

Abstract
Myeloid cells (macrophages, monocytes, dendritic cells, and granulocytes) survey the body for signs of infection and tissue damage and regulate tissue homeostasis, organogenesis, and immunity. They express receptors that initiate the inflammatory response, send signals that alter the vascular and cytokine milieu, and oversee the recruitment, differentiation, and activation of other myeloid and adaptive immune cells. Their activation must therefore be tightly regulated, optimized for maximal innate-immune protection with a minimum of collateral tissue damage or disorganization. In this review we discuss what it means for myeloid cells to become activated, with emphasis on the receptors and signaling molecules important for the recognition of pathogen- and damage-associated molecular patterns. We also outline how these signals are regulated by the steric properties of proteins, by adhesive and cytoskeletal interactions, and by negative feedback to keep inflammation in check and support healthy tissue development and homeostasis. Throughout the text we highlight recent publications and reviews that illustrate key elements of myeloid-cell regulation and direct readers therein for a comprehensive bibliography.
Keywords: Innate immune system, Macrophage, Autoimmune disease, Inflammation, Src-family kinase (SFK), Lyn, tumor microenvironment, myeloid-cell polarization, ITAM-coupled receptor, feedback and regulatory signaling
Graphical Abstract
Short Abstract
Myeloid cells (macrophages, monocytes, dendritic cells, and granulocytes) survey tissues for signs of infection and damage and regulate tissue homeostasis, organogenesis, and immunity. They express receptors that initiate the inflammatory response, alter the vascular and cytokine milieu, and oversee the recruitment, differentiation, and activation of other myeloid and adaptive immune cells. Their activation must therefore be tightly regulated, optimized for maximal protection with minimum collateral tissue damage. Here we discuss what it means for myeloid cells to become activated, with emphasis on receptors and signaling molecules recognizing pathogen- and damage-associated molecular patterns. We outline how these signals are regulated by steric properties of proteins, adhesive and cytoskeletal interactions, and negative feedback to keep inflammation in check and support healthy tissue development and homeostasis. Throughout the text we highlight recent publications and reviews that illustrate key elements of myeloid-cell regulation and direct readers therein for a comprehensive bibliography.
Introduction
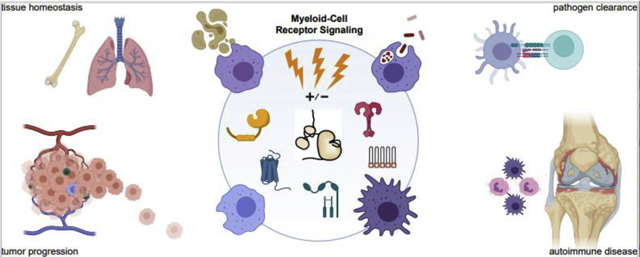
Myeloid cells populate nearly every space in the body. They are first responders, sensing infection and tissue damage, killing pathogens, and regulating innate and adaptive immune functions. As part of this interplay, they direct immune-cell recruitment and differentiation, mediate antigen presentation, and control the amplitude and kinetics of the inflammatory response [1]. Myeloid cells also have critical roles in tissue architecture, remodeling extracellular matrix [2], guiding organ development [3], and directing vascularization [4] (Figure 1A). Mechanisms regulating the quality, strength, and duration of myeloid-cell activation are therefore critically important.
Figure 1. Effects and mediators of classical myeloid-cell activation.
(A) Examples of myeloid-cell functions. (B) Prominent receptor pathways (representative ligands above, receptors below) controlling myeloid-cell activation. Parentheses highlight ITAM coupling. (C) Myeloid-lineage cell types and representative diseases driven by their dysregulated activation. Figures created with BioRender.com and Adobe Software.
Due to their pleiotropic responsiveness to a vast array of stimuli, the spectrum of “myeloid-cell activation” encompasses many distinct functional programs. Activation is broadly defined as a response to pathogen- or damage-associated molecular patterns (PAMPS, DAMPS), opsonized particles, or cytokines that results in an acute functional outcome. Generally, receptor ligation triggers a kinase cascade (within seconds); rearrangement of the actin cytoskeleton, phagocytosis, degranulation, and release of reactive oxygen, nitric, oxide, calcium, and other second-messengers (within minutes); and expression/secretion of cytokines and polarization-specific proteins (within hours).
“Classical” pro-inflammatory activation occurs via Toll-like Receptors (TLRs), Complement Receptors (CRs), intracellular Immunoreceptor Tyrosine-based Activation Motifs (ITAMs), and receptors for cytokines such as interferon (IFN)-γ (Figure 1B, left). Inflammatory responses have distinct features determined by the myeloid-cell subtype, the tissue milieu, and the receptor ligands presented by the pathogen. For example, gram-positive bacteria activate TLR2 on macrophages, whereas gram-negative bacteria activate TLR2+TLR4, producing tailored inflammatory responses with distinct cytokine profiles [5].
“Alternative” activation is defined as the myeloid response to interleukins (ILs) (e.g. IL-4, −13, −5, −33) that induce anti-helminth or tissue-repair functions, depending on other environmental cues [4]. The functional results of these cues vary but include metabolic changes that promote oxidative phosphorylation in macrophages [6], degranulation of eosinophils and mast cells, and tissue reorganization [7].
Inflammatory polarization
Tissue- and perturbation-specific signals alter gene transcription to bias activation of specific signaling pathways, immediate cellular functions, and transcriptional programs [8,9]. For instance, macrophage exposure to IFN-γ, Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF), or Tumor Necrosis Factor (TNF) increases subsequent sensitivity to pathogens and alters myeloid-cell and lymphocyte recruitment, differentiation, polarization, and activation [10]; C-C motif chemokine ligand (CCL)2 and macrophage inflammatory protein (MIP)-1 induce similar responses through G-Protein-Coupled Receptors (GPCRs) [11] (Figure 1B, right). PAMPs and DAMPs are dually activating and polarizing. For example, ligation of the hemi-ITAM-containing receptor Dectin-1 by cell-wall β-glucans induces phagocytosis and TNF production (acute, classical activation) [12], and increases transcription of inducible nitric oxide synthase (iNos) and other pro-inflammatory factors to increase the sensitivity to future activation [13].
