

Abstract
Gastric cancers can develop even after Helicobacter pylori (H. pylori) eradication in 0.2–2.9% cases per year. Since H. pylori is reported to directly activate or inactivate cancer-related pathways, molecular profiles of gastric cancers with current and past H. pylori infection may be different. Here, we aimed to analyze whether profiles of point mutation and gene amplification are different between the two groups. Current or past infection by H. pylori was determined by positive or negative amplification of H. pylori jhpr3 gene by PCR, and past infection was established by the presence of endoscopic atrophy. Among the 90 gastric cancers analyzed, 55 were with current infection, and 35 were with past infection. Target sequencing of 46 cancer-related genes revealed that 47 gastric cancers had 68 point mutations of 15 different genes, such as TP53 (36%), KRAS (4%), and PIK3CA (4%) and that gene amplification was present for ERBB2, KRAS, PIK3CA, and MET among the 26 genes assessed for copy number alterations. Gastric cancers with current and past infection had similar frequencies of TP53 mutations (38% and 31%, respectively; p = 0.652) and oncogene activation (20% and 29%, respectively; p = 0.444). Gastric cancers with current and past infection had comparable profiles of genetic alterations.
Subject terms: Next-generation sequencing, Gastric cancer, Oncogenes, Cancer genomics, Gastric cancer, Gastric cancer
Introduction
Helicobacter pylori (H. pylori) is almost the exclusive cause of gastric cancers1,2, and H. pylori-triggered chronic inflammation is deeply involved in gastric carcinogenesis3–6. At the molecular level, aberrant DNA methylation is strongly induced by H. pylori infection-triggered chronic inflammation long before cancer development7,8. Aberrant DNA methylation of promoter CpG islands can suppress various tumor-suppressor genes, such as CDKN2A encoding p16 and CDH1 encoding E-cadherin9,10. Genetic alterations are also induced by H. pylori infection-triggered inflammation. Up-regulated AID, which encodes cytidine deaminase, and accumulation of TP53 mutation in gastric mucosa inflamed by H. pylori is well known11. Accumulation of both epigenetic and genetic alterations in gastric mucosa is associated with increased cancer risk, forming a field for cancerization12.
Despite the presence of the field, H. pylori eradication has the benefit of preventing gastric cancers13–16. Eradication therapy has been covered by public health insurance since 2013 in Japan, and 1,400,000 or more healthy people with H. pylori infection are treated with the therapy every year17. However, even after H. pylori eradication, gastric cancer develops at an incidence of 0.15–0.67% per year in healthy individuals18, and metachronous gastric cancers develop at an incidence of 1.4–2.9% per year in gastric cancer patients who underwent endoscopic submucosal dissection14,19,20. The presence of a field for cancerization suggests that molecular profiles in gastric cancers with current and past H. pylori infection are the same. At the same time, H. pylori itself can enhance pro-oncogenic signaling pathways involved in the proliferation and differentiation of cells, mainly mediated by CagA1,3. This suggests the possibility that different signaling pathways can be active between gastric cancers with current and past H. pylori infection.
Here, we aimed to analyze whether genetic alterations, namely point mutations and gene amplifications, are the same or different between gastric cancers with current and past H. pylori infection.
Results
52% of all cancers had somatic point mutations of cancer-related genes
Among the 90 gastric cancers (Supplementary Figure S1), current H. pylori infection was detected in 55 cancers, and 35 cancers were considered to have had past infection. Target sequencing of 46 cancer-related genes was conducted for the 90 gastric cancers, and 47 cancers (52%) had 68 somatic point mutations of 15 different genes (TP53, KRAS, PIK3CA, ERBB2, FBXW7, SMAD4, CTNNB1, ERBB4, PTPN11, SMARCB1, BRAF, GNAS, NOTCH1, NRAS, and PTEN) (Tables 1 and 2). Among the 68 mutations, 66 were missense mutations and 2 were nonsense mutations. TP53 was most frequently mutated (32 of the 90 gastric cancers, 36%). KRAS, PIK3CA, ERBB2, FBXW7, SMAD4, CTNNB1, ERBB4, PTPN11, and SMARCB1 were mutated in multiple gastric cancers (Fig. 1). 5, 5, 3, and 2 mutations of KRAS, PIK3CA, ERBB2 and CTNNB1 were observed in 4, 4, 3, and 2 gastric cancers, respectively (5, 3, 3, and 2 hotspot mutations, respectively) (Tables 1 and 2). These results showed that 12% of gastric cancers had activating point mutations of oncogenes (Table 3).
Table 1.
