

ABSTRACT
RNA molecules function as messenger RNAs (mRNAs) that encode proteins and noncoding transcripts that serve as adaptor molecules, structural components, and regulators of genome organization and gene expression. Their function and regulation are largely mediated by RNA binding proteins (RBPs). Here we present RNA proximity labelling (RPL), an RNA-centric method comprising the endonuclease-deficient Type VI CRISPR-Cas protein dCas13b fused to engineered ascorbate peroxidase APEX2. RPL discovers target RNA proximal proteins in vivo via proximity-based biotinylation. RPL applied to U1 identified proteins involved in both U1 canonical and noncanonical functions. Profiling of poly(A) tail proximal proteins uncovered expected categories of RBPs and provided additional evidence for 5ʹ-3ʹ proximity and unexplored subcellular localizations of poly(A)+ RNA. Our results suggest that RPL allows rapid identification of target RNA binding proteins in native cellular contexts, and is expected to pave the way for discovery of novel RNA–protein interactions important for health and disease.
KEYWORDS: Type VI Crispr-Cas (Cas13), engineered soybean ascorbate peroxidase APEX2, proximity-dependent biotinylation, RNA proximity labelling (RPL), RNA proximal proteins, RNA-centric method, RRNA–proteininteractions, poly(A) tail, RNA binding proteins (RBPs), U1 snRNA RBPS, noncoding RNAs (ncRNAs)
Introduction
RNA molecules include both messenger RNAs (mRNAs) that encode proteins and noncoding RNAs (ncRNAs) such as adaptor tRNAs and long noncoding RNAs (lncRNAs). While only ~2% of the human genome encodes mRNAs [1,2], up to 80% of the human genome encodes ncRNAs [3,4], including lncRNAs that are widely considered as a large group of potential regulators [5–7]. However, most lncRNAs remain uncharacterized, largely due to the technical challenges in determining the function of transcripts for which the canonical genetic code does not apply [8].
The function and regulation of RNA transcripts are mediated by other molecules they associate with, particularly RNA binding proteins (RBPs) [9–12]. Discovery of the interacting proteins for a given transcript plays a pivotal role in unveiling its function. Mechanistic studies of lncRNAs can be achieved by applying methods such as antisense probe-based ChIRP [13] or RAP [14] to enrich a transcript of interest and its associated proteins, through hybridization and purification after crosslinking via UV light or chemical crosslinking agents. However, crosslinkers such as formaldehyde also crosslink pprotein–proteininteractions, which may lead to false-positive associations [15]. UV has very low crosslinking efficiency, necessitating large numbers of cells (~100-800 million) be used as input [14,16], which may not be feasible for slow-growing model systems such as primary cell cultures. Moreover, UV-crosslinking induces RNA modifications [17] that can alter the binding affinity of RNA to certain RBPs [18] and impair downstream protein analysis [19]. An alternative approach, tagging of endogenous RNA, requires genetic manipulation and may interfere with endogenous RNA function [20]. Therefore, methods to discover endogenous RNA interacting proteins without genetic manipulation are needed.
We developed the RPL (RNA proximity labelling) method to identify in vivo target RNA proximal proteins without crosslinking or genetic manipulation. First, we profiled proteins proximal to U1, recovering RBPs related to U1 canonical and noncanonical roles. Second, we catalogued the universe of poly(A) tail proximal proteins in a mammalian cell. In addition to retrieving expected categories of RBPs, poly(A) tail proximal proteins provided additional support for the poly(A)+ RNA 5ʹ-3ʹ proximity [21,22]. Poly(A) tail proximal proteins also provided evidence that poly(A)+ transcripts likely reside in a more diverse array of subcellular localizations than previously appreciated. Thus, the RPL method can be readily applied to discover target RNA binding partners in vivo and is expected to pave the way to novel insights into the role of lncRNAs in health and disease.
Materials and methods
Plasmids and cloning
pC0046-EF1a-PspCas13b-NES-HIV was a gift from Dr. Feng Zhang (Addgene plasmid number: 103,862). pCMV-dPspCas13b-FLAG-APEX2-HA (RPL plasmid) was constructed by replacing ADAR2DD-delta-984-1090 in pC0053-CMV-dPspCas13b-GS-ADAR2DD (E488Q)-delta-984-1090 (a gift from Dr. Feng Zhang, Addgene plasmid number: 103,869) with FLAG-APEX2-HA subcloned from pcDNA3-APEX2-NES (a gift from Dr. Alice Ting, Addgene plasmid number: 49,386) using the following primers: dPspCas13b-For: 5ʹTACCCATACGATGTTCCAGATTACGCTTAAGCGGCCGCTCGAGTC3ʹ,
dPspCas13b-Rev: 5ʹGTCGTCATCCTTGTAGTCGGATCCCAGTGTCAGTCTTTCAAG3ʹ,
FLAG-APEX2-HA-For: 5ʹGACTACAAGGATGACGACG3ʹ,
FLAG-APEX2-HA-Rev: 5ʹTGGAACATCGTATGGGTACTGCAGGGCATCAGCAAAC3ʹ.
PCR was performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs, Catalogue number: M0491L). PCR fragments were assembled using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Catalogue number: E2621S) according to manufacturer’s instructions. The following spacer sequences were used to express gRNAs using pC0043-PspCas13b crRNA backbone (a gift from Dr. Feng Zhang, Addgene plasmid number: 103,854):
NTC (scrambled): ATGTCTTCCTGGGACGAAGACAA,
U1-11-30: ATCATGGTATCTCCCCTGCCAGGTAAGTAT,
U1-2101-130: CAAATTATGCAGTCGAGTTTCCCACATTTG,
U1-3108-137: ACTACCACAAATTATGCAGTCGAGTTTCCC,
Poly(A): TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT,
Poly(U): AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA.
The sequences of all constructs have been confirmed using Sanger sequencing.
Transfection and In vivo proximity-dependent biotinylation
For validation of U1 gRNAs in directing the RPL protein to target U1, HEK293T cells were seeded into 12-well plates and transfected with 1.5 µg the RPL plasmid and 0.5 µg Cas13b gRNAs (NTC, U1-1, U1-2, U1-3) while ~80% confluency using Lipofectamine 3000 (Thermo Fisher Scientific, Catalogue number: L3000015). For RIP experiments, HEK293T cells were seeded into 6-well plates and transfected with 2.5 µg the RPL plasmid and 1.5 µg Cas13b gRNAs while ~80% confluency using Lipofectamine 3000. For proximity-dependent biotinylation, HEK293T cells were seeded into a 150 mm plate and transfected with 25 µg the RPL plasmid and 15 µg Cas13b gRNAs (NTC, U1-1, U1-2, U1-3, poly[A], poly[U]) while ~80% confluency using Lipofectamine 3000. HEK293T cells were incubated with 25 mL of DMEM media containing 25 µL of 500 mM biotin-phenol (Iris Biotech, Catalogue number: LS-3500.1000) in DMSO for 30 min at 37°C 24 h post transfection. Cells were then treated with 1 mM hydrogen peroxide (H2O2) (Sigma Aldrich, Catalogue number: H1009) for 1 min on a horizontal shaker at room temperature. The labelling solution was then aspirated and cells were washed twice with 25 mL of quencher solution (10 mM sodium azide, Sigma Aldrich, Catalogue number: S2002-5 G; 10 mM sodium ascorbate, Sigma Aldrich, Catalogue number: PHR1279-1 G; and 5 mM Trolox, Sigma Aldrich, Catalogue number: 238,813–1 G) in DPBS (Thermo Fisher Scientific, Catalogue number: 14,040,182). Cells were washed three times with 15 mL of DPBS and were pelleted by centrifugation at 1,500 g for 5 min at 4°C. Cell pellets were snap frozen and stored at −80°C.
Streptavidin enrichment of biotinylated proteins
Cell pellets from two 150 mm plates of transfected HEK293T cells were lysed in 2 mL cell lysis buffer (10 mM HEPES, pH7.5 by KOH, 150 mM NaCl, 0.1% NP-40, 5 mM EGTA, 5 mM Trolox, 10 mM Sodium ascorbate acid, 10 mM Sodium azide, 1 mM PMSF). Streptavidin magnetic beads (Thermo Fisher Scientific, Catalogue number: 88,817) were washed twice with cell lysis buffer and 3.5 mg of each whole cell lysate sample were incubated with 100 µL magnetic bead slurry with rotation for 2 h at room temperature. After enrichment, the flowthrough was removed and beads were washed with 2 × 1 mL cell lysis buffer, 1 mL 1 M KCl, 1 mL 0.1 M Na2CO3, 1 mL of 2 M urea in 10 mM Tris-HCl (pH 8.0), and again with 2 × 1 mL cell lysis buffer. Biotinylated proteins were then eluted by boiling the magnetic beads in 30 µL 4 × Laemmli sample buffer (Bio-Rad, Catalogue number: 1,610,747) supplemented with 20 mM DTT and 2 mM biotin.
LC-MS/MS and label-free quantitative mass spectrometry proteomic analysis
The streptavidin-enriched proteins were profiled using label-free quantitative mass spectrometry as previously described [23] at Cedars-Sinai Medical Center Biomarker Discovery Platform Core.
Data analysis for RNA proximal proteins
Data were first filtered to exclude non-human proteins and proteins that were detected in only one or none of the U1 replicates or poly(A) replicates. Then, proteins detected with two or greater unique peptides were subjected to log2 transformation. Only the top gene name was kept from multiple candidates. Since U1 has compact structure in the pre-B complex and its size is much smaller than the biotinylating range of APEX2, experiments using three U1 gRNAs (U1-1, U1-2, U1-3) were considered as technical replicates to compare with nontargeting controls (NTC1, scrambled; NTC2, targeting poly[A]; NTC3, targeting poly[U]). Moderated t-test with a paired design was used to compare the log2-transformed iBAQ values between U1 and NTC or between poly(A) and poly(U) using limma package [24]. p values were adjusted using the Benjamini-Hochberg (BH) method [25] for multiple comparisons. Proteins with p < 0.05 were considered statistically significant. There are 226 U1 proximal proteins with p < 0.05, log2FC > 2, FDR < 0.25 and 786 poly(A) tail proximal proteins with BH-adjusted p < 0.05, log2FC > 2.