Due to their near-ubiquity and myriad functions, inappropriately activated myeloid cells underlie drive many pathologies (Figure 1C). Chronic inflammatory activation, often resulting from a feedback cycle of increased signaling amplitude downstream of ITAMs [14], TLRs [15], or CRs [16] and increased myeloid-cell infiltration of tissues, drives autoimmune, allergic, and other inflammatory diseases [17–19]. Megakaryocytes, platelets, and neutrophils have been particularly highlighted in the recent literature. Defects in megakaryocyte autophagy reduce platelet function in immune thrombocytopenic purpura [20]. Megakaryocytes can also mediate inflammatory exchange between neutrophils and platelets through emperipolesis, a cell-engulfment process [21]. Neutrophils and macrophages drive diseases such as RA and endometriosis via classical inflammatory activation and by the formation of extracellular traps. Dysregulation of these myeloid cells also drives the cytokine storm and respiratory disruption that accompany the most severe forms of sepsis [22], influenza [23], and COVID-19 [24,25].
Alternative polarization
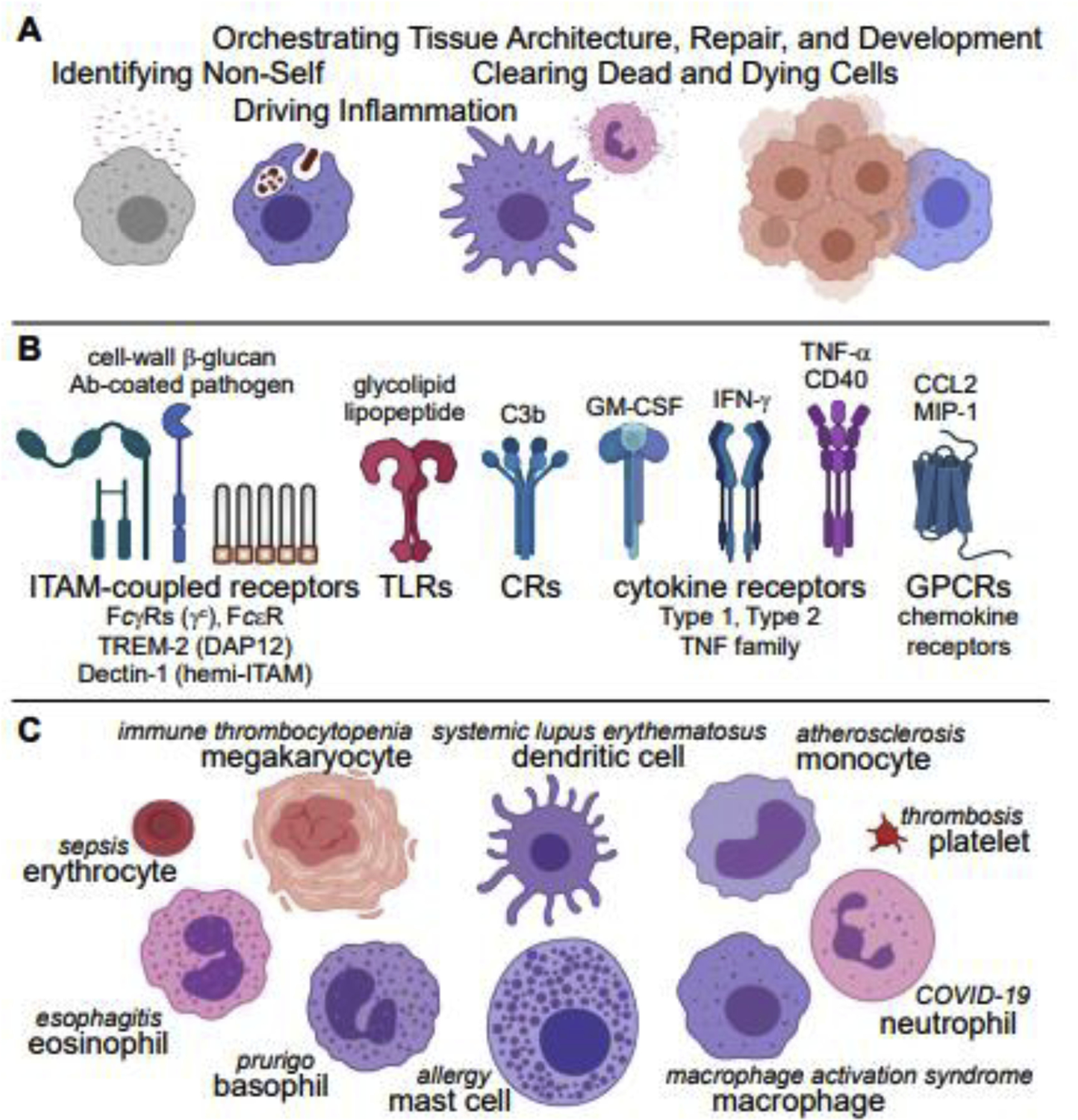
Myeloid cells can be alternatively polarized to repair or remodel tissue, suppress or end the antimicrobial response, and antagonize the effects of inflammatory cytokines [4]. The combined effects of tissue- and context-specific signals on different types of myeloid cells, however, are complex and defy simple categorization. As with inflammatory polarization, immunosuppressive or tissue-remodeling polarization may be effected through multiple receptor pathways [4] (Figure 2A). The effects of these signals depend on receptor availability, cell type, and other factors in the tissue. IL-4 and IL-13, for instance, can trigger anti-inflammatory signaling, leading to reduced production of IL-1β and TNF by macrophages, but they are also components of type 2 inflammation, driving host defense against parasites. Detection of neutrophil apoptotic cell debris can comprise a second signal to induce a tissue-repair polarization in macrophages during wound healing [26,27].
Figure 2. Polarization alters myeloid-cell activation and function.
(A) Receptor signaling pathways that can decrease inflammatory signaling and mediate tissue-repair, remodeling, and immunosuppressive polarities. (B) Effects of polarization on myeloid-cell activation.
