

Abstract
This Letter details our efforts to develop novel tricyclic M4 PAM scaffolds with improved pharmacological properties. This endeavor involved a “tie-back” strategy to replace the 3-amino-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide core which lead to the discovery of two novel tricyclic cores: a 7,9-dimethylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine core and 2,4-dimethylthieno[2,3-b:5,4-c’]dipyridine core. Both tricyclic cores displayed low nanomolar potency against the human M4 receptor.
Keywords: M4, Muscarinic acetylcholine receptor, Positive Allosteric modulator (PAM), Structure Activity Relationship (SAR)
Graphical Abstract
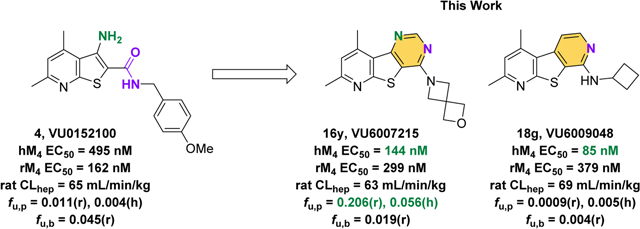
Muscarinic acetylcholine receptor subtype 4 (M4) positive allosteric modulators (PAMs) continue to be important drug targets as novel treatments for various neurological disorders such as Parkinson’s disease,1 Huntington’s disease,2 and schizophrenia (both the positive and negative symptom clusters).3–7 Classically, M4 PAMs possess a β-amino carboxamide moiety as a key pharmacophore (circled in 2, Figure 1) which was believed to be essential for M4 PAM activity.8–14 As such chemotypes have been plagued with poor solubility, varying degrees of P-gp efflux, and potency differences across species, efforts have been made to develop M4 PAM chemotypes devoid of the β-amino carboxamide moiety.3,14–26 Clinical studies featuring xanomeline, a M1/M4 preferring agonists that lacks the β -amino carboxamide moiety, have validated targeting the muscarinic cholinergic system as a method for treating the psychosis and behavioral disturbances observed in both Alzeheimer’s and schizophrenia patients.23,24 However, the lack of receptor subtype selectivity resulted in undesired side effects which ultimately lead to a discontinuation of clinical development. An effort to circumvent these adverse events led to the development of KarXT which is currently undergoing clinical trials. KarXT is a treatment in which xanomeline is co-administered with a pan-selective peripheral muscarinic acetylcholine receptor antagonist (trospium) to minimize the adverse side effects of xanomeline.25 Additional work in the field led to the identification of a selective M4 PAM which lacked the β-amino carboxamide moiety. Not only was this PAM efficacious in preclinical assays but had fewer and less severe adverse cholinergic-related side effects than observed in rats treated with the nonselective M4 agonist xanomeline.26 This indicates that receptor-subtype-selective M4 PAMs could potentially improving safety profiles. In fact, CVL-231 (a selective M4 PAM) is currently undergoing clinical trials.27,28
Figure 1.
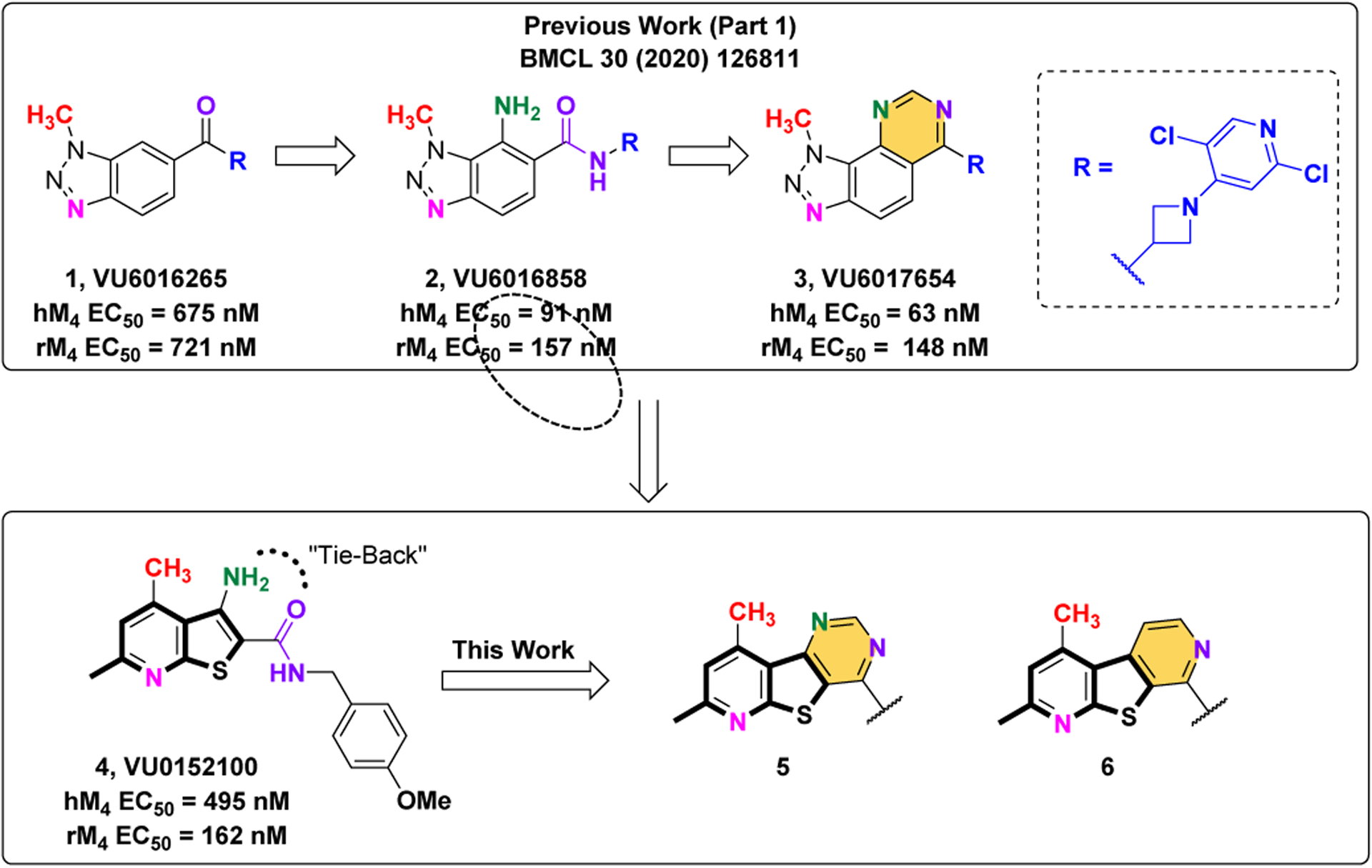
