

Abstract
Late-onset Alzheimer disease (LOAD) is highly polygenic, with a heritability estimated between 40 and 80%, yet risk variants identified in genome-wide studies explain only ~8% of phenotypic variance. Due to its increased power and interpretability, genetically regulated expression (GReX) analysis is an emerging approach to investigate the genetic mechanisms of complex diseases. Here, we conducted GReX analysis within and across 51 tissues on 39 LOAD GWAS data sets comprising 58,713 cases and controls from the Alzheimer’s Disease Genetics Consortium (ADGC) and the International Genomics of Alzheimer’s Project (IGAP). Meta-analysis across studies identified 216 unique significant genes, including 72 with no previously reported LOAD GWAS associations. Cross-brain-tissue and cross-GTEx models revealed eight additional genes significantly associated with LOAD. Conditional analysis of previously reported loci using established LOAD-risk variants identified eight genes reaching genome-wide significance independent of known signals. Moreover, the proportion of SNP-based heritability is highly enriched in genes identified by GReX analysis. In summary, GReX-based meta-analysis in LOAD identifies 216 genes (including 72 novel genes), illuminating the role of gene regulatory models in LOAD.
Subject terms: Genomics, Molecular neuroscience
Introduction
Late-onset Alzheimer’s disease (LOAD) is the most common neurodegenerative disease in the world, occurring in >35% of individuals age 85 years and older [1], and it is the sixth most common cause of death in the US. LOAD has a substantial genetic component, with heritability estimated to be between 40 and 80% [2–4]. Despite this high heritability and numerous genome-wide studies conducted to date, known LOAD-associated single nucleotide polymorphisms (SNPs) explain only 30.62% of the genetic variance [5], with the majority of risk attributed to variants within APOE [5, 6]. Much of the SNP-based heritability of LOAD remains unmapped to causal genetic factors; this so-called “missing heritability”, is common to many complex diseases [7]. Recently developed methods leveraging transcriptomic data have powered the identification of novel genetic risk factors, increasing the proportion of heritability explained by known loci and expanding our understanding of the biological processes underlying disease, such as cardiometabolic diseases and neuropsychiatric traits [8–10].
In the most recent large-scale meta-analysis of LOAD GWAS a total of 25 genes were identified, including five novel genes, IQCK, ACE, ADAM10, ADAMTS1, and WWOX [11]. Another recent meta-analysis, maximized the sample size by including potential AD cases based on parental AD status, identifying 13 novel loci [12]. Although these genes have been associated with LOAD by the position of GWAS-identified SNPs, understanding and interpreting the biological implications of LOAD associations is a difficult challenge due to our limited knowledge of the underlying mechanisms. Among the 1073 GWAS-identified LOAD variants, only 2% are located in gene coding regions, and most do not have a clear molecular function for LOAD [11]. Gene expression studies provide an opportunity to investigate an important class of molecular mediators and their consequent effects on disease outcomes. Gene expression is regulated by complex mechanisms involving various factors, e.g., genetic variation, age, and environmental stimuli. The importance of gene expression is demonstrated by the enrichment of GWAS-identified loci in expression quantitative trait loci (eQTLs) [13]. As previously estimated, over 70% of LOAD variant have the potential to regulate the gene expression across all tissues [11]. A previous transcriptome study in human brain tissues has identified 207 differentially expressed genes [14, 15]. In addition to identifying more novel LOAD genes, transcriptome-informed studies offer a potential genetic mechanism of disease. Previously, rs1057233 was reported as a CELF1 LOAD locus [16], but a further gene expression study suggests it may cause LOAD via affecting SPI1 expression instead of CELF1 [17]. Therefore, investigating the association between gene expression and traits of interest can be an effective way to understand the functional mechanism of GWAS loci.
Although studying gene expression has improved our knowledge about the genetic mechanism of LOAD, the difficulty of collecting LOAD relevant tissues historically has limited the sample sizes available for analysis, and the complexity of gene expression regulation is also heavily affected by environment and other uncontrollable factors. Genetically regulated expression (GReX) association is an emerging method to test the association of gene expression, imputed using common genetic variants, with phenotype. Since genetic influence on phenotype is lifelong, GReX reduces noise from temporal and environmental factors, and instead identifies the direct impact of genetic effects that influence gene expression on trait [18]. Also, the imputed GReX is based on genetic variants, so we can leverage the sample size of previous genetic LOAD studies to increase the statistical power of this gene-based analysis. Moreover, GReX aggregates genetic variants into a gene-level functional unit, based on gene expression. As a result, GReX results are intrinsically related to function in contrast to single variant GWAS where the results are often found in intergenic regions and the relevant gene remains unclear. The gene-level test also has the effect of increasing the statistical power by reducing multiple testing burden.
In this study, we imputed tissue-specific GReX using Predixcan for 12,162 LOAD cases and 13,614 healthy controls from 31 epidemiological studies of LOAD. PrediXcan trains models of gene expression in a transcriptome reference panel linked to whole-genome data, such as the Genotype Tissue Expression Project (GTEx) and leverages the models to impute GReX in independent genomic data [18]. In addition, S-PrediXcan was applied for another eight LOAD studies (8451 cases and 24,044 controls) for which individual genotype data were not available. S-PrediXcan was developed to use GWAS summary statistics instead, and estimates the gene-trait association based on these statistics and reference linkage disequilibrium structure [19]. With both SNP-based and GWAS summary statistic-based approaches, data on a total of 58,713 individuals were examined in this study. Although summary-based GReX has been successfully applied in various traits [20–22], the fact that the allele frequency and linkage disequilibrium data come from an external population may reduce power to detect gene-trait associations, and its effects have not been determined. Therefore, we compared the sensitivity of genotype-based and summary statistic-based GReX methods to quantify the benefit of using an internal LD structure. In addition to tissue-specific GReX analysis, cross-tissue GReX has been proposed to increase the power to detect gene-trait associations [23]. Due to the high correlation of gene expression across tissues [24], cross-tissue GReX models extract the co-expression across selected tissues, thereby allowing for the identification of more genes by aggregating their effects, especially for the identification of genes with modest signals in several tissues that do not meet single-tissue significance thresholds. Therefore, we also implemented two cross-tissue models, one including all brain tissues and one including all available tissues. Finally, to quantify the improvement of GReX approaches over traditional GWAS, we estimated the proportion of heritability captured by our identified LOAD genes, demonstrating that the major component of LOAD SNP-based heritability is captured by this approach.
Subjects and methods
ADGC Subjects
The ADGC GWAS data comprised 31 studies with different sample sizes and ratios of controls to cases (Supplementary Table 1). Seven waves of subjects were selected from the National Institute on Aging (NIA) Alzheimer’s Disease Centers (ADCs), including the Adult Changes in Thought (ACT) study [25], the Alzheimer Disease Neuroimaging Initiative (ADNI) study [26], the National Institute on Aging Late-Onset Alzheimer’s Disease Family (NIA-LOAD) study [27], the Mayo Clinic Jacksonville (MAYO), the Multi Institutional Research of Alzheimer Genetic Epidemiology (MIRAGE) Study [28], Oregon Health and Science University (OHSU), the Rush University Religious Orders Study/Memory and Aging Project (ROS/MAP) [29], the Translational Genomics Research Institute series 2 (TGEN2) [30], the University of Miami/Case Western Reserve University/Mt. Sinai School of Medicine (UM/CWRU/MSSM), University of Pittsburgh (UPitt) [31], two waves from Washington University (WASHU), the Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer’s disease (GenADA) [28], the Universitätsklinikum des Saarlandes (UKS), and the Netherlands Brain Bank (NBB) [32], Biomarkers of Cognitive Decline Among Normal Individuals: the BIOCARD cohort (BIOCARD), Chicago Health and Aging Project (CHAP2), Einstein Aging Study (EAS), Mayo Clinic (RMAYO), Washington Heights-Inwood Community Aging Project (WHICAP). A detailed description of inclusion and exclusion criteria has been provided in previous studies [11, 32, 33].
Genotyping was done with either Illumina or Affymetrix high-density SNP microarrays. The criteria for minimal call rate and minor allele frequency (MAF) were 0.95 and 0.02 for the Illumina chip, and 0.98 and 0.01 for Affymetrix. Genotype imputation was conducted using the HRC r1.1 as the reference panel and minimac3 for imputation on the University of Michigan Imputation Server [34]. Variants with MAF > 0.05 and imputation score (R2 for MaCH/Minimac3 > 0.5) were included in further analyses. A total of 25,776 samples of European ancestry, comprising 12,162 cases and 13,614 controls, were carried forward in the ADGC analysis [16].
Imputing tissue-specific expression using PrediXcan models
Tissue-specific GReX in ADGC was imputed using PrediXcan with publicly-available gene expression imputation models built-in reference transcriptome data sets [18]. In total, 51 tissue-specific models were used, including the whole blood model for DGN [35], the dorsolateral prefrontal cortex (DLPFC) model from the CommonMind Consortium [21, 36], and another 49 tissues models from the Genotype-Tissue Expression (GTEx) project (version v8) [37, 38]. DGN and DLPFC models were trained using an elastic net approach, and the other models from GTEx v8 used multivariate adaptive shrinkage (MASHR) [38]. These PrediXcan models leveraged cross-tissue information in the model building step by applying a Bayesian methodology, the multivariate adaptive shrinkage model (MASHR), which allows for sparse effects and correlation in effect sizes across tissues [38].