Cellular fractionation
Cells were fractionated as previously described with slight modifications [16]. Six million HEK293T cells were treated with plasma membrane lysis buffer (10 mM Tris-HCl, pH 7.5, 0.15% NP-40, 150 mM NaCl) on ice for 4 min after homogenization by flicking. Lysates were loaded onto a 24% sucrose cushion (24% RNase-free sucrose in plasma membrane lysis buffer) using large orifice tips, and centrifuged at 15,000 g for 10 min at 4°C. The supernatant (cytoplasmic fraction) was retained, and the pellet (nuclear fraction) was washed with 1 × PBS/1 mM EDTA and resuspended in 200 μL of 1 × PBS/1 mM EDTA. Fractionation efficiency was validated by western blot using antibodies against β-tubulin (Sigma Aldrich, Catalogue number: T8328, 1:2,000) as cytoplasmic marker and U1-70k (EMD Millipore, Catalogue number: 05–1588, 1:1,000) as nuclear marker.
RNA immunoprecipitation (RIP)
RIP was performed as previously described with slight modifications [16]. Twelve microlitre Dynabeads Protein A (Thermo Fisher Scientific, Catalogue number: 10001D) or Dynabeads Protein G (Thermo Fisher Scientific, Catalogue number: 10004D) were washed with 200 μL HBS (150 mM NaCl, 10 mM HEPES, pH7.5 by KOH) and incubated with 2 μg antibody (anti-HA, Santa Cruz, Catalogue number: sc-7392; anti-GTF2F2, Developmental Studies Hybridoma Bank, Catalogue number: PCRP-GTF2F2-1B3; anti-KPNB1, Bethyl Laboratories, Catalogue number: A300-482A; anti-AMOT, Bethyl Laboratories, Catalogue number: A303-305A; rabbit IgG isotype, Thermo Fisher Scientific, Catalogue number: 10,500 C; mouse IgG isotype, Santa Cruz, Catalogue number: sc-2025) in the presence of 80 μL HBS buffer at room temperature for 1 h. Eight million HEK293T cells were lysed with 800 μL cell lysis buffer (HBS, 0.1% NP-40, 5 mM EGTA, supplemented with 1 × protease inhibitor cocktail [Roche, Catalogue number: 11,873,580,001], 1 × PhosSTOP protease inhibitor cocktail [Roche, Catalogue number: 4,906,837,001], 1 mM PMSF [Sigma Aldrich, Catalogue number: 93,482], and 50 U Superase-in [Ambion, Catalogue number: AM2696]) at 4°C for 1 h. Cell debris and insoluble proteins were removed by centrifugation at 4°C, 12,000 g for 10 min. Supernatants were incubated with specific antibody-conjugated or isotype control IgG-conjugated Dynabeads at 4°C for 1 h. The Dynabeads were then washed 3 times with wash buffer (HBS, 0.1% NP-40) and aliquoted into two halves. Proteins associated with half of the Dynabeads were eluted with 22 μL 4 × Laemmli sample buffer (Bio-Rad, Catalogue number: 1,610,747) by boiling at 95°C for 5 min. RNA was extracted from the other half of Dynabeads using TRIzol LS (Thermo Fisher Scientific, Catalogue number: 10,296,028).
RT-qPCR
M-MLV reverse transcriptase (Promega, Catalogue number: M5301) and random hexamers (Promega, Catalogue number: C1181) were used for reverse transcription of RNA extracted from RIP experiments. Gene expression was quantified by RT-qPCR using iQ SYBR Green supermix (Bio-Rad, Catalogue number: 170–8886). The relative gene expression was calculated using the 2−ΔΔCt method and normalized to GAPDH. Five nanograms cDNA was used for RT-qPCR analysis on CFX96 Touch Real-Time PCR Detection System (Bio-Rad) using the following primer pairs:
U1-RT-For: 5ʹCCAGGGCGAGGCTTATCCATT3ʹ, U1-RT-Rev: 5ʹGCAGTCCCCCACTACCACAAAT3ʹ;
U2-RT-For: TTCTCGGCCTTTTGGCTAAG; U2-RT-Rev: CTCCCTGCTCCAAAAATCCA;
U6-RT-For: GCTTCGGCAGCACATATACTAAAAT; U6-RT-Rev: CGCTTCACGAATTTGCGTGTCAT;
5.8S-RT-For: GGTGGATCACTCGGCTCGT; 5.8S-RT-Rev: GCAAGTGCGTTCGAAGTGTC;
18S-RT-For: 5ʹCAGCCACCCGAGATTGAGCA3ʹ, 18S-RT-Rev: 5ʹTAGTAGCGACGGGCGTGTG3ʹ;
28S-RT-For: CCCAGTGCTCTGAATGTCAA; 28S-RT-Rev: AGTGGGAATCTCGTTCATCC;
GAPDH-RT-For: 5ʹTGCCAAATATGATGACATCAAGAA3ʹ; GAPDH-RT-Rev: 5ʹGGAGTGGGTGTCGCTGTTG3ʹ.
Enriched KEGG pathways in RNA proximal proteins and comparison of RNA proximal proteins with proteins from different gene ontology (GO) terms
Analysis of KEGG pathways enriched in RNA proximal proteins was conducted using SRTING [26]. Lists of human proteins were retrieved (04/13/2020) from QuickGO (https://www.ebi.ac.uk/QuickGO/) via searching corresponding GO terms and selecting ‘Homo sapiens (9606)’ under Taxon, except P-bodies and stress granule, which were both curated using data summarized from Wikipedia (04/24/2020) (https://en.wikipedia.org/wiki/P-bodies, https://en.wikipedia.org/wiki/Stress_granule). The venn diagrams were generated using online tools (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Western blot
Protein samples were run on 4–20% gradient precast protein gel (Bio-Rad, Catalogue number: 456–1096) and transferred onto PVDF membrane (Bio-Rad, Catalogue number: 1,704,157). After 1 h blocking, membranes were incubated with anti-FLAG (Santa Cruz, Catalogue number: sc-166,384, 1:1,000), anti-HA (Santa Cruz, Catalogue number: sc-7392, 1:1,000), anti-AMOT (Bethyl Laboratories, Catalogue number: A303-305A, 1:1000); anti-KPNB1 (Bethyl Laboratories, Catalogue number: A300-482A, 1:2000); anti-GTF2F2, Developmental Studies Hybridoma Bank, Catalogue number: PCRP-GTF2F2-1B3, 1:50), anti-biotin (Santa Cruz, Catalogue number: sc-57,636, 1:1,000), or anti- β-actin (Santa Cruz, Catalogue number: sc-47,778, 1:2,000) at 4°C overnight. Membranes were washed three times with Tris-buffered saline containing 0.2% Tween 20 (TBST) before incubating with HRP-conjugated secondary antibody at room temperature for 2 h. Clean-Blot IP detection reagent (Thermo Fisher Scientific, Catalogue number: 21,230) was used for blotting immunoprecipitated samples except GTF2F2. Then the membranes were incubated briefly with ECL Western Blotting Substrate (Thermo Fisher Scientific, Catalogue number: 32,106) after three times of wash with TBST. The membranes were exposed to HyBlot Autoradiography Film (Denville Scientific, Catalogue number: E3018).
Distance calculation
The distances between U1 snRNA and its proximal proteins identified by RPL in the pre-B complex structure (PDB ID: 6QX9) were measured using PyMOL [27]. The maximum distance from U1 snRNA (nucleotide 1) to the distant residues of U1 proximal proteins was used to estimate the actual distance (D1). Since no structure is currently available for PspCas13b, the structure of PbuCas13b (PDB ID: 6DTD) was used to infer the distance between U1 proximal proteins and APEX2 in the RPL protein. The average distances between gRNA (nucleotide 1, 12, and 23 of spacer) and the C-terminus of PbuCas13b, where the APEX2 was fused to, were measured (D2). The inferred distances between APEX2 and U1 proximal proteins were then calculated as absolute values of the differences between D1 and D2.
Data availability
Raw images for western blots and raw and processed mass spectrometry data are included as supporting files.
Results
Design and development of the RPL method
Inspired by applications of the RNA-targeting Type VI CRISPR-Cas systems [28–31] and proximity-dependent labelling with engineered soybean ascorbate peroxidase [32,33] or biotin ligase [34–36], we designed RPL, an RNA-centric approach based on a fusion protein of endonuclease-deficient Cas13 (dCas13) and proximity labelling enzyme APEX2 (Figure 1(a)). The fusion protein can be directed to target RNA with a sequence-specific guide RNA (gRNA). In the presence of hydrogen peroxide (H2O2), APEX2 in the fusion oxidizes substrate biotin-phenol (BP) into short-lived biotin-phenoxyl radicals which covalently react with electron-rich amino acids (e.g. tyrosine) on proteins within the biotinylating range of the fusion protein (Figure 1(a)). The biotinylated proteins, which will include proteins that bind to the target RNA directly or indirectly and potentially other proteins present within the biotinylating range, can be readily enriched using streptavidin beads and profiled by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure 1(a)).
Figure 1.