List of somatic mutations in the 55 gastric cancers with current H. pylori infection.
| Sample | Gene | Coverage | Variant allele frequency | Nucleotide change | Amino acid change |
|---|---|---|---|---|---|
| B-GC1 | TP53 | 3157 | 30.6 | c.818G>A | p.Arg273His |
| B-GC3 | SMAD4 | 9192 | 13.2 | c.1525T>G | p.Trp509Gly |
| TP53 | 4942 | 30.3 | c.857A>T | p.Glu286Val | |
| B-GC8 | No mutation | ||||
| B-GC11 | No mutation | ||||
| B-GC12 | No mutation | ||||
| B-GC14 | No mutation | ||||
| B-GC15 | CTNNB1a | 4389 | 79.7 | c.101G>A | p.Gly34Glu |
| B-GC16 | TP53 | 5638 | 31.6 | c.818G>A | p.Arg273His |
| B-GC17 | NRAS | 5643 | 29.2 | c.34G>T | p.Gly12Cys |
| FBXW7 | 6577 | 28.4 | c.1514G>A | p.Arg505His | |
| FBXW7 | 4646 | 21.9 | c.1394G>A | p.Arg465His | |
| B-GC19 | TP53 | 6252 | 14.8 | c.818G>A | p.Arg273His |
| B-GC22 | TP53 | 5911 | 38.0 | c.844C>T | p.Arg282Trp |
| B-GC23 | No mutation | ||||
| B-GC27 | ERBB4 | 9636 | 25.1 | c.1817A>G | p.Lys606Arg |
| B-GC33 | TP53 | 4987 | 63.1 | c.743G>A | p.Arg248Gln |
| B-GC34 | No mutation | ||||
| B-GC35 | No mutation | ||||
| B-GC37 | No mutation | ||||
| B-GC38 | No mutation | ||||
| B-GC39 | No mutation | ||||
| B-GC52 | No mutation | ||||
| B-GC56 | No mutation | ||||
| B-GC63 | SMAD4 | 1269 | 34.8 | c.1082G>A | p.Arg361His |
| B-GC64 | No mutation | ||||
| B-GC66 | PIK3CAa | 618 | 15.7 | c.1624G>A | p.Glu542Lys |
| KRASa,b | 336 | 22.3 | c.34G>A | p.Gly12Ser | |
| KRASa,b | 333 | 18.0 | c.35G>A | p.Gly12Asp | |
| B-GC70 | No mutation | ||||
| B-GC71 | TP53 | 1066 | 66.8 | c.659A>G | p.Tyr220Cys |
| B-GC73 | No mutation | ||||
| B-GC74 | TP53 | 1520 | 55.6 | c.853G>A | p.Glu285Lys |
| B-GC75 | TP53 | 602 | 51.3 | c.536A>G | p.His179Arg |
| B-GC77 | TP53 | 566 | 43.8 | c.404G>A | p.Cys135Tyr |
| B-GC80 | TP53 | 546 | 43.4 | c.536A>G | p.His179Arg |
| FBXW7 | 1063 | 46.8 | c.1393C>T | p.Arg465Cys | |
| B-GC81 | No mutation | ||||
| B-GC83 | GNAS | 222 | 12.6 | c.2531G>A | p.Arg844His |
| B-GC85 | No mutation | ||||
| B-GC86 | TP53 | 1664 | 59.1 | c.818G>T | p.Arg273Leu |
| B-GC87 | TP53 | 569 | 31.6 | c.388C>G | p.Leu130Val |
| B-GC88 | TP53 | 675 | 52.6 | c.524G>A | p.Arg175His |
| ERBB4 | 988 | 53.7 | c.719G>A | p.Gly240Glu | |
| B-GC90 | TP53 | 1833 | 20.7 | c.818G>A | p.Arg273His |
| B-GC92 | No mutation | ||||
| B-GC95 | No mutation | ||||
| B-GC96 | PIK3CAa | 806 | 12.3 | c.1633G>A | p.Glu545Lys |
| B-GC97 | No mutation | ||||
| B-GC98 | No mutation | ||||
| S2 | TP53 | 496 | 34.1 | c.581T>G | p.Leu194Arg |
| S4 | TP53 | 438 | 74.2 | c.581T>G | p.Leu194Arg |
| S13 | TP53 | 70 | 15.7 | c.478A>G | p.Met160Val |
| ERBB2a | 482 | 23.9 | c.2264T>C | p.Leu755Ser | |
| S17 | No mutation | ||||
| S19 | No mutation | ||||
| S20 | No mutation | ||||
| S21 | No mutation | ||||
| S22 | No mutation | ||||
| S23 | TP53 | 565 | 67.8 | c.537T>A | p.His179Gln |
| S36 | TP53 | 1142 | 34.9 | c.524G>A | p.Arg175His |
| S43 | TP53 | 239 | 74.9 | c.1024C>T | p.Arg342Ter |
| S124 | No mutation |
aActivated oncogene mutation.
bThese mutations did not exist on the same allele.
