent with the cell viability and clonogenic survival assays, IF staining of Ki-67, a marker of cell proliferation, was significantly reduced in cells treated with the combination of olaparib and entinostat (Fig. 4A).
Figure 4. Olaparib combined with entinostat decreases proliferation, alters HR machinery and increases apoptosis in HR-proficient SKOV-3 cells.
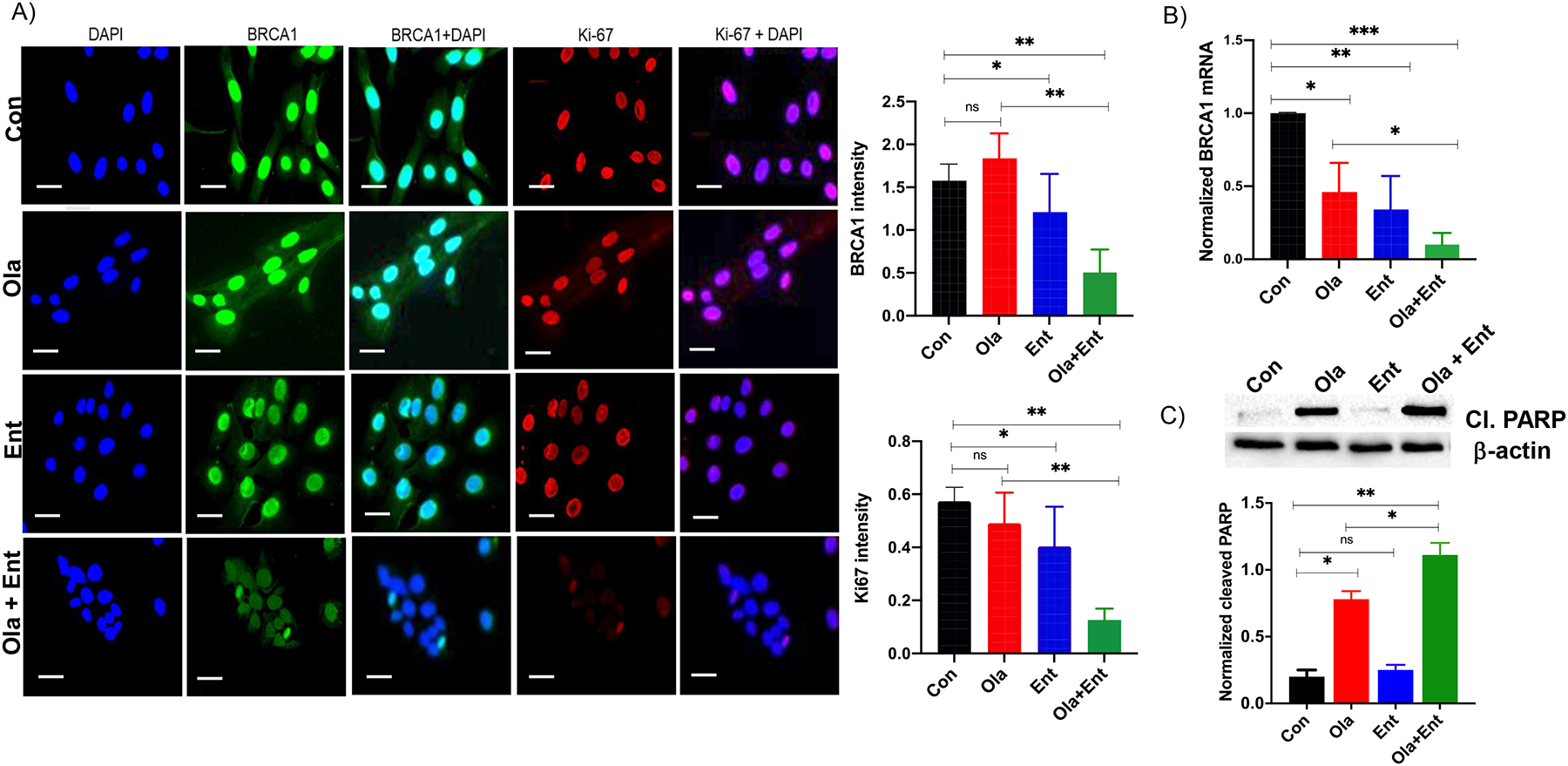
(A). Immunofluorescence showing that Ola+Ent significantly reduces BRCA1 and Ki-67 expression levels in SKOV-3 cells. DAPI was used as nuclear stain (40X 100 micron scale bar). Quantification of BRCA1 and Ki-67 indicated significant downregulation in cells treated with Ola+Ent (*p < 0.05, **p<0.005 Students T-test). (B) BRCA1 transcript levels showed significant downregulation in all treatment groups, with enhanced downregulation in Ola+Ent (*p < 0.012; **p<0.0036; ***p<0.0005, one-way ANOVA; Sidak’s multiple comparison test) indicating that BRCA1 expression is affected markedly at the mRNA levels compared to protein levels. (C) Western blot analysis of SKOV-3 cells treated with Ola+Ent as described for SRB assay showing increased cleaved PARP expression in olaparib and Ola+Ent treated cells. Quantification showed significant upregulation of cleaved PARP, a marker of cell death (*p< 0.05; **p<0.005, Students T-test). The antibody used was specific for fragments of PARP1. [Con – Control; Ola – Olaparib; Ent – Entinostat].
