

Abstract
Oncogenic mutations in the KRAS gene are found in 30–50% of colorectal cancers (CRC), and recent findings have demonstrated independent and nonredundant roles for wild-type and mutant KRAS alleles in governing signaling and metabolism. Here, we quantify proteomic changes manifested by KRAS mutation and KRAS allele loss in isogenic cell lines. We show that the expression of KRASG13D upregulates aspartate metabolizing proteins including PCK1, PCK2, ASNS, and ASS1. Furthermore, differential expression analyses of transcript-level data from CRC tumors identified the upregulation of urea cycle enzymes in CRC. We find that expression of ASS1 supports colorectal cancer cell proliferation and promotes tumor formation in vitro. We show that loss of ASS1 can be rescued with high levels of several metabolites.
Keywords: aspartate, colorectal cancer, metabolomics, mutant KRAS, quantitative proteomics, urea cycle
Introduction
Oncogenic mutations in the KRAS gene are found in over 30% of colorectal adenocarcinomas, yet KRAS remains a notoriously difficult therapeutic target [1]. At the molecular level, the majority of missense mutations in KRAS occur in codons 12 and 13, leading to constitutive signaling through the small GTPase protein that the KRAS gene encodes. These activating mutations drive proliferative signaling programs independent of upstream input from growth factor receptors [2]. Both codon 12 and codon 13 encode neighboring glycine residues (Gly12 and Gly13); however, mutations in these sites do not yield equivalent signaling outputs [3]. For example, cultured colorectal cancer cells with a KRASG12V or KRASG13D mutation exhibit distinct phosphoproteome profiles [4]. Moreover, in primary colorectal cancer (CRC), patients with a Gly13 mutation have a worse prognosis than patients with a Gly12 mutation, even though Gly13 mutations seem to have a lower transforming potential [5]. The differences manifested by independent KRAS mutations can also be seen in the response to chemotherapy. It has been reported that individuals with Glycine-13 mutations respond better to therapies targeting the epidermal growth factor receptor (EGFR) than individuals with Glycine-12 mutations in metastatic colorectal cancer [6]. Critically, the amino acid that is substituted for Glycine-12 or Glycine-13 can also dictate possible therapeutic interventions. For example, KRASG12C-mutant cancers can be targeted by electrophilic compounds that irreversibly bond to the mutated cysteine residue [7]. This has led to a promising phase I clinical trial against KRASG12C-mutant colorectal cancer [8]. However, to date, specific interventions targeting KRASG12D and KRASG13D remain more elusive. Beyond individual mutations, it is also appreciated that allele-specific alterations impact cellular functions downstream of mutant KRAS [9,10]. Cells heterozygous for mutant KRAS exhibit an altered feedback loop to drive mitogen-activated protein kinase (MAPK) signaling [9]. Moreover, colorectal cancer cells only expressing KRAS−/G13D are distinctly susceptible to the inhibition of oxidative phosphorylation and mitochondrial translation [11].
To confound the biological complexity arising from distinct KRAS mutations, CRC is a heterogeneous disease as a whole. Nonetheless, KRAS mutation and upregulation of metabolic genes are defining features of one molecular subtype of CRC and recent work has highlighted the role of cellular metabolism, and specifically glycolysis, in CRC [11–14]. However, more research is needed to describe allele- and mutation-specific changes in KRAS signaling and downstream effectors in metabolism and tumor progression.
Therefore, here we took a multi-omics approach to profile the effect of KRAS allelic loss and mutation in a colorectal adenocarcinoma cell line isogenic for mutant KRAS. Through quantitative proteomics, we identified major protein-level changes in response to loss of expression of KRASWT or KRASG13D from heterozygous KRASWT/G13D parental cells. Cells expressing KRASG13D upregulated several aspartate metabolizing proteins including ASS1, ASNS, PCK1, and PCK2. Interestingly, ASS1 (argininosuccinate synthase) is transcriptionally upregulated in primary gastric, epithelial, ovarian, lung, and colorectal cancers relative to matched normal tissue, and limited evidence suggests its upregulation at the protein level in colorectal cancers [15–17]. To further support our findings from cell lines, we show that ASS1, PCK1, PCK2, ASL, and NOS1 are upregulated at the transcript level in KRAS-mutant primary CRC. We further show that ASS1 is required for colorectal cancer cell growth and supports tumor formation in clonogenic assays. Using metabolomics, isotope labeling and rescue experiments with cells and tumor spheroids, we find that ASS1 supports cell growth through the production of TCA cycle intermediates. This integrated analysis provides further insight into KRAS-specific biology and highlights mutation-dependent metabolic alterations and effectors of KRAS.
Results
KRAS allelic alteration and mutation induce significant proteomic alterations
To understand allele- and mutation-specific proteomic alterations, we performed label-free quantitative proteomics on whole cell extracts from DLD-1 KRASWT/G13D, KRASWT/−, and KRAS−/G13D cells in biological triplicate (Fig. 1A). A total of 3818 protein groups were identified in common to all cell lines at a 1% protein-level false discovery rate (FDR) cutoff (Fig. 1B, Data S1). For quantitative comparisons, integrated intensity values were quantile-normalized prior to log-transformation [18], and examined compared with log intensity-adjusted P-values. Relative to KRASWT/G13D cells, KRASWT/− cells differently upregulate a small set of proteins including the autophagic regulator SQSTM1 by ~ 3-fold (Fig. 2A). Relative to KRASWT/G13D cells, KRAS−/G13D cells upregulate several proteins with documented roles in late-stage colorectal adenocarcinomas and tumor metastasis, including CXCL5 [19], MT-ND1 [20], and HSPH1 [21] (Fig. 2B, Data S1). Upregulation of these proteins in KRAS−/G13D cells is consistent with previous reports that the expression of only the mutant oncogene may represent a more advanced disease state [13,22,23]. Relative to KRASWT/− hemizygous cells, several metabolic enzymes are upregulated in the KRASWT/G13D cell line by ~6-fold, including cytosolic and mitochondrial phosphoenolpyruvate carboxykinases (PCK1 and PCK2, respectively), ASS1, and asparagine synthetase (ASNS) (Fig. 2A). PCK1 and ASNS are known regulators of CRC cell proliferation and anabolic metabolism and are upregulated by mutant KRAS in models of CRC [24–26]. Our proteomic findings further support for their role in mutant KRAS-regulated metabolic processes. By label-free quantitative proteomics, we also identified a 3.25-fold reduction in ASS1 in KRAS−/G13D cells relative to KRASWT/G13D cells; however, with a P-value of 0.47, this finding is not statistically significant at our cutoff (P < 0.05). Nonetheless, KRAS-dependent differences in ASS1 protein levels could be demonstrated by western blot (Fig. 2C). Fewer significant differences were observed between the KRASWT/− and KRAS−/G13D (Data S1).
Fig. 1.
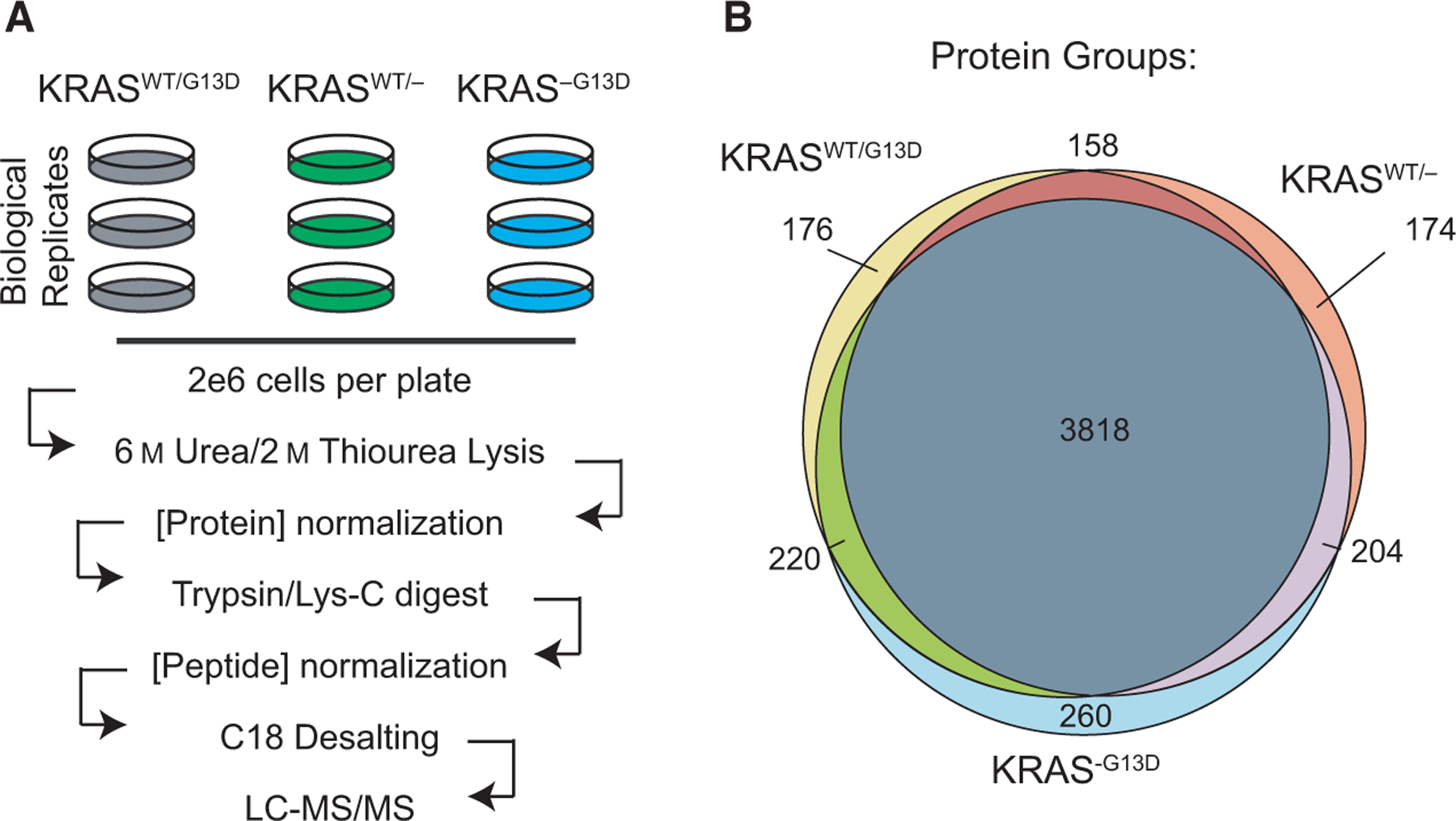
Quantitative proteomic workflow. (A) Experimental design and workflow for quantitative label-free proteomics. (B) Venn diagram for protein-group level identifications for all isogenic cell lines.