Table 2.
List of somatic mutations in the 35 gastric cancers with past H. pylori infection.
| Sample | Gene | Coverage | Variant allele frequency | Nucleotide change | Amino acid change |
|---|---|---|---|---|---|
| B-GC2 | No mutation | ||||
| B-GC6 | TP53 | 504 | 22.4 | c.524G>A | p.Arg175His |
| B-GC9 | No mutation | ||||
| B-GC13 | TP53 | 4255 | 21.1 | c.380C>T | p.Ser127Phe |
| TP53 | 5126 | 18.7 | c.376T>C | p.Tyr126His | |
| B-GC18 | No mutation | ||||
| B-GC21 | No mutation | ||||
| B-GC25 | TP53 | 3216 | 13.0 | c.535C>T | p.His179Tyr |
| B-GC26 | No mutation | ||||
| B-GC28 | No mutation | ||||
| B-GC30 | No mutation | ||||
| B-GC41 | ERBB2a | 4450 | 42.9 | c.2434G>A | p.Val812Ile |
| NOTCH1 | 5375 | 28.5 | c.4723G>C | p.Val1575Leu | |
| PIK3CA | 2109 | 32.5 | c.1031T>G | p.Val344Gly | |
| PIK3CA | 3448 | 12.2 | c.2091G>A | p.Met697Ile | |
| B-GC43 | No mutation | ||||
| B-GC45 | TP53 | 5152 | 85.9 | c.814G>A | p.Val272Met |
| B-GC46 | No mutation | ||||
| B-GC47 | No mutation | ||||
| B-GC50 | No mutation | ||||
| B-GC51 | No mutation | ||||
| B-GC53 | No mutation | ||||
| B-GC55 | TP53 | 3195 | 42.7 | c.637C>T | p.Arg213Ter |
| B-GC58 | KRASa | 664 | 28.2 | c.35G>A | p.Gly12Asp |
| B-GC60 | No mutation | ||||
| B-GC61 | TP53 | 1025 | 24.8 | c.742C>T | p.Arg248Trp |
| TP53 | 1263 | 30.8 | c.565G>A | p.Ala189Thr | |
| TP53 | 649 | 25.3 | c.523C>T | p.Arg175Cys | |
| PTPN11 | 1566 | 31.0 | c.214G>A | p.Ala72Thr | |
| FBXW7 | 844 | 30.0 | c.1393C>T | p.Arg465Cys | |
| PIK3CAa | 337 | 23.7 | c.3140A>G | p.His1047Arg | |
| B-GC62 | TP53 | 1062 | 25.6 | c.659A>G | p.Tyr220Cys |
| B-GC68 | BRAF | 1593 | 13.1 | c.1406G>C | p.Gly469Ala |
| B-GC72 | SMAD4 | 1347 | 10.3 | c.1081C>T | p.Arg361Cys |
| TP53 | 1290 | 26.7 | c.536A>G | p.His179Arg | |
| B-GC78 | TP53 | 1057 | 24.1 | c.818G>A | p.Arg273His |
| SMARCB1 | 603 | 21.9 | c.1129C>T | p.Arg377Cys | |
| B-GC82 | No mutation | ||||
| B-GC91 | TP53 | 1305 | 37.2 | c.844C>T | p.Arg282Trp |
| B-GC99 | PTEN | 1370 | 27.2 | c.752G>T | p.Gly251Val |
| S5 | KRASa | 1626 | 54.4 | c.38G>A | p.Gly13Asp |
| SMARCB1 | 50 | 56 | c.1130G>A | p.Arg377His | |
| S6 | TP53 | 2077 | 24.7 | c.820G>C | p.Val274Leu |
| S12 | ERBB2a | 24,516 | 63.8 | c.2264T>C | p.Leu755Ser |
| S31 | KRASa | 1979 | 56.6 | c.35G>T | p.Gly12Val |
| PTPN11 | 7391 | 56.8 | c.182A>G | p.Asp61Gly | |
| S40 | No mutation | ||||
| S47 | CTNNB1a | 4591 | 33.7 | c.121A>G | p.Thr41Ala |
aActivated oncogene mutation.
Figure 1.