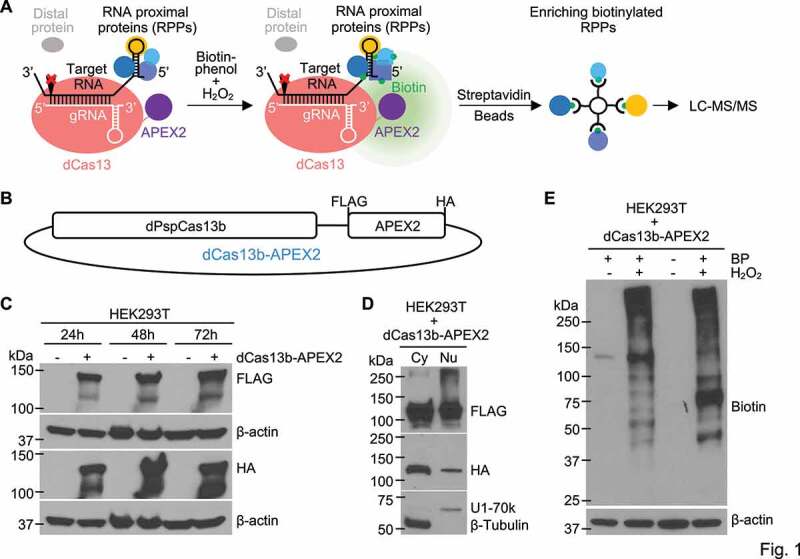
Designing and developing the RPL method. (a) Schematic illustration of the RPL workflow. A sequence-specific gRNA directs dCas13-APEX2 to target RNA and APEX2 in the fusion biotinylates target RNA proximal proteins in vivo in the presence of biotin-phenol and H2O2. Biotinylated RNA proximal proteins are then enriched using streptavidin beads and analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). (b) Diagram of the fusion protein dPspCas13b-FLAG-APEX2-HA (dCas13b-APEX2, or the RPL protein) expression construct. (c) Expression validation of the RPL protein by western blot. HEK293T cells transfected with or without the RPL plasmid were harvested 24 h-72 h post transfection and whole cell lysates were blotted with an anti-FLAG or anti-HA antibody. (d) The RPL protein is expressed in both the cytoplasm and the nucleus. HEK293T cells transfected with the RPL plasmid for 24 h were fractionated into cytoplasmic (Cy) and nuclear (Nu) fractions. Fractionation efficiency was evaluated by blotting for cytoplasmic protein β-Tubulin and nuclear protein U1-70k. (e) Validation of enzymatic activity of APEX2 in the RPL protein. HEK293T cells transfected with the RPL plasmid were treated with different combinations of biotin-phenol (BP) and H2O2. Proximity labelling was performed using different batches of HEK293T cells for lane 2 and lane 4. Whole cell lysates were blotted with anti-biotin antibody. β-actin in (C) and (E) was used as loading control
To construct the fusion protein, Cas13b was used for its high efficacy in RNA knockdown with minimal off-target effect [28] and high specificity in RNA labelling[37]. APEX2 was chosen as proximity labelling enzyme for its fast kinetics and high activity [32]. Catalytically dead Cas13b from Prevotella sp. P5-125 (dPspCas13b) [28] was fused to APEX2, with FLAG and HA tags incorporated (Figure 1(b)). The expression of the fusion protein dCas13b-APEX2 (from hereon in called the RPL protein) was confirmed by western blot using an anti-FLAG or anti-HA antibody (Figure 1(c)). The subcellular localization of the RPL protein was examined when ectopically expressed in HEK293T cells. Efficient separation between cytoplasmic and nuclear fractions was confirmed by blotting for cytoplasmic marker β-Tubulin and nuclear marker U1-70k. The RPL protein was detected in both the cytoplasmic and nuclear fractions (Figure 1(d)). To test if the peroxidase activity of APEX2 is maintained in the RPL protein, HEK293T cells were treated with BP and H2O2 24 h post transfection with the RPL plasmid. The detection of biotinylated proteins required both BP and H2O2, indicating that APEX2 in the RPL protein retains peroxidase activity (Figure 1(e)). The results also suggest that endogenous biotinylated proteins are rare in HEK293T cells and efficient biotinylation was not triggered by endogenous H2O2. These data indicate that the RPL protein has intact peroxidase activity and can be applied to target both cytoplasmic and nuclear transcripts.
Design and validation of gRNAs targeting U1 snRNA
To test our approach, we applied PRL to catalogue the U1 snRNA proximal proteins. The
U1 snRNA was selected for three reasons: (1) its high abundance [38], (2) structures of the human U1 small nuclear ribonucleoprotein (snRNP) and spliceosome are available [39–41], and (3) interacting proteins in both U1 snRNP [42] and spliceosome [43] have been well documented.
Since Cas13b targets only single-stranded RNA [28,44], three gRNAs (U1-1, U1-2, and U1-3) targeting single-stranded regions of U1 were designed based on its structure in the pre-B complex [39] (Figure 2(a)). We first tested whether U1 gRNAs direct wild-type PspCas13b to U1 and cleave it by measuring U1 expression in HEK293T cells cotransfected with plasmid expressing wild-type PspCas13b and plasmid expressing U1 gRNA or scrambled nontargeting control (NTC) gRNA at a 1:1 molar ratio. The expression of U1 was significantly lower in U1 gRNA-transfected cells compared with NTC gRNA-transfected cells (Figure 2(b)). The expression of a group of nontargets with a wide range of abundance was not affected (Figure 2(b)), except U2, which may be caused by Cas13b collateral activity [45] since U1 and U2 are in close contact during spliceosome assembly. The result indicated that U1 gRNAs can specifically direct PspCas13b to U1. We then tested if U1 gRNAs deliver the RPL protein to U1 using RNA immunoprecipitation. Since the U6 promoter is slightly stronger than the CMV promoter in HEK293T cells [46], a 1:2 molar ratio between the RPL plasmid (CMV promoter) and gRNA expressing plasmid (U6 promoter) was used to limit non-specific targeting due to excess RPL protein. The RPL protein was efficiently retrieved by anti-HA but not isotype control IgG (Figure 2(c)). Analysis of immunoprecipitated RNA showed that anti-HA pulled down ~5 times more RNA than control (Figure 2(d)), certifying RNA binding activity of the RPL protein. Although there is no significant difference among the amount of RNA pulled down by the RPL protein with NTC or U1 gRNAs (Figure 2(d)), U1 gRNAs significantly enriched U1 for ~2-3-fold compared with NTC gRNA (Figure 2(e)). The fact that much more abundant 18S was not enriched (Figure 2(e)) suggests that U1 gRNAs specifically direct the RPL protein to U1.
Figure 2.
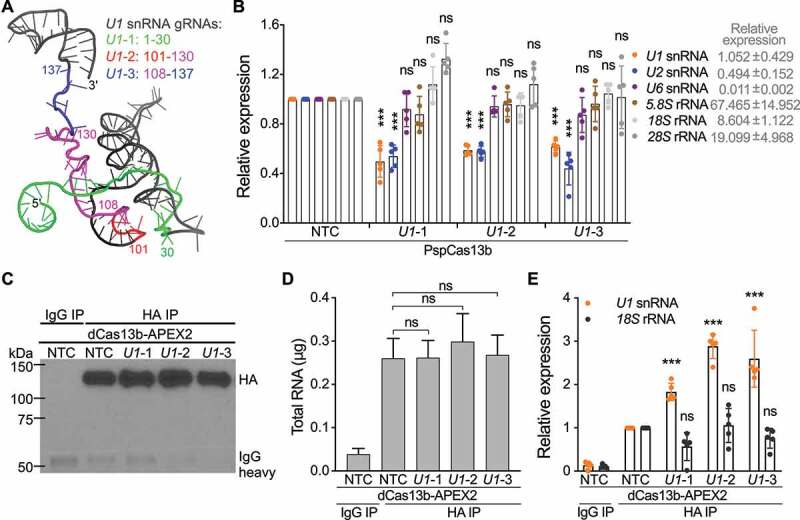
Designing and validating gRNAs targeting U1 snRNA. (a) Based on the structure of U1, three gRNAs with spacers targeting U1 nucleotides (nt) 1–30 (U1-1), 101–130 (U1-2), and 108–137 (U1-3) were designed. Cartoon representation of U1 (PDB ID: 6QX9, pre-B complex) is coloured in black, 1–30 in green, 101–107 in red, 108–130 in magenta, and 131–137 in blue. (b) The expression of U1 snRNA was significantly downregulated in U1 gRNA-transfected cells. HEK293T cells were cotransfected with plasmid expressing wild-type PspCas13b and plasmid expressing U1 or NTC gRNA (1:1 molar ratio). The expression of U1 or a group of nontargets was quantified by RT-qPCR and normalized to GAPDH. (c) Confirmation of pulldown of the RPL protein using western blot. HEK293T cells were cotransfected with the RPL plasmid and plasmid expressing U1 or NTC gRNA (1:2 molar ratio). Anti-HA antibody or isotype control IgG were used to immunoprecipitate the RPL protein. (d) The amount of total RNA extracted from immunoprecipitation experiments. (e) U1 gRNAs specifically directed the RPL protein to U1. The expression of U1 snRNA and nontarget 18S rRNA was quantified by RT-qPCR and normalized to GAPDH. Data shown in (b), (d) and (e) are mean ± SD from 5 independent experiments. ***p < 0.001, ns, not significant. Student’s t test
RPL-MS identified U1 RBPs related to U1 canonical and noncanonical roles
We next enriched proteins proximal to U1 using RPL. U1 has a compact structure in the pre-B complex [39] (Figure 2(a)) and its size (<10 nm in diameter) is much smaller than the biotinylating range of APEX2 (~20-40 nm or larger in diameter) [33,47,48], so we considered experiments performed using our three U1 gRNAs as technical replicates. We profiled streptavidin-enriched biotinylated proteins with LC-MS/MS (RPL-MS). Using label-free intensity-based absolute quantification (iBAQ) values to measure enrichment in U1 gRNA sample relative to protein amounts in the NTC gRNA sample, U1 RPL-MS identified 226 proteins (p < 0.05 and log2 fold change [FC] > 2, false discovery rate [FDR] <0.25, Benjamini-Hochberg method). No proteins were enriched in NTC gRNA transfected cells using the same criteria (Figure 3(a), Table S1). U1 proximal proteins included known direct U1 binding partners (e.g. SNRNP70, also known as U1-70k) [42] and proteins in the spliceosome (e.g. SNRPA1 and SNRPB2) [43] that interact with U1 indirectly (Figure 3(a)). We verified the enrichment of U1-70k using western blot and found that it was significantly enriched ~2-fold by all three U1 gRNAs (Figure 3(b)), consistent with the RPL-MS results (Figure 3(a)). Analysis of KEGG pathways [26] enriched in U1 proximal proteins showed that ‘Spliceosome’ is the most significantly enriched pathway (FDR < 10−[8]) (Figure 3(c)), which aligns with U1 canonical role in spliceosome assembly, suggesting that RPL enriches the most relevant proteins to target RNA. Indeed, proteins proximal to U1 included 99 splicing
Figure 3.