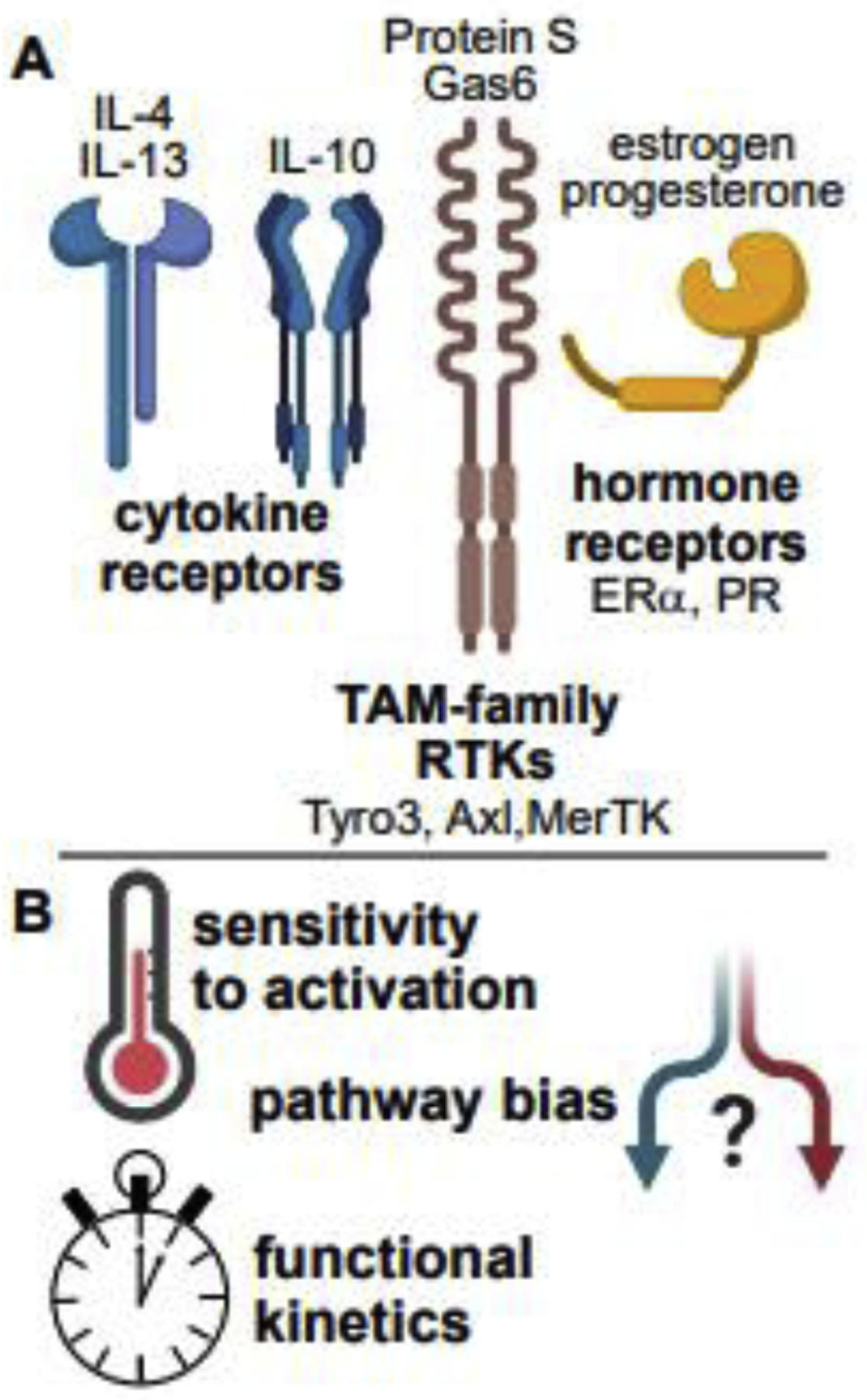
The effects of inflammatory and tissue-remodeling polarization may be shared or distinct. For instance, either IFN-γ or IL-4 increases upstream and downstream kinase phosphorylation in macrophages responding to fungal cell wall [12], but these cytokines have different functional outcomes with respect to phagocytosis and transcription—how specificity is achieved is complex [28] and incompletely understood. Combinatorial effects of tissue- and danger-specific inputs yield a spectrum of polarization states. Via coordinated changes in receptor, effector, and negative-regulator expression, initial triggering and signal amplitudes are sensitized or desensitized to bias pathway activation. For example, the activation of T-cells by dendritic cells via antigen presentation, induces an increase in expression of ligands for Tyro/Axl/Mer (TAM-family) receptor tyrosine kinases on myeloid cells. Ligation of these kinases then restrains inflammatory activation of dendritic cells [29].
Polarization also time-regulates the inflammatory response, mediating shifts from anti-pathogen to tissue-repair functionality (Figure 2B). While simultaneous detection of intact pathogen and apoptosis mediates an antimicrobial response [30,31], the detection only of apoptosis and tissue signals such as IL-4 induces healing and inflammation resolution [26]. Therefore, a time- and environment-dependent shift from inflammatory to alternative polarization after a tissue insult ensures that pathogen elimination is accompanied by repair of damaged tissue and return to homeostasis [32,33].
ITAM size sensors
ITAMs and hemi-ITAMs trigger myeloid-cell activation through the Src-family kinases (SFKs) and Syk kinase [34]. Unlike T cells, which can be activated by as few as 4–6 high-affinity T-cell receptor (TCR)/peptide/MHC interactions [35], most myeloid cells must interact closely with intact pathogens that ligate receptors over a large surface area to enable phagocytosis and minimize the toxic effects of inflammation, degranulation, and reactive oxygen. Macrophages, dendritic cells, and neutrophils sense the valency of receptor interactions as a proxy for the size of an interacting particle, functioning as sensors of stimulus size: high-valency ligands such as those presented by an intact pathogen (μm-scale) ligate receptors through highly multimeric cell-wall components or opsonins, initiating an antimicrobial response and inflammatory polarization. Low-valency (nm-scale) receptor ligation [36] or intracellular kinase activation alone [37] is not fully activating (Figure 3A). The probability of cell activation via ligation of ITAM-coupled receptors varies according to cell type, polarization, and receptor identity. Mast cells, for example, have a lower triggering threshold than macrophages [38], with the potential to signal through lower-valency ligation of FcεR [39,40]; mechanisms of mast-cell regulation are discussed later.
Figure 3. Mechanisms regulating myeloid-cell activation.