Fig. 2.
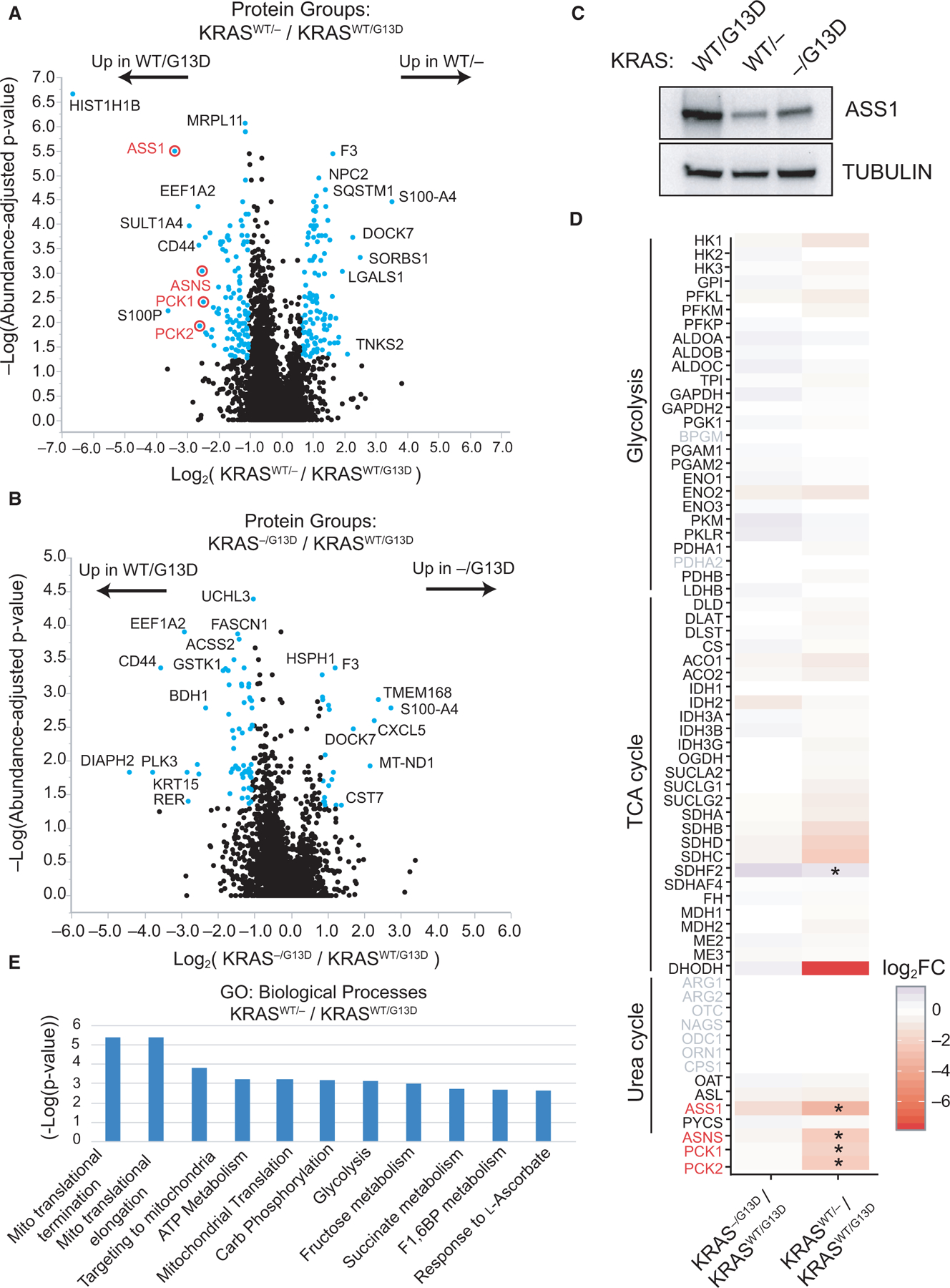
Proteomic alterations manifested by KRAS mutation and allelic imbalance. Label-free proteomics data were compared between (A) KRASWT/G13D and KRASWT/− cells and (B) KRASWT/G13D and KRAS−/G13D DLD-1 cell lines (n = 3). Proteins differentially expressed more than twofold are highlighted in blue. (C) Peptide-level results were validated by western blot to ASS1 in the three cellular contexts. (D) The fold change in major metabolic enzymes in glycolysis, TCA cycle, and urea cycle is shown for each quantitative analysis. Significant changes (P < 0.05) are marked (*), and gene symbols not identified by proteomics are in gray. (E) Proteins with fold change ≥ 2 (KRASWT/G13D/KRASWT/−) cell lines were subjected to Gene Ontology analysis to determine enrichment in the GO term of biological processes. Fold enrichment and −log10(P-value) for each term are provided. Three independent biological replicates of each cell line were used for proteomic experiments.
Given that one consensus molecular subtype of colorectal cancer (CMS3) is defined by KRAS mutations and metabolic dysregulation [4], we wanted to further examine proteomic changes in central metabolism. Therefore, for each cell line the protein fold change relative to the parental cell line was plotted for enzymes of glycolysis, the TCA cycle, and the urea cycle (Fig. 2D). For central metabolic enzymes, only ASS1, SDHF2 (succinate dehydrogenase complex assembly factor 2), PCK1, PCK2, and ASNS are differentially expressed. DHODH, an enzyme in de novo pyrimidine biosynthesis, was identified in KRASWT/G13D and KRAS−/G13D cells but was not detected in KRASWT/− cells. Interestingly, a previous investigation demonstrated that DHODH activity is required for mutant KRAS cell growth and it represents a synthetic lethal vulnerability in KRAS-mutant cancers [27]. Together with PCK1, DHODH drives CRC metastasis to the liver [26]. Overall, while only a fraction of proteins have significantly altered expression between isogenic cell lines, many of have acknowledged functional roles in CRC, and more remain to be studied in greater detail.
To further examine KRAS-dependent changes at a pathway level, proteins upregulated over twofold in KRASWT/G13D cells relative to KRASWT/− cells were subjected to gene ontology enrichment to determine upregulated biological processes [28]. This analysis identified mitochondrial processes including mitochondrial transcriptional termination (P = 3.8e-6), mitochondrial transcriptional elongation (P = 4.1e-6), and targeting to mitochondria (P = 1.6e-4) as upregulated due to expression of KRASG13D (Fig. 2E). This pathway-level analysis supports recent work highlighting mitochondrial translation and oxidative phosphorylation as promoters of KRAS-mutant CRC cell growth [11].
Urea cycle transcripts are upregulated in primary colorectal cancers
We next assessed whether ASS1 is also upregulated in colorectal adenocarcinoma in vivo and turned to clinical histology data from The Human Protein Atlas (HPA) (https://www.proteinatlas.org/ENSG00000130707-ASS1/) [29]. Histological data from normal and primary cancer tissue demonstrate that ASS1 stains strongly in primary cancers and is significantly upregulated at the protein level in CRC relative to normal colorectal tissue (Fig. 3A). This finding is independently supported by a more in-depth investigation of the clinical histology of ASS1 across ~ 600 samples [29,30]. In this previous study by Alexandrou et al., identified positive expression of ASS1 in 80% of CRC cases examined by clinical histology [30]. Unfortunately, the KRAS mutation status of the 600 clinical samples and HPA data is not available. Yet, Alexandrou et al. also noted that cell lines with low, basal ASS1 expression re-express ASS1 in response to arginine-deprivation monotherapy as a common acquired resistance mechanism. Moreover, all cultured cell lines with low ASS1 expression also only expressed wild-type KRAS [30].
Fig. 3.