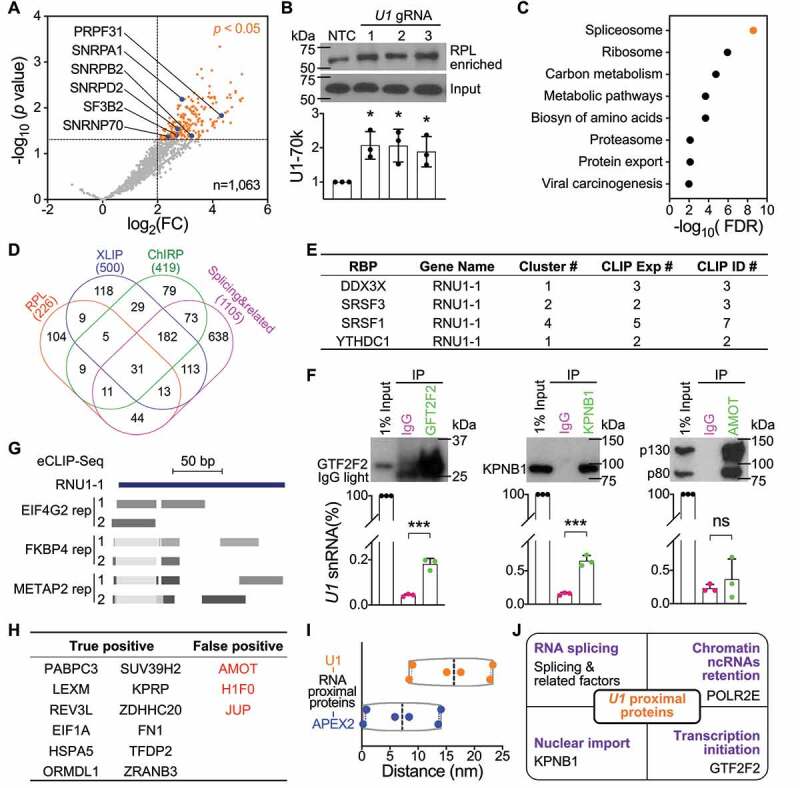
U1 RNA Proximity Labelling-Mass Spectrometry. (a) U1 proximal proteins revealed by RPL-MS include known U1 RBPs. Volcano plot shows U1/NTC iBAQ ratio (fold change, FC) of protein quantification in U1 gRNA cells compared with NTC gRNA cells. RPL-MS enriched 226 U1 proximal proteins (orange dots, p < 0.05 and log2 FC > 2, FDR < 0.25, Benjamini-Hochberg method). Each dot represents the average value from experiments using three U1 gRNAs. Blue dots represent proteins from the pre-B spliceosome complex. (b) U1 direct RBP U1-70k was enriched by RPL. HEK293T cells transfected with the RPL plasmid and plasmid expressing U1 gRNA or NTC gRNA were treated with BP and H2O2. U1-70k from whole cell lysates (Input) or streptavidin-enriched biotinylated proteins (RPL enriched) was blotted and representative result is shown on the top panel. Quantifications are mean ± SD from 3 independent experiments. *p < 0.05, Student’s t test. (c) KEGG pathways significantly enriched in U1 proximal proteins, identified using STRING. (d) Comparison of U1 proximal proteins enriched by RPL, U1 interactors identified by ChIRP-MS, U1 interactors identified by XLIP-MS using anti-U1A and/or anti-U1-70k antibody, and splicing & related proteins. Numbers listed below are total number of proteins from each group. (e) List of 4 U1 proximal proteins with CLIP-Seq data supporting their association of U1 found in ENCORI. (f) Validation of 3 U1 proximal proteins as U1 RBPs via RNA immunoprecipitation. Top panel are western blots of IP samples using corresponding antibody or isotype control IgG. Lower panel are RT-PCR results of IP samples showing the relative expression of U1. Data shown are mean ± SD from 3 independent experiments. ***p < 0.001, ns, not significant. Student’s t test. (g) eCLIP-Seq data for EIF4G2 and METAP2 were performed in K562 cells and for FKBP4 was performed in HepG2 cells. Reads mapped to minus strand of chr1:149,224,044–149,224,238 (hg19) are shown in dense mode for both replicates. (h). Estimation of false-positive rate for the RPL method. A list of 15 binding proteins uniquely for lncRNA UCA1 but not for U1 as true negatives for U1. Proteins highlighted in red are false-positive proteins that do not associate with U1. (i). Inferred maximum distances between U1 or APEX2 in the RPL protein and 6 U1 proximal proteins present in the pre-B complex shown in (A). (j). Summary of U1 proximal proteins related to U1 canonical and noncanonical functions
and related factors [49], 56 proteins found by U1 ChIRP-MS [13], and 58 proteins revealed by XLIP-MS using an anti-U1A and/or anti-U1-70k antibody [50] (Figure 3(d)). The binding between U1 and four U1 proximal proteins that were also revealed by ChIRP-MS and/or XLIP-MS was further supported by crosslinking immunoprecipitation sequencing (CLIP-Seq) data as shown in ENCORI [51] (Figure 3(e)).
For the novel U1 proximal proteins identified, we generated additional experimental evidence supporting their interactions with U1. Three candidates with different rankings based on log2(FC) and p value were chosen for validation using RNA immunoprecipitation: GTF2F2 (also previously identified by ChIRP-MS), U1 nuclear import factor KPNB1 (recovered by RPL but not by XLIP-MS nor ChIRP-MS) and AMOT (an U1 proximal protein identified only by RPL). All three proteins were efficiently pulled down by the corresponding antibody (Figure 3(f)). GTF2F2 and KPNB1 enrich significantly more U1 compared with isotype control IgG but not AMOT (Figure 3(f)), indicating that GTF2F2 and KPNB1 are true U1 RBPs but not AMOT, consistent with our previous notion that AMOT does not bind to U116. We then turned to enhanced CLIP sequencing (eCLIP-Seq) data [52] from the ENCORE project. Among the 104 unique U1 proximal proteins (Figure 3(d)), there were 3 RBPs with eCLIP-Seq data available (EIF4G2, METAP2, and FKBP4), all of which have reads mapped to U1 (Figure 3(g)), confirming that they are true U1 RBPs. The positive rate for novel U1 proximal proteins examined to be U1 RBPs is estimated to be 75% (3/4). Next, a group of 15 proteins that are candidate RBPs uniquely associated with lncRNA UCA1 [16] but not with U1 [13,16,50] were used as true negatives for U1 to estimate false-positive rate of the RPL method (Figure 3(h)). Three proteins including AMOT were significantly enriched in U1 proximal proteins, while the other twelve were not (Figure 3(h)). We estimate
that the ffalse-positiverate for the RPL method to be around 20% (3/15). It should be noted that both estimations are based on a small proportion of the data and additional experimental evidence will be required to accurately determine the specificity of the method.
We then used the 6sixRBPs occurring in the pre-B complex [39] that were enriched by U1 RPL-MS (Figure 3(a)) to infer the biotinylating range for the RPL protein, recognizing that accuracy may be impaired by the dynamic participation of U1 snRNA in the pre-B complex [53]. The inferred distances between APEX2 in the RPL protein and those six RBPs are all smaller than 15 nm and the average is 7.2 nm (Figure 3(i)), suggesting that APEX2 biotinylates proteins within 15 nm. The inferred distances between U1 and those six RBPs range from 8.4 nm to 23.3 nm with an average of 16.4 nm (Figure 3(i)), suggesting that RPL can biotinylate proximal proteins within ~25 nm of target RNA.
Proximal proteins identified by U1 RPL-MS also included previously reported U1 interactor RNA polymerase II [54,55] (Figure 3(j)), which is required for a noncanonical function of U1 in chromatin retention of ncRNAs [56,57]. The interaction between GTF2F2 and U1 (Figure 3(f)) may relate to its role in regulation of transcription initiation [58,59] (Figure 3(j)). This role likely extends beyond, as U1 RPL retrieved a total of 15 proteins involved in chromatin remodelling, DNA modification, histone modification, and transcription (Table S1) [60,61]. These data indicate that RPL enables efficient identification of validated U1 RBPs associated with both U1 canonical and noncanonical functions.
RPL-MS recovered expected categories of proteins for poly(A) tails
To further test the generality of RPL, we applied it to poly(A) tails, which are adenosines added to the 3ʹ ends of the majority of eukaryotic mRNAs and many lncRNAs in the absence of template [62–65]. Poly(A) tails play a critical role in mRNA translation and stability [66] and their removal triggers mRNA decapping and decay [67–69]. Although the 5ʹ and 3ʹ ends of pre-translational mRNAs [70] and actively deadenylating mRNAs [71] are distant (Figure 4(a), 5ʹ-3ʹ distal model), the physical distances between the two ends of diverse RNAs are incredibly close regardless of their nature [21,22] (Figure 4(a), 5ʹ-3ʹ proximal model). Since oligomers of 30 nt poly(U) rarely occur in the human transcriptome [72,73], poly(U)-targeting gRNA, or poly(U) gRNA, was used as negative control. The RPL plasmid was cotransfected with plasmid expressing poly(A) or poly(U) gRNA into HEK293T cells at a 1:2 molar ratio and RPL was performed. Using label-free iBAQ values to measure enrichment of proteins in poly(A) gRNA transfected samples relative to the poly(U) gRNA transfected samples, RPL-MS enriched 786 proteins as poly(A) tail proximal proteins (Benjamini-Hochberg-adjusted p < 0.05 and log2FC > 2). No proteins were enriched in the negative control (Figure 4(b), Table S2). Poly(A) tail proximal proteins included 7 poly(A) binding proteins, 15 3ʹUTR binding proteins, 10 5ʹUTR binding proteins, and 1 cap binding protein (Figure 4(b), Table S3), all of which are known to associate with poly(A) tails. Retrieval of proteins from both 5ʹ and 3ʹ ends by RPL within a small radius provided additional evidence for poly(A)+ RNA 5ʹ-3ʹ proximity [21,22]. Poly(A) tail proximal proteins include 117 proteins associated with U1, which were most enriched in the ‘Spliceosome’ pathway. The majority of poly(A) tail proximal proteins are unique to poly(A) tail RPL-MS and contain the majority of known categories of proteins associated with poly(A) tails (Figure 4(c)). At least 48% of poly(A) tail proximal proteins were poly(A)+ RBPs [74–78] (Figure 4(d)). In theory, poly(A) gRNA can direct the RPL protein to any transcript with a poly(A) tail no shorter than 30 nt (Figure 4(a)), including transcripts actively undergoing polyadenylation, readenylation, deadenylation, or translation. Indeed, poly(A) tail RPL-MS enriched five cleavage and polyadenylation factors for poly(A)+ RNA (Figure 4(e)) but none of the factors unique for poly(A)− RNA (e.g. SLBP and ZNF473) [79] (Table S3). Moreover, poly(A) tail proximal proteins included three exosome proteins [80], 2 deadenylase complex proteins [81], and decapping factor EDC3 [82] (Figure 4(e), Table S3). Importantly, 20 translation initiation factors, 15 translation elongation factors, 70 ribosomal subunits, and 18 tRNA ligases were also
Figure 4.