BRCA1 gene expression levels, measured by qPCR, was significantly suppressed in cells treated with the combination of drugs (Fig. 4B). We also measured cleaved PARP, a marker of apoptosis, by Western blot. Entinostat potentiated the effects of olaparib-induced apoptosis as observed by significantly enhanced level of cleaved PARP expression in combination compared to olaparib alone. (Fig. 4C), consistent with results showing enhanced suppression of HR repair, increased DNA damage, and reduced cell viability. The HR pathway is involved in replication fork protection through BRCA1/2 and RAD51 (53,54). Thus, a loss in BRCA1 function suppresses replication fork progression and enhances olaparib-induced DNA damage in cells with BRCA1 mutations (55–57). Because entinostat suppresses BRCA1, we tested the effects of entinostat on replication fork progression using DNA fiber assays. As predicted, entinostat, alone or combined with olaparib, significantly reduced replication fork progression compared to controls, whereas olaparib had no effect (Fig. 5A–5B).
Figure 5. Entinostat slows replication fork progression in HR-proficient SKOV-3 cells.
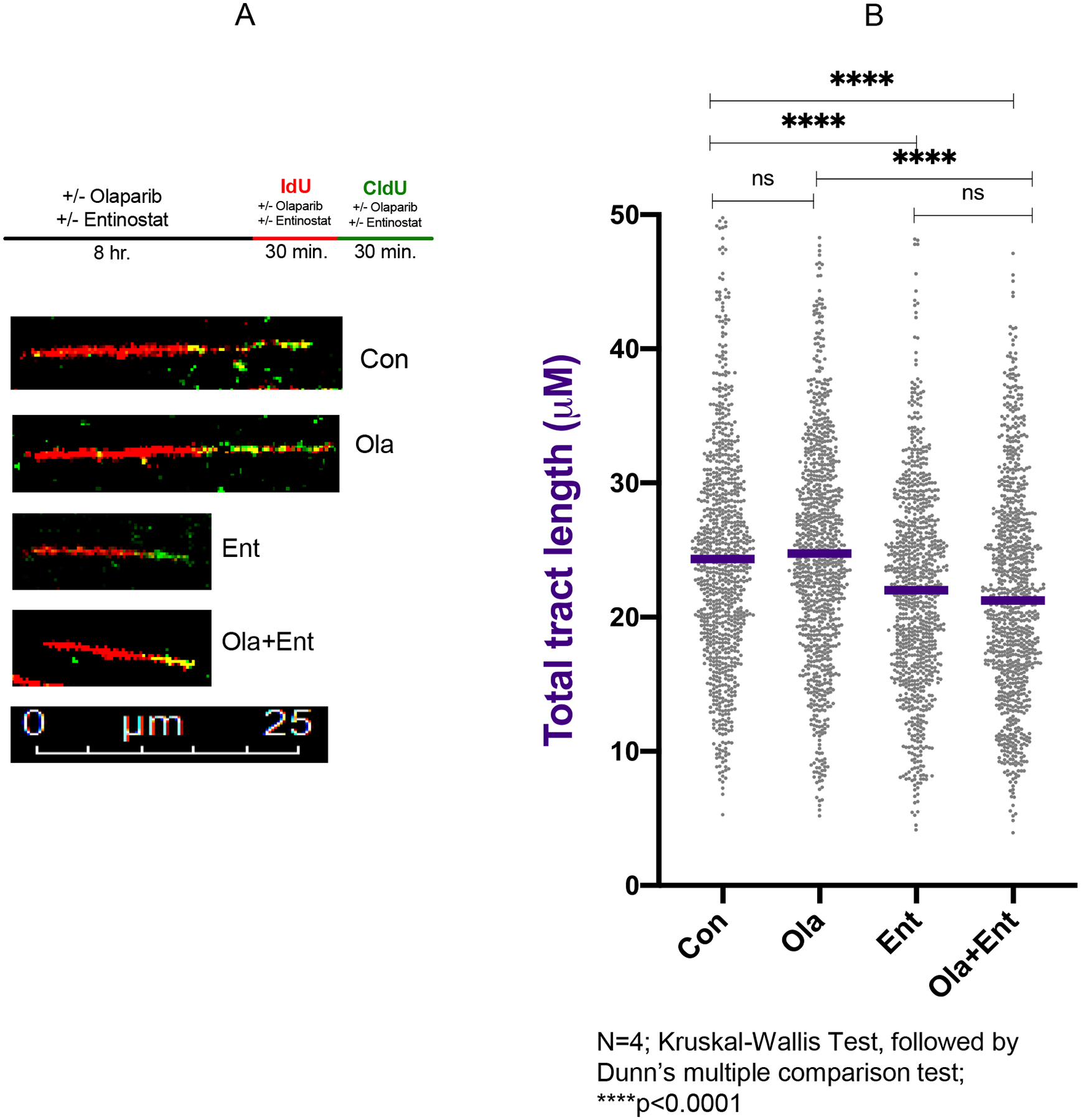
A) Experimental scheme for DNA fiber assays and representative images for each condition. B) Dot plot and median values (purple line) of total tract lengths (IdU+CldU) upon mock treatment or treatment with Ola (10 μM), Ent(0.5 μM), or combination of Ola+Ent. Ent and Ola+Ent combination treatment lead to decreased fork progression relative to Con and Ola treated SKOV-3 cells (n=4; ****P<0.0001; Kruskal-Wallis test followed by Dunn’s multiple comparison test). [Con – Control; Ola – Olaparib; Ent – Entinostat].
