

Abstract
Methods for site-selective labeling of long, native RNAs are needed for studying mRNA biology and future therapies. Current approaches involve engineering RNA sequences, which may alter folding, or are limited to specific sequences or bases. Here, we describe a versatile strategy for mRNA conjugation via a novel DNA tiling approach. The method, TRAIL, exploits a pool of “protector” oligodeoxynucleotides to hybridize and block the mRNA, combined with an “inducer” DNA that extrudes a reactive RNA loop for acylation at a predetermined site. Using TRAIL, an azido-acylimidazole reagent was employed for labeling and controlling RNA for multiple applications in vitro and in cells, including analysis of RNA-binding proteins, imaging mRNA in cells, and analysis and control of translation. The TRAIL approach offers an efficient and accessible way to label and manipulate RNAs of virtually any length or origin without altering native sequence.
Keywords: DNA tiling, Site-selective labelling, mRNA, Acylation, Translation switching
Graphical Abstract
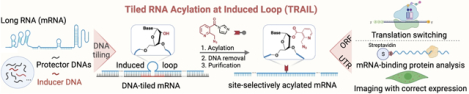
INH: A general method that enables site-localized acylation of mRNA at virtually any determined sites is described. The TRAIL method utilizes a novel DNA tiling strategy by hybridizing a pool of inexpensive “protector” DNAs to block mRNA and combining with an “inducer” DNA to extrude a reactive RNA loop for acylation. This approach can be employed for both mRNA labeling and control for multiple applications.
Introduction
Messenger RNAs (mRNAs) encoding proteins control gene expression, which operate under regulation of numerous non-coding RNAs and scaffold binding proteins[1]. Study of these mRNA related controlling mechanisms are important to human disease, as dysfunctional gene expression is found to be directly connected to cancers, autoimmunity, neurodegenerative diseases, and others[1d, 2]. Recent advances make mRNAs become a spotlight in new therapies. Researchers are pursuing disease-related mRNAs as targets for downregulating expression of specific genes[3] and are developing mRNA as vaccines against SARS-CoV-2 or other diseases, as well as protein-encoding agents for protein replacement and cellular reprogramming[4]. For these reasons, gaining increased understanding of mRNA structures, functions, dynamics and interactions are important both for basic biology and for future therapeutic development, and the development of general methodological tools for mRNA manipulation is expected to facilitate this research.
Labeling and conjugation of mRNAs constitutes one of the most important strategies to study mRNA natural properties and interactions, and while multiple approaches have been developed, they remain limited in versatility. Bioengineering-based approaches are widely employed by fusing the mRNA of interest to recognition domains for proteins or small molecules. Such strategies have involved addition of aptamer sequences (e.g. broccoli aptamer)[5], of stem-loop structures (e.g. MS2-MCP system)[6], or other designed structures[7] downstream of the stop-codon, enabling the binding of fluorescent reporters for visualization or installing pull-down handles (e.g., biotin) for interaction studies[8]. A risk of such strategies is that they involve the addition of extensive non-native sequences into the mRNA, which can alter its biology by changing the secondary structure and modulating native RNA-protein interactions.
For these reasons, methods for mRNA conjugation that do not alter the native sequence are desirable, and covalent labeling with small molecules offers the advantage of potentially being less perturbing to mRNA biology. One approach for internal labeling of RNA is incorporation of nucleotides during transcription; metabolic labeling with certain modified nucleotides based on endogenous synthesis is useful for intracellular mRNA labeling[9], but it does not allow for site-localized RNA modification. A related in vitro labeling strategy relies on co-transcription of fluorophore-labeled nucleotides[10] or azide-/alkyne-functionalized nucleotides[11], which can be further modified with bioorthogonal reactive probes. This also leads to general labeling of the mRNA with little control of the location within the RNA strand; in addition, larger modifications on bases (such as Cy5-UTP) can impede both transcription and translation efficiency[10a]. Labeling at the extreme ends of mRNAs is possible, via 5’-cap analogues combined with click chemistry[12] or poly(A) polymerase incorporated of modified ATP derivatives on 3’-poly(A) tails [13]. However, post-transcriptional internal labeling of mRNA presents a much greater challenge. One recent development involves ribozyme-assisted labeling by selectively catalyzing the reaction of a targeted adenosine with N6-modified ATP or non-natural nucleotide analogues[14]; while elegant, the approach is limited to adenosines and cannot be reversed. Another method utilizes promiscuous mRNA methyltransferases and their modified substrates for tag-free internal RNA labeling and caging, but is limited to specific consensus sequences[15]. Classical RNA-reactive chemistries tend to exhibit low yields and usually cannot be programmed to specific sites; examples include photocrosslinking with psoralens, which are selective only for double-stranded regions[16], dimethylsulfate, which generally reacts at unpaired cytosines and adenines[17], and stochastic reaction with 2’-OH acylating reagents that form bonds at 2′-OH groups of unpaired nucleotides[18]. An ideal method could enable internal labeling of mRNAs at specific, predetermined internal sites without limitations to specific bases or sequences, and without the need for modification of native sequence. To our knowledge, no such method is yet known.
Our approach for development of methods for internal conjugation and labeling of RNAs involves acylation at 2’-OH groups. RNA acylation with selected acylimidazole reagents has been reported to functionalize RNAs in high stoichiometric yields by esterification of 2’-OH groups, enabling efficient labeling and caging of RNAs at random sites in unpaired regions of RNA[19]. Recent site-random 2’-OH biotinylating agents have also been reported based on isatoic anhydride structure[20]. To broaden the utility of acylation-based modification, we recently introduced a method for labeling short RNAs, in which a complementary oligodeoxynucleotide generally blocks 2’-OH groups in duplex structure, while a remaining gap or loop in the RNA can be selectively acylated[21]. This approach was limited to short RNAs due to the limitations in the length of synthetic DNAs (typically ~150–200nt). Long RNAs (>200nt), however, display greater challenges in selective modification due to the more intricate structures and functions from diverse folding, base pairing and 3D interactions compared with short RNAs. As mRNAs are commonly ~600–2000nt or longer, new strategies for conjugation are required.