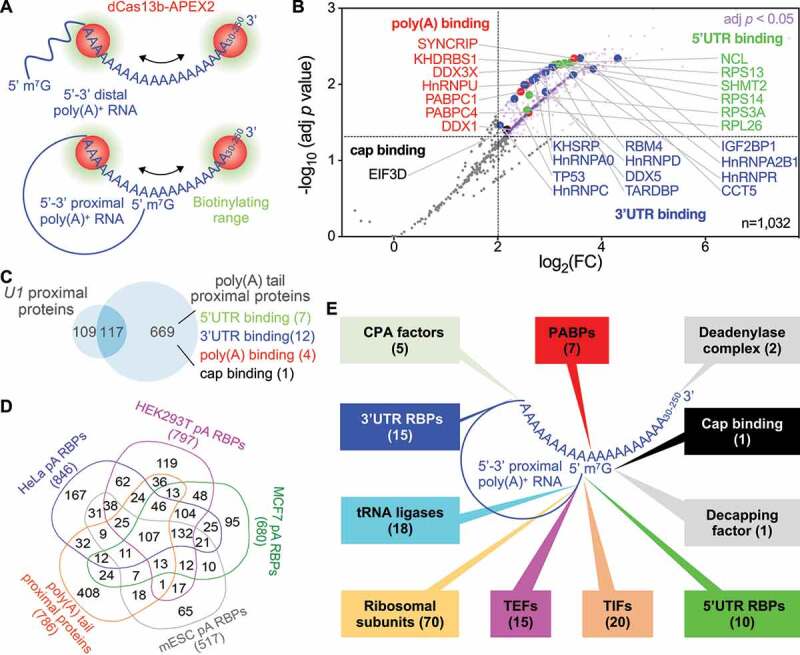
RPL-MS analysis of poly(A) tail proximal proteins in HEK293T cells. (a) Application of RPL to poly(A) tails. In the presence of gRNA, the RPL protein (dCas13b-APEX2) is directed to poly(A) tails ranging from 30 nt up to ~250 nt. In the 5ʹ-3ʹ distal model, RPL will detect poly(A) binding proteins and 3ʹUTR binding proteins that bind proximal to poly(A) tail within the range of biotinylation. In the 5ʹ-3ʹ proximal model, RPL will also identify cap-binding proteins and 5ʹUTR binding proteins that bind proximal to the cap and lie within the biotinylating range. (b) Volcano plot shows RPL-labelled proteins in HEK293T cells. For each protein, the poly(A)/poly(U) iBAQ ratio reflects the enrichment of identified protein in poly(A) gRNA cells compared with poly(U) gRNA transfected cells. RPL-MS identified 786 proteins (light purple dots) as significantly enriched (Benjamini-Hochberg-adjusted p < 0.05 and log2FC > 2). Each data point represents the average value from biological triplicates. Red dots represent poly(A) binding proteins, blue dots 3ʹUTR binding proteins, green dots 5ʹUTR binding proteins, and the black dot denotes a cap binding protein. (c) Venn diagram shows the comparison between poly(A) tail and U1 RPL experiments. KEGG pathways enriched in shared proteins (FDR < 0.01). Authentic categories of proteins associated with poly(A) tail are indicated and number of proteins are shown in brackets. (d) Venn diagram shows the comparison of proteins associated with poly(A)+ RNA in different cells. Numbers below each group represent the size. (e) Summary of expected categories of poly(A) tail proximal proteins that support 5ʹ-3ʹ proximity and the role of poly(A) tail in mRNA translation. Each category of proteins points to a location/region of poly(A)+ RNA where they most likely associate with when identified by RPL. PABPs, poly(A) binding proteins; CPA, cleavage and polyadenylation; TIFs, translation initiation factors; TEFs, translation elongation factors
identified by poly(A) tail RPL-MS (Figure 4(e), Table S3), putatively supporting a model that the poly(A) tail recruits translation initiation factors to initiate translation at the 5ʹ end like their viral counterparts [79,83]. Moreover, poly(A) tail RPL-MS revealed 12 proteins involved in degradation of AU-rich element-containing mRNAs and 66 nonsense-mediated decay proteins (including 58 ribosomal subunits) [84–86] (Table S3), further suggesting that RPL enables efficient discovery of most relevant and validated RBPs proximal to poly(A) tails.
Poly(A) tail RPL-MS expands the repertoire of subcellular compartments occupied by polyadenylated transcripts
Poly(A) tails are important for RNA nuclear export [87] via the nuclear pore complex [88]. This is further supported by the presence of 90 mRNA processing factors, 20 mRNA nuclear export proteins, and 13 nuclear pore complex proteins in poly(A) tail RPL-MS (Figure 5(a)). Poly(A) tail proximal proteins included eight tRNA processing factors, five tRNA nuclear export factors [89], three pri-miRNA processing factors, and two pre-miRNA export factors [90–92] (Table S3), supporting that their processing is coupled with export [93,94]. tRNA and pre-miRNA nuclear export factors were expected since their precursors are also polyadenylated [95,96]. Poly(A) tail RPL-MS recovered 27 proteins involved in mRNA transport (including zipcodes binding protein IGF2BP1) and 48 microtubule proteins, and 139 plasma membrane proteins that are used by mRNAs to direct subcellular localization [97,98] (Figure 5(a)), possibly suggesting a role for the poly(A) tail in RNA subcellular localization.
Figure 5.
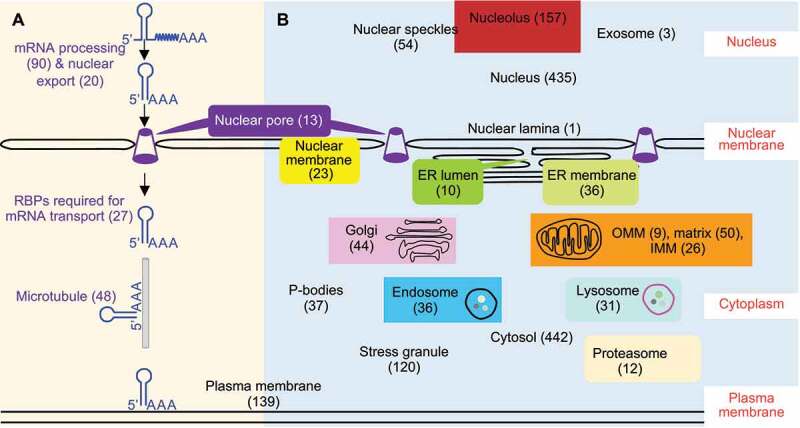
Poly(A) tail proximal proteins included proteins involved in subcellular localization of RNA. (a) RPL-MS revealed proteins involved in RNA processing, nuclear export, transport, and subcellular localization for poly(A) tail proximal proteins. (b) A putative subcellular localization map of poly(A)+ RNA built upon subcellular localization of poly(A) tail proximal proteins. Poly(A) tail proximal proteins were compared with proteins extracted from corresponding GO terms. Numbers in brackets represent the size of each category of proteins. A full list of proteins in each category can be found in Table S3
Many of the poly(A) tail proximal proteins we identified are known to have high fidelity to discrete localization patterns in vivo. We reasoned that we could leverage this knowledge to construct a putative localization map for poly(A)+ RNA across different subcellular compartments (Figure 5(b), Table S3). The results are generally consistent with previous reports that both mRNAs and noncoding RNAs have multiple subcellular localizations [47,99–101] and also support the presence of mRNAs in P-bodies, stress granule, and the exosome [80,102]. Interestingly, poly(A) tail RPL-MS also identified marker proteins of the endosome, lysosome, proteasome, and Golgi apparatus, indicative of expanded subcellular localizations for poly(A)+ RNA (Figure 5(b), Table S3). Discovery of lysosomal and proteasomal proteins associated with poly(A) tails is consistent with the existence of an RNA degradation pathway ‘RNautophagy’ in the lysosome [103] and degradation function of proteasomes for AU-rich element-containing mRNAs [85,86]. The identification of endosomal proteins supports emerging evidence that late endosomes can be used by mRNAs as a platform for translation [104]. Poly(A) tail proximal proteins included Golgi marker cis-Golgi matrix protein GOLGA2 [105] (Table S3), which has recently been annotated as an RBP by multiple groups [106–108], suggesting that Golgi may be an unexplored subcellular location for poly(A)+ RNA. More experimental data are needed to determine which specific transcripts are associated with GOLGA2 in the Golgi apparatus and the biological significance of those interactions.
Discussion
RPL: an RNA-centric approach for identification of RRNA–protein interactions in living cells
We present an RNA-centric method, RPL, for cataloguing proteins proximal to transcripts of interest and evaluate the approach in two distinct contexts–first interrogating a specific ncRNA U1 and second surveying the heterogenous group of poly(A)+ RNA in living human cells. Both analyses demonstrated that RPL enables efficient discovery of functionally relevant RBPs for target transcripts. Compared with alternative methods, RPL needs no crosslinking or sonication, requires far fewer cells (~20-40 million vs ~100-800 million) and involves no genetic manipulation, which may interfere with target RNA functions [20]. The short pulse of labelling potentially permits RPL to be applied to study dynamic RRNA–proteininteractions. Recently, APEX2 has also been reported to biotinylate proximal nucleic acids [47,48,109], suggesting that RPL could be applied to identify RNA and DNA in addition to proteins proximal to the target RNA (together as ‘RNA proximitome’) within living cells.
RPL may also permit the identification of novel RNA–proteins interactions, as shown by the recovery and validation of novel U1 proximal proteins. We noted that KPNB1 was identified as a U1 interactor by RPL but not by ChIRP or iRAP methods [13,16], suggesting that RPL allows to detect transient and/or weak interacting proteins [110,111] usually missed by existing antisense probe-based methods due to their harsh conditions.
During the preparation of our manuscript, similar strategies using different fusion proteins of endonuclease-deficient Cas13 protein (dLwaCas13a, dPspCas13b, and dRfxCas13d) and proximity labelling enzyme (APEX2, BioID2, BASU, and PafA) were reported [112–115]. Applications of these methods together with ours to both mRNAs and ncRNAs with wide range of abundance (~102 – 106 copies/cell) demonstrate that these methods have broad potential to identify functional relevant RBPs for diverse transcripts.
Proteins recovered by RPL are expected to include three classes of proteins: proteins that directly bind to target RNA, proteins that are associated with target RNA indirectly via pprotein–proteininteractions, and proteins just present within the biotinylating range. More biological replicates (3) are expected to help enrich the first two groups of RBPs and reduce non-interacting ffalse-positivehits that are not expected to be enriched repeatedly. In addition, an optimal molar ratio between the fusion protein and gRNA, which enables efficient proximity-based biotinylation and prevents non-specific labelling due to excess fusion protein, is crucial for separating signal from noise. When possible, lower expression of the fusion protein, coupled with carefully selected negative control gRNAs, are expected to reduce the background signal. A validated set of gRNAs that can specifically direct the fusion protein to the target RNA with low off-target activity is also key. As complementarity between the gRNA spacer and targeted region as well as local RNA accessibility aisessential for RNA targeting [28,29,31,44], general principles for gRNA designing can provide critical help in choosing spacer sequence and length for gRNA aiming at single-stranded regions of the target RNA [116,117]. Validation of interacting proteins using protein-centric methods such as RNA immunoprecipitation or cross-linking immunoprecipitation in concert with RT-PCR or sequencing will provide crucial orthogonal evidence for newly discovered RRNA–proteininteractions.