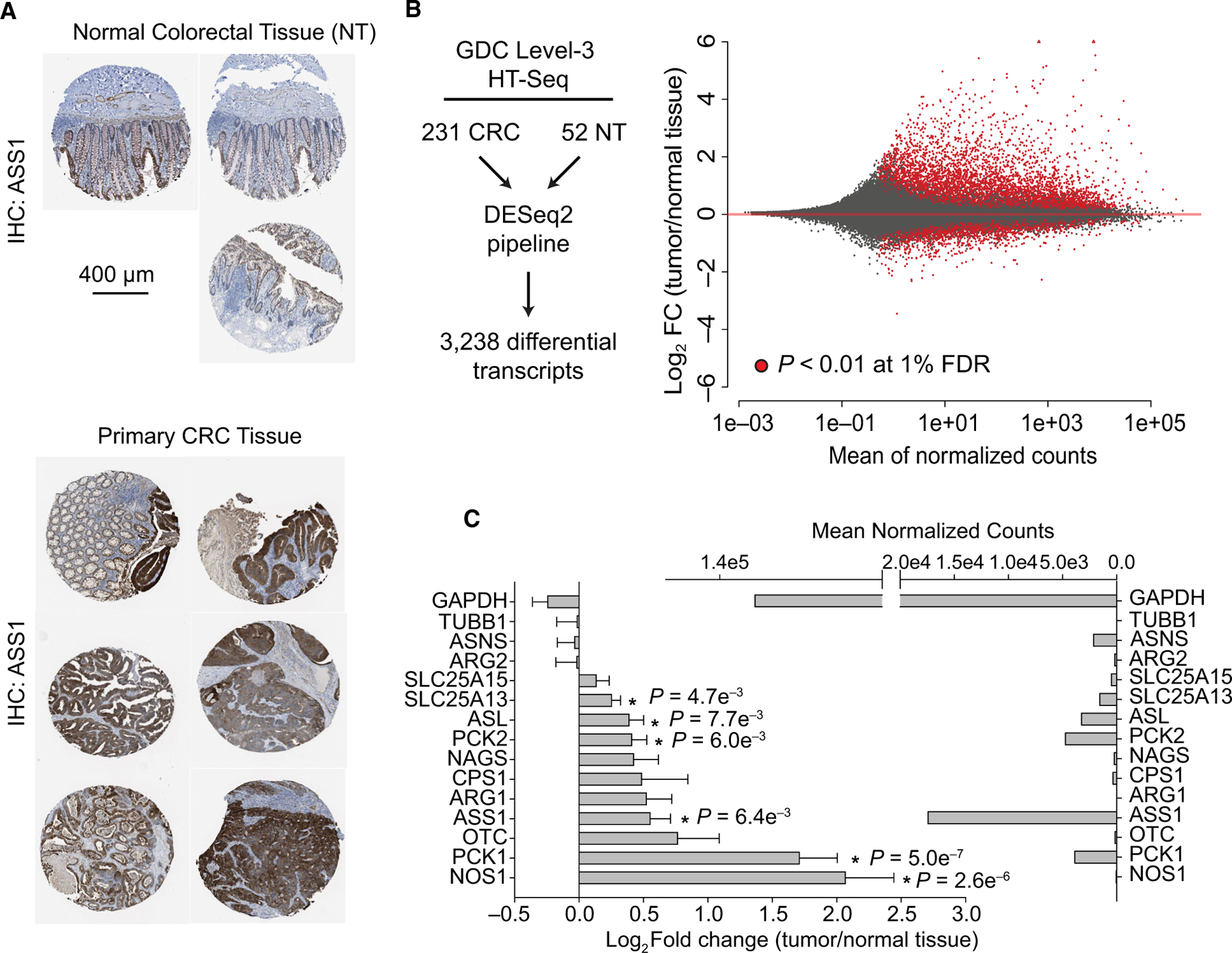
The metabolic landscape of CRC includes upregulation of urea cycle enzymes. (A) Immunochemistry against ASS1 protein for normal colorectal tissue samples and primary colorectal adenocarcinomas. A scale bar measuring 400 microns is provided for IHC images. (B) Differential expression analysis was performed using the DESeq2 pipeline on solid primary colorectal tumors (CRC, n = 231) and normal tissue (NT, n = 52) from clinical HTSeq data from the Genomic Data Commons. A Bland–Altman plot is shown with significantly changing transcripts (P < 0.01 at 1% FDR) provided in red. (C) Urea cycle enzymes and control enzymes (GAPDH and TUBB1) are shown. The log2 fold change for urea cycle enzymes and housekeeping genes was calculated, and p-values are presented at a 1% FDR cutoff. The mean normalized counts for each gene are shown. Error bars represent the standard deviation from the mean. Image credits: The Human Protein Atlas [29].
To gain more insight into the molecular features of KRAS-mutant CRC, we also compared level-3 HTSeq data from 231 KRAS-mutant colorectal tumors to 52 normal colorectal tissue samples using the NIH Genome Data Commons (http://portal.gdc.cancer.gov/) (Fig. 3B,C, Data S2) [31]. This analysis utilized HTseq data across all CRC subtypes, and clinical stages and mutant samples represent 37 different mutations in KRAS, with KRASG12D, KRASG12V, KRASG13D, and KRASG12C representing 27%, 21%, 17%, and 7% of cases, respectively. Differential expression analysis was performed using DESeq2 following library normalization [32], revealing 3238 transcripts from 2460 genes as differentially expressed (P < 0.01, at 1% FDR cutoff) in primary CRC relative to normal colorectal tissue. Of all differentially expressed transcripts, 1523 were upregulated by more than twofold, while 94 transcripts are downregulated by more than twofold (Fig. 3B, Data S2). Transcripts from the PI3K/AKT pathway, metabolic, and calcium signaling pathways were significantly upregulated at the transcript level (Table 1, Data S2). For transcripts, encoding enzymes participating in the urea cycle, PCK1, NOS1, SLC25A13, PCK2, and ASS1 are upregulated in primary colorectal cancer tumors relative to control colorectal tissue samples and are expressed across several orders of magnitude (Fig. 3C, Data S2).
Table 1.
Upregulated genes in the PI3K/AKT pathway identified by RNA-seq differential expression analysis.
| Ensemblgene ID | UniProt accession | Gene name |
|---|---|---|
| ENSG00000177455 | P15391 | CD19 |
| ENSG00000127588 | Q9P2W3 | GNG13 |
| ENSG00000162188 | P63215 | GNG3 |
| ENSG00000176533 | O60262 | GNG7 |
| ENSG00000167414 | Q9UK08 | GNG8 |
| ENSG00000040199 | Q6ZVD8 | PHLPP2 |
| ENSG00000100721 | P56279 | TCL1A |
| ENSG00000060566 | Q68CJ9 | CREB3L3 |
| ENSG00000181072 | P08172 | CHRM2 |
| ENSG00000136457 | O15335 | SLRR4A |
| ENSG00000139219 | P02458 | COL2A1 |
| ENSG00000197565 | Q14031 | COL4A6 |
| ENSG00000172752 | A8TX70 | COL6A5 |
| ENSG00000070193 | O15520 | FGF10 |
| ENSG00000129682 | Q92913 | FGF13 |
| ENSG00000066468 | P21802 | FGFR2 |
| ENSG00000131482 | P35575 | G6PT |
| ENSG00000112964 | P10912 | GHR |
| ENSG00000160712 | P08887 | IL6R |
| ENSG00000101680 | P25391 | LAMA1 |
| ENSG00000124253 | P35558 | PCK1 |
| ENSG00000162409 | P54646 | AMPK |
| ENSG00000156475 | Q00005 | PPP2R2B |
| ENSG00000189056 | P78509 | RELN |
| ENSG00000118515 | O00141 | SGK1 |
| ENSG00000168477 | P22105 | TNXB |
| ENSG00000113296 | P35443 | THBS4 |
| ENSG00000165197 | O43915 | VEGFD |
To assess correlation of gene expression between the same 231 KRAS-mutant primary CRC cases used for differential expression analyses, FPKM-UQ data from primary tumors were examined for 196 enzymes in glycolysis, the TCA cycle, nucleotide metabolism, the urea cycle, and commonly differentially regulated proteins and regulators such as P53, KRAS, and MYC. Following correlative clustering and statistical analyses, we found that ASS1 expression strongly and significantly correlates with several genes including P53 and argininosuccinate lyase (ASL) (Fig. 4A, Data S3 and S4). Co-expression of ASS1 and ASL is not surprising as the two enzymes catalyze neighboring reactions in the de novo arginine biosynthesis pathway and urea cycle [33,34]. However, given that our proteomic investigations were performed in P53 mutant cells and P53 mutations are common in CRC, it may be interesting for future clinical studies to resolve specific programs driven by KRAS/P53 double mutants [35].
Fig. 4.
