

Abstract
Targeting cereblon (CRBN) is currently one of the most frequently reported proteolysis-targeting chimera (PROTAC) approaches, owing to favorable drug-like properties of CRBN ligands, immunomodulatory imide drugs (IMiDs). However, IMiDs are known to be inherently unstable, readily undergoing hydrolysis in body fluids. Here we show that IMiDs and IMiD-based PROTACs rapidly hydrolyze in commonly utilized cell media, which significantly affects their cell efficacy. We designed novel CRBN binders, phenyl glutarimide (PG) analogues, and showed that they retained affinity for CRBN with high ligand efficiency (LE >0.48) and displayed improved chemical stability. Our efforts led to the discovery of PG PROTAC 4c (SJ995973), a uniquely potent degrader of bromodomain and extra-terminal (BET) proteins that inhibited the viability of human acute myeloid leukemia MV4–11 cells at low picomolar concentrations (IC50 = 3 pM; BRD4 DC50 = 0.87 nM). These findings strongly support the utility of PG derivatives in the design of CRBN-directed PROTACs.
Keywords: Phenyl Glutarimide, PG, PROTAC, Cereblon, IMiD
Graphical abstract

IMiD-based PROTACs rapidly hydrolyze in commonly utilized cell media, which significantly affects their cell efficacy. We designed novel BET PROTACs based on phenyl glutarimide (PG) and showed that they retained affinity for CRBN and displayed improved chemical stability. To demonstrate the utility of PG-based PROTACs we developed SJ995973 (4c), a uniquely potent degrader of BET proteins.
1. INTRODUCTION
Deconvolution of thalidomide’s mechanism of action[1] over half a century after its first and infamous launch on the market has led to the recent emergence of a novel chemical biology concept: Targeted Protein Degradation (TPD).[2] TPD is widely seen as a game-changing paradigm that shows promise of having a profound and lasting effect on basic chemical biology research as well as drug discovery.[3] There are two distinct approaches to design small molecules capable of inducing TPD, namely Molecular Glues (MGs)[4],[5] and Proteolysis-Targeting Chimeras (PROTACs).[6],[7] Thalidomide and structurally related immunomodulatory imide drugs (IMiDs) pomalidomide and lenalidomide are the “original” molecular glues, which retrospectively provided the mechanistic and clinical validation for this approach.[8] PROTACs are molecules rationally designed to bring the desired target protein in close proximity to an E3 ubiquitin ligase to induce protein ubiquitination, followed by its proteasomal degradation.[6] A typical PROTAC consists of three distinct structural motifs: one binding to the protein of interest, another binding to an E3 ligase complex, and a linker tethering them together.[9] Early proof-of-concept PROTACs reported by Crews et. al. was based on a phosphopeptide inhibitor of NF-κBα (IκBα) designed to recruit the protein of interest to the E3 ligase complex SCF-βTRCP.[10] The breakthrough for the field came in 2015 when the Crews[11] and Bradner[12] laboratories independently reported thalidomide-based PROTACs ARV825 and dBET1 targeting BRD4, a member of a small family of Bromodomain and Extra Terminal (BET) proteins.[13] Both these PROTACs showed CRBN-dependent degradation of the BRD4 protein in cancer cells, and importantly, more potent inhibition of cell proliferation and apoptosis induction than their corresponding BRD4 inhibitors. For example, dBET1 displayed a superior in vitro and in vivo inhibitory effect on acute myeloid leukemia (AML) MV4–11 cells proliferation compared to the parent BRD4 inhibitor JQ1.[12] BRD4 protein degradation was observed in MV4–11 cells after only a 2-hour incubation with dBET1 at 100 nM. Interestingly, a partial recovery in BRD4 protein abundance was observed after 24 hours and attributed to compound instability.[12] It is established that thalidomide is inherently unstable and undergoes racemization and hydrolysis in bodily fluids, the rate of which accelerates with increasing pH.[14] Thus, at pH 7, 7.4 and 8, thalidomide has half-lives to hydrolysis of 11, 5 and 1.25 hours, respectively.[15] Thalidomide contains four carbonyl moieties that display different pH-dependent susceptibilities to hydrolysis. The main route of thalidomide hydrolysis at pH 6–8 is cleavage of the phthalimido ring producing α-(carboxybenzamido) glutarimide.[15] This hydrolysis product with an intact glutarimide moiety is also one of the major thalidomide metabolites in human plasma.[14],[15]
Interestingly, we have been able to find only a limited number of reports in the peer-reviewed literature describing IMiD modifications and reporting their structure activity relationship (SAR). Crews et. al. described thalidomide analogues with various substitutions around the phthalimide phenyl ring, which showed comparable CRBN affinity to pomalidomide, but only a few were able to recruit and degrade IKZF1.[16] In 2015 Hagner and colleagues at Celgene reported CC-122 (avadomide),[17] an IMiD derivative with the phthalimide replaced with a quinazolinone moiety, which is currently in Phase 2 clinical trials in patients with relapsed/refractory B-cell malignancies (NCT03310619). Celgene and more recently our group have also reported IMiD derivatives that potently and selectively degrade GSPT1 protein.[5],[18],[19] CC-90009 is the first and currently the only GSPT1 degrader that entered the clinic and has recently successfully completed Phase 1 clinical trials in patients with relapsed or refractory AML (NCT02848001).[20] In terms of PROTAC design, the only alternative to commonly used lenalidomide and pomalidomide scaffolds was reported by Kim et. al. who developed JQ1-based BET PROTACs using TD-106, an aza-analogue of CC-122, as a CRBN-directing warhead.[21] However, given the recent interest in the field of targeted protein degradation and the utility of E3 ligase ligands for PROTAC applications, a broad range of patents have been reported describing alternative CRBN ligands. Although a review of the patent literature is outside of the scope of this article, it is relevant to cite some of the patents from C4 Therapeutics published during the course of our studies. In their work, a multitude of phthalimide and glutarimide replacements, including heteroaromatic and aromatic alternatives to phthalimide were described as novel CRBN binders.[22],[23] However, as common for patent literature, broader properties of these analogues such as chemical stability, physicochemical properties, degradation potential, cellular activity and relative comparison to the classical IMiDs were not disclosed.