Here, we report a versatile strategy for site-selective acylation of mRNA, enabling both labeling and control. The method, Tiled RNA Acylation at Induced Loops (TRAIL), exploits a pool of inexpensive complementary DNAs tiling and blocking the mRNA except for at a predetermined site that induces a reactive RNA loop. We show that mRNA can be efficiently acylated at selective loop sites in different regions (untranslated region (UTR), poly(A) tail or open reading frame (ORF)), and we characterize the adducts’ effects on mRNA translation. We find that precise acylation at sites within the ORF region enables the suppression of mRNA expression and leads to specific ribosome stalling. If desired, the acylated mRNA activity can be effectively restored by phosphine treatment to recover the translation ability. For labeling without affecting translation, we find that localized acylation at UTR sites or poly(A) tails retains the translation capacity allowing for mRNA labeling with fluorophores or affinity handles. We expect that the TRAIL approach will be broadly applicable to many long RNAs to facilitate the study of function and interaction, and should be accessible to a wide range of chemistry and biology laboratories.
Results and Discussion
Design of DNA-tiled mRNA acylation at induced loops (TRAIL).
To expand the capability of site-localized acylation to long RNAs such as mRNAs, we considered applying our recent loop-inducing strategy, which can localize reactivity to programmed sites within short RNAs[21]. The challenge for long RNAs is how to block reaction all along the RNA sequence while at the same time inducing a loop at one site; here we considered the use of a pool of complementary “protector” DNA oligonucleotides binding side-by-side (tiling) along the RNA (Figure 1). In principle, the protector DNAs tiled along the mRNA results in nicked duplexes, which have been demonstrated to remain largely stacked and stable at nicked sites[21–22], and we hypothesized that this stacking at the junctions might be sufficient to block reactivity in a long RNA. While this approach requires a number of DNAs for one long RNA (e.g., 15–25 oligodeoxynucleotides at 60mer length to cover 1000–1500nt RNA lengths), such DNAs are now routinely and rapidly available at low cost. In this way, we expected that the majority of mRNA might be protected in RNA/DNA duplex structures, leaving highly reactive 2’-OH groups of mRNA that are exposed in the specific loop site extruded by the inducer DNA. Subsequent to this structure induction, addition of a water-soluble acylating agent (e.g. the azide-containing reagent NAI-N3 [19b]) should effect the covalent conjugation. For isolation, we hypothesized that the DNAs might be removed via one-step digestion with DNase, and the conjugated RNA purified with a commercial RNA clean-up column. Several questions regarding this strategy remained unknown: How well do commercial oligonucleotides protect an mRNA when tiled in this fashion? Does the folded structure of a long RNA block the binding of complementary DNAs? How efficiently can the labeling occur, and how large an induced loop is ideal for achieving high conjugation yields in a long RNA? Do mRNAs undergo degradation during these steps? Can the complementary DNAs be degraded efficiently enough to remove any antisense RNA-suppressive activity? And finally, how does localized acylation affect mRNA function? Experiments were directed to these issues.
Figure 1.
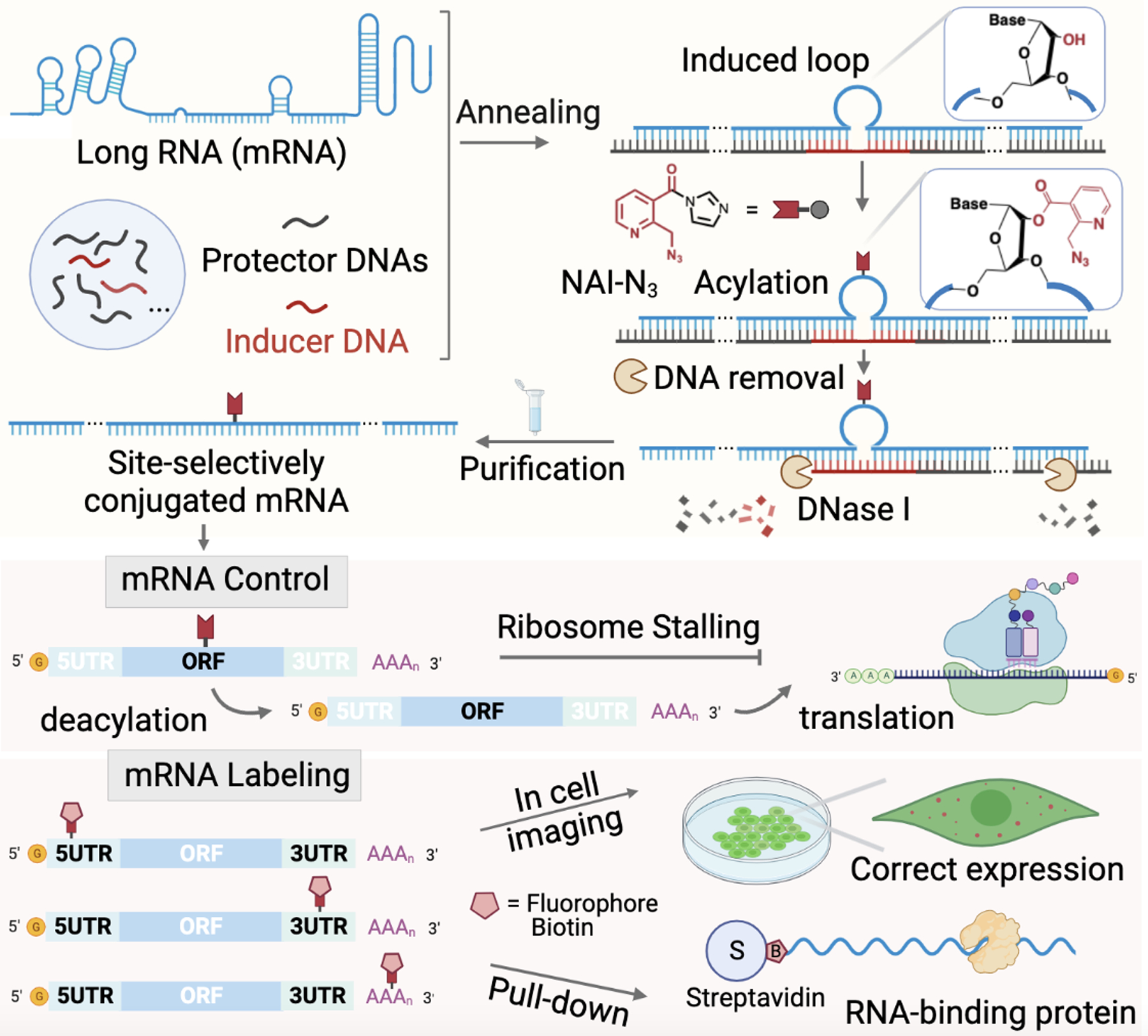
Schematic outline of the TRAIL approach for site-localized mRNA acylation, illustrating applications in mRNA labeling and control. mRNA (blue) is tiled/covered with protector DNAs (black) to cover most of mRNA, except for a reactive loop extruded by an inducer DNA (red). The 2’-OH groups in loop site can then selectively react with an acylating reagent, such as NAI-N3, yielding a site-localized mRNA conjugate after one-step DNA digestion and column purification. The azide group on the adduct can be further utilized for addition of a fluorophore or biotin label, or can be removed by Staudinger reduction to switch on biological activity.
