

Abstract
The Hedgehog (Hh) family of lipid-modified signaling proteins directs embryonic tissue patterning and postembryonic tissue homeostasis, and dysregulated Hh signaling drives familial and sporadic cancers. Hh ligands bind to and inhibit the tumor suppressor Patched and allow the oncoprotein Smoothened (SMO) to accumulate in cilia, which in turn activates the GLI family of transcription factors. Recent work has demonstrated that endogenous cholesterol and oxidized cholesterol derivatives (oxysterols) bind and modulate SMO activity. Here we discuss the myriad sterols that activate or inhibit the Hh pathway, with emphasis on endogenous 24(S),25-epoxycholesterol and 3β,5α-dihydroxycholest-7-en-6-one, and propose models of sterol regulation of SMO. Synthetic inhibitors of SMO have long been the focus of drug development efforts. Here, we discuss the possible utility of steroidal SMO ligands or inhibitors of enzymes involved in sterol metabolism as cancer therapeutics.
Keywords: medulloblastoma, basal cell carcinoma, Sonic hedgehog, sterol-sensing domain, oncogene
1. Introduction
Discovered in Drosophila melanogaster, the Hedgehog (Hh) pathway plays an essential and conserved role in tissue development and homeostasis across species [1]. Genetic and biochemical studies, first in Drosophila and later in vertebrates, revealed that Hh proteins (Sonic hedgehog, Desert hedgehog and Indian hedgehog in mammals) are secreted with two covalently attached lipid moieties: a palmitoyl moiety at the N terminus and a cholesterol molecule at the C terminus. Secreted Hh proteins act on distant cells by diffusing through the extracellular environment with the aid of proteins that shield the hydrophobic lipids [2]. The Patched receptor (Ptc in flies and PTCH1 or PTCH2 in vertebrates) on recipient cells interacts with Hh proteins, which relieves suppression of Smoothened (SMO) and activates the glioma-associated oncogene family of transcription factors (Ci in flies and GLI in vertebrates; Fig. 1). Although core Hh pathway components are conserved across species, mechanisms of Hh signal transduction have diverged between flies and mammals [3]. First, canonical mammalian Hh signaling requires a microtubule-based organelle called the primary cilium, and many Hh pathway components, such as PTCH, SMO, and GLI, dynamically localize to cilia depending on pathway activation [4]. Second, SMO activation in mammals requires cellular sterols, which is demonstrated by loss of Hh pathway activation after depletion of sterols from the plasma membrane [5]. Although recent structural studies using X-ray crystallography and cryo-electron microscopy (cryo-EM) have provided insights into mechanisms of PTCH regulation of SMO in mammals (reviewed in [2]), the precise role of cholesterol and its metabolites remains poorly understood and controversial due to the lack of tools to specifically manipulate endogenous sterols. However, misactivation of Hh signaling causes both familial and sporadic cancers [6]. Thus, understanding the role of lipid metabolites and the regulation of these compounds in the Hh pathway is vital for improving our knowledge of basic mechanisms of Hh signal transduction, and represents an unexplored avenue for novel therapeutics.
Fig. 1.
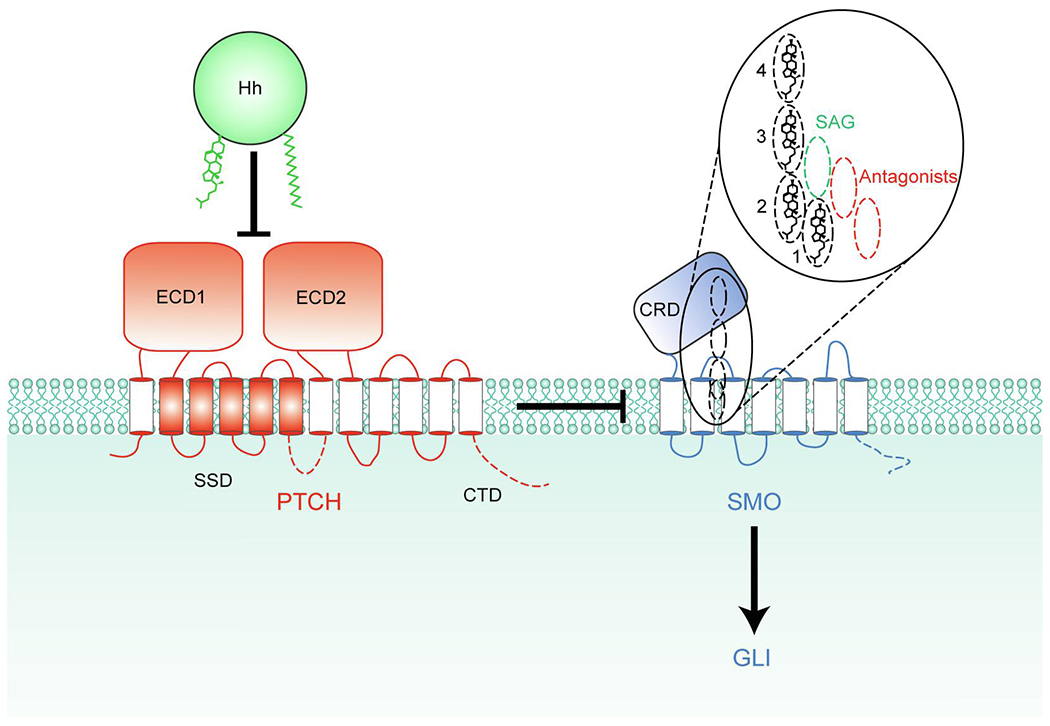
PTCH inhibits SMO by an unknown mechanism. Binding of Hh inhibits PTCH, allowing SMO activation. The 4 partially overlapping sites within SMO that can be occupied by sterols are depicted as dashed ovals, and their vertical positions compared to the synthetic SMO agonist (SAG) and various antagonists are shown in the inset. Sterols in all 4 sites within SMO have the same orientation (hydroxyl group facing towards the extracellular side and the side chain facing towards the intracellular side) potentially allowing sterols to move between these sites without flipping. The intracellular domains of PTCH1 and SMO missing from the structures, including the C-terminal domain (CTD) of PTCH1, are indicated by dashed lines. ECD1 and ECD2, extracellular domains 1 and 2; SSD, sterol-sensing domain; CRD, cysteine-rich domain.
2. (A)steroid impact on SMO
SMO is a Frizzled (FZD) family G protein-coupled receptor (GPCR) and contains a seven-transmembrane (7TM) domain and an extracellular cysteine-rich domain (CRD; Fig. 1). In all other FZD family receptors, the CRD binds to the Wnt family of lipid-modified signaling proteins, where a palmitoleate moiety on Wnt is directly involved in CRD binding [7]. Compared to all other known GPCRs, the 7TM domains of FZDs have a narrow pocket that is not amenable to binding small molecules and is considered to be undruggable [8,9]. Hh ligands are also covalently modified by lipids, but Hh proteins bind to PTCH instead of SMO and the role of SMO CRD is controversial. These observations raise a fundamental question: how does PTCH regulate SMO activity? PTCH is a 12TM transporter-like protein (see Section 4 below), and one alluring hypothesis is that PTCH regulates the abundance of a small molecule regulating SMO activity. In support of that hypothesis, SMO has at least 4 different small molecule binding sites, one in CRD and three within the 7TM domain, that have been structurally and biochemically shown to bind cholesterol, several oxysterols, and numerous synthetic agonists and antagonists of Hh pathway [2,10–13] (Fig. 1). The first small molecules identified to bind SMO were the steroidal plant compounds cyclopamine and jervine [14]. Fluorescent cyclopamine derivatives were shown to bind the 7TM domain, and used to find other small molecules that would compete for binding at the same site [15]. This approach led to the discovery of several synthetic small molecule antagonists, including vismodegib, sonidegib and the cyclopamine derivative saridegib, that are either approved as cancer therapeutics or are currently under investigation in clinical trials [16]. More recently, the SMO CRD and 7TM domains were shown to bind a partially overlapping set of sterols (see Fig. 3). Currently, 24(S),25-epoxycholesterol holds the record for the number of sites it has been shown to bind within SMO. Two molecules of 24(S),25-epoxycholesterol are observed to simultaneously bind sites 2 and 3 in one cryo-EM structure [12], and both we [11] and Qi et al. [12] showed biochemically that it can also bind CRD (site 4, Fig. 1). Residue N521 of SMO interacts with the epoxy tail of 24(S),25-epoxycholesterol bound at site 2, and when this residue is mutated, SMO can no longer be activated by 24(S),25-epoxycholesterol. It remains to be determined if this mutant can be activated by other sterols or Shh. In sum, SMO is regulated by a divergent mechanism compared to other FZD GPCRs, and it remains to be understood how endogenous SMO ligands mediate PTCH regulation of SMO.
Fig. 3.
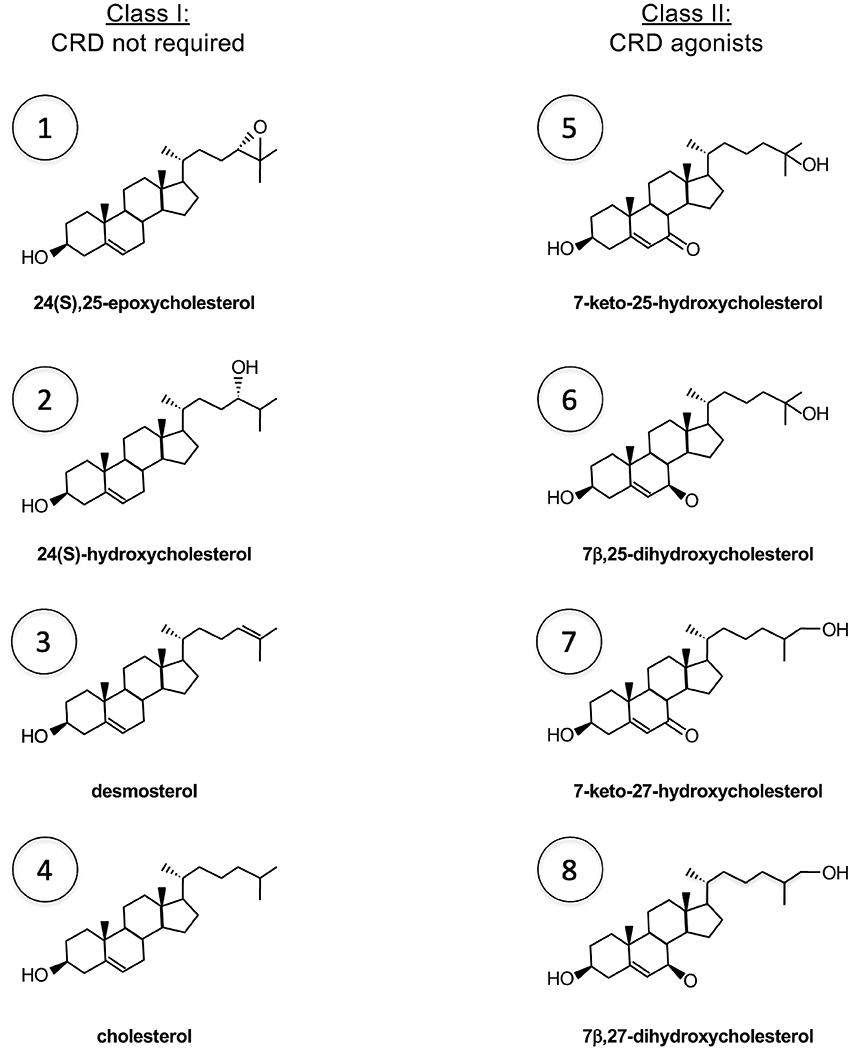
Sterol agonists of SMO. Class I sterols do not require CRD as they can activate SMOΔCRD when delivered as MCD inclusion complexes, although cholesterol and 24(S),25-epoxycholesterol can also bind CRD. Class II sterols only bind SMO CRD and can activate full-length SMO but not SMOΔCRD regardless of whether they are added from ethanol stocks or as MCD complexes. When a class I and a class II sterol are added together from ethanol stocks, a synergistic (more than additive) effect is observed. See Fig. 4 for biosynthesis of individual sterols (indicated by numbers).