Profiles of genetic alterations in 90 gastric cancers. Genetic alterations of 46 cancer-related genes were analyzed by next-generation target sequencing. Among the 90 gastric cancers, 47 cancers had 68 somatic point mutations of 15 different genes, such as TP53, KRAS, and PIK3CA. Ten cancers had gene amplification of one of ERBB2, KRAS, PIK3CA, and MET. Gastric cancers in individual groups and genes analyzed were aligned in the order of the number of total mutations and mutation frequency, respectively. Black and red boxes show somatic point mutations and gene amplifications, respectively. Gastric cancers with current and past infection had comparable profiles of somatic point mutations and gene amplifications.
Table 3.
Molecular profiles in 90 gastric cancers.
| Characteristic | H. pylori infection status | p value | |
|---|---|---|---|
| Current | Past | ||
| N (%) | N (%) | ||
| Oncogene point mutations in hotspots | |||
| Yes | 4 (7.3) | 7 (20.0) | 0.100 |
| No | 51 (92.7) | 28 (80.0) | |
| Gene amplification of oncogenes | |||
| Yes | 7 (12.7) | 3 (8.6) | 0.735 |
| No | 48 (87.3) | 32 (91.4) | |
| Oncogene activation (either or both point mutations in hotspots and gene amplification) | |||
| Yes | 11 (20.0) | 10 (28.6) | 0.444 |
| No | 44 (80.0) | 25 (71.4) | |
Regarding SNPs observed in gastric cancer patients, their frequencies were compared between gastric cancer patients and healthy Japanese people in datasets of the Tohoku Medical Megabank Organization (ToMMo 4.7 K JPN). SNPs of PIK3CA (p.Glu707Lys) and KDR (p.Gln472His) were more frequent in gastric cancer patients than in healthy Japanese people (p < 2.2 × 10–16 and p = 0.001, respectively; Bonferroni-corrected significance level = 0.003) (Supplementary Table S1). Pathogenicity of these SNPs was assessed using the Catalogue of Somatic Mutations in Cancer (COSMIC) database. PIK3CA (p.Glu707Lys) and KDR (p.Gln472His) were registered as “Pathogenic” and “Neutral”, respectively. Therefore, PIK3CA (p.Glu707Lys) could be a germline mutation that confers tumor predisposition. 16 other SNPs did not give Bonferroni-corrected statistical significance considering multiple testing.
ERBB2, KRAS, PIK3CA, and MET were amplified
Gene amplifications were analyzed for 26 cancer-related genes. Among the 90 gastric cancers, 10 cancers had gene amplification of one of ERBB2, KRAS, PIK3CA, and MET (Figs. 1, 2, Supplementary Table S2). ERBB2 was most frequently amplified (6 of the 90 gastric cancers, 7%), and KRAS (2 cancers, 2%), PIK3CA (1 cancer, 1%), and MET (1 cancer, 1%) followed. Combined with somatic hotspot mutations, ERBB2 was activated in 9 of the 90 gastric cancers (10%), KRAS was in 6 (7%), and PIK3CA was in 4 (4%). These results showed that 23% of gastric cancers had genetic activation of known oncogenes (Table 3).
Figure 2.

Gene amplification analysis of cancer-related genes. Gene amplification of 26 cancer-related genes was evaluated by utilizing reading depth of individual genes. For an individual sample, reading depths of 160 amplicons were plotted in a panel. Each amplicon was expected to be on a regression line calculated from all amplicons, but amplicons of the amplified gene were outlying. ERBB2 was amplified in 3 gastric cancers; KRAS was amplified in 2 cancers; and PIK3CA and MET were amplified in one cancer. Open circles show the amplicon of amplified genes. Black circles show that of all the other genes.
Molecular profiles were similar between gastric cancers with current and past H. pylori infection
To analyze whether molecular profiles between gastric cancers with current and past H. pylori infection are different, frequencies of the somatic point mutations and gene amplifications were compared between the two groups. Both groups had similar frequencies of TP53 mutations (38% and 31% in gastric cancers with current and past infection, respectively; p = 0.652), KRAS mutations (2% and 9%; p = 0.295), and PIK3CA mutations (4% and 6%; p = 0.641) (Fig. 3a). As for gene amplifications, gastric cancers with current and past infection also had similar frequencies of ERBB2 amplification (9% and 3%; p = 0.398) and KRAS amplification (2% and 3%; p = 1.000) (Fig. 3b, Table 3). These results showed that gastric cancers with current and past infection had comparable profiles of genetic alterations.
Figure 3.