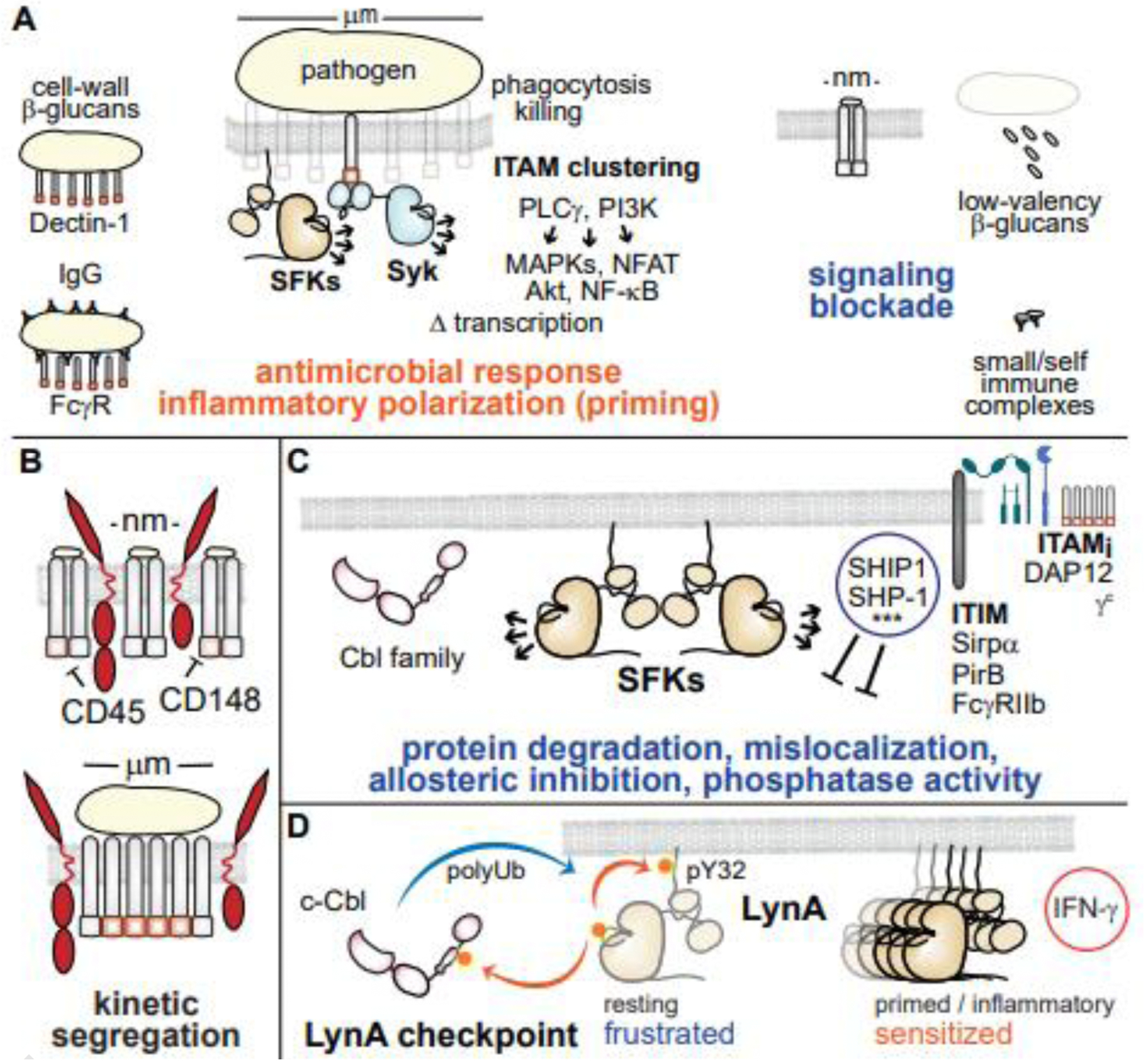
(A) ITAM-coupled receptors in macrophages and dendritic cells function as size sensors to correctly identify intact pathogen cells and initiate an antimicrobial response. (B) The kinetic segregation model of signaling at a phagocytic synapse. (C) Negative-feedback pathways suppressing myeloid-cell activation, including recruitment of phosphatases and (***) negative-regulatory functions of adaptor proteins such as Grb2 and the Dok family. (D) Rapid, selective degradation and environment/cell-dependent expression of LynA and Cbl tune myeloid-cell sensitivity to activation.
One component of the ITAM particle-size sensor involves kinetic protection of ITAM/signalosome phosphorylation. Inspired by earlier studies of the T-cell synapse [41], the kinetic segregation model applied to myeloid cells postulates that the rigid/glycosylated extracellular domains of the tyrosine phosphatases CD45 and CD148 are sterically excluded from the phagocytic synapse, protecting activating phosphorylation of ITAMs, SFKs, and other molecules (Figure 3B). Lower-valency ITAM nanoclusters lack the requisite steric occlusion and are thus reversed before signal propagation [36,42,43].
At the other extreme, if phagocytosis is frustrated in an encounter with an overly large foreign object (e.g. fungal hypha), ITAM/hemi-ITAM signaling and subsequent production of reactive oxygen act as molecular timers, swapping classical activation for extracellular trap formation [44,45]. Inflammation associated with neutrophil extracellular traps (NETs) is also a hallmark of chronic inflammation, production of anti-nuclear antibodies, and autoimmune disease [46].
Regulation by actin and integrin barriers
Activation of myeloid cells by pathogen-associated ligands (via Dectin-1) and antibodies (via FcRs) is regulated by constraints on lateral diffusion. Hyaluronan fences interact with CD44 pickets, anchored intracellularly to cortical actin filaments. While diffusion in two dimensions within the resulting actin corrals is relatively unrestricted, diffusion between corrals is limited. Spontaneous formation of higher-order receptor clusters is blocked, protecting against amplification of spurious or stochastic initiating signals [47]. With sufficiently robust receptor activation (straddling multiple corrals and/or with high-affinity/low-off-rate ligand-receptor interactions), the activities of SFKs and Syk initiate remodeling of cortical actin, reorganizing corrals, relieving constraints on lateral diffusion, enabling higher-order receptor clustering, and forming a phagocytic cup [48].
Integrins contribute to receptor clustering and activation by stabilizing interactions between myeloid and target cells via interactions with complement and cell-wall β-glucans. This close-contact region favors formation of new interactions and rebinding of high-off-rate interacting partners, so the two cell surfaces zip together. Positive feedback through integrins during phagocytosis enhances receptor binding and kinetic segregation [42]. This is especially important when a myeloid cell interacts with non-diffusible components of a pathogen cell wall: a μm-scale cluster of ITAM-coupled receptors need not be continuous but may be punctate on the nanoscale, with integrins mediating interstitial interactions. Integrin-triggered signaling can also potentiate activation of other receptors, such as TLRs [49,50].
