

SUMMARY
The protein tau has been implicated in many brain disorders. In animal models, tau reduction suppresses epileptogenesis of diverse causes and ameliorates synaptic and behavioral abnormalities in various conditions associated with excessive excitation-inhibition (E/I) ratios. However, the underlying mechanisms are unknown. Global genetic ablation of tau in mice reduces the action potential (AP) firing and E/I ratio of pyramidal cells in acute cortical slices without affecting the excitability of these cells. Tau ablation reduces the excitatory inputs to inhibitory neurons, increases the excitability of these cells, and structurally alters their axon initial segments (AISs). In primary neuronal cultures subjected to prolonged overstimulation, tau ablation diminishes the homeostatic response of AISs in inhibitory neurons, promotes inhibition, and suppresses hypersynchrony. Together, these differential alterations in excitatory and inhibitory neurons help explain how tau reduction prevents network hypersynchrony and counteracts brain disorders causing abnormally increased E/I ratios.
Graphical Abstract
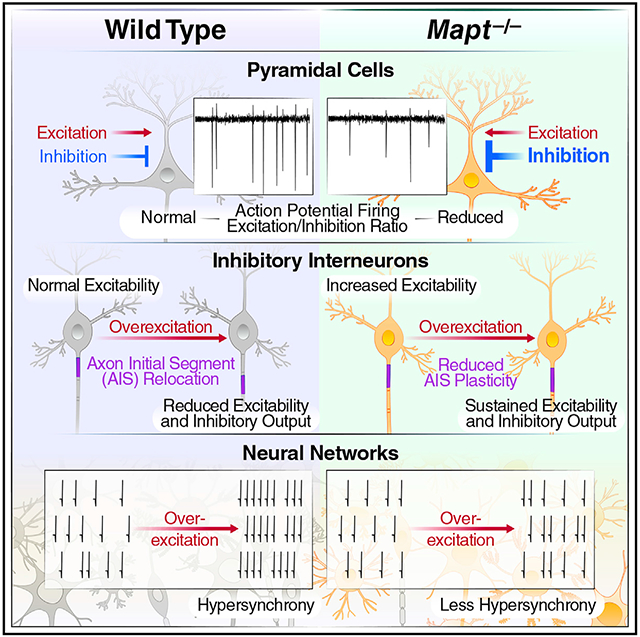
In brief
Tau reduction prevents epileptogenesis, but underlying mechanisms are uncertain. Chang et al. show that tau ablation decreases the baseline activity of excitatory neurons and modulates the intrinsic excitability and axon initial segments of inhibitory neurons, promoting network inhibition. In combination, these effects counteract network hypersynchrony and diseases causing excitation/inhibition imbalance.
INTRODUCTION
Tau has been studied primarily in relation to Alzheimer’s disease (AD) and other neurodegenerative disorders associated with the intraneuronal accumulation of microscopically visible tau aggregates (Goedert and Spillantini, 2019; Chang et al., 2021). However, tau may also enable or promote disorders in which aberrant neural network activities are not associated with obvious tau pathology, including autism, epilepsies, stress-induced depression, and stroke (Chang et al., 2021). Highlighting the diseaseenabling role of tau, reducing brain levels of physiological, nonaggregated wild-type (WT) tau prevents or reduces neural network and behavioral dysfunctions in relevant experimental models (Roberson et al., 2007, 2011; Ittner et al., 2010; Holth et al., 2013; Gheyara et al., 2014; Tai et al., 2020). The mechanisms are unknown.
Epilepsy may worsen other brain disorders (Kanner, 2016; Vossel et al., 2016). For example, cognitive functions decline faster in AD patients with nonconvulsive epileptiform activity than in those without (Vossel et al., 2016). Epileptiform activity may also contribute to cognitive impairments and behavioral alterations in disease models (Sanchez et al., 2012; Gheyara et al., 2014; Palop and Mucke, 2016; Tai et al., 2020). For example, suppressing epileptiform activity reduced synaptic and cognitive deficits in mice simulating aspects of AD (Sanchez et al., 2012).
Genetic ablation or reduction of tau can prevent nonconvulsive epileptiform activity in AD-related mouse models (Roberson et al., 2007, 2011). Tau reduction also made mice more resistant to chemically induced seizures (Roberson et al., 2007). These observations have been confirmed and expanded upon in other models with epileptic activity (Chang et al., 2021), including models of AD (Ittner et al., 2010; Roberson et al., 2011), autism (Holth et al., 2013; Gheyara et al., 2014; Tai et al., 2020), and stroke (Bi et al., 2017). Because tau reduction can diminish epileptic activity of such diverse causes, it may alter neurons in ways that broadly diminish the probability of epilepsy-promoting processes such as excitation/inhibition (E/I) imbalance, hyperexcitability, and hypersynchronization (Goldberg and Coulter, 2013).
The results of the current study suggest that tau reduction counteracts epilepsy and related conditions by differentially modulating the functions of excitatory and inhibitory neurons in ways that, together, reduce the E/I ratio of neural networks.
RESULTS
Tau ablation reduces AP firing and E/I ratio in excitatory neurons
To investigate effects of tau ablation on the activity of excitatory neurons, we recorded spontaneous action potential (sAP) firing of pyramidal cells (PCs) in layer 5 of the somatosensory cortex (L5) in acute cortical slices from WT and tau-deficient (Mapt−/−) mice. We focused on this brain region because it is involved in epilepsy and AD (Polack et al., 2007; Verret et al., 2012; Tai et al., 2014), and the prominent morphological differences between PCs and interneurons in this region make it easy to visually distinguish them for recordings.
The sAP frequency was lower in Mapt−/− slices (Figures 1A and 1B). The minimal currents to trigger single APs (rheobase current) and the AP thresholds, amplitudes, and half-widths were similar in Mapt−/− and WT slices (Figures 1C-1E; Table S1), as were frequency-current intensity (F-I) curves of PCs (Figures 1F and 1G).
Figure 1. Tau ablation reduces sAP firing and E/I ratio of PCs but does not affect their excitability.

sAP firing, AP thresholds, input-output responses, and spontaneous (s) and miniature (m) EPSCs and IPSCs were recorded in PCs in acute slices of somatosensory cortex from 24- to 28-day-old WT and Mapt−/− mice. See Table S1 for additional measures.
(A) Representative traces of sAPs recorded by cell-attached patch-clamp.
(B) Quantitation of sAP frequencies. The graph on the right shows cumulative probability curves of sAP frequencies binned at 2 Hz.
(C) Representative APs induced by minimal current injection.
(D and E) Quantitations of AP rheobase currents (D) and thresholds (E).
(F) Representative AP firing patterns induced by a 200-pA current injection.
(G) AP frequency in response to increasing current injections (F-I curves).
(H) Representative PSC traces recorded from WT (top) and Mapt−/− (bottom) cells.
(I–L) Quantitations of sIPSC (I), mIPSC (J), sEPSC (K), and mEPSC (L) frequencies.