Discussion
PARPi cause synthetic lethality and confer substantial clinical benefit in HR-deficient BRCA1/2 mutated ovarian cancer (4,6,50). New treatments are needed to improve the efficacy of PARPi in HR-proficient ovarian cancer. In this study, we show that entinostat can be used to convert HR-proficient ovarian cancer cells to BRCA-like, HR-deficient phenotypes by causing contextual synthetic lethality in the presence of olaparib. We have shown that entinostat enhances the anti-tumor efficacy of olaparib in vitro and in vivo. In addition, entinostat potentiates olaparib-induced DNA damage, measured by γ-H2AX and comet assays, in part by repressing BRCA1 expression and replication fork progression (both surrogates of HR repair). These results support our hypothesis that entinostat potentiates the effects of olaparib in HR-proficient ovarian cancer by increasing olaparib-induced DSBs through decreased HR gene expression and perturbed replication fork progression, which leads to irreparable DSBs and ultimately, cell death (Fig. 6). Reduced fork progression could be due to HR-deficient cells removing DNA damage at replication forks less efficiently or due to the loss of BRCA1 reducing protection of replication forks from nuclease activity. Moreover, the collision of replication forks with the incoming DNA damage or cleavage of the forks by junction-specific nucleases can lead to increased DSB accumulation and genome instability (58,59). The events leading to and following slower replication fork progression will be explored in future experiments.
Figure 6. Graphical summary indicating the mechanism for entinostat-induced olaparib sensitivity in HR-proficient cells.