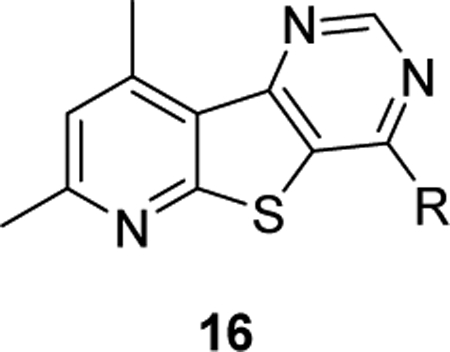
Exploration of novel tricyclic cores as M4 PAMs. We utilized the same “tie-back” strategy to mask the β-amino carboxamide moiety (dotted circle), which contributed to the discovery of VU6017654 (3), to develop two unique M4 PAM tricyclic chemotypes: a 7,9-dimethylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine core (5) and a 2,4-dimethylthieno[2,3-b:5,4-c’]dipyridine core (6).
Our laboratory previously described two structurally distinct 5,6,6-tricyclic scaffolds devoid of the β-amino carboxamide moiety that afforded potent and CNS penetrant M4 PAMs.17 In an effort to identify additional novel M4 PAM chemotypes, we employed a “tie-back” strategy in order to mask the β-amino carboxamide moiety of one of our early lead M4 PAM compounds, VU0152100.10–11 When designing this new scaffold, we elected to keep the 4,6-dimethylthieno[2,3-b]pyridine core intact while masking the β-amino carboxamide as a pyrimidine moiety, a strategy utilized in the development of VU6017654 (3). This resulted in the discovery of a novel M4 PAM chemotype containing a 7,9-dimethylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine core, 5. Further exploration revealed a second novel, tricyclic M4 PAM chemotype containing a 2,4-dimethylthieno[2,3-b:5,4-c’]-dipyridine core, 6. This body of work details the development of these two novel M4 PAM chemotypes.
The synthesis of core 5 began with the commercially available amine 7, which was first condensed with formamide followed by formamidine acetate salt to afford the pyrimidone intermediate 9 (Scheme 1). Treatment of pyrimidone 9 with POCl3 gave chloride 11 which readily underwent nucleophilic aromatic substitution with a variety of primary and secondary amines as well as alcohols to yield desired analogs 16 and 19. For this exercise, we chose to evaluate benzyl amines and amino azetidines that proved to be successful in our past endeavors to afford potent M4 PAMs.15–17,29
Scheme 1.
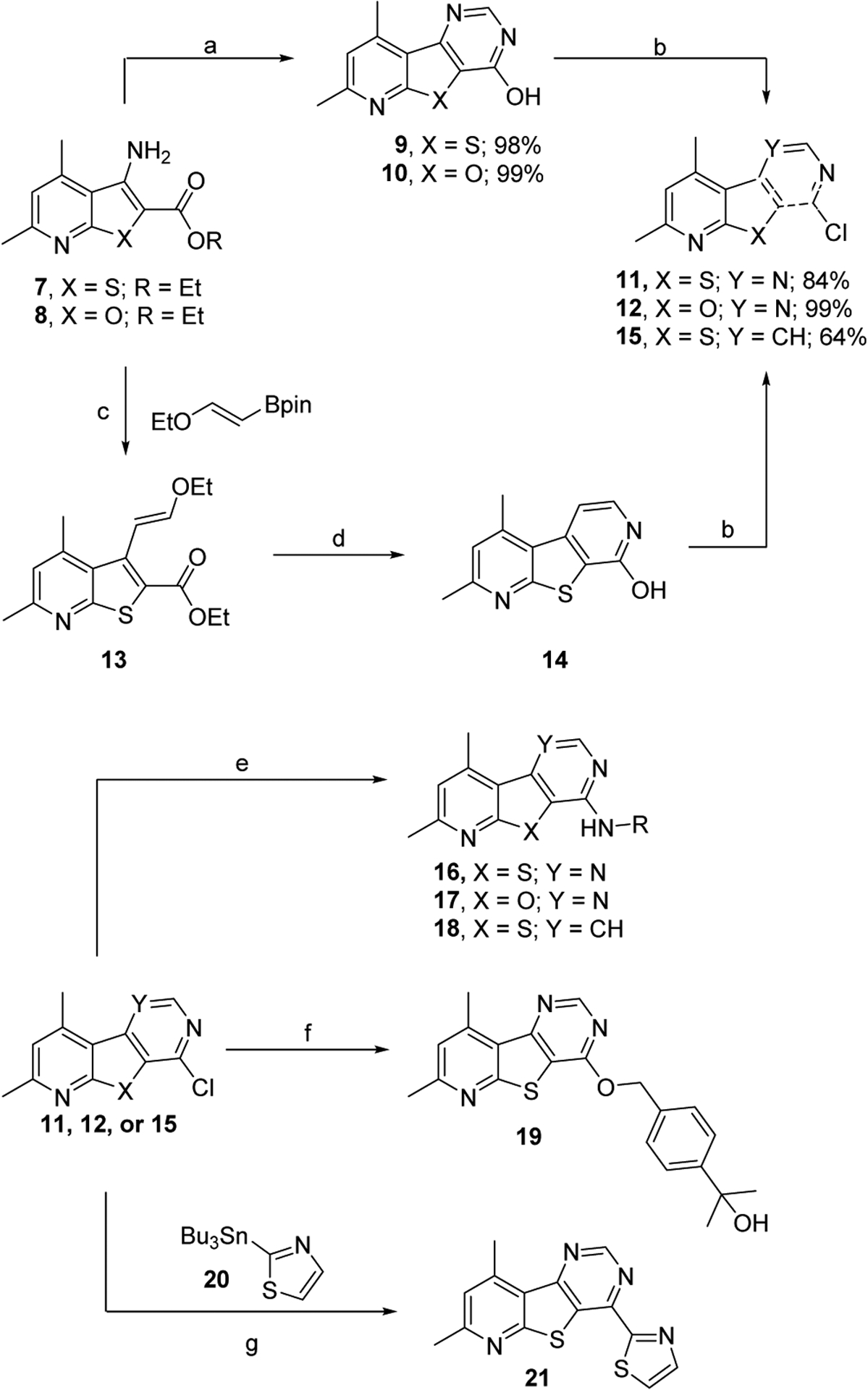
Synthesis of M4 PAM analogs. Reagents and conditions: (a) i. HCONH2, 150 °C; ii. AcOH • HN=CHNH2, 150 °C; (b) POCl3, 120 °C, 18h; (c) i. CuBr2, tBuONO, CH3CN, 60 °C, 56%; ii. Cs2CO3, Pd(dppf)Cl2, dioxane/H2O (10:1), 90 °C, 18h, 99%; (d) i. TFA, 100 °C, 4h, 98%; ii. NH4OH, 100 °C, 48h, 98%; (e) amine, DIEA, NMP, 120 °C, 21–88%; (f) i. Methyl (4-hydroxymethyl)benzoate, NaH, THF, 0 °C, 17%; ii. MeMgBr, THF, 0 °C, 38%; (g) 20, Pd(PPh3)4, 1,4-dioxane, μW 120 °C, 0.5h, 10%.