(M) Ratios of (sEPSC frequency × amplitude) over (sIPSC frequency × amplitude).
n = 43–48 cells (from 4 mice) per genotype for sAPs; 2–4 cells per slice and 3–4 slices per mouse (B) were analyzed. n = 16–17 cells (from 3 mice) per genotype for APs; 1 cell per slice and 5–6 slices per mouse (D, E, and G) were analyzed. n = 14–17 cells (from 3 mice) per genotype for PSCs; 1 cell per slice and 4–6 slices per mouse (I–M) were analyzed. *p < 0.05, **p < 0.01 by unpaired two-tailed Student’s t test (I) or Mann-Whitney test (B and M). In all figures, differences without asterisk(s) were not statistically significant. See Table S4 for all other p values and STAR methods for an explanation of box-and-whisker plots (B, D, E, and I–M). Values in (G) are means ± SEM.
The frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) in PCs was higher in Mapt−/− than WT slices, but spontaneous excitatory postsynaptic current (sEPSC) frequencies were comparable (Figures 1H, 1I, and 1K; Table S1). Miniature IPSCs (mIPSCs) and EPSCs (mEPSCs) were unchanged in tau-deficient slices (Figures 1J and 1L; Table S1). The E/I ratio in PCs was lower in Mapt−/− slices (Figure 1M). In dentate granule cells of 4-to-6-month-old Mapt−/− mice, increases in sIPSC and AP-driven IPSC frequencies were more subtle (Roberson et al., 2011), possibly due to differences in brain regions and mouse ages. Taken together, these results suggest that tau ablation reduces the sAP firing and E/I ratio of PCs, enhances AP-dependent inhibitory input onto PCs, and has much less, if any, impact on the excitability of PCs.
Tau ablation increases intrinsic excitability of inhibitory interneurons
Next, we examined whether global tau reduction elevates the intrinsic excitability of interneurons, identified by morphology and fast spiking (FS) firing patterns. Biocytin injection and immunostaining (Figure S1) confirmed that the FS neurons in L5 we recorded from expressed parvalbumin (PV), consistent with previous observations (Naka and Adesnik, 2016; Gouwens et al., 2019, 2020). The rheobase current and AP threshold were lower in FS neurons from Mapt−/− than WT mice (Figures 2A-2C); AP amplitudes and half-widths were comparable (Table S2). F-I curves differed in that AP frequencies of FS cells were higher in Mapt−/− slices at relatively low (100–250-pA) levels of current injection (Figures 2D and 2E).
Figure 2. Tau ablation increases the excitability of interneurons but decreases the excitatory inputs they receive.

AP thresholds, input-output responses, and EPSCswere recorded from fast-spiking (FS) neurons in acute slices of somatosensory cortex from 24- to 28-day-old WT and Mapt−/− mice. See Table S2 for additional measures.
(A) Representative individual APs induced by minimal current injection.
(B and C) Quantitations of AP rheobase currents (B) and thresholds (C).
(D) Representative AP firing patterns induced by a 200-pA current injection.
(E) AP frequency in response to increasing current injections (F-I curves).
(F) Representative traces of sEPSCs and mEPSCs in FS cells.
(G and H) Quantitations of sEPSC (G) and mEPSC (H) frequencies.
n = 25–28 cells (from 6–7 mice) per genotype for APs; 1 cell per slice and 4–6 slices per mouse (B, C, and E) were analyzed. n = 20–21 cells (from 5–6 mice) per genotype for EPSCs; 1 cell per slice and 3–5 slices per mouse (G and H) were analyzed. #p = 0.09, *p < 0.05, ***p < 0.001 by unpaired, two-tailed t test (B, C, and G), unpaired, two-tailed t test for areas under the curves (E), or Mann-Whitney test (H). Values in (E) are means ± SEM.
mEPSC frequency in FS cells was lower in Mapt−/− slices, and the sEPSC frequency showed a trend in the same direction (Figures 2G and 2H; Table S2), suggesting lower excitatory inputs onto FS cells in Mapt−/− slices, which might increase interneuron excitability by homeostatic mechanisms (Kuba et al., 2010; Grubb et al., 2011). The increased excitability of interneurons could contribute to the higher frequency of sIPSCs and lower E/I ratio of PCs in Mapt−/− slices (Figures 1I and 1M).
Tau ablation alters the AIS of interneurons
To explore mechanisms of increased excitability of FS cells in Mapt−/− mice, we examined the axon initial segment (AIS), which strongly influences the intrinsic excitability of neurons (Grubb and Burrone, 2010; Kuba et al., 2010; Grubb et al., 2011; Bender and Trussell, 2012) and can be altered by the accumulation of abnormal tau species (Hatch et al., 2017; Sohn et al., 2019). The AISs of PV cells and PCs in L5 were identified by immunostaining coronal brain sections from Mapt−/− and WT mice for ankyrin G (AnkG) (Figure 3A). In Mapt−/− mice, the AISs of PV cells were longer and showed a right shift toward greater lengths along most of the cumulative probability curve (Figure 3B). AIS-soma distances were shorter in Mapt−/− mice and showed a left shift toward reduced distances (Figure 3C). The AISs of PCs were shorter and farther from the soma in Mapt−/− than WT mice, but only the difference in length was significant (Figures 3D and 3E).
Figure 3. Tau ablation preferentially modulates AISs of inhibitory neurons.

In (A)–(E), coronal sections of somatosensory cortex from 23- to 25-day-old WT and Mapt−/− mice were co-immunostained for the AIS marker AnkG and for PV and analyzed by confocal microscopy.
(A) Representative images depicting AISs (green) of PV cells (red) and PCs (not labeled). In the inset, dashed lines indicate the soma boundary, and arrowheads indicate the start and end of AISs. Scale bars: 20 μm, 10 μm (inset).
(B–E) AIS length (B and D) and AIS-soma distance (C and E) in PV cells (B and C) and PCs (D and E). Cumulative probability curves binned at 1 μm for AIS length and at 0.5 μm for AIS-soma distance are shown to the right of box-and-whisker plots.
(F–H) At DIV13–14, primary hippocampal neuronal cultures from P0–1 WT and Mapt−/− mice were or were not exposed to 15 mM KCl (10 mM added plus 5 mM in medium) for 4 or 16h; fixed; immunostained for markers of the AIS (AnkG, white), excitatory cells (CaMKII, cytoplasmic, green), including granule cells (PROX1, nuclear, white), and interneurons (GAD67, cytoplasmic, red); and analyzed by confocal microscopy.
(F) Representative images depicting AISs of GABAergic interneurons. Arrowheads mark the start and end of AISs and dashed lines the soma boundary. Scale bar: 20 μm.
(G and H) AIS-soma distance (G) and AIS length (H) in GABAergic cells. Cumulative probability curves binned at 2 μm are shown on the right. For AIS measurements in granule cells, see Figures S2A-S2C.
n = 83–92 AISs (from 4 mice) per genotype; 8–12 AISs per section and 2 sections per mouse (B and C) were analyzed. n = 68–84 AISs for AIS length and 55–56 AISs for AIS-soma distance (from 4 mice) per genotype; 7–14 AISs per section and 2 sections per mouse (D and E) were analyzed. n = 24–42 AISs (from 3 mice) per genotype and treatment; 7–14 AISs per well and 1 well per mouse (G and H) were analyzed. #p < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired, two-tailed t test (B–D) or two-way ANOVA and Holm-Sidak test (G and H).
Since neuronal excitability correlates with AIS length and proximity to the soma (Grubb et al., 2011), these data are broadly consistent with the differential effects of tau ablation on FS cells and PCs in our electrophysiological experiments.