Performance of site-selective mRNA acylation via tiling.
To explore the protector/inducer approach for mRNA reaction, we first determined the DNA tiling effect on mRNA function. Although previous studies reported that a single nick does not result in high levels of acylation of a short RNA[21], the combination of many nicks might add to a substantial amount of acylation in a long RNA, which together could block mRNA expression. The new strategy also raised the issue of DNA removal, as digested DNA fragments remaining might promote mRNA silencing due to antisense effects. As a platform to test these issues, we chose a 996nt mRNA encoding green fluorescence protein (GFP). To monitor RNA function after DNA tiling, we compared the in vitro translation efficiency of untreated mRNA (UT) with mRNA purified from fully complementary DNAs (FC) via DNA tiling, in this case with 16 oligodeoxynucleotides of 30–60nt length for the UTR and ORF regions, and one oligodeoxynucleotide of 100–150nt for the poly(A) tail. This allowed us to test DNA removal steps, enzyme sources and amounts, and clean-up columns, resulting in optimal DNase treatment conditions and choice of clean-up column (Figure S1). Under the optimized conditions, the translation efficiency of the mRNA after being hybridized with fully complementary tiling (FC mRNA) is found to be almost identical to untreated mRNA (UT) (Figure 2a). Interestingly, it was necessary to fully remove the DNAs, as incomplete digestion caused some loss of translation efficiency (Figure S1).
Figure 2.
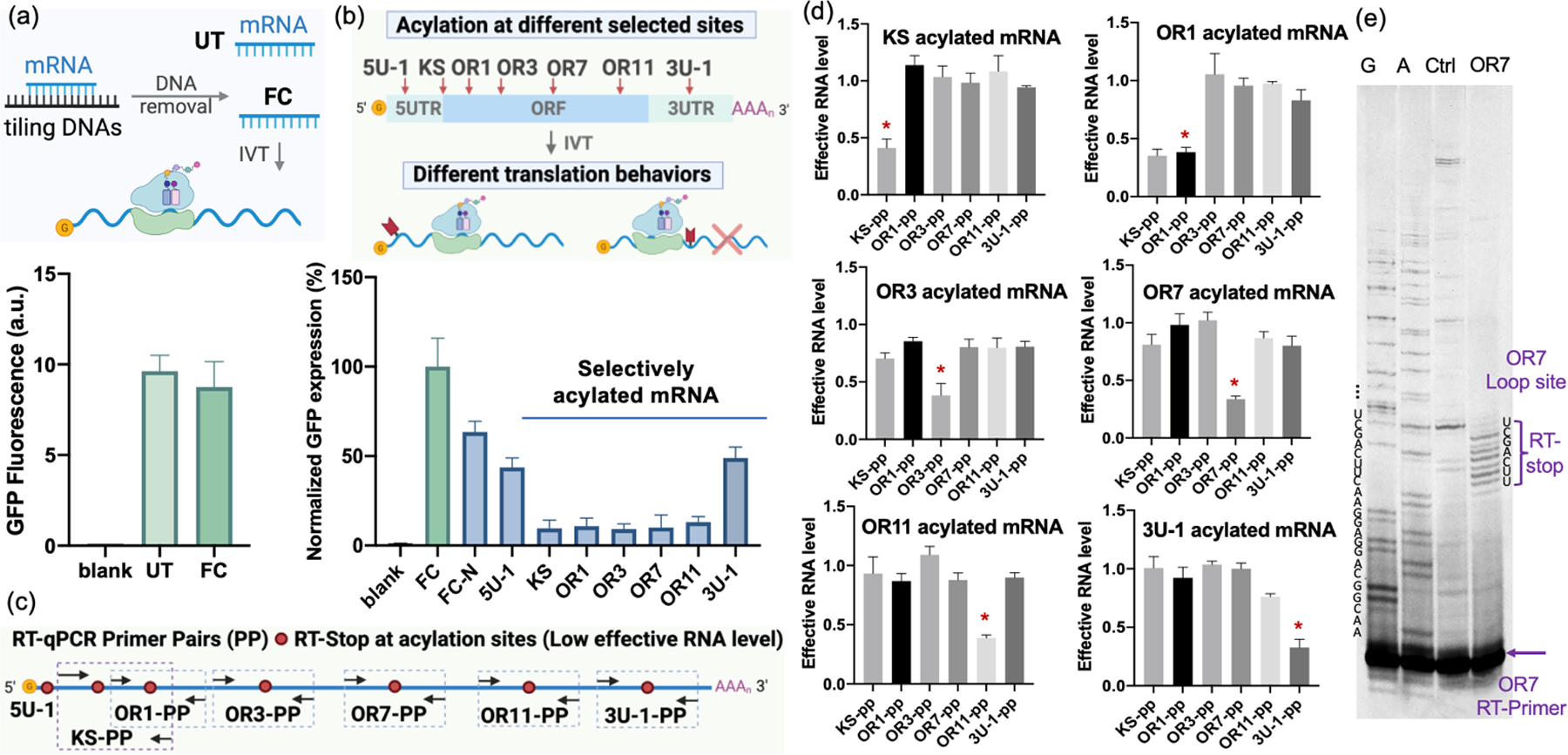
Translation behavior and analysis of site-selectively acylated mRNA via TRAIL. (a) In vitro translation (IVT) comparison of untreated mRNA (UT) and mRNA purified from full complementation via DNA tiling (FC) to evaluate DNA tiling and removal efficacy. Blank: IVT without mRNA as a negative control. (b) In vitro translation of selectively acylated mRNA (5U-1, KS, OR1, OR3, OR7, OR11, 3U-1) at different sites from 5’UTR to 3’UTR, showing strong suppression of translation at KS and ORF sites. FC was employed as a positive control and its GFP expression level was normalized to 100%. FC_N: FC reacted with 50mM NAI-N3. (c) Schematic of RT-qPCR primer pair designs for each acylated mRNA site. (d) RT-qPCR of selectively acylated mRNA. Specific site-acylated mRNA cannot be effectively amplified by corresponding primer pairs resulting in low effective RNA level, but can be efficiently amplified by unrelative primer pairs, establishing site selectivity of acylation. Intended acylation sites are marked by “ * ”. (e) PAGE analysis of RT stops for site-selectively acylated mRNA at OR7 loop-induced site, showing the degree of loop selectivity relative to protected RNA surrounding the site, as well as multiple acylations occurring in the loop site.