Firth regression
Maximum likelihood estimation from conventional logistic regression may suffer from bias due to the presence of rare events, such as in extremely unbalanced data sets. For this reason, we used Firth logistic regression for GReX association testing, which is less sensitive to bias due to case–control imbalance [39]. To test the association between imputed gene expression level and disease status, logistic Firth regression was performed within each study using R package, logistf, with covariate adjustment for sex, age (age-at-onset for cases and age-at-last-exam or age-at-death for controls), and principal components analysis (PCA) to correct for population structure [40] (from 2 to 4 components for each dataset, based on total variance explained, as described in Naj et al. [33]).
Conditional analyses in known LOAD-associated regions
Because our PrediXcan results validated many previously reported LOAD regions, for all PrediXcan significant genes located 10 Mb upstream or downstream of a known LOAD-risk SNP reported in Naj et al. [41], we conducted conditional analyses two ways. Firstly, we adjusted for the nearby previously reported SNP by including the additive genotype for each SNP as a covariate in the Firth regression model. Secondly, we similarly adjusted for GReX of the gene reported for the known SNP to characterize independent residual effects. The known risk SNPs included 29 total SNPs located in 28 genes (Supplementary Table 2) [41]. If the previously known SNP was not available in the set of high-quality imputed variants in ADGC, a tag SNP was chosen, prioritizing SNPs in strongest linkage disequilibrium with the risk SNP (r2 > 0.6 based on the 1000 Genomes Project CEU reference population). No SNPs with r2 > 0.6 were available in most studies for eight SNPs of interest, and thus we did not perform SNP-adjusted conditional analysis for rs75932628 (TREM2), rs11218343 (SORL1), rs74615166 (TRIP4), rs138190086 (ACE), rs8093731 (DSG2), rs145999145 (PLD3), rs63750847 (APP), and rs7412 (APOE) (though data was available for the other APOE ε2/ε3/ε4 haplotype SNP, rs429358). Conditional analyses were performed using logistic Firth regression in R, adjusting for the same covariates used in the primary analysis, as well as the dosage of the LOAD-risk SNP or the tissue-specific GReX of the gene reported for the known SNP.
Cross-tissue analyses
To evaluate genetic effects across tissues, we used multivariate logistic Firth regression. We considered two different cross-tissue analyses: (1) cross-all tissues, combining expression data from all available GTEx tissues (Supplementary Table 3), and (2) cross-brain tissues, combining expression data from all available brain tissues in GTEx (Supplementary Table 3). Only genes with prediction models in at least five (cross-all) or three (cross-brain) tissues were included in these analyses. Because we expect correlation in expression across tissues, to avoid collinearity, we computed principal components for each gene using all available predicted gene expression levels across (1) all tissues and (2) brain tissues only. Principal components sufficient to explain >80% of variance were carried forward in association tests. We conducted Firth regression including covariate adjustment for sex, age-at-onset/age-at-last-exam, SNP-based principal components (i.e., genetic ancestry) for the reduced model, Mreduced, and, in addition, the expression-based principal components for the full model, Mfull. Significance was tested by comparing Mfull and Mreduced using a likelihood ratio test:
Here and are the likelihood for the full and reduced models, respectively. This likelihood ratio statistic has an asymptotic χ2 distribution under the null hypothesis. We calculated the p-value using a χ2 test in which the number of degrees of freedom is equal to the number of principal components, which varies by gene.
IGAP subjects
The International Genomics of Alzheimer’s Project (IGAP) data used here comprised eight European GWAS from seven studies with LOAD data: four prospective cohort studies from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium [42], including the Atherosclerosis Risk in Communities (ARIC) Study [43], Cardiovascular Health Study (CHS) [44], the Framingham Heart Study (FHS) [45], and two waves of subjects from the Rotterdam Study (RS) [46]; the Genetic and Environmental Risk in Alzheimer’s Disease (GERAD) Consortium [47]; the European Alzheimer’s Disease Initiative (EADI) [48], and the Bonn Study [49, 50]. A previous meta-analysis of these and other LOAD GWAS has described sampling and phenotyping procedures as well as demographic characteristics for each of these studies [11, 16]. Genotyping and association analysis methods are also detailed in Kunkle et al. [11]. Summary statistics from each study were cleaned to remove low frequency and low imputation quality using the same inclusion thresholds used for ADGC (MAF < 0.01 and R2 < 0.5).
S-PrediXcan and S-MultiXcan
S-PrediXcan [19] infers GReX-trait association from GWAS summary statistics using the same expression prediction models as for PrediXcan, as well as linkage disequilibrium data from a reference population (here 1000 Genomes Project), which captures covariance of variants in the prediction model. Summary statistics from the IGAP GWAS were pruned to remove variants with low frequency (MAF < 0.01) and low imputation quality (R2 < 0.5) and supplied as input to S-PrediXcan. We inferred cross-tissue GReX-trait association based on summary statistics in IGAP using S-MultiXcan. S-MultiXcan extracts cross-tissue GReX principal components, and evaluates statistical significance by assessing model fitness using an F-test. The same criteria for allele frequency and imputation quality were applied. Two cross-tissue models were implemented, cross-all GTEx tissues and cross-GTEx-brain tissues.
Meta-analysis and multiple testing correction
We performed fixed-effects meta-analysis of the single-tissue and the cross-tissue results, separately, across studies (Supplementary Table 1). Let and be the z-score for a gene and sample size respectively from study i. The weighted z-score statistic is defined as:
This statistic is distributed as a standard normal distribution, , under the null hypothesis, yielding a p-value. We used METAL [51] for the single-tissue results and the metap R package [52] implementation for the cross-tissue model.
We applied the Benjamini–Hochberg method for study-wide multiple testing correction [53]. The total number of tests for discovery was 733,844 (from single-tissue and cross-tissue analyses). We used B–H adjusted p-value < 0.05 as the significance cutoff, corresponding to an original study-wide p-value significance threshold of p = 1.02 × 10-4.
Causal inference
We conducted the FOCUS to clarify the importance of genes within the same linkage disequilibrium region [54]. We tested all the multivariate adaptive shrinkage models from GTEx v8, and the linkage disequilibrium region was determined by the European ancestry population in 1000 Genomes. Only the regions containing GWAS p-value <1 × 10−5 were used for the fine mapping. The LOAD GWAS was produced with all ADGC studies. We used the marginal posterior inclusion probability (PIP) to determine the gene’s importance, and used Spearman’s rank correlation test to compare it with the Z-score from our GReX association test in ADGC. Furthermore, we used Mendelian randomization to distinguish the SNPs’ effect on LOAD whether through gene expression or not. The same ADGC’s GWAS was used, and the eQTLs associations were from GTEx v.8. We applied the median-based Mendelian randomization (mr_median) in the R package, “MendelianRandomization”, on our identified LOAD genes [55, 56].
Sensitivity analyses
To facilitate a direct comparison of the PrediXcan and S-PrediXcan approaches, we completed an S-PrediXcan analysis using the same ADGC data/samples used for the PrediXcan analysis. Summary statistics from the ADGC GWAS performed above were pruned to remove variants with low frequency (MAF < 0.01) and low imputation quality (R2 < 0.5) and supplied as input to S-PrediXcan.
To evaluate the performance of individual tissue-specific models, we used the true positive rate of the association test from GReX analysis in each tissue, using the method described in Storey et al. [57].
Heritability analyses
We used linkage disequilibrium score regression (as implemented in LDSC [58]) to estimate the heritability of LOAD and the proportion of heritability attributable to our identified LOAD genes. LDSC estimates heritability based on the relationship between GWAS summary statistics and linkage disequilibrium. To quantify the overall increase in heritability explained in our study, we used LDSC in summary statistics from a previous LOAD GWAS [16]. We calculated the proportion of heritability explained by all genes identified in our study, conducting an LDSC analysis including only SNPs within 1 Mb of our identified genes, and compared this to the previous estimate of heritability. We also estimated heritability explained by the APOE region alone, including only SNPs within 10 Mb of APOE, and compared this to the estimated heritability outside of the APOE region, including all SNPs within 1 Mb of our identified genes but not within 10 Mb of APOE. Finally, we calculated heritability from different tissue-specific findings, including SNPs within 1 Mb of our findings from (1) only whole blood and (2) only brain tissues.