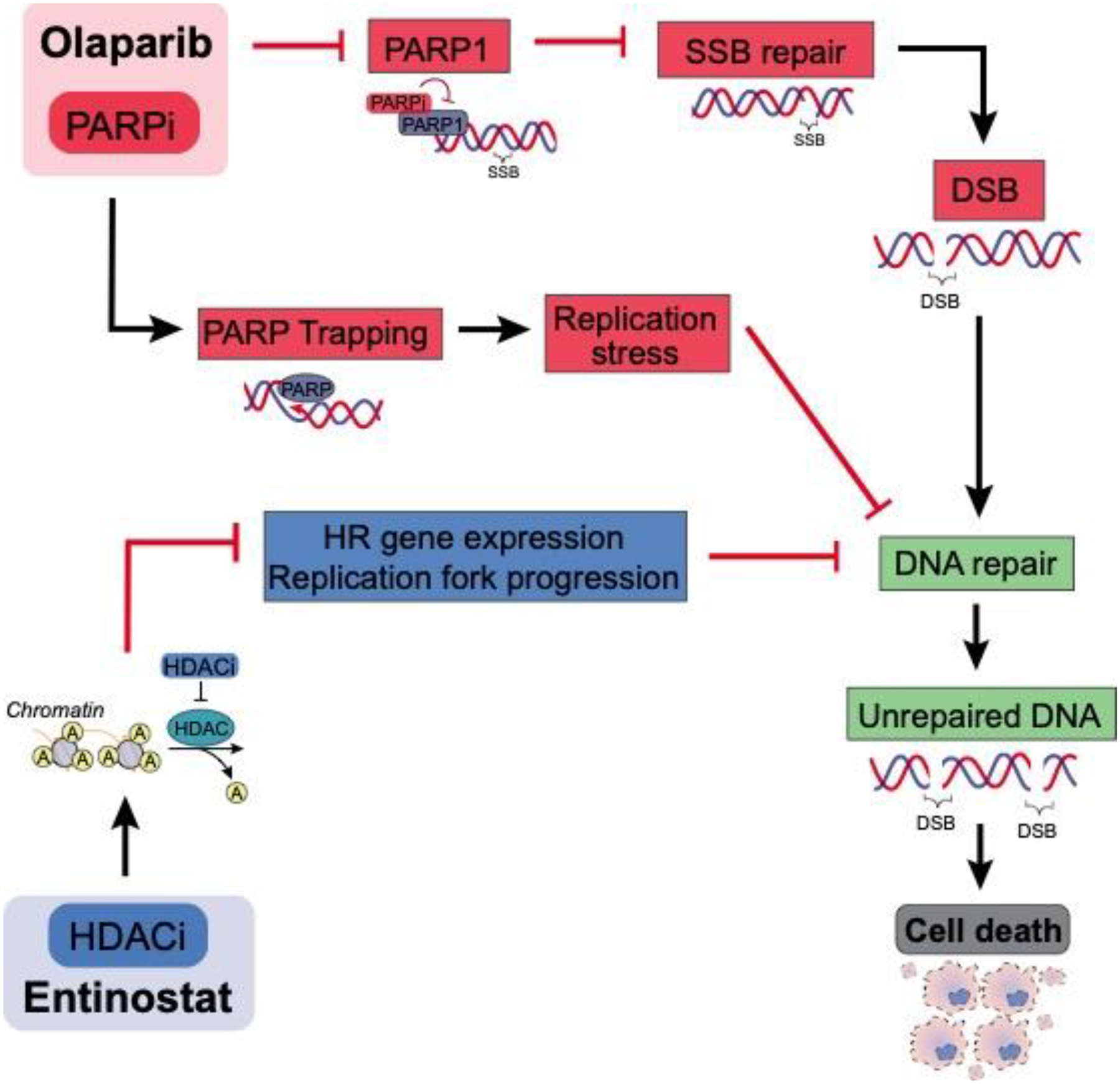
As described, entinostat causes breakdown of HR-DNA repair pathway, whereas olaparib inactivates PARP, crippling the cell’s potential to repair SSBs. This in turn leads to accumulation of DSBs, which the cell is unable to repair due to defective HR repair pathway, accumulating unrepaired DNA ultimately resulting in cell death.