Next, we investigated inducing reaction at specific sites in the RNA by replacing one of the protector DNAs with an inducer DNA designed to induce a site-localized loop. Inducers were 30–60nt in length and were designed to bind to the mRNA directly abutting the adjacent protectors, but omitting 3–7 nucleotides of mRNA complementarity at a central site, thus inducing an RNA bulge loop. We tested loops induced at seven sites in the mRNA from 5’UTR along to 3’UTR (5U-1, KS, OR1, OR3, OR7, OR11, 3U-1) (Figure 2b). The constructed mRNAs were then reacted with NAI-N3 under optimized conditions (50 mM NAI-N3 at 37 °C for 4 h, Figure S2a, S2b) followed by DNA removal. For in vitro translation, we used FC mRNA as a positive control with its GFP expression level normalized to 100%. The mRNAs acylated at different sites showed strikingly different translation abilities (Figure 2b). The 5’UTR-acylated mRNA exhibited ~50% expression of the positive control while the ORF-acylated mRNA only displayed ~10% expression, suggesting strong blocking of the ribosome. Testing the effects of different induced loop sizes, we compared 3nt-loop and 7nt-loop induced at the same ORF region of the mRNA (Figure S2c). The mRNA acylated with a 7nt-loop in the ORF showed more efficient inhibition of translation compared with 3nt-loop cases, likely due to higher numbers of acyl groups. Comparison of the broad effects of DNA tiling with acylation revealed a small to moderate degree of suppression: the FC-RNA exposed to NAI-N3 (FC-N) showed a ~30–40% decrease in GFP signal relative to untreated RNA (Figure 2b, Figure S2a), which might be attributed to a low level of background acylation in fully duplex regions or at nicked sites.
To further evaluate the selectivity of the localized reaction strategy, we probed acylated mRNAs both with quantitative PCR (RT-qPCR) and by reverse transcriptase (RT) primer extension analyzed by denaturing polyacrylamide gel electrophoresis (PAGE). We designed six primer pairs for RT-qPCR to probe each acylation site (except 5U-1 as its location is very near the 5’-cap) (Figure 2c). The PCR results revealed that acylating mRNA between the corresponding primer pairs led to clearly low amplification, with effective RNA levels of ca. 0.3–0.4 relative to untreated mRNA, as RT enzymes are known to be blocked by this acylation[23]. Importantly, relatively high effective RNA levels (near 1.0, similar to UT) were found with off-target primer pairs, showing effective DNA-mediated protection at sites away from the induced loop (Figure 2d, Figure S3). Similarly, PAGE gel analysis showed truncation of cDNAs from RT primer extension at acylated mRNA sites at near-nucleotide resolution (Figure 2e, S4). It also indicated multiple acylations occurring in the induced 7nt-loop of mRNA, consistent with the acylation pattern in short RNAs (Figure S5)[21]. Tests with this short RNA model showed an average of 1–2 acyl groups in 5nt-loop site and 2–3 groups in a 7nt-loop site (Figure S5) under our mRNA reaction conditions.
Importantly, the TRAIL method resulted in high-yield local acylation of mRNA totaling ~60–70% in a given loop according to RT-qPCR (Figure 2d) and ~70% based on the semi-quantitative gel analysis of acylated and further biotinylated mRNA before and after streptavidin bead pull-down (Figure S6). Overall, the experiments revealed that mRNA can be efficiently acylated at selectively induced loop sites in high yield by simply tiling a set of unmodified and inexpensive protector DNAs on the target, combining them with selected loop-inducer DNAs. Importantly, the data also showed that acylation at some sites in an mRNA can strongly inhibit translation; this is to our knowledge the first observation that 2’-O-acylation blocks translation. The results suggest further use in controlling RNA expression (see below).
Site-directed control of mRNA translation via TRAIL-mediated acylation at ORF.
Encouraged by the different translation behaviors of selectively acylated mRNAs, we next evaluated whether it is possible to take advantage of this site-specific modification method for switchable blocking (caging) and subsequent activation of translation. Caging RNAs with labile modifications can be activated by chemicals or by light, and is used widely in short synthetic RNAs in which modifications can be placed at precise positions[24]. Caging much longer messenger RNAs, which could be broadly useful in studies of gene expression (such as in embryonic development) is considerably more difficult, due in large part to the challenges of modifying such long RNAs. One report described photolabile diazo groups that react with mRNA at random phosphates but generate unstable RNA phosphotriester linkages that lead to strand degradation[25]. Among the most effective current approaches to this problem involves engineering multiple enzyme recognition sequences (such as tRNA transglycosylase[26]) into the mRNA; limitations of such an approach are that it cannot readily be incorporated into the coding sequence of the RNA, and that the added sequences may alter mRNA folding and interactions. Thus the development of a strategy for caging mRNA at any position (including coding regions) and without the need for the addition of nonnative sequences is desirable.
We first explored the possible control of translation by acylation at specific sites in the ORF region, using the NAI-N3 reagent, which is chemically reversible by phosphine treatment[19b]. We tested the effects of both single ORF sites as well as combined acylation sites (Figure 3a; 1 loop samples: KS, OR1, OR3, OR7, OR11; 2 loops samples: KS/OR1, OR1/3, OR3/7, OR7/11, OR1/7; 3 loops samples: KS/OR1/3, OR3/7/11). The data show that in vitro GFP expression of acylated mRNAs was efficiently suppressed to 5–15% of that of non-acylated mRNA (FC) and could be successfully recovered after reversal to 50–70% for single-loop cases, 40%−50% for 2-loop cases and 30–40% for 3-loop cases after adding 3 mM water-soluble phosphine (TPPMS or THPP) at 37 °C for 3 h (Figure 3b, Figure S7). There were similar increases (~4–7-fold turn-on) of GFP expression for all the loop samples, and the data revealed that multi-acylated mRNAs led to somewhat stronger repression but also yielded less activation, presumably due to incomplete or slow loss of some of the acyl groups. Notably, the translation of randomly acylated mRNA (SS, no protector or inducer DNAs) was completely inhibited, but this RNA yielded no recovery after phosphine treatment (Figure 3b), which establishes that the use of protector DNAs for localized acylation is critical. To confirm the biological viability of the approach, we tested the same caged RNAs for translation in HeLa cells after transfection and subsequent treatment by phosphine. The results show that the ORF-acylated GFP mRNAs could also be controlled in HeLa cells, giving a 2–3-fold increase in GFP signal after phosphine addition (Figure S8).