ITIMs, inhibitory ITAMs, protein modification, and the LynA rheostat
Negative-regulatory processes limit myeloid-cell signaling in magnitude and duration. Lipid and tyrosine phosphatases (e.g. SHIPs, SHPs) and negative-regulatory adaptor proteins (e.g. Grb2, Doks) are recruited to immunoreceptor tyrosine inhibitory motifs (ITIMs, e.g. Sirpα, PirB) [51] or monophosphorylated inhibitory ITAMs (ITAMis) [52] to suppress inflammatory signaling (Figure 3C).
Activated SFKs within receptor complexes also phosphorylate and activate Cbl-family E3 ubiquitin ligases, which then monoubiquitinate or polyubiquitinate nearby targets. Ubiquitin-modification of signaling components may directly block their activity, flag them for degradation, or trigger receptor internalization [53]. SFK-mediated phosphorylation and activation of c-Cbl also feeds back to downregulate all the SFKs via a slow (half-life 10+ min) process of polyubiquitination and degradation [37].
In addition to its more promiscuous negative-regulatory functions, c-Cbl mediates the rapid, selective degradation of the SFK LynA, the longer of two Lyn splice variants [34,37,38]. In macrophages, phosphorylation of LynA at tyrosine 32 (pY32) [38,54] targets LynA for rapid degradation (half-life 1 min), causing a signaling blockade downstream of PLCγ and PI3K [37] (Figure 3D). LynA and c-Cbl expression are regulated by cell type [38] and inflammatory polarization [37], lending context specificity to this signaling checkpoint and regulating each cell’s sensitivity to activation. For example, in resting macrophages high expression of c-Cbl and low expression of LynA block cell activation in the absence of low-valency receptor ligation. Inflammatory polarization with IFN-γ upregulates LynA, overcoming the signaling checkpoint and sensitizing macrophage activation [37]. Mast cells, in contrast, express little c-Cbl, increasing steady-state accumulation of LynA protein and sustaining LynA activation during signaling [38]. This sensitivity to SFK-mediated signaling may underlie the exquisite sensitivity of mast-cell FcεRs to low-valency receptor ligands [39,40]. The cell- and environment-specific expression level of LynA and c-Cbl therefore comprise a coordinated signaling rheostat that tunes the intensity and longevity of the LynA response in a cell-type- and environment-specific manner.
In contrast, the shorter splice form of Lyn kinase, LynB, has the dominant role in preventing autoimmune disease. In a recent study using CRISPR/Cas9-generated LynA and LynB isoform-specific knockout mice [55], LynB knockout mice preferentially develop the lupus disorder observed in total Lyn knockout mice [56]. Together, these observations suggest that the dual positive and negative functions of Lyn kinase shared unequally by the two splice forms, LynA and LynB, respectively [34,37,38,55].
Spotlight on macrophage activation and disease
Macrophages are particularly plastic cells, with myriad polarization and tissue-specific states. Their sensitivity to environmental cues, which induce a spectrum of substates with tissue-remodeling (Figure 4A) and/or inflammatory (Figure 4B) functionalities, is reflected in their many roles in systemic and tissue-specific disease. In autoimmune disease, macrophages promote immune-cell infiltration, release inflammatory cytokines (e.g. TNF, IL-1, IL-6) and drive a feedback cycle of chronic inflammation (Figure 4C). In macrophage activation syndrome (MAS), for example, systemic overproduction of IFN-γ [57], chronic elevation of IL-18 [58], and/or sustained activation of TLRs [59] drives excessive and sustained release of inflammatory cytokines and dysregulates phagocytosis [60]. Macrophages are among the cells found in the presumably sterile synovia of RA and juvenile idiopathic arthritis (JIA) patients [61]. In atherosclerosis, low-density lipoprotein induces foamy macrophage differentiation, dysregulation, and necrosis [62]. Proliferating intima-resident macrophages are then gradually replaced by infiltrating monocytes; a combination of inflammatory and tissue-resident functions then drives plaque progression [63]. Chronic obstructive pulmonary disorder (COPD) and acute respiratory distress syndrome (ARDS) are linked to downregulation of Programmed Death Ligand (PDL) proteins in the lung, resulting in hyperactivation of alveolar macrophages [64,65]. Tumor cells establish a microenvironment in which the cytokine milieu may evoke tissue-remodeling as well as inflammatory functions in macrophages (Figure 4D), including matrix reorganization [66], support for metastasis, and T-cell suppression [67,68]. Deleterious activation of macrophages by abnormal production or responsiveness to either pro- or anti-inflammatory stimuli breaks tissue homeostasis and promotes disease.
Figure 4. Polarization and differentiation regulate macrophage activation in healthy and diseased tissues.