Results
We included two sources of data in this study. First, we utilized imputed genotype data from Alzheimer’s Disease Genetics Consortium (ADGC). ADGC contains 31 LOAD epidemiological studies with 12,162 LOAD cases and 13,614 controls (Supplementary Table 1). Genome-wide GReX were imputed by PrediXcan with the whole blood model from Depression Susceptibility Genes and Networks (DGN), dorsolateral prefrontal cortex model from CommonMind Consortium (DLPFC), and 49 tissue-specific models from GTEx v8. Second, we also analyzed GWAS summary results from the International Genomics of Alzheimer’s Project (IGAP) after excluding samples overlapping with ADGC, comprising another eight LOAD studies with 8451 cases and 24,044 controls. We applied S-PrediXcan to these summary statistics to identify gene-level associations with LOAD. Across both data sets, a total of 23,625 unique genes across 51 tissue-specific models were tested. To determine study-wide significance, we used the Benjamini–Hochberg procedure to control the false discovery rate. Genes with an adjusted p-value of <0.05 were considered significant (p-value < 1.02 × 10-4 across 733,844 tests).
In the 51 single-tissue models, we identified 1459 tissue-specific GReX from 208 unique genes significantly associated with LOAD (Fig. 1, Table 1, and Supplementary Table 4). The strongest gene association identified, TOMM40, falls within a large region of 43 significantly associated genes encompassing APOE and spanning 10 Mb in either direction (lead signals for TOMM40 were observed in GTEx V8 skin tissues, p-value = 1.28 × 10−600). After adjusting for known LOAD SNPs nearby, 11 unique genes maintained their significance, including LILRA5 from the APOE region, TMEM163 from the BIN1 region, SIK2 from the SORL1 region, and another 8 genes from the CELF1 region (Table 2). TOMM40 was not statistically significant after conditional analysis. In addition, 72 genes are novel LOAD genes that are neither reported in previous GWAS [59] (Table 1) nor within 10 Mb of 31 well-known LOAD SNPs from Naj et al. [41] (Supplementary Table 2).
Fig. 1. Miami plot of tissue-specific GReX association tests with LOAD from meta-analysis of AGDC and IGAP (top) or ADGC-only (bottom).

The green region labels the ±10 Mb of known AD loci, the blue dot indicates the novel gene, which locates outside the known AD region (green) and is never reported in previous GWAS, and the brown dot in ADGC-only meta-analysis suggests the gene holds significance in conditional analysis. P-values less than 1 × 10−15 were truncated (pink dot) to improve the clarity of peaks in other, less significant regions.
Table 1.
Strongest tissue-specific associations of novel LOAD genes.
| ADGC & IGAP | ADGC | IGAP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genes | Tissue | N | Z | p-value | N | Z | p-value | N | Z | p-value |
| TRIT1 | Brain amygdala | 58,713 | 4.615 | 3.92E-06 | 25,776 | 3.025 | 2.49E-03 | 32,937 | 3.487 | 4.89E-04 |
| TSPAN14 | Cells cultured fibroblasts | 58,624 | 4.572 | 4.83E-06 | 25,687 | 2.224 | 2.61E-02 | 32,937 | 4.136 | 3.54E-05 |
| LRRC8D | Testis | 58,713 | −4.493 | 7.02E-06 | 25,776 | −3.599 | 3.19E-04 | 32,937 | −2.815 | 4.88E-03 |
| SHARPIN | Colon transverse | 58,713 | 4.472 | 7.76E-06 | 25,776 | 3.426 | 6.13E-04 | 32,937 | 2.94 | 3.29E-03 |
| MYCL | Artery coronary | 58,713 | −4.437 | 9.13E-06 | 25,776 | −2.947 | 3.21E-03 | 32,937 | −3.316 | 9.12E-04 |
| CLUAP1 | Cells cultured fibroblasts | 56,140 | 4.333 | 1.47E-05 | 24,133 | 3.948 | 7.89E-05 | 32,007 | 2.31 | 2.09E-02 |
| LTBP2 | Prostate | 58,713 | −4.314 | 1.60E-05 | 25,776 | −2.677 | 7.44E-03 | 32,937 | −3.392 | 6.94E-04 |
| RERE | Heart atrial appendage | 58,713 | −4.249 | 2.15E-05 | 25,776 | −1.44 | 1.50E-01 | 32,937 | −4.399 | 1.09E-05 |
| ARHGEF10L | Artery tibial | 58,684 | −4.198 | 2.69E-05 | 25,747 | −2.698 | 6.97E-03 | 32,937 | −3.218 | 1.29E-03 |
| TMEM14EP | DGN-WB | 58,713 | 4.155 | 3.25E-05 | 25,776 | 3.344 | 8.27E-04 | 32,937 | 2.59 | 9.60E-03 |
| SSBP4 | Esophagus muscularis | 58,713 | −4.142 | 3.44E-05 | 25,776 | −3.937 | 8.24E-05 | 32,937 | −2.048 | 4.06E-02 |
| MTDH | Adipose subcutaneous | 58,449 | 4.094 | 4.24E-05 | 25,512 | 3.338 | 8.42E-04 | 32,937 | 2.515 | 1.19E-02 |
| LRRC25 | Artery tibial | 58,713 | −4.081 | 4.48E-05 | 25,776 | −2.8 | 5.11E-03 | 32,937 | −2.972 | 2.96E-03 |
| TRIB3 | Breast mammary tissue | 58,713 | 4.059 | 4.94E-05 | 25,776 | 3.234 | 1.22E-03 | 32,937 | 2.558 | 1.05E-02 |
| AC007228.9 | Muscle skeletal | 56,813 | −4.056 | 5.00E-05 | 23,876 | −2.445 | 1.45E-02 | 32,937 | −3.245 | 1.17E-03 |
| SCARB1 | Muscle skeletal | 58,713 | 4.041 | 5.33E-05 | 25,776 | 2.576 | 1.00E-02 | 32,937 | 3.116 | 1.83E-03 |
| EXOSC10 | Skin not sun-exposed suprapubic | 58,713 | 4.027 | 5.64E-05 | 25,776 | 1.648 | 9.95E-02 | 32,937 | 3.92 | 8.86E-05 |
| YDJC | Brain putamen basal ganglia | 58,713 | −4.01 | 6.08E-05 | 25,776 | −3.581 | 3.43E-04 | 32,937 | −2.186 | 2.88E-02 |
| VPS13D | Brain cortex | 58,713 | −4.004 | 6.22E-05 | 25,776 | −1.297 | 1.95E-01 | 32,937 | −4.199 | 2.68E-05 |
| ADA | DLPFC | 58,713 | 3.994 | 6.51E-05 | 25,776 | 2.25 | 2.45E-02 | 32,937 | 3.342 | 8.32E-04 |
| AP000553.3 | Nerve tibial | 58,713 | 3.993 | 6.52E-05 | 25,776 | 3.487 | 4.89E-04 | 32,937 | 2.247 | 2.46E-02 |
| CCDC116 | Brain spinal cord cervical c-1 | 58,713 | 3.993 | 6.52E-05 | 25,776 | 3.487 | 4.89E-04 | 32,937 | 2.247 | 2.46E-02 |
| MVB12B | Artery aorta | 58,713 | −3.993 | 6.52E-05 | 25,776 | −2.155 | 3.12E-02 | 32,937 | −3.425 | 6.14E-04 |
| NBAT1 | Artery aorta | 58,713 | −3.973 | 7.09E-05 | 25,776 | −2.623 | 8.71E-03 | 32,937 | −2.984 | 2.84E-03 |