To further test whether tau regulates activity-dependent AIS plasticity, we analyzed AISs in primary hippocampal neurons from WT and Mapt−/− mice at DIV13–14 before and after prolonged depolarization induced by KCl, which elicits robust AIS adaptations in such cultures (Grubb et al., 2011; Evans et al., 2013, 2015). KCl increased the AIS-soma distance in GABAergic interneurons but did so less rapidly and to a lesser extent in Mapt−/− cultures (Figures 3F and 3G). At baseline, AISs were longer in interneurons from Mapt−/− mice, and KCl decreased this difference (Figure 3H). AIS-soma distances and AIS lengths did not significantly differ between excitatory PROX1-positive granule cells from Mapt−/− and WT mice (Figures S2A-S2C). Consistent with previous findings (Evans et al., 2013, 2015; Sohn et al., 2019; Booker et al., 2020), NaCl treatment did not alter AISs in inhibitory or excitatory neurons (Figures S2D-S2G), indicating that the AIS changes after KCl treatment resulted from prolonged depolarization rather than from osmotic stress. These findings further support our conclusion that global tau ablation modulates AIS plasticity more strongly in inhibitory than in excitatory neurons.
Tau ablation promotes inhibition and suppresses network hypersynchrony
To examine whether the changes in AIS plasticity of interneurons in tau-deficient primary cultures are associated with alterations in excitability and inhibitory output, we expressed the GABA sensor iGABASnFR (Marvin et al., 2019) in primary cultures of hippocampal neurons. Neuronal cultures from Mapt−/− and WT mice were stimulated with KCl and exposed to increasing numbers of electrical field stimuli to evoke GABA release and obtain input-output curves (Figures 4A and S3).
Figure 4. Tau ablation supports inhibition and prevents network hypersynchrony after stimulation of neuronal cultures.

In (A) and (B), primary hippocampal neuronal cultures from P0–1 WT and Mapt−/− mice were transduced on DIV4 to express iGABASnFR. At DIV13–14, cultures were or were not exposed to 15 mM KCl (10 mM added plus 5 mM in medium) for 4 or 16 h, followed by incubation in conditioned medium (without KCl addition) for 1 h. Fluorescence changes indicating GABA release were then triggered by electrical field stimulation (1 ms, 90 mA, 100 Hz, and 5 pulses) and monitored by fluorescence microscopy.
(A) Representative traces of fluorescence changes in neuronal somas triggered by electrical field stimulation.
(B) GABA release (areas under signal traces). See Figure S3 for input-output curves.
In (C)–(F), spontaneous activity in neuronal cultures recorded by MEA after KCl treatment and incubation in conditioned medium as in (A) and (B) is shown.
(C) Representative traces from single-channel MEA recordings showing spontaneous spiking in neuronal cultures at baseline (0 h KCl).
(D) Spike frequencies per channel. Individual values were calculated by averaging spike frequencies recorded from all 16 channels per MEA well.
(E) Raster plots from cultures before and after 4 h KCl treatment.
(F) Global synchrony index obtained from the phase synchronization matrix of all pairs of electrodes (see STAR Methods). See Table S3 for additional measures.
n = 64–84 cells (from 5 mice) per genotype and treatment; 7–29 cells per coverslip and 1–2 coverslips per mouse (B) were analyzed. n = 16–18 wells (from 8–9 mice) per genotype and treatment; 2 wells were analyzed per mouse (D and F). *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA and Holm-Sidaktest (D and F) or permutation test with Holm-Sidak correction (B). Table S4 provides p values for all comparisons.
Before KCl treatment, evoked GABA release was slightly higher in Mapt−/− cultures (Figure 4B). After 4 h of KCl exposure, GABA release was maintained in Mapt−/− cultures but declined in WT cultures (Figure 4B). These differences are consistent with the reduced AIS plasticity in GABAergic neurons of Mapt−/− mice (Figure 3G) and could help maintain inhibition after network excitation. With prolonged KCl exposure (16 h), the GABA release decreased in WT and Mapt−/− cultures (Figure 4B), possibly as a result of increases in interneuron AIS-soma distances (Figure 3G). In all KCl conditions, inhibitory output was higher in Mapt−/− cultures (Figure S3).
We found no evidence of tau-dependent changes in inhibitory vesicle content (mIPSC amplitude; Table S1), inhibitory vesicles released by APs (sIPSC amplitude; Table S1), number of inhibitory synapses on excitatory cells (mIPSC frequency; Figures 1J, S4A, and S4B), or proportion of inhibitory neurons in cultures (Figures S4C and S4D). Thus, the enhanced input-output responses of GABA release in tau-deficient neuronal cultures (Figures 4A, 4B, and S3) likely reflect increased excitability of interneurons, consistent with our findings in tau-deficient slices (Figures 2A-2E). Collectively, these results suggest that global tau ablation reduces the activity-dependent AIS plasticity in interneurons and enhances their excitability and inhibitory output.
Next, we assessed AP generation and network synchrony before and after KCl treatment in primary hippocampal cultures by microelectrode array (MEA) recordings (Figures 4C and 4E). At baseline and after 4 h of KCl exposure, spike frequency was lower in Mapt−/− than WT cultures (Figure 4D). For both single-channel and network bursts, the frequency and number of spikes per burst were lower in Mapt−/− cultures at baseline and after KCl exposure, but only the difference in the frequency of network bursts at baseline reached statistical significance (Table S3).
To assess the synchrony of neuronal activities, we computed a global synchrony index (GSI) based on the phase synchronization matrix and eigenvalue decomposition. At baseline, GSIs did not differ in WT and Mapt−/− cultures (Figure 4F; Table S4). However, KCl significantly increased the GSI only in WT cultures (Figures 4E and 4F; Table S4). Thus, tau reduction may counteract network hypersynchrony, at least in part, by preventing adaptive reductions in the excitability of interneurons after network stimulations.
DISCUSSION
In this study, global tau ablation decreased spontaneous spiking of excitatory neurons and increased their inhibitory inputs. Tau ablation also increased the excitability of inhibitory neurons and modulated homeostatic responses of the AISs in these cells. At the neural network level, tau ablation reduced the hypersynchrony induced by overexcitation. Thus, global tau ablation affects excitatory pyramidal cells and inhibitory interneurons differentially. The combined changes tau ablation elicits in these cell types should reduce the E/I ratio of neural networks, providing a plausible cellular/network mechanism by which tau reduction counteracts diseases with aberrant increases in E/I ratios. Indeed, tau reduction prevents or reduces epileptiform activity, behavioral abnormalities, and/or premature death in models of AD, autism, depression, epilepsy, and stroke (Chang et al., 2021)—all of which can promote E/I imbalance and network hyperexcitability (Rakhade and Jensen, 2009; Tuchman et al., 2010; Mertens et al., 2015; Staley, 2015; Palop and Mucke, 2016; Frere and Slutsky, 2018; Duman et al., 2019; Sohal and Rubenstein, 2019).
A tau-targeting antisense oligonucleotide (ASO) approach (DeVos et al., 2017) has advanced into a clinical trial for early AD (ClinicalTrials.gov identifier NCT03186989). An important objective of tau-lowering strategies in tauopathies is to block the intraneuronal accumulation of abnormal tau aggregates, a pathological hallmark (DeVos et al., 2017; Goedert and Spillantini, 2019; VandeVrede et al., 2020). However, together with other findings (Chang et al., 2021), our results suggest that tau reduction could also be beneficial by suppressing aberrant neural network activities that are enabled by nonaggregated tau.