Figure 3.
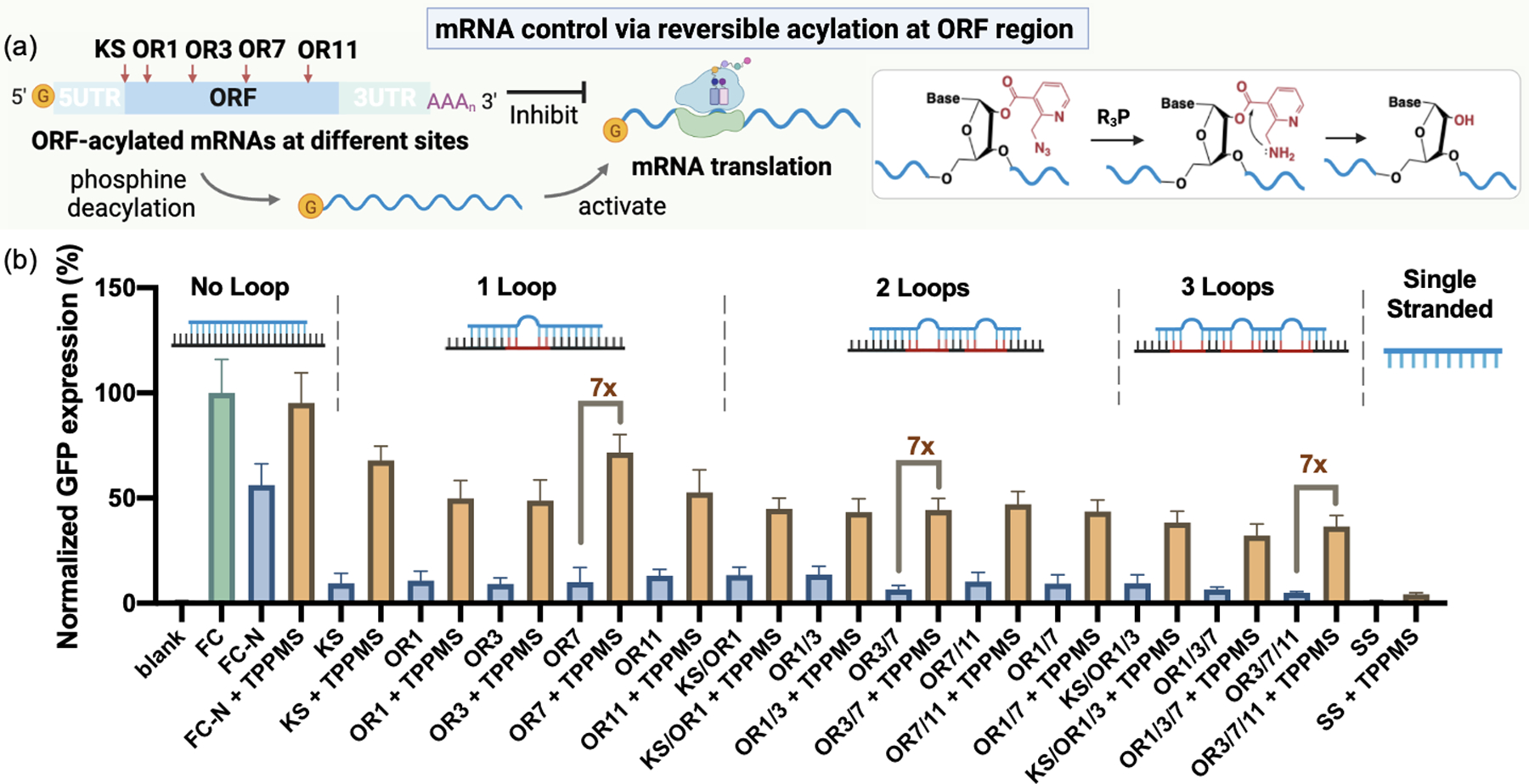
(a) Schematic of mRNA control by precise acylation at different sites of ORF. (b) In vitro translation of ORF-acylated mRNAs and deacylated mRNA restored with 7-fold increases in activity by treating with 3mM TPPMS. Samples were tested with no loop structures (FC, FC-N), 1-loop acylation (KS, OR1, OR3, OR7, OR11), 2-loop acylation (KS/OR1, OR1/3, OR3/7, OR7/11, OR1/7), 3-loop acylation (KS/OR1/3, OR1/3/7, OR3/7/11) and single-stranded random acylation (SS). The GFP expression level of FC was normalized to 100%. Error bar represents mean ± SD.
To test the generality of this approach in locally modifying mRNAs, we applied TRAIL-mediated acylation to a second, considerably longer mRNA, encoding firefly luciferase (FLuc, 1929nt). We annealed 32 unpurified protector/inducer DNAs of 30–150nt length and tested the expression of FLuc mRNA by a luciferase assay (Figure S9a). Although increased annealing temperature modestly lowered FLuc mRNA activity to ~55–65% of untreated mRNA, it showed improved DNA protection of this long mRNA (Figure S9b). The selectively acylated FLuc mRNA revealed strikingly different translation ability, similar to that seen with GFP mRNA (Figure S9c). The ORF-acylated RNA was suppressed to 5–10% expression levels, while UTR- and poly(A) tail-acylated RNA showed much less suppression. After TPPMS treatment, the expression of ORF-acylated FLuc mRNA increased by 5–6-fold (Figure S9c). Overall, the results suggest that the TRAIL method can be widely applicable.
Selective ribosome stalling induced by locally acylated mRNA at ORF.
Having established conditions that yield efficient suppression of translation by localized acylation, we then proceeded to test whether the ORF-acylated mRNA could induce ribosome stalling at varied acylation sites. Ribosome stalling is manifested by the local accumulation of ribosomes at specific codon positions of mRNAs. It is an important cue for recognition of defective mRNA, and helps maintain protein homeostasis via ribosome-associated quality control and ribosome recycling[27]. The ability to control ribosome stalling at pre-selected sites could serve as a useful tool to study these mechanisms. To test this possibility, we employed localized acylation to stall translation, and detected the resulting truncated peptides by western blotting with an anti-puromycin antibody (Figure 4a). ORF-acylated mRNAs (KS, OR1, OR3, OR5, OR7, OR9, OR11) were optimized for polypeptide detection by in vitro translation for 1.5 h at 25 °C followed by addition of 1 mM puromycin, which causes the release of the peptides (Figure S10). We successfully detected stalled peptides with sizes correlated to downstream ORF acylation sites of GFP mRNA ranging from OR5 to OR11 (Figure 4b). Interestingly, the mRNA with acylated sites at the upstream end of ORF (OR1, OR3) did not show a clear stalled peptide although they displayed efficient translation inhibition similar to that seen for downstream sites. For the stalled sites we found that treatment by phosphines, which remove the blocking acyl groups, resulted in disappearance of the stalled peptides (Figure 4c), and the above in vitro translation experiments show that the fluorescence of full-length GFP is restored, further confirming the localized stalling and its reversibility (Figure 3b).