Select analogs 16 & 19 were screened against the human M4 (hM4) receptor to determine potency with results highlighted in Table 1. It became very clear that the amine “tail” greatly influences potency. It was noted that minor changes, such as a fluoro-substitution on the phenyl ring (16m vs. 16n), led to a ~2.5-fold decrease in potency. Moreover, removal of one fluorine from the trifluoromethoxy group of 16d (inactive) afforded analog 16e (EC50 = 4 μM), which reestablished activity at hM4. Another intriguing finding was observed when the amine linker (16m; hM4 EC50 = 910 nM) was exchanged for an ether linker (19; hM4 EC50 = 8.7 μM) resulting in a 9.6-fold loss of potency.
Table 1.
Structures and activities for analogs 16 & 19.
| ||
|---|---|---|
| Cmpd | R Group | hM4 EC50 (nM)a [%ACh Max] |
| 16a |
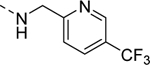
|
Inactive |
| VU6008258 | ||
| 16b |
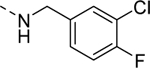
|
Inactive |
| VU6008243 | ||
| 16c |
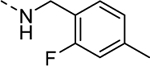
|
Inactive |
| VU6008261 | ||
| 16d |
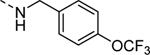
|
Inactive |
| VU6008266 | ||
| 16e |
|
4,010 |
| VU6008279 | [45] | |
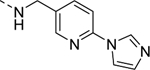
| 16f |
|
1,560 |
| VU6008248 | [34] | |
| 16g |
|
1,110 |
| VU6008250 | [47] | |
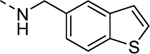
| 16h |
|
Inactive |
| VU6008255 | ||
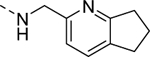
| 16i |
|
2,560 |
| VU6008257 | [30] | |
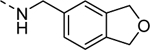
| 16j |
|
1,140 |
| VU6008256 | [41] | |
| 16k |
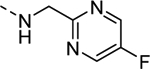
|
>10,000 |
| VU6008254 | [30] | |
| 16l |
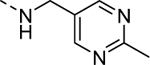
|
1,400 |
| VU6008283 | [54] | |
| 16m |
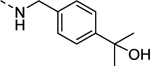
|
910 |
| VU6008284 | [76] | |
| 16n |
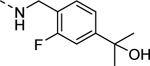
|
2,320 |
| VU6010076 | [74] | |
| 16o |
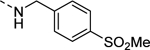
|
293 |
| VU6008280 | [42] | |
| 16p |
|
6,550 |
| VU6008452 | [52] | |
| 16q |
|
341 |
| VU6007221 | [67] | |
| 16r |
|
1,640 |
| VU6008278 | [61] | |
| 19 |
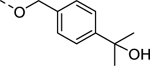
|
8,740 |
| VU6010153 | [40] | |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine, n =1 independent experiment in triplicate.
Noting the importance of the amine “tail”, we next elected to explore small aliphatic amines (Table 2) as well as small carbon-linked groups (Table 3) which were synthesized in accordance with Scheme 2. Briefly, commercially available 2-mercapto-4,6-dimethyl nicotinonitrile (22) was condensed with various α-bromo ketones in a Gewald-type reaction to afford thieno[2,3-b] pyridines 23. As previously described in Scheme 1, condensation with formamide afforded final compounds 24. The piperidine intermediate 24e could be further transformed via nucleophilic aromatic substitutions (26), HATU-assisted amide formation (28), or reductive amination (30). Finally, the synthesis of biaryl analog 21 was accomplished utilizing intermediate 11 in a Stille cross-coupling reaction as described in Scheme 1.
Table 2.