| RABEP1 | Adipose subcutaneous | 58,713 | 3.973 | 7.10E-05 | 25,776 | 2.779 | 5.46E-03 | 32,937 | 2.846 | 4.42E-03 |
| DDX41 | Brain putamen basal ganglia | 52,443 | 3.972 | 7.12E-05 | 23,391 | 3.442 | 5.77E-04 | 29,052 | 2.249 | 2.45E-02 |
| SDS | Brain frontal cortex BA9 | 58,713 | −3.959 | 7.54E-05 | 25,776 | −2.147 | 3.18E-02 | 32,937 | −3.386 | 7.10E-04 |
| UBE2L3 | DGN-WB | 58,713 | −3.956 | 7.61E-05 | 25,776 | −3.584 | 3.38E-04 | 32,937 | −2.112 | 3.47E-02 |
| HARS1 | DGN-WB | 58,713 | −3.955 | 7.64E-05 | 25,776 | −2.201 | 2.77E-02 | 32,937 | −3.334 | 8.57E-04 |
| AREL1 | Brain cerebellar hemisphere | 58,713 | 3.948 | 7.90E-05 | 25,776 | 3.037 | 2.39E-03 | 32,937 | 2.584 | 9.77E-03 |
| MFSD2A | Minor salivary gland | 58,713 | 3.946 | 7.96E-05 | 25,776 | 2.959 | 3.09E-03 | 32,937 | 2.65 | 8.04E-03 |
| FKTN | Brain caudate basal ganglia | 58,713 | 3.944 | 8.02E-05 | 25,776 | 1.11 | 2.67E-01 | 32,937 | 4.284 | 1.84E-05 |
| MT1G | Brain frontal cortex BA9 | 58,713 | 3.941 | 8.13E-05 | 25,776 | 0.741 | 4.58E-01 | 32,937 | 4.605 | 4.12E-06 |
| ARHGEF3 | DLPFC | 58,713 | 3.934 | 8.34E-05 | 25,776 | 3.067 | 2.16E-03 | 32,937 | 2.54 | 1.11E-02 |
| FOLR2 | Minor salivary gland | 58,351 | 3.918 | 8.92E-05 | 25,414 | 4.577 | 4.71E-06 | 32,937 | 1.195 | 2.32E-01 |
| TRAM1 | Esophagus gastroesophageal junction | 58,713 | −3.912 | 9.14E-05 | 25,776 | −3.57 | 3.57E-04 | 32,937 | −2.066 | 3.89E-02 |
| NTRK1 | Artery tibial | 58,713 | 3.886 | 1.02E-04 | 25,776 | 2.197 | 2.80E-02 | 32,937 | 3.245 | 1.17E-03 |
| CLUL1 | Brain caudate basal ganglia | 58,713 | −3.827 | 1.30E-04 | 25,776 | −1.158 | 2.47E-01 | 32,937 | −4.085 | 4.42E-05 |
| ACP1 | Testis | 58,713 | −3.802 | 1.43E-04 | 25,776 | −3.889 | 1.01E-04 | 32,937 | −1.636 | 1.02E-01 |
| L3MBTL2 | Heart atrial appendage | 58,713 | 3.783 | 1.55E-04 | 25,776 | 4.126 | 3.69E-05 | 32,937 | 1.4 | 1.61E-01 |
| CHADL | Heart left ventricle | 58,713 | −3.773 | 1.62E-04 | 25,776 | −4.201 | 2.66E-05 | 32,937 | −1.321 | 1.87E-01 |
| OSER1 | DLPFC | 58,713 | −3.753 | 1.75E-04 | 25,776 | −3.947 | 7.92E-05 | 32,937 | −1.52 | 1.29E-01 |
| EP300 | Lung | 58,713 | 3.715 | 2.03E-04 | 25,776 | 4.318 | 1.58E-05 | 32,937 | 1.141 | 2.54E-01 |
| INPPL1 | Ovary | 58,351 | 3.686 | 2.28E-04 | 25,414 | 4.354 | 1.34E-05 | 32,937 | 1.081 | 2.80E-01 |
| PATJ | Esophagus mucosa | 58,713 | 3.683 | 2.30E-04 | 25,776 | 1.138 | 2.55E-01 | 32,937 | 3.911 | 9.21E-05 |
| RILPL2 | Lung | 57,358 | −3.621 | 2.94E-04 | 24,421 | −0.259 | 7.95E-01 | 32,937 | −4.555 | 5.25E-06 |
| SH3YL1 | Adrenal gland | 58,713 | 3.616 | 2.99E-04 | 25,776 | 3.909 | 9.27E-05 | 32,937 | 1.37 | 1.71E-01 |
| COLCA2 | Stomach | 58,713 | 3.616 | 3.00E-04 | 25,776 | 4.206 | 2.60E-05 | 32,937 | 1.106 | 2.69E-01 |
| ALKAL2 | Brain substantia nigra | 58,713 | 3.583 | 3.40E-04 | 25,776 | 3.898 | 9.69E-05 | 32,937 | 1.335 | 1.82E-01 |
| GDAP1L1 | Artery tibial | 58,713 | 3.575 | 3.51E-04 | 25,776 | 4.141 | 3.46E-05 | 32,937 | 1.11 | 2.67E-01 |
| ECE1 | Heart left ventricle | 58,713 | −3.574 | 3.52E-04 | 25,776 | −0.686 | 4.93E-01 | 32,937 | −4.164 | 3.12E-05 |
| NAV3 | Esophagus gastroesophageal junction | 58,713 | −3.567 | 3.61E-04 | 25,776 | −0.991 | 3.22E-01 | 32,937 | −3.887 | 1.02E-04 |
| NPC2 | Brain hippocampus | 58,713 | −3.467 | 5.26E-04 | 25,776 | −0.133 | 8.94E-01 | 32,937 | −4.511 | 6.44E-06 |
| MAP3K7 | Pancreas | 58,713 | −3.43 | 6.04E-04 | 25,776 | −0.721 | 4.71E-01 | 32,937 | −3.941 | 8.11E-05 |
| RBCK1 | Testis | 58,713 | 3.389 | 7.02E-04 | 25,776 | 0.108 | 9.14E-01 | 32,937 | 4.429 | 9.46E-06 |
| PHB2 | Brain cortex | 58,713 | −3.171 | 1.52E-03 | 25,776 | −0.378 | 7.06E-01 | 32,937 | −3.9 | 9.62E-05 |
| EMG1 | Skin sun-exposed lower leg | 58,713 | 3.138 | 1.70E-03 | 25,776 | 0.183 | 8.55E-01 | 32,937 | 4.029 | 5.61E-05 |
| AC026367.3 | Cells cultured fibroblasts | 58,713 | −3.099 | 1.94E-03 | 25,776 | −4.513 | 6.40E-06 | 32,937 | −0.146 | 8.84E-01 |
| TBC1D1 | Cells cultured fibroblasts | 58,713 | 3.057 | 2.23E-03 | 25,776 | 0.161 | 8.72E-01 | 32,937 | 3.94 | 8.16E-05 |
| RABEPK | Brain substantia nigra | 35,698 | 3.013 | 2.58E-03 | 19,912 | 0.428 | 6.69E-01 | 15,786 | 4.051 | 5.10E-05 |
| AC026367.2 | Brain frontal cortex BA9 | 58,713 | −2.985 | 2.84E-03 | 25,776 | −4.364 | 1.28E-05 | 32,937 | −0.124 | 9.01E-01 |
| ZNF295−AS1 | Minor salivary gland | 58,713 | −2.882 | 3.96E-03 | 25,776 | −3.91 | 9.23E-05 | 32,937 | −0.389 | 6.98E-01 |
| KAT6B | Cells cultured fibroblasts | 58,624 | 2.842 | 4.49E-03 | 25,687 | 3.928 | 8.58E-05 | 32,937 | 0.323 | 7.47E-01 |
| SPATA6L | Brain cortex | 58,713 | 2.832 | 4.63E-03 | 25,776 | 4.27 | 1.95E-05 | 32,937 | 0.003 | 9.98E-01 |
| SUDS3 | Esophagus mucosa | 58,713 | 2.733 | 6.28E-03 | 25,776 | 4.169 | 3.07E-05 | 32,937 | −0.039 | 9.69E-01 |
| TMCC3 | Brain cortex | 58,713 | −2.687 | 7.22E-03 | 25,776 | −4.329 | 1.50E-05 | 32,937 | 0.243 | 8.08E-01 |
| HSPA2 | Colon sigmoid | 58,175 | 2.521 | 1.17E-02 | 25,238 | 4.071 | 4.68E-05 | 32,937 | −0.213 | 8.31E-01 |
| RRS1 | Heart left ventricle | 58,713 | −2.152 | 3.14E-02 | 25,776 | −3.921 | 8.84E-05 | 32,937 | 0.595 | 5.52E-01 |
| TRHDE | Uterus | 58,537 | −2.11 | 3.48E-02 | 25,600 | −3.886 | 1.02E-04 | 32,937 | 0.612 | 5.40E-01 |
| LINC02664 | Artery tibial | 58,624 | 2.036 | 4.17E-02 | 25,687 | 4.385 | 1.16E-05 | 32,937 | −1.156 | 2.48E-01 |
| ADAMTSL3 | Brain cortex | 58,713 | 2.02 | 4.34E-02 | 25,776 | 3.949 | 7.84E-05 | 32,937 | −0.797 | 4.25E-01 |
| SH3GLB1 | Adrenal gland | 58,713 | 1.89 | 5.88E-02 | 25,776 | 3.91 | 9.25E-05 | 32,937 | −0.935 | 3.50E-01 |
False discovery rate adjusted genome-wide significance: p-value < 1.02 × 10−4.
Table 2.
The genes with significant effect independent to nearby known LOAD SNPs.