The main strength of this study is the use of multiple preclinical models of HR-proficient ovarian cancer, including SKOV-3, OVCAR-3 and primary cultures of patient derived high-grade serous ovarian cancer cells. In addition, the use of primary cancer cell lines could serve as rapid personalized models to predict the efficacy of drug combinations in parallel with clinical investigation. Although SKOV-3 cells do not represent high-grade serous ovarian cancer, these results suggest that the combination of olaparib and entinostat could be used to treat other types of HR-proficient epithelial ovarian cancer, for example, clear cell. The effects seen in high-grade serous HR-proficient CCNE1 amplified OVCAR-3 and primary ovarian cancer cells that were validated in the PDX model are encouraging because CCNE1 amplified tumors have a poor prognosis and are a chemotherapy-resistant subset of HR-proficient tumors (14). More importantly, suppression of BRCA1 pharmacologically or by siRNA knock down is synthetically lethal in CCNE1 gain or amplified tumors (18–21,60,61). We have shown that CCNE1/cyclin E protein expression is associated with platinum resistance and thus, could be used as a molecular biomarker for tumors with poor prognosis (14). Therefore, we will stratify patients with HR-proficient tumors enrolled in the clinical trial of entinostat and olaparib by CCNE1 amplification status. Our mouse studies also suggest that this drug combination is well tolerated. We closely monitored behavioral changes throughout the study and found no obvious toxic effects of the drugs on mouse behavior, mouse weights, or gross inspection of organs.
Additional mechanistic studies are needed to understand the precise role of HDACi in potentiating the effects of PARPi. The sensitivity of BRCA-deficient tumors to PARPi may involve single stranded DNA replication gaps rather than unrepaired DSBs (62). Inhibition of Class I HDACs is associated with DNA damage through repression of DNA repair and replication (63–65). However, the relationship between PARP1 and HDACs 1/2/3 is an area of ongoing investigation. Precise mechanisms and differences in the suppression of BRCA1 at translational and transcriptional levels suggest cooperative effects of HDACi and PARPi on promoter and enhancer activity (unpublished data). We are also investigating the role of HDACi-PARPi combinations on the tumor microenvironment in HR-proficient ovarian cancer.
Since there are no other clinical trials using the combination of olaparib and entinostat at this time (66), these results along with our ongoing clinical trial have potential implications for PARPi resistant tumors and for other cancers. Finally, these results provide additional preclinical support for the clinical trial of entinostat and olaparib in HR-proficient ovarian cancer and suggest potential benefit even for poor prognosis, chemotherapy-resistant, HR-proficient, CCNE1 amplified subtypes.
Supplementary Material
Research highlights.
Entinostat, a selective HDAC1/2 inhibitor enhances olaparib efficacy in preclinical models of HR-proficient ovarian cancer.
Entinostat combined with olaparib decreases proliferation and clonogenicity in HR-proficient ovarian cancer cells.
Entinostat combined with olaparib significantly decreases peritoneal tumor spread in SKOV-3 xenograft mouse model.
Entinostat combined with olaparib significantly improves survival in CCNE1 amplified HGSOC PDX model.
Entinostat combined with olaparib increases DNA damage and decreases HR repair in HR-proficient ovarian cancer cells.
Acknowledgments
This research was funded in part by NCI R01CA243511(DK), NCI R21CA210210(DK), NCI P30CA168524(DK), NIGMS P20 GM130423(AKG), and a gift from the Janet Burros Memorial Foundation (AKG). SP was supported by the Ovarian Cancer Research Alliance Ann and Sol Schreiber Mentored Investigator Award (600245). The work in AV lab is supported by NIH grant R01CA237263, by DOD BRCP Expansion Award BC191374, and by the Siteman Cancer Center at Washington University in St. Louis. EC is supported by the NCI fellowship F30CA254215. Graphical summary (Fig. 6) was created with help from InPrint at Washington University in St. Louis. Primary Illustrator Astrid Rodriguez Velez, and Advising designer Amanda Dicks.
Conflict of interest/Disclosures:
Dr. Khabele: Grant support from Astra-Zeneca and Deciphera, and speaker’s Bureau for Astra Zeneca. Dr. Crispens: Grant support from Astra-Zeneca. Dr. Fuh: Grant support from Merck and is on advisory board of Genentech, Immunogen, GSK, Myriad and Aravive. Dr. Godwin: Co-founder of Sinochips Diagnostics and received support from Biovica and VITRAC Therapeutics, LLC. Remaining authors have no COI or disclosures.