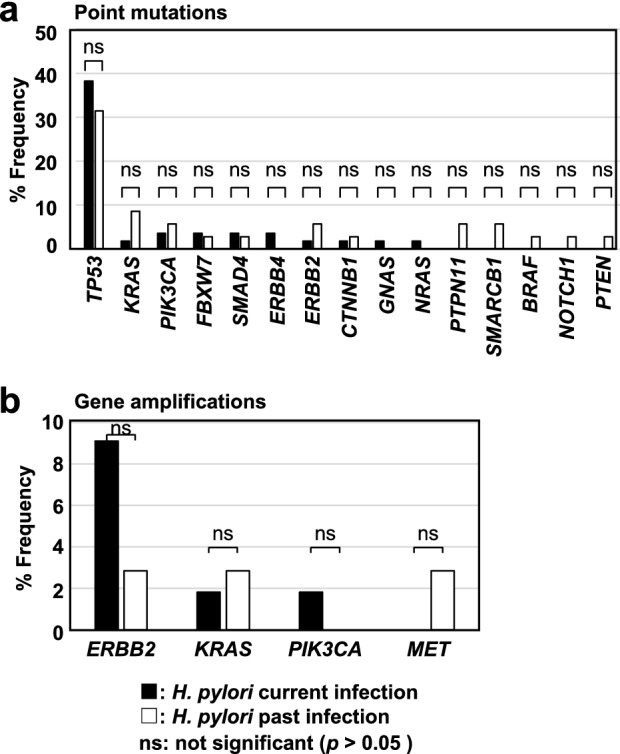
Frequency of point mutations and gene amplification in gastric cancers with current and past H. pylori infection. (a) Frequency of point mutations. Mutation frequencies of TP53, KRAS, and PIK3CA were similar between gastric cancers with current H. pylori infection (TP53, 38%; KRAS, 2%; and PIK3CA, 4%) and those with past infection (TP53, 31%; KRAS, 9%; and PIK3CA, 6%). Black and white bars show frequencies in gastric cancers with current and past H. pylori infection, respectively. (b) Frequency of gene amplification of ERBB2, KRAS, PIK3CA, and MET. The frequency was similar between gastric cancers with current H. pylori infection (ERBB2, 9%; and KRAS, 2%) and those with past infection (ERBB2, 3%; and KRAS, 3%). Black and white bars show frequencies in gastric cancers with current and past H. pylori infection, respectively.
Discussion
Gastric cancers with current and past H. pylori infection had comparable profiles of genetic alterations, namely somatic point mutations and gene amplification. Even when activation of known oncogenes, such as ERBB2 and PIK3CA, by either a point mutation or gene amplification was analyzed, both groups had similar frequencies. Since genetic activation of these genes has been clinically utilized in molecular targeted therapy21,22, it was considered that similar therapeutic strategies can be applicable for both gastric cancers with current and past infection.
It is known that H. pylori can directly activate oncogenic pathways, such as the MEK-ERK pathway and WNT pathway, and inactivate tumor-suppressive pathways, such as the p53 pathway, by injecting CagA into epithelial cells3. Therefore, it was considered that the alteration mechanisms of cancer-related signaling pathways might be different between gastric cancers with current infection and those with past infection. However, both groups had similar frequencies of alterations of genes involved in these cancer-related pathways. This suggested that direct activation or inactivation of cancer-related pathways by H. pylori has limited influence on genetic alterations.
Approximately 47% and 46% of gastric cancers with current H. pylori infection and past infection, respectively, had no genetic alterations of known cancer-related genes. In such gastric cancers, repression of tumor-suppressive pathways, such as cell cycle regulation and the p53 pathway, and activation of oncogenic pathways, such as the WNT pathway, are known to be frequently caused by epigenetic alterations, namely aberrant DNA methylation23. Therefore, it was considered that epigenetic alterations might be important in both gastric cancers with current H. pylori infection and past infection.
Somatic point mutations were analyzed by next-generation target sequencing, which covered 190 regions of 46 cancer-related genes. Although this panel covered almost all of the mutation hot spots of oncogenes, such as KRAS, PIK3CA, and CTNNB1, it covered limited regions of tumor-suppressor genes, such as TP53 (55.3%), CDH1 (7.5%), and MLH1 (2.6%). In addition, this panel did not cover several genes known to be mutated in 10% or more of gastric cancers, such as ARID1A, CREBBP, ERBB3, SMARCA4, and TGFBR2. Gene amplification was analyzed for 26 genes, including both oncogenes and tumor-suppressor genes, but was detected only in oncogenes, supporting the methodological validity. Approximately 9% of gastric cancers are known to be affected by Epstein-Barr virus (EBV), but EBV infection status was not analyzed in this study. EBV-positive gastric cancers are reported to have recurrent mutations of PIK3CA, ARID1A, and BCOR and amplifications of JAK2, PD-L1, and PD-L224.