| Unadjusted model | Adjusted for SNP | Adjusted for gene | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Known LOAD SNP | Genes | Tissue | N | Z | p-value | Z | p-value | Z | p-value |
| APOE (rs429358); APOE (rs7412); CD33 (rs3865444) | LILRA5 | Lung | 21,924 | −5.301 | 1.15E-07 | −4.903 | 9.43E-07 | −5.358 | 8.43E-08 |
| BIN1 (rs6733839) | TMEM163 | Thyroid | 25,776 | −4.163 | 3.14E-05 | −4.177 | 2.95E-05 | −4.141 | 3.46E-05 |
| CELF1 (rs10838725) | C1QTNF4 | Brain cerebellar hemisphere | 25,776 | 4.397 | 1.10E-05 | 4.055 | 5.01E-05 | 4.115 | 3.87E-05 |
| FAM180B | Artery coronary | 25,776 | −4.504 | 6.67E-06 | −4.13 | 3.64E-05 | −4.317 | 1.58E-05 | |
| KBTBD4 | Kidney cortex | 25,776 | 3.996 | 6.45E-05 | 3.911 | 9.19E-05 | 3.803 | 0.0001431 | |
| MTCH2 | Skin sun-exposed lower leg | 25,776 | −4.245 | 2.19E-05 | −3.969 | 7.23E-05 | −3.783 | 0.000155 | |
| NDUFS3 | Cells cultured fibroblasts | 25,776 | 4.261 | 2.03E-05 | 3.899 | 9.64E-05 | 4.323 | 1.54E-05 | |
| PSMC3 | Cells cultured fibroblasts | 25,776 | −4.263 | 2.01E-05 | −3.89 | 1.00E-04 | −4.3 | 1.71E-05 | |
| PTPRJ | Brain cortex | 25,776 | 4.356 | 1.32E-05 | 4.274 | 1.92E-05 | 3.834 | 0.0001259 | |
| RAPSN | Stomach | 25,776 | 4.519 | 6.23E-06 | 4.205 | 2.61E-05 | 4.39 | 1.13E-05 | |
| SLC39A13 | Adipose visceral omentum | 25,776 | −4.279 | 1.88E-05 | −3.903 | 9.49E-05 | −4.771 | 1.84E-06 | |
| SORL1(rs11218343) | SIK2 | Brain cerebellar hemisphere | 25,776 | 4.462 | 8.14E-06 | 4.44 | 9.00E-06 | NA | NA |
In addition to the tissue-specific models, we performed two cross-tissue analyses: cross-brain, including the 14 brain tissues in GTEx, and cross-all tissues, including all 49 GTEx tissues across organ systems. In the cross-all and cross-brain tissue models, 34 genes were significantly associated with LOAD; APOE was the most strongly associated gene in both models (p-value = 4.3 × 10−364 in the cross-all tissue model and p-value = 1.0 × 10−373 in the cross-brain model, Fig. 2 and Table 3). Notably, eight genes identified in the cross-tissue analyses were not significantly associated with tissue-specific models, including ACOT8, ASPG, CNN2, CTSA, ABP1, ISOC2, PLTP, and ZSWIM1.
Fig. 2. Miami plot of cross-all GTEx tissue (blue dots, top) and cross-brain tissues model (gray dots, bottom).

p-values less than 1 × 10−15 (pink dots) were truncated to improve the clarity of peaks in other, less significant regions.
Table 3.
The significant genes in cross-tissue models.
| All GTEx tissues | GTEx brain tissues | |||||||
|---|---|---|---|---|---|---|---|---|
| Genes | N | Tissues | PCs | p-value | N | Tissues | PCs | p-value |
| APOE | 58,713 | 36.92 | 5.74 | 4.36E-364 | 58,713 | 9.72 | 1.59 | 1.00E-373 |
| NECTIN2 | 58,713 | 45.18 | 8.23 | 9.22E-198 | 58,713 | 9.18 | 4.33 | 4.13E-75 |
| APOC1 | 58,713 | 16.15 | 4.56 | 1.14E-165 | 19,246 | 3.00 | 2.00 | 4.02E-53 |
| APOC4 | 58,713 | 9.90 | 3.13 | 1.49E-73 | 58,713 | 4.00 | 1.21 | 1.65E-88 |
| BCAM | 58,713 | 45.05 | 5.74 | 9.62E-20 | 57,358 | 12.00 | 2.00 | 8.11E-02 |
| APOC2 | 58,713 | 47.28 | 4.95 | 2.90E-13 | 58,713 | 12.79 | 3.00 | 4.14E-06 |
| BIN1 | 58,713 | 46.00 | 8.13 | 7.28E-11 | 58,713 | 13.00 | 4.92 | 7.03E-10 |
| TRAPPC6A | 58,713 | 26.97 | 3.05 | 7.01E-09 | 58,713 | 6.54 | 2.59 | 9.60E-10 |
| ABCA7 | 57,358 | 44.32 | 7.68 | 2.94E-08 | 57,358 | 10.79 | 2.61 | 1.82E-06 |
| CNN2 | 57,358 | 42.00 | 8.39 | 3.33E-08 | 57,358 | 12.34 | 3.79 | 3.91E-02 |
| CLU | 58,713 | 23.00 | 5.56 | 2.80E-07 | 25,776 | 3.00 | 2.00 | 7.16E-03 |
| ZNF296 | 58,713 | 45.00 | 5.51 | 5.04E-07 | 58,713 | 10.00 | 2.21 | 2.13E-11 |
| CLPTM1 | 58,713 | 31.18 | 3.87 | 8.41E-07 | 58,713 | 8.38 | 2.97 | 1.49E-08 |
| EXOC3L2 | 58,713 | 29.92 | 4.18 | 2.87E-06 | 58,713 | 4.00 | 2.28 | 3.53E-03 |
| CEACAM19 | 58,713 | 49.00 | 5.59 | 3.50E-06 | 58,713 | 13.00 | 1.05 | 4.97E-06 |
| SLC39A13 | 58,713 | 47.00 | 3.77 | 1.45E-05 | 58,713 | 12.00 | 2.00 | 3.57E-04 |
| ASPG | 58,713 | 41.33 | 7.00 | 2.22E-05 | 58,713 | 11.51 | 3.64 | 8.72E-02 |
| CTSA | 58,713 | 19.10 | 4.67 | 2.50E-05 | 58,713 | 4.44 | 2.00 | 1.37E-02 |
| ACP2 | 58,713 | 49.00 | 3.18 | 2.59E-05 | 58,713 | 13.00 | 2.03 | 3.86E-06 |
| PICALM | 58,713 | 41.00 | 5.00 | 3.72E-05 | 58,713 | 8.00 | 4.00 | 2.56E-05 |
| PSMC3 | 58,713 | 44.00 | 3.05 | 4.54E-05 | 58,713 | 11.00 | 2.00 | 4.62E-06 |
| ZSWIM1 | 58,713 | 32.00 | 4.49 | 6.49E-05 | 58,713 | 9.00 | 2.00 | 4.71E-03 |
| FABP1 | 58,713 | 43.00 | 2.85 | 9.56E-05 | 58,713 | 13.00 | 2.62 | 7.91E-02 |
| ACOT8 | 58,713 | 47.74 | 3.18 | 1.05E-04 | 58,713 | 12.90 | 2.26 | 6.21E-05 |
| APH1B | 58,713 | 47.59 | 4.41 | 1.49E-04 | 58,713 | 11.79 | 3.03 | 1.28E-06 |
| MS4A4E | 58,713 | 29.97 | 2.87 | 1.69E-04 | 25,776 | 3.00 | 1.00 | 5.62E-06 |
| CR1 | 58,713 | 25.03 | 3.03 | 3.16E-04 | 58,713 | 9.21 | 1.41 | 1.55E-06 |
| PVR | 58,713 | 48.00 | 7.54 | 3.77E-04 | 58,713 | 12.00 | 2.92 | 7.14E-05 |
| PLTP | 58,713 | 44.41 | 6.23 | 1.38E-03 | 58,713 | 13.00 | 4.28 | 8.58E-06 |
| SCARA3 | 58,713 | 36.21 | 8.00 | 3.97E-03 | 58,713 | 9.00 | 4.64 | 2.73E-05 |
| DMWD | 58,713 | 39.95 | 5.23 | 5.22E-03 | 58,713 | 6.00 | 2.00 | 1.11E-02 |
| SYMPK | 58,713 | 38.72 | 6.18 | 1.17E-01 | 58,713 | 7.00 | 2.03 | 8.97E-03 |
| EML2 | 58,713 | 30.56 | 9.15 | 1.87E-01 | 57,358 | 7.21 | 3.71 | 5.91E-02 |
| ISOC2 | 57,358 | 38.21 | 3.11 | 9.12E-01 | 56,328 | 7.84 | 2.38 | 8.84E-01 |
To assess causal inference, we used the GReX fine-mapping method FOCUS to identify the most likely causal genes, testing 34 genome regions across 49 tissues. The PIP was used to evaluate the potential of our observed GReX association representing a causal effect, and it was strongly correlated with the Z-score from our GReX association tests (rho = 0.758). Among the tested GReX signals, 152 from 21 unique genes were considered likely causal (PIP > 0.9, Supplementary Table 5). We also used Mendelian randomization to examine if the association from eQTL to LOAD is mediated by gene expression. Among 1395 identified LOAD-related GReX, 556 signals from 107 unique genes reached statistical significance (Benjamini & Hochberg FDR-adjusted p-value < 0.05, Supplementary Table 6), indicating that the effect of these eQTLs on disease status is through their impact on gene expression.
Furthermore, we compared the sensitivity between PrediXcan and S-PrediXcan in ADGC (Fig. 3). Overall, we observed a high correlation between their log-transformed p-values (r2 = 0.90). However, 66 signals were significant (FDR-adjusted p < 0.05) only in PrediXcan, and 47 were only identified in S-PrediXcan, highlighting the subtle change in power due to slight differences between the in-sample and reference sample linkage disequilibrium patterns (Supplementary Fig. 1). We also used the true positive rate to evaluate the sensitivity of each tissue-specific model (Fig. 4). The brain tissues had a similar true positive rate (π1, see “Subjects and methods” section) compared to other tissues (mean = 0.039 in 14 brain tissues and 0.041 in others, p-value = 0.736), and the true positive rate was not significantly correlated with the sample size for model building (r2 = 0.144, p-value = 0.311).