Figure 4.
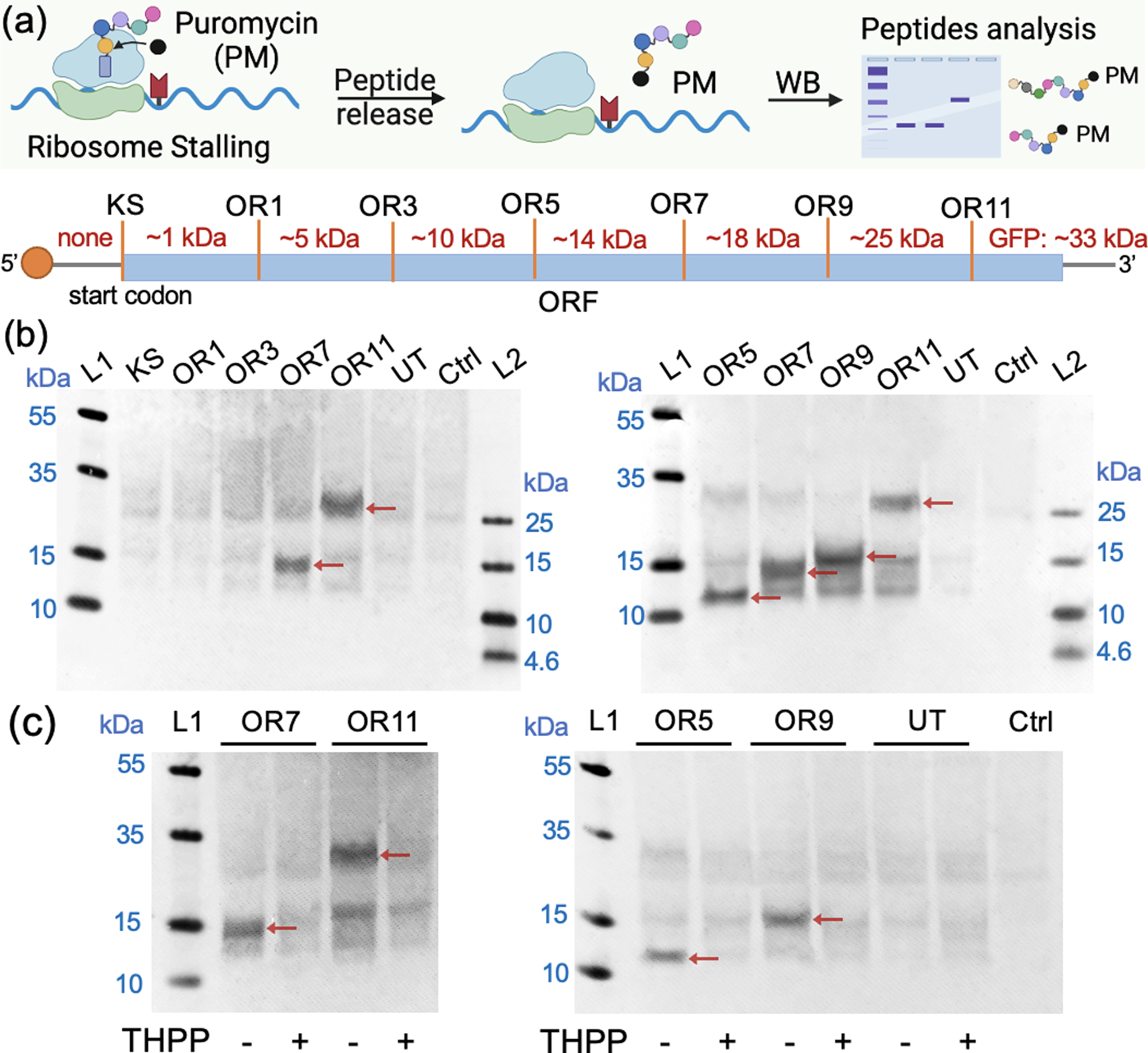
(a) Schematic of selective ribosome stalling induced by ORF-acylated mRNAs. Stalled peptides were released by reacting with puromycin and analysis by anti-puromycin antibody via western blot. The molecular weight of predicted stalled peptides are shown. (b) Western-blot analysis of stalled peptides from each acylated mRNA and untreated mRNA (UT) after 1.5h incubation in wheat-germ extract and later 1h reaction with 1mM puromycin at 25 °C. 0.5M KOAc was added along with puromycin to prevent the dissociation of ribosomes[27]. Ctrl: blank sample without adding mRNA. (c) Ribosome stalling comparison of each acylated mRNA with and without phosphine treatment via western blot. 3mM THPP was used to remove the acylation of mRNA, and the stalled peptides disappeared after deacylation. The arrows in the figure point to the stalled peptides of expected lengths.
Pull-down of RNA-binding protein with UTR-biotinylated mRNA.
An important application of RNA conjugation is identification of associated proteins via biotinylation[28]. For mRNAs, this would be ideally done at a site that does not affect biological activity. Since the above acylation at UTR sites retained mRNA translation ability, we next applied TRAIL for UTR acylation and biotinylation, and tested its ability to identify an associated cellular protein. Since the acylating agent NAI-N3 contains an azide functional group, this enables biotinylation via reaction with strained cyclooctyne reagents. To test this, we installed a biotin handle in 5’UTR via TRAIL, reacted with a biotinylating agent (Figure 5a), and tested a pull-down experiment with a known mRNA-associated protein (poly(A) binding protein (PABP))[29] (Figure 5b). We conducted the pull-down assay both from cell lysates and in live cells by transfection with biotinylated mRNA. The Western-blot results showed that PABP could be successfully pulled down by the 5’UTR-biotinylated mRNA in both ways, while controls with azido-mRNA lacking biotin or untreated mRNA showed no protein band (Figure 5c).