Enzymatic oxidation products of cholesterol are one class of compounds that are of particular interest in regulating SMO activity. Historically thought of as intermediate metabolites in bile acid or steroid hormone biosynthesis, oxysterols are now known to act as important intracellular and intercellular signaling molecules [17]. A precedent for a GPCR with oxysterols as bona fide ligands is EBI2 [18,19]. EBI2 does not have a large extracellular domain, and presumably, oxysterols bind to the orthosteric ligand binding pocket found within the 7TM domain of most GPCRs. The most potent EBI2 agonist is 7α,25-dihydroxycholesterol, with an affinity (Kd) of ~1 nM, which is close to the endogenous concentration of oxysterols. Depletion of endogenous 7α,25-dihydroxycholesterol due to genetic deficiencies in either oxysterol 7α-hydroxylase (Cyp7b1) or cholesterol 25-hydroxylase (Ch25h) demonstrates a physiological requirement of 7α,25-dihydroxycholesterol for activating EBI2 [20]. Thus, SMO activity too may be modulated by an endogenous sterol and PTCH may act to regulate the availability of this endogenous ligand. However, it is unclear whether the putative substrate of PTCH is an agonist (activator) or antagonist (inhibitor) of SMO (green or red arrows, respectively, Fig. 2). Regardless of the identity of the endogenous SMO ligand, one interpretation is that the four ligand binding sites of SMO constitute a continuous tunnel, and the CRD facilitates access or removal of sterols that bind to the SMO 7TM domain. The role of the CRD in responding to Hh stimulation, however, would have to be auxiliary rather than obligatory as PTCH can still inhibit the activity of SMO constructs lacking the CRD [10,21].
Fig. 2.
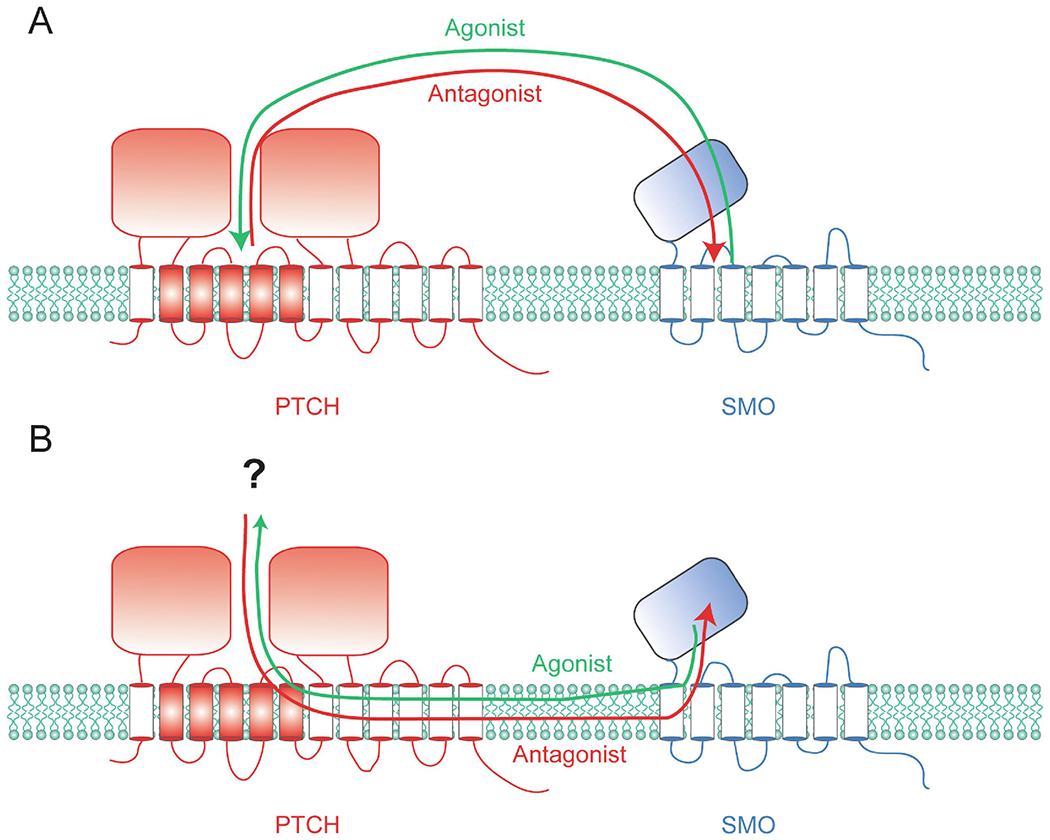
Models for PTCH regulation of SMO. PTCH likely inhibits SMO by either removing an agonist from SMO (green arrows) or transferring an antagonist to SMO (red arrows). It is unclear whether the exchange between PTCH and SMO occurs between the extracellular domains (A) or transmembrane domains (B). The latter case implicates the existence of a sterol carrier protein that cooperates with PTCH (shown with a question mark).
3. Good sterol, bad sterol
There are three plausible models for how PTCH may regulate SMO: (1) PTCH inhibits SMO by reducing the accessibility of (ciliary) cholesterol, (2) PTCH removes an oxysterol agonist from SMO, and (3) SMO requires cholesterol but PTCH inhibits SMO via a sterol antagonist (Table 1). Model 1 is the prevailing model and posits that cholesterol is necessary and sufficient for SMO activation, and PTCH reduces the accessibility of cholesterol to SMO [22] (green arrows, Fig. 2). Cholesterol is observed around the 7TM bundle of many GPCR structures and, in some cases, modulates their activity by increasing binding affinities for orthosteric ligands [23,24]. In the case of oxytocin receptor, this was shown using methyl-β-cyclodextrin (MCD), a macrocyclic compound that selectively extracts sterols from the plasma membrane [24]. MCD was similarly used to demonstrate a sterol requirement for SMO function [5]. Moreover, cholesterol is present in large concentrations to act as a signaling molecule as it constitutes close to 50% of lipids in the plasma membrane [25], and two studies using toxin-based cholesterol sensors, such as perfringolysin O (PFO), suggested PTCH affects cholesterol accessibility to SMO (Model 1). One of these studies proposed that PTCH flips cholesterol from the inner membrane to the outer membrane when expressed heterologously in HEK293 cells [26]. Another study using a mutant version of PFO and other sensors suggested that physiological levels of PTCH reduces cholesterol accessibility in only the ciliary membrane [27]. However, the general conclusions of the former study has been challenged for the quantitative accuracy of PFO [28]. Moreover, there are other putative cholesterol transporters, and likely ciliary receptors that are cholesterol-sensitive, raising the question of how PTCH could inhibit SMO specifically with such a mechanism. It is unlikely that PTCH removes ligands directly from SMO for two reasons. First, PTCH and SMO do not physically interact [29], nor colocalize, as they dynamically segregate in distinct domains within the ciliary membrane [30]. Second, one PTCH molecule is estimated to catalytically inhibit ~50 molecules of SMO [29], which also raises the question of the ultimate fate of the ligands that would be removed from SMO. In particular, cholesterol is insoluble and could not leave the membrane freely, and no protein has been implicated to cooperate with PTCH by serving as a sink for cholesterol. Thus, although the prevailing model suggests PTCH regulates SMO by regulating cholesterol accessibility, there remain fundamental open questions that need to be answered to support this model.
Table 1.
Arguments for and against different models by which PTCH regulates SMO.
| Model1: PTCH reduces accessibility of (ciliary) cholesterol for SMO |
| Pro: Unless a different sterol or steroidal detergent is added during purifications, the sterol-like densities seen in PTCH1 and SMO structures are likely cholesterol. |
| Con: |
| - Alteration of cholesterol distribution by PTCH would have pleiotropic effects. |
| - SMO activity would be affected by various other cholesterol transporters. |
| - An ultimate acceptor for cholesterol removed away from SMO is required. |
|
|
| Model 2: PTCH removes an oxysterol agonist from SMO |
| Pro: specific |
| Con: |
| - affinity << abundance (except 24(S),25-hydroxycholesterol, see text) |
| - no genetic evidence (does not apply for 24(S),25-hydroxycholesterol, see text) |
|
|
| Model 3: SMO requires cholesterol but PTCH inhibits SMO via a sterol antagonist |
| Pro: |
| - specific |
| - evidence for a positive effect of MCD under certain conditions |
| - can explain non-cell-autonomous inhibition of SMO by PTCH |
| Con: hard to genetically disentangle a negative effect of 7-dehydrocholesterol derivatives from the positive effect of cholesterol |
Vikas Daggubati: Writing- Original draft preparation, Writing- Reviewing and Editing. David R. Raleigh: Writing- Reviewing and Editing. Navdar Sever: Conceptualization, Writing- Original draft preparation, Writing- Reviewing and Editing, Supervision.
First, the reagents used to study the cholesterol’s role in Hh signaling are imperfect. Both MCD and PFO are promiscuous tools, as the former is known to extract sterols other than cholesterol and the latter is known to bind to diverse sterols, including desmosterol and sitosterol [31–33]. In addition to 24(S),25-epoxycholesterol mentioned above, the search for putative endogenous oxysterols that modulate SMO activity led to the discovery of isomers of EBI2 ligands, 7-keto-25-hydroxycholesterol, 7-keto-27-hydroxycholesterol, 7β,25-dihydroxycholesterol and 7β,27-dihydroxycholesterol, as CRD agonists [10,11,34] (Figs. 3 and 4). Therefore, a second possibility is that the physiologic substrate of PTCH is not cholesterol, but another sterol that is a specific regulator of SMO (Model 2). Indeed, sterols such as 24-ketocholesterol and 24(S),25-epoxycholesterol were both detected by targeted mass spectrometry of lipids extracted from purified PTCH1 protein [35], and found to be enriched in cilia purified from sea urchins [11]. There are also precedents for a distinct sterol composition in eukaryotic cilia or flagella compared to the rest of the plasma membrane. For instance, in spermatozoa of some species, desmosterol is found in large amounts (in some cases exceeding cholesterol) and is exclusively confined to the flagella, which are specialized cilia [36–40]. However, the effective concentration of oxysterols required to activate SMO is orders of magnitude higher than their endogenous levels. Second, unlike mice lacking enzymes that catalyze distal steps of the cholesterol biosynthesis pathway, triple-knockout mice deficient in cholesterol 24-hydroxylase (Cyp46a1), Ch25h and sterol 27-hydroxylase (Cyp27a1) do not show developmental defects associated with reduced Hh signaling [41]. These mice have dramatically reduced levels of side-chain oxysterols, but some oxysterols may still be synthesized by redundant enzymes or non-enzymatic reactions.