Structures and activities for analog 16 containing small aliphatic amines.
| ||
|---|---|---|
| Cmpd | R Group | hM4 EC50 (nM)a [%ACh Max] |
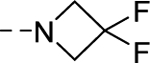
| 16s |
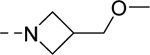
|
762 |
| VU6007214 | [50] | |
| 16t |
|
456 |
| VU6007218 | [69] | |
| 16u |
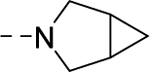
|
453 |
| VU6007216 | [75] | |
| 16v |
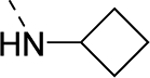
|
359 |
| VU6007220 | [76] | |
| 16w |
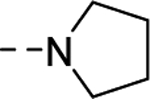
|
277 |
| VU6007219 | [67] | |
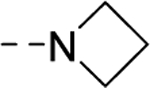
| 16x |
|
172 |
| VU6007217 | [59] | |
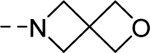
| 16y |
|
144 |
| VU6007215 | [84] | |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine, n =1 experiment performed in in triplicate.
Table 3.
Structures and activities for C-linked analogs 21, 26, 28, & 30.
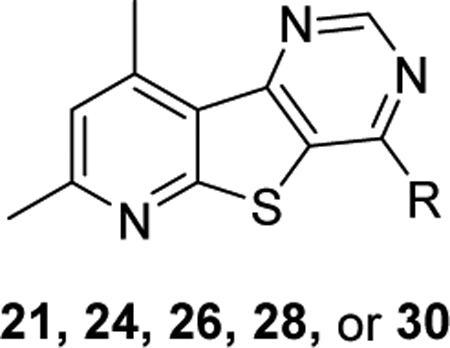
| ||
|---|---|---|
| Cmpd | R Group | hM4 EC50 (nM)a [%ACh Max] |
| 24a |
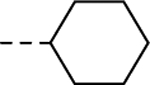
|
6,790 |
| VU6008673 | [71] | |
| 24b |
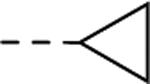
|
3,900 |
| VU6008479 | [62] | |
| 24c | --CH3 | 2,820 |
| VU6008362 | [58] | |
| 24d |
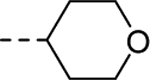
|
152 |
| VU6008676 | [99] | |
| 24e |
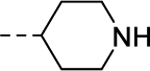
|
6,110 |
| VU6009094 | [44] | |
| 28a |
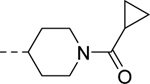
|
620 |
| VU6009098 | [62] | |
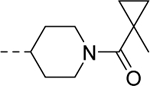
| 28b |
|
234 |
| VU6009100 | [72] | |
| 30 |
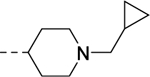
|
Inactive |
| VU6009198 | ||
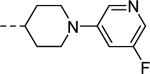
| 26 |
|
Inactive |
| VU6009197 | ||
| 21 |
|
3,410 |
| VU6007227 | [62] | |
Calcium mobilization assays with hM4/Gqi5-CHO cells or performed in the presence of an EC20 fixed concentration of acetylcholine, n =1 independent experiment performed in triplicate.
Scheme 2.
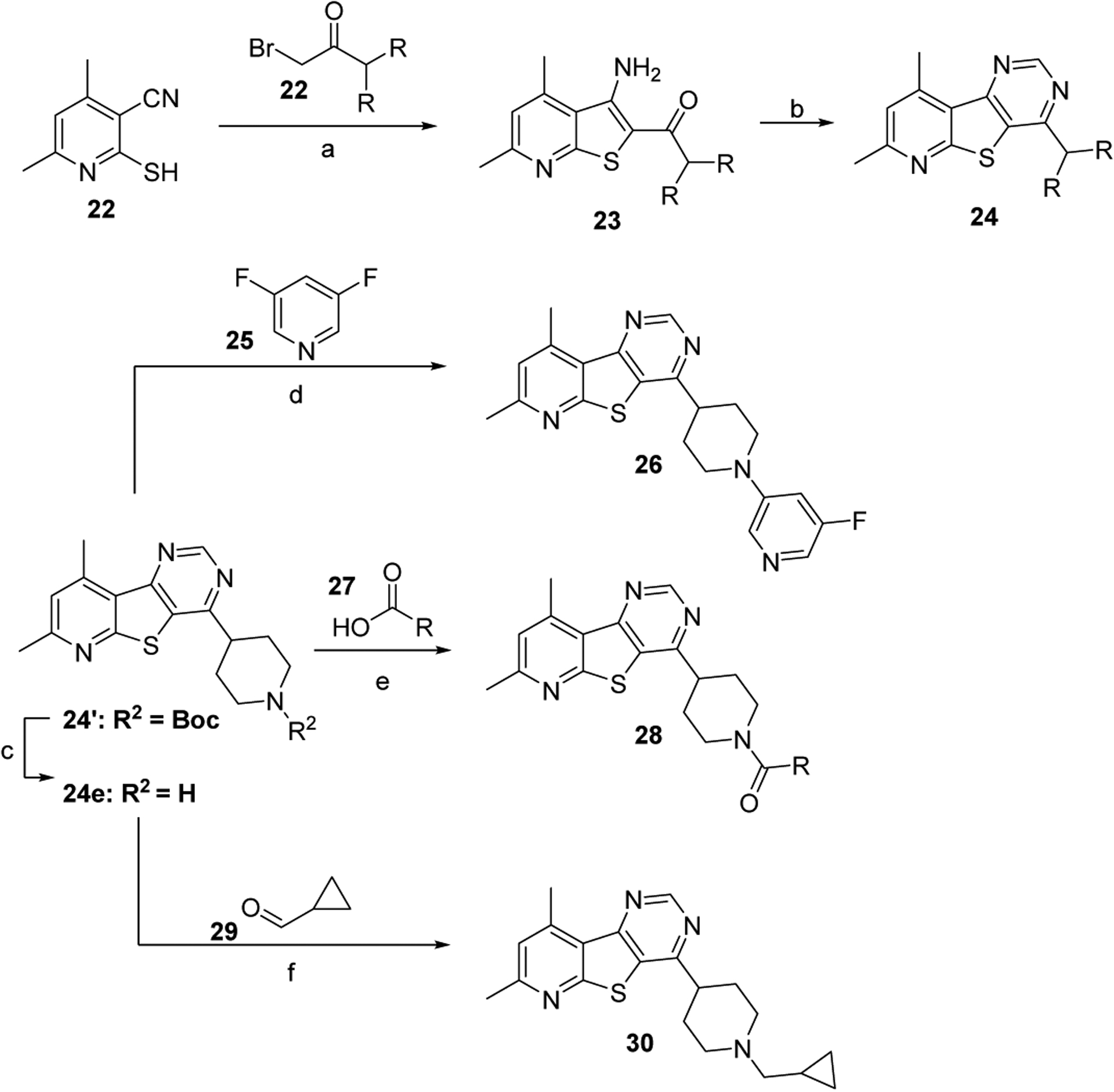
Synthesis of M4 PAM analogs. Reagents and conditions: (a) 22, KOH, IPA, 105 °C, 10–74%; (b) i. HCONH2, 150 °C; ii. AcOH • HN=CHNH2, 150 °C, 62 – 95%; (c) HCl, 1,4-dioxane/H2O (1:1), 79%; (d) 25, DIEA, NMP, 120 °C, 72h, 92%; (e) 27, HATU, DIEA, DMF, 54–62%; (f) 29, STAB. AcOH, DCM, 18h, 95%.