References
- 1.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuh K, Mullen M, Blachut B, Stover E, Konstantinopoulos P, Liu J, et al. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol Oncol 2020 [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps17. [DOI] [PubMed] [Google Scholar]
- 4.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012;366:1382–92 [DOI] [PubMed] [Google Scholar]
- 5.Mirza MR, Pignata S, Ledermann JA. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol 2018;29:1366–76 [DOI] [PubMed] [Google Scholar]
- 6.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 2018;379:2495–505 [DOI] [PubMed] [Google Scholar]
- 7.Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1274–84 [DOI] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A 1995;92:2298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997;88:265–75 [DOI] [PubMed] [Google Scholar]
- 11.Zhou Q, Huang J, Zhang C, Zhao F, Kim W, Tu X, et al. The bromodomain containing protein BRD-9 orchestrates RAD51-RAD54 complex formation and regulates homologous recombination-mediated repair. Nat Commun 2020;11:2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer S, Zhang S, Yucel S, Horn H, Smith SG, Reinhardt F, et al. Genetically defined syngeneic mouse models of ovarian cancer as tools for the discovery of combination immunotherapy. Cancer Discov 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 2020;11:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersen S, Wilson AJ, Hirst J, Roby KF, Fadare O, Crispens MA, et al. CCNE1 and BRD4 co-amplification in high-grade serous ovarian cancer is associated with poor clinical outcomes. Gynecol Oncol 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lepage CC, Palmer MCL, Farrell AC, Neudorf NM, Lichtensztejn Z, Nachtigal MW, et al. Reduced SKP1 and CUL1 expression underlies increases in Cyclin E1 and chromosome instability in cellular precursors of high-grade serous ovarian cancer. Br J Cancer 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhn E, Wang TL, Doberstein K, Bahadirli-Talbott A, Ayhan A, Sehdev AS, et al. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod Pathol 2016;29:1254–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karst AM, Jones PM, Vena N, Ligon AH, Liu JF, Hirsch MS, et al. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res 2014;74:1141–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etemadmoghadam D, Weir BA, Au-Yeung G, Alsop K, Mitchell G, George J, et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc Natl Acad Sci U S A 2013;110:19489–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson AJ, Sarfo-Kantanka K, Barrack T, Steck A, Saskowski J, Crispens MA, et al. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol Oncol 2016;143:143–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson AJ, Lalani AS, Wass E, Saskowski J, Khabele D. Romidepsin (FK228) combined with cisplatin stimulates DNA damage-induced cell death in ovarian cancer. Gynecol Oncol 2012;127:579–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol Oncol 2014;133:599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 2008;30:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010;18:436–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res 2010;70:4470–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett 2009;280:125–33 [DOI] [PubMed] [Google Scholar]
- 26.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014;124:30–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khabele D. The therapeutic potential of class I selective histone deacetylase inhibitors in ovarian cancer. Front Oncol 2014;4:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bandolik JJ, Hamacher A, Schrenk C, Weishaupt R, Kassack MU. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int J Mol Sci 2019;20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimbrel EA, Davis TN, Bradner JE, Kung AL. In vivo pharmacodynamic imaging of proteasome inhibition. Mol Imaging 2009;8:140–7 [PubMed] [Google Scholar]
- 31.Wilson AJ, Stubbs M, Liu P, Ruggeri B, Khabele D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol Oncol 2018;149:575–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55 [DOI] [PubMed] [Google Scholar]
- 33.Hirst J, Pathak HB, Hyter S, Pessetto ZY, Ly T, Graw S, et al. Licofelone Enhances the Efficacy of Paclitaxel in Ovarian Cancer by Reversing Drug Resistance and Tumor Stem-like Properties. Cancer Res 2018;78:4370–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 2004;26:249–61 [DOI] [PubMed] [Google Scholar]
- 35.Pu X, Wang Z, Klaunig JE. Alkaline Comet Assay for Assessing DNA Damage in Individual Cells. Curr Protoc Toxicol 2015;65:3 12 1–3 1 [DOI] [PubMed] [Google Scholar]