Fig. 4.
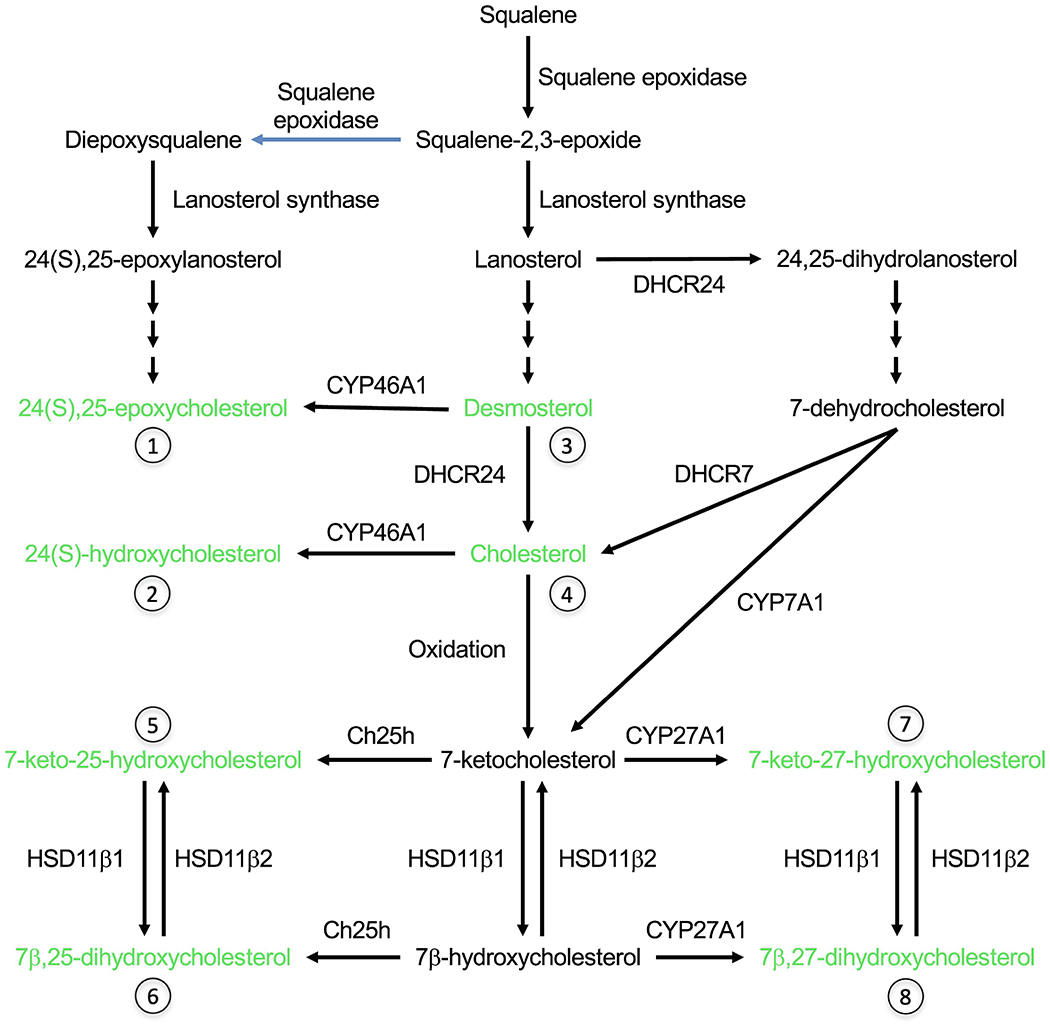
Biosynthesis of cholesterol and the sterol agonists of SMO (shown in green). 7-ketocholesterol can be formed either by non-enzymatic oxidation of cholesterol or enzymatically from 7-dehydrocholesterol. See Fig. 3 for the structures of sterols indicated with numbers.
24(S),25-epoxycholesterol is exceptional with respect to both arguments against Model 2. First, the levels of endogenous 24(S),25-epoxycholesterol range from 0.1-1 % of cholesterol, which corresponds in liver to 10-30 μM, well within its effective concentration for SMO activation in vitro [42,43]. Second, 24(S),25-epoxycholesterol can be synthesized either from desmosterol by CYP46A1, or by a shunt pathway that does not require any additional enzyme besides those required for cholesterol synthesis [43,44] (Fig. 4). In fact, compared to this shunt pathway, cholesterol synthesis requires an additional enzyme, 24-dehydrocholesterol reductase (DHCR24), that converts desmosterol to cholesterol. In a CRISPR screen, Kinnebrew et al. [27] used the finding that loss of Dhcr24 blocks Hh signaling to argue that cholesterol rather than 24(S),25-epoxycholesterol regulates SMO. However, they were not able to rescue Hh signaling in Dhcr24−/− cells by exogenous cholesterol, in contrast to the successful rescue in mutant cells deficient in other enzymes such as 7-dehydrocholesterol reductase (DHCR7). The lack of response to Shh in Dhcr24−/− cells is surprising because desmosterol itself can support SMO activation [45], and clear Hh-related phenotypes are not observed in the human disease desmosterolosis caused by mutations in DHCR24 [46]. This is in contrast to mutations in DHCR7 that cause Smith-Lemli Opitz syndrome (SLOS) with a subset of the developmental abnormalities observed in mice and humans with Shh mutations, such as holoprosencephaly [47]. One possibility is that desmosterol has an adverse but indirect effect on Hh signaling, for instance by disrupting lipid rafts, which cannot be alleviated by excess cholesterol [48]. Thus, further work needs to be done to test the involvement of endogenous 24(S),25-epoxycholesterol in Hh signaling. One way to accomplish this would be by adding diepoxysqualene, the committed precursor of 24(S),25-epoxycholesterol, and/or manipulating lanosterol synthase activity. As lanosterol synthase has a higher affinity for diepoxysqualene than monoepoxysqualene, partial inhibition of this enzyme has been shown to divert the metabolic flux from cholesterol biosynthesis towards the shunt pathway [49–51].
A third possibility is that SMO activity requires cholesterol (or another oxysterol agonist) but PTCH inhibits SMO via a sterol antagonist (Model 3, Table 1). A sterol requirement for SMO function has been demonstrated using two experimental protocols: (i) by acute treatment with a high concentration of MCD followed by chronic treatment with 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors, where HMG CoA reductase catalyzes the rate-limiting step of cholesterol synthesis, or (ii) by chronic treatment with a lower concentration of MCD [5]. Using the first protocol, 100 μM cholesterol added in ethanol (after MCD treatment) had been shown previously to rescue the response to Shh, indicating that cholesterol has access to SMO under these conditions but was not sufficient to activate SMO in the absence of Shh [10]. MCD can also be used as a delivery vehicle by solubilizing cholesterol in the form of an inclusion complex. Using the second protocol, the ability of MCD-cholesterol (100-250 μM final cholesterol concentration) to activate Hh pathway in the absence of any other agonist has been used to propose that cholesterol is not only necessary but also sufficient to activate SMO [45,52]. This is a misinterpretation based on the assumption that MCD is an inert delivery vehicle. An inclusion complex of MCD with a sterol effectively exchanges the included sterol with cellular sterols. In other words, MCD-cholesterol not only increases the cholesterol content of the plasma membrane but also extracts other sterols. MCD is only needed to deliver cholesterol to intracellular locations such as the endoplasmic reticulum (ER). It is well established that cholesterol added from an ethanol stock can rescue the growth of cholesterol auxotrophic cell lines, indicating that it can at least be incorporated into the plasma membrane where it can serve its essential functions [53]. The fact that cholesterol in MCD, but not in ethanol, activates Hh pathway in the absence of Shh suggests that MCD could extract both positive and negative sterols. Since positive sterols are required for SMO activity, the net effect of empty MCD treatment is inhibition. When an MCD-cholesterol complex is used, the negative sterols are still extracted but the excess cholesterol allows SMO to be fully active. These data give rise to the possibility that PTCH regulates SMO, at least in part, via a sterol antagonist (Model 3, Table 1). Further support for an endogenous SMO antagonist came from a study demonstrating non-cell-autonomous inhibition of Hh response by PTCH from neighboring cells [54]. PTCH-expressing cells displayed an elevated Hh pathway inhibitory activity when the Dhcr7 gene was mutated, consistent with the idea that 7-dehydrocholesterol or a derivative inhibits SMO. As PTCH cannot deplete sterols from other cells, this suggests that PTCH exports a SMO antagonist. Further, we showed that the activity of the synthetic SMO agonist (SAG) (Fig. 1) is potentiated by MCD, but not β-cyclodextrin or hydroxypropyl-β-cyclodextrin, even though all three cyclodextrins can extract cholesterol and inhibit Shh stimulation [55]. The ability of different cyclodextrins to potentiate SAG correlates instead with their ability to extract 7-dehydrocholesterol.
Consistent with the idea that MCD extracts a SMO inhibitor, some oxysterols (class I), but not others (class II), are more efficacious in stimulating SMO activity when delivered as MCD complexes (Fig. 3). This is unlikely to be related to efficiency of delivery as most oxysterols are positional isomers with similar physicochemical properties, such as solubility that is orders of magnitude higher than cholesterol. Rather, the difference between the two classes of sterols seems to be their binding site on SMO. Class I sterols act on SMO 7TM domain (even though some of them can also bind CRD) and have no effect when added in ethanol but can fully activate Hh pathway when delivered as an MCD complex. In contrast, class II sterols, which act on SMO CRD, are not more potent when delivered in MCD vs in ethanol. Further, a synergistic effect is seen when a class I sterol and a class II sterol are added together from ethanol stocks at low micromolar concentrations, indicating class I sterols do not need MCD to access their binding site within SMO 7TM domain [10,56]. Rather, as in the case of MCD-cholesterol complex, MCD extraction of an endogenous antagonist permits class I sterols to activate SMO. On the other hand, CRD agonists (class II oxysterols) apparently can activate SMO despite the presence of the putative antagonist.