Select analogs 16s-y, 21, 24, 26, 28, and 30 were screened against human M4 (hM4) to determine potency with results highlighted in Tables 2 & 3. Selecting small amines as our “tail” groups (16s-y) proved most advantageous as the majority of analogs in Table 2 possess hM4 PAM functional potencies less than 500 nM, with three analogs displaying EC50’s < 300 nM: 16w (hM4 EC50 = 277 nM), 16x (hM4 EC50 = 172 nM), and 16y (hM4 EC50 = 144 nM).
Conversely, similar small carbon-linked tail groups resulted in a loss of hM4 functional potency (Table 3): 21 (hM4 EC50 = 3.4 μM), 24a (hM4 EC50 = 6.8 μM), 24b (hM4 EC50 = 3.9 μM), 24c (hM4 EC50 = 2.8 μM). Most notably, the addition of a hydrogen bond acceptor into the cyclohexyl ring of analog 24a to give the tetrahydropyran analog 24d (hM4 EC50 = 152 nM), resulted in a nearly 45-fold increase in hM4 functional potency. Comparison of 24d to the corresponding piperidine analog 24e (hM4 EC50 = 6.1 μM) once again resulted in a loss of hM4 functional potency (~40-fold). To investigate the importance of a hydrogen bond acceptor at this position, we further investigated the piperidine tail by eliminating its ability to serve as a hydrogen bond donor. This exercise generated amides (28a & 28b), small aliphatic C-linked piperidines (30), as well as aryl-linked piperidines (26). Interestingly, converting piperidine 24e into amides 28a (hM4 EC50 = 620 nM) and 28b (hM4 EC50 = 234 nM) led to an increase in functional M4 potency by ~10-fold and 26-fold, respectively. Removing the carbonyl of analog 28a to afford analog 30 resulted in a complete loss of M4 potency, further supporting our theory of the importance of a hydrogen bond acceptor at this position. Similarly, a complete loss of activity was observed when the piperidine tail of 24e was capped with 3-fluoropyridine (26). This loss of activity could be attributed to several factors including the large size of the C-linked “tail”, the position/orientation of the electron lone pair of the pyridine, and/or the effects of the fluorine substituent on the basicity of the pyridine.
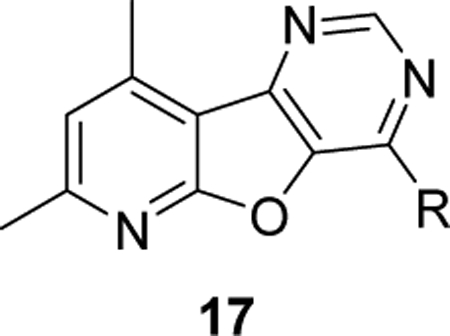
After extensive exploration of the amine tail, we turned our attention toward accessing the importance of the sulfur of the 7,9-dimethylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine core. To do so, we exchanged the sulfur with an oxygen to generate a 7,9-dimethylpyrido[3’,2’:4,5]furo[3,2-d]pyrimidine core, 17. Similar to the synthesis 16, the synthesis of core 17 began by condensing formamide with ethyl 3-amino-4,6-dimethylfuro[2,3-b]pyridine-2-carboxylate (8) to afford the pyrimidone intermediate 10 (Scheme 1). Treatment of pyrimidone 10 with POCl3 gave chloride 12 which readily underwent nucleophilic aromatic substitution with a variety of primary and secondary amines to afford desired analogs 17 in moderate to good yields.
Functional hM4 potencies for analogs 17 were determined and the results highlighted in Table 4. Of the compounds evaluated, none possessed sub-micromolar potencies. In fact, we observed complete loss of activity in several analogs (17a, 17d, 17e, and 17f). Directly comparing analog 16m (hM4 EC50 = 910 nM) to 17e (inactive), 16w (hM4 EC50 = 277 nM) to 17a (inactive), and 16v (hM4 EC50 = 359 nM) to 17c (hM4 EC50 = 1.59 μM; 4.4-fold potency decrease) further illustrates the significance of the sulfur atom in the tricyclic ring.
Table 4.