Our findings raise interesting possibilities that could help guide studies to identify molecular mechanisms by which tau reduction differentially affects the functions of excitatory and inhibitory neurons. These differential effects may reflect differences in the functions or biological activities of tau within the respective cell types, which could be mediated by different tau species or by interactions of similar tau species with cell-type-specific molecules and pathways. Alternatively, the most critical cell-autonomous effects of tau ablation might occur in one cell type and affect the other cell type indirectly through changes in synaptic inputs or network activity. If the primary effects occur in excitatory neurons, in which global tau ablation reduced sAP firing, the increased excitability and enhanced inhibitory output of interneurons might represent homeostatic responses to reduced excitatory inputs and, possibly, overall changes in neural network activity. If the primary effects of tau ablation occur in interneurons, the reduced activity of excitatory cells might result indirectly from increased inhibition.
The altered AIS plasticity in tau-deficient interneurons is interesting, as tau is closely associated with the AIS. Tau interacts with the microtubule end-binding protein 3 (Sayas et al., 2015; Sohn et al., 2019), which binds to and stabilizes the scaffolding protein AnkG (Leterrier et al., 2011; Fréal et al., 2016), raising the possibility that tau ablation affects the AIS of interneurons directly. However, AIS modifications also form part of the homeostatic responses to altered synaptic inputs (Kuba et al., 2010; Grubb et al., 2011) and could result indirectly from effects of tau ablation on excitatory cells. Indeed, FS cells in L5 are predominantly driven by local PCs (Naka and Adesnik, 2016) and may use AIS modifications to adapt to reduced inputs from tau-deficient excitatory cells. Furthermore, tau is expressed not only in neurons, but also in astrocytes, microglia, and oligodendrocytes (Zhang et al., 2014, 2016; Saunders et al., 2018). Experimental models allowing for the selective ablation of tau in specific cell types are needed to ascertain the functions of tau in different brain cells and their roles in neural network effects of tau reduction in health and disease.
These uncertainties notwithstanding, the combined effects of tau reduction on excitatory and inhibitory neurons we identified should counteract aberrant increases in E/I ratios and network hyperexcitability, consistent with the beneficial effects of tau reduction in diverse models of epilepsy and other conditions in which epilepsy is a comorbidity (Chang et al., 2021). The conditions tau reduction can prevent or diminish in animal models have diverse causal triggers, ranging from mutations in genes encoding sodium or potassium channels to the pharmacological blockade of GABAA receptors, stroke, and neural accumulation of metabolites derived from the amyloid precursor protein (Chang et al., 2021). Since these abnormalities promote E/I imbalance and hyperexcitability (Rakhade and Jensen, 2009; Mertens et al., 2015; Staley, 2015; Palop and Mucke, 2016; Frere and Slutsky, 2018; Duman et al., 2019; Sohal and Rubenstein, 2019), the reduced E/I ratio in tau-deficient brains may explain the broad protection that tau-lowering strategies provide in models of diverse brain disorders.
Baseline neural network activity can affect the ability of pathogenic factors to elicit hyperexcitability. For example, amyloid-β (Aβ) dimers elicit hyperactivity in vivo but not in ex vivo brain slices with lower levels of neuronal activity; elevating baseline activity allowed Aβ-induced neuronal hyperactivity to emerge in the slices (Zott et al., 2019). Reduced sAP firing of excitatory cells in tau-deficient brains could help explain the suppression of epileptiform activity after tau reduction in human amyloid precursor protein transgenic mice (Roberson et al., 2007, 2011; Ittner et al., 2010). These findings may be relevant to AD, which is characterized by abnormal accumulation of Ab and tau and often causes subclinical epileptiform activity (Vossel et al., 2016; Lam et al., 2020).
The detection of seizures or epileptiform activity in transgenic mice overexpressing WT or mutant human tau in neurons (García-Cabrero et al., 2013; Wheeler et al., 2015; Maeda et al., 2016) seems broadly consistent with our findings, as does the finding that human tau overexpression in interneurons impairs inhibitory functions (Zheng et al., 2020). Overexpression of human tau in glutamatergic forebrain neurons has yielded evidence for both increased (Crimins et al., 2012; Shimojo et al., 2020) and decreased (Menkes-Caspi et al., 2015; Busche et al., 2019) neuronal activity. Several experimental variables may account for these discrepancies, including spurious effects of overexpressing tau with strong heterologous promoters.
In conclusion, the physiological activity of both excitatory and inhibitory neurons partly depends on tau, and tau reduction differentially modulates the functions of these cells in ways that should have beneficial effects in diseases that aberrantly increase neural E/I ratios. Studies are needed to fully elucidate how tau contributes to the functions of different brain cells and to further explore the molecular mechanisms and therapeutic potential of tau-lowering strategies in different pathological contexts.
LIMITATIONS OF THE STUDY
A limitation of all tau studies, including ours, is that the consequences of tau modulation were investigated in select brain regions, cell populations, or neural circuits. It could be informative to assess the extent to which tau-related observations can be generalized across the central nervous system. We recorded from and analyzed the AISs of many neurons per genotype but from a limited number of mice. Thus, our findings must be validated in larger cohorts. Lastly, our study focused on the genetic ablation of tau. From a therapeutic perspective, it will be interesting to explore whether partial reduction of tau in Mapt+/− mice causes alterations similar to those in Mapt−/− mice and to determine whether such changes can be elicited at different ages in WT mice and disease models by treatment with tau-targeting ASOs or tau-lowering small-molecule drugs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lennart Mucke (lennart.mucke@gladstone.ucsf.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon reasonable request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type (WT) C57BL/6J mice (No. 000664) and tau knockout (Mapt−/−) mice on the C57BL/6J background (No. 007251) were obtained from The Jackson Laboratory. WT mice were bred with each other to generate male and female WT controls for experiments. WT mice were also bred with homozygous Mapt−/− mice to generate heterozygous Mapt+/− mice. Mapt+/− mice from different pairs of sires and dams were crossed to generate Mapt−/− breeders. Mapt−/− mice from different pairs of sires and dams were then bred with each other to generate male and female Mapt−/− mice for experiments. Mice were housed in groups of 2–5 per cage on a 12-h light/dark cycle with ad libitum access to food (PicoLab Rodent Diet 20, 5053; LabDiet) and water. All experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Primary neuronal cultures
The following procedures were done on individual newborn mouse pups without pooling tissues or cells from different mice. Hippocampi of newborn (P0–P1) WT and Mapt−/− mice were dissected in ice-cold Earle’s balanced salt solution lacking CaCl2, MgSO4, and phenol red (GIBCO, 14155-063) and digested with papain (Worthington, LK003178) in the same solution for 18 min at 37°C. The digestion was stopped by adding low-ovomucoid solution containing 0.15% bovine serum albumin (Sigma-Aldrich, A8806), 0.15% trypsin inhibitor (GIBCO, R007100) in DPBS, and deoxyribonuclease I (Sigma-Aldrich, D5025) for 5 min. Tissues were triturated in fresh low-ovomucoid solution with a fire-polished Pasteur pipette, debris was removed with a 70-μm nylon strainer, and the cells were spun at 1000 rpm for 10 min. Cell pellets were resuspended in 37°C culture medium containing Neurobasal A (GIBCO, 12349015), 2% B27 (GIBCO, 17504044), 0.5 mM GlutaMAX (GIBCO, 35050061), and penicillin/streptomycin (GIBCO, 15140122).