Fig. 3. Scatter plot of log-transformed p-values from SNP-based (PrediXcan) and summary statistic (S-PrediXcan) GReX approach.
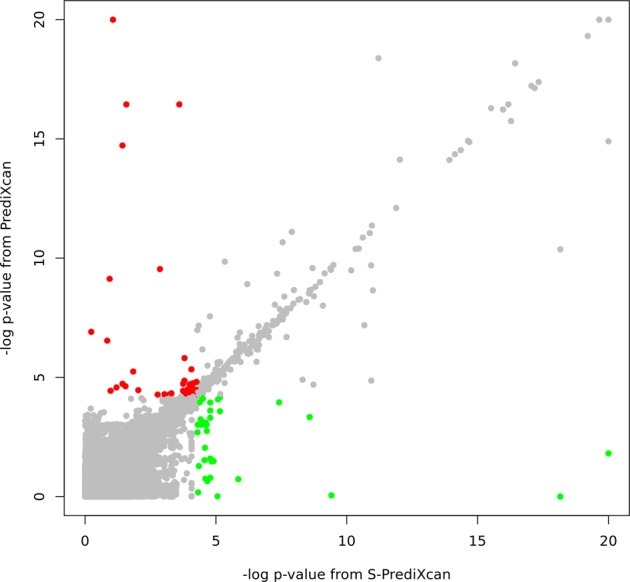
Each dot shows the gene-level p-value from a SNP-based approach (PrediXcan, y-axis) and summary statistic approach (S-PrediXcan, x-axis). Color indicates that the signal (tissue-specific GReX) was only identified in PrediXcan (red, FDR < 0.05) or only in S-PrediXcan (green, FDR < 0.05).
Fig. 4. Scatter plot of sample size used for model building against the true positive rate.
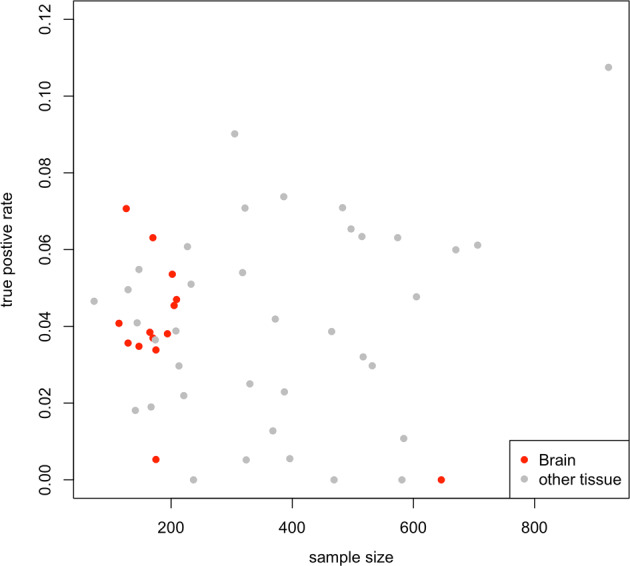
X-axis represents the number of subjects used in PrediXcan model building and Y-axis represents the true positive rate for each model in our analysis. Color represents tissue source (red for brain tissues and gray for other tissues).
We calculated LOAD heritability using summary statistics from a previous LOAD GWAS [16]. Overall genome-wide heritability from this study was 6.8%. We then estimated several partitions of heritability based on our GReX findings. This resulted in three primary findings. First, the 216 identified LOAD genes contain only 7.1% of the SNPs used for heritability estimation, but explain 60.9% of heritability, representing a significant enrichment (p-value = 1.3E–04, Table 4). Among these genes, implicated genes in the APOE region (comprising 0.5% of SNPs) explain 16.1% of heritability, and the genes outside of this region (comprising 6.6% of SNPs) explain 44.8%. Second, we estimated the heritability from the brain and blood tissues alone. The brain tissue models identified more LOAD genes (117 genes) than blood tissues (62 genes) and explained slightly higher heritability (47.4% in the brain vs. 43.1% in the blood). Third, we estimated heritability using the 22 LOAD SNPs which were previously used to generate an optimal polygenic risk score model for LOAD [60] and SNPs in the surrounding ± 5 Kb regions; these SNPs explained 16.8% of heritability.
Table 4.
Partitioned of the SNP-based heritability of LOAD.
| Category | SNPs (%) | h2 (%) | SE | Enrichmenta | SE | p-value |
|---|---|---|---|---|---|---|
| ALL SNPs | 100.0 | 100.0 | 0.1% | 1 | ||
| AD genes | 7.1 | 60.9 | 11.4% | 8.62 | 1.61 | 1.3E–04 |
| APOE region | 0.5 | 16.1 | 12.7% | 32.02 | 25.18 | 0.2474 |
| Non-APOE region | 6.6 | 44.8 | 6.5% | 6.83 | 0.99 | 1.9E–19 |
| Brain tissue | 4.5 | 47.4 | 10.1% | 10.63 | 2.27 | 3.9E–04 |
| Blood tissue | 2.7 | 43.1 | 12.1% | 15.91 | 4.46 | 4.7E–03 |
| Previous GWAS | 0.008 | 16.8 | 7.4% | 2039.62 | 900.05 | 0.0189 |
The “All SNPs” category used all available SNPs to estimate the heritability of LOAD, and defines 100% of the SNP-based heritability; AD genes used all SNPs located within ±1 Mb of our 216 identified AD genes; previous GWAS used 22 SNPs, which were used to generate an optimal polygenic risk score model [60], and their surrounding SNPs within ±5 Kb.
aEnrichment = Proportion of heritability/proportion of SNPs
Discussion
To further understand the genetic mechanisms of LOAD, we performed functionally oriented association analyses targeting tissue-specific and cross-tissue effects on LOAD-risk. We applied GReX models in a total of 51 tissues to 20,613 LOAD cases and 37,658 controls. As a result, 72 novel genes and 136 genes from known LOAD regions were identified after study-wide multiple test correction (FDR < 0.05). In addition, we applied both cross-brain-tissues and cross-GTEx models and we found eight additional genes significantly associated with LOAD. Secondly, we compared the SNP-based GReX approach to the summary statistic-based approach, and demonstrated the benefit of SNP-based methods, which leverage the internal LD structure of the empirical data instead of an external panel. Finally, we estimated the proportion of LOAD SNP-based heritability that can be captured by our identified LOAD genes to represent the improvement of GReX analysis over traditional GWAS. Our results suggest over 60% of LOAD heritability can be captured by our 216 identified LOAD genes.
Most previously described LOAD genes have been identified through GWAS, and we observe that for eight known genes, APOE, BIN1, CLU, PTK2B, CR1, MS4A2, PICALM, IQCK, and TREML2, their genetically regulated expression levels were significantly associated with LOAD status as well. Several genes close to these known genes were also significantly associated with LOAD. For instance, results for TOMM40 in GTEx skin tissues were the strongest association signals in all analyses; this strong effect observed in skin tissues may be due to shared genetic regulation between skin and brain tissues and increased power due to more available expression data in skin tissues. Notably, skin biopsies have been suggested as an approach to detect dysregulated or abnormal protein levels in Alzheimer’s disease between healthy participants, participants with LOAD, and participants with dementia due to other causes [61]. TOMM40 encodes Tom40 protein, Translocase of the Outer Mitochondrial Membrane, 40 kD, and is located in a region in high linkage disequilibrium with APOE. Association of TOMM40 variants with LOAD has been observed in previous genetic studies [62], and several studies indicate that the effect of TOMM40 is independent of APOE [63–65]. Lower expression of TOMM40 in blood has also been reported in LOAD cases [66], with a consistent direction of effect with our results in the DGN whole blood model. Furthermore, we adjusted for the known LOAD variants representing the APOE ε2/ε3/ε4 haplotype reported in Naj et al. [41] in conditional analysis of this locus, and results indicate that the effect of the genetically regulated expression on LOAD was driven mostly by the index variants at APOE (rs7412, rs429358, rs145999145, and rs3865444). We used FOCUS to clarify the causal inference of identified genes from known LOAD regions, and TOMM40 was also identified as a likely causal gene in many tissues, e.g., muscle, brain cortex and pituitary gland. In addition, our Mendelian randomization results also suggest that the effect of eQTLs of APOE on LOAD were not significantly mediated by the expression of APOE in many tissues, including both skin and muscle tissues. These results suggest that previously identified APOE SNPs may impact the risk of LOAD through coregulation of the expression of nearby genes, such as TOMM40. We observed a similar pattern throughout our results, indicating that previously identified LOAD SNPs may co-regulate the expression of several nearby genes rather than only the nearest gene. After adjusting the nearby known LOAD SNPs, we still can identify 11 unique genes that retained significance, including PTPRJ from the CELF1 region. PTPRJ encodes a protein of the family of tyrosine phosphatase, and it has been reported as a shared genetic factor of LOAD and major depression disorder [67].