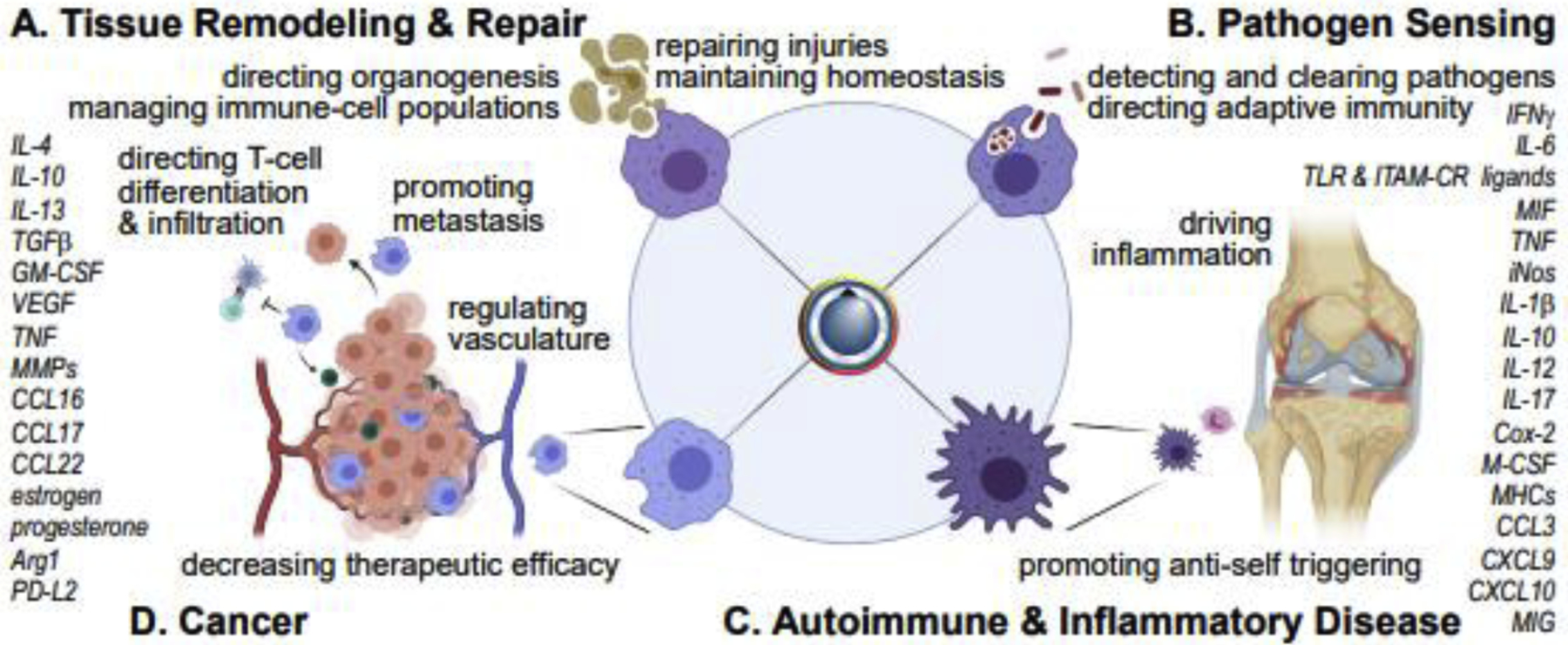
(A) Functions and signaling components driving tissue remodeling/repair polarization. (B) Functions and signaling components driving an inflammatory, antimicrobial polarization. (C) Chronic inflammation is driven by pathological inflammatory activation of macrophages. (D) Macrophages can become polarized in service of tumors. Cytokines, signaling pathways, and transcriptional changes are heterogeneous and, in many cases, cannot be cleanly binned into pro- vs. anti-inflammatory.
The developmental origins of tissue macrophages add another layer of pathway bias and functional heterogeneity. Tissues are initially seeded by yolk-sac progenitors, which give rise to resident macrophages in the brain (microglia), liver (Kupffer cells), and other sites [69–71]. A second wave of hematopoiesis from the fetal liver gives rise to other tissue macrophages (e.g. intestine, lung) and circulating monocytes [70], which may also repopulate tissues [72]. Tissue signals imprint newly differentiated macrophages with site-specific activation profiles [73], restraining the inflammatory response during routine clearance of apoptotic cells [74]. However, these signals become dysregulated when embryonically derived macrophages drive fibrosis and cancer [75]. Recruitment and replacement of tissue-resident macrophages following tissue injury can also confer protection from bacterial infections, as newly differentiated macrophages may retain epigenetic traits from their monocyte precursors that facilitate rapid production of cytokines after a bacterial encounter [76]. Tissues with resident macrophages “paralyzed” by previous pathogen exposure [77] are thus supplemented with new, potentially pro-inflammatory macrophages. However, unrestrained inflammation, as in malarial infection [59], can drive monocyte differentiation into red-blood cell phagocytes that drive pathologic anemia. While the seeding of tissues by circulating monocytes and macrophage programming by the tissue microenvironment add flexibility to tissue homeostasis, both processes may be dysregulated in disease.
Concluding remarks
Myeloid cells are highly diverse, with a complex blend of overlapping abilities and cell-specific functions. Regulation of oxidative killing, phagocytosis, degranulation, and cytokine secretion is complex and context-specific. Nevertheless, patterns of regulatory modalities emerge as general principles: polarization by cytokines and tissue signals, receptor diffusion barriers, and intracellular regulation of receptor activation, with the SFKs and Lyn standing out as a central regulatory node. Crosstalk between receptor pathways confers higher-order regulation, sensitizing or frustrating interacting cascades. Diseases such as lupus and cancer be accompanied by the subversion of these regulatory features. Mechanistic research defining how myeloid cells integrate positive- and negative-regulatory signals from tissues, immune-cells, and multiple receptors to bias or sensitize their myriad signaling pathways will be the key to understanding the processes of homeostasis and disease and modulating myeloid-cell function with ever-increasing specificity to the benefit of research and therapeutics.
Highlights.
Myeloid-cell regulation optimizes innate/adaptive immunity and tissue health.
Competing positive and negative signaling biases and tunes myeloid activation.
Src-family kinase (e.g. Lyn) signaling is regulated by environment and cell type.
Dysregulated myeloid activation drives autoimmunity, cancer, and cytokine storm.
Defining context-specific myeloid regulatory mechanisms may yield novel therapies.
Acknowledgements
Research in the Freedman Lab is supported by National Institutes of Health awards R01AR073966 (TSF), R03AI130978 (TSF), T32CA009138 (JTG), and T32DA007097 (BFB) and by American Cancer Society - Kirby Foundation postdoctoral fellowship PF-21-068-01-LIB (JTG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: none
References
• of special interest •• of outstanding interest
- 1.Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL: The Myeloid Cell Compartment-Cell by Cell. Annu Rev Immunol 2019, 37:269–293. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Chaffee TS, LaRue RS, Huggins DN, Witschen PM, Ibrahim AM, Nelson AC, Machado HL, Schwertfeger KL: Tissue-resident macrophages promote extracellular matrix homeostasis in the mammary gland stroma of nulliparous mice. Elife 2020, 9:e57438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady NJ, Farrar MA, Schwertfeger KL: STAT5 deletion in macrophages alters ductal elongation and branching during mammary gland development. Dev Biol 2017, 428:232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M: Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 2013, 229:176–185. [DOI] [PubMed] [Google Scholar]
- • 5.Gottschalk RA, Dorrington MG, Dutta B, Krauss KS, Martins AJ, Uderhardt S, Chan W, Tsang JS, Torabi-Parizi P, Fraser ID, et al. : IFN-mediated negative feedback supports bacteria class-specific macrophage inflammatory responses. Elife 2019, 8:e46836. [DOI] [PMC free article] [PubMed] [Google Scholar]; Macrophages integrate signaling following ligation of multiple TLRs by gram-positive and gram-negative bacteria, highlighting the prominent role of Type I Interferons in limiting the production of inflammatory cytokines following interactions with gram-negative bacteria.