Structures and activities for analog 17.
| ||
|---|---|---|
| Cmpd | R Group | hM4 EC50 (nM)a [%ACh Max] |
| 17a |
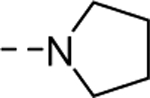
|
Inactive |
| VU6008413 | ||
| 17b |
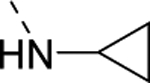
|
2,600 |
| VU6008838 | [64] | |
| 17c |
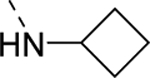
|
1,590 |
| VU6008833 | [44] | |
| 17d |
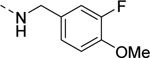
|
Inactive |
| VU6008822 | ||
| 17e |
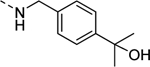
|
Inactive |
| VU6009080 | ||
| 17f |
|
Inactive |
| VU6008669 | ||
| 17g |
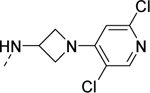
|
1,490 |
| VU6008806 | [49] | |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine, n =1 independent experiment performed in triplicate.
Finally, we evaluated the relevance of the pyrimidine nitrogen at the 5-position by synthesizing analogs 18 (Scheme 1). These analogs were synthesized from commercially available amine 7, which underwent a Sandmeyer reaction to give the corresponding bromide. The bromide intermediate was transformed into intermediate 13 via a Suzuki cross-coupling reaction. Cyclization of 13 to form the pyrone followed by exchange with ammonia afforded the pyridone 14, which was chlorinated via treatment with POCl3 to give intermediate 15 in moderate to good yields. Chloride 15 then underwent nucleophilic aromatic substitution with varies amines to give analogs 18.
Analogs 18 were screened against hM4 to determine potency with results highlighted in Table 5. It was immediately apparent that this modification was not detrimental to hM4 activity. In fact, several analogs displayed increased potencies. For instance, direct comparisons between 16m (hM4 EC50 = 910 nM) and 18b (hM4 EC50 = 327 nM) resulted in a ~2.8-fold increase in potency. Similarly, comparing 16p (hM4 EC50 = 6.55 μM) and 18c (hM4 EC50 = 150 nM) resulted in a ~44-fold increase in potency. Likewise, this trend was also observed when analyzing 16v (hM4 EC50 = 359 nM) and 18g (hM4 EC50 = 85 nM) which resulted in a ~4-fold increase in functional hM4 potency.
Table 5.
Structures and activities for analog 18.
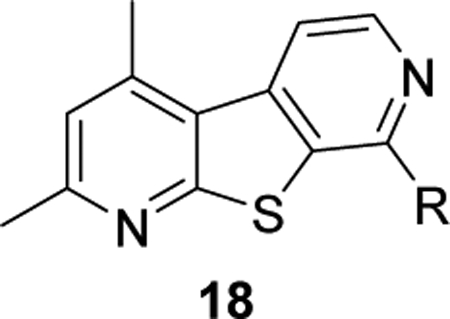
| ||
|---|---|---|
| Cmpd | R Group | hM4 EC50 (nM)a [%ACh Max] |
| 18a |
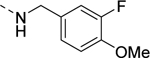
|
>10,000 |
| VU6010206 | [90] | |
| 18b |
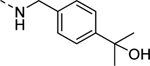
|
327 |
| VU6010225 | [101] | |
| 18c |
|
150 |
| VU6010205 | [80] | |
| 18d |
|
233 |
| VU6008889 | [98] | |
| 18e |
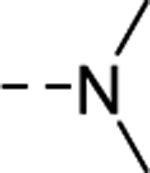
|
202 |
| VU6009049 | [82] | |
| 18f |
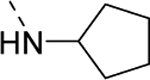
|
124 |
| VU6009203 | [71] | |
| 18g |
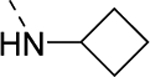
|
85 |
| VU6009048 | [90] | |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine, n =1 independent experiment performed in triplicate.
Of these compounds, 16o, 16w, 16x, 16y, 28b, 18b, 18d, and 18g were advanced into a battery of in vitro DMPK assays and our standard rat plasma:brain level (PBL) cassette paradigm (Table 6).18 These compounds were chosen to move forward based on their M4 potency (EC50 < 350 nM) as well as their chemical diversity across subseries. In regard to physicochemical properties, these analogs all possessed molecular weights less than 450 Da possessed molecular weights less than 450 Da with 16o, 16w, and 16x having the most attractive CNS xLogP values (2.79 – 3.39).30–31 All analogs tested displayed high predicted hepatic clearance in at least one species based on microsomal CLint data (rat CLheps > 56 mL/min/kg). While analogs 16o and 28b displayed moderate human predicted hepatic clearance based on microsomal CLint data (human CLheps of 11–12 mL/min/kg) all other analogs tested displayed high human predicted hepatic clearance based on microsomal CLint data (human CLhep > 15 mL/min/kg).
Table 6.
In vitro DMPK and rat PBL data for select analogs 16, 18 & 28.