Here, we postulated that in addition to its intrinsic hydrolytic instability, the electron-withdrawing phthalimide moiety also activates the glutarimide ring toward hydrolysis. Thus, we hypothesized that replacing the phthalimide with an aromatic group will generate analogues with improved stability towards hydrolysis while not affecting their CRBN binding affinity. This study details the design, synthesis, physicochemical, pharmacological, and protein degradation properties of phenyl glutarimide (PG)-based BET PROTACs, and compares them to corresponding IMiD analogues.
2. RESULTS AND DISCUSSION
2.1. Novel Cereblon Binders and PROTAC Design
Our goal was to discover novel CRBN binders with improved chemical stability and ligand efficiency and use them to design novel BET PROTACs with improved degradation efficiency. We first investigated the previously reported co-crystal structure of thalidomide in complex with CRBN-DDB1 obtained from the Protein Data Bank (PDB code: 4CI1).[24] This structure revealed that the phthalimide portion of the molecule is not buried within the binding pocket, but is exposed on the protein surface, stacked against the aliphatic face of Pro354, with one of its carbonyl groups engaged in a water-mediated hydrogen bond to His359 (Figure 1A). To investigate how a PG analogue may fit in this pocket, the bound thalidomide (PDB: 4CI1) was modified manually to structure of 2a (Figure 1B) using Builder tool in MOE,[25] which was then minimized while keeping the protein and water molecules fixed. The generated PG structure (Figure 1A) showed the phenyl bound deeply in the pocket in a position suggestive of a more productive interaction with Pro354, which may compensate for loss of the phthalimide water-mediated hydrogen bond to His359 in thalidomide.
Figure 1.

Structures of cereblon binders and related PROTACs. (A) Surface representation of G. gallus cereblon (gray: carbon; blue: nitrogen) in complex with thalidomide 1a (green sticks), PDB: 4CI1, model structure of phenyl glutarimide 2a (magenta sticks), and water molecule (red sphere). (B) Chemical structures of immunomodulatory drugs (IMiDs), phenyl glutarimide (PG), their analogues and related PROTACs. (C) Small molecule crystal structure of the active (R)-2b enantiomer (yellow) superimposed over the (S)-2b enantiomer (faded ball-and-stick model).
As per this model, the phenyl analogue would not only retain CRBN affinity but also display improved chemical stability due to replacing the hydrolysis-prone phthalimide moiety and reducing activation of the conserved glutarimide ring. To test this hypothesis, we synthesized PG 2a, and its analogues containing methoxy and amino groups that are commonly used as PROTAC linker attachment points 2b and 2c, respectively. We also synthesized a range of JQ1-based PG and IMiD-derived PROTACs, including CLICK-chemistry derivatives 3a-d and analogues of dBET1 (4a-d), as outlined in Figure 1B. Modeling of PG complexed with CRBN (Figure 1A) suggested that positions 3- and 4- on the PG phenyl group provide vectors toward the solvent, making them suitable sites to anchor a linker to JQ1 for designing BET-directed PG-PROTACs. Synthetic schemes and methods are provided in the Supporting Information.
2.2. Binding Affinity, Cell Media Stability and Physicochemical Properties
To assess the binding of PG-based analogues and PROTACs to CRBN, we used a fluorescence polarization assay using a Cy5-conjugated lenalidomide as a fluorescent probe. Consistent with previous reports,[26] in this assay lenalidomide 1b and pomalidomide 1c showed slightly higher affinities for CRBN than thalidomide 1a (Table 1, Supp. Figure 1A). The PG analogues displayed binding affinities in a range similar to IMiDs. The PG itself (2a) and its 4-methoxy analogue 2b showed slightly lower affinity compared to thalidomide (IC50 values of 2.19 μM, 3.15 μM and 1.28 μM respectively). Interestingly, similar to amino group containing IMiDs 1b and 1c, 4-amino PG analogue 2c displayed a higher CRBN affinity (IC50 = 0.123 μM) than its parent compound 2a (IC50 = 2.19 μM). The improvement in affinity was rationalized by molecular dynamics studies of 2c bound to CRBN, which showed a productive NH···π hydrogen-bonded arrangement between 2c aniline and His353 (Supp. Figure 2A). Given their similar CRBN affinities but smaller sizes when compared to IMiDs, PG analogues had higher ligand efficiency (LE= 0.48–0.64) than IMiDs (LE= 0.38–0.45; Table 1).[27] Other physicochemical properties of PG analogues, such as aqueous solubility and cell permeability, were moderate to high, similar to those of classical IMiDs (Table 1). Metabolic stability of PG derivatives measured in mouse and human liver microsomes was generally high (t1/2 >15 h), except for MeO-PG 2b that similar to the MeO-IMiD analogue 1d displayed shorter half-life in mouse liver microsomes, with t1/2 >1.99 h and 3.79 h, respectively (Table 1).
Table 1.