- 6.Saha S, Shalova IN, Biswas SK: Metabolic regulation of macrophage phenotype and function. Immunol Rev 2017, 280:102–111. [DOI] [PubMed] [Google Scholar]
- 7.de Kouchkovsky DA, Ghosh S, Rothlin CV: Negative Regulation of Type 2 Immunity. Trends Immunol 2017, 38:154–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okabe Y, Medzhitov R: Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014, 157:832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernandez-Calvo G, Khoyratty TE, van Grinsven E, Gonzalez-Hernandez S, Nicolas-Avila JA, et al. : Co-option of Neutrophil Fates by Tissue Environments. Cell 2020, 183:1282–1297 e1218. [DOI] [PubMed] [Google Scholar]
- 10.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA: IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol 2011, 12:231–238. [DOI] [PubMed] [Google Scholar]
- 11.Ruytinx P, Proost P, Van Damme J, Struyf S: Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front Immunol 2018, 9:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Underhill DM, Rossnagle E, Lowell CA, Simmons RM: Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 2005, 106:2543–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M, Luo F, Ding C, Albeituni S, Hu X, Ma Y, Cai Y, McNally L, Sanders MA, Jain D, et al. : Dectin-1 Activation by a Natural Product beta-Glucan Converts Immunosuppressive Macrophages into an M1-like Phenotype. J Immunol 2015, 195:5055–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben Mkaddem S, Benhamou M, Monteiro RC: Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front Immunol 2019, 10:811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •• 15.Stanbery AG, Newman ZR, Barton GM: Dysregulation of TLR9 in neonates leads to fatal inflammatory disease driven by IFN-gamma. Proc Natl Acad Sci U S A 2020, 117:3074–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]; Disrupting normal TLR9 regulation leads to IFN-γ-driven inflammation and autoimmunity, particularly when this disruption occurs early in life. This work highlights the importance of regulating the myeloid response to self nucleic acids during organ development and tissue reorganization.
- 16.Teirila L, Heikkinen-Eloranta J, Kotimaa J, Meri S, Lokki AI: Regulation of the complement system and immunological tolerance in pregnancy. Semin Immunol 2019, 45:101337. [DOI] [PubMed] [Google Scholar]
- 17.Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA: Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A 2013, 110:E3311–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito Y, Satoh T, Takayama K, Miyagishi C, Walls AF, Yokozeki H: Basophil recruitment and activation in inflammatory skin diseases. Allergy 2011, 66:1107–1113. [DOI] [PubMed] [Google Scholar]
- 19.Olivera A, Beaven MA, Metcalfe DD: Mast cells signal their importance in health and disease. J Allergy Clin Immunol 2018, 142:381–393. [DOI] [PubMed] [Google Scholar]
- 20.Sun RJ, Shan NN: Megakaryocytic dysfunction in immune thrombocytopenia is linked to autophagy. Cancer Cell Int 2019, 19:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunin P, Bouslama R, Machlus KR, Martinez-Bonet M, Lee PY, Wactor A, Nelson-Maney N, Morris A, Guo L, Weyrich A, et al. : Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. Elife 2019, 8:e44031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson HL, Brodsky IE, Mangalmurti NS: The Evolving Erythrocyte: Red Blood Cells as Modulators of Innate Immunity. J Immunol 2018, 201:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT: Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 2011, 179:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Li Q, Yin Y, Zhang Y, Cao Y, Lin X, Huang L, Hoffmann D, Lu M, Qiu Y: Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front Immunol 2020, 11:2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabo PA, Dogra P, Gray JI, Wells SB, Connors TJ, Weisberg SP, Krupska I, Matsumoto R, Poon MML, Idzikowski E, et al. : Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54:797–814 e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, Weinstein JS, Licona-Limon P, Schmid ET, Pelorosso F, et al. : Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356:1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P: Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358:111–116. [DOI] [PubMed] [Google Scholar]
- 28.Zhan Y, Lew AM, Chopin M: The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front Immunol 2019, 10:2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrera Silva EA, Chan PY, Joannas L, Errasti AE, Gagliani N, Bosurgi L, Jabbour M, Perry A, Smith-Chakmakova F, Mucida D, et al. : T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 2013, 39:160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torchinsky MB, Garaude J, Martin AP, Blander JM: Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature 2009, 458:78–82. [DOI] [PubMed] [Google Scholar]
- 31.Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM: Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 2011, 474:385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, Roers A, Eming SA: Differential roles of macrophages in diverse phases of skin repair. J Immunol 2010, 184:3964–3977. [DOI] [PubMed] [Google Scholar]
- 33.Rothlin CV, Hille TD, Ghosh S: Determining the effector response to cell death. Nat Rev Immunol 2021, 21:292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brian BF, Freedman TS: The Src-family Kinase Lyn in Immunoreceptor Signaling. Endocrinology 2021, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J: T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc Natl Acad Sci U S A 2011, 108:9089–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, Bose N, Chan AS, Magee AS, Danielson ME, et al. : Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature 2011, 472:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freedman TS, Tan YX, Skrzypczynska KM, Manz BN, Sjaastad FV, Goodridge HS, Lowell CA, Weiss A: LynA regulates an inflammation-sensitive signaling checkpoint in macrophages. eLife 2015, 4:e09183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •• 38.Brian BF, Jolicoeur AS, Guerrero CR, Nunez MG, Sychev ZE, Hegre SA, Saetrom P, Habib N, Drake JM, Schwertfeger KL, et al. : Unique-region phosphorylation targets LynA for rapid degradation, tuning its expression and signaling in myeloid cells. eLife 2019, 8:e46043. [DOI] [PMC free article] [PubMed] [Google Scholar]; Phosphorylation of LynA at position Y32 (lacking the alternatively spliced LynB) flags it for polyubiquitination by c-Cbl and rapid degradation. Cell- and environment-specific regulation of LynA and c-Cbl expression therefore comprise a rheostatic tuning mechanism, in which the resulting dose and longevity of activating LynA helps to set the sensitivity of myeloid cells to activation.