A search for putative endogenous antagonists led to the discovery of oxidized derivatives of 7-dehydrocholesterol such as 3β,5α-dihydroxycholest-7-en-6-one (DHCEO), which accumulates in SLOS [55,57] (Fig. 5). DHCEO is formed in a three-step process from 7-dehydrocholesterol that parallels a known biotransformation of cholesterol. Although the initial product 5,6-epoxycholesterol does not require enzyme activity, it is converted by cholesterol-5,6-epoxide hydrolase (ChEH) to cholestane-3β,5α,6β-triol, which is then transformed into 6-oxo-cholestan-3β,5α-diol by 11β-hydroxysteroid dehydrogenase type 2 (HSD11β2), the same enzyme that interconverts CRD agonists in addition to converting cortisol to cortisone [11,58] (Figs. 4 and 5). Interestingly, ChEH is a complex between DHCR7 and emopamil-binding protein (EBP), which is another enzyme involved in cholesterol biosynthesis [59]. Therefore, in the absence of DHCR7, the analogous step in DHCEO biosynthesis must be carried out by one of the other known epoxide hydrolase enzymes [60]. Although there is evidence for an enzymatic basis for the last step of DHCEO synthesis as well [61], it is not yet known if it is also carried out by HSD11β2. In various cultured cell and animal models, both cholesterol deficiency and 7-dehydrocholesterol accumulation have been shown to contribute to SLOS phenotype [62–65]. The current standard of care for SLOS is cholesterol supplementation, which also reduces accumulation of cholesterol precursors via feedback inhibition [66]. A combined supplementation of cholesterol with antioxidants that block the formation of abnormal oxysterols is currently under investigation in clinical trials [67]. Indeed, DHCEO accumulates to a concentration of ~3.5 μM in tissues of SLOS patients, but it is not clear whether the 100-fold lower concentration of DHCEO in normal tissues would be sufficient to regulate SMO activity [57].
Fig. 5.
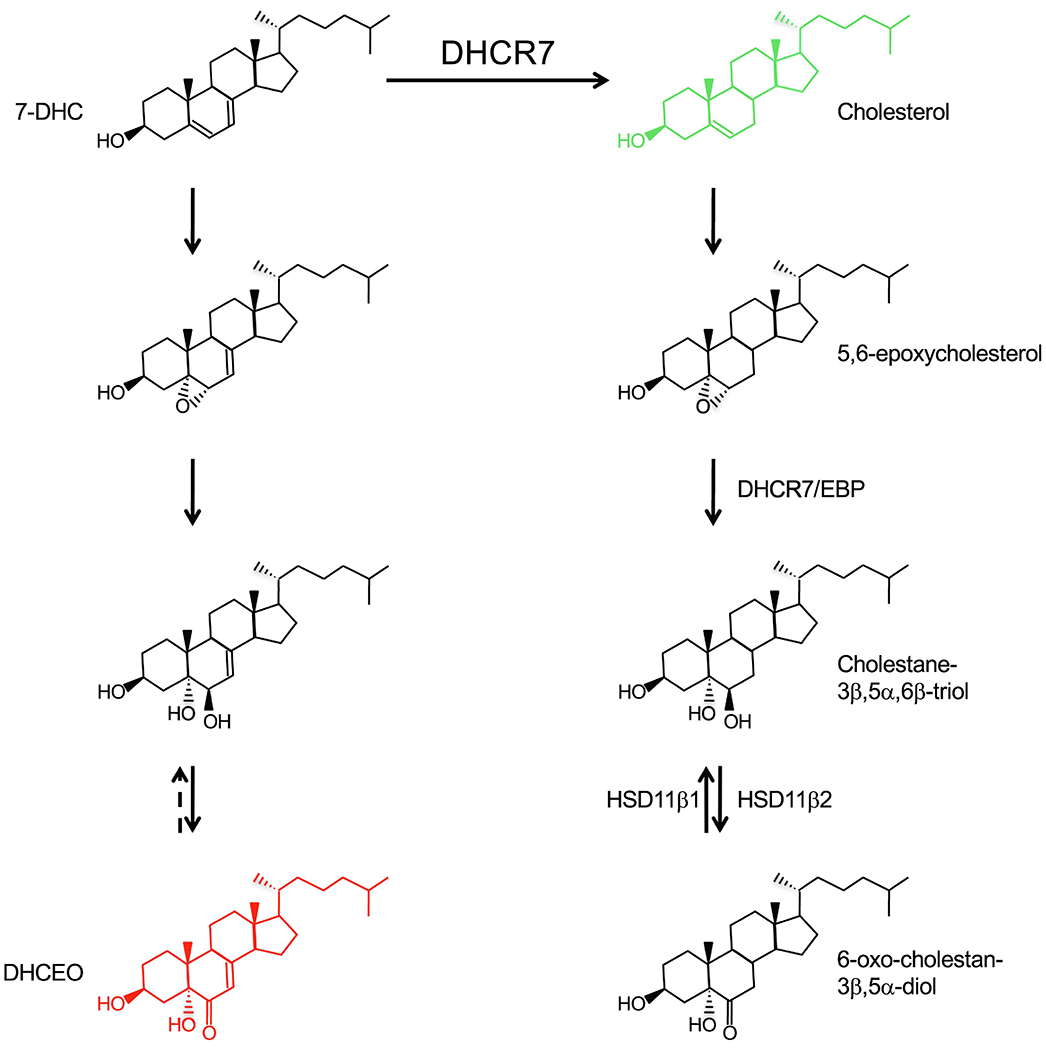
Formation mechanism of the SMO antagonist DHCEO. Besides cholesterol, which has a positive effect, none of the other sterols shown affect Hh pathway (unpublished observations). Whether the last step of DHCEO synthesis is reversible and which enzymes are responsible are not known.
Altogether, how SMO is regulated by sterols to activate the downstream Hh pathway remains unclear. Here we posit three models that all have varying levels of data that support or undermine their plausibility. However, these models are not mutually exclusive; i.e., an antagonist might displace positive sterols bound to SMO, or PTCH might remove an agonist from SMO in addition to providing the antagonist. As monitoring and manipulating endogenous lipids remain technically challenging, both technical innovations and novel insights are necessary to elucidate how sterols regulate SMO.
4. The origin story of a super family
The idea that PTCH acts as a transporter for a small molecule that modulates SMO activity is based on homology between PTCH and bacterial resistance, nodulation, division (RND) family of 12TM transporters [68], a family of antiporters that pump substrates out while importing protons down their concentration gradient (Fig. 6). The substrates of bacterial RND proteins range from metal ions to various xenobiotics, including sterol-like molecules called hopanoids [68]. Besides PTCH, Dispatched, Niemann-Pick C1 (NPC1) disease protein, NPC1-like 1 (NPC1L1) and the poorly characterized Patched-related (Ptr) proteins [69] are all eukaryotic members of the RND family. Further, all these proteins contain a five-transmembrane domain termed the sterol-sensing domain (SSD) that is conserved in three other proteins besides the RND family: the sterol sensor SCAP and two enzymes within the cholesterol biosynthetic pathway, HMG CoA reductase and DHCR7. Understanding how these proteins function may shed light on how PTCH regulates SMO.
Fig. 6.
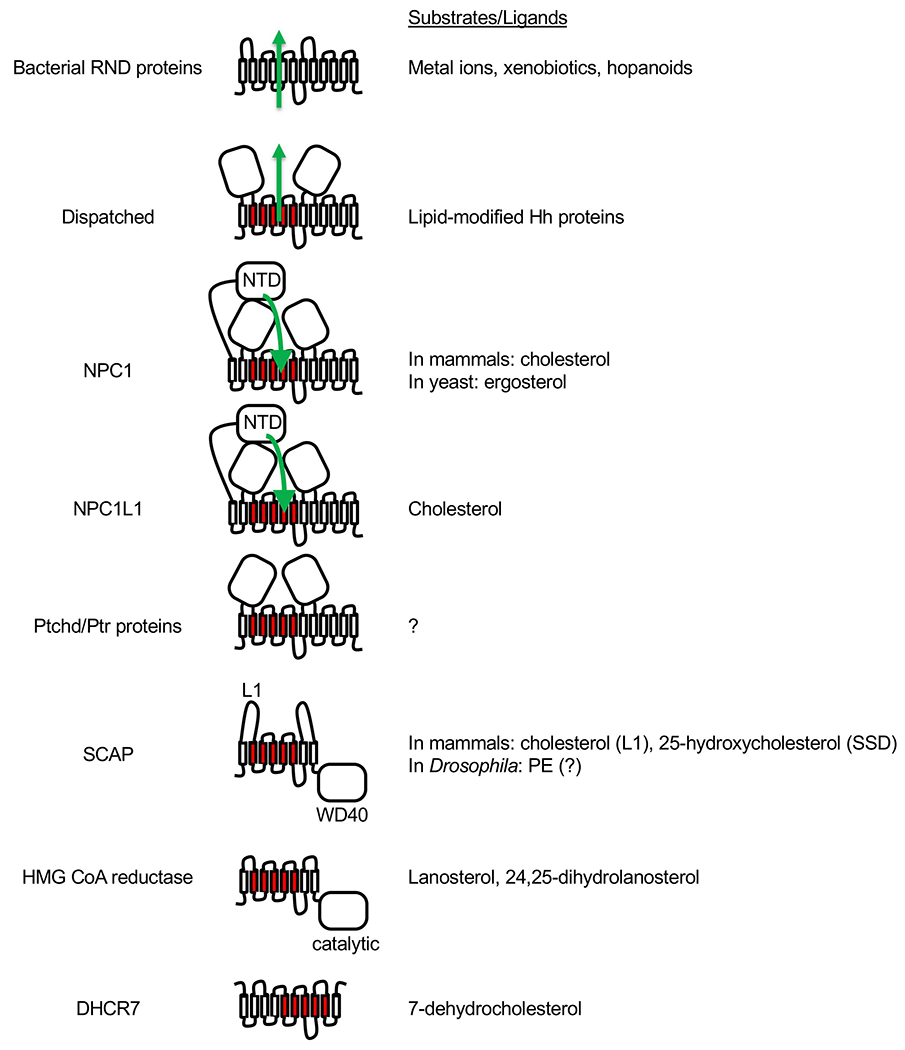
Bacterial RND proteins and eukaryotic SSD family members. For all proteins, the extracellular or luminal side faces up, and the cytosolic side faces down. The five TM domains that constitute the SSD are shown in red. Dispatched, NPC1, NPC1L1 and Ptchd/Ptr proteins are thought to function as transporters similar to the bacterial RND proteins, and the direction of substrate transport (where known) is indicated with green arrows. NPC1 and NPC1L1 each has an additional N-terminal domain (NTD). In the case of NPC1, cholesterol or ergosterol is first transferred from the luminal NPC2 protein (not shown) to the NTD and then travels through a tunnel between the other two large luminal domains towards the SSD. SCAP and HMG CoA reductase each has 8 TM domains and an additional cytosolic domain. In the case of SCAP, the cytosolic WD40 domain binds sterol regulatory element-binding proteins (SREBPs). Binding of either cholesterol to loop 1 (L1) or of 25-hydroxycholesterol to SSD of mammalian SCAP causes retention of SCAP-SREBP complex in the ER. Drosophila SREBP pathway responds to phosphatidylethanolamine (PE) instead of sterols but direct binding of PE to Drosophila SCAP has not been demonstrated. In the case of HMG CoA reductase, the cytosolic domain is necessary and sufficient for its catalytic activity, which is inhibited by statins. The membrane domain mediates ER-associated degradation of HMG CoA reductase in the presence of excess sterols. Lanosterol and 24,25-dihydrolanosterol have been shown to induce HMG CoA reductase degradation without affecting SREBP pathway. The topology of DHCR7 is not certain as it is predicted to contain 9 or 10 TMs, but its SSD is presumably the catalytic site where 7-dehydrocholesterol binds as a substrate.