CRBN binders: Affinity, Stability and Physicochemical Properties
| Cmpd | CRBN IC50 (μM) | LE | t1/2 (h) | Sol. (μM) | CaCo2 Papp (nm/s) | ||||
|---|---|---|---|---|---|---|---|---|---|
| MV4–11a | HD-MB03a | PBS | mLM | hLM | |||||
| 1a (thal) | 1.282 ±0.883 | 0.43 | 3.3 ±0.4 | 1.8 ± 0.1 | 5.4 ±0.2 | 3.99 ±0.01 | 4.55 ±0.61 | 61.5 ±1.5 | 349.1 ±14.6 |
| 1b (lena) | 0.699 ±0.428 | 0.45 | 11. 7 ±6.2 | 7.6 ±0.6 | 33.6 ± 2.5 | >24 | >24 | 110.8 ±6.2 | 40.0 ±10.2 |
| 1c (poma) | 0.400 ±0.319 | 0.45 | 12.2 ±0.5 | 5.4 ±0.7 | 32.5 ±1.0 | 7.8 ±0.69 | >24 | 84.6 ±2.3 | 360.2 ±13.9 |
| 1d | 1.889 ±1.288 | 0.38 | 5.9 ±0.2 | 2.4 ±0.1 | 12.2 ±0.6 | 3.79 ±0.31 | 8.08 ±1.78 | 55.7 ±1.6 | 162.0 ±79.7 |
| 1e | > 25 | NA | <0.1 | <0.1 | 0.3 ±0.03 | 0.19 ±0.03 | 0.18 ±0.04 | 100 ± 6.8 | 23.4 ±1.7 |
| 2a (PG) | 2.191 ±1.608 | 0.57 | >24 | 20.8 ±0.8 | >48 | >24 | >24 | 76.2 ± 0.1 | 215.5 ±65.4 |
| 2b | 3.158 ±0.248 | 0.48 | >24 | 19.7 ±0.1 | >48 | 1.99 ±0.23 | >24 | 89.0 ± 1.8 | 818.0 ±251.4 |
| 2c | 0.123 ±0.066 | 0.64 | >24 | 16.4 ±0.3 | >48 | 23.19 ±1.63 | 15.66 ±4.78 | 148.5 ± 4.7 | 157.2 ±9.4 |
Half-life in cell media; Data represent the average of 2 ≥ independent determinations; ± Deviation represents standard error of the mean; LE: Ligand efficiency;15 Sol: Aqueous solubility. PBS: Phosphate-buffered saline; mLM: mouse liver microsomes; hLM: human liver microsomes; CaCo2 Papp: cell permeability measured in human colon adenocarcinoma cell line.
The PG analogues were synthesized and tested as racemates, and we were therefore interested to determine if the (R)-configuration was preferred for CRBN binding, as our modeling studies suggested (Figure 1A). For that, optically pure enantiomers of 2b were obtained by chiral supercritical fluid chromatography (SFC) separation (enantiomeric excess (ee) >99%), and their absolute configuration was determined by small molecule X-ray crystallography (Figure 1C, and Supp. Figure 3). The (R)-2b proved to be the preferred enantiomer with a CRBN IC50 of 1.41 μM, whereas (S)-2b was inactive (IC50 >25 μM). Thalidomide and its clinically used analogues can rapidly undergo racemization at the chiral carbon of the 3-aminoglutarimide moiety in bodily fluids and water.[28] Hence, IMiDs are produced and sold as racemates. Our chiral stability studies in cell media showed that PG analogues also undergo rapid chiral inversion. We observed a 10% inversion in MV4–11 cell media and 21% inversion in HD-MB03[29] cell media after 24 hours of incubation in (R)-2b at 37°C, and 13% and 24%, respectively, for the (S)-2b enantiomer (Supp. Figure 4). Chiral instability was observed even in DMSO stock solutions stored at room temperature, with approximately 7% inversion over two weeks. Therefore, all compounds in this study were synthesized and tested as racemates.
Next, we measured the chemical stability of CRBN binders in cell culture media. We focused particularly on media for AML cell line MV4–11, since this was one of the most commonly reported cell lines for evaluation of PROTAC anti-proliferative effects.[12],[30],[31],[32] In this assay, compounds were incubated at 37°C in cell media (pH = 7.4), and the percentage remaining was measured by liquid chromatography-mass spectrometry (LCMS) at five time points over 24 hours. Under these conditions, thalidomide 1a degraded rapidly, showing t1/2 of only 3.3 hours (Table 1). After 6 hours, only 20 % of the thalidomide remained, and at 24 hours it was below limits of detection (Supp. Figure 5A). Lenalidomide 1b contains a hydrolytically more stable isoindolinone moiety, and thereby displayed slightly better stability, with t1/2 of 11.7 hours. Pomalidomide 1c was most stable of the three IMiDs, with t1/2 of 12.2 hours. Its higher stability to hydrolysis can be rationalized by the electron donating effect of the amino group that reduces nucleophilic reactivity of the phthalimide moiety. In contrast, PG derivatives 2a and 2b displayed very little degradation, with half-lives over 24 hours (Table 1). As we originally hypothesized, the phthalimide group in IMiDs is not only a hydrolytic liability itself, but also activates the glutarimide ring toward hydrolysis. Consequently, its replacement with an aromatic moiety in PG analogues resulted in enhanced chemical stability. A similar trend in culture media was seen for the medulloblastoma cell line HD-MB03 (Supp. Figure 5B).[29] However, possibly due to slightly higher basicity of this cell media (pH = 7.7), the rate of hydrolysis was higher. Thus, after a 24-hour incubation, levels of all tested IMiDs were below detection. Stabilities of PG and its analogues were also affected, but they still displayed respectable half-lives of approximately 20 hours. Similarly, PG analogues displayed considerably greater stability in phosphate-buffered saline (PBS) compared to IMiDs (Table 1).
Interestingly, all PROTACs in this study showed higher CRBN binding affinities compared to their parent CRBN binders (Table 2 and Supp. Figure 1B), possibly because the linker formed additional interactions at the protein surface. Indeed, a PG analogue 2d with a shortened linker attachment displayed a 6-fold higher affinity compared to the parent MeO-PG 2b, with CRBN IC50 values of 0.540 μM and 3.158 μM, respectively. Molecular modeling studies pointed to a hydrogen bond between the ligand amide group and His353, in a similar fashion to 2c, that may account for the improved affinity (Supp. Figure 2B). A similar increase in the CRBN affinity of dBET1 compared to thalidomide has been recently reported by others.[33] We found this “linker effect” even more pronounced in the context of BRD2 and BRD4 binding. For example, CLICK chemistry based PROTACs 3a-d showed two orders of magnitude higher affinity compared to JQ1 (IC50 = 4.76 ± 1.56 nM) in the BRD2 (BD2) TR-FRET assay (Supp. Figure 6A). Given this observation, we generated a crystal structure of PROTAC 3b bound to the BD2 domain of the BRD2 protein (PDB: 6WWB, Supp. Figure 6B). Compared to the previously reported JQ1-BD2 BRD2 complex (PDB: 3ONI), the structure of PROTAC 3b showed several additional interactions with the protein compared to JQ1, which might rationalize its higher BRD affinity (Supp. Figure 6C). Taken together, these data suggest that in addition to connecting two protein binding moieties, PROTAC linkers can also productively engage in binary complex formation.