However, we also identified 72 novel independent genes that are >10 MB from previously reported signals, including TRIT1, TSPAN14, and MTDH. TRIT1 encodes TRNA Isopentenyltransferase 1, which is targeted to mitochondria and modified the transfers RNA. TRIT1 has been known as a tumor suppressor for several forms of cancer [68], and its pleiotropy between LOAD and major depressive disorder [67] or cardiovascular risk factor has been previously noted [69]. TSPAN14 is a member of tetraspanins, a family of compact and glycosylated transmembrane proteins. Previous research has reported tetraspanins’ function on the precursor of amyloid-beta (Aβ) peptide, amyloid precursor protein, and their potential roles in the development of LOAD [70]. In addition, MTDH encodes metadherin, which is also known as astrocyte elevated gene-1 protein (AEG-1). The role of AEG-1 in tumor progression and neurodegeneration, especially HIV-induced dementia, has been previously reported [71, 72]. The GReX-based analysis has been applied in several previous small LOAD studies [20, 73, 74]. With our larger sample size and updated models, we successfully replicated their findings of known LOAD genes, and identified additional novel genes (Supplementary Table 4).
To explore which tissues contribute the most significant effects to LOAD-risk, we also evaluated the true positive rate for each tissue-specific model using the methods outlined in Storey et al. [57]. Our findings suggest that non-brain tissues, particularly whole blood, are important for detecting GReX effects on LOAD and may imply the relevance of non-brain tissue in LOAD pathology. Given the difficulty of characterizing transcriptomic effects in brain tissue, the utility of alternate, accessible tissues in understanding LOAD pathogenesis will be key to future studies.
Motivated by the extensive observed shared genetic regulation of gene expression across tissues [37], we conducted cross-tissue analyses in addition to tissue-specific analyses. If a gene is tissue-specific in its regulation, then a tissue-specific model rather than a cross-tissue model, should have a better power to detect its effect. However, if patterns of expression suggest that a gene has shared regulation across many tissues and effects are small but consistent across many tissues that may have small sample sizes in GTeX (e.g., brain tissues), then a cross-tissue model would be expected to have greater power than a single-tissue model. A total of 34 genes were identified in our cross-tissue analyses, eight of which were only identified in cross-tissue analyses, while 188 were only identified by single-tissue analysis. These results indicate that cross-tissue analysis can detect additional significant associations at genes where the effects are consistent across tissues, because cross-tissue and tissue-specific analyses may provide complementary information.
Moreover, we compared the use of SNP-based GReX analysis, PrediXcan, and summary statistic-based GReX analysis, S-PrediXcan, in the same dataset and found that the SNP-based approach identified more significant signals. Since summary statistic-based GReX prediction approaches rely on an external linkage disequilibrium panel, the reduced significance of the summary statistic-based method may be caused by differences in linkage disequilibrium between the external panel and the population in which the summary statistics were derived. Though these differences in linkage disequilibrium are likely subtle, our findings suggest that although the shifts in p-values were not large, for borderline signals, S-PrediXcan may have slightly reduced power to detect true effects. We note, however, that careful simulation will be needed to fully characterize the impact of differences in linkage disequilibrium on the power of these approaches, which is beyond the scope of the present study.
To further validate our findings, we estimated the proportion of LOAD heritability that can be explained by the identified genes. Our results indicate that our 216 LOAD genes explain a significantly enriched proportion of heritability (enrichment = 8.62, p-value = 1.3 × 10−4, Table 4). For comparison, we also calculated heritability explained by the 22 LOAD SNP loci (±5 kb) included in a recent LOAD polygenic risk score model [60]. Although the loci containing the 22 SNPs used in LOAD PGRS are strongly enriched, our identified LOAD gene set explains more than three times the SNP-based heritability of LOAD, demonstrating the benefit of applying the functionally oriented approach to investigate the pathology of LOAD. Furthermore, we compared the proportion of explained heritability by the identified genes from brain tissue and blood tissue. Even though the brain is considered to be the causal tissue for LOAD, the blood tissues can capture similar heritability with brain tissues, and it may emphasize the importance of non-brain tissue in LOAD again. Also, a previous study demonstrated that the blood transcriptome can well predict the genes’ expression in other tissues [75], and it may offer another explanation about our highly explained LOAD heritability from blood.
There are several limitations of the present study. The gene expression prediction models are primarily based on GTEx data, and our power to detect associations is limited somewhat by tissue-specific sample sizes in GTEx. In particular, limited sample sizes for the brain tissues in GTEx reduced our ability to identify associations between LOAD-risk and genes with tissue-specific expression in the brain. To mitigate this limitation, we included models built in the CommonMind data. Also, some genes have expression patterns dependent on age [76], and so our predicted genetically regulated expression profile may reflect the age of the GTEx reference samples instead of the advanced ages associated with LOAD, limiting our power to detect such effects. Furthermore, our analysis examines the association of predicted genetically regulated expression, rather than directly measured expression levels. These imputed levels may differ from true, measured expression, which is affected by other environmental or temporal factors [77]. Although we may not perfectly impute the true gene expression levels or capture the effect of gene-environment, the models of genetically regulated expression that we applied here offer an opportunity to investigate age- and environment-independent effects of gene regulation on LOAD. Finally, our study populations are entirely European ancestry, limiting our ability to detect some loci with effects that vary by population and the generalizability of our results to other populations. Further study in individuals of non-European ancestry is needed to ensure that this limitation does not persist and contributes to health disparities.
In conclusion, analyzing genetic data from 20,613 LOAD cases and 37,658 controls from 39 epidemiological studies of LOAD, we profiled the genetically regulated expression genome-wide in 51 diverse tissues and expression data from GTEx, CommonMind, and DGN using SNP-based and summary statistics-based GReX approaches. Many of our significant gene-based associations fall within 10 Mb distance of previously described loci, suggesting co-regulation by GWAS-identified SNPs. In addition, we identify 72 novel genes from either tissue-specific analysis or cross-tissue analysis. Together, the 216 LOAD genes we identify explain over 60% of the SNP-based heritability of LOAD in these data. While the role of genes with differentially regulated expression levels in LOAD progression is still unclear, and more functionally oriented genetic studies of LOAD are needed, these findings highlight the power of expression prediction approaches to identify novel genes.
Supplementary information
Acknowledgements
H-HC, LEP, JS, YZ, AK, OV, WB, ACN, and JEB were supported by funding from the National Institute of Aging, National Institutes of Health, R01AG061351. ERG is supported by the National Institutes of Health (NIH) grants: NHGRI R35HG010718, NHGRI R01HG011138, NIA AG068026, and NIGMS R01GM140287.
Author contributions
Conception and study design: JEB, ACN, WB, and ERG; data preparation: JS, YZ, AK, and OV; data analysis: H-HC and LEP; manuscript drafting: H-HC; manuscript editing: H-HC, LEP, JEB, ACN, WB, and ERG.
Data availability
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
All code is available on GitHub https://github.com/belowlab/LOAD-GReX/.
Competing interests
The authors declare no competing interests.
Ethics approval
This study was reviewed and approved as exempt by the institutional review board at Vanderbilt University Medical Center under IRB# 181206.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A full list of members and their affiliations appears in the Supplementary Information.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-021-01677-0.