Cells from individual newborn mice were plated on poly-D-lysine–coated 96-well plates (Corning, 356640) at a density of 40,000 cells/well for immunocytochemistry, and on poly-D-lysine/laminin–coated 8-mm coverslips (Warner Instruments, 64-0701) in 48-well plates at a density of 80,000 cells/well for GABA imaging. For MEA experiments, cells were resuspended in a mixed-medium containing 90% of the above culture medium and 10% conditioned medium (Neurobasal A, 2% B27,0.5 mM GlutaMAX, 10% fetal bovine serum) obtained from WT mouse glial cultures at DIV14, and plated into 0.05% polyethylenimine– and poly-D-lysine/laminin–coated MEA recording wells (Automate Scientific, MED-RG515A) with a cloning cylinder (Corning, 3166-8) at a density of 150,000 cells/well. Cells were grown in 5% CO2 in a humidified incubator at 37°C. Half the medium was replaced weekly. For immunocytochemistry, cells that had or had not been treated with KCl or NaCl were fixed immediately after the treatment. For GABA imaging and MEA recordings, cultures were first treated with KCl. The medium containing added KCl was then replaced with conditioned medium saved before the treatment, and the recordings were done 1 h later.
METHOD DETAILS
Preparation of acute cortical slices
Coronal slices of somatosensory cortex were prepared from male and female P24–28 mice. Mice were anesthetized with isoflurane and decapitated. Brains were immediately removed and immersed in ice-cold oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF), containing (in mM) 92 N-methyl-D-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 sodium ascorbate, 3 sodium pyruvate, 12 N-acetyl-L-cysteine, 0.5 CaCl2, and 10 MgSO4 (pH 7.35, 305 mOsm). Slices (400 μm thick) were cut with a vibrating microtome (HM650V, Thermo Scientific) and immediately transferred to a chamber containing slicing ACSF (see above) at 37°C for 10 min. Slices were then transferred to a second chamber with recovery ACSF, containing (in mM) 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 sodium ascorbate, 3 sodium pyruvate, 12 N-acetyl-L-cysteine, 2 CaCl2, and 2 MgSO4 (pH 7.35, 305 mOsm) for 45 min at room temperature, and then to a third chamber with standard recording ACSF containing (in mM) 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 12.5 glucose, 2 CaCl2, and 2 MgSO4 (pH 7.35, 315 mOsm), or active ACSF (to increase spontaneous neuronal activity) containing (in mM) 124 NaCl, 3.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 12.5 glucose, 1 CaCl2, and 0.25 MgSO4 (pH 7.35, 310 mOsm) for at least 30 min at room temperature. For recording, slices were placed in a recording chamber, which was mounted on an Olympus BX51WI microscope equipped with infrared differential interference contrast optics (900 nm) and perfused with oxygenated recording ACSF at a flow rate of 2 ml/min at room temperature. Chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Electrophysiology
Intrinsic excitability measurements
Whole-cell recordings were made from individual neurons located within layer 5 of the somatosensory cortex. Pyramidal neurons and interneurons were identified under infrared differential interference contrast optics. Patch pipettes pulled from borosilicate glass (World Precision Instruments, 140N-15) with a tip resistance of 4–6 MU were filled with internal solution, containing (in mM) 20 KCl, 100 K-gluconate, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, 0.2 EGTA, and 0.2% biocytin (pH 7.35, 295 mOsm). While the membrane potential of neurons was held at −70 mV, single APs were elicited by injecting current of the minimal amplitude required to elicit an AP. After several single APs were recorded, stimulating currents of different amplitudes were applied to measure the frequency of APs elicited by current stimulation to generate F-I curves. Data were acquired with a MultiClamp 700B amplifier (Molecular Devices) and WinLTP 2.20b software (University of Bristol) at a sampling rate of 40 kHz. Neurons whose membrane potential was higher than −55 mV and recordings whose series resistance exceeded 25 MΩ were excluded from analysis for quality control purposes. FS cells exhibiting AP adaption upon stimulation, as indicated by increasing inter-spike intervals, were excluded from analysis because their firing patterns were inconsistent with those of typical PV cells (Gouwens et al., 2019, 2020).
AP characteristics were extracted by scripts custom-written in MATLAB (The Math Works). The AP threshold was defined as the voltage at which the first derivative (dV/dt) of the AP waveform reached 20 mV/ms. The AP amplitude was calculated as the peak amplitude relative to the threshold. The AP half-width was defined as the width of the AP (in ms) at half amplitude. The AP afterhyperpolarization amplitude was calculated as the depth of the after-discharge potential (in mV) relative to the threshold.
Postsynaptic current recordings
EPSCs and IPSCs of pyramidal neurons were measured by whole cell recordings in standard ACSF. Patch pipettes were filled with an internal solution, containing (in mM) 9 CsCl, 123 CsMeSO3, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, 5 Na-phosphocreatine, 1 QX314, 1 EGTA, and 0.2% biocytin (pH 7.35, 295 mOsm). sEPSC were recorded at a holding voltage of −70 mV, at which there are no net currents through GABAA receptors. sIPSCs were isolated by recording at a holding potential of 0 mV, the reversal potential of AMPA receptor (AMPAR)- and NMDA receptor (NMDAR)-mediated currents. Slices were then perfused with recording ACSF containing 1 μM tetrodotoxin (Abcam ab120055) for the recording of mEPSCs and mIPSCs.
To record EPSCs from fast-spiking interneurons, patch pipettes were filled with an internal solution, containing (in mM) 9 KCl, 120 K-gluconate, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, 1 EGTA, and 0.2% biocytin (pH 7.35, 295 mOsm). To confirm that cells were indeed fast-spiking, APs were elicited with a 200 pA current at a membrane potential held at −70 mV in current clamp. sEPSCs and mEPSCs were then recorded as described above. Chemicals were purchased from Sigma-Aldrich unless stated otherwise. For each cell, > 200 EPSCs and IPSCs were detected and analyzed using Stimfit 0.15.8 (Guzman et al., 2014).
Spontaneous action potential recordings
A cell-attached voltage-clamp with loose-seal configuration was used to measure sAP spiking at a holding potential of 0 mV. Patch pipettes with a tip resistance of 1.5–2.5 MΩ were filled with active ACSF (see above), and the resistance of the seal ranged from 50–200 MΩ. Recordings were made for 3 min after a stable seal resistance was achieved. Numbers of APs were analyzed with Clampfit 10.4.