Table 2.
PROTACs: Affinity, Stability and Physicochemical Properties
| Cmpd | CRBNa IC50 (μM) | t1/2 (h) | Sol. (μM)a | Caco-2 Papp (nm/s)a | ||
|---|---|---|---|---|---|---|
| MV4–11c | HD-MB03c | PBS | ||||
| 3a | 0.017 ±0.007 | 3.8 ±0.5 | 1.9 ± 0.3 | 10.9 ±0.7 | 50.8 ±1.3 | 17.0 ±23.0 |
| 3b | 0.004 ±0.001 | 16.2 ±1.5 | 5.2 ± 0.1 | >48 | 32.3 ±1.4 | 2.2 ±3.3 |
| 3c | 0.036 ±0.016 | >24 | 20.0 ±0.6 | >48 | 60.4 ±1.7 | 21.4 ±33.2 |
| 3d | 0.092 ±0.026 | >24 | >24 | >48 | 56.8 ±1.8 | 3.2 ±1.0 |
| 4a (dBET1) | 0.034 ±0.016 | 4.2 ±0.8 | 2.1 ±0.3 | 12.3 ±0.6 | 53.5 ±0.3 | 11.6 ±4.7 |
| 4b | 0.058 ±0.012 | >24 | 15.4 ±0.0 | >48 | 78.5 ±6.6 | 16.6 ±2.3 |
| 4c (SJ995973) | 0.017 ±0.007 | >24 | 16.8 ±1.2 | >48 | 78.3 ±1.2 | 14.3 ±1.8 |
| 4d | 0.137 ±0.043 | >24 | 14.4 ±1.1 | >48 | 65.3 ±2.1 | 1.0 ±0.9 |
| 4e | 0.149 ±0.031 | >24 | 13.8 ±0.7 | >48 | 65.3 ±1.3 | 9.7 ±0.2 |
Data represent the average of 2 ≥ independent determinations. ± Deviation represents standard error of the mean;
CRBN Fluorescence Polarization assay;
Time-resolved fluorescence energy transfer (TR-FRET) assay;
Half-life in cell media;
PBS: Phosphate-buffered saline; Sol: Aqueous solubility; CaCo2 Papp: cell permeability measured in human colon adenocarcinoma cell line.
Having demonstrated that PG-PROTACs retained CRBN and BET protein affinities, we investigated their chemical stabilities. The PG-based PROTACs showed considerably higher stability in both MV4–11 and HD-MB03 cell culture media than the corresponding IMiD-based PROTACs (Table 2 and Supp. Figure 5). Particularly striking was the difference between 4a (dBET1) and its PG-analogues 4b and 4c, with half-lives in MV4–11 media of 4.2 hours and >24 hours, respectively. A similar difference was observed between IMiD- and PG-based CLICK PROTACs such as 3a and 3c, with t1/2 of 3.7 hours and >24 hours, respectively. The same stability trend occurred in PBS, where PG-PROTACs displayed approximately an order of magnitude longer half-lives than IMiD-PROTACs.
Although all PROTACs in this study showed moderate to high aqueous solubilities, similar to JQ1 (52.1 μM), their permeabilities in Caco-2 cells were predictably low (Table 2). Interestingly, oxygen-linked PROTACs in both IMiD and PG series, 3a and 3c, showed up to 10-fold higher permeability than their nitrogen-linked analogues, which was likely the consequence of an additional hydrogen bond donor in 3b and 3d. However, it was reassuring that the previously reported and well-studied IMiD PROTAC 4a showed very similar Caco-2 permeability to its direct PG-analogues 4b and 4c (11.6 nm/s, 16.6 nm/s and 14.3 nm/s, respectively). These data support the hypothesis that replacing IMiDs in CRBN-directed PROTACs with PGs would yield improved chemical stability without compromising binding affinity or physicochemical properties.
We also evaluated metabolic stability of 4a and 4c in mouse liver microsomes. Both PROTACs showed rapid clearance, 4a t1/2 = 0.13 ± 0.01 h and 4c t1/2 = 0.05 ± 0.01 h, which is probably not surprising considering the poor stability of the parent inhibitor itself (JQ1 t1/2 = 0.8 ± 0.05 h). It is worth pointing out that plasma stabilities of these two PROTACs were aligned with their chemical stabilities: 4a t1/2 = 3.27 ± 0.25 h and 4b t1/2 = 22.89 ± 1.27 h.
2.4. Cell Efficacy and Protein Degradation of PROTACs
We next evaluated the antiproliferative activity of PROTACs in MV4–11 and HD-MB03 cell lines (Table 3 and Supp. Figure 7). Cells were treated with compounds over 3 days, and their viability was assessed using the CellTiter-Glo assay kit (Promega). The IC50 values of compounds were determined by the proprietary software Robust Investigation of Screening Experiments (RISE), developed in house on the Pipeline Pilot platform (Biovia, v. 17.2.0). Consistent with previous reports, 4a exhibited high MV4–11 cytotoxicity with IC50 of 6.1 nM.[12] All PG PROTACs displayed an even greater efficacy in this cell line. For example, 3b and 3c showed cytotoxicity similar to that of IMiD analogues 3a and 3b, and over a 30-fold improvement in IC50 compared to 4a. Particularly striking was the several orders of magnitude greater efficacy of PG-PROTACs 4b and 4c compared to their direct IMiD analogue 4a (Figure 1B). In fact, the MV4–11 IC50 value of 3 pM 4c (SJ995973) is one of the most potent BET PROTACs reported in literature (Figure 2A).[34] The lower potency of PG regioisomers 4d and 4e suggest that the C3-linker attachment in this case is suboptimal compared to the C4- position in 4b and 4c. Although HD-MB03 cells were less sensitive to BET PROTACs (Table 3), the rank order was very similar to that in the MV4–11 cell line. PG PROTAC 4c was still by far the most potent compound, displaying >600-fold greater cytotoxicity compared to 4a (HD-MB03 IC50 = 1.83 nM and 1,237.80 nM, respectively), as shown in Figure 2B.