Limitations and directions for improvement
Although the 1 minute treatment with 1 mM H2O2 used in the RPL method did not cause
enrichment of oxidative RBPs (such as PNPT1, SF3B4, and DAZAP1) in our two applications, such high concentration of H2O2 may cause oxidative stress and necrosis to the cells [118] and may preclude the application of RPL to systems sensitive to oxidative stress and cell harm. An alternative option is to use biotin ligase in the fusion protein instead of APEX2 to avoid introducing oxidative damage.
The large size (~1130 kDa of the RPL protein and other similar fusion proteins may pose steric hindrance to restrict access to the target RNA and may increase the biotinylating range and reduce specificity. This could potentially limit the application to high-resolution mapping of RNA functional domains [119] for some transcripts. In such scenarios, smaller Cas13 proteins by structure-guided truncations may be more appropriate [120]. Alternatively, the CIRTS strategy could be applied to assemble a smaller gRNA-dependent RNA proximity labelling enzyme [121].
Another limitation is that RPL and similar tools may not be as efficient as antisense probe-based methods. This is exemplified by the discrepancy in recovery of U1 snRNA direct binding proteins in U1 snRNP: antisense probe-based methods ChIRP or iRAP identified eight or six U1 snRNA direct binding proteins while RPL recovered only 313[16]. Likely, this could be improved by increasing the number of replicates to increase the power of the method, as seven U1 snRNA direct binding proteins were detected by U1 gRNAs but four of these were not enriched relative to the scrambled control. In addition, using gRNAs with higher specificity and lower off-target effect and/or controllable proximity labelling enzymes (such as split APEX2 [122] and/or chemical inducers of dimerization [123]) are potential ways to improve sensitivity by increasing the signal-to-noise-ratio. The lower efficacy of RPL in part could be due to the requirement for electron-rich amino acids like tyrosine to be exposed on the surface and within the biotinylating range [33]. The same limitation also applies to other proximity labelling enzymes including BioID and its relatives and PafA, which all favour lysine as labelling substrate [124,125]. Another possible reason is that the RPL protein can only access single-stranded regions of target RNA [28,44] and it has to compete with the RBPs bound to the target transcript [117].
Perspectives
Since both Cas13s and proximity labelling are very active research areas, further optimization and refinements of RPL and similar methods are expected. We anticipate that with further improvements RPL and similar methods will be widely applied to identify in vivo interacting proteins and associated nucleic acids of diverse categories of RNAs including both mRNAs and ncRNAs in multiple cell types and organisms. RPL and related technologies are expected to be particularly helpful in discovering transient and/or weak interactors of target RNAs. Combined with existing hybridization-based purification methods, RPL and similar tools will allow identification of a more complete interacting proteome of target RNAs, which facilitate their functional characterization and mechanistic dissection. Use of these tools together with protein-centric methods [126,127] and annotation of RNA structure [128,129] will shed light on the mechanisms and regulation of lncRNA functions, RRNA–proteininteractions, RRNA–RNAinteractions, RNA-DNA/chromatin interactions, RNA functional domains, and binding specificities for RBPs.
Supplementary Material
Acknowledgments
This work was supported by the Ovarian Cancer Research Alliance (Ann and Sol Schreiber Mentored Investigator Award 458799 to X.L.) and the National Cancer Institute (K99/R00-CA184415 and R01-CA207456 to K.L.), the Cedars-Sinai Samuel Oschin Comprehensive Cancer Institute (Cancer Biology Grant 231433 to K.L. and J.J.B.). We thank Drs. Wei Yang and Bo Zhou at the Cedars-Sinai Medical Center Biomarker Discovery Platform Core for label-free quantitative mass spectrometry analysis. We also thank anonymous reviewers for their insightful comments and suggestions.
Funding Statement
This work was supported by the Cedars-Sinai Medical Center [Cancer Biology Program Discovery Fund-213433]; National Cancer Institute [K99/R00-CA184415]; National Cancer Institute [R01-CA207456]; Ovarian Cancer Research Alliance [Ann and Sol Schreiber Mentored Investigator Award-458799].
Disclosure statement
The authors declare no competing interests.
Author contributions
X.L conceived the project, designed and performed all experiments, and analysed the data. R.I.C. and M.A.S.F. analysed the data. J.J.B. contributed reagents. K.L. supervised the project. X.L. and K.L. wrote the manuscript with input from all authors.
Supplemental data
Supplemental data for this article can be accessed here.
References
- [1].International human genome sequencing consortium. Initial sequencing and analysis of the human genome. Nature [Internet] 2001. [cited 2020 Jul 6]; 409:860–921. Available from. http://www.nature.com/articles/35057062.11237011 [Google Scholar]
- [2].Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. Internet] 2001. [cited 2020 Jul 6]; 291:1304–1351. Available from.(5507):https://www.sciencemag.org/lookup/doi/10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- [3].Berretta J, Morillon A.. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep Internet] 2009. [cited 2020 May 12]; 10(9):973–982. Available from. https://onlinelibrary.wiley.com/doi/abs/10.1038/embor.2009.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature [Internet] 2012. 4897414: 101–108Available from. http://www.ncbi.nlm.nih.gov/pubmed/22955620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Batista PJ, Chang HY.. Long noncoding RNAs: cellular address codes in development and disease. Cell Internet] 2013. [cited 2020 May 13]; 152(6):1298–1307. Available from. https://linkinghub.elsevier.com/retrieve/pii/S0092867413002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Iyer MK, Niknafs YS, Malik R, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. Internet] 2015. [cited 2020 May 13]; 47(3): 199–208. Available from. http://www.nature.com/articles/ng.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang L, Froberg JE, Lee JT. Long noncoding RNAs: fresh perspectives into the RNA world. Trends Biochem Sci Internet] 2014. [cited 2020 May 13]; 39(1): 35–43. Available from.https://linkinghub.elsevier.com/retrieve/pii/S0968000413001710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell Internet] 2018. [cited 2020 May 13]; 172(3): 393–407. Available from.https://linkinghub.elsevier.com/retrieve/pii/S0092867418300485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol Internet] 2002. [cited 2020 May 12]; 3(3):195–205. Available from. http://www.nature.com/articles/nrm760 [DOI] [PubMed] [Google Scholar]
- [10].Glisovic T, Bachorik JL, Yong J, et al. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582(14):1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hentze MW, Castello A, Schwarzl T, et al. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol Internet] 2018. [cited 2020 May 12]; 19(5): 327–341. Available from http://www.nature.com/articles/nrm.2017.130 [DOI] [PubMed] [Google Scholar]
- [12].Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol Internet] 2007. [cited 2020 May 12]; 8(6): 479–490. Available from http://www.nature.com/articles/nrm2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chu C, Zhang QC, Da Rocha ST, et al., Systematic discovery of Xist RNA binding proteins. Cell. Internet] 2015; 161(2): 404–416. Available from. https://www.ncbi.nlm.nih.gov/pubmed/25843628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McHugh CA, Chen CK, Chow A, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. Internet] 2015; 521(7551): 232–236. Available from http://www.ncbi.nlm.nih.gov/pubmed/25915022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Panhale A, Richter FM, Ramírez F, et al. CAPRI enables comparison of evolutionarily conserved RNA interacting regions. Nat Commun Internet] 2019. [cited 2020 May 13]; 10(1): 2682. Available from http://www.nature.com/articles/s41467-019-10585–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin X, Spindler TJ, De Souza Fonseca MA, et al. Super-enhancer-associated LncRNA UCA1 interacts directly with AMOT to activate YAP target genes in epithelial ovarian cancer. iScience. 2019;17:242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wurtmann EJ, Wolin SL. RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol. 2009;44(1):34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jj B, Cowing-Zitron C, Nakatsuji T, et al. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med Internet] 2012. [cited 2020 Jul 28]; 18(8):1286–1290. Available from.http://www.nature.com/articles/nm.2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Urdaneta EC, Beckmann BM. Fast and unbiased purification of RNA-protein complexes after UV cross-linking. Methods [Internet] 2019. [cited 2020 May 13];:S1046202318304869. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1046202318304869 [DOI] [PubMed]
- [20].Laprade H, Querido E, Smith MJ, et al. Imaging of telomerase RNA reveals a recruitment-retention model for telomere elongation. Mol Cell Internet] 2020. [cited 2020 Aug 28]; 79(1): 115–126.e6. Available from https://linkinghub.elsevier.com/retrieve/pii/S1097276520303063 [DOI] [PubMed] [Google Scholar]
- [21].Lai W-JC, Kayedkhordeh M, Cornell EV, et al. mRNAs and lncRNAs intrinsically form secondary structures with short end-to-end distances. Nat Commun. 2018;9(1):4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Leija-Martínez N, Casas-Flores S, Cadena-Nava RD, et al. The separation between the 5′-3′ ends in long RNA molecules is short and nearly constant. Nucleic Acids Res Internet] 2014. [cited 2020 Aug 18]; 42(22): 13963–13968. Available from http://academic.oup.com/nar/article/42/22/13963/2411719/The-separation-between-the-53-ends-in-long-RNA [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou B, Wang Y, Yan Y, et al. Low-Background Acyl-biotinyl exchange largely eliminates the coisolation of non-s-acylated proteins and enables DeepS-acylproteomic analysis. Anal Chem Internet] 2019. [cited 2019 Sep 27]; 91(15): 9858–9866. Available from https://pubs.acs.org/doi/10.1021/acs.analchem.9b01520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol Internet] 2004. [cited 2020 Aug 14]; 3(1): 1–25. Available from.https://www.degruyter.com/view/j/sagmb.2004.3.issue-1/sagmb.2004.3.1.1027/sagmb.2004.3.1.1027.xml [DOI] [PubMed] [Google Scholar]
- [25].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. Internet] [cited 2020 Aug 14] Available from [Google Scholar]
- [26].Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Schrodinger LLC. The PyMOL molecular graphics system. Version 1.8. 2015. [Google Scholar]