Immunofluorescence staining
Male and female mice were anesthetized with Avertin (2,2,2-tribromoethanol, 250 mg/kg body weight) and transcardially perfused with 0.9% NaCl solution. Brains were drop-fixed in 1% paraformaldehyde in phosphate-buffered saline (PBS) for 16 h at 4°C, washed three times with PBS, and immersed in 30% sucrose solution at 4°C overnight. Coronal brain sections (50-μm thick) were cut with a microtome (Leica SM2000R) and mounted on Superfrost Plus glass slides (Thermo Fisher) for immunostaining at room temperature. Brain sections were first incubated in PBS with 0.25% Triton X-100 for 30 min, in 10% normal goat serum in PBS for 1 h, and in primary antibodies against AnkG (NeuroMab, 75-146, diluted 1:500) or PV (Swant, PV27 or PV235, diluted 1:1000) overnight. Goat anti-mouse IgG2a Alexa 488, anti-rabbit Alexa 647, or anti-mouse IgG1 Alexa 647 (Invitrogen, diluted 1:500) were used as secondary antibodies.
For staining involving biocytin-filled cells, brain slices were fixed in 4% paraformaldehyde in PBS for 16 h at 4°C, washed three times with PBS, incubated in PBS with 0.25% Triton X-100 for 1 h, 10% normal goat serum in PBS for 1 h, and primary antibodies against PV (Swant, PV235, diluted 1:1000) overnight. Goat anti-mouse IgG1 Alexa 488 and streptavidin Alexa 594 (Invitrogen, diluted 1:500) were used as secondary antibodies.
Z stack images (1 μm/frame, 512 × 512 pixels) of immunostained sections and slices were obtained with a Zeiss LSM880 confocal microscope using a 20X objective and an additional 2X zoom. Eight z stack images of layer V in somatosensory cortex were taken per brain section, and two brain sections were imaged per mouse. Images were reconstructed with the Imaris software package (Oxford Instruments) to reveal the 3-D orientation of the AIS. Imaris was also used to measure the length of the AIS and the distance from the soma to the beginning of the AIS. In brief, the surface module in Imaris was used to identify individual AISs as an object based on a set of thresholding parameters, which were consistently applied to all relevant images. The length of the objects identified as AISs was automatically measured by the software. The distance from the start of an AIS object to the boundary of a cell was measured manually.
Primary neuronal cultures from male and female mice were fixed on DIV14 with 4% paraformaldehyde for 20 min at room temperature followed by 3 washes with PBS. Cells were then incubated in PBS with 0.25% Triton X-100 for 10 min, in 10% normal goat serum in PBS for 1 h at room temperature, and in 2% normal goat serum in PBS containing primary mouse antibodies against AnkG (NeuroMab, 75-147; 1:500), CaMKII (Millipore, 05-532; 1:1000), GAD67 (Millipore, MAB5406; 1:1000), and PROX1 (Abnova, M02; 1:1000), or against CaMKII, PROX1, and Vgat (Synaptic Systems, 131004; 1:1000), at 4°C overnight. Goat anti-mouse IgG1 Alexa 488, anti-mouse IgG2a Alexa 546, and anti-mouse IgG2b Alexa 647 (Invitrogen; 1:1000) were used as secondary antibodies. The same anti-mouse IgG2b (Alexa 647) was used for the detection of anti-AnkG and anti-PROX1 because the distinct subcellular distribution of the corresponding antigens (in AIS versus nucleus, respectively) makes it easy to distinguish between the resulting immunoreactivities. Z stack images were taken with a Zeiss LSM880 confocal microscope and analyzed with Imaris as described above.
GABA imaging
Primary neuronal cultures from male and female mice were transduced on DIV4 with AAV1 encoding iGABASnFR (Marvin et al., 2019) (Addgene, 112159-AAV1; 50,000 viral genomic copies/cell). Imaging was done at DIV13–14 in oxygenated standard ACSF at room temperature. A stimulus isolator (World Precision Instrument, A365) connected to a field stimulation chamber (Warner Instruments, RC-49MFS) was used to deliver field stimuli of 1 ms, 90 mA, and a frequency of 100 Hz. Images were acquired at 20 Hz with a Hamamatsu CMOS camera (ORCA-Flash4.0, C11440) on an Olympus BX51WI fluorescence microscope with a 40 × 0.8 numerical aperture objective (LUMPLFLN40XW) and a Lambda XL light source (Sutter Instrument). Data were analyzed with MetaFluor for Olympus 7.7.9, ImageJ 2.1.0/1.53c, and MATLAB R2016a. Briefly, the difference (DF) of the fluorescence intensities of a cell before and after stimulation was normalized to the intensity before stimulation with background subtraction (F) to obtain ΔF/F for further analysis.
MEA recordings
Recordings from primary neuronal cultures from male and female mice were obtained at DIV13–14 in standard ACSF for five 2-min periods with the MED64-Quad II system (Automate Scientific) at a sampling rate of 20 kHz using band-pass filters of 0.1 and 10 kHz. Traces were filtered with a Bessel lowpass filter of 1 kHz and spikes were detected with the threshold set to 5 times the standard deviation of the baseline noise, using the spike_frequency_analysis_filter package in Mobius analysis software (Automate Scientific). Bursts in single channels were detected by analyzing the extracted spikes with the Mobius offline toolkit (Automate Scientific) and defined as beginning with ≥2 spikes that are ≤100 ms apart, containing ≥10 spikes in total, and lasting ≥100 ms. To quantify network bursts, the extracted spikes from each channel were binned at 10 ms and the “array-wide” spike frequency was calculated from the sum of spike frequencies at a given time point across all 16 channels. Network bursts were defined by the “Schmitt trigger” method using high (1 kHz) and low (0.5 kHz) thresholds to detect array-wide spike frequencies.