Table 3.
PROTACs: Viabilities of cancer cells
| Compound | CRBN warhead | MV4–11 IC50 (nM)a | HD-MB03 IC50 (nM)a |
|---|---|---|---|
| 2a | PG | >7,000 | >7,000 |
| 3a | IMiD | 0.222 ± 0.075 | 710.45 ± 90.16 |
| 3b | IMiD | 0.208 ± 0.031 | 105.00 ± 10.40 |
| 3c | PG | 0.199 ± 0.002 | 334.55 ± 72.76 |
| 3d | PG | 0.133 ± 0.058 | 102.78 ± 59.83 |
| 4a (dBET1) | IMiD | 6.100 ± 1.559 | 1,237.80 ± 261.25 |
| 4b | PG | 0.060 ± 0.021 | 13.22 ± 3.42 |
| 4c (SJ995973) | PG | 0.003 ± 0.001 | 1.83 ± 0.35 |
| 4d | PG | 20.076 ± 7.252 | 379.65 ± 61.02 |
| 4e | PG | 1.147 ± 0.413 | 64.85 ± 1.54 |
| 4f | Inact-PG | 1,282.160 ± 456.725 | >10 μM |
| JQ1 | - | 82.960 ± 10.315 | 265.20 ± 62.59 |
Antiproliferative effects of compounds assessed after 3 days of incubation, using the CellTiter-Glo assay kit (Promega). Data represent the average of ≥ 2 independent determinations. ± Deviation represents standard error of the mean.
Figure 2.

Effect of treatment on cell viability with increasing concentrations of 4a (●) and 4c (■) over 3 days in (A) MV4–11 cells, and (B) HD-MB03 cells. Data represent the average of three independent determinations. Error bars represent standard error of the mean.
Lower sensitivity to BET protein degradation is potentially a consequence of MYC gene amplification, which characterizes this patient derived Group 3 medulloblastoma cell line.[29] Further, our chemical stability studies suggested that rapid degradation in this cell media may also, at least in part, affect PROTACs HD-MB03 activity. Indeed, extending the incubation time from 3 days to 7 days rendered 4a (t1/2 = 2.1 hours in HD-MB03 media) practically inactive in the HD-MB03 cell line (IC50 >4 μM), whereas under the same conditions the more stable PG PROTAC 4c (t1/2 = 17 h) retained high efficacy (IC50 = 58.4 nM; Supp. Table 4).
To demonstrate the CRBN dependency of 4c, we synthesized an inactive N- methylated analogue 4f as a negative control (CRBN IC50 >50 μM). As expected, compound 4f showed dramatically reduced activity, with IC50 values of 1,282 nM against MV4–11 and >10 μM against HDMB03 cells, confirming the CRBN dependence of the cellular efficacy of 4c (Table 3).
To induce TPD, PROTACs must bring E3 ligase in close proximity to the intended target. To observe this process experimentally, we employed a proximity-based AlphaScreen assay that could generate high signal amplification in response to PROTAC-induced ternary complex formation between GST-BRD4 and His-CRBN-DDB1 within an energy transfer distance of up to 200 nm. In this assay, PG-PROTACs yielded the characteristic bell-shaped profile over a range of concentrations, very similar to 4a (Figure 3A). Luminescence emission increased at lower concentrations of PROTAC as the ratio of ternary complex was increasing, and decreased at higher concentrations due to saturation of CRBN and BRD4 binding sites, the so-called “hook effect”.[35] As expected, JQ1 itself showed no signal in this assay. Consistent with AlphaScreen data, compounds 3a, 3b and 4a-c induced BRD2 and BRD4 protein degradation after a 4-hour incubation at 0.1 μM, resulting in suppression of the MYC protein expression (Figure 3B).
Figure 3.

PROTACs ternary complex formation and protein degradation in MV4–11. (A) AlphaScreen assay data (n = 3): Compound-induced ternary complex formation of His-CRBN-DDB1 and GST-BRD4 (49–170). (B) Immunoblot for BRD4, BRD2 and MYC proteins after 4-hour pretreatment of MV4–11 cells with DMSO, compounds 3a, 3b and 4a-c at a concentration of 100 nM. GAPDH was used as loading control. Dotted lines indicate division between different regions of the same gel (unmodified blots are shown in the Supporting Information). (C) Immunoblot for BRD4 and BRD2 in MV4–11 cells after treatment with 100 nM of 4a and 4c over the indicated time course (0–48 hours). (D) Graphic representation of the BRD4 protein level changes relative to GAPDH over the indicated time course after treatment with 100 nM of 4a and 4c. Tmax is the time by which compound induced maximum degradation (data from immunoblot in Figure 3C). The BRD4/α-Tubulin ratio at time 0 = 100%. (E) Immunoblot for BRD4 in MV4–11 cells after treatment with 100 nM of 4a in presence and absence of the proteasome inhibitor MG132, and CRBN inactive analogue 4f. (F) Concentration dependent BRD4 protein degradation in MV4–11 by 4a and 4c, after a 12-hour treatment, and (G) after a 24-hour treatment. DC50 is the compound concentration at which 50% of target protein is degraded. DC50 values were calculated using quantified band intensities from the immunoblotting analysis in Supp. Figure 8. Data represent the average of ≥2 independent determinations. Error bars represent standard error of the mean.