| Property | 16o VU6008280 |
16w VU6007219 |
16x VU6007217 |
16y VU6007215 |
28b VU6009100 |
18b VU6010225 |
18d VU6008889 |
18g VU6009048 |
|---|---|---|---|---|---|---|---|---|
| MW | 398.5 | 284.38 | 270.35 | 312.39 | 380.51 | 377.5 | 283.39 | 283.39 |
| xLogP | 3.39 | 3.24 | 2.79 | 1.85 | 3.75 | 4.89 | 3.69 | 3.92 |
| TPSA | 84.8 | 41.9 | 41.9 | 51.1 | 59 | 58 | 29 | 37.8 |
| In vitro PK parameters | ||||||||
| CLINT (mL/min/kg), rat | 176 | 1896 | 741 | 594 | 367 | 334 | 2098 | 3859 |
| CLHEP (mL/min/kg), rat | 50 | 68 | 64 | 63 | 59 | 58 | 68 | 69 |
| CLINT (mL/min/kg), human | 28 | 134 | 296 | 129 | 25 | 50 | 161 | 345 |
| CLHEP (mL/min/kg), human | 12 | 18 | 20 | 18 | 11 | 15 | 19 | 20 |
| Rat fuplasma | 0.024 | 0.054 | 0.071 | 0.206 | 0.017 | 0.005 | 0.016 | 0.009 |
| Human fuplasma | 0.002 | 0.016 | 0.021 | 0.056 | 0.017 | 0.001 | 0.003 | 0.005 |
| Rat fubrain | 0.011 | 0.004 | 0.009 | 0.019 | 0.011 | 0.003 | 0.002 | 0.004 |
| Brain Distribution (0.25 h) (SD Rat; 0.2 mg/kg IV) | ||||||||
| Kp, brain:plasma | 0.45 | 5.36 | 6.49 | 4.47 | 0.54 | 2.27 | 37.1 | 10.4 |
| Kpuu, brain:plasma | 0.21 | 0.65 | 0.27 | 0.40 | 0.35 | 1.16 | 4.87 | 4.18 |
Compounds within the tricycle series of core 6 (18b, 18d, & 18g) were, in general, highly bound to plasma protein (rat fus plasma 0.005 – 0.016; rat fus brain 0.002 – 0.004; human fus plasma 0.001 – 0.005). By contrast, compounds within the tricyclic series of core 5 (16o, 16w, 16x, 16y, 28b) exhibited reduced protein binding profiles (rat fus plasma 0.017 – 0.21; rat fus brain 0.004 – 0.019; human fus plasma 0.002 – 0.056). Interestingly, compounds 16y and 28b displayed the best overall protein binding profiles (rat fus 0.017 – 0.21; rat fus brain 0.032 – 0.053; human fus 0.011 – 0.019); however, these analogs were not progressed forward due to their high predicted hepatic clearance in rat and/or human. Analogs 16o (rat brain:plasma Kp = 0.45, Kp,uu = 0.21) and 16x (rat brain:plasma Kp = 6.49, Kp,uu = 0.27) proved to have low CNS distribution of unbound drug, while analogs 16w, 16y and 28b displayed moderate CNS distribution (rat Kp,uus = 0.35 – 0.65). On the other hand, analogs 18d (rat brain:plasma Kp = 37.1, Kp,uu = 4.87) and 18g (rat brain:plasma Kp = 10.4, Kp,uu = 4.18) displayed excellent CNS penetration; however, these compounds suffered from low fraction unbound.
In summary, a scaffold hopping exercise utilizing a “tie-back” strategy based on M4 PAM 3 proved to be a successful strategy in converting an early lead compound, VU0152100, into potent tricyclic M4 PAM analogs devoid of the classic β-amino carboxamide moiety. Analogs within the tricycle series containing the 2,4-dimethylthieno[2,3-b:5,4-c’]dipyridine core (6) gave many potent M4 PAMs (Table 5; hM4 EC50 = 85–327 nM); however, these analogs displayed very poor fraction unbound in regards to brain and plasma protein (fus < 0.01) as well as high human (CLheps ≥ 15 mL/min/kg) and rat (CLheps ≥ 60 mL/min/kg) predicted hepatic clearance. Thus, additional analogs of core 6 were not pursued further. Directing our effort toward the 7,9-dimethylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine core (5) showed benzyl amine linked analogs generally provide only moderately potent analogs (Table 1). Changing the linker to small aliphatic amine linkers provided several potent M4 PAMs (Table 2; hM4 EC50 = 144–453 nM). On further evaluation, this subseries of analogs generally exhibited improved plasma protein binding profiles. Unfortunately, the small aliphatic amine linker analogs suffered from very high human (CLheps ≥ 18 mL/min/kg) and rat (CLheps ≥ 62 mL/min/kg) predicted hepatic clearance and further work on this subseries was abandoned. Further exploration revealed that carbon-linked groups (Table 3) showed improved brain and plasma fus when compared to the 2,4-dimethylthieno[2,3-b:5,4-c’]dipyridine core (6) as previously discussed; however, due to poor human and rat predicted hepatic clearance, further work on this subset of analogs was not pursued.
Our efforts to mask the β-amino carboxamide moiety of an early lead M4 PAM compound, VU0152100, resulted in the discovery of two new tricyclic cores (5 & 6) which provided potent M4 PAM analogs. This endeavor also provided analogs VU6007215 & VU6009048, which display 3.5- fold and 6-fold increase in human M4 activity, respectively, when compared to parent compound VU0152100. Moreover, VU6007215 exhibits a reduced protein binding profile in relation to the parent compound VU0152100. In comparison to the previously reported VU6017654 (rat fus plasma 0.007; rat fus brain 0.003; human fus plasma 0.021), VU6007215 shows a much-improved protein binding profile (rat fus plasma 0.206; rat fus brain 0.019; human fus plasma 0.056). Additionally, VU6007215 displayed higher CNS distribution of unbound drug (rat brain:plasma Kp = 4.47, Kp,uu = 0.40) than that of VU6017654 (rat brain:plasma Kp = 0.25, Kp,uu = 0.11); however, a ~2-fold loss in hM4 and rM4 functional potency was observed with VU6007215. Although this excursion did not deliver PAMs with DMPK profiles to warrant advancement as development candidates, it did garner insights for future scaffold designs toward that goal. These refinements will be reported in due course.