- 39.Andrews NL, Pfeiffer JR, Martinez AM, Haaland DM, Davis RW, Kawakami T, Oliver JM, Wilson BS, Lidke DS: Small, mobile FcepsilonRI receptor aggregates are signaling competent. Immunity 2009, 31:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felce JH, Sezgin E, Wane M, Brouwer H, Dustin ML, Eggeling C, Davis SJ: CD45 exclusion- and cross-linking-based receptor signaling together broaden FcepsilonRI reactivity. Sci Signal 2018, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis SJ, van der Merwe PA: The kinetic-segregation model: TCR triggering and beyond. Nat Immunol 2006, 7:803–809. [DOI] [PubMed] [Google Scholar]
- 42.Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, Hinz B, van der Merwe PA, Das R, Grinstein S: Integrins Form an Expanding Diffusional Barrier that Coordinates Phagocytosis. Cell 2016, 164:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakalar MH, Joffe AM, Schmid EM, Son S, Podolski M, Fletcher DA: Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets. Cell 2018, 174:131–142 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V: Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 2014, 15:1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warnatsch A, Tsourouktsoglou TD, Branzk N, Wang Q, Reincke S, Herbst S, Gutierrez M, Papayannopoulos V: Reactive Oxygen Species Localization Programs Inflammation to Clear Microbes of Different Size. Immunity 2017, 46:421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, Choi JH, Choi Y, Shim S, Lyu IS, et al. : Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev 2017, 16:1160–1173. [DOI] [PubMed] [Google Scholar]
- 47.Freeman SA, Vega A, Riedl M, Collins RF, Ostrowski PP, Woods EC, Bertozzi CR, Tammi MI, Lidke DS, Johnson P, et al. : Transmembrane Pickets Connect Cyto- and Pericellular Skeletons Forming Barriers to Receptor Engagement. Cell 2018, 172:305–317 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaumouille V, Farkash Y, Jaqaman K, Das R, Lowell CA, Grinstein S: Actin cytoskeleton reorganization by Syk regulates Fcgamma receptor responsiveness by increasing its lateral mobility and clustering. Dev Cell 2014, 29:534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 49.Ma J, Abram CL, Hu Y, Lowell CA: CARD9 mediates dendritic cell-induced development of Lyn deficiency-associated autoimmune and inflammatory diseases. Sci Signal 2019, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Linking Lyn and CARD9 activation downstream of the integrin CD11b to the development of TLR-induced autoimmunity, highlighting the distinct architecture of these signaling pathways in dendritic cells and macrophages.
- 50.Lim TJF, Bunjamin M, Ruedl C, Su IH: Talin1 controls dendritic cell activation by regulating TLR complex assembly and signaling. J Exp Med 2020, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 51.Morrissey MA, Kern N, Vale RD: CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity 2020, 53:290–302 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]; CD47 directs Sirpα to the immune synapse to prevent phagocytosis by recruiting phosphatases to integrin-mediated activation of the Src-family kinases. Exclusion of inhibitory receptors from the phagocytic synapse is necessary for phagocytosis.
- 52.Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, Launay P, Benhamou M, Blank U, Daugas E, et al. : Lyn and Fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun 2017, 8:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lutz-Nicoladoni C, Wolf D, Sopper S: Modulation of Immune Cell Functions by the E3 Ligase Cbl-b. Front Oncol 2015, 5:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brian BF, Guerrero CR, Freedman TS: Immunopharmacology and Quantitative Analysis of Tyrosine Kinase Signaling. Curr Protoc Immunol 2020, 130:e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brian BF, Sauer ML, Ruis BL, Auger JL, E. SS, Swanson WL, Nunez MG, Moriarity BS, Lowell CA, Binstadt BA, et al. : Splice-specific lyn knockout mice reveal a dominant function of LynB in preventing autoimmunity. bioRxiv 2021. [Google Scholar]
- 56.Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR: Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 1995, 83:301–311. [DOI] [PubMed] [Google Scholar]
- 57.Jordan MB, Hildeman D, Kappler J, Marrack P: An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004, 104:735–743. [DOI] [PubMed] [Google Scholar]
- 58.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, Schiffrin EJ, Foell D, Grom AA, Ammann S, et al. : Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131:1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •• 59.Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, Gessay G, Whalen E, Mason M, Presnell SR, et al. : Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 2019, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]; A novel mechanism by which chronic signaling via endosomal TLR7 and TLR9 leads to the differentiation of a unique population of disease-causing macrophages: inflammatory hemophagocytes.
- 60.Bracaglia C, Prencipe G, De Benedetti F: Macrophage Activation Syndrome: different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J 2017, 15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hunter PJ, Nistala K, Jina N, Eddaoudi A, Thomson W, Hubank M, Wedderburn LR: Biologic predictors of extension of oligoarticular juvenile idiopathic arthritis as determined from synovial fluid cellular composition and gene expression. Arthritis Rheum 2010, 62:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flynn MC, Pernes G, Lee MKS, Nagareddy PR, Murphy AJ: Monocytes, Macrophages, and Metabolic Disease in Atherosclerosis. Front Pharmacol 2019, 10:666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams JW, Zaitsev K, Kim KW, Ivanov S, Saunders BT, Schrank PR, Kim K, Elvington A, Kim SH, Tucker CG, et al. : Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol 2020, 21:1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morrell ED, Wiedeman A, Long SA, Gharib SA, West TE, Skerrett SJ, Wurfel MM, Mikacenic C: Cytometry TOF identifies alveolar macrophage subtypes in acute respiratory distress syndrome. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vasudevan S, Vasquez JJ, Chen W, Aguilar-Rodriguez B, Niemi EC, Zeng S, Tamaki W, Nakamura MC, Arjomandi M: Lower PDL1, PDL2, and AXL Expression on Lung Myeloid Cells Suggests Inflammatory Bias in Smoking and Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol 2020, 63:780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, Shenoy A, Bassat E, Halpern Z, Geiger T, et al. : Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med 2016, 213:2315–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez H, Hagerling C, Werb Z: Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 2018, 32:1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liyasova MS, Ma K, Lipkowitz S: Molecular pathways: cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res 2015, 21:1789–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, Shi H, Zeng Y, Liu C, He J, et al. : Deciphering human macrophage development at single-cell resolution. Nature 2020, 582:571–576. [DOI] [PubMed] [Google Scholar]
- 70.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. : Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. : Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ginhoux F, Guilliams M: Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44:439–449. [DOI] [PubMed] [Google Scholar]
- 73.Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, Ego KM, Bruni CM, Deng Z, Schlachetzki JCM, et al. : Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51:655–670 e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM: Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 2017, 47:913–927 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. : Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47:323–338 e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, Mack M, Beinke S, Wack A: Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol 2020, 21:145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roquilly A, Jacqueline C, Davieau M, Molle A, Sadek A, Fourgeux C, Rooze P, Broquet A, Misme-Aucouturier B, Chaumette T, et al. : Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol 2020, 21:636–648. [DOI] [PubMed] [Google Scholar]