Eradication of H. pylori is known to prevent the progression of gastric atrophy or intestinal metaplasia (IM)25, and almost all patients with gastric cancers are known to have gastric atrophy or IM. Actually, also in this study, most patients with past H. pylori infection had atrophy (Supplementary Figure S1). Although information on clinical history will improve the data quality, we consider that the number of patients with H. pylori eradicated before the development of gastric atrophy or IM would be small.
In conclusion, gastric cancers with current H. pylori infection and those with past infection had comparable profiles of genetic alterations.
Methods
Clinical samples
Surgically resected and fresh-frozen samples of 96 pairs of gastric cancers and corresponding non-cancerous tissues were obtained from the National Cancer Center Biobank. Twenty-one pairs of gastric cancers and corresponding non-cancerous tissues were collected for our previous study23, and also used for this study. This study was approved by the Institutional Review Boards of the National Cancer Center, Japan (2012-305 and 2018-024), and written informed consents were obtained from all the patients. All methods were carried out in accordance with relevant guidelines and regulations. Genomic DNA was extracted from gastric cancers and corresponding non-cancerous tissues by the phenol/chloroform method.
Analysis of H. pylori infection status
The infection status of H. pylori was determined by detection of PCR products specific for H. pylori jhpr3 gene and endoscopic gastric atrophy. Sensitivity and specificity for H. pylori detection by PCR test, urea breath test and serology test are reported to be > 95% and > 95%, 95.9% and 95.7%, and 76–84% and 79–90%, respectively26. Therefore, the reliability of a PCR test can be considered to be comparable with the other two methods. To avoid false-negative results in PCR, the quality of genomic DNA extracted from non-cancerous tissues was first evaluated by measuring the copy number of RPPH1 using quantitative PCR (qPCR) with primers listed in Supplementary Table S327. Among the 117 samples, 110 samples had 1,000 copies or more in 10 ng of genomic DNA, and were qualified for the evaluation of H. pylori infection status.
The presence of H. pylori was evaluated by qPCR using primers specific to the jhpr3 gene of H. pylori8 (Supplementary Table S4) and 100 ng of genomic DNA from non-cancerous tissues. Samples with successful amplification of the jhpr3 gene in two independent experiments were regarded as H. pylori-positive, and those in neither experiment were regarded as negative. Samples with one positive and one negative result were excluded from the entire analysis. Among the 110 samples, 59 samples were H. pylori-positive, 36 samples were -negative, and 15 samples were excluded. Endoscopic gastric atrophy was evaluated according to the endoscopic atrophic-border scale described by Kimura and Takemoto28. Fifty-seven of 59 H. pylori-positive samples had gastric atrophy (current infection), and 35 of 36 H. pylori-negative samples had gastric atrophy (past infection). These 92 samples (57 samples with current infection and 35 samples with past infection) were used for next-generation target sequencing. Clinicopathological characteristics, sex and pathology classification were not different among the two groups, but patients with past infection were slightly older (p = 0.033) (Supplementary Table S5).
Next-generation target sequencing
Next-generation target sequencing was conducted using an Ion AmpliSeq Cancer Panel Kit (Thermo Fisher Scientific, Waltham, MA), as described previously23,29. The sequence library was prepared by a multiplex PCR, which amplified 190 regions of 46 cancer-related genes. The library DNA was loaded onto an Ion PI Chip v3 (Thermo Fischer Scientific) or Ion 318 Chip v2 (Thermo Fischer Scientific) using Ion Chef (Thermo Fischer Scientific), and was sequenced using an Ion Proton sequencer (Thermo Fischer Scientific) or an Ion PGM sequencer (Thermo Fischer Scientific). The sequences obtained were mapped onto the human reference genome hg19 with Torrent Suite 5.0 (Thermo Fischer Scientific). An amplicon with 50 reads or less was considered to have low coverage, and two samples with 10% or more amplicons of low coverage were excluded from the analysis. Finally, 55 samples with current infection and 35 samples with past infection were used for mutation and amplification analysis. A variant call was conducted using CLC Genomics Workbench 20.0 (Qiagen, Hilden, Germany) with the following criteria; (i) with an allele frequency of 10% or more, (ii) not in homopolymers with 3 bp or more, (iii) present in both forward and reverse reads, and (iv) with a non-synonymous amino acid change. Sequence variations registered in dbSNP Build 137 were considered as SNPs, and were excluded before Sanger sequencing.
