

Abstract
The enzymes involved in H2S homeostasis regulate its production from sulfur-containing amino acids and its oxidation to thiosulfate and sulfate. Two gatekeepers in this homeostatic circuit are cystathionine β-synthase (CBS), which commits homocysteine to cysteine, and sulfide quinone oxidoreductase (SQOR), which commits H2S to oxidation via a mitochondrial pathway. Inborn errors at either locus affect sulfur metabolism, increasing homocysteine derived H2S synthesis in the case of CBS deficiency, and reducing complex IV activity in the case of SQOR deficiency. In this review, we focus on structural perspectives on the reaction mechanisms and regulation of these two enzymes, which are key to understanding H2S homeostasis in health and its dysregulation and potential targeting in disease.
Hydrogen sulfide has dual effects in mammalian energy metabolism, functioning as a respiratory poison and a respiratory substrate for the electron transport chain (ETC). Unlike its reputation as a noxious environmental toxin, the systematic study of the cellular capacity to biosynthesize H2S in mammals has been studied more recently. Of the three enzymes ascribed roles in H2S biogenesis [1], two, cystathionine β-synthase (CBS) and γ-cystathionase (CSE), constitute the transsulfuration pathway while the third, 3-mercaptopyruvate sulfurtransferase (cytoplasmic MPST1 and mitochondrial MPST2), is involved in cysteine catabolism (Figure 1) [2–5]. H2S is not a product of the canonical reactions catalyzed by CBS and CSE but rather, a consequence of their substrate and reaction promiscuities [6]. Cystathionine, produced by CBS, is converted to cysteine by CSE, which in turn, is a substrate for the H2S producing reactions catalyzed by both CBS and CSE [2, 3]. While large gaps remain in our understanding of the relative output of H2S versus cysteine, it is estimated that the predominant cellular transsulfuration flux yields cysteine [7]. The concentrations of CBS versus CSE and of their substrates, influence their relative contributions to H2S synthesis in a tissue-specific manner [8]. MPST is presumably activated under conditions of cysteine excess, although its regulation remains to be fully elucidated [5].
Figure 1. Enzymes involved in H2S homeostasis.
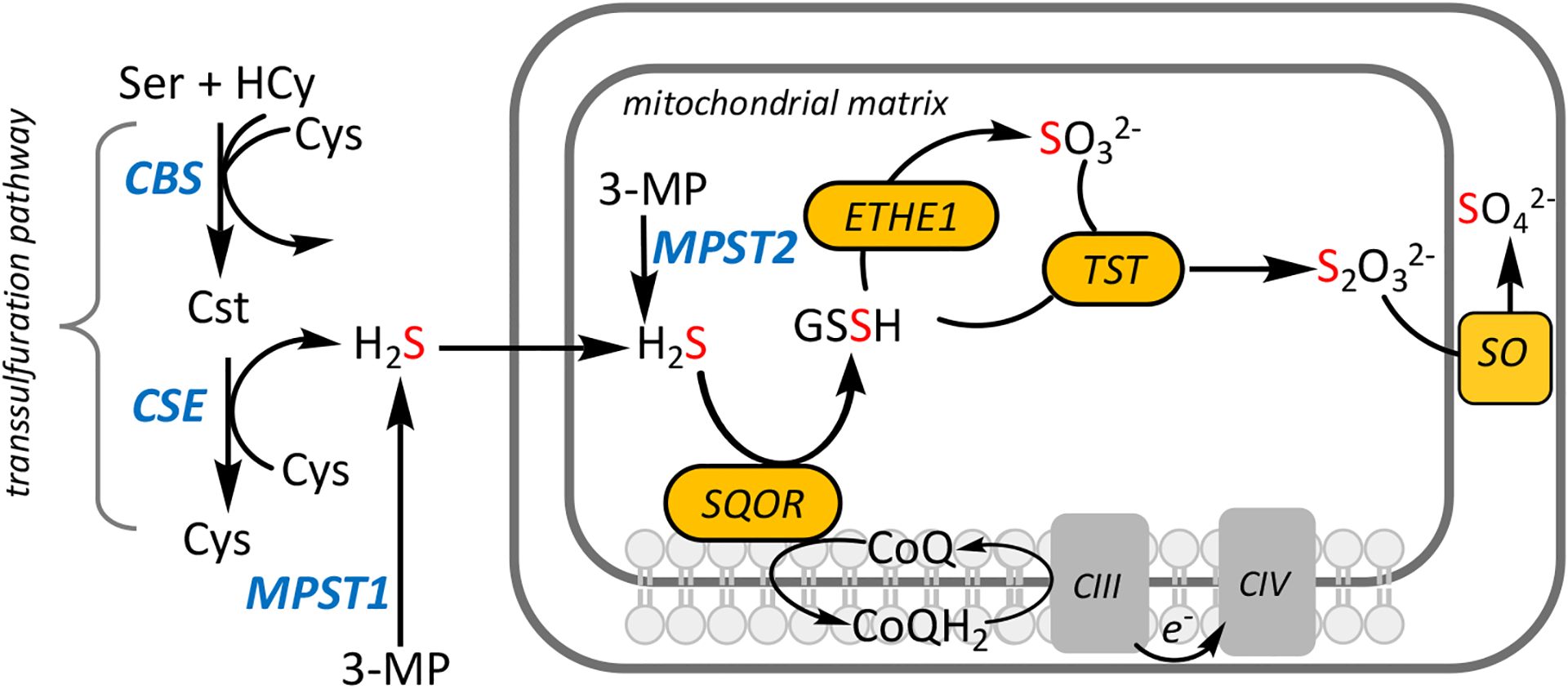
H2S biogenesis is catalyzed by three enzymes (blue): CBS, CSE and MPST while H2S catabolism is catalyzed by enzymes in the mitochondrial sulfide oxidation pathway (yellow). ETHE1, TST and SO denote persulfide dioxygenase, rhodanese and sulfite oxidase, respectively. CIII, CIV, 3-MP, Hcy and Cst denote complexes III and IV, 3-mercaptopyruvate, homocysteine and cystathionine, respectively.
While H2S biogenesis piggybacks on other housekeeping functions, a dedicated pathway exists for sulfide oxidation in mammals and includes sulfide quinone oxidoreductase (SQOR) [9], a persulfide dioxygenase (ETHE1) [10], rhodanese (or TST) [11] and sulfite oxidase [12] (Figure 1). The products of this mitochondrial pathway are thiosulfate and sulfate [13]. Electrons released during sulfide oxidation enter the ETC at the level of complex III (from SQOR via CoQ) and complex IV (from sulfite oxidase via cytochrome c) and establish sulfide as an inorganic substrate for oxidative phosphorylation in mammals [14]. SQOR deficiency presents clinically as Leigh disease and reduced complex IV activity [15]. The wide prevalence of the sulfide oxidation pathway across cell and tissue types suggests an important role for H2S in cellular metabolism.