From this point we focused on benchmarking our novel PG-PROTAC 4c against the previously reported and well-characterized IMiD-based analogue 4a (dBET1). We first investigated the kinetics of protein degradation induced by these two compounds in MV4–11 cells. At a concentration of 100 nM both 4a and 4c achieved >90% depletion of BRD2 and BRD4 proteins after a 4-hour incubation (Figures 3C and 4D). We then evaluated whether BRD4 degradation by 4c is mediated by the proteasome. Since PROTAC mechanism of action involves ubiquitination of protein of interest followed by its proteasomal degradation, we subjected MV4–11 cells to proteasome inhibitor MG132 and 4c over 4 hours. As anticipated, immunoblotting analysis showed that BRD4 degradation was abrogated in the presence of MG132, which is consistent with previously reported mechanistic studies with other PROTACs (Figure 3E). Furthermore, we found that the treatment of MV4–11 cells with 4f, a CRBN-inactive analogue of 4c, did not affect BRD4 protein levels, indicating the CRBN-dependent mechanism of 4c cytotoxicity (Figure 3E).
Figure 4.
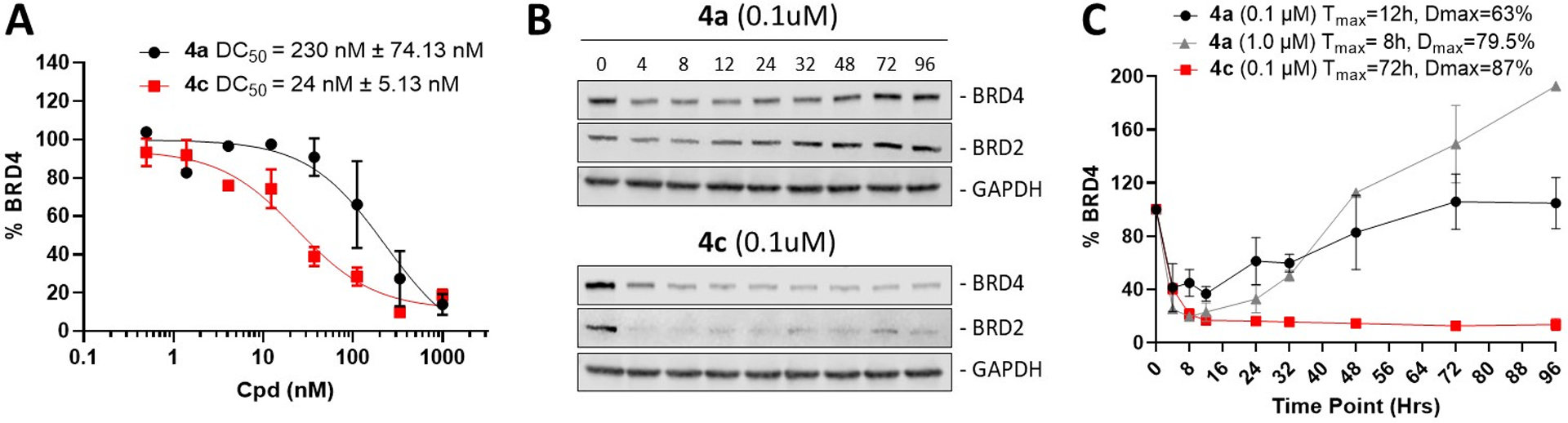
BRD4 protein degradation in HD-MB03 cell line. (A) Concentration-dependent BRD4 degradation after a 12-hour treatment of HD-MB03 cells with 4a and 4c. DC50 is the compound concentration at which 50% of target protein is degraded. DC50 values were calculated using quantified band intensities from immunoblot analysis (Supp. Figure 10). (B) BRD2 and BRD4 immunoblot analysis in HD-MB03 cells after treatment with 0.1 μM of 4a, and 4c over the indicated time course (0–96 hours). The same images are shown in Supplementary Figure 11. (C) Graphic representation of BRD4 protein level changes relative to GAPDH over the indicated time course after treatment with 1 μM, 0.1 μM of 4a, and 0.1 μM of 4c. Tmax is the time at which compound induces maximum degradation (Dmax) of BRD4 protein, based on data from immunoblot in Supp. Figure 11. The BRD4/GAPDH ratio at time zero was 100%. Data represent the average of three independent determinations. Error bars represent standard error of the mean.
To determine concentration-dependent effects of the two PROTACs on BRD4 protein stability, MV4–11 cells were treated over 12 hours with increasing concentrations of 4a or 4c, and then assayed for endogenous levels of BRD4 by immunoblotting. Under these conditions 4a displayed a half maximum degradation concentration (DC50) of 5.27 nM, and 4c showed a 6-fold higher degradation potency, with a DC50 of 0.87 nM (Figure 3F and Supp. Figure 8A). After a 24-hour incubation the difference in BRD4 degradation efficacy between the two PROTACs increased to 15-fold (Figure 3G, and Supp. Figure 8B). Neither of the two PROTACs degraded classical IMiD neosubstrates, such as IKZF1 and CK1α (Supp. Figure 9).
Next, we investigated protein degradation profiles of these two PROTACs in the HD-MB03 cell line. Consistent with their lower stability in this cell media, both 4a (t1/2 2.1 h) and 4c (t1/2 17.0 h) yielded reduced levels of BRD4 protein degradation in HD-MB03 cells, with DC50 values of 230 nM and 24 nM, respectively (Figure 4A and Supp. Figure 10).