Sanger sequencing
Genomic regions where a sequence variation was found were amplified using 20 ng of genomic DNA (gastric cancers and corresponding non-cancerous tissues) and primers listed in Supplementary Table S6. The PCR products were purified by a DNA Clean and Concentrator-5 Kit (Zymo Research, Irvine, CA), and were sequenced by using a BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fischer Scientific) and 3730xl DNA Analyzer (Thermo Fischer Scientific). Sequence variations detected only in gastric cancers were considered as a somatic point mutation. Hotspot mutations were defined using information registered in COSMIC. Namely, a pathogenic mutation at the specific base position whose frequency was 5% or more of all the mutations in a specific gene was defined as a hotspot mutation. Among the 154 variations detected in 72 gastric cancers (newly analyzed cases in this study) by a next-generation sequencer, 101 variations were confirmed by Sanger sequencing (54 and 47 were somatic mutations and SNPs, respectively).
Analysis of SNPs
Six sequence variations registered in dbSNP Build 137 and twelve sequence variations confirmed as a SNP by Sanger sequencing were considered as SNPs (Supplementary Table S1). The frequencies of identified SNPs in gastric cancer patients (cases in this study) and healthy Japanese people in datasets of the Tohoku Medical Megabank Organization (ToMMo 4.7K JPN) were compared by the Fisher’s exact test.
Analysis of gene amplification
Gene amplification was analyzed using a next-generation sequencer since copy number variations (CNVs) detected by next-generation sequencers are now known to be accurately confirmed by Multiplex ligation-dependent probe amplification (MLPA), the gold-standard method to evaluate CNVs (Specificity 100%)30. Gene amplification of 26 genes (ABL1, APC, ATM, CDH1, EGFR, ERBB2, ERBB4, FBXW7, FGFR2, FGFR3, FLT3, KDR, KIT, KRAS, MET, PDGFRA, PIK3CA, PTEN, RB1, RET, SMAD4, SMARCB1, SMO, STK11, TP53, and VHL), which had three PCR amplicons or more, was analyzed, as described previously23. For an individual sample, reading depths of 160 amplicons of the 26 genes in the sample (y-axis) and in all the samples (average, x-axis) were plotted. The amplicons were expected to be on a regression line, but amplicons of an amplified gene were outlying. The ratio of the slope of a specific gene to that of the all genes was calculated, and genes with a ratio of three or more were defined as amplified genes. Since the next-generation target sequencing of 74 gastric cancers newly collected in this study was conducted in two sequencing runs, there were two background average reading depths (Supplementary Tables S7 and S8). The origins of gastric cancer samples with gene amplification (from our previous study or new in this study) are noted (Supplementary Table S2).
Statistical analysis
The Fisher’s exact test was used to analyze categorical variables and the Mann–Whitney U test was used to analyze quantitative variables. p < 0.05 was considered to be statistically significant.
Supplementary Information
Acknowledgements
The authors are grateful to Department of Biobank and Tissue Resources, National Cancer Center Research Institute for providing fresh-frozen samples of gastric cancers and corresponding non-cancerous tissues. This study was supported by AMED JP21ck0106552 and JP21gm1310006.
Author contributions
S.T., H.T., S.Y., S.M., and T.U. conceived and designed the study. S.S. and T.A. helped to collect clinical samples. Y.Y. and T.Y. collected clinical information. S.T., H.T., S.Y., and T.U. performed data extraction and analyzed the data. S.T., H.T., and T.U. wrote the manuscript. All authors critically revised and approved the final version of the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-02761-7.