- 36.Qian G, Yu T. Nanosecond Electric Pulses Induce Early and Late Phases of DNA Damage and Cell Death in Cisplatin-Resistant Human Ovarian Cancer Cells. Biomed Res Int 2018;2018:4504895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karakashev S, Zhu H, Yokoyama Y, Zhao B, Fatkhutdinov N, Kossenkov AV, et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep 2017;21:3398–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson AJ, Saskowski J, Barham W, Khabele D, Yull F. Microenvironmental effects limit efficacy of thymoquinone treatment in a mouse model of ovarian cancer. Mol Cancer 2015;14:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson AJ, Saskowski J, Barham W, Yull F, Khabele D. Thymoquinone enhances cisplatin-response through direct tumor effects in a syngeneic mouse model of ovarian cancer. J Ovarian Res 2015;8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 2013;20:347–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A. DNA Fiber Analysis: Mind the Gap! Methods Enzymol 2017;591:55–82 [DOI] [PubMed] [Google Scholar]
- 42.Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell 2020;77:461–74 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Essel KG, Behbakht K, Lai T, Hand L, Evans E, Dvorak J, et al. PARPi after PARPi in epithelial ovarian cancer. Gynecol Oncol Rep 2021;35:100699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Della Corte L, Foreste V, Di Filippo C, Giampaolino P, Bifulco G. Poly (ADP-ribose) polymerase (PARP) as target for the treatment of epithelial ovarian cancer: what to know. Expert Opin Investig Drugs 2021:1–12 [DOI] [PubMed] [Google Scholar]
- 45.Thorsell AG, Ekblad T, Karlberg T, Low M, Pinto AF, Tresaugues L, et al. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J Med Chem 2017;60:1262–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore KN, Mirza MR, Matulonis UA. The poly (ADP ribose) polymerase inhibitor niraparib: Management of toxicities. Gynecol Oncol 2018;149:214–20 [DOI] [PubMed] [Google Scholar]
- 47.Sun K, Mikule K, Wang Z, Poon G, Vaidyanathan A, Smith G, et al. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models. Oncotarget 2018;9:37080–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim DS, Camacho CV, Kraus WL. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp Mol Med 2021;53:42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson AJ, Holson E, Wagner F, Zhang YL, Fass DM, Haggarty SJ, et al. The DNA damage mark pH2AX differentiates the cytotoxic effects of small molecule HDAC inhibitors in ovarian cancer cells. Cancer Biol Ther 2011;12:484–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer 2016;60:49–58 [DOI] [PubMed] [Google Scholar]
- 51.Gourley C, Balmana J, Ledermann JA, Serra V, Dent R, Loibl S, et al. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J Clin Oncol 2019;37:2257–69 [DOI] [PubMed] [Google Scholar]
- 52.Konstantinopoulos PA, Matulonis UA. Targeting DNA Damage Response and Repair as a Therapeutic Strategy for Ovarian Cancer. Hematol Oncol Clin North Am 2018;32:997–1010 [DOI] [PubMed] [Google Scholar]
- 53.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011;145:529–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012;22:106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schoonen PM, Kok YP, Wierenga E, Bakker B, Foijer F, Spierings DCJ, et al. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol Oncol 2019;13:2422–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wurster S, Hennes F, Parplys AC, Seelbach JI, Mansour WY, Zielinski A, et al. PARP1 inhibition radiosensitizes HNSCC cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget 2016;7:9732–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu H, Wei M, Xu J, Hua J, Liang C, Meng Q, et al. PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Mol Cancer 2020;19:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thangavel S, Mendoza-Maldonado R, Tissino E, Sidorova JM, Yin J, Wang W, et al. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol 2010;30:1382–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carvajal-Maldonado D, Byrum AK, Jackson J, Wessel S, Lemacon D, Guitton-Sert L, et al. Perturbing cohesin dynamics drives MRE11 nuclease-dependent replication fork slowing. Nucleic Acids Res 2019;47:1294–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther 2007;6:795–801 [DOI] [PubMed] [Google Scholar]
- 61.Wilson AJ, Cheng YQ, Khabele D. Thailandepsins are new small molecule class I HDAC inhibitors with potent cytotoxic activity in ovarian cancer cells: a preclinical study of epigenetic ovarian cancer therapy. J Ovarian Res 2012;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, et al. Replication Gaps Underlie BRCA-deficiency and Therapy Response. Cancer Res 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang B, Lyu J, Yang EJ, Liu Y, Wu C, Pardeshi L, et al. Class I histone deacetylase inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells. Acta Pharm Sin B 2020;10:615–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kiweler N, Wunsch D, Wirth M, Mahendrarajah N, Schneider G, Stauber RH, et al. Histone deacetylase inhibitors dysregulate DNA repair proteins and antagonize metastasis-associated processes. J Cancer Res Clin Oncol 2020;146:343–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stengel KR, Hiebert SW. Class I HDACs Affect DNA Replication, Repair, and Chromatin Structure: Implications for Cancer Therapy. Antioxid Redox Signal 2015;23:51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.National Institutes of Health (U.S.), National Library of Medicine (U.S.), United States. Food and Drug Administration. ClinicalTrials.gov. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.