Dispatched exports lipid-modified Hh proteins to the extracellular acceptor SCUBE [70,71]. NPC1 and NPC1L1 facilitate cholesterol transport across the lysosomal membrane and the intestinal epithelial cell apical membrane, respectively, in a direction that is topologically opposite to that of bacterial RND proteins. If NPC1 also uses a transmembrane proton gradient as an energy source, this would mean it acts as a symporter in contrast to its bacterial homologs. The N-terminal luminal domain of NPC1, which is distantly related to the SMO CRD [72], contains the initial binding site for cholesterol and receives cholesterol from the soluble luminal protein NPC2 and is postulated to deliver it to its SSD [73,74]. Consistent with this hypothesis, cryo-EM structures of NPC1 [75,76] revealed sterols within a tunnel that connects the N-terminal domain to the SSD. In contrast, yeast NPC1 and NPC2 homologs transport ergosterol, the primary sterol in yeast, which has a ring structure identical to that of 7-dehydrocholesterol and was captured within a tunnel between the luminal domain and the SSD of yeast NPC1 [75,77].
SSDs found within paralogous as well as orthologous proteins can bind to different sterols and even phospholipids. In the case of SCAP, biochemical experiments have shown that cholesterol binds to a luminal loop rather than the SSD [78]. A recent cryo-EM structure, however, has shown that 25-hydroxycholesterol is sandwiched between the SCAP SSD and the 6TM protein Insig, and binds to the opposite surface of SSD compared to the sterol-like densities seen in PTCH1 structures [79]. In Drosophila, there is no Insig homolog, and the SCAP homolog is thought to sense phosphatidylethanolamine rather than sterols [80]. In mammals, Insig can also bind to the SSD within the regulatory domain of HMG CoA reductase in a sterol-dependent manner [81]. Although there is no direct evidence for sterol binding to HMG CoA reductase SSD, lanosterol and 24,25-dihydrolanosterol can induce Insig-dependent HMG CoA reductase degradation without affecting SCAP activity, suggesting differences in sterol specificity for binding to the two SSD proteins [82]. In the case of DHCR7, although the sequence similarity to SSDs of other proteins is low, the corresponding domain presumably serves as the binding site for the substrate 7-dehydrocholesterol.
A ‘Patched domain-containing protein’ called Ptchd3 was found to be localized to the sperm midpiece [83] but, as this gene is non-essential for viability and fertility [84,85], whether it is involved in regulating flagellar sterol composition is not clear. In Caenorhabditis elegans, there are 3 PTCH homologs and 24 Ptr proteins, which is perplexing because Hh pathway does not exist in this organism due to the absence of Hh and SMO orthologs [69]. Further, C. elegans does not synthesize cholesterol and only needs small amounts of it from dietary sources. Knockdown experiments revealed that these proteins function in multiple aspects of C. elegans development [86] but it is unlikely that all of them transport cholesterol. It remains to be seen if they transport different lipids, or act in different intracellular locations or cell types.
Several structures of truncated PTCH1 variants alone or in complex with either recombinant (unmodified) or native (lipid-modified) Shh have been determined by cryo-EM [87–91]. Despite significant differences between their oligomeric states or binding interfaces between PTCH1 and Shh, these structures support a model in which PTCH controls the accessibility of a sterol (perhaps cholesterol itself) to SMO, as revealed by sterols identified within a putative hydrophobic tunnel between its extracellular domains and SSD [13,22], similar to the tunnel found in mammalian and yeast NPC1 homologs. PTCH can accommodate sterols other than cholesterol as revealed by cryo-EM structures that show bound cholesterol hemisuccinate or the steroidal detergent glyco-diosgenin with two relatively large maltose moieties [92,93]. Regardless of the exact transport mechanism of PTCH, binding of Shh may inhibit the transport activity of PTCH. In support, the palmitate and cholesterol moieties of native Shh bind to the extracellular domains of PTCH1 in such a way to either prevent the binding of a PTCH1 substrate or block its movement through the tunnel [13]. In particular, the palmitate moiety occupies a site similar to one in NPC1 occupied by the small molecule inhibitor itraconazole [76]. However, earlier biochemical data indicated that unmodified and lipid-modified Shh can bind PTCH with the same affinity even though palmitoylation dramatically increases Shh potency in most functional assays [94]. Surprisingly, structures of PTCH1 in complex with unmodified Shh showed that PTCH1 binds to the opposite surface of Shh when compared to lipid-modifed Shh. In fact, one lipid-modified Shh molecule can bind two PTCH1 molecules using the two different interfaces [89,90]. Although inhibition of Shh palmitoylation has dramatic effects on embryonic development, two observations suggest that the binding mode of unmodified Shh may also be physiologically relevant. First, recombinant (unmodified) Shh induces the differentiation of floor plate cells and motor neurons in chick neural plate explants with the same potency (2-25 nM) as native Shh [95]. Second, in cultured cell assays where unmodified Shh is inactive, introducing two hydrophobic isoleucine residues to its N terminus results in a substantial increase in its potency [96]. This Shh variant still binds PTCH1 in a manner identical to unmodified Shh [87]. It is possible that this binding mode still inhibits the transport activity of PTCH without the palmitate group penetrating deep inside the putative tunnel, but this would not explain why unmodified Shh can inhibit PTCH only in some contexts but not others. A more intriguing possibility is that PTCH may have two distinct activities depending on the cell type or developmental context (see below), that are inhibited by the two distinct binding modes of Shh. In summary, several structures of PTCH and its homology to RND proteins and NPC1 suggest that it functions, at least in part, as a transporter regulating the levels of a sterol lipid, but further work is necessary to uncover the identity of the substrate and the direction of transport. In this regard, PTCH1 was recently shown to use the transmembrane potassium gradient instead [97], but it remains unclear whether PTCH1 acts as a symporter or an antiporter.
SMO regulation likely evolved from a FZD-like mechanism where it was regulated by an extracellular ligand (a lipid or lipidated protein) that directly bound to its CRD to its current state, where the ability of SMO CRD to bind sterols can be seen as auxiliary, if not vestigial [98]. It is important to note that Drosophila SMO (dSmo) is unresponsive to cyclodextrin, sterols, and all synthetic ligands of mammalian SMO, and importantly, the structure of its CRD does not display a pocket that can accommodate sterols [99]. When dSmo was targeted to cilia by fusing it with the mammalian SMO C-terminal tail, the resulting chimeric protein was constitutively active suggesting that mammalian PTCH cannot repress dSmo [21]. This construct was also uninhibited by sterol depletion, consistent with the idea that dSmo is not regulated by a sterol agonist. Surprisingly, however, human PTCH1 expressed in fly cells can suppress dSmo activity as monitored by posttranslational modifications of downstream components [100]. One possible explanation for this discrepancy is that dSmo is regulated by an antagonist that does not exist in mammalian cells; but mammalian PTCH is more promiscuous and can transport this putative dSmo antagonist when expressed in fly cells. If the proposed model of PTCH function as a transporter is conserved in evolution, the same endogenous regulator of SMO would seem likely to operate in both vertebrate and fly systems because divergence of the effector small molecule would require the unlikely co-evolution of substrate specificity for PTCH transport and of binding specificity for SMO. It would be interesting to see if Drosophila Ptc targeted to cilia in mammalian cells could suppress mammalian SMO. Structure-function analyses, however, suggest that there may be fundamental differences in PTCH functions between flies and vertebrates. For instance, mutations in SSD confer dominant negative activity to Drosophila Ptc but does not detectably alter the function of mammalian PTCH [101]. Similarly, the poorly conserved cytoplasmic C-terminal domain (CTD) of Ptc is essential in Drosophila and its deletion creates a dominant-negative protein that blocks wild-type Ptc function in vivo [102]. In contrast, the CTD of mammalian PTCH1, which is deleted in the constructs used for all published structural investigations, seems to be required in a tissue-specific manner. This was revealed by a spontaneous mouse mutant called mesenchymal dysplasia (mes) caused by a deletion of part of the CTD [103]. On the one hand, mes homozygotes exhibit normal spinal cord development in contrast to Ptch1−/− homozygotes, which die around day 10 of embryonic development with severe neural tube defects [104]. This indicates that CTD is dispensable in early embryogenesis. On the other hand, mes mutants exhibit pleiotropic Hh-associated phenotypes including excess skin, increased body weight and preaxial polydactyly. These observations support the idea that, depending on the tissue or cell type, PTCH1 may suppress SMO activity by two different mechanisms, one that is CTD-dependent and another that is CTD-independent, where Drosophila Ptc mainly uses the first mechanism. Another potential difference between Drosophila and vertebrate systems was revealed with the discovery of a negative regulator of the Drosophila Hh pathway, called Target of Wingless (Tow), that seems to act between Ptc and Smo [105]. No such factor has been identified in vertebrates.
5. Oncogenic Hh signaling: SMO on steroids
Dysregulated Hh signaling is known to drive multiple cancers. This was first appreciated through patients with Gorlin’s syndrome, a rare genetic condition caused by heterozygous inactivating mutations in negative regulators of the Hh pathway, who are predisposed to developing basal cell carcinomas (BCCs), medulloblastomas, and rhabdomyosarcomas (RMSs) [106–108]. Hh pathway misactivation also underlies sporadic BCCs, the most common cancer in the United States [109–111], approximately one-third of sporadic medulloblastomas, one of the most common malignant brain tumors in children [112,113], and a subset of sporadic RMSs, the most common pediatric soft tissue sarcoma [114–116]. Aberrant Hh signaling has been also implicated in several other malignancies, including pancreatic ductal adenocarcinoma, lung cancer, and prostate cancer, but the mechanistic connections linking Hh signaling to these tumors are incompletely understood [6]. Understanding how Hh signaling drives tumor growth across cancer subtypes is fundamental, as small molecules modulating the Hh pathway represent a powerful therapy modality.