Immunoblot time-course analysis in HD-MB03 cells revealed marked depletion of BRD4 protein levels within the first few hours of treatment by 4a at a concentration of 0.1 μM (Figure 4B, 4C and Supp. Figure 11). However, the initial loss of BRD4 protein was followed by a fast rebound resulting in a full recovery by the 50-hour time-point. A similar rebound was observed even after treating cells with concentration of 4a as high as 1 μM. Interestingly, at this concentration, BRD4 levels continued to rise, and reached 200 % of pre-treatment protein levels after 96 hours (Figure 4C). This observation is consistent with previously reported BRD4 protein accumulation induced by small molecule inhibitors such as JQ1.[11] Together, these finding suggest that although degradation product(s) of 4a lost their PROTAC effect, they still displayed BRD4 inhibition properties at the higher concentration. In contrast, the chemically more stable PG-PROTAC 4c achieved a similar Dmax at a 10-fold lower concentration (100 nM) than 4a and sustained that level of BRD4 protein degradation throughout the duration of the experiment (Figures 4B and 4C). These data provide further insights on the stark difference observed in potencies between these two analogues in HD-MB03 cells.
3. SUMMARY AND CONCLUSION
The emergence of the PROTAC paradigm in recent years has gained considerable interest across industry and academia as a novel therapeutic modality having potential multiple advantages over classical inhibitors, including dosing, duration, selectivity, and drug resistance. However, these are early days and the PROTAC critical design elements are still being elucidated. The momentous elucidation of thalidomide’s mechanism of action inspired the use of IMiDs in designing CRBN-directed PROTACs. Targeting CRBN is currently one of the most frequently reported PROTAC design approaches, owing to IMiDs’ favorable drug-like properties, available structural information and synthetic feasibility.[36] However, IMiDs are also known to be inherently unstable, readily undergoing hydrolysis in body fluids.[14],[15] We show here that IMiDs and IMiD-based PROTACs rapidly hydrolyze in PBS, and in relatively mild and widely utilized cell media. Although effects of hydrolysis on cellular potency are often masked by the PROTAC catalytic mechanism of action, in some cases they can be extensive enough to diminish or even completely obliterate cellular effects of PROTACs. For example, since BET bromodomain inhibition has an anti-proliferative effect in MYC driven medulloblastoma,[37] we evaluated BET PROTACs in HD-MB03 cells[29] derived from tumor material of a patient with MYC amplified Group 3 medulloblastoma, which has a particularly poor prognosis. However, we found that IMiD-based BET-PROTACs such as 3a and 4a rapidly hydrolyze in the HD-MB03 cell media, with t1/2 values <3h, and consequently show poor activity in HD-MB03 cell viability assay.
To address intrinsic stability issues with IMiDs and expand the options available for developing CRBN-directing PROTACs, we designed phenyl glutarimide (PG) as an alternative CRBN-binding chemotype. The phenyl group of PG was intended not only to replace the hydrolysis-prone IMiD functionalities, but also to reduce activation of the conserved glutarimide ring toward hydrolysis. We found that PG derivatives retained CRBN affinity with high ligand efficiencies (LE > 0.48) and displayed greatly improved chemical stability compared to classical IMiDs. Consequently, PG-based BET PROTACs such as 4b-e showed high anti-proliferative efficacy in both MV4–11 and HD-MB03 cell lines. To the best of our knowledge, 4c (SJ995973) displayed one of the highest cellular potency amongst BET PROTACs reported in literature, with an MV4–11 IC50 value of 3 pM, and BRD4 DC50 = 0.87 nM (Dmax = 99%).
In summary, we showed that PG is an alternative cereblon binder that offers several advantages over classical IMiDs used in PROTAC design, including chemical stability, smaller size and TPSA, greater ligand efficiency and synthetic feasibility.
Supplementary Material
4. ACKNOWLEDGEMENTS
We are grateful for the support of the ALSAC and St. Jude Children’s Research Hospital, and would like to thank the patients, their families, and the staff at our institution. We thank the compound management team at the department of Chemical Biology & Therapeutics for performing general QC and compound plate reformatting for screening. We also thank the protein production facility at St. Jude Children’s Research Hospital for expression and purification of 6X-His-CRBN-DDB1 protein and Sivaraja Vaithiyalingam at St. Jude Children’s Research Hospital for QC of the protein. The HD-MB03 cell line was a gift from Dr. Till Milde, German Cancer Research Center (DKFZ), Heidelberg, Germany. We are also grateful to Dana Farmer at St. Jude Children’s Research Hospital for helping with HD-MB03 cell culture. Funding sources: NCI P01 CA096832 and Core Grant P30 CA021765 (to M.F.R), and ALSAC and 1R35GM142772-01 (to MF).