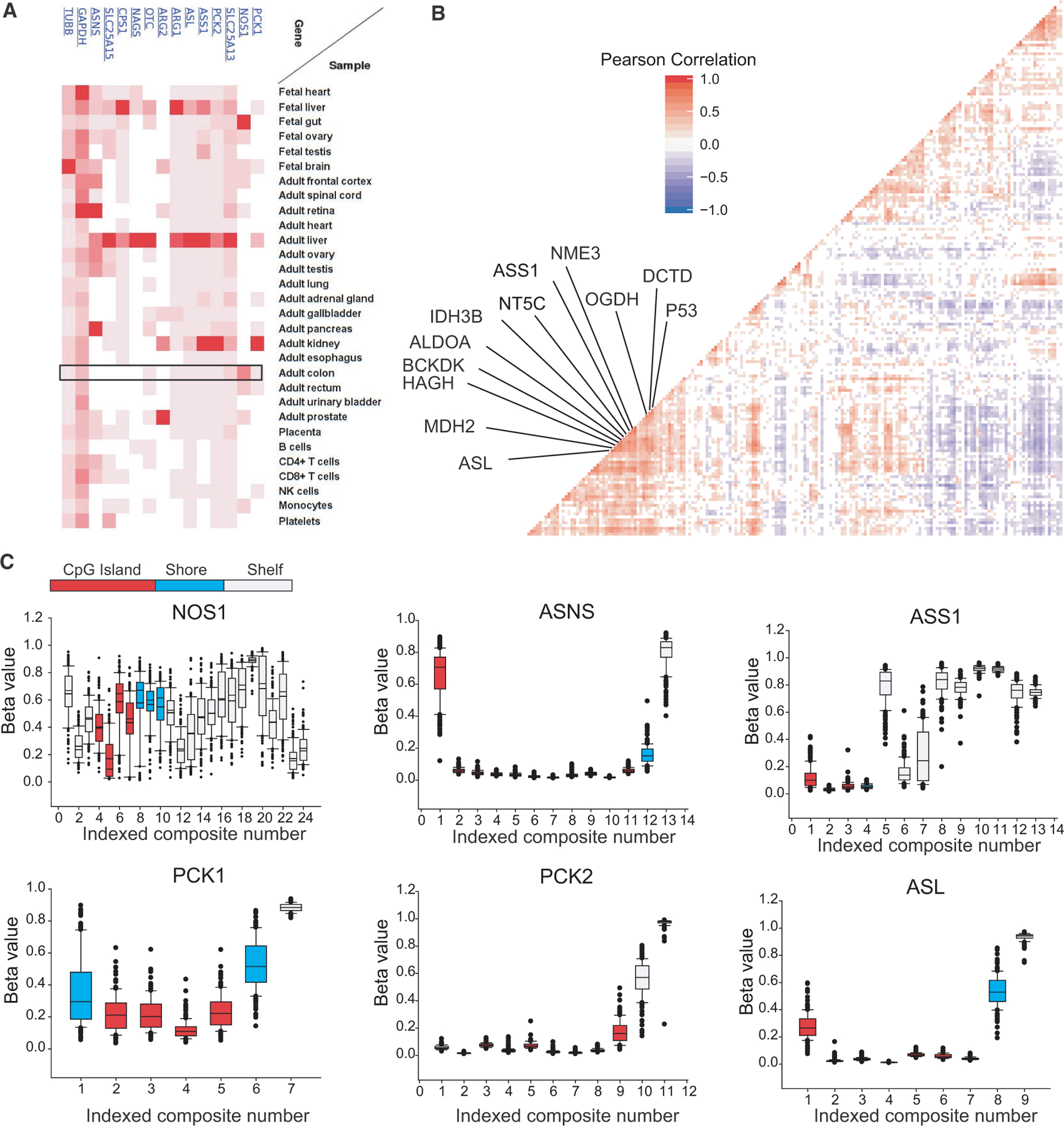
Transcript-level correlations and urea cycle enzyme methylation in CRC. (A) Expression of ASS1 and urea cycle enzymes at the protein level from [57]. (B) Correlation plot of FPKM-UQ reads from metabolic genes and commonly up- and downregulated genes from colorectal adenocarcinomas (n = 231). Significant correlations are colored and nearest neighbors to ASS1 shown. (C) Given chromosomal instability/methylation phenotypes (CIMP) in primary CRC, level-3 promotor methylation array data for all available CRC cases were examined for genes identified as central to CRC growth, and additionally NOS1. Methylation beta values are shown for genomic distances relative to transcription start sites. Composites represent unique genomic locations for which beta values were calculated. The promoter region is colored relative to the distance to the transcription start site. Error bars represent 95% confidence intervals.
While our transcript-level analysis provides additional support for the upregulation of the urea cycle in CRC, one consensus molecular subtype of CRC is clinically characterized by a CpG island methylator phenotype (CIMP) which acts to epigenetically repress transcription [36]. Provided that our RNA-seq investigations did not discriminate against CIMP-subtype CRC samples, and the promoter methylation status of ASS1 and ASL strongly regulates transcription of these genes in several cancers [37–39], promoter methylation for ASS1, ASL, NOS1, PCK1, and PCK2 transcriptional start sites (TSS) were examined in 351 colorectal tumors with level-3 data from GDC. Relative methylation values were examined across distinct genomic loci for each gene. Overall, low levels of methylation were identified for the promoter CpG island regions of ASS1, ASL, PCK1, and PCK2, (average β-value < 0.1), suggesting that these genes are not epigenetically repressed in primary CRC. Interestingly however, even though NOS1 is upregulated by ~ 4-fold in primary CRC by our RNA-seq studies, two promoter CpG island regions exhibit average β-values of ~ 0.5, indicating that the primary tumors cells used examined in the methylation array are highly variable for NOS1 methylation (Fig. 4B).
Argininosuccinate synthase is required for colorectal cancer cell growth
Motivated by datasets from primary CRC samples and isogenic cell lines, we investigated the role of ASS1 in colorectal cells. DLD-1 cells isogenic for KRAS were transduced with lentivirus to one of two doxycycline-inducible shRNAs to separate ASS1-exon targeting regions or to a doxycycline-inducible scrambled control. Following selection and generation of stable cell lines, shRNA expression was induced with doxycycline for 48 h. ASS1 levels were significantly reduced with either ASS1-targeting shRNA sequence relative to the scrambled control (Fig. 5A). To confirm that doxycycline alone is not responsible for observed differences in cell growth, nontransduced isogenic DLD-1 cell lines were independently treated with doxycycline to establish an in cell IC50 for doxycycline at 48 h (Fig. 5B). In line with recent reports for other tetracyclines [40], the calculated in cell IC50 after 4 days was 20.0 μM, 22.7 μM, and 20.5 μM for KRASWT/G13D, KRASWT/−, and KRAS−/G13D cell lines, respectively. Next, proliferation was monitored in lentivirally transduced cells with and without shASS1 induction with concentrations up to 500 ng·mL−1 of doxycycline across 8 days. Additionally, for each KRAS background, cells not infected with lentivirus were provided 1000 ng·mL−1 doxycycline as a control condition (Fig. 5C). Overall, knockdown of ASS1 significantly reduced cellular proliferation, independent of KRAS mutation status, and nontransduced cells administered 1000 ng·mL−1 doxycycline were not observed to have defects in cell growth relative to shASS1 cells given DMSO as a control (Fig. 5C).
Fig. 5.
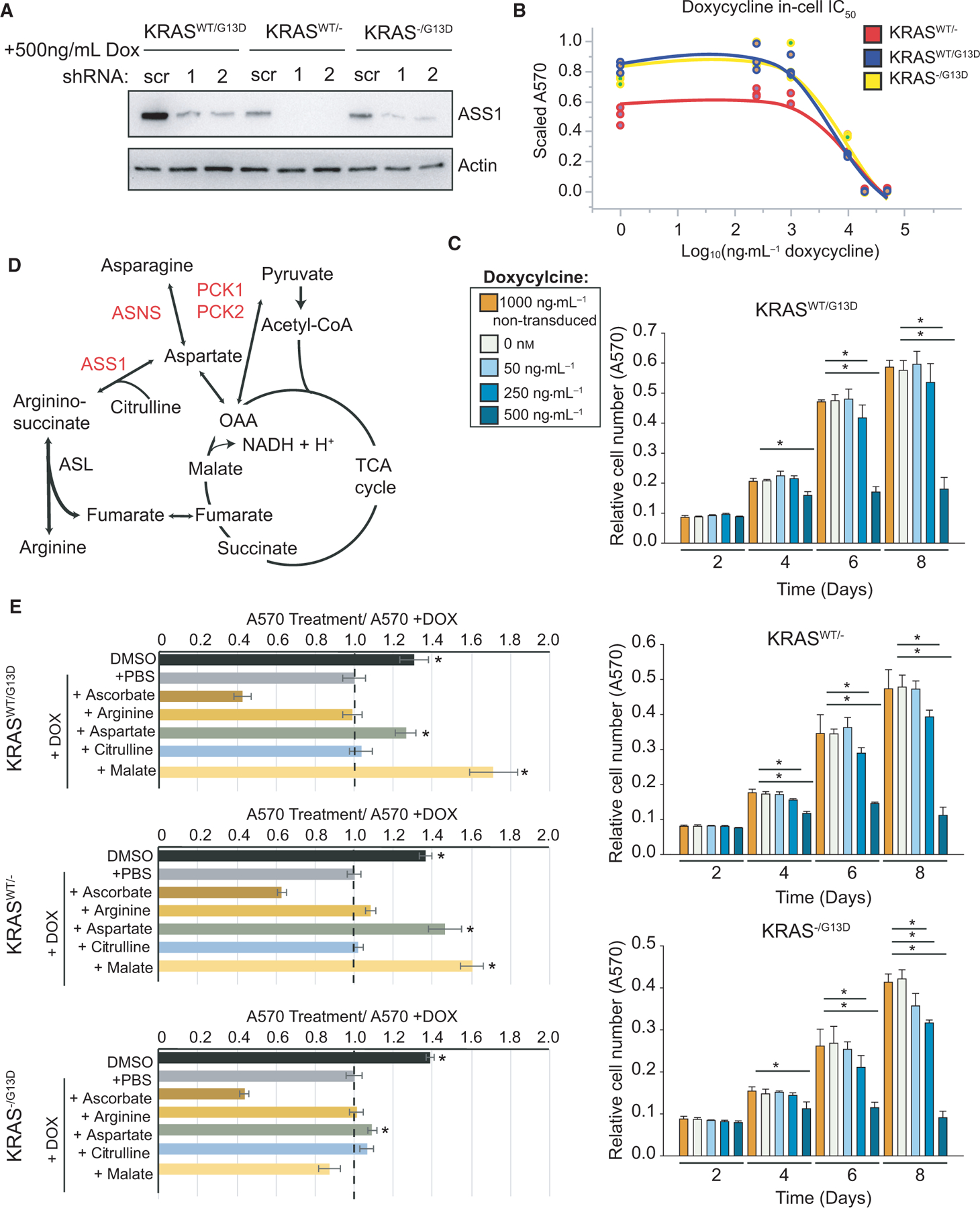
ASS1 is required for DLD-1 cell growth. (A) Knockdown of ASS1 was established with two shRNA sequences and validated by western blot in DLD-1 cell lines relative to actin after 48 h of doxycycline induction. (B) In cell IC50, curves for cell lines lacking doxycycline-inducible shRNA provided high concentrations of doxycycline for 48 h. (C) Metabolic network map of central metabolism and urea cycle. (D) Cell proliferation was tracked in two-dimensional culture by crystal violet staining following doxycycline administration at 2, 4, 6, and 8 days (n = 6). (E) Metabolic rescue experiments by supplementation with 1 mM of exogenous metabolites (n = 3). For all subfigures, *P < 0.01 and error bars represent the standard deviation from the mean.