Acknowledgments
We thank the NIH for funding via the NIH Roadmap Initiative 1X01 MH077607 (C.M.N.), the Molecular Libraries Probe Center Network (U54MH084659 to C.W.L.) and U01MH087965 (Vanderbilt NCDDG). We also thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Shen W, Plotkin JL, Francardo V, et al. M4 Muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L-DOPA-induced dyskinesia. Neuron. 2015;88:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pancani T, Foster DJ, Moehle MS, et al. Allosteric activation of M4 muscarinic receptors improve behavioral and physiological alteration in early symptomatic YAC128 mice. Proc Natl Acad Sci USA. 2015;112:14078–14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bridges TM, LeBois EP, Hopkins CR, et al. The Antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect. 2010;23:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster DJ, Wilson JM, Wess J, et al. Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of Dopamine release. Neuron. 2016;91:1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones CK, Byun N, Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of Schizophrenia. Neuropsychopharmacology. 2012;37:16–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrell M, Roth BL. Allosteric Antipsychotics: M4 Muscarinic potentiators as novel treatments for Schizophrenia. Neuropsychopharmacology. 2010;35:851–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruse AC, Kobilka BK, Gautam D, et al. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat Rev Drug Discov.2014;13: 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan WY, McKinzie DL, Bose S, et al. Allosteric modulation of the muscarinic M4 receptor as an approach to treating Schizophrenia. Proc Natl Acad Sci USA. 2008;105:10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leach K, Loiacono RE, Felder CC, et al. Molecular Mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brady A, Jones CK, Bridges TM, et al. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharm Exp Ther. 2008;327;941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byun NE, Grannan M, Bubser M, et al. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology. 2014;39:1578–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bubser M, Bridges TM, Dencker D, et al. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem Neurosci. 2014;5:920–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melancon BJ, Wood MR, Noetzel MJ, et al. Optimization of M4 positive allosteric modulators (PAMs): The discovery of VU0476406, a non-human primate in vivo tool compound for translational pharmacology. Bioorg Med Chem Lett. 2017;27:2296–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood MR, Noetzel J, Melancon BH, et al. Discovery of VU0467485/AZ13713945: An M4 PAM evaluated as a preclinical candidate for the treatment of Schizophrenia. ACS Med Chem Lett. 2017;8:233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Temple KJ, Engers JL, Long MF, et al. Discovery of a novel 3,4-dimethylcinnoline carboxamide M4 positive allosteric modulator (PAM) chemotype via scaffold hopping. Bioorg Med Chem Lett. 2019;29:126678. [DOI] [PubMed] [Google Scholar]
- 16.Temple KJ, Engers JL, Long MF, et al. Discovery of a novel 2,3-dimethylimidazo[1,2-a]pyrazine-6-carboxamide M4 positive allosteric modulator (PAM) chemotype. Bioorg Med Chem Lett. 2020;30:126812. [DOI] [PubMed] [Google Scholar]
- 17.Temple KJ, Long MF, Engers JL, et al. Discovery of structurally distinct tricyclic M4 positive allosteric modulator (PAM) chemotypes. Bioorg Med Chem Lett. 2020;30:126811. [DOI] [PubMed] [Google Scholar]
- 18.Tarr JC, Wood MR, Noetzel MJ, et al. Challenges in the development of an M4 PAM preclinical candidate: The discovery, SAR, and in vivo characterization of a series of 3-aminoazetidine-derived amides. Bioorg Med Chem Lett.2017;27:2990–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le U, Melancon BJ, Bridges TM, et al. Discovery of a selective M4 positive allosteric modulator based on 3-amino-thieno[2,3- b]pyridine-2-carboxamide scaffold: Development of ML253, a potent and brain penetrant compound that is active in a preclinical model of schizophrenia. Bioorg Med Chem Lett. 2013;23:346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kennedy JP, Bridges TM, Gentry PR, et al. Synthesis of Structure-Activity Relationships of Allosteric Potentiators of the M4 Muscarinic Acetylcholine Receptor. ChemMedChem. 2009;4:1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salovich JM, Vinson PN, Sheffler DJ. Discovery of N-(4-methoxy-7-methylbenzo[d]thiazol—2-yl)isonicotinamide, ML293, as a novel, selective and brain penetrant positive allosteric modulator of the muscarinic 4 (M4) receptor. Bioorg Med Chem Lett. 2012;22:5084–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith E, Peter C, Niswender CM. Application of Parallel Multiparametric Cell-Based FLIPR Detection Assays for the Identification of Modulators of the Muscarinic Acetylcholine Receptor 4 (M4). Biomol Screening. 2015;20:858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bodick NC, Offen WW, Levey AI, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behaviorl symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–473. [DOI] [PubMed] [Google Scholar]
- 24.Shekhar A, Potter WZ, Lightfoot J, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psyciatry. 2018;165:1033–1039. [DOI] [PubMed] [Google Scholar]
- 25.Brannan SK, Sawchak S, Miller AC, et al. Muscarinich Cholinergic Receptor Agonist and Peripheral Antagonist for Schizophrenia. N Engl J Med. 2021;384:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuber JW, Harrison ST, Mulhearn J, et al. Discovery, Optimization, and Biological Characterization of 2,3,6-Trisubstituted Pyridine-Containing M4 Positive Allosteric Modulators. ChemMedChem. 2019:14:943–951. [DOI] [PubMed] [Google Scholar]
- 27.https://clinicaltrials.gov/ct2/show/NCT04136873. A Multiple Ascending Dose Trial of CVL-231 in Subjects with Schizophrenia. Date of Access: 10/11/2021
- 28.https://clinicaltrials.gov/ct2/show/NCT04787302?term=CVL-231. PET Trial to Evaluate Target Occupancy of CVL-231 on Brain Receptors Following Oral Dosing. Date of Access: 10/11/2021
- 29.Long MF, Engers JL, Chang S, et al. Discovery of a novel 2,4-dimethylquinolin-6-carboxamide M4 positive allosteric modulator (PAM) chemotype via scaffold hopping. Bioorg Med Chem Lett. 2017;27:4999–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rankovic Z CNS Drug Design: Balancing Physiochemical Properties for Optimal Brain Exposure. J Med Chem. 2015;58:2584–2608. [DOI] [PubMed] [Google Scholar]
- 31.Wager TT, Hou X, Verhoest PR, et al. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem Neurosci. 2016;7:767–775. [DOI] [PubMed] [Google Scholar]