- [28].Cox DBT, Gootenberg JS, Abudayyeh OO, et al. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550(7675):280–284. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yan WX, Chong S, Zhang H, et al. Cas13d Is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-Domain-containing accessory protein. Mol Cell. 2018;70(2):327–339.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Konermann S, Lotfy P, Brideau NJ, et al. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018;173(3):665–676.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ss L, Jd M, Kj K, et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12(1):51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].H-w R, Zou P, Nd U, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science Internet] 2013. [cited 2020 Mar 30]; 339(6125): 1328–1331. Available from. https://www.sciencemag.org/lookup/doi/10.1126/science.1230593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tc B, Ja B, Ad S, et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 2018;36(9):880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim DI, Jensen SC, Noble KA, et al. An improved smaller biotin ligase for BioID proximity labeling. MBoC Internet] 2016. [cited 2020 Jul 21]; 27(8): 1188–1196. Available from. https://www.molbiolcell.org/doi/10.1091/mbc.E15–12–0844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Roux KJ, Kim DI, Raida M, et al. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol Internet] 2012. [cited 2020 Mar 30]; 196(6):801–810. Available from.https://rupress.org/jcb/article/196/6/801/36934/A-promiscuous-biotin-ligase-fusion-protein [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang L-Z, Wang Y, Li S-Q, et al. Dynamic Imaging of RNA in living cells by CRISPR-Cas13 systems. Mol Cell Internet] 2019. [cited 2020 May 14]; 76(6): 981–997.e7. Available fromhttps://linkinghub.elsevier.com/retrieve/pii/S1097276519308020 [DOI] [PubMed] [Google Scholar]
- [38].Gesteland RF. The RNA world: the nature of modern RNA suggests a prebiotic RNA world. New York: Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- [39].Charenton C, Wilkinson ME, Nagai K. Mechanism of 5ʹ splice site transfer for human spliceosome activation. Science Internet] 2019. [cited 2020 Apr 26]; 364(6438): 362–367. Available fromhttps://www.sciencemag.org/lookup/doi/10.1126/science.aax3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pomeranz Krummel DA, Oubridge C, Leung AKW, et al. Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution. Nature [Internet] 2009. [cited 2020 May 1]; 458:475–480. Available from: http://www.nature.com/articles/nature07851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Weber G, Trowitzsch S, Kastner B, et al. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J Internet] 2010. [cited 2020 May 1]; 29(24):4172–4184. Available from http://emboj.embopress.org/cgi/doi/10.1038/emboj.2010.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stark H, Dube P, Lührmann R, et al. Arrangement of RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature. 2001;409(6819):539–542. [DOI] [PubMed] [Google Scholar]
- [43].Zhou Z, Licklider LJ, Gygi SP, et al. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419(6903):182–185. [DOI] [PubMed] [Google Scholar]
- [44].Smargon AA, Cox DBT, Pyzocha NK, et al. Cas13b Is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell. 2017;65(4):618–630.e7. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gootenberg JS, Abudayyeh OO, Kellner MJ, et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science Internet] 2018. [cited 2020 Jul 25]; 360(6387: 439–444. Available fromhttps://www.sciencemag.org/lookup/doi/10.1126/science.aaq0179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lebbink RJ, Lowe M, Chan T, et al. Polymerase II promoter strength determines efficacy of microRNA adapted shRNAs. PLoS ONE. 2011;6(10):e26213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fm F, Han S, Kr P, et al. Atlas of subcellular RNA localization revealed by APEX-Seq. Cell. 2019;178(2):473–490.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Padrón A, Iwasaki S, Ingolia NT. Proximity RNA labeling by APEX-Seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol Cell Internet] 2019. [cited 2019 Aug 26]; 75(4): 875–887.e5. Available from.https://linkinghub.elsevier.com/retrieve/pii/S1097276519305854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cvitkovic I, Jurica MS. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013;41(D1):D132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].So BR, Di C, Cai Z, et al. Complex of U1 snRNP with Cleavage and Polyadenylation factors controls telescripting, regulating mRNA transcription in human cells. Mol Cell Internet] 2019. [cited 2020 Apr 30]; 76(4): 590–599.e4. Available from. https://linkinghub.elsevier.com/retrieve/pii/S1097276519306239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Li J-H, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucl Acids Res Internet] 2014. [cited 2020 Aug 8]; 42(D1): D92–7. Available from.https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkt1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Van Nostrand EL, Pratt GA, Yee BA, et al. Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol. Internet] 2020. [cited 2021 Jan 18]; 21(1):90. Available from.https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-01982–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhan X, Yan C, Zhang X, et al. Structures of the human pre-catalytic spliceosome and its precursor spliceosome. Cell Res Internet] 2018. [cited 2020 Dec 17]; 28(12): 1129–1140. Available from http://www.nature.com/articles/s41422-018-0094–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Spiluttini B, Gu B, Belagal P, et al. Splicing-independent recruitment of U1 snRNP to a transcription unit in living cells. J Cell Sci Internet] 2010. [cited 2020 May 2]; 123(12): 2085–2093. Available from http://jcs.biologists.org/cgi/doi/10.1242/jcs.061358 [DOI] [PubMed] [Google Scholar]
- [55].Yu Y, Reed R. FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP. Proc Natl Acad Sci USA Internet] 2015. [cited 2020 May 3]; 112(28): 8608–8613. Available from. http://www.pnas.org/lookup/doi/10.1073/pnas.1506282112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yin Y, Lu JY, Zhang X, et al. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature. 2020;580(7801):147–150. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Azam S, Hou S, Zhu B, et al. Nuclear retention element recruits U1 snRNP components to restrain spliced lncRNAs in the nucleus. RNA Biol Internet] 2019. [cited 2020 Dec 28]; 16(8): 1001–1009. Available fromhttps://www.tandfonline.com/doi/full/10.1080/15476286.2019.1620061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Damgaard CK, Kahns S, Lykke-Andersen S, et al. A 5′ splice site enhances the recruitment of basal transcription initiation factors in vivo. Mol Cell Internet] 2008. [cited 2020 May 3]; 29(2): 271–278. Available from.https://linkinghub.elsevier.com/retrieve/pii/S1097276508000075 [DOI] [PubMed] [Google Scholar]
- [59].Kwek KY, Murphy S, Furger A, et al. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat Struct Biol. Internet] 2002. [cited 2020 May 3]; Available from http://www.nature.com/doifinder/10.1038/nsb862 [DOI] [PubMed] [Google Scholar]
- [60].Huang Y, Guo Q, Ding X-P WX. Mechanism of long noncoding RNAs as transcriptional regulators in cancer. RNA Biol. Internet] 2020. [cited 2020 Apr 7]:1–13. Available from. https://www.tandfonline.com/doi/full/10.1080/15476286.2019.1710405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li X, Fu X-D. Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet [Internet] 2019 [cited 2020 Apr 27]; 20:503–19. Available from: http://www.nature.com/articles/s41576-019-0135-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. Internet] 2012; 22(9):1775–1789. Available from.http://www.ncbi.nlm.nih.gov/pubmed/22955988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. Internet] 2009; 458(7235):223–227. Available from http://www.ncbi.nlm.nih.gov/pubmed/19182780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tian B. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res Internet] 2005. [cited 2020 Apr 6]; 33(1): 201–212. Available from https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gki158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yang L, Duff MO, Graveley BR, et al. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011;12(2):R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dreyfus M, Régnier P. The poly(A) tail of mRNAs. Cell Internet] 2002. [cited 2019 Aug 21]; 111(5): 611–613. Available from.https://linkinghub.elsevier.com/retrieve/pii/S0092867402011376 [DOI] [PubMed] [Google Scholar]
- [67].Muhlrad D, Decker CJ, Parker R. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5’-->3’ digestion of the transcript Genes Dev. 1994;8(7):855–866. [DOI] [PubMed] [Google Scholar]
- [68].Norbury CJ. a case of the tail wagging the dog. Nat Rev Mol Cell Biol. 2013;14(10):643–653. [DOI] [PubMed] [Google Scholar]
- [69].Yamashita A, Chang T-C, Yamashita Y, et al. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol. 2005;12(12):1054–1063. [DOI] [PubMed] [Google Scholar]
- [70].Metkar M, Ozadam H, Lajoie BR, et al. Higher-Order organization principles of pre-translational mRNPs. Mol Cell Internet] 2018. [cited 2020 Jul 4]; 72(4):715–726.e3. Available from. https://linkinghub.elsevier.com/retrieve/pii/S1097276518307834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chen C-YA, Shyu A-B. Mechanisms of deadenylation-dependent decay: mechanisms of deadenylation-dependent decay. Internet] 2011. [cited 2020 Aug 18]; 2:167–183. Available from 2 WIREs RNA. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chang H, Lim J, Ha M, et al. TAIL-seq: genome-wide determination of Poly(A) tail length and 3′ end modifications. Mol Cell Internet] 2014. [cited 2019 Aug 21]; 53(6): 1044–1052. Available fromhttps://linkinghub.elsevier.com/retrieve/pii/S109727651400121X [DOI] [PubMed] [Google Scholar]
- [73].Lim J, Kim D, Lee Y, et al. Mixed tailing by TENT4A and TENT4B shields mRNA from rapid deadenylation. Science Internet] 2018. [cited 2019 Aug 21]; 361(6403):701–704. Available from.http://www.sciencemag.org/lookup/doi/10.1126/science.aam5794 [DOI] [PubMed] [Google Scholar]
- [74].Baltz AG, Munschauer M, Schwanhäusser B, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. Internet] 2012. [cited 2019 Sep 10]; 46(5): 674–690. Available from https://linkinghub.elsevier.com/retrieve/pii/S1097276512004376. [DOI] [PubMed] [Google Scholar]
- [75].Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149(6):1393–1406. . [DOI] [PubMed] [Google Scholar]
- [76].Kwon SC, Yi H, Eichelbaum K, et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013;20(9):1122–1130. [DOI] [PubMed] [Google Scholar]
- [77].Milek M, Imami K, Mukherjee N, et al. DDX54 regulates transcriptome dynamics during DNA damage response. Genome Res. 2017;27(8):1344–1359. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gilmartin GM. Eukaryotic mRNA 3’ processing: a common means to different ends. Genes Dev. 2005;19(21):2517–2521. [DOI] [PubMed] [Google Scholar]
- [79].Truniger V, Miras M, Aranda MA. Structural and functional diversity of plant Virus 3ʹ-Cap-Independent Translation Enhancers (3ʹ-CITEs). Front Plant Sci. 2017;8:2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chlebowski A, Lubas M, Jensen TH, et al. RNA decay machines: the exosome. Biochim Biophys Acta Gene Regul Mech Internet] 2013. [cited 2019 Sep 19]; 1829(6–7):552–560. Available from. https://linkinghub.elsevier.com/retrieve/pii/S1874939913000163 [DOI] [PubMed] [Google Scholar]
- [81].Collart MA. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip Rev RNA. 2016;7(4):438–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mugridge JS, Coller J, Gross JD. Structural and molecular mechanisms for the control of eukaryotic 5′–3′ mRNA decay. Nat Struct Mol Biol Internet] 2018. [cited 2020 Aug 21]; 25(12):1077–1085. Available fromhttp://www.nature.com/articles/s41594-018-0164–z [DOI] [PubMed] [Google Scholar]