Loss of argininosuccinate synthase alters intracellular metabolism
ASS1 catalyzes the rate-limiting step in de novo arginine biosynthesis to generate argininosuccinate from aspartate and citrulline, which is rapidly converted to arginine and fumarate by argininosuccinate lyase (ASL) [41]. However, our previous studies where an ASS1-dependent growth defect was observed were all performed in tissue culture media replete with 1 mm arginine. Therefore, the ASS1-dependent growth phenotype we observed suggests that the role of ASS1 in colorectal cancer cells is not strictly to support de novo arginine production. As an alternate hypothesis, ASS1 expression may promote the funneling of its product fumarate into the TCA cycle and downstream production of NADH and H+. In effect, this would drive oxidative phosphorylation and support redox balance inside CRC cells (Fig. 5D) [42]. Another alternate hypothesis arises from the reverse reaction for ASS1 to convert argininosuccinate to aspartate and citrulline. However, this explanation is unlikely, given an optimal reaction at pH 6.6 and the requirement of a two order-of-magnitude higher concentration of argininosuccinate, relative to product metabolites to drive the reaction [41]. Therefore, to further understand how ASS1 activity supports CRC cell metabolism, we performed rescue experiments by supplementing ASS1 knockdown cell lines with exogenous metabolites. For each KRAS isogenic cell line, 1 mM arginine, malate, aspartate, or citrulline was added to replete RPMI media with 10% dialyzed FBS. In addition, cells were independently treated with 1 mM sodium ascorbate as a positive control for metabolically induced cell death [43]. Cell proliferation was assessed by crystal violet staining and subsequent absorption at 570 nm over 4 days (Fig. 5E). In all isogenic cell lines, 1 mm argi-nine could not rescue cell number. Moreover, KRAS−/G13D cells could not be rescued by exogenous metabolite treatment, suggesting that KRAS−/G13D cells lack a direct compensatory mechanism to overcome loss of ASS1 in 4 days in cell culture or that higher concentrations of metabolites are required to fulfill metabolic demands and rescue these cells (Fig. 5E). However, for KRASWT/G13D and KRASWT/− cells, supplementation with aspartate or malate could rescue cell proliferation.
The finding that aspartate can rescue cells in 2D cell culture is unexpected given that aspartate is a direct substrate of ASS1. Often the knockdown of an enzyme results in the accumulation of its substrate and the other direct substrate of ASS1, citrulline, could not rescue cells [44]. However, the rescue effect of malate and aspartate on ASS1 knockdown cells may be explained as a function of the malate-aspartate shuttle. In highly metabolic cancer cells, this shuttle is crucial to transfer reducing equivalents into mitochondria and support energy production. Through the shuttle, aspartate is converted to oxaloacetate and subsequently to malate and NAD+ in the intermembrane space of mitochondria. The cytoplasmic NAD+ pool supports NAD-dependent glycolytic reactions while malate shuttles electrons into the mitochondrial matrix by crossing the inner mitochondrial membrane [45]. Mitochondrial malate dehydrogenase then oxidizes malate to oxaloacetate, NADH, and H+. The NADH and H+ generated by this shuttle enter the electron transport chain to produce ATP. In the context of KRAS-mutant cancer cells, ensuring ATP production and redox balance is critical for cellular homeostasis and proliferation.
While our studies support a functional role for ASS1 outside of de novo arginine production, cell metabolism in two-dimensional culture systems may not replicate the complex physiology and metabolism of three-dimensional tumors in vivo, as solid tumors are challenged by nutrient availability, waste generation, and hypoxia [46,47]. Therefore, we investigated the function of ASS1 in a tumor spheroid model. An equal number of DLD-1 cells in each KRAS isogenic cell line were dissociated to single cells and separately seeded at low density in 0.35% agarose and cultured under standard conditions with and without knockdown of ASS1. After 4 weeks in culture, significantly smaller tumor spheroids formed following loss of ASS1 (Fig. 6A). Combining biological triplicate data across all cell lines, loss of ASS1 led to a 10-fold reduction in the number of tumors above 100 μm in diameter (Fig. 6B). In many cases, clonogenic expansion of DLD-1 cells with reduction in ASS1 yielded poorly aggregated cell masses approximately 50 μm in diameter and with internal density relative to control tumor spheroids. Cells expressing mutant KRAS exhibited over fivefold more tumor spheroids above 100 μm in diameter relative to DLD-1WT/− cells, demonstrating the strong oncogenic drive of mutant KRAS (Fig. 6B). Nonetheless, loss of ASS1 led to similar levels of tumor growth retardation in DLD-1WT/− and DLD-1−/G13D cells (Fig. 6B).
Fig. 6.
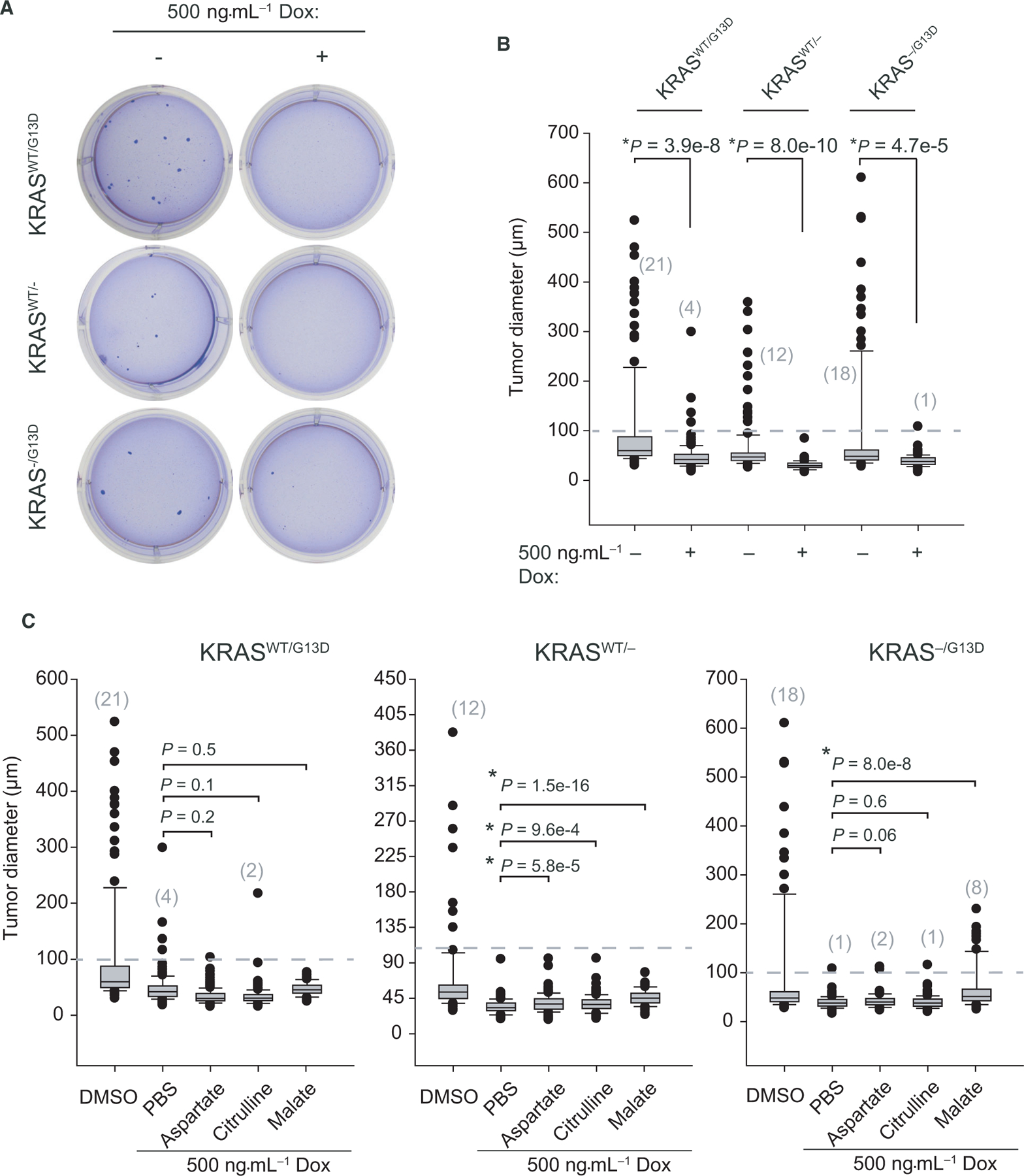
Loss of ASS1 can be partially rescued by exogenous malate. (A) Representative wells from low-density agarose clonogenic tumor formation assays after four weeks are shown. (B) Tumor images were acquired to assess tumor number and tumor size in each cell line. (C) Diameter of crystal violet stained tumor spheroids, measured, and quantified using imagej (> 100 tumors/condition, n = 3). (D) Rescue experiments with supplementation by 1 mM exogenous metabolites (> 100 tumors/condition, n = 3). Error bars represent the standard deviation from the mean.