In this regard, several synthetic small-molecule inhibitors of the Hh pathway have been developed and are approved for use in cancer patients, including the SMO inhibitors vismodegib and sonidegib. These inhibitors have been extensively studied in the treatment of medulloblastoma and BCC [117–119], leading the FDA approval of vismodegib and sonidegib for metastatic or locally advanced BCC, and vismodegib for use in recurrent medulloblastoma. Responses to SMO inhibitors are variable, and resistance to molecular monotherapy is common in Hh-associated cancers. Both BCC and medulloblastoma can regress below the threshold of detection by visual, histopathological, or radiological examination, but many tumors can recur after developing resistance mechanisms [120]. Point mutations in SMO [121] and misactivation of the PI3K pathway [122] underlie resistance to SMO inhibitors in Hh-associated medulloblastoma, and similar mutations in SMO [123] and non-canonical activation of the Hh pathway [124] underlies resistance to SMO inhibitors in BCC. Moreover, these therapies cause myriad adverse side effects, such as nausea, muscle cramps, loss of taste, weight loss, and alopecia, leading to medical non-compliance [125]. Pediatric medulloblastoma patients who are treated with SMO inhibitors also have short stature and other medical problems due to premature and irreversible growth plate fusion from systemic Hh pathway inhibition [126]. Considering the resistance mechanisms to Hh pathway inhibition in cancer, and the adverse side effects of these medications, there is an urgent, unmet need for new therapies to treat Hh-associated cancers.
With respect to modulating oncogenic Hh signaling for therapeutic purposes, naturally occurring oxysterols, including the antagonist DHCEO, have modest affinities to SMO that are typically in the micromolar range. There are also many other oxysterol receptors with diverse ligand-binding specificities that may cause pleiotropic effects, especially if a high concentration is needed to inhibit oncogenic Hh signaling. A different approach to inhibit Hh pathway would be to deprive SMO of its endogenous agonists. Given that both SMO and PTCH are likely capable of binding diverse sterols, it is possible that there is a different endogenous regulator of SMO depending on cell type, tissue, developmental or oncogenic context. In addition, overproduction of an abnormal oxysterol agonist of SMO by tumor cells can conceivably drive constitutive SMO activity. Regardless, there is a positive role for sterols for the activation of Hh pathway. Inhibition of HMG CoA reductase by statins indeed repress Hh signaling and proliferation in medulloblastoma cells [127]. Statins further synergize with vismodegib in inhibiting medulloblastoma growth. This is a promising strategy given that point mutations in SMO that could give rise to vismodegib resistance would not necessarily circumvent the positive requirement for sterols, and statins are used safely by millions of people as cholesterol-lowering drugs.
Recently, HSD11β2 was identified as a Hh target gene and a novel therapeutic target in Hh-associated medulloblastoma [11]. HSD11β2 is an oxysterol synthase that produces SMO-activating lipids to drive Hh signaling in development and medulloblastoma, and inhibition of HSD11β2 using a small molecule derived from black licorice, carbenoxolone, blocks the Hh pathway and the growth of Hh-associated medulloblastoma [128] (Fig. 4). Recently, HSD11β2 enzymatic activity was shown to be inhibited by the antifungal drugs itraconazole and posaconazole [129]. These drugs were originally known as inhibitors of lanosterol demethylase (CYP51) but have since been shown to also interfere with intracellular sterol trafficking, at least in part, by inhibiting NPC1 [130] (see above) and oxysterol-binding protein family [131]. A combination of these mechanisms can explain SMO inhibition observed with these drugs [132].
Moreover, recent work has identified that CDK6 is a direct transcriptional target of oncogenic Hh signaling, and CDK6 antagonists such as abemaciclib and palbociclib are effective treatments in preclinical models of Hh-associated medulloblastoma and are currently under investigation in clinical trials [128]. Nevertheless, genetically engineered mouse models of Hh-associated medulloblastoma treated with carbenoxolone or CDK6 inhibitors ultimately succumb to their tumors [11,128]. These data suggest that Hh-associated cancers can also develop resistance mechanisms to therapies blocking non-canonical Hh pathway activators or effectors. Genome-wide CRISPR screens and transcriptomic profiling of Hh-associated medulloblastomas lacking Cdk6 reveals loss of CDK6 leads to ER stress, activating the unfolded protein response and inducing enzymes producing SMO-activating lipids, such as HSD11β2 and DHCR7, that sustain oncogenic Hh signaling [133]. Consistently, combination molecular therapy with abemaciclib and carbenoxolone blocks the growth of Hh-associated medulloblastoma and prolongs survival more than molecular monotherapy [133]. Thus, preclinical strategies targeting lipids that activate the Hh pathway in conjunction with effectors of oncogenic Hh signaling overcome resistance mechanisms and may improve outcomes in patients with Hh-associated cancer.
6. Conclusion
The structures of PTCH1 and SMO provide insights into putative substrate/ligand binding sites and confirm the similarity between PTCH and other eukaryotic RND proteins, but they have not clarified the identity of endogenous substrates or ligands, or the direction of PTCH transport of those molecules. Future studies directed at elucidating endogenous lipids that bind PTCH and SMO would benefit from native mass spectrometry, which has emerged within the last decade as a complementary technique to probe protein-lipid interactions [134]. The exact mechanism of PTCH action will likely require reconstitution of its transport activity in a cell-free system. With respect to Hh signaling and cancer, multiple open questions remain that will require further investigation: (1) What other human malignances are driven by misactivated Hh signaling? (2) Why do some tumors respond well to targeted Hh inhibitors, whereas other tumors rapidly develop resistance to monotherapy? (3) Will oxysterol synthase inhibitors, such as carbenoxolone translate to the clinic? Answering these questions will not only further our understanding of Hh signaling and cancer but will also lead to improved cancer therapies.
Acknowledgements
D.R.R. is the recipient of an NIH grant (K08 CA212279).
Abbreviations
- BCC
basal cell carcinoma
- ChEH
cholesterol-5,6-epoxide hydrolase
- Ci
Cubitus interruptus
- CRD
cysteine-rich domain
- CTD
C-terminal domain
- cryo-EM
cryo-electron microscopy
- Cyp
cytochrome P450
- DHCEO
3β,5α-dihydroxycholest-7-en-6-one
- DHCR7
7-dehydrocholesterol reductase
- DHCR24
24-dehydrocholesterol reductase
- dSmo
Drosophila Smoothened
- EBP
emopamil-binding protein
- ECD
extracellular domain
- ER
endoplasmic reticulum
- FZD
Frizzled
- GPCR
G protein-coupled receptor
- Hh
Hedgehog
- HMG CoA
3-hydroxy-3-methylglutaryl coenzyme A
- HSD11β2
11β-hydroxysteroid dehydrogenase type 2
- MCD
methyl-β-cyclodextrin
- mes
mesenchymal dysplasia
- NPC1
Niemann-Pick C1
- NPC1L1
Niemann-Pick C1-like 1
- PE
phosphatidylethanolamine
- PFO
perfringolysin O
- Ptc
Patched
- PTCH
Patched homolog
- Ptchd
Patched domain
- Ptr
Patched-related
- RMS
rhabdomyosarcoma
- RND
resistance, nodulation, division
- SAG
Smoothened agonist
- SLOS
Smith-Lemli-Opitz syndrome
- SMO
Smoothened
- SREBP
sterol regulatory element-binding protein
- SSD
sterol-sensing domain
- Tow
Target of Wingless
- 7TM
seven-transmembrane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that no conflict of interest exists.
References
- [1].Ingham PW. From Drosophila segmentation to human cancer therapy. Development 2018;145. doi: 10.1242/dev.168898. [DOI] [PubMed] [Google Scholar]
- [2].Qi X, Li X. Mechanistic Insights into the Generation and Transduction of Hedgehog Signaling. Trends Biochem Sci 2020;45:397–410. doi: 10.1016/j.tibs.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 2006;133:3–14. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- [4].Kopinke D, Norris AM, Mukhopadhyay S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin Cell Dev Biol 2021;110:89–103. doi: 10.1016/j.semcdb.2020.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet 2003;33:508–13. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- [6].Raleigh DR, Reiter JF. Misactivation of Hedgehog signaling causes inherited and sporadic cancers. J Clin Invest 2019;129:465–75. doi: 10.1172/JCI120850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science 2012;337:59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang S, Wu Y, Xu T-H, de Waal PW, He Y, Pu M, et al. Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 2018;560:666–70. doi: 10.1038/s41586-018-0447-x. [DOI] [PubMed] [Google Scholar]
- [9].Tsutsumi N, Mukherjee S, Waghray D, Janda CY, Jude KM, Miao Y, et al. Structure of human Frizzled5 by fiducial-assisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. Elife 2020;9. doi: 10.7554/eLife.58464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, et al. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell 2013;26:346–57. doi: 10.1016/j.devcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Raleigh DR, Sever N, Choksi PK, Sigg MA, Hines KM, Thompson BM, et al. Cilia-Associated Oxysterols Activate Smoothened. Mol Cell 2018;72:316–327.e5. doi: 10.1016/j.molcel.2018.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qi X, Friedberg L, De Bose-Boyd R, Long T, Li X. Sterols in an intramolecular channel of Smoothened mediate Hedgehog signaling. Nat Chem Biol 2020;16:1368–75. doi: 10.1038/s41589-020-0646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kowatsch C, Woolley RE, Kinnebrew M, Rohatgi R, Siebold C. Structures of vertebrate Patched and Smoothened reveal intimate links between cholesterol and Hedgehog signalling. Curr Opin Struct Biol 2019;57:204–14. doi: 10.1016/j.sbi.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 2002;16:2743–8. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA 2002;99:14071–6. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Khatra H, Bose C, Sinha S. Discovery of hedgehog antagonists for cancer therapy. Curr Med Chem 2017;24:2033–58. doi: 10.2174/0929867324666170316115500. [DOI] [PubMed] [Google Scholar]
- [17].Brown AJ, Sharpe LJ, Rogers MJ. Oxysterols: From physiological tuners to pharmacological opportunities. Br J Pharmacol 2020. doi: 10.1111/bph.15073. [DOI] [PubMed] [Google Scholar]
- [18].Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature 2011;475:519–23. doi: 10.1038/nature10226. [DOI] [PubMed] [Google Scholar]