However, there is scant information on the quantitative significance of H2S production and the cellular contexts in which it is regulated. In this review, we focus on CBS, which commits homocysteine to transsulfuration or H2S synthesis, and SQOR, which commits H2S to oxidation, emphasizing structural perspectives that could be important for regulation.
Structural basis of H2S synthesis by CBS
CBS catalyzes the condensation of serine and homocysteine, generating cystathionine and water (Figure 2a). Alternatively, CBS catalyzes the condensation of cysteine and homocysteine, forming cystathionine and H2S, or eliminates H2S from one or two moles of cysteine to generate pyruvate and lanthionine, respectively. CBS is heavily regulated [16–19] and its dysfunction leads to homocystinuria, an inborn error of metabolism. The increased excretion by homocystinuric patients of homolanthionine [20], a product of the CSE-catalyzed condensation of two moles of homocysteine [2], points to a role for CBS in regulating H2S levels systemically. CBS and CSE can also generate cysteine persulfides from cystine [21, 22].
Figure 2.
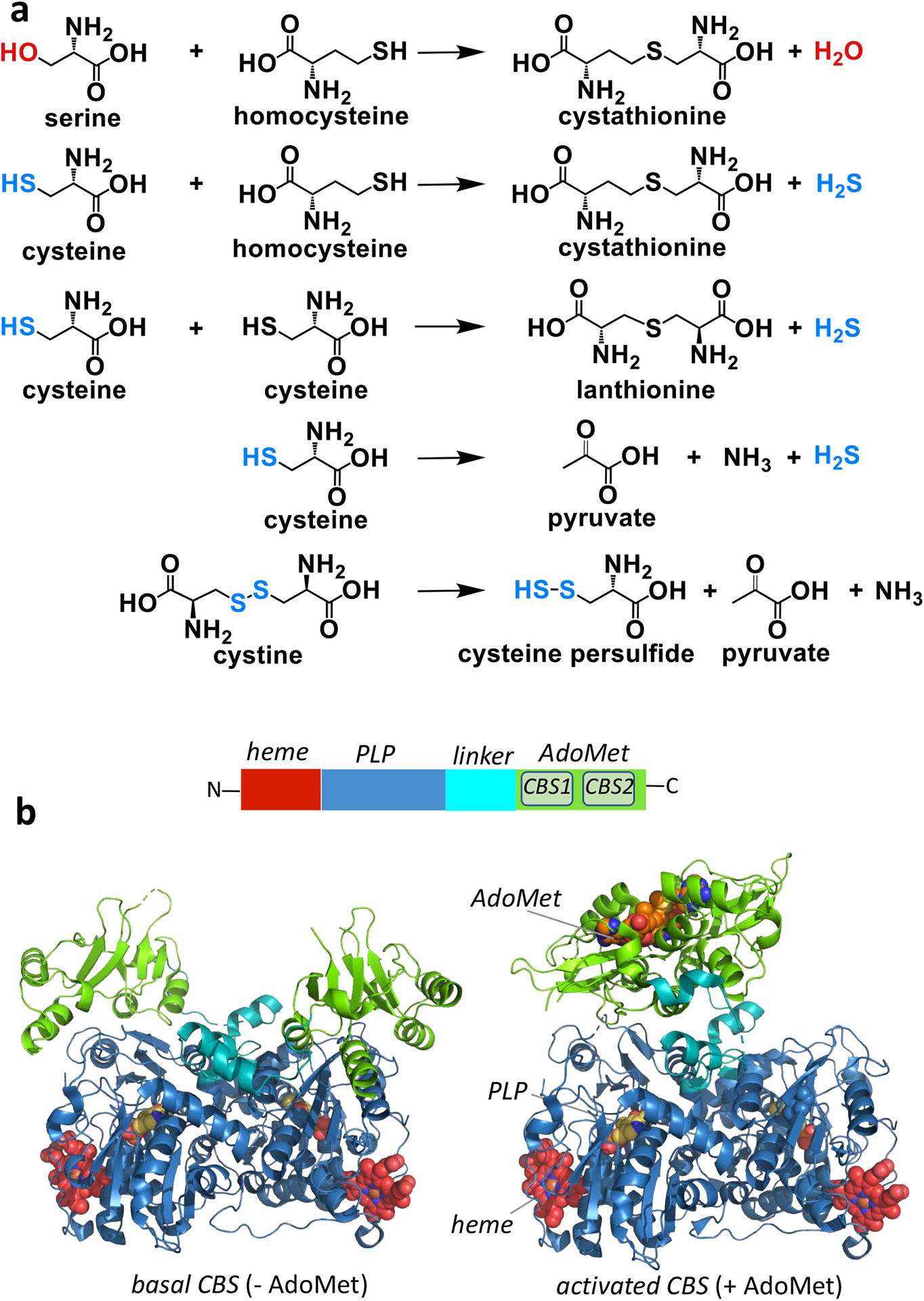
Structure and functions of CBS. (a) Reactions catalyzed by CBS including the canonical transsulfuration reaction utilizing serine, the H2S generating reactions utilizing cysteine (± homocysteine) and the persulfide generating reaction utilizing cystine. (b) Domain organization of human CBS (upper) and the structures of CBS captured in a basal (PDB: 4COO, left) and an activated (PDB: 4PCU right) conformation. The domains are colored as in the key on top and CBS1 and CBS2 refer to the two CBS domains within the C-terminal regulatory segment that binds AdoMet.
Crystal structures of CBS [23–27] have provided a framework for understanding its reaction mechanism and allosteric regulation by AdoMet and heme. CBS is a modular enzyme [28] in which the N- and C-terminal regulatory domains bind heme and S-adenosylmethionine (AdoMet) respectively and sandwich a catalytic core that binds pyridoxal 5´-phosphate (PLP) (Figure 2b). The 8-fold α/β barrel motif of the catalytic core is very similar to other members of the β family of PLP enzymes [27, 29, 30].Two distinct CBS conformations are seen in the absence (basal) and presence (activated) of AdoMet (Figure 2b).
The postulated reaction mechanism for CBS invokes classical β-elimination chemistry [31–33] and begins with an internal aldimine between PLP and Lys-119 (Figure 3a). The PLP cofactor is anchored via Interactions between its phosphate moiety and a glycine rich loop (256Gly-260Thr) and hydrogen bonds with Ser-349 and Asn-149 (Figure 3b) [23]. Binding of serine or cysteine leads to formation of the corresponding external aldimine and is followed by proton abstraction from Cα resulting in a stabilized carbanion (Figure 3a). The carbanion intermediate was captured in a 1.70 Å crystal structure of Drosophila CBS with electrostatic stabilization provided by the ε-nitrogen of Lys-88 (corresponding to human Lys-119) at a distance of 2.1 Å from Cα, [23]. Their high homology (68%) and sequence identity (51%) suggest that the Drosophila CBS is likely to be an excellent model for the human enzyme.