References
- 1.Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778–83. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raiha I, Kaprio J, Koskenvuo M, Rajala T, Sourander L. Alzheimer’s disease in Finnish twins. Lancet. 1996;347:573–8. doi: 10.1016/s0140-6736(96)91272-6. [DOI] [PubMed] [Google Scholar]
- 3.Gatz M, Pedersen NL, Berg S, Johansson B, Johansson K, Mortimer JA, et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci. 1997;52:M117–125. doi: 10.1093/gerona/52a.2.m117. [DOI] [PubMed] [Google Scholar]
- 4.Pedersen NL, Posner SF, Gatz M. Multiple-threshold models for genetic influences on age of onset for Alzheimer disease: findings in Swedish twins. Am J Med Genet. 2001;105:724–8. doi: 10.1002/ajmg.1608. [DOI] [PubMed] [Google Scholar]
- 5.Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:e13–200.e220. doi: 10.1016/j.neurobiolaging.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridge PG, Mukherjee S, Crane PK, Kauwe JS, Alzheimer’s Disease Genetics Consortium. Alzheimer’s disease: analyzing the missing heritability. PLoS ONE. 2013;8:e79771. [DOI] [PMC free article] [PubMed]
- 7.So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet Epidemiol. 2011;35:310–7. doi: 10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- 8.Petty LE, Highland HM, Gamazon ER, Hu H, Karhade M, Chen HH, et al. Functionally oriented analysis of cardiometabolic traits in a trans-ethnic sample. Hum Mol Genet. 2019;28:1212–24. doi: 10.1093/hmg/ddy435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362:eaat8127. [DOI] [PMC free article] [PubMed]
- 10.Gamazon ER, Zwinderman AH, Cox NJ, Denys D, Derks EM. Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat Genet. 2019;51:933–40. doi: 10.1038/s41588-019-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su L, Chen S, Zheng C, Wei H, Song X. Meta-analysis of gene expression and identification of biological regulatory mechanisms in Alzheimer’s disease. Front Neurosci. 2019;13:633. doi: 10.3389/fnins.2019.00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen M, Kachadoorian M, Quicksall Z, Zou F, Chai HS, Younkin C, et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res Ther. 2014;6:39. doi: 10.1186/alzrt268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052–61. doi: 10.1038/nn.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–8. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerring ZF, Lupton MK, Edey D, Gamazon ER, Derks EM. An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:43. doi: 10.1186/s13195-020-00611-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardiñas AF, et al. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat Genet. 2019;51:659–74. doi: 10.1038/s41588-019-0364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Roige S, Fontanillas P, Elson SL, Research T, Pandit A, Schmidt EM, et al. Genome-wide association study of delay discounting in 23,217 adult research participants of European ancestry. Nat Neurosci. 2018;21:16–18. doi: 10.1038/s41593-017-0032-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barbeira AN, Pividori M, Zheng J, Wheeler HE, Nicolae DL, Im HK. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019;15:e1007889. doi: 10.1371/journal.pgen.1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GTEx Consortium. et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–46. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 26.Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R, National Institute on Aging Late-Onset Alzheimer’s Disease Family Study Group. Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: implication of additional loci. Arch Neurol. 2008;65:1518–26. doi: 10.1001/archneur.65.11.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green RC, Cupples LA, Go R, Benke KS, Edeki T, Griffith PA, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA. 2002;287:329–36. doi: 10.1001/jama.287.3.329. [DOI] [PubMed] [Google Scholar]
- 29.Bennett DA, Schneider JA, Buchman AS, Mendes de Leon C, Bienias JL, Wilson RS. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology. 2005;25:163–75. doi: 10.1159/000087446. [DOI] [PubMed] [Google Scholar]
- 30.Reiman EM, Webster JA, Myers AJ, Hardy J, Dunckley T, Zismann VL, et al. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron. 2007;54:713–20. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamboh MI, Minster RL, Demirci FY, Ganguli M, Dekosky ST, Lopez OL, et al. Association of CLU and PICALM variants with Alzheimer’s disease. Neurobiol Aging. 2012;33:518–21. doi: 10.1016/j.neurobiolaging.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373–84. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–7. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C, et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffman GE, Bendl J, Voloudakis G, Montgomery KS, Sloofman L, Wang YC, et al. CommonMind Consortium provides transcriptomic and epigenomic data for Schizophrenia and Bipolar Disorder. Sci Data. 2019;6:180. doi: 10.1038/s41597-019-0183-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–30. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbeira AN, Bonazzola R, Gamazon ER, Liang Y, Park Y, Kim-Hellmuth S, et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021;22:49. doi: 10.1186/s13059-020-02252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma C, Blackwell T, Boehnke M, Scott LJ. Go TDi. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet Epidemiol. 2013;37:539–50. doi: 10.1002/gepi.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 41.Naj AC, Schellenberg GD. Alzheimer’s Disease Genetics C. Genomic variants, genes, and pathways of Alzheimer’s disease: an overview. Am J Med Genet B Neuropsychiatr Genet. 2017;174:5–26. doi: 10.1002/ajmg.b.32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.THE ARIC INVESTIGATORS. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 44.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–76. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 45.Dawber TR, Kannel WB. The Framingham study. An epidemiological approach to coronary heart disease. Circulation. 1966;34:553–5. doi: 10.1161/01.cir.34.4.553. [DOI] [PubMed] [Google Scholar]
- 46.Hofman A, Breteler MM, van Duijn CM, Janssen HL, Krestin GP, Kuipers EJ, et al. The Rotterdam Study: 2010 objectives and design update. Eur J Epidemiol. 2009;24:553–72. doi: 10.1007/s10654-009-9386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 49.Luck T, Riedel-Heller SG, Kaduszkiewicz H, Bickel H, Jessen F, Pentzek M, et al. Mild cognitive impairment in general practice: age-specific prevalence and correlate results from the German study on ageing, cognition and dementia in primary care patients (AgeCoDe) Dement Geriatr Cogn Disord. 2007;24:307–16. doi: 10.1159/000108099. [DOI] [PubMed] [Google Scholar]
- 50.Jessen F, Wolfsgruber S, Wiese B, Bickel H, Mösch E, Kaduszkiewicz H, et al. AD dementia risk in late MCI, in early MCI, and in subjective memory impairment. Alzheimers Dement. 2014;10:76–83. doi: 10.1016/j.jalz.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 51.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dewey M. metap: meta-analysis of significance values. 2018.
- 53.Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Stat. 2000;25:60–83. [Google Scholar]
- 54.Mancuso N, Freund MK, Johnson R, Shi H, Kichaev G, Gusev A, et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat Genet. 2019;51:675–82. doi: 10.1038/s41588-019-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowden J, Davey, Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yavorska OO, Burgess S. Mendelian randomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–9. doi: 10.1093/ije/dyx034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–35. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leung YY, Valladares O, Chou YF, Lin HJ, Kuzma AB, Cantwell L, et al. NIAGADS: the NIA genetics of Alzheimer’s disease data storage site. Alzheimer’s Dement. 2016;12:1200–3. [Google Scholar]
- 60.Zhang Q, Sidorenko J, Couvy-Duchesne B, Marioni RE, Wright MJ, Goate AM, et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat Commun. 2020;11:4799. doi: 10.1038/s41467-020-18534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mukhamedyarov MA, Rizvanov AA, Yakupov EZ, Zefirov AL, Kiyasov AP, Reis HJ, et al. Transcriptional analysis of blood lymphocytes and skin fibroblasts, keratinocytes, and endothelial cells as a potential biomarker for Alzheimer’s disease. J Alzheimers Dis. 2016;54:1373–83. doi: 10.3233/JAD-160457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10:375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li G, Bekris LM, Leong L, Steinbart EJ, Shofer JB, Crane PK, et al. TOMM40 intron 6 poly-T length, age at onset, and neuropathology of AD in individuals with APOE epsilon3/epsilon3. Alzheimers Dement. 2013;9:554–61. doi: 10.1016/j.jalz.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cruchaga C, Nowotny P, Kauwe JS, Ridge PG, Mayo K, Bertelsen S, et al. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol. 2011;68:1013–9. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Payton A, Sindrewicz P, Pessoa V, Platt H, Horan M, Ollier W, et al. A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in nonpathologic aging. Neurobiol Aging. 2016;39:217 e211–217. doi: 10.1016/j.neurobiolaging.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 66.Mise A, Yoshino Y, Yamazaki K, Ozaki Y, Sao T, Yoshida T, et al. TOMM40 and APOE gene expression and cognitive decline in Japanese Alzheimer’s disease subjects. J Alzheimers Dis. 2017;60:1107–17. doi: 10.3233/JAD-170361. [DOI] [PubMed] [Google Scholar]
- 67.Lutz MW, Sprague D, Barrera J, Chiba-Falek O. Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder. Transl Psychiatry. 2020;10:88. doi: 10.1038/s41398-020-0769-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spinola M, Galvan A, Pignatiello C, Conti B, Pastorino U, Nicander B, et al. Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene. 2005;24:5502–9. doi: 10.1038/sj.onc.1208687. [DOI] [PubMed] [Google Scholar]
- 69.Broce IJ, Tan CH, Fan CC, Jansen I, Savage JE, Witoelar A, et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropathol. 2019;137:209–26. doi: 10.1007/s00401-018-1928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seipold L, Saftig P. The emerging role of tetraspanins in the proteolytic processing of the amyloid precursor protein. Front Mol Neurosci. 2016;9:149. doi: 10.3389/fnmol.2016.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emdad L, Sarkar D, Su ZZ, Lee SG, Kang DC, Bruce JN, et al. Astrocyte elevated gene-1: recent insights into a novel gene involved in tumor progression, metastasis and neurodegeneration. Pharm Ther. 2007;114:155–70. doi: 10.1016/j.pharmthera.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Noch EK, Khalili K. The role of AEG-1/MTDH/LYRIC in the pathogenesis of central nervous system disease. Adv Cancer Res. 2013;120:159–92. doi: 10.1016/B978-0-12-401676-7.00006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhattacharya A, Li Y, Love MI. MOSTWAS: multi-omic strategies for transcriptome-wide association studies. PLoS Genet. 2021;17:e1009398. doi: 10.1371/journal.pgen.1009398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagpal S, Meng X, Epstein MP, Tsoi LC, Patrick M, Gibson G, et al. TIGAR: an improved Bayesian tool for transcriptomic data imputation enhances gene mapping of complex traits. Am J Hum Genet. 2019;105:258–66. doi: 10.1016/j.ajhg.2019.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basu M, Wang K, Ruppin E, Hannenhalli S. Predicting tissue-specific gene expression from whole blood transcriptome. Sci Adv. 2021;7:eabd6991. [DOI] [PMC free article] [PubMed]
- 76.Yang J, Huang T, Petralia F, Long Q, Zhang B, Argmann C, et al. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci Rep. 2015;5:15145. doi: 10.1038/srep15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buil A, Brown AA, Lappalainen T, Viñuela A, Davies MN, Zheng HF, et al. Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat Genet. 2015;47:88–91. doi: 10.1038/ng.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
All code is available on GitHub https://github.com/belowlab/LOAD-GReX/.