- [19].Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature 2011;475:524–7. doi: 10.1038/nature10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity 2012;37:535–48. doi: 10.1016/j.immuni.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol 2013;9:557–64. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Radhakrishnan A, Rohatgi R, Siebold C. Cholesterol access in cellular membranes controls Hedgehog signaling. Nat Chem Biol 2020;16:1303–13. doi: 10.1038/s41589-020-00678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Waltenspühl Y, Schöppe J, Ehrenmann J, Kummer L, Plückthun A. Crystal structure of the human oxytocin receptor. Sci Adv 2020;6:eabb5419. doi: 10.1126/sciadv.abb5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 1995;34:13784–93. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- [25].Das A, Goldstein JL, Anderson DD, Brown MS, Radhakrishnan A. Use of mutant 125I-perfringolysin O to probe transport and organization of cholesterol in membranes of animal cells. Proc Natl Acad Sci USA 2013;110:10580–5. doi: 10.1073/pnas.1309273110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang Y, Bulkley DP, Xin Y, Roberts KJ, Asarnow DE, Sharma A, et al. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018;175:1352–1364.e14. doi: 10.1016/j.cell.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kinnebrew M, Iverson EJ, Patel BB, Pusapati GV, Kong JH, Johnson KA, et al. Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. Elife 2019;8. doi: 10.7554/eLife.50051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Courtney KC, Fung KY, Maxfield FR, Fairn GD, Zha X. Comment on “Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol”. Elife 2018;7. doi: 10.7554/eLife.38493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002;418:892–7. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- [30].Weiss LE, Milenkovic L, Yoon J, Stearns T, Moerner WE. Motional dynamics of single Patched1 molecules in cilia are controlled by Hedgehog and cholesterol. Proc Natl Acad Sci USA 2019;116:5550–7. doi: 10.1073/pnas.1816747116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sokolov A, Radhakrishnan A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J Biol Chem 2010;285:29480–90. doi: 10.1074/jbc.M110.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Savinov SN, Heuck AP. Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis? Toxins (Basel) 2017;9. doi: 10.3390/toxins9120381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nelson LD, Johnson AE, London E. How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J Biol Chem 2008;283:4632–42. doi: 10.1074/jbc.M709483200. [DOI] [PubMed] [Google Scholar]
- [34].Abdel-Khalik J, Hearn T, Dickson AL, Crick PJ, Yutuc E, Austin-Muttitt K, et al. Bile acid biosynthesis in Smith-Lemli-Opitz syndrome bypassing cholesterol: Potential importance of pathway intermediates. J Steroid Biochem Mol Biol 2021;206:105794. doi: 10.1016/j.jsbmb.2020.105794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Qi X, Liu H, Thompson B, McDonald J, Zhang C, Li X. Cryo-EM structure of oxysterol-bound human Smoothened coupled to a heterotrimeric Gi. Nature 2019;571:279–83. doi: 10.1038/s41586-019-1286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin DS, Connor WE, Wolf DP, Neuringer M, Hachey DL. Unique lipids of primate spermatozoa: desmosterol and docosahexaenoic acid. J Lipid Res 1993;34:491–9. [PubMed] [Google Scholar]
- [37].Mourvaki E, Cardinali R, Roberti R, Dal Bosco A, Castellini C. Desmosterol, the main sterol in rabbit semen: distribution among semen subfractions and its role in the in vitro spermatozoa acrosome reaction and motility. Asian J Androl 2010;12:862–70. doi: 10.1038/aja.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Awano M, Kawaguchi A, Morisaki M, Mohri H. Identification of cholesta-7,24-dien-3 beta-ol and desmosterol in hamster cauda epididymal spermatozoa. Lipids 1989;24:662–4. doi: 10.1007/BF02535086. [DOI] [PubMed] [Google Scholar]
- [39].Zalata A, Hassan A, Christophe A, Comhaire F, Mostafa T. Cholesterol and desmosterol in two sperm populations separated on Sil-Select gradient. Int J Androl 2010;33:528–35. doi: 10.1111/j.1365-2605.2009.00961.x. [DOI] [PubMed] [Google Scholar]
- [40].Sion B, Grizard G, Boucher D. Quantitative analysis of desmosterol, cholesterol and cholesterol sulfate in semen by high-performance liquid chromatography. J Chromatogr A 2001;935:259–65. doi: 10.1016/s0021-9673(01)01105-0. [DOI] [PubMed] [Google Scholar]
- [41].Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab 2007;5:73–9. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Spencer TA, Gayen AK, Phirwa S, Nelson JA, Taylor FR, Kandutsch AA, et al. 24(S),25-Epoxycholesterol. Evidence consistent with a role in the regulation of hepatic cholesterogenesis. J Biol Chem 1985;260:13391–4. [PubMed] [Google Scholar]
- [43].Brown AJ. 24(S),25-epoxycholesterol: a messenger for cholesterol homeostasis. Int J Biochem Cell Biol 2009;41:744–7. doi: 10.1016/j.biocel.2008.05.029. [DOI] [PubMed] [Google Scholar]
- [44].Goyal S, Xiao Y, Porter NA, Xu L, Guengerich FP. Oxidation of 7-dehydrocholesterol and desmosterol by human cytochrome P450 46A1. J Lipid Res 2014;55:1933–43. doi: 10.1194/jlr.M051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, et al. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife 2016;5. doi: 10.7554/eLife.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dias C, Rupps R, Millar B, Choi K, Marra M, Demos M, et al. Desmosterolosis: an illustration of diagnostic ambiguity of cholesterol synthesis disorders. Orphanet J Rare Dis 2014;9:94. doi: 10.1186/1750-1172-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kelley RL, Roessler E, Hennekam RC, Feldman GL, Kosaki K, Jones MC, et al. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am J Med Genet 1996;66:478–84. doi:. [DOI] [PubMed] [Google Scholar]
- [48].Vainio S, Jansen M, Koivusalo M, Róg T, Karttunen M, Vattulainen I, et al. Significance of sterol structural specificity. Desmosterol cannot replace cholesterol in lipid rafts. J Biol Chem 2006;281:348–55. doi: 10.1074/jbc.M509530200. [DOI] [PubMed] [Google Scholar]
- [49].McCrae C, Dzgoev A, Ståhlman M, Horndahl J, Svärd R, Große A, et al. Lanosterol synthase regulates human rhinovirus replication in human bronchial epithelial cells. Am J Respir Cell Mol Biol 2018;59:713–22. doi: 10.1165/rcmb.2017-0438OC. [DOI] [PubMed] [Google Scholar]
- [50].Hubler Z, Friedrich RM, Sax JL, Allimuthu D, Gao F, Rivera-León AM, et al. Modulation of lanosterol synthase drives 24,25-epoxysterol synthesis and oligodendrocyte formation. Cell Chem Biol 2021. doi: 10.1016/j.chembiol.2021.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rowe AH, Argmann CA, Edwards JY, Sawyez CG, Morand OH, Hegele RA, et al. Enhanced synthesis of the oxysterol 24(S),25-epoxycholesterol in macrophages by inhibitors of 2,3-oxidosqualene:lanosterol cyclase: a novel mechanism for the attenuation of foam cell formation. Circ Res 2003;93:717–25. doi: 10.1161/01.RES.0000097606.43659.F4. [DOI] [PubMed] [Google Scholar]
- [52].Huang P, Nedelcu D, Watanabe M, Jao C, Kim Y, Liu J, et al. Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 2016;166:1176–1187.e14. doi: 10.1016/j.cell.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xu F, Rychnovsky SD, Belani JD, Hobbs HH, Cohen JC, Rawson RB. Dual roles for cholesterol in mammalian cells. Proc Natl Acad Sci USA 2005;102:14551–6. doi: 10.1073/pnas.0503590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Roberts B, Casillas C, Alfaro AC, Jägers C, Roelink H. Patched1 and Patched2 inhibit Smoothened non-cell autonomously. Elife 2016;5. doi: 10.7554/eLife.17634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sever N, Mann RK, Xu L, Snell WJ, Hernandez-Lara CI, Porter NA, et al. Endogenous B-ring oxysterols inhibit the Hedgehog component Smoothened in a manner distinct from cyclopamine or side-chain oxysterols. Proc Natl Acad Sci USA 2016;113. doi: 10.1073/pnas.1604984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J Biol Chem 2007;282:8959–68. doi: 10.1074/jbc.M611741200. [DOI] [PubMed] [Google Scholar]
- [57].Xu L, Mirnics K, Bowman AB, Liu W, Da J, Porter NA, et al. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol Dis 2012;45:923–9. doi: 10.1016/j.nbd.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].de Medina P, Diallo K, Huc-Claustre E, Attia M, Soulès R, Silvente-Poirot S, et al. The 5,6-epoxycholesterol metabolic pathway in breast cancer: Emergence of new pharmacological targets. Br J Pharmacol 2020. doi: 10.1111/bph.15205. [DOI] [PubMed] [Google Scholar]
- [59].Long T, Hassan A, Thompson BM, McDonald JG, Wang J, Li X. Structural basis for human sterol isomerase in cholesterol biosynthesis and multidrug recognition. Nat Commun 2019; 10:2452. doi: 10.1038/s41467-019-10279-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Morisseau C Role of epoxide hydrolases in lipid metabolism. Biochimie 2013;95:91–5. doi: 10.1016/j.biochi.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xu L, Korade Z, Rosado DA, Liu W, Lamberson CR, Porter NA. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J Lipid Res 2011;52:1222–33. doi: 10.1194/jlr.M014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gaoua W, Chevy F, Roux C, Wolf C. Oxidized derivatives of 7-dehydrocholesterol induce growth retardation in cultured rat embryos: a model for antenatal growth retardation in the Smith-Lemli-Opitz syndrome. J Lipid Res 1999;40:456–63. [PubMed] [Google Scholar]
- [63].Blassberg R, Macrae JI, Briscoe J, Jacob J. Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum Mol Genet 2016;25:693–705. doi: 10.1093/hmg/ddv507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Korade Z, Xu L, Shelton R, Porter NA. Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome. J Lipid Res 2010;51:3259–69. doi: 10.1194/jlr.M009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Pfeffer BA, Xu L, Porter NA, Rao SR, Fliesler SJ. Differential cytotoxic effects of 7-dehydrocholesterol-derived oxysterols on cultured retina-derived cells: Dependence on sterol structure, cell type, and density. Exp Eye Res 2016;145:297–316. doi: 10.1016/j.exer.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Svoboda MD, Christie JM, Eroglu Y, Freeman KA, Steiner RD. Treatment of Smith-Lemli-Opitz syndrome and other sterol disorders. Am J Med Genet C Semin Med Genet 2012;160C:285–94. doi: 10.1002/ajmg.c.31347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fliesler SJ . Antioxidants: The Missing Key to Improved Therapeutic Intervention in Smith-Lemli-Opitz Syndrome? Hereditary Genet 2013;2:119. doi: 10.4172/2161-1041.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, et al. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol 1999;1:107–25. [PubMed] [Google Scholar]
- [69].Zhong Y, Gu LJ, Sun XG, Yang SH, Zhang XH. Comprehensive analysis of patched domain-containing genes reveals a unique evolutionary pattern. Genet Mol Res 2014;13:7318–31. doi: 10.4238/2014.February.13.11. [DOI] [PubMed] [Google Scholar]
- [70].Chen H, Liu Y, Li X. Structure of human Dispatched-1 provides insights into Hedgehog ligand biogenesis. Life Sci Alliance 2020;3. doi: 10.26508/lsa.202000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cannac F, Qi C, Falschlunger J, Hausmann G, Basler K, Korkhov VM. Cryo-EM structure of the Hedgehog release protein Dispatched. Sci Adv 2020;6:eaay7928. doi: 10.1126/sciadv.aay7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bazan JF, de Sauvage FJ. Structural ties between cholesterol transport and morphogen signaling. Cell 2009;138:1055–6. doi: 10.1016/j.cell.2009.09.006. [DOI] [PubMed] [Google Scholar]