Figure 5.
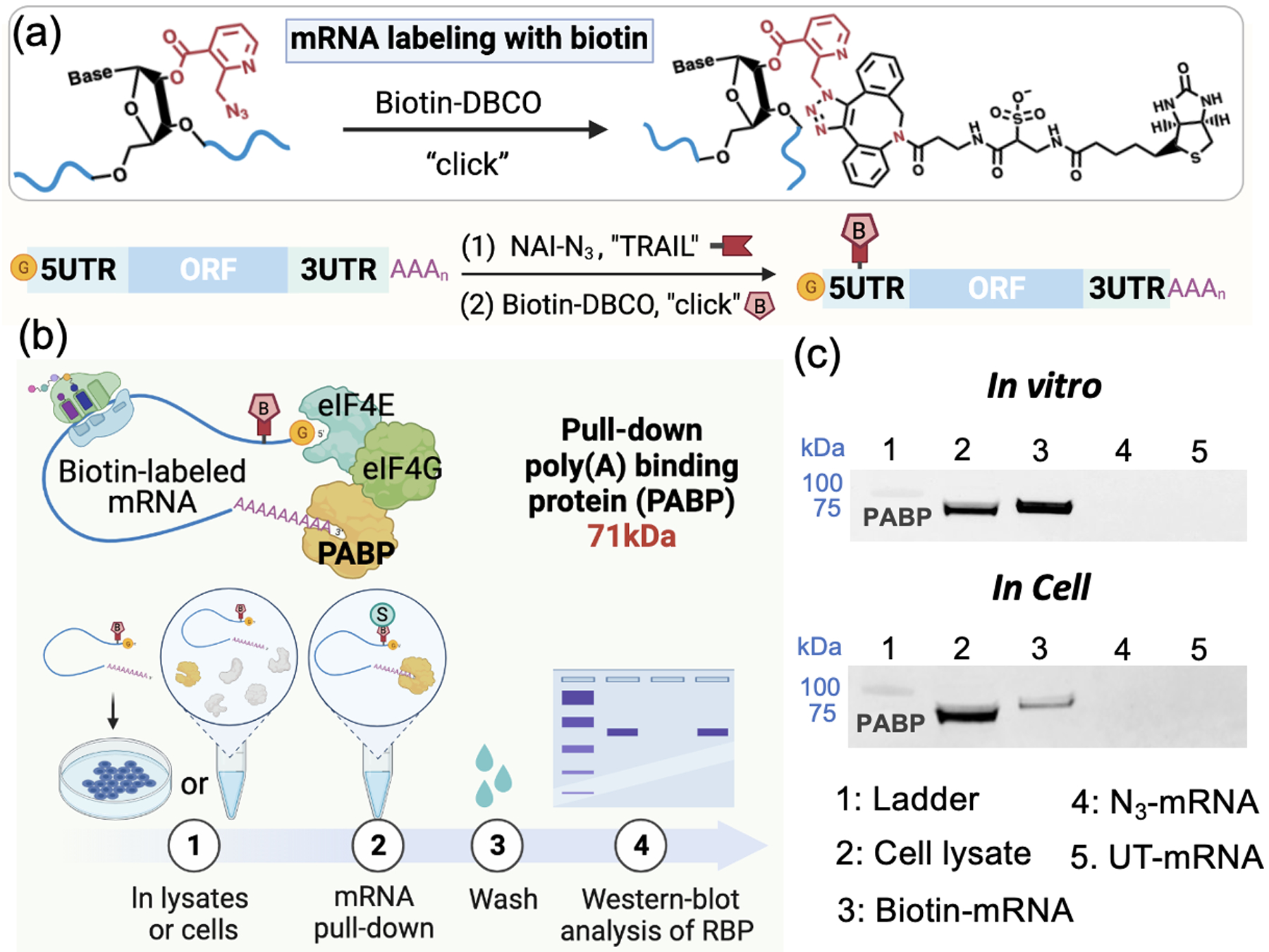
(a) Schematic of generating selectively biotinylated mRNA via TRAIL followed by click chemistry. (b) Schematic of the pull-down assay for poly(A)-binding protein (PABP) via biotinylated mRNA in vitro and in cells. (c) Detection of the protein PABP (71 kDa), to test the pulldown capacity of biotin-labeled mRNA in vitro and in cells by Western blot.
Visualization and translation of site-selectively fluorophore-labeled mRNA in cell culture.
Next, we applied this labeling method to visualize mRNA and the translation of the mRNA in living cells by conjugating a fluorophore to the target mRNAs[10b]. Acylated mRNAs with azide groups were first reacted with TAMRA-DBCO (Figure S11a) and success of the labeling was confirmed by agarose gel (Figure S11b). The labeling sites were tested by evaluating the in vitro translation efficiency of TAMRA-labeled mRNAs at different UTR sites and poly(A) tails. The results indicated a range of 30%−60% expression relative to non-acylated mRNA (Figure S11c), which is better than a widely used fluorophore-labeled commercial mRNA (~20% translation capacity reported in the literature[10b]).
In order to enable three-color cellular imaging, GFP mRNA was labeled with Cy5 which emits fluorescence at ca. 670 nm; the blue channel was used for Hoechst 33342 (nucleus), the green channel for GFP (translation efficiency), and the red channel for the Cy5-labeled mRNA. Transfection of Cy5 labeled mRNAs into HeLa cells showed bright red punctate images which corresponds to the transfected mRNA (Figure 6a, 6c; Z-stack imaging in Supplementary video 1,2). Cells transfected with Cy5 dye alone or Cy5 mock-treated mRNA without acylation showed negligible fluorescence as expected (Figure S12). To evaluate the intracellular translation efficiency depending on the labeling sites in mRNA, Cy5 was labeled at different sites in mRNA such as 5’UTR (5U-0_Cy5, 5U-1_Cy5, 5U-2_Cy5), ORF (KS_Cy5, OR1_Cy5, OR3_Cy5, OR7_Cy5, OR11_Cy5), 3’UTR (3U-1_Cy5, 3U-2_Cy5) and poly(A) tail (p(A)) (Figure 6b). We observed efficient GFP expression in the cases of UTR-labeled and poly(A) tail labeled mRNAs (fluorescence intensity ratio of GFP/mRNA is ~2.2–4.8; GFP/Hoechst is ~1.0–1.9), while minimal expression levels were observed in ORF-labeled mRNAs (GFP/mRNA ratio is ~0.6–1.0; GFP/Hoechst ratio is ~0.2–0.5; Figure 6c, S13, S14), as expected from the above results showing that acylation blocks translation in the ORF. The observed green fluorescence establishes that the Cy5-mRNAs labeled in the UTR region or poly(A) tail can be functionally processed by cellular ribosomes. Moreover, the fluorophores could be installed in several sites of mRNA in UTR region to enhance the single-molecule brightness; and the multiple-tagged mRNA showed similarly effective translation as single-site labeled mRNA(Figure S15). Overall, the results document the success of a method for localized fluorescent mRNA tagging that does not prevent translation.