References
- 1.Amieva M, Peek RM., Jr Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology. 2016;150:64–78. doi: 10.1053/j.gastro.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uemura N, et al. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi-Kanemitsu A, Knight CT, Hatakeyama M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell Mol. Immunol. 2020;17:50–63. doi: 10.1038/s41423-019-0339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan P, Yeoh KG. Genetics and molecular pathogenesis of gastric adenocarcinoma. Gastroenterology. 2015;149:1153–1162. doi: 10.1053/j.gastro.2015.05.059. [DOI] [PubMed] [Google Scholar]
- 5.Ushijima T, Hattori N. Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin. Cancer Res. 2012;18:923–929. doi: 10.1158/1078-0432.CCR-11-2011. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Wei B, Hu B. Chronic inflammation and long-lasting changes in the gastric mucosa after Helicobacter pylori infection involved in gastric cancer. Inflamm. Res. 2021 doi: 10.1007/s00011-021-01501-x. [DOI] [PubMed] [Google Scholar]
- 7.Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: Aberrant DNA methylation pathway. Gastric Cancer. 2017;20:8–15. doi: 10.1007/s10120-016-0650-0. [DOI] [PubMed] [Google Scholar]
- 8.Niwa T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- 9.Llorca-Cardenosa MJ, et al. Epigenetic changes in localized gastric cancer: The role of RUNX3 in tumor progression and the immune microenvironment. Oncotarget. 2016;7:63424–63436. doi: 10.18632/oncotarget.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Usui G, et al. DNA methylation and genetic aberrations in gastric cancer. Digestion. 2021;102:25–32. doi: 10.1159/000511243. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu T, et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology. 2014;147:407–417. doi: 10.1053/j.gastro.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 12.Takeshima H, Ushijima T. Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. NPJ Precis. Oncol. 2019;3:7. doi: 10.1038/s41698-019-0079-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi IJ, et al. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N. Engl. J. Med. 2018;378:1085–1095. doi: 10.1056/NEJMoa1708423. [DOI] [PubMed] [Google Scholar]
- 14.Fukase K, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392–397. doi: 10.1016/S0140-6736(08)61159-9. [DOI] [PubMed] [Google Scholar]
- 15.Lee YC, et al. Association between Helicobacter pylori eradication and gastric cancer incidence: A systematic review and meta-analysis. Gastroenterology. 2016;150:1113–1124. doi: 10.1053/j.gastro.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 16.Liou JM, et al. Systematic review with meta-analysis: 10-or 14-day sequential therapy vs 14-day triple therapy in the first line treatment of Helicobacter pylori infection. Aliment. Pharm. Ther. 2016;43:470–481. doi: 10.1111/apt.13495. [DOI] [PubMed] [Google Scholar]
- 17.Tsuda M, et al. Effect on Helicobacter pylori eradication therapy against gastric cancer in Japan. Helicobacter. 2017;22:1. doi: 10.1111/hel.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Take S, et al. Risk of gastric cancer in the second decade of follow-up after Helicobacter pylori eradication. J. Gastroenterol. 2020;55:281–288. doi: 10.1007/s00535-019-01639-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae SE, et al. Effect of Helicobacter pylori eradication on metachronous recurrence after endoscopic resection of gastric neoplasm. Am. J. Gastroenterol. 2014;109:60–67. doi: 10.1038/ajg.2013.404. [DOI] [PubMed] [Google Scholar]
- 20.Mori G, et al. Incidence of and risk factors for metachronous gastric cancer after endoscopic resection and successful Helicobacter pylori eradication: Results of a large-scale, multicenter cohort study in Japan. Gastric Cancer. 2016;19:911–918. doi: 10.1007/s10120-015-0544-6. [DOI] [PubMed] [Google Scholar]
- 21.Bang YJ, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 22.Shitara K, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 2020;382:2419–2430. doi: 10.1056/NEJMoa2004413. [DOI] [PubMed] [Google Scholar]
- 23.Yoda Y, et al. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer. 2015;18:65–76. doi: 10.1007/s10120-014-0348-0. [DOI] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research, N Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyokawa T, Suwaki K, Miyake Y, Nakatsu M, Ando M. Eradication of Helicobacter pylori infection improved gastric mucosal atrophy and prevented progression of intestinal metaplasia, especially in the elderly population: A long-term prospective cohort study. J. Gastroenterol. Hepatol. 2010;25:544–547. doi: 10.1111/j.1440-1746.2009.05995.x. [DOI] [PubMed] [Google Scholar]
- 26.Sabbagh P, et al. Diagnostic methods for Helicobacter pylori infection: Ideals, options, and limitations. Eur. J. Clin. Microbiol. Infect. Dis. 2019;38:55–66. doi: 10.1007/s10096-018-3414-4. [DOI] [PubMed] [Google Scholar]
- 27.Ueda S, et al. Influence of degree of DNA degradation in formalin-fixed and paraffin-embedded tissue samples on accuracy of genome-wide DNA methylation analysis. Epigenomics. 2021;13:565–576. doi: 10.2217/epi-2020-0431. [DOI] [PubMed] [Google Scholar]
- 28.Kimura K, Takemoto T. An endoscopic recognition of the atrophic border and its significance in chronic gastritis. Endoscopy. 1969;1:87–97. doi: 10.1055/s-0028-1098086. [DOI] [Google Scholar]
- 29.Kim JG, et al. Comprehensive DNA methylation and extensive mutation analyses reveal an association between the CpG island methylator phenotype and oncogenic mutations in gastric cancers. Cancer Lett. 2013;330:33–40. doi: 10.1016/j.canlet.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 30.Schenkel LC, et al. Clinical next-generation sequencing pipeline outperforms a combined approach using sanger sequencing and multiplex ligation-dependent probe amplification in targeted gene panel analysis. J. Mol. Diagn. 2016;18:657–667. doi: 10.1016/j.jmoldx.2016.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.