- [73].Gong X, Qian H, Zhou X, Wu J, Wan T, Cao P, et al. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 2016;165:1467–78. doi: 10.1016/j.cell.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G. Structure of human Niemann-Pick C1 protein. Proc Natl Acad Sci USA 2016. doi: 10.1073/pnas.1607795113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Qian H, Wu X, Du X, Yao X, Zhao X, Lee J, et al. Structural Basis of Low-pH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell 2020;182:98–111.e18. doi: 10.1016/j.cell.2020.05.020. [DOI] [PubMed] [Google Scholar]
- [76].Long T, Qi X, Hassan A, Liang Q, De Brabander JK, Li X. Structural basis for itraconazole-mediated NPC1 inhibition. Nat Commun 2020;11:152. doi: 10.1038/s41467-019-13917-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Winkler MBL, Kidmose RT, Szomek M, Thaysen K, Rawson S, Muench SP, et al. Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 2019;179:485–497.e18. doi: 10.1016/j.cell.2019.08.038. [DOI] [PubMed] [Google Scholar]
- [78].Motamed M, Zhang Y, Wang ML, Seemann J, Kwon HJ, Goldstein JL, et al. Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J Biol Chem 2011;286:18002–12. doi: 10.1074/jbc.M111.238311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Yan R, Cao P, Song W, Qian H, Du X, Coates HW, et al. A structure of human Scap bound to Insig-2 suggests how their interaction is regulated by sterols. Science 2021. doi: 10.1126/science.abb2224. [DOI] [PubMed] [Google Scholar]
- [80].Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, Rawson RB. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 2002;296:879–83. doi: 10.n26/science.1071124. [DOI] [PubMed] [Google Scholar]
- [81].Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 2003;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- [82].Song B-L, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab 2005;1:179–89. doi: 10.1016/j.cmet.2005.01.001. [DOI] [PubMed] [Google Scholar]
- [83].Fan J, Akabane H, Zheng X, Zhou X, Zhang L, Liu Q, et al. Male germ cell-specific expression of a novel Patched-domain containing gene Ptchd3. Biochem Biophys Res Commun 2007;363:757–61. doi: 10.1016/j.bbrc.2007.09.047. [DOI] [PubMed] [Google Scholar]
- [84].González Morales SR, Liu C, Blankenship H, Zhu G-Z. Mouse Ptchd3 is a non-essential gene. Gene X 2020;5:100032. doi: 10.1016/j.gene.2020.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ghahramani Seno MM, Kwan BYM, Lee-Ng KKM, Moessner R, Lionel AC, Marshall CR, et al. Human PTCHD3 nulls: rare copy number and sequence variants suggest a non-essential gene. BMC Med Genet 2011;12:45. doi: 10.1186/1471-2350-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zugasti O, Rajan J, Kuwabara PE. The function and expansion of the Patched- and Hedgehog-related homologs in C. elegans. Genome Res 2005;15:1402–10. doi: 10.1101/gr.3935405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Qi C, Di Minin G, Vercellino I, Wutz A, Korkhov VM. Structural basis of sterol recognition by human hedgehog receptor PTCH1. Sci Adv 2019;5:eaaw6490. doi: 10.1126/sciadv.aaw6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Qi X, Schmiege P, Coutavas E, Wang J, Li X. Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature 2018;560:128–32. doi: 10.1038/s41586-018-0308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Qi X, Schmiege P, Coutavas E, Li X. Two Patched molecules engage distinct sites on Hedgehog yielding a signaling-competent complex. Science 2018;362. doi: 10.1126/science.aas8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Qian H, Cao P, Hu M, Gao S, Yan N, Gong X. Inhibition of tetrameric Patched1 by Sonic Hedgehog through an asymmetric paradigm. Nat Commun 2019;10:2320. doi: 10.1038/s41467-019-10234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gong X, Qian H, Cao P, Zhao X, Zhou Q, Lei J, et al. Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 2018;361. doi: 10.1126/science.aas8935. [DOI] [PubMed] [Google Scholar]
- [92].Zhang Y, Lu W-J, Bulkley DP, Liang J, Ralko A, Han S, et al. Hedgehog pathway activation through nanobody-mediated conformational blockade of the Patched sterol conduit. Proc Natl Acad Sci USA 2020;117:28838–46. doi: 10.1073/pnas.2011560117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rudolf AF, Kinnebrew M, Kowatsch C, Ansell TB, El Omari K, Bishop B, et al. The morphogen Sonic hedgehog inhibits its receptor Patched by a pincer grasp mechanism. Nat Chem Biol 2019;15:975–82. doi: 10.1038/s41589-019-0370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem 1998;273:14037–45. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- [95].Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, et al. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 1995;81:445–55. doi: 10.1016/0092-8674(95)90397-6. [DOI] [PubMed] [Google Scholar]
- [96].Taylor FR, Wen D, Garber EA, Carmillo AN, Baker DP, Arduini RM, et al. Enhanced potency of human Sonic hedgehog by hydrophobic modification. Biochemistry 2001;40:4359–71. doi: 10.1021/bi002487u. [DOI] [PubMed] [Google Scholar]
- [97].Petrov K, Wierbowski BM, Liu J, Salic A. Distinct cation gradients power cholesterol transport at different key points in the hedgehog signaling pathway. Dev Cell 2020;55:314–327.e7. doi: 10.1016/j.devcel.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hausmann G, von Mering C, Basler K. The hedgehog signaling pathway: where did it come from? PLoS Biol 2009;7:e1000146. doi: 10.1371/joumal.pbio.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Byrne EFX, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 2016;535:517–22. doi: 10.1038/naturel8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].De Rivoyre M, Ruel L, Varjosalo M, Loubat A, Bidet M, Thérond P, et al. Human receptors patched and smoothened partially transduce hedgehog signal when expressed in Drosophila cells. J Biol Chem 2006;281:28584–95. doi: 10.1074/jbc.M512986200. [DOI] [PubMed] [Google Scholar]
- [101].Hime GR, Lada H, Fietz MJ, Gillies S, Passmore A, Wicking C, et al. Functional analysis in Drosophila indicates that the NBCCS/PTCH1 mutation G509V results in activation of smoothened through a dominant-negative mechanism. Dev Dyn 2004;229:780–90. doi: 10.1002/dvdy.10499. [DOI] [PubMed] [Google Scholar]
- [102].Johnson RL, Milenkovic L, Scott MP. In vivo functions of the patched protein: requirement of the C terminus for target gene inactivation but not Hedgehog sequestration. Mol Cell 2000;6:467–78. doi: 10.1016/sl097-2765(00)00045-9. [DOI] [PubMed] [Google Scholar]
- [103].Makino S, Masuya H, Ishijima J, Yada Y, Shiroishi T. A spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of patched (ptc). Dev Biol 2001;239:95–106. doi: 10.1006/dbio.2001.0419. [DOI] [PubMed] [Google Scholar]
- [104].Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- [105].Ayers KL, Rodriguez R, Gallet A, Ruel L, Thérond P. Tow (Target of Wingless), a novel repressor of the Hedgehog pathway in Drosophila. Dev Biol 2009;329:280–93. doi: 10.1016/j.ydbio.2009.02.037. [DOI] [PubMed] [Google Scholar]
- [106].Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- [107].Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- [108].Smith MJ, Beetz C, Williams SG, Bhaskar SS, O’Sullivan J, Anderson B, et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 2014;32:4155–61. doi: 10.1200/JCO.2014.58.2569. [DOI] [PubMed] [Google Scholar]
- [109].Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet 2016;48:398–406. doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- [110].Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998;391:90–2. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- [111].Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- [112].Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol 2018;20:iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017;31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom L-G, Toftgård R, Undén AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol 2006;208:17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- [115].Zibat A, Missiaglia E, Rosenberger A, Pritchard-Jones K, Shipley J, Hahn H, et al. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010;29:6323–30. doi: 10.1038/onc.2010.368. [DOI] [PubMed] [Google Scholar]
- [116].Pressey JG, Anderson JR, Crossman DK, Lynch JC, Barr FG. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer 2011;57:930–8. doi: 10.1002/pbc.23174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012;366:2171–9. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol 2015;33:2646–54. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Raleigh DR, Algazi A, Arron ST, Neuhaus IM, Yom SS. Induction Hedgehog pathway inhibition followed by combined-modality radiotherapy for basal cell carcinoma. Br J Dermatol 2015;173:544–6. doi: 10.1111/bjd.13748. [DOI] [PubMed] [Google Scholar]
- [120].Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015;27:342–53. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Yao CD, Haensel D, Gaddam S, Patel T, Atwood SX, Sarin KY, et al. AP-1 and TGFß cooperativity drives non-canonical Hedgehog signaling in resistant basal cell carcinoma. Nat Commun 2020; 11:5079. doi: 10.1038/s41467-020-18762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- [126].Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017;8:69295–302. doi: 10.18632/oncotarget.20619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Gordon RE, Zhang L, Peri S, Kuo Y-M, Du F, Egleston BL, et al. Statins Synergize with Hedgehog Pathway Inhibitors for Treatment of Medulloblastoma. Clin Cancer Res 2018;24:1375–88. doi: 10.1158/1078-0432.CCR-17-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Raleigh DR, Choksi PK, Krup AL, Mayer W, Santos N, Reiter JF. Hedgehog signaling drives medulloblastoma growth via CDK6. J Clin Invest 2018;128:120–4. doi: 10.1172/JCI92710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Beck KR, Bächler M, Vuorinen A, Wagner S, Akram M, Griesser U, et al. Inhibition of 11β-hydroxysteroid dehydrogenase 2 by the fungicides itraconazole and posaconazole. Biochem Pharmacol 2017;130:93–103. doi: 10.1016/j.bcp.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Trinh MN, Lu F, Li X, Das A, Liang Q, De Brabander JK, et al. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc Natl Acad Sci USA 2017;114:89–94. doi: 10.1073/pnas.1619571114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Strating JRPM, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep 2015;10:600–15. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Chen B, Trang V, Lee A, Williams NS, Wilson AN, Epstein EH, et al. Posaconazole, a Second-Generation Triazole Antifungal Drug, Inhibits the Hedgehog Signaling Pathway and Progression of Basal Cell Carcinoma. Mol Cancer Ther 2016;15:866–76. doi: 10.1158/1535-7163.MCT-15-0729-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Daggubati V, Hochstetler J, Bommireddy A, Choudhury A, Krup AL, Choksi PK, et al. Smoothened-activating lipids drive resistance to CDK4/6 inhibition in Hedgehog-associated medulloblastoma cells and preclinical models. J Clin Invest 2021. doi: 10.1172/JCI141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Bolla JR, Agasid MT, Mehmood S, Robinson CV. Membrane Protein-Lipid Interactions Probed Using Mass Spectrometry. Annu Rev Biochem 2019;88:85–111. doi: 10.1146/annurev-biochem-013118-111508. [DOI] [PubMed] [Google Scholar]