5. REFERNCES
- [1].Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H, Science 2010, 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- [2].Kostic M, Jones LH, Trends Pharmacol. Sci 2020, 41, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chamberlain PP, Hamann LG, Nat. Chem. Biol 2019, 15, 937–944. [DOI] [PubMed] [Google Scholar]
- [4].Chamberlain PP, Cathers BE, Drug Discov. Today Technol 2019, 31, 29–34. [DOI] [PubMed] [Google Scholar]
- [5].Nishiguchi G, Keramatnia F, Min J, Chang Y, Jonchere B, Das S, Actis M, Price J, Chepyala D, Young B, McGowan K, Slavish PJ, Mayasundari A, Jarusiewicz JA, Yang L, Li Y, Fu X, Garrett SH, Papizan JB, Kodali K, Peng J, Pruett Miller SM, Roussel MF, Mullighan C, Fischer M, Rankovic Z, J. Med. Chem 2021, 64, 7296–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schapira M, Calabrese MF, Bullock AN, Crews CM, Nat. Rev. Drug Discov 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- [7].Chang Y, Min J, Jarusiewicz J, Actis M, Bradford SY-C, Mayasundari A, Yang L, Chepyala D, Alcock LJ, Roberts KG, Nithianantham S, Maxwell D, Rowland L, Larsen R, Seth A, Goto H, Imamura T, Akahane K, Hansen B, Pruett-Miller SM, Paietta EM, Litzow MR, Qu C, Yang JJ, Fischer M, Rankovic Z, Mullighan CG, Blood 2021, blood.2020006846. [DOI] [PMC free article] [PubMed]
- [8].Sievers QL, Petzold G, Bunker RD, Renneville A, Słabicki M, Liddicoat BJ, Abdulrahman W, Mikkelsen T, Ebert BL, Thomä NH, Science 2018, 362, eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Paiva S-L, Crews CM, Curr. Opin. Chem. Biol 2019, 50, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ, Proc. Natl. Acad. Sci 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, Crews CM, Chem. Biol 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang C-Y, Qin C, Bai L, Wang S, Drug Discov. Today Technol 2019, 31, 43–51. [DOI] [PubMed] [Google Scholar]
- [14].Schumacher H, Smith RL, Williams RT, Br. J. Pharmacol. Chemother 1965, 25, 338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schumacher H, Smith RL, Williams RT, Br. J. Pharmacol. Chemother 1965, 25, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Burslem GM, Ottis P, Jaime‐Figueroa S, Morgan A, Cromm PM, Toure M, Crews CM, ChemMedChem 2018, 13, 1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hagner PR, Man H-W, Fontanillo C, Wang M, Couto S, Breider M, Bjorklund C, Havens CG, Lu G, Rychak E, Raymon H, Narla RK, Barnes L, Khambatta G, Chiu H, Kosek J, Kang J, Amantangelo MD, Waldman M, Lopez-Girona A, Cai T, Pourdehnad M, Trotter M, Daniel TO, Schafer PH, Klippel A, Thakurta A, Chopra R, Gandhi AK, Blood 2015, 126, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lopez-Girona A, Lu G, Rychak E, Mendy D, Lu C-C, Rappley I, Fontanillo C, Cathers BE, Daniel TO, Hansen J, Blood 2019, 134, 2703–2703. [Google Scholar]
- [19].Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu C-C, Miller K, Fang W, Wang N-Y, Nguyen D, Houston J, Carmel G, Tran T, Riley M, Nosaka L, Lander GC, Gaidarova S, Xu S, Ruchelman AL, Handa H, Carmichael J, Daniel TO, Cathers BE, Lopez-Girona A, Chamberlain PP, Nature 2016, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- [20].Uy GL, Minden MD, Montesinos P, DeAngelo DJ, Altman JK, Koprivnikar J, Vyas P, Fløisand Y, Belén Vidriales M, Gjertsen BT, Esteve J, Buchholz TJ, Couto S, Fan J, Hanna B, Li L, Pierce DW, Hege K, Pourdehnad M, Zeidan AM, Blood 2019, 134, 232–232. [Google Scholar]
- [21].Kim SA, Go A, Jo S-H, Park SJ, Jeon YU, Kim JE, Lee HK, Park CH, Lee C-O, Park SG, Kim P, Park BC, Cho SY, Kim S, Ha JD, Kim J-H, Hwang JY, Eur. J. Med. Chem 2019, 166, 65–74. [DOI] [PubMed] [Google Scholar]
- [22].Phillips A, Nasveschuk C, Henderson J, Liang Y, He M, Lazarski K, Veits G, Vora H. C3-Carbon linked glutarimide degronimers for target protein degradation. WO2017197046A1, 2017. [Google Scholar]
- [23].Nasveschuk C, Zeid R, Yin N, Jackson KL, Veits GK, Moustakim M, Yap JL. Compounds for targeted degradation of BRD9. WO2021178920A1, 2021. [Google Scholar]
- [24].Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith REJ, Harper JW, Jenkins JL, Thomä NH, Nature 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2019.
- [26].Chessum NEA, Sharp SY, Caldwell JJ, Pasqua AE, Wilding B, Colombano G, Collins I, Ozer B, Richards M, Rowlands M, Stubbs M, Burke R, McAndrew PC, Clarke PA, Workman P, Cheeseman MD, Jones K, J. Med. Chem 2018, 61, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hopkins AL, Groom CR, Alex A, Drug Discov. Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- [28].Reist M, Carrupt P-A, Francotte E, Testa B, Chem. Res. Toxicol 1998, 11, 1521–1528. [DOI] [PubMed] [Google Scholar]
- [29].Milde T, Lodrini M, Savelyeva L, Korshunov A, Kool M, Brueckner LM, Antunes ASLM, Oehme I, Pekrun A, Pfister SM, Kulozik AE, Witt O, Deubzer HE, J. Neurooncol 2012, 110, 335–348. [DOI] [PubMed] [Google Scholar]
- [30].Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang C-Y, Liu L, McEachern D, Przybranowski S, Wang M, Stuckey J, Meagher J, Bai L, Chen Z, Lin M, Yang J, Ziazadeh DN, Xu F, Hu J, Xiang W, Huang L, Li S, Wen B, Sun D, Wang S, J. Med. Chem 2018, 61, 6685–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Burslem GM, Song J, Chen X, Hines J, Crews CM, J. Am. Chem. Soc 2018, 140, 16428–16432. [DOI] [PubMed] [Google Scholar]
- [32].Mu X, Bai L, Xu Y, Wang J, Lu H, Biochem. Biophys. Res. Commun 2020, 521, 833–839. [DOI] [PubMed] [Google Scholar]
- [33].Lin W, Li Y, Min J, Liu J, Yang L, Lee RE, Chen T, Bioconjug. Chem 2020, 31, 2564–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang C-Y, Liu L, McEachern D, Przybranowski S, Wang M, Stuckey J, Meagher J, Bai L, Chen Z, Lin M, Yang J, Ziazadeh DN, Xu F, Hu J, Xiang W, Huang L, Li S, Wen B, Sun D, Wang S, J. Med. Chem 2018, 61, 6685–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gadd MS, Testa A, Lucas X, Chan K-H, Chen W, Lamont DJ, Zengerle M, Ciulli A, Nat. Chem. Biol 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Edmondson SD, Yang B, Fallan C, Bioorg. Med. Chem. Lett 2019, 29, 1555–1564. [DOI] [PubMed] [Google Scholar]
- [37].Venataraman S, Alimova I, Harris P, Birks DK, Balakrishnan I, Remke M, Taylor MD, Handler M, Foreman NK, Vibhakar R, Oncotarget 2014, 5, DOI 10.18632/oncotarget.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.