Figure 6.
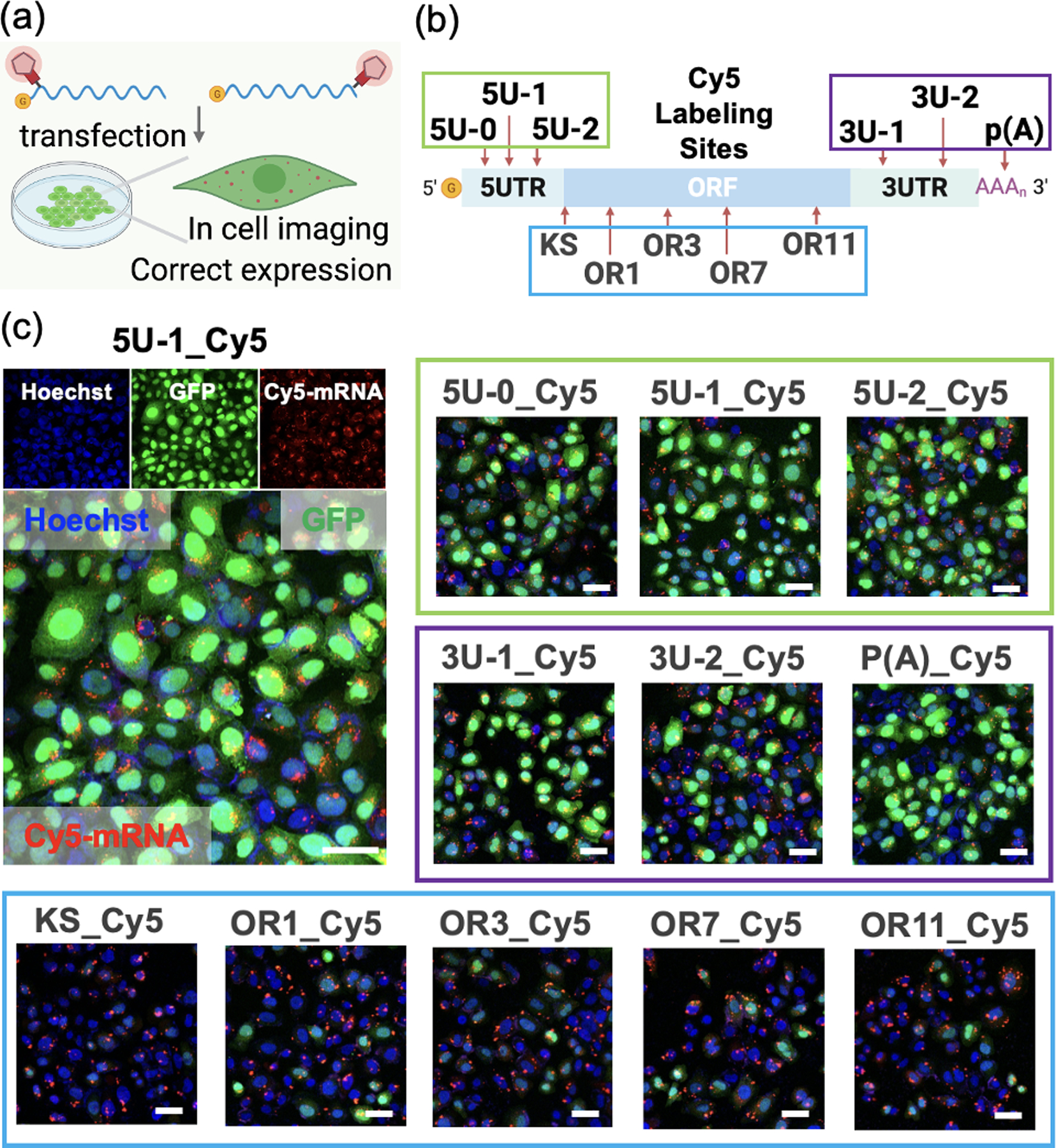
Imaging and translation of Cy5-labeled mRNA (labeled via TRAIL) transfected into HeLa cells. The translation was measured by the fluorescence intensity of GFP protein, and mRNA was visualized by labeled Cy5 dye (red) using confocal microscopy after 4h incubation. (a) Schematics of Cy5-labeled mRNA transfection into cells. (b) Schematics of the Cy5 labeling sites in the mRNA. (c) Images of cells transfected with Cy5-labeled mRNAs at different sites from 5UTR (5U-0_Cy5, 5U-1_Cy5, 5U-2_Cy5), ORF (KS_Cy5, OR1_Cy5, OR3_Cy5, OR7_Cy5, OR11_Cy5), 3UTR (3U-1_Cy5, 3U-2_Cy5) to poly(A) tail (p(A)). Blue: nuclei stained by Hoechst 33342; Green: translated GFP protein; Red: Cy5-labeled mRNA. Scale bar: 50 μm.
As a further test of tracking protein-mRNA interactions by TRAIL labeling, we monitored Cy5-labeled mRNA and its translation levels in the presence/absence of stress granules (Figure 7a). Stress granules (SGs) are phase-separated membraneless organelles in the cytoplasm, which consist of mRNAs and RNA-binding proteins formed in response to stress, with reported connections to human diseases[30]. The SGs were induced by sodium arsenite, providing oxidative stress, and were visualized with a primary antibody for G3BP1 protein, which is documented to initiate stress granule formation[30a]. We found that the expression level of GFP in the presence of stress granules was significantly reduced regardless of the labeling site (Figure 7b, Figure S16), and the signal from Cy5-labeled mRNA (red) was colocalized to G3BP1-labeled SGs (blue) after sodium arsenite treatment (Figure 7c, 7d; Z-stack imaging in supporting video 3). These observations are consistent with previous reports showing that a fraction of cytoplasmic mRNAs localize to SGs upon their assembly, and that association with SGs is correlated with suppressed translation[31]. Overall, we conclude that TRAIL can be used to fluorescently label an mRNA without strong interference to translation or to association with subcellular compartments.
Figure 7.