Figure 3. CBS reaction mechanism and structure of two key intermediates.
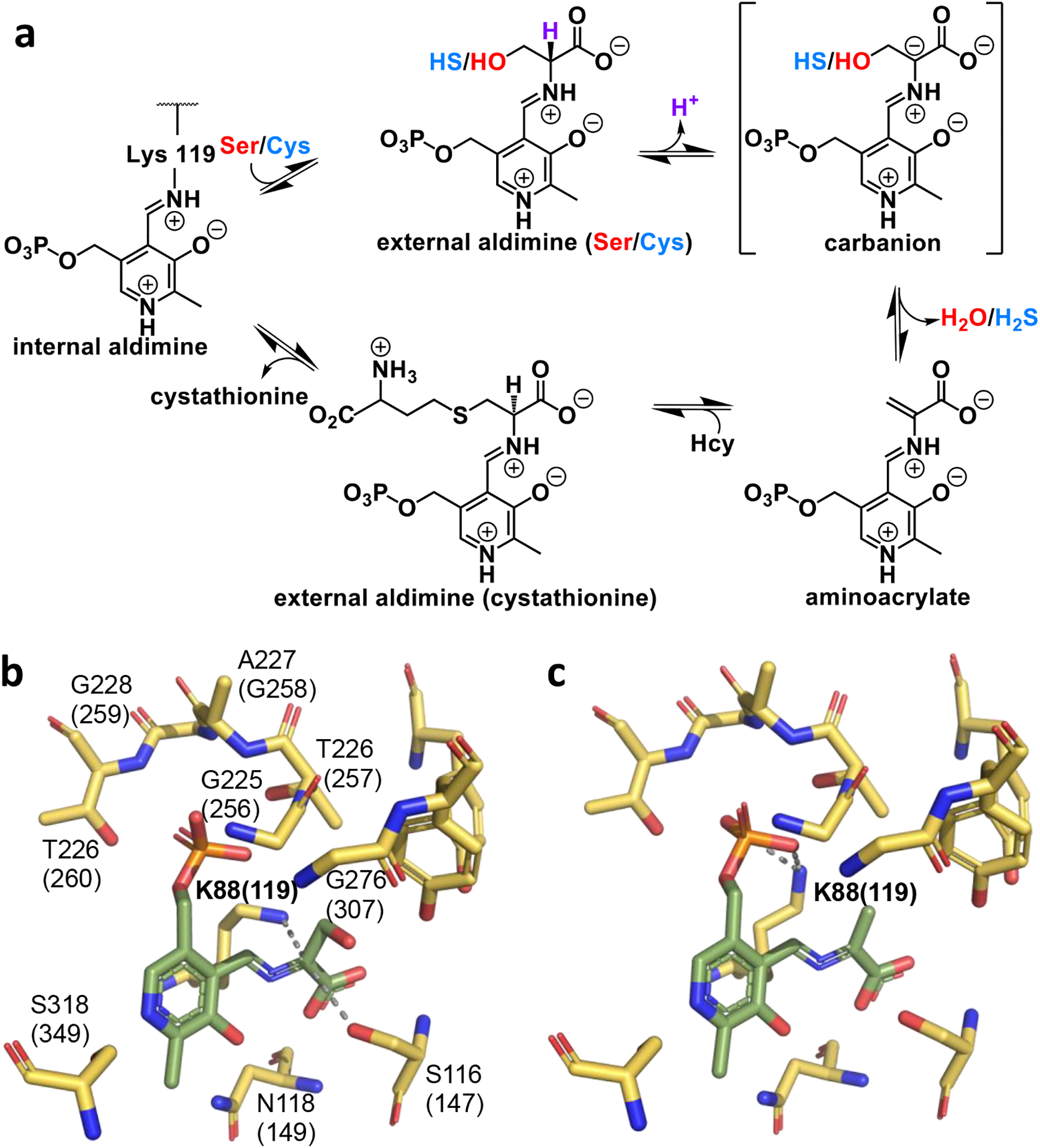
(a) Postulated reaction mechanism for CBS-catalyzed H2O or H2S elimination. (b,c) Close-up of the structures of full-length Drosophila CBS in which a carbanion (PDB:3PC4) and an aminoacrylate (PDB: 3PC3) intermediate are seen. The residues and numbering in human CBS are shown in parentheses. Lys-88 (corresponding to Lys-119 in human CBS) is 2.5 Å from Cα providing electrostatic stabilization of the carbanion intermediate (b) but is rotated away and engaged in a hydrogen-bonding interaction with a phosphate oxygen in the aminoacrylate species (c). The PLP bound intermediates are shown in green.
The carbanion is converted to an aminoacrylate intermediate following β-elimination of water (from serine) or H2S (from cysteine) (Figure 3a). The 1.55 Å crystal structure of the aminoacrylate intermediate revealed that the side chain of Lys-88 is rotated away from Cα and engaged in a hydrogen bonding interaction with a phosphate oxygen [23] (Figure 3c). Since the heme cofactor obscures PLP-associated spectral changes, difference stopped-flow spectroscopy was used to monitor the internal aldimine (λmax = 406 nm) and its conversion to the aminoacrylate intermediate (λmax = 465 nm) [23]. The aminoacrylate was also observed in the heme-less variant of human CBS, which is less stable [34], and in yeast CBS, which lacks the heme cofactor [35]. The catalytic cycle is completed upon addition of homocysteine, forming an external aldimine with cystathionine, followed by a transchiffization reaction and product release.
In H2S generating reactions in which one or two moles of cysteine are involved (Figure 2a), water or cysteine add to the aminoacrylate intermediate forming the external aldimine with serine or lanthionine, respectively. Serine can subsequently undergo α,β-elimination to form pyruvate and ammonia [3].
Structural basis of heme-dependent regulation of CBS
The heme in CBS is housed in a hydrophobic pocket comprising residues 50–67 (Figure 4a) [26, 27]. The heme iron (Fe2+/Fe3+) is coordinated by His-65 and Cys-52 while the latter is also engaged in an ionic interaction with Arg-166 located at one end of an α-helix, which connects to the PLP pocket at the other end [36, 37]. The heme is tightly bound but was released from CBS crystals under reducing conditions following addition of CO [38]. The heme is not essential for activity since the heme-less variant lacking the N -terminal 69 residues retains ~40% of wild-type CBS activity [34]. The CO-induced heme loss from CBS suggests that a ligand-induced protein destabilization could be deployed as a regulatory strategy.
Figure 4. Regulatory domains in CBS.
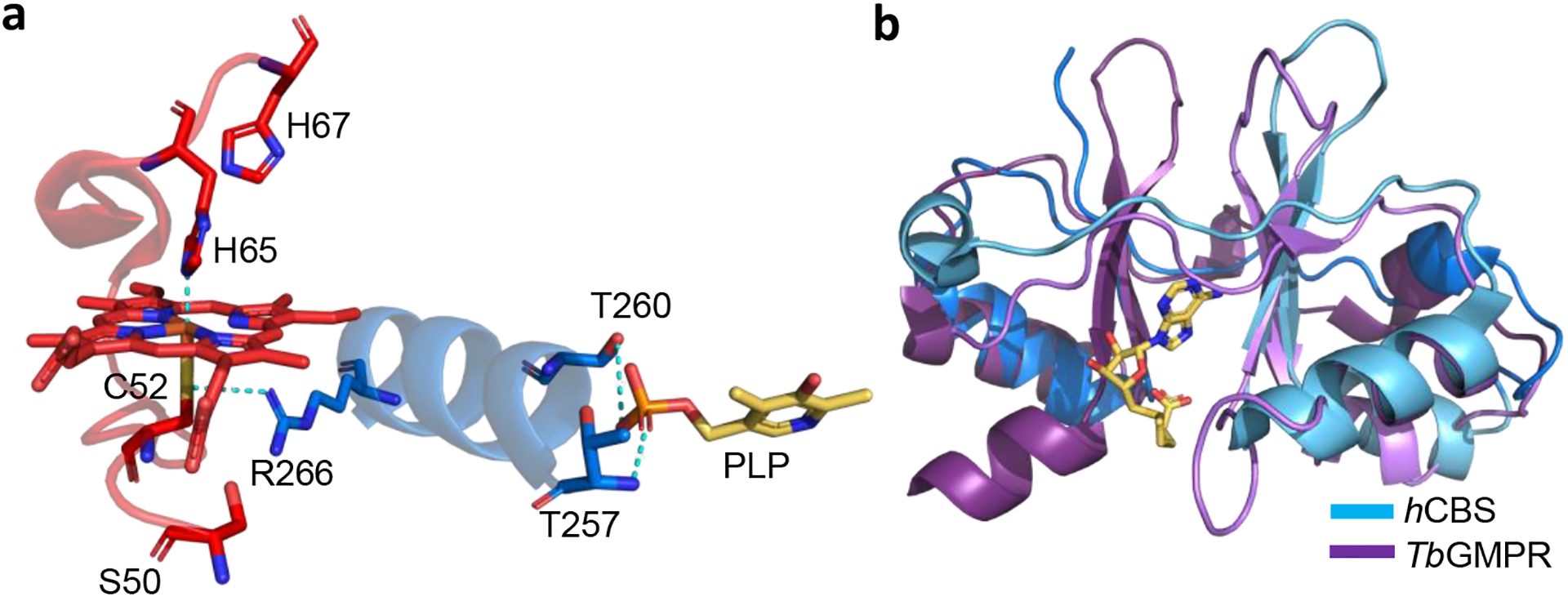
(a) Close-up showing the connection between the heme and PLP pockets in the structure of human CBS. Residues comprising the hydrophobic heme pocket are shown in red. His-65 and Cys-52 serve as the heme ligands while Cys-52 is also involved in an electrostatic interaction with Arg-266. (b) Overlay of the CBS domains in human CBS (PDB: 4PCU, blue) and T. brucei GMP reductase (PDB: 6JL8, purple). AdoMet bound to human CBS is shown in yellow stick display.
Changes in the heme coordination environment influence CBS activity, although the context in which heme-dependent regulation is physiologically important is not entirely clear [39]. Binding of CO to ferrous CBS displaces Cys-52, forming a 6-corodinate species in which the His-65 ligand is retained [40]. On the other hand, NO• displaces His-65, which is followed by loss of Cys-52 coordination and formation of a 5-coordinate ferrous-nitrosyl species [41]. Both CO and NO• binding are correlated with inhibition of CBS activity [41, 42], which is presumed to arise from loss of the interaction between Cys-52 and Arg-266 and is correlated with a tautomeric shift from the active ketoeneamine to the inactive enolimine form of PLP [36, 37]. Air exposure leads to recovery of active 6-coordinate ferric CBS. Surprisingly, despite the >50 Å distance separating them, AdoMet increases the affinity of heme for NO• (2-fold) and CO (5-fold) and sensitizes CBS to inhibition [43]. AdoMet also increases the rate constant for CO binding by ~10-fold and for NO• by 1.5-fold [43]. Hence, in the interplay between the N- and C-terminal regulatory domains, heme trumps AdoMet as an allosteric regulator. The structural basis for the long-range allosteric communication from the AdoMet-bound C-terminal and the N-terminal heme domain is not understood.
The structural basis for AdoMet-dependent regulation of CBS
CBS is a molecular jumping jack alternating between the lateral and overhead extension of its C-terminal regulatory side arms as it transitions from the basal to the activated state in response to AdoMet (Figure 2b). The C-terminal domain houses a tandem repeat of the Bateman module, better known as the CBS domain [44], which extends between residues 412–471 (CBS1) and 472–538 (CBS2) [45]. In the basal conformation, the C-terminal domain of one subunit sits atop the catalytic core of the other subunit and impedes substrate access to the active site, lowering activity (Figure 2b). Upon AdoMet binding, the regulatory domains dimerize above the active site, increasing substrate access and is correlated with a 2 to 4-fold increase in activity for the canonical and the H2S-generating reactions [3, 46]. Full-length human CBS is prone to aggregation and it is likely that structural elements in the C-terminal domain are responsible for this behavior, since its truncation leads to a well-behaved CBS dimer [47]. A stable dimer is also formed upon deletion of a 10-residue loop (516–525) in the CBS2 domain in full-length human CBS [24]. Residues 513–519 are postulated to act as a hook, locking two dimers into a tetramer.
The CBS domains in the Trypanosoma brucei GMP reductase play a role in its oligomerization [48]. In the presence of GMP, the enzyme exhibits the native octameric state while in the presence of ATP, it separates into two tetramers, which is accompanied by a 1.6-fold decrease in activity [48]. Deletion of the CBS domain leads to formation of the tetrameric enzyme, which is however, inactive. Given the similarities between the CBS domains of GMP reductase and CBS (Figure 4b), it is plausible that this region is similarly involved in ligand induced oligomeric changes in human CBS with potential regulatory implications.
Sulfide quinone oxidoreductase gates H2S clearance
SQOR belongs to the flavin disulfide reductase superfamily [49] and is anchored to the inner mitochondrial membrane via two amphipathic C-terminal helices, while the remainder of the protein faces the matrix [50, 51]. SQOR catalyzes the committing step in the sulfide oxidation pathway, converting H2S to a persulfide while concomitantly reducing CoQ (Figure 5). The overall reaction proceeds via two half reactions. In the first, sulfide adds into the cysteine trisulfide, forming persulfides on Cys-201 and Cys-379. In the second half-reaction, electrons from FADH2 are transferred to CoQ, which then connects to the ETC [52]. Bacterial and human SQORs differ in important details. While the oxidized sulfur exits the human enzyme as a persulfide product after each catalytic cycle [13, 53, 54], bacterial SQORs successively add sulfide, building polysulfide chains [55–57]. Human SQOR resembles flavocytochrome c sulfide dehydrogenase in its active site architecture and its catalytic mechanism, and, following addition of sulfide into the cysteine disulfide, the sulfane sulfur is transferred to a second equivalent of sulfide to form HSSH, while the electrons are transferred to cytochrome c [58].
Figure 5. Proposed catalytic mechanism for SQOR.
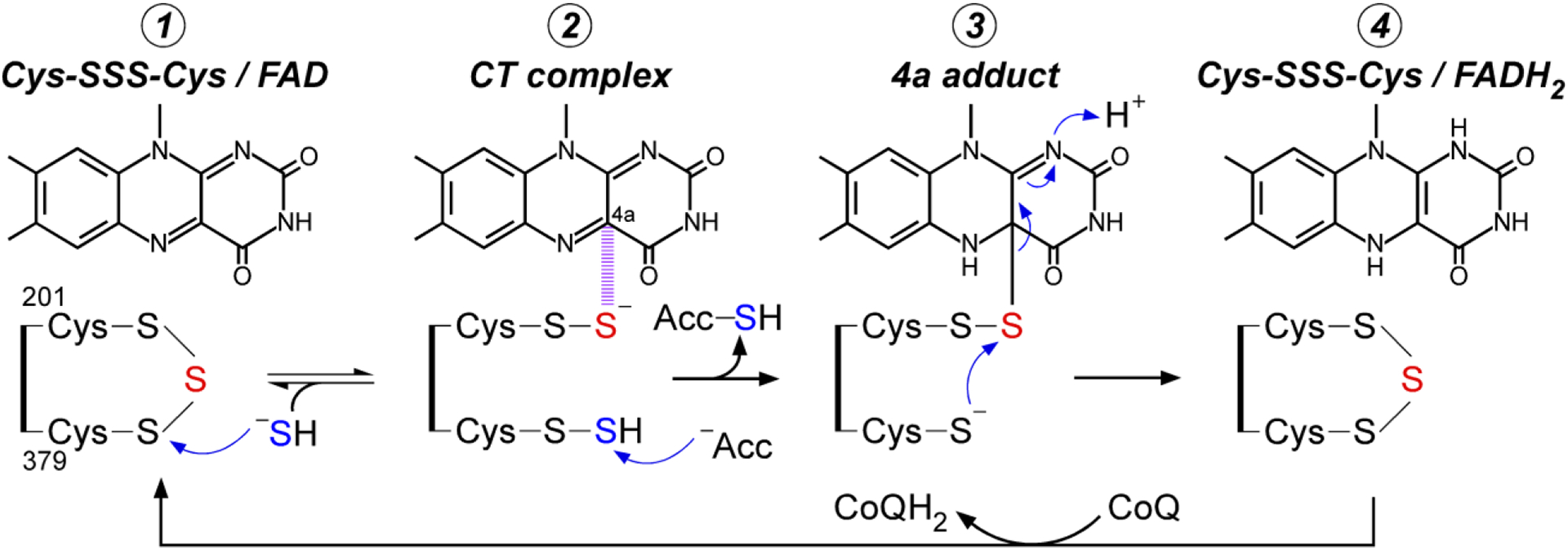
The bridging sulfur of the cysteine trisulfide is shown in red and the sulfur undergoing oxidation is shown in blue. The dashed magenta lines denote a charge transfer (CT) complex between the Cys-201 persulfide and the flavin C4a, which leads to formation of the 4a adduct followed by FADH2.
The overall structure of SQOR exhibits the typical characteristics of the flavin disulfide reductase superfamily, including tandem Rossmann fold repeats that accommodate a non-covalently bound FAD and two redox-active cysteines (Figure 6a). The first Rossmann fold binds the ADP moiety of FAD and presents much of the negative charge to the matrix-facing domain of the enzyme. The second Rossmann fold has electropositive patches that presumably facilitate interaction with membrane phospholipid head groups. Unexpectedly, the crystal structures of human SQOR revealed a cysteine trisulfide (Figure 6a, inset), the first reported in a flavin disulfide reductase family member [50, 51]. The structures revealed a large electropositive cavity for substrate entry, leading to the re face of FAD while CoQ was bound to the si face at the end of a hydrophobic tunnel that presumably leads to the membrane.
Figure 6. Structure of human SQOR.
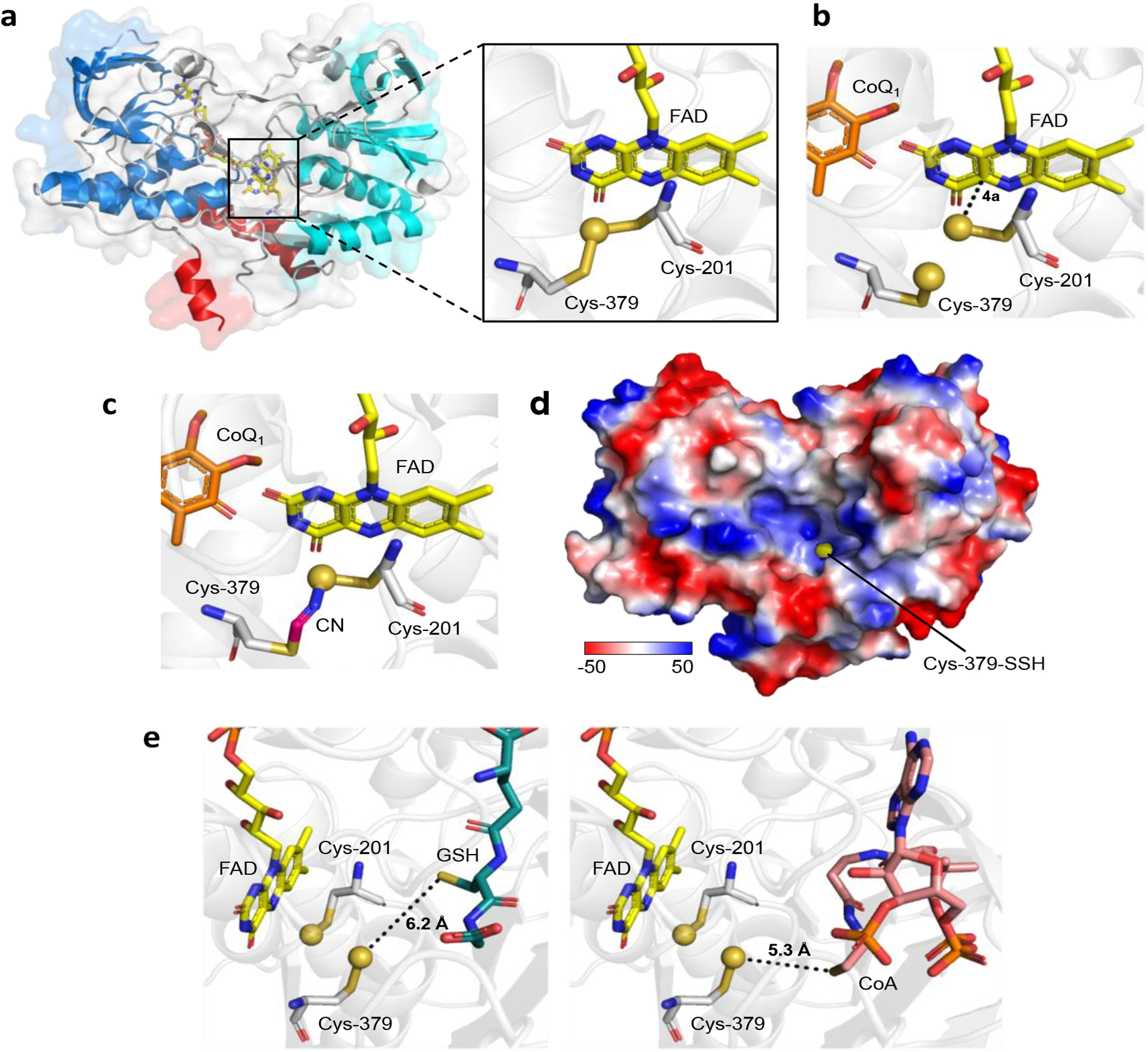
(a) Overall structure of native SQOR (PDB ID: 6OI5), with the first and second Rossmann fold domains shown in blue and cyan, respectively, and membrane-anchoring helices shown in in red. The inset is a close-up of the active site showing the cysteine trisulfide. (b) SQOR co-crystallized with CoQ (orange sticks; PDB ID: 6OIB), contains a persulfide pair, corresponding to [2] in Figure 5. (c) The SQOR Cys-379 N-(201Cys-disulfanyl)-methanimido thioate intermediate generated upon addition of cyanide (CN; PDB ID: 6WH6). (d) Electrostatic surface potential map showing the solvent exposure of Cys-379-SSH at the bottom of the electropositive cavity. (e) Models of physological acceptors docked in the SQOR cavity. The distances between the Cys-379 persulfide and the thiol moieties of GSH (left panel, teal sticks) and CoA (right panel, pink sticks) are noted. The sulfane sulfurs in all panels are shown as yellow spheres.
A cysteine trisulfide catalyzes H2S oxidation
The cysteine trisulfide in SQOR exposes the Sγ of Cys-379 to bulk solvent while Cys-201 lies within a 9-residue loop that shields the remainder of the active site [50, 51]. Molecular dynamic simulations predict a greater electrophilic character at Cys-379 and susceptibility to nucleophilic attack by sulfide [59]. The Cys-379-SSH and Cys-201-SSH persulfide pair formed upon addition of sulfide to the trisulfide was observed in an SQOR structure (Figure 6b) [50]. The Cys-201-SSH is 3 Å from C4a in FAD, forming an unusually intense charge transfer complex centered at 675 nm [53, 60]. Protrusion of the Cys-379-SSH out of the active site facilitates its transfer to a thiophilic acceptor, reforming the resting trisulfide. Addition of sulfide presumably led to the in crystallo reduction of CoQ and led to its relocation to the entrance of the hydrophobic tunnel [50].
The presence of the cysteine trisulfide in SQOR was confirmed by its sensitivity to cyanolysis [50, 59]. Cyanide initially adds into the trisulfide to form a charge transfer complex, which slowly collapses to form a Cys-379 N-(201Cys-disulfanyl)-methanimido thioate intermediate (Figure 6c) [59]. Attack by a second cyanide extracts the sulfane sulfur, yielding thiocyanate. Post-cyanolysis, SQOR is inactive and thermally unstable and harbors a Cys-379 N-(201Cys-sulfanyl)-methanimido thioate species. The bridging intermediate preserves the oxidation state at both cysteines, and as a result, sulfide addition can reinstall the bridging sulfur and regenerate the cysteine trisulfide. Based on these findings, a mechanism for the initial formation of the cysteine trisulfide in vivo has been proposed [9], but remains to be tested.
SQOR accommodates a diversity of substrates
The large electropositive cavity leading to the SQOR active site confers a remarkable degree of substrate promiscuity (Figure 6d). While a similar opening is present in flavocytochrome c sulfide dehydrogenase [58], the active site is capped in bacterial SQORs and accommodates the growing polysulfide product [55–57]. Sulfite, an efficient sulfane sulfur acceptor [53], is proposed to dock in close proximity to Cys-379, with Ser-343 and Ala-202 providing hydrogen bonding interactions [51]. The docked structures of GSH and CoA reveal that their thiol moieties are oriented toward Cys-379 (Figure 6e) [50, 51].
Both the size and the pKa of the sulfane sulfur acceptors influence their catalytic efficiency. The larger acceptors GSH and CoA (pKa = 9.2 and 9.6, respectively), exhibit an ~10 to 20-fold lower kcat/KM versus methanethiol (pKa = 10.4), which in turn, has a 6-fold lower kcat/KM than sulfite (pKa = 7.2) [50, 61]. The direct addition of some acceptors, like methanethiol and sulfite traps the enzyme in a dead-end complex, which decays slowly and regenerates active SQOR [61]. The slow kon values for the acceptors and/or their low intracellular abundance likely reduce their cellular relevance, although pathological conditions, e.g., sulfite oxidase deficiency [62], could promote adventitious reactions.