The global synchrony index of the neuronal network within a given MEA chamber was calculated based on previously described approaches. Briefly, a phase synchronization matrix was constructed for all channels in an MEA chamber by assigning an instantaneous phase to each channel and calculating circular variances of phase differences between pairs of channels (Patel et al., 2012). The eigenvalues of this matrix were cauculated, and a surrogate matrix was created by randomizing the instantaneous phase of each channel with amplitude-adjusted Fourier transform, and eigenvalues of this surrogate matrix were calculated. This randomization was repeated multiple times to obtain the mean and standard deviation of the surrogate eigenvalues. The global synchrony index was calculated from the maximal eigenvalue of the phase synchronization matrix and the mean and standard deviation of the surrogate eigenvalues (Li et al., 2007). A custom Python script was developed and run in NeuroExplorer (Nex Technologies) to obtain global synchrony indices and is available from https://github.com/NeuroExplorer/Scripts/blob/master/Analysis_SyncIndex.py.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Most sets of data are displayed as box-and-whisker plots, in which the lower and upper ends of the boxes represent the 25th and 75th quartile of each distribution, respectively. The horizontal line in each box represents the median. The ends of the whiskers terminate at the farthest points that are within 1.5 times the inter-quartile range (difference between upper and lower ends of the box). The numbers immediately following “n =” in the figure legends were used for statistical analyses, based on typical approaches used in the published literature for the respective types of experiments. For example, numbers of cells were used as the biological “n” for electrophysiological recordings from slices and cultures, consistent with previous reports (Grubb and Burrone, 2010; Polepalli et al., 2017; Frega et al., 2019; Yamamuro et al., 2020), while numbers of AISs (corresponding to individual cells) were used for AIS characterizations, similar to Grubb and Burrone (2010), Evans et al. (2013, 2015), Sohn et al. (2019), and Pan-Vazquez et al. (2020), and numbers of culture wells for MEA recordings, as reported by Black et al. (2017), Sohn et al. (2019), and Tukker et al. (2020). See Table S4 for a complete list of statistical comparisons based on numbers of AISs, cells, or mice. The permutation test was done in RStudio V1.2.1335 (https://www.rstudio.com/). All other statistical analyses were done with Prism software version 8.00 (GraphPad Software). For each untransformed and log-transformed dataset, we first used the D’Agostino-Pearson test to ascertain whether all groups were distributed normally. If so, parametric tests were used. If not, nonparametric tests were used. For normally distributed data, the F-test was used to determine whether the variances among groups were comparable. If so, the unpaired, two-tailed Student’s t test was used for datasets with one independent variable and two-way ANOVA, followed by Holm-Sidak post hoc test for pairwise comparisons, for datasets with two independent variables. For nonparametric data, the Mann-Whitney test was used for datasets with one independent variable and the permutation test with Holm-Sidak correction for multiple comparisons for datasets with two independent variables. For comparisons of F-I curves and input-output curves obtained by AP recording or fluorescence imaging after repeated stimulation of cells, areas under the curves were compared by unpaired, two-tailed t test or permutation test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-Vgat | Synaptic Systems | Cat#131004; RRID: AB_887873 |
| Mouse anti-AnkG | NeuroMab | Cat#75-146; RRID: AB_10673030 |
| Mouse anti-AnkG | NeuroMab | Cat#75-147; RRID: AB_10675130 |
| Mouse anti-CaMKII | MilliporeSigma | Cat#05-532; RRID: AB_309787 |
| Mouse anti-GAD67 | MilliporeSigma | Cat#MAB5406; RRID: AB_2278725 |
| Mouse anti-PROX1 | Abnova | Cat#H00005629-M02; RRID: AB_606848 |
| Mouse anti-PV | Swant Inc. | Cat#235; RRID: AB_10000343 |
| Rabbit anti-PV | Swant Inc. | Cat#PV27; RRID: AB_2631173 |
| Bacterial and virus strains | ||
| pAAV.hSynap.iGABASnFR | Marvin et al., 2019 | Addgene 112159-AAV1 |
| Chemicals, peptides, and recombinant proteins | ||
| Papain | Worthington | Cat#LK003178 |
| Tetrodotoxin | Abcam | Cat#ab120055 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129X1-Mapttm1Hnd/J | The Jackson Laboratory | 007521 |
| Mouse: C57BL/6J | The Jackson Laboratory | 000664 |
| Software and algorithms | ||
| Algorithm for global synchrony Index | This paper | https://github.com/NeuroExplorer/Scripts/blob/master/Analysis_SyncIndex.py |
| ImageJ 2.1.0/1.53c | ImageJ | RRID: SCR_003070 |
| Imaris 9.6 | Oxford Instruments | RRID: SCR_007370 |
| MATLAB R2016a | MathWorks | RRID: SCR_001622 |
| MED64 Mobius | Alpha MED Scientific Inc. | https://www.med64.com/products/med64-mobius-software/ |
| MED64 Mobius Offline Toolkit | Alpha MED Scientific Inc. | https://www.med64.com/products/med64-mobius-software/mobius-offline-toolkit/ |
| MetaFluor for Olympus 7.7.9 | Olympus | https://www.olympus-lifescience.com/en/software/metamorph/features/ |
| NeuroExplorer 5 | Nex Technologies | RRID: SCR_001818 |
| pCLAMP | Molecular Devices | RRID: SCR_011323 |
| Prism 8.0 | GraphPad | RRID: SCR_002798 |
| RStudio V1.2.1335 | RStudio | RRID: SCR_000432 |
| Stimfit 0.15.8 | Guzman et al., 2014 | RRID: SCR_016050 |
| WinLTP 2.20b | WinLTP Ltd. | RRID: SCR_008590 |
Highlights.
Tau ablation reduces baseline activity and E/I ratio of excitatory neurons
Tau ablation preferentially increases the excitability of inhibitory neurons
Tau ablation modulates the AISs of inhibitory neurons
Reduced neuronal activity and enhanced inhibition counteract network hypersynchrony
ACKNOWLEDGMENTS
We thank Roger Nicoll for review and discussion of the manuscript, Chao Tai and Lei Zhu for advice on electrophysiological methods, Yu-Wei Wu for advice on GABA imaging, Kelli Lauderdale and Jorge Palop for advice on MEA recordings, Alexander Kirillov from NeuroExplorer for development of the GSI python script, the Bioinformatics Core and the Histology and Light Microscopy Core of the Gladstone Institutes for technical assistance, Stephen Ordway for editorial review, and Randi Mott and Essie Kimball for administrative assistance. This work was supported by U.S. National Institutes of Health grant MH115679 and gifts from the Ray and Dagmar Dolby Family Fund and the Tau Consortium to L.M. C.-W.C. was supported by a Research Fellowship from the Alzheimer’s Association and M.D.E. by an Alan Kaganov Scholarship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109855.
DECLARATION OF INTERESTS
L.M. has received research funding from Cure Network Dolby Acceleration Partners (CNDAP) and has served on CNDAP’s board of managers. He has also served on the scientific advisory boards of Arvinas, Biogen, and Dolby Family Ventures and has consulted for Eisai and Sangamo Therapeutics. L.M. is a coinventor on patents held by the Gladstone Institutes that focus on tau reduction as a strategy to block neural network dysfunction.