- [83].Simon AE, Miller WA. 3′ Cap-independent translation enhancers of plant viruses. Annu Rev Microbiol. 2013;67(1):21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chang Y-F, Imam JS, Wilkinson MF. The nonsense-mediated Decay RNA surveillance pathway. Annu Rev Biochem Internet] 2007. [cited 2019 Sep 19]; 76(1): 51–74. Available from. http://www.annualreviews.org/doi/10.1146/annurev.biochem.76.050106.093909 [DOI] [PubMed] [Google Scholar]
- [85].Laroia G. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science. 1999;284(5413):499–502. [DOI] [PubMed] [Google Scholar]
- [86].Laroia G, Sarkar B, Schneider RJ. Ubiquitin-dependent mechanism regulates rapid turnover of AU-rich cytokine mRNAs. Proc Natl Acad Sci USA. 2002;99(4):1842–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Huang Y, Carmichael GG. Role of polyadenylation in nucleocytoplasmic transport of mRNA. Mol Cell Biol Internet] 1996. [cited 2019 Aug 21]; 16(4): 1534–1542. Available from;http://mcb.asm.org/lookup/doi/10.1128/MCB.16.4.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Okamura M, Inose H, Masuda S. RNA export through the NPC in Eukaryotes. Genes (Basel) Internet] 2015. [cited 2020 May 14]; 6(1): 124–149. Available from. http://www.mdpi.com/2073-4425/6/1/124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kruse C, Willkomm DK, Grünweller A, et al. Export and transport of tRNA are coupled to a multi-protein complex. Biochem J. 2000;346(1):107–115. [PMC free article] [PubMed] [Google Scholar]
- [90].Bohnsack MT, Czaplinski K, Gorlich D. 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10(2):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lund E, Güttinger S, Calado A, et al. Nuclear export of microRNA precursors. Science. 2004;303(5654):95–98. [DOI] [PubMed] [Google Scholar]
- [92].Yi R, Qin Y, Ig M, et al. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;175(24):212–223.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;10(5):376–385. [DOI] [PubMed] [Google Scholar]
- [94].Köhler A, Exporting HE. RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol. 2007;8(10):761–773. [DOI] [PubMed] [Google Scholar]
- [95].Cai X. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA Internet] 2004. [cited 2020 Apr 6]; 10(12): 1957–1966. Available fromhttp://www.rnajournal.org/cgi/doi/10.1261/rna.7135204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kadaba S. Nuclear RNA surveillance in Saccharomyces cerevisiae: trf4p-dependent polyadenylation of nascent hypomethylated tRNA and an aberrant form of 5S rRNA. RNA. 2006;12(3):508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Holt CE, Bullock SL. Subcellular mRNA localization in animal cells and why it matters. Science Internet] 2009. [cited 2019 Jul 31]; 326(5957): 1212–1216. Available from.http://www.sciencemag.org/lookup/doi/10.1126/science.1176488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mofatteh M, Bullock SL. SnapShot: subcellular mRNA Localization. Cell. 2017;169(1):178–178.e1. [DOI] [PubMed] [Google Scholar]
- [99].Blower MD. Molecular Insights into intracellular RNA localization [Internet]. In: International review of cell and molecular biology. Elsevier; [2013. cited 2020 Aug 26]. p. 1–39. Available from https://linkinghub.elsevier.com/retrieve/pii/B9780124076990000017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Carlevaro-Fita J, Johnson R. Global positioning system: understanding long noncoding RNAs through subcellular localization. Mol Cell. 2019;73(5):869–883. [DOI] [PubMed] [Google Scholar]
- [101].Wilk R, Hu J, Blotsky D, et al. Diverse and pervasive subcellular distributions for both coding and long noncoding RNAs. Genes Dev. 2016;30(5):594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Decker CJ, P-bodies PR. and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol Internet] 2012. [cited 2020 Mar 11]; 4(9): a012286–a012286. Available from.http://cshperspectives.cshlp.org/lookup/doi/10.1101/cshperspect.a012286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Fujiwara Y, Furuta A, Kikuchi H, et al. Discovery of a novel type of autophagy targeting RNA. Autophagy. Internet] 2013. [cited 2020 Apr 18]; 9(3):403–409. Available from.http://www.tandfonline.com/doi/abs/10.4161/auto.23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Cioni J-M, Lin JQ, Holtermann AV, et al. Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell. Internet] 2019. [cited 2020 Aug 26]; 176(1–2): 56–72.e15. Available from https://linkinghub.elsevier.com/retrieve/pii/S0092867418315551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Munro S. The golgin coiled-coil proteins of the Golgi Apparatus. Cold Spring Harb Perspect Biol. Internet] 2011. [cited 2020 Sep 3]; 3:a005256–a005256. Available from. http://cshperspectives.cshlp.org/lookup/doi/10.1101/cshperspect.a005256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Caudron-Herger M, Rusin SF, Adamo ME, et al. R-DeeP: proteome-wide and quantitative identification of RNA-dependent proteins by density gradient ultracentrifugation. Mol Cell. 2019;75(1):184–199.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Queiroz RML, Smith T, Villanueva E, et al. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat Biotechnol. 2019;37(2):169–178. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Trendel J, Schwarzl T, Horos R, et al. The human RNA-binding proteome and its dynamics during translational arrest. Cell. 2019;176(1–2):391–403.e19. [DOI] [PubMed] [Google Scholar]
- [109].Zhou Y, Wang G, Wang P, et al. Expanding APEX2 substrates for proximity-dependent labeling of nucleic acids and proteins in living cells. Internet] 2019. [cited 2019 Aug 26]; 58(34): 11763–11767. Available from Angew Chem Int Ed. . [DOI] [PubMed] [Google Scholar]
- [110].Branon T, Han S, Beyond immunoprecipitation: TA. Exploring new interaction spaces with proximity biotinylation. Biochemistry Internet] 2017. [cited 2020 Apr 7]; 56(26): 3297–3298. Available from. https://pubs.acs.org/doi/10.1021/acs.biochem.7b00466 [DOI] [PubMed] [Google Scholar]
- [111].Roux KJ, Kim DI, Burke B. BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci. 2013;74(1):19.23.1–19.23.14. [DOI] [PubMed] [Google Scholar]
- [112].Han S, Zhao BS, Myers SA, et al. RNA–protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc Natl Acad Sci USA. Internet] 2020. [cited 2020 Aug 24];:202006617. Available from. http://www.pnas.org/lookup/doi/10.1073/pnas.2006617117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Li Y, Liu S, Cao L, et al. CBRPP: a new RNA-centric method to study RNA-protein interactions. Mol Biol. Internet] ; 2020. [cited 2020 May 14]. Available from http://biorxiv.org/lookup/doi/10.1101/2020.04.09.033290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Yi W, Li J, Zhu X, et al. CRISPR-assisted detection of RNA–protein interactions in living cells. Nat Methods. Internet] 2020. [cited 2020 Jun 22]; Available from.17(7):685–688. http://www.nature.com/articles/s41592-020-0866–0 [DOI] [PubMed] [Google Scholar]
- [115].Zhang Z, Sun W, Shi T, et al. Capturing RNA–protein interaction via CRUIS. Nucleic Acids Res . gkaa143. 2020. cited 2020 Mar 15 Internet] Available from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkaa143/5788206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Bandaru S, Tsuji MH, Shimizu Y, et al. Structure-based design of gRNA for Cas13. Sci Rep Internet] 2020. [cited 2020 Aug 24]; 10(1): 11610. Available from. http://www.nature.com/articles/s41598-020-68459–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wessels -H-H, Méndez-Mancilla A, Guo X, et al. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat Biotechnol Internet] 2020. [cited 2020 May 19]; 38(6):722–727. : Available from.;http://www.nature.com/articles/s41587-020-0456–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Clément M-V PS, Pervaiz S. Intracellular superoxide and hydrogen peroxide concentrations: a critical balance that determines survival or death. Redox Rep Internet. 2001. cited 2020 Aug 22; 64: 211–214Available from. http://www.tandfonline.com/doi/full/10.1179/135100001101536346 [DOI] [PubMed] [Google Scholar]
- [119].Quinn JJ, Ilik IA, Qu K, et al. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat Biotechnol Internet] 2014. [cited 2020 Aug 30]; 32(9): 933–940. Available from.http://www.nature.com/articles/nbt.2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhang C, Konermann S, Brideau NJ, et al. Structural basis for the RNA-guided ribonuclease activity of CRISPR-Cas13d. Cell Internet] 2018. [cited 2020 Apr 26]; 175(1): 212–223.e17. Available from.https://linkinghub.elsevier.com/retrieve/pii/S0092867418311735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Rauch S, He E, Srienc M, et al. Programmable RNA-guided RNA effector proteins built from human parts. Cell Internet] 2019. [cited 2019 Jun 24]; 178:122–134.e12. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867419306208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Han Y, Branon TC, Martell JD, et al. Directed evolution of split APEX2 peroxidase. ACS Chem Biol Internet] 2019. [cited 2020 Dec 26]; 14(4): 619–635. Available from. https://pubs.acs.org/doi/10.1021/acschembio.8b00919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Shekhawat SS, Ghosh I. Split-protein systems: beyond binary protein–protein interactions. Curr Opin Chem Biol. Internet] 2011. [cited 2020 Dec 26]; 15:789–797. Available from. https://linkinghub.elsevier.com/retrieve/pii/S136759311100161X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Liu Q, Zheng J, Sun W, et al. A proximity-tagging system to identify membrane protein–protein interactions. Nat Methods Internet] 2018. [cited 2020 Jul 20]; 15(9): 715–722. Available from http://www.nature.com/articles/s41592-018-0100–5 [DOI] [PubMed] [Google Scholar]
- [125].Samavarchi-Tehrani P, Samson R, Gingras A-C. Proximity dependent biotinylation: key enzymes and adaptation to proteomics approaches. Mol Cell Proteomics Internet] 2020. [cited 2020 Aug 13]; 19(5): 757–773. Available fromhttp://www.mcponline.org/lookup/doi/10.1074/mcp.R120.001941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Licatalosi DD, Mele A, Fak JJ, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. Internet] 2008. [cited 2020 May 31]; 456(7221): 464–469. Available from http://www.nature.com/articles/nature07488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Van Nostrand EL, Pratt GA, Shishkin AA, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods. Internet] 2016. [cited 2020 May 31]; 13(6):508–514. Available from. http://www.nature.com/articles/nmeth.3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Spitale RC, Flynn RA, Zhang QC, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. Internet] 2015. [cited 2020 May 31]; 519(7544): 486–490. Available from. http://www.nature.com/articles/nature14263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sun L, Fazal FM, Li P, et al. RNA structure maps across mammalian cellular compartments. Nat Struct Mol Biol Internet] 2019. [cited 2020 May 31]; 26(4): 322–330. Available from http://www.nature.com/articles/s41594-019-0200–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw images for western blots and raw and processed mass spectrometry data are included as supporting files.