We next evaluated the ability of exogenous metabolites to rescue tumor spheroid size and number in our 3D culture model (Fig. 6C). In this setting, cells face different metabolic and environmental pressures compared with 2D culture studies, including hypoxia which is known to reduce intracellular aspartate levels [48]. Following knockdown of ASS1, cells were provided with exogenous aspartate, citrulline, or malate every other day. Aspartate and citrulline had no significant effect on rescuing tumor size across cell lines relative to ASS1 expressing controls. However, high levels of exogenous malate could partially rescue tumor spheroid volume in KRASWT/− and KRAS−/G13D cells (Fig. 6C). The ability of aspartate to rescue cells in our 2D model but not a 3D model can be explained by the poor cell permeability of aspartate and the complex, hypoxic state of tumor spheroids [49]. Reduced nutrient, ATP, and O2 levels alter the allocation and partitioning of resources to fuel anabolic tumor cell proliferation.
Finally, we examined how loss of ASS1 alters central metabolism with metabolomics. Loss of ASS1 in DLD-1 isogenic cell lines was induced with 100 ng·mL−1 doxycycline for 4 days, and then, two million cells were plated for control and shASS1 conditions for each cell line. For cells provided 100 ng·mL−1 doxycycline, the cell number is not significantly different between growing and knockdown cells. A limited knockdown of ASS1 is observed at this concentration of doxycycline, yet indirect or confounding effects on metabolism from cell death are mitigated (Fig. 7A). Unlabeled, semi-targeted metabolomics identified ASS1 knockdown-dependent decreases in intracellular downstream products argininosuccinate, arginine, succinate, and fumarate. No net accumulation of aspartate was observed for ASS1 knockdown conditions (Fig. 7B). As noted by previous publications, the proteogenic amino acid is utilized by additional biochemical pathways to support tumor cell metabolism [48]. A lack of strong difference in the KRASWT/G13D cell line can be explained by limited knockdown in this cell line, which has high basal expression (Fig 7A). Separately, central carbon metabolism was also examined by flux studies. Cells were fed full RMPI, replete with U13C6-glucose and 10% dialyzed FBS to isotopically label intracellular metabolite pools, and metabolite isotopologue enrichment was identified by LC-MS. At steady state, upper glycolysis was labeled with heavy glucose, and nearly 100% the pyruvate pool is labeled in each cell line, with no significant difference between cell lines (Fig. 7C). However, an examination of TCA cycle intermediates demonstrates that 13C-labeling of citrate/isocitrate is significantly reduced in shASS1 cells, independent of KRAS mutation status (Fig. 7C). Further, flux to aspartate and argininosuccinate are significantly altered upon loss of ASS1, with quantitatively less glucose-derived carbon entering these metabolite pools at steady state (Fig. 7D). Reduced TCA cycle flux can be explained by loss of ASS1 impinging on mitochondrial metabolism. A reduction in TCA cycle flux limits cofactor generation and the production of NADH and H+ needed to drive oxidative phosphorylation. Moreover, lower flux of glucose-derived carbon into M + 2 aspartate suggests that under ASS1 knockdown conditions, significantly fewer carbons are funneled into aspartate then remain in the TCA cycle.
Fig. 7.
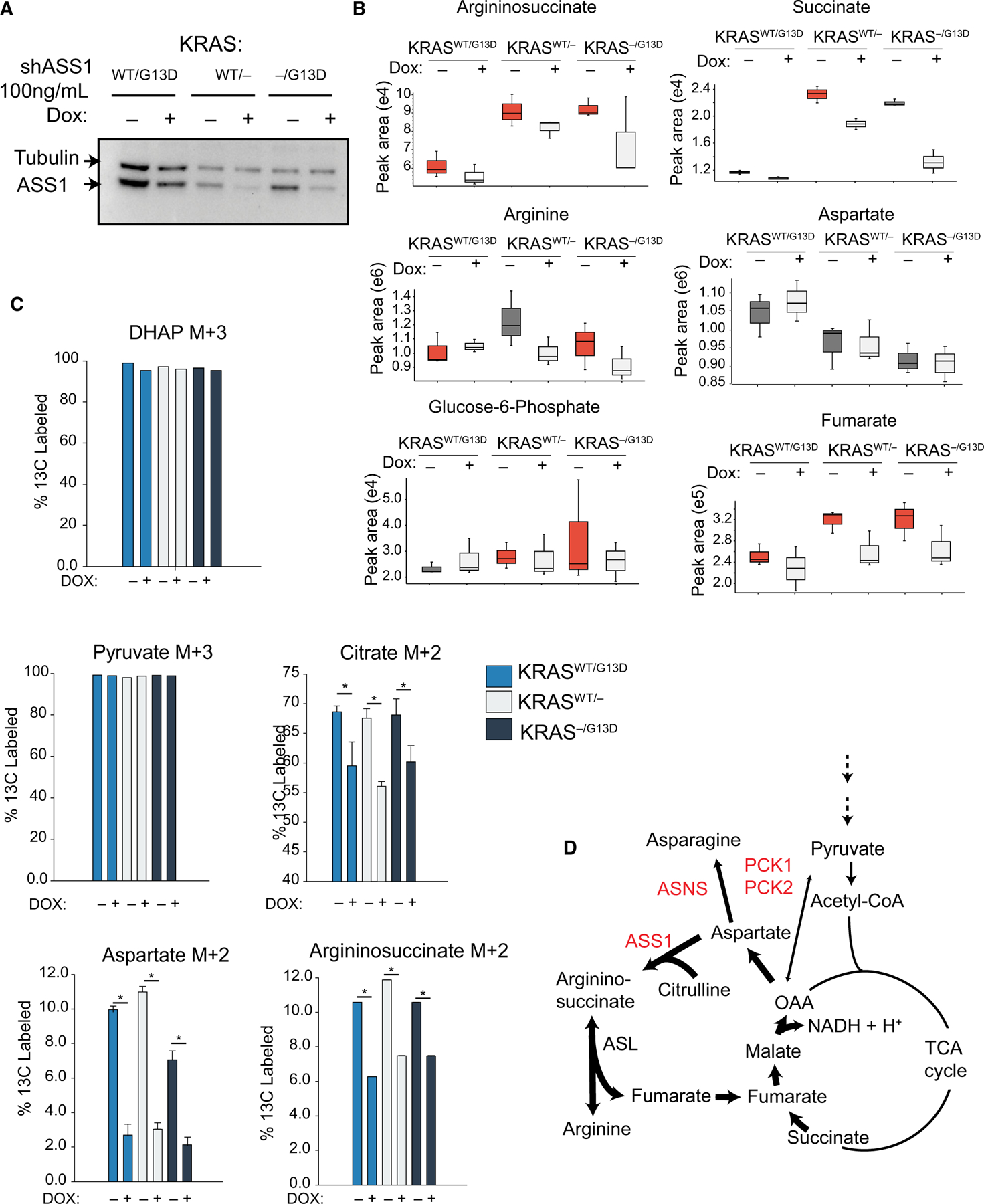
Metabolic rewiring in CRC due to loss of ASS1. (A) Western blot to ASS1 and tubulin to show relative knockdown of ASS1 in metabolomics experiments. (B) Polar metabolites from DLD-1 cell lines +/− shASS1 were extracted and identified by (−) ESI LC-HRMS/MS. Samples were run in biological triplicate on an Orbitrap ID-X (n = 3). (C) Isotopologue enrichment DLD-1 cells labeled to steady state with U13C-glucose (n = 3). (D) Metabolic map showing ASS1, ASNS, PCK1, and PCK2 positioned between the TCA and urea cycles. Error bars represent the standard deviation from the mean.