REFERENCES
- Bender KJ, and Trussell LO (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci 35, 249–265. [DOI] [PubMed] [Google Scholar]
- Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, Chua SW, van der Hoven J, Lee WS, Müller J, et al. (2017). Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun 8, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BJ, Atmaramani R, and Pancrazio JJ (2017). Spontaneous and evoked activity from murine ventral horn cultures on microelectrode arrays. Front. Cell. Neurosci 11, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker SA, Simões de Oliveira L, Anstey NJ, Kozic Z, Dando OR, Jackson AD, Baxter PS, Isom LL, Sherman DL, Hardingham GE, et al. (2020). Input-output relationship of CA1 pyramidal neurons reveals intact homeostatic mechanisms in a mouse model of fragile x syndrome. Cell Rep. 32, 107988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, Kamath TV, Carlson GA, Nelken I, and Hyman BT (2019). Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci 22, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Shao E, and Mucke L (2021). Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371, eabb8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimins JL, Rocher AB, and Luebke JI (2012). Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 124, 777–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Sanacora G, and Krystal JH (2019). Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 102, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Sammons RP, Lebron S, Dumitrescu AS, Watkins TB, Uebele VN, Renger JJ, and Grubb MS (2013). Calcineurin signaling mediates activity-dependent relocation of the axon initial segment. J. Neurosci 33, 6950–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Dumitrescu AS, Kruijssen DLH, Taylor SE, and Grubb MS (2015). Rapid modulation of axon initial segment length influences repetitive spike firing. Cell Rep. 13, 1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fréal A, Fassier C, Le Bras B, Bullier E, De Gois S, Hazan J, Hoogenraad CC, and Couraud F (2016). Cooperative interactions between 480 kDa ankyrin-G and EB proteins assemble the axon initial segment. J. Neurosci 36, 4421–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frega M, Linda K, Keller JM, Gümüş-Akay G, Mossink B, van Rhijn JR, Negwer M, Klein Gunnewiek T, Foreman K, Kompier N, et al. (2019). Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat. Commun 10, 4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere S, and Slutsky I (2018). Alzheimer’s disease: From firing instability to homeostasis network collapse. Neuron 97, 32–58. [DOI] [PubMed] [Google Scholar]
- García-Cabrero AM, Guerrero-López R, Giráldez BG, Llorens-Martín M, Avila J, Serratosa JM, and Sánchez MP (2013). Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis 58, 200–208. [DOI] [PubMed] [Google Scholar]
- Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, and Mucke L (2014). Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol 76, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, and Spillantini MG (2019). Ordered assembly of tau protein and neurodegeneration. In Tau Biology. Advances in Experimental Medicine and Biology, Takashima A, Wolozin B, and Buee L, eds. (Springer; ). [DOI] [PubMed] [Google Scholar]
- Goldberg EM, and Coulter DA (2013). Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci 14, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Berg J, Lee C, Jarsky T, Ting J, Sunkin SM, Feng D, Anastassiou CA, Barkan E, et al. (2019). Classification of electrophysiological and morphological neuron types in the mouse visual cortex. Nat. Neurosci 22, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T, Alfiler L, Baker K, Barkan E, Berry K, et al. (2020). Integrated morpho-electric and transcriptomic classification of cortical GABAergic cells. Cell 183, 935–953.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, and Burrone J (2010). Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Shu Y, Kuba H, Rasband MN, Wimmer VC, and Bender KJ (2011). Short- and long-term plasticity at the axon initial segment. J. Neurosci 31, 16049–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman SJ, Schlögl A, and Schmidt-Hieber C (2014). Stimfit: quantifying electrophysiological data with Python. Front. Neuroinform 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch RJ, Wei Y, Xia D, and Götz J (2017). Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 133, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, and Noebels JL (2013). Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci 33, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. [DOI] [PubMed] [Google Scholar]
- Kanner AM (2016). Management of psychiatric and neurological comorbidities in epilepsy. Nat. Rev. Neurol 12, 106–116. [DOI] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, and Ohmori H (2010). Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature 465, 1075–1078. [DOI] [PubMed] [Google Scholar]
- Lam AD, Sarkis RA, Pellerin KR, Jing J, Dworetzky BA, Hoch DB, Jacobs CS, Lee JW, Weisholtz DS, Zepeda R, et al. (2020). Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology 95, e2259–e2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Vacher H, Fache MP, d’Ortoli SA, Castets F, Autillo-Touati A, and Dargent B (2011). End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc. Natl. Acad. Sci. USA 108, 8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cui D, Jiruska P, Fox JE, Yao X, and Jefferys JG (2007). Synchronization measurement of multiple neuronal populations. J. Neurophysiol 98, 3341–3348. [DOI] [PubMed] [Google Scholar]
- Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, et al. (2016). Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 17, 530–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Shimoda Y, Magloire V, Leite M, Kawashima T, Jensen TP, Kolb I, Knott EL, Novak O, Podgorski K, et al. (2019). A genetically encoded fluorescent sensor for in vivo imaging of GABA. Nat. Methods 16, 763–770. [DOI] [PubMed] [Google Scholar]
- Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, and Stern EA (2015). Pathological tau disrupts ongoing network activity. Neuron 85, 959–966. [DOI] [PubMed] [Google Scholar]
- Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S, et al. ; Pharmacogenomics of Bipolar Disorder Study (2015). Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka A, and Adesnik H (2016). Inhibitory circuits in cortical layer 5. Front. Neural Circuits 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, and Mucke L (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci 17, 777–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan-Vazquez A, Wefelmeyer W, Gonzalez Sabater V, Neves G, and Burrone J (2020). Activity-dependent plasticity of axo-axonic synapses at the axon initial segment. Neuron 106, 265–276.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Ventre SC, and Meaney DF (2012). Dynamic changes in neural circuit topology following mild mechanical injury in vitro. Ann. Biomed. Eng 40, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, and Charpier S (2007). Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J. Neurosci 27, 6590–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polepalli JS, Wu H, Goswami D, Halpern CH, Südhof TC, and Malenka RC (2017). Modulation of excitation on parvalbumin interneurons by neuroligin-3 regulates the hippocampal network. Nat. Neurosci 20, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, and Jensen FE (2009). Epileptogenesis in the immature brain: emerging mechanisms. Nat. Rev. Neurol 5, 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, and Mucke L (2007). Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu G-Q, et al. (2011). Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci 31, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu G-Q, Palop JJ, and Mucke L (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 109, E2895–E2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayas CL, Tortosa E, Bollati F, Ramírez-Ríos S, Arnal I, and Avila J (2015). Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J. Neurochem 133, 653–667. [DOI] [PubMed] [Google Scholar]
- Shimojo M, Takuwa H, Takado Y, Tokunaga M, Tsukamoto S, Minatohara K, Ono M, Seki C, Maeda J, Urushihata T, et al. (2020). Selective disruption of inhibitory synapses leading to neuronal hyperexcitability at an early stage of tau pathogenesis in a mouse model. J. Neurosci 40, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, and Rubenstein JLR (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn PD, Huang CT, Yan R, Fan L, Tracy TE, Camargo CM, Montgomery KM, Arhar T, Mok SA, Freilich R, et al. (2019). Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104, 458–470.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K (2015). Molecular mechanisms of epilepsy. Nat. Neurosci 18, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Abe Y, Westenbroek RE, Scheuer T, and Catterall WA (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 111, E3139–E3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Chang CW, Yu GQ, Lopez I, Yu X, Wang X, Guo W, and Mucke L (2020). Tau reduction prevents key features of autism in mouse models. Neuron 106, 421–437.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman R, Cuccaro M, and Alessandri M (2010). Autism and epilepsy: historical perspective. Brain Dev. 32, 709–718. [DOI] [PubMed] [Google Scholar]
- Tukker AM, Bouwman LMS, van Kleef RGDM, Hendriks HS, Legler J, and Westerink RHS (2020). Perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) acutely affect human α1-β2γ2L GABAA receptor and spontaneous neuronal network function in vitro. Sci. Rep 10, 5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandeVrede L, Boxer AL, and Polydoro M (2020). Targeting tau: Clinical trials and novel therapeutic approaches. Neurosci. Lett 731, 134919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, et al. (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol 80, 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JM, McMillan PJ, Hawk M, Iba M, Robinson L, Xu GJ, Dombroski BA, Jeong D, Dichter MA, Juul H, et al. (2015). High copy wildtype human 1N4R tau expression promotes early pathological tauopathy accompanied by cognitive deficits without progressive neurofibrillary degeneration. Acta Neuropathol. Commun 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamuro K, Bicks LK, Leventhal MB, Kato D, Im S, Flanigan ME, Garkun Y, Norman KJ, Caro K, Sadahiro M, et al. (2020). A prefrontal-paraventricular thalamus circuit requires juvenile social experience to regulate adult sociability in mice. Nat. Neurosci 23, 1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Li HL, Tian N, Liu F, Wang L, Yin Y, Yue L, Ma L, Wan Y, and Wang JZ (2020). Interneuron accumulation of phosphorylated tau impairs adult hippocampal neurogenesis by suppressing GABAergic transmission. Cell Stem Cell 26, 331–345.e6. [DOI] [PubMed] [Google Scholar]
- Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, Sakmann B, Walsh DM, and Konnerth A (2019).A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 365, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon reasonable request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.