Conclusions
The structures of CBS and SQOR, two important gatekeepers of cellular H2S homeostasis, have revealed many surprises and provided a framework for interrogating their regulation. The role of the heme and the mechanism of long-range interaction between the N- and C-terminal domains in CBS to exert allosteric control, are poorly understood. Furthermore, the high KM values for the CBS substrates raise the question as to whether other metabolites or proteins modulate substrate affinities and bring them within physiologically relevant ranges. The novel trisulfide configuration in SQOR helps explain the enhanced rate of the sulfide addition reaction but raises questions as to how the cofactor is installed and what the source of the bridging sulfur is.
Finally, the cavernous entrance and solvent exposure of the electrophilic sulfur in the trisulfide cofactor, begs the question as to how adventitious side reactions are averted. Continued insights into the enzymology of H2S homeostasis will be important for shaping rational targeting efforts.
Acknowledgement
This work was supported in part by the grants from the National Institutes of Health (GM130183 to RB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Disclosure Statement
RB is a paid member of the scientific advisory board of Apneo Therapeutics and owns equity in the company.
References and Recommended Readings
•Special interest
••Outstanding interest
- [1].Kabil O, Vitvitsky V, Banerjee R. Sulfur as a signaling nutrient through hydrogen sulfide. Ann Rev Nutr 2014; 34:171–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chiku T, Padovani D, Zhu W et al. H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J Biol Chem 2009; 284:11601–11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Singh S, Padovani D, Leslie RA et al. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem 2009; 284:22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yadav PK, Yamada K, Chiku T et al. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J Biol Chem 2013; 288:20002–20013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yadav PK, Vitvitsky V, Carballal S et al. Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically-relevant concentrations. J Biol Chem 2020; 295:6299–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Banerjee R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr Opin Chem Biol 2017; 37:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kabil O, Yadav V, Banerjee R. Heme-dependent metabolite switching regulates H2S synthesis in response to ER stress. J Biol Chem 2016; 291:16418–16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kabil O, Vitvitsky V, Xie P, Banerjee R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal 2011; 15:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Landry AP, Ballou DP, Banerjee R. Hydrogen sulfide oxidation by sulfide quinone oxidoreductase. Chembiochem 2021; 22:949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kabil O, Banerjee R. Characterization of Patient Mutations in Human Persulfide Dioxygenase (ETHE1) Involved in H2S Catabolism. J Biol Chem 2012; 287:44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Libiad M, Sriraman A, Banerjee R. Polymorphic variants of human rhodanese exhibit differences in thermal stability and sulfur transfer kinetics. J Biol Chem 2015; 290:23579–23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kisker C, Schindelin H, Pacheco A et al. Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell 1997; 91:973–983. [DOI] [PubMed] [Google Scholar]
- [13].Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 2008; 275:3352–3361. [DOI] [PubMed] [Google Scholar]
- [14].Goubern M, Andriamihaja M, Nubel T et al. Sulfide, the first inorganic substrate for human cells. FASEB J 2007; 21:1699–1706. [DOI] [PubMed] [Google Scholar]
- [15].Friederich MW, Elias AF, Kuster A et al. Pathogenic variants in SQOR encoding sulfide:quinone oxidoreductase are a potentially treatable cause of Leigh disease. J Inherit Metab Dis 2020; 43:1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••Reports the first hereditary mutations in SQOR that present as Leigh disease
- [16].Niu WN, Yadav PK, Adamec J, Banerjee R. S-glutathionylation enhances human cystathionine beta-synthase activity under oxidative stress conditions. Antioxid Redox Signal 2015; 22:350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kabil O, Weeks CL, Carballal S et al. Reversible Heme-Dependent Regulation of Human Cystathionine beta-Synthase by a Flavoprotein Oxidoreductase. Biochemistry 2011; 50:8261–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kabil O, Zhou Y, Banerjee R. Human cystathionine beta-synthase is a target for sumoylation. Biochemistry 2006; 45:13528–13536. [DOI] [PubMed] [Google Scholar]
- [19].Kriebitzsch C, Verlinden L, Eelen G et al. 1,25-dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3-E1 cells by direct regulation of cystathionine beta-synthase. J Bone Miner Res 2011; 26:2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Perry TL, Hansen S, MacDougall L. Homolanthionine excretion in homocystinuria. Science 1966; 152:1750–1752. [DOI] [PubMed] [Google Scholar]
- [21].Ida T, Sawa T, Ihara H et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A 2014; 111:7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yadav PK, Martinov M, Vitvitsky V et al. Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J Am Chem Soc 2016; 138:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Koutmos M, Kabil O, Smith JL, Banerjee R. Structural basis for substrate activation and regulation by cystathionine beta-synthase domains in cystathionine beta-synthase. Proc Natl Acad Sci USA 2010; 107:20958–20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ereno-Orbea J, Majtan T, Oyenarte I et al. Structural basis of regulation and oligomerization of human cystathionine beta-synthase, the central enzyme of transsulfuration. Proc Natl Acad Sci U S A 2013; 110:E3790–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ereno-Orbea J, Majtan T, Oyenarte I et al. Structural insight into the molecular mechanism of allosteric activation of human cystathionine beta-synthase by S-adenosylmethionine. Proc Natl Acad Sci U S A 2014; 111:E3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Meier M, Janosik M, Kery V et al. Structure of human cystathionine beta-synthase: a unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J 2001; 20:3910–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Taoka S, Lepore BW, Kabil O et al. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry 2002; 41:10454–10461. [DOI] [PubMed] [Google Scholar]
- [28].Taoka S, Widjaja L, Banerjee R. Assignment of enzymatic functions to specific regions of the PLP-dependent heme protein cystathionine beta-synthase. Biochemistry 1999; 38:13155–13161. [DOI] [PubMed] [Google Scholar]
- [29].Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5’-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem 1994; 219:953–960. [DOI] [PubMed] [Google Scholar]
- [30].Christen P, Mehta PK. From cofactor to enzymes. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Chem Rec 2001; 1:436–447. [DOI] [PubMed] [Google Scholar]
- [31].Drewe WF Jr., Dunn MF. Detection and identification of intermediates in the reaction of L-serine with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry 1985; 24:3977–3987. [DOI] [PubMed] [Google Scholar]
- [32].Gallagher DT, Gilliland GL, Xiao G et al. Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure 1998; 6:465–475. [DOI] [PubMed] [Google Scholar]
- [33].Rabeh WM, Cook PF. Structure and mechanism of O-acetylserine sulfhydrylase. J Biol Chem 2004; 279:26803–26806. [DOI] [PubMed] [Google Scholar]
- [34].Evande R, Ojha S, Banerjee R. Visualization of PLP-bound intermediates in hemeless variants of human cystathionine beta-synthase: evidence that lysine 119 is a general base. Arch Biochem Biophys 2004; 427:188–196. [DOI] [PubMed] [Google Scholar]
- [35].Taoka S, Banerjee R. Stopped-flow kinetic analysis of the reaction catalyzed by the full length yeast cystathionine beta synthase. J. Biol. Chem 2002; 277:22421–22425. [DOI] [PubMed] [Google Scholar]
- [36].Weeks CL, Singh S, Madzelan P et al. Heme regulation of human cystathionine beta-synthase activity: insights from fluorescence and Raman spectroscopy. J Am Chem Soc 2009; 131:12809–12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Singh S, Madzelan P, Stasser J et al. Modulation of the heme electronic structure and cystathionine beta-synthase activity by second coordination sphere ligands: The role of heme ligand switching in redox regulation. J Inorg Biochem 2009; 103:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bruno S, Schiaretti F, Burkhard P et al. Functional properties of the active core of human cystathionine beta-synthase crystals. J Biol Chem 2001; 276:16–19. [DOI] [PubMed] [Google Scholar]
- [39].Singh S, Madzelan P, Banerjee R. Properties of an unusual heme cofactor in PLP-dependent cystathionine beta-synthase. Nat. Prod. Rep 2007; 24:631–639. [DOI] [PubMed] [Google Scholar]
- [40].Green EL, Taoka S, Banerjee R, Loehr TM. Resonance Raman characterization of the heme cofactor in cystathionine beta-synthase. Identification of the Fe-S(Cys) vibration in the six-coordinate low-spin heme. Biochemistry 2001; 40:459–463. [DOI] [PubMed] [Google Scholar]
- [41].Taoka S, Banerjee R. Characterization of NO binding to human cystathionine [beta]-synthase:Possible implications of the effects of CO and NO binding to the human enzyme. J Inorg Biochem 2001; 87:245–251. [DOI] [PubMed] [Google Scholar]
- [42].Puranik M, Weeks CL, Lahaye D et al. Dynamics of Carbon Monoxide Binding to Cystathionine beta-Synthase. J Biol Chem 2006; 281:13433–13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vicente JB, Colaco HG, Sarti P et al. S-Adenosyl-l-methionine Modulates CO and NO* Binding to the Human H2S-generating Enzyme Cystathionine beta-Synthase. J Biol Chem 2016; 291:572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 1997; 22:12–13. [DOI] [PubMed] [Google Scholar]
- [45].Ereno-Orbea J, Oyenarte I, Martinez-Cruz LA. CBS domains: Ligand binding sites and conformational variability. Arch Biochem Biophys 2013; 540:70–81. [DOI] [PubMed] [Google Scholar]
- [46].Finkelstein JD, Kyle WE, Martin JL, Pick AM. Activation of cystathionine synthase by adenosylmethionine and adenosylethionine. Biochem Biophys Res Commun 1975; 66:81–87. [DOI] [PubMed] [Google Scholar]
- [47].Kery V, Poneleit L, Kraus JP. Trypsin cleavage of human cystathionine beta-synthase into an evolutionarily conserved active core: structural and functional consequences. Arch Biochem Biophys 1998; 355:222–232. [DOI] [PubMed] [Google Scholar]
- [48].Imamura A, Okada T, Mase H et al. Allosteric regulation accompanied by oligomeric state changes of Trypanosoma brucei GMP reductase through cystathionine-beta-synthase domain. Nat Commun 2020; 11:1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Argyrou A, Blanchard JS. Flavoprotein disulfide reductases: advances in chemistry and function. Progress in nucleic acid research and molecular biology 2004; 78:89–142. [DOI] [PubMed] [Google Scholar]
- [50].Landry AP, Moon S, Kim H et al. A catalytic trisulfide in human sulfide quinone oxidoreductase catalyzes coenzyme A persulfide synthesis and inhibits butyrate oxidation. Cell Chem Biol 2019; 26:1515–1525 e1514. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••Presents in crystallo snapshots of the SQOR reaction coordinate, biochemically confirming the cysteine trisulfide as the active form of the enzyme, and characterizes CoA as a physiological acceptor.
- [51].Jackson MR, Loll PJ, Jorns MS. X-Ray Structure of Human Sulfide:Quinone Oxidoreductase: Insights into the Mechanism of Mitochondrial Hydrogen Sulfide Oxidation. Structure 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Reports the first structure of human SQOR but assigns the cysteine trisulfide as the inactive form of the enzyme.
- [52].Goubern M, Andriamihaja M, Nubel T et al. Sulfide, the first inorganic substrate for human cells. FASEB Journal 2007; 21:1699–1706. [DOI] [PubMed] [Google Scholar]
- [53].Jackson MR, Melideo SL, Jorns MS. Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 2012; 51:6804–6815. [DOI] [PubMed] [Google Scholar]
- [54].Libiad M, Yadav PK, Vitvitsky V et al. Organization of the human mitochondrial sulfide oxidation pathway. J Biol Chem 2014; 289:30901–30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brito JA, Sousa FL, Stelter M et al. Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 2009; 48:5613–5622. [DOI] [PubMed] [Google Scholar]
- [56].Cherney MM, Zhang Y, Solomonson M et al. Crystal structure of sulfide:quinone oxidoreductase from Acidithiobacillus ferrooxidans: insights into sulfidotrophic respiration and detoxification. Journal of Molecular Biology 2010; 398:292–305. [DOI] [PubMed] [Google Scholar]
- [57].Marcia M, Ermler U, Peng G, Michel H. The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proceedings of the National Academy of Sciences of the United States of America 2009; 106:9625–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen ZW, Koh M, Van Driessche G et al. The structure of flavocytochrome c sulfide dehydrogenase from a purple phototrophic bacterium. Science 1994; 266:430–432. [DOI] [PubMed] [Google Scholar]
- [59].Landry AP, Moon S, Bonanata J et al. Dismantling and rebuilding the trisulfide cofactor demonstrates Its essential role in human sulfide quinone oxidoreductase. J Am Chem Soc 2020; 142:14295–14306. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••Establishes the catalytic competency of the SQOR cysteine trisulfide upon reversibly removing and reinstalling the bridging sulfur.
- [60].Mishanina TV, Yadav PK, Ballou DP, Banerjee R. Transient Kinetic Analysis of Hydrogen Sulfide Oxidation Catalyzed by Human Sulfide Quinone Oxidoreductase. J Biol Chem 2015; 290:25072–25080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Landry AP, Ballou DP, Banerjee R. Modulation of catalytic promiscuity during hydrogen sulfide oxidation. ACS Chem Biol 2018; 13:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shih VE, Abroms IF, Johnson JL et al. Sulfite oxidase deficiency. Biochemical and clinical investigations of a hereditary metabolic disorder in sulfur metabolism. N. Engl. J. Med 1977; 297:1022–1028. [DOI] [PubMed] [Google Scholar]