Discussion
KRAS mutations are found in a large number of cancers, yet direct inhibition of the small GTPase remains elusive in the clinical setting. Therefore, metabolic alterations and signaling programs driven by distinct KRAS genotypes remain under intense investigation. Here, we expanded on the known proteomic alterations driven by mutant KRAS. Through quantitative proteomics, we identified significant upregulation of asparagine synthase (ASNS), phosphoenolpyruvate carboxykinases 1 and 2 (PCK1 and PCK2), and argininosuccinate synthase 1 (ASS1) in cells harboring KRASG13D. Our data provide further evidence to existing studies that show how rewiring of aspartate and asparagine metabolism supports CRC cell proliferation, viability, and metastasis [24–26]. Upregulation of ASS1 in KRAS-mutant CRC cells is interesting given distinct expression patterns of ASS1 across cancer types [10,12]. The downregulation of ASS1 in breast and renal cancers has led to the clinical evaluation of arginine auxotrophy as a cancer treatment [50,51]. Classically, defects in ASS1 cause type I citrullinemia [52], yet the role of the protein requires greater examination in KRAS-mutant cancer settings. In the present study, ASS1 is upregulated in primary colorectal cancers at the protein level and we further demonstrated that urea cycle enzymes are upregulated in KRAS-mutant primary CRC. We found that knockdown of ASS1 reduced cell growth in DLD-1 cells and reduced tumor spheroid size in 3D culture studies. The ASS1-dependent growth phenotype can be partially rescued in 2D and 3D culture by supplementation with exogenous malate and aspartate but not with arginine, indicating that ASS1 does not function strictly to support de novo arginine biosynthesis, but its other product, fumarate, is utilized to support the metabolic demands of CRC cells. From metabolic tracer studies, we find that loss of ASS1 rewires metabolism to reduce glucose-derived carbon into the TCA cycle and subsequently to form aspartate. Similar to other studies, our data identified a key role for aspar-tate metabolism in fueling cancer cell growth [48].
From our studies, it is clear that cooperation between wild-type KRAS and KRASG13D alleles effects the expression of specific proteins leading to a unique phenotype. Wild-type KRAS retains a functional role in these cells as noted by a distinct proteomic profile observed for cells expressing only KRASG13D. Moreover, the fact that in 2D cell culture, exogenous metabolites cannot rescue KRAS−/G13D cells points to a distinct or inflexible metabolism in cells lacking wild-type KRAS. Interestingly, there is strong evidence that distinct KRAS mutations have distinct functional impacts on metabolism and transforming potential [5,11]. Unfortunately, we lack direct evidence in our study to compare our findings to CRC cells with KRASG12D, KRASG12C, or KRASG12V mutations. More mutation-resolved data would be helpful as Gly13 mutations are observed one-fourth as frequently as Gly12 mutations in CRC [2]. In more frequently mutated KRASG12D cells, glutaminolysis is promoted by the oncoprotein, yet in our system, an aspartate metabolism signature and not a glutamine metabolism signature was observed [53]. Therefore, it is unclear if our observations translate to other KRAS mutations. However, our transcript-level analysis of KRAS-mutant primary CRC did not distinguish between specific KRAS mutants, yet ASS1, PCK1, PCK2, and NOS1 are all upregulated. Therefore, dysregulation of the urea cycle represents a more common metabolic phenotype in CRC that demands further dissection and more attention.
In conclusion, our data point to KRASG13D-specific proteomic alterations and ASS1 as an important regulator of CRC metabolism. It would be interesting to perform a wider panel of functional studies for distinct KRAS mutations and specifically target the urea cycle. Additionally, it would be of interest to examine the therapeutic potential of ASS1-specific inhibitors. Metabolic resistance mechanisms may thwart this option, yet concomitant alterations in the urea cycle may represent a strategy to induce synthetic lethality in CRC.
Materials and methods
Proteomic and metabolomic methods can be found in Supplementary Material.
Clinical HTseq and Illumina 450 methylation array data analyses
All clinical data were downloaded from TCGA/GDC (https://portal.gdc.cancer.gov). Level-3 HTseq count data and FPKM-UQ data were downloaded for all COAD primary solid tumor samples, KRAS-mutant COAD primary solid tumor samples, and COAD normal solid tissue samples. All HTseq count files were processed in the DESeq2 R package as previously described [32]. Briefly, count files were first subjected to library normalization and gene-level dispersion estimates were calculated prior to differential expression analysis. Average counts, log2(fold change), fold change standard error, Wald statistic (fold change / standard error), p-values, and FDR-corrected p-values (pAdj) were exported at a 1% FDR cutoff. Level-3 Illumina HumanMethylation450 array data for 351 primary colorectal cancer cases (COAD) were downloaded from the TCGA’s DNA Methylation Liftover Pipeline [54]. For all 351 case files, data corresponding to unique array probes (composite elements) mapping to each gene were extracted. Methylation levels [β-value = methylated/ (methylated + unmethylated)] were plotted as a function of the distance from the transcriptional start site, as specified from the mapping in original array files. Data were downloaded in the Winter of 2017.
Statistics
All error bars are presented as +/− standard deviation from the mean value. Statistical analyses were performed in R3.5.1 or jmp13 (SAS) software.
Cell culture and generation of shASS1 cell lines
DLD-1 cell lines were acquired from Horizon Discovery (Hinxton, UK) and propagated in RMPI 1640 media supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Waltham, MA, USA). Cell lines were short-tandem repeat (STR) validated. The following shRNA sequences were ordered from IDT for shASS1: #1 CCCATCCTTTACCATGCTCATT, #2 CTCAGGCTGAAGGAATATCAT and sh-Scrambled: CCTAAGGTTAAGTCGCCCTCG. Subcloning and lentiviral production of tetracycline-inducible shRNA was conducted as described [55]. Linear oligonucleotide sequences were annealed and ligated into the Tet-pLKO-puro backbone (Addgene, #21915) with T4 ligase (NEB #0202) and amplified in Stbl3 E. coli (Invitrogen, Waltham, MA, USA). Lentivirus was packaged in HEK293T cells (ATCC CRL-11268) with cotransfection of tet-puro-pLKO with psPAX2 and pMD2.G (Addgene #12260, #12256) by the calcium phosphate method. DLD-1 cell lines were transduced with polybrene (Sigma, St. Louis, MO, USA) and shASS1-lentivirus. Stably expressing DLD-1 cell was grown in media containing dialyzed FBS and selected with 1 μg·mL−1 puromycin (Fisher, Waltham, MA, USA). Optimal shRNA induction was tested experimentally and finally induced by 500 ng·mL−1 doxycycline (Fisher).
Proliferation and IC50 assays, and metabolite administration
Cells were plated at an equal cell number in triplicate and fed fresh media daily. At the time of collection, cells were stained for 25 min with 0.1% (w/v) crystal violet in 5% MeOH. Cells were air-dried, and absorbance at 570 nm was read on a BioTek Take5 plate reader, taking, and averaging 25 independent readings per well. All exogenous metabolites were purchased from Sigma and resuspended in H2O and sterile filtered through 0.2-μm PES filters (Sigma) prior to media supplementation.
Tumor spheroid assays
Tumor spheroid assays were conducted as previously described [56]. A base layer of sterile 0.9% (w/v) agarose (Fisher) diluted in RPMI media was plated in 6- or 12-well plates. Cells were diluted 1:2 in sterile 0.7% (w/v) agarose (Sigma) and plated at 15 000 cells/well of each plate to a final concentration of 1X RPMI. Cells were fed fresh full media and doxycycline every other day for four weeks prior to staining with 0.005% (w/v) crystal violet in 4% formaldehyde, made fresh in phosphate-buffered saline, pH 7.4.
Immunoblotting
Proteins from lysates were resolved on AnyKD SDS/PAGE (Bio-Rad, Hercules, CA, USA) at 100 V for ~60 min and transferred to 0.2-μm PVDF membranes at 200 mA for 1.2 h at 4 °C. Membranes were blocked in 5% (w/v) nonfat dry milk in TBS-T for 1 h at room temperature and then incubated in primary antibodies in 5% BSA (Sigma) overnight at 4 °C. All primary antibodies were diluted to 1:1000 in TBS-T. Antibodies were purchased from Cell Signaling Technology to ASS1 (#70720), actin (#4970), and tubulin (#2146). Membranes were washed three times with TBS-T and incubated with corresponding secondary antibody (Millipore, Burlington, MA, USA) in TBS-T at a dilution of 1:10 000 for 1 h. Membranes were washed three times with TBS-T before visualization with enhanced chemiluminescence (Millipore) on a ChemiDocs machine (Bio-Rad).
Supplementary Material
Acknowledgments
This research was carried out with support from the National Resource for Translational and Developmental Proteomics under Grant P41 GM108569 from the National Institute of General Medical Sciences (NLK) and supported by the Sherman Fairchild Foundation. PFD acknowledges a NSF Fellowship under award number 2015210477, and the authors thank Emily Bojas and Henry Rodriguez for support in accessing proteomic data and isogenic cell lines from the National Cancer Institute (Office of Cancer Clinical Proteomics Research), National Institutes of Health, under Contract No. HHSN26 1200800001E.
Abbreviations
- ASS1
argininosuccinate synthase
- CRC
colorectal cancer
- GDC
genome data commons
- MS
mass spectrometry
Footnotes
Conflict of interest
Ioanna Ntai is an employee of Thermo Fisher Scientific.
Data accessibility statement
References
- 1.Tan C & Du X (2012) KRAS mutation testing in metastatic colorectal cancer. World J Gastroenterol 18, 5171–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serebriiskii IG, Connelly C, Frampton G, Newberg J, Cooke M, Miller V, Ali S, Ross JS, Handorf E, Arora S et al. (2019) Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun 10, 3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunelli L, Caiola E, Marabese M, Broggini M & Pastorelli R (2014) Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget. 5, 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammond DE, Mageean CJ, Rusilowicz EV, Wickenden JA, Clague MJ & Prior IA (2015) Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J Proteome Res 14, 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yokota T (2012) Are KRAS/BRAF mutations potent prognostic and/or predictive biomarkers in colorectal cancers? Anticancer Agents Med Chem 12, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kishiki T, Ohnishi H, Masaki T, Ohtsuka K, Ohkura Y, Furuse J, Sugiyama M & Watanabe T (2014) Impact of genetic profiles on the efficacy of anti-EGFR antibodies in metastatic colorectal cancer with KRAS mutation. Oncol Rep 32, 57–64. [DOI] [PubMed] [Google Scholar]
- 7.Ostrem JM, Peters U, Sos ML, Wells JA & Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS et al. (2020) KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med 383, 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young A, Lou D & McCormick F (2013) Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov 3, 112–123. [DOI] [PubMed] [Google Scholar]
- 10.McFall T, Diedrich JK, Mengistu M, Littlechild SL, Paskvan KV, Sisk-Hackworth L, Moresco JJ, Shaw AS & Stites EC (2019) A systems mechanism for KRAS mutant allele-specific responses to targeted therapy. Sci Signal 12, 8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin TD, Cook DR, Choi MY, Li MZ, Haigis KM & Elledge SJ (2017) A role for mitochondrial translation in promotion of viability in K-Ras mutant cells. Cell Rep 20, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P et al. (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21, 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgess MR, Hwang E, Mroue R, Bielski CM, Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L et al. (2017) KRAS allelic imbalance enhances fitness and modulates MAP kinase dependence in cancer. Cell 168, 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA et al.(2019) Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szlosarek PW, Grimshaw MJ, Wilbanks GD, Hagemann T, Wilson JL, Burke F, Stamp G & Balkwill FR (2007) Aberrant regulation of argininosuccinate synthetase by TNF-alpha in human epithelial ovarian cancer. Int J Cancer 121, 6–11. [DOI] [PubMed] [Google Scholar]
- 16.Wu MS, Lin YS, Chang YT, Shun CT, Lin MT & Lin JT (2005) Gene expression profiling of gastric cancer by microarray combined with laser capture microdissection. World J Gastroenterol 11, 7405–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T & Szlosarek PW (2010) Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer 126, 2762–2772. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S et al. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, Liu D, Zheng M, Sun J, Feng H et al. (2017) Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3beta/beta-catenin pathways. Mol Cancer 16, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallace L, Mehrabi S, Bacanamwo M, Yao X & Aikhionbare FO (2016) Expression of mitochondrial genes MT-ND1, MT-ND6, MT-CYB, MT-COI, MTATP6, and 12S/MT-RNR1 in colorectal adenopolyps. Tumour Biol 37, 12465–12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh HJ, Kim JH, Lee TH, Park HE, Bae JM, Lee HS & Kang GH (2017) Dominant high expression of wild-type HSP110 defines a poor prognostic subgroup of colorectal carcinomas with microsatellite instability: a whole-section immunohistochemical analysis. APMIS 125, 1076–1083. [DOI] [PubMed] [Google Scholar]
- 22.Kong G, Chang YI, Damnernsawad A, You X, Du J, Ranheim EA, Lee W, Ryu MJ, Zhou Y, Xing Y et al. (2016) Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia 30, 1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu CC, Qiu W, Juang CS, Mansukhani MM, Halmos B & Su GH (2017) Mutant allele specific imbalance in oncogenes with copy number alterations: Occurrence, mechanisms, and potential clinical implications. Cancer Lett 384, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montal ED, Dewi R, Bhalla K, Ou L, Hwang BJ, Ropell AE, Gordon C, Liu WJ, DeBerardinis RJ, Sudderth J et al. (2015) PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol Cell 60, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toda K, Kawada K, Iwamoto M, Inamoto S, Sasazuki T, Shirasawa S, Hasegawa S & Sakai Y (2016) Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 18, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, Janjigian YY, Paty PB, Saltz LB, Kingham TP, Loo JM et al. (2019) PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife. 8, 52135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, Menguy I, Schreiber I, Perrault C, Vougier S et al. (2018) Dependence on the pyrimidine biosynthetic enzyme DHODH is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem Biol. 25, 705–717. [DOI] [PubMed] [Google Scholar]
- 28.da Huang W, Sherman BT & Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 29.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 30.Alexandrou C, Al-Aqbi SS, Higgins JA, Boyle W, Karmokar A, Andreadi C, Luo JL, Moore DA, Viskaduraki M, Blades M et al. (2018) Sensitivity of colorectal cancer to arginine deprivation therapy is shaped by differential expression of urea cycle enzymes. Sci Rep 8, 12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA & Staudt LM (2016) Toward a shared vision for cancer genomic data. N Engl J Med 375, 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love MI, Huber W & Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart JM, Segal E, Koller D & Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. [DOI] [PubMed] [Google Scholar]
- 34.Husson A, Brasse-Lagnel C, Fairand A, Renouf S & Lavoinne A (2003) Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. Eur J Biochem 270, 1887–1899. [DOI] [PubMed] [Google Scholar]
- 35.Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV & Lane DP (1990) p53 mutations in colorectal cancer. Proc Natl Acad Sci USA 87, 7555–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA & Zali MR (2013) The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol Bed Bench. 6, 120–128. [PMC free article] [PubMed] [Google Scholar]
- 37.Delage B, Luong P, Maharaj L, O’Riain C, Syed N, Crook T, Hatzimichael E, Papoudou-Bai A, Mitchell TJ, Whittaker SJ et al. (2012) Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis 3, e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Syed N, Langer J, Janczar K, Singh P, Lo Nigro C, Lattanzio L, Coley HM, Hatzimichael E, Bomalaski J, Szlosarek P et al. (2013) Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis 4, e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Worthley DL & Leggett BA (2010) Colorectal cancer: molecular features and clinical opportunities. Clin Biochem Rev. 31, 31–38. [PMC free article] [PubMed] [Google Scholar]
- 40.Mortison JD, Schenone M, Myers JA, Zhang Z, Chen L, Ciarlo C, Comer E, Natchiar SK, Carr SA, Klaholz BP et al. (2018) Tetracyclines modify translation by targeting key human rRNA substructures. Cell Chem Biol. 25, 1506–1518, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaheen N, Kobayashi K, Terazono H, Fukushige T, Horiuchi M & Saheki T (1994) Characterization of human wild-type and mutant argininosuccinate synthetase proteins expressed in bacterial cells. Enzyme Protein 48, 251–264. [DOI] [PubMed] [Google Scholar]
- 42.Hanse EA, Ruan C, Kachman M, Wang D, Lowman XH & Kelekar A (2017) Cytosolic malate dehydrogenase activity helps support glycolysis in actively proliferating cells and cancer. Oncogene 36, 3915–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho S, Chae JS, Shin H, Shin Y, Song H, Kim Y, Yoo BC, Roh K, Cho S, Kil EJ et al. (2018) Hormetic dose response to L-ascorbic acid as an anti-cancer drug in colorectal cancer cell lines according to SVCT-2 expression. Sci Rep 8, 11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER & Martinou JC (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96. [DOI] [PubMed] [Google Scholar]
- 45.Spinelli JB & Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinshaw DC & Shevde LA (2019) The tumor microenvironment innately modulates cancer progression. Cancer Res 79, 4557–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimmelman AC & White E (2017) Autophagy and tumor metabolism. Cell Metab 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, Papagiannakopoulos T, Molina H, Snuderl M, Lewis CA et al. (2018) Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol 20, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, Lau AN, Elmiligy S, Malstrom S, Lewis CA et al. (2018) Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 20, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng CT, Qi Y, Wang YC, Chi KK, Chung Y, Ouyang C, Chen YR, Oh ME, Sheng X, Tang Y et al. (2018) Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun Biol. 1, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ochocki JD, Khare S, Hess M, Ackerman D, Qiu B, Daisak JI, Worth AJ, Lin N, Lee P, Xie H et al. (2018) Arginase 2 suppresses renal carcinoma progression via biosynthetic cofactor pyridoxal phosphate depletion and increased polyamine toxicity. Cell Metab 27, 1263–1280, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi K, Jackson MJ, Tick DB, O’Brien WE & Beaudet AL (1990) Heterogeneity of mutations in argininosuccinate synthetase causing human citrullinemia. J Biol Chem 265, 11361–11367. [PubMed] [Google Scholar]
- 53.White E (2013) Exploiting the bad eating habits of Ras-driven cancers. Genes Dev 27, 2065–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou W, Laird PW & Shen H (2017) Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 45, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C et al. (2009) Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504. [DOI] [PubMed] [Google Scholar]
- 56.Dunlop EA, Johnson CE, Wiltshire M, Errington RJ & Tee AR (2017) Targeting protein homeostasis with nelfinavir/salinomycin dual therapy effectively induces death of mTORC1 hyperactive cells. Oncotarget. 8, 48711–48724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S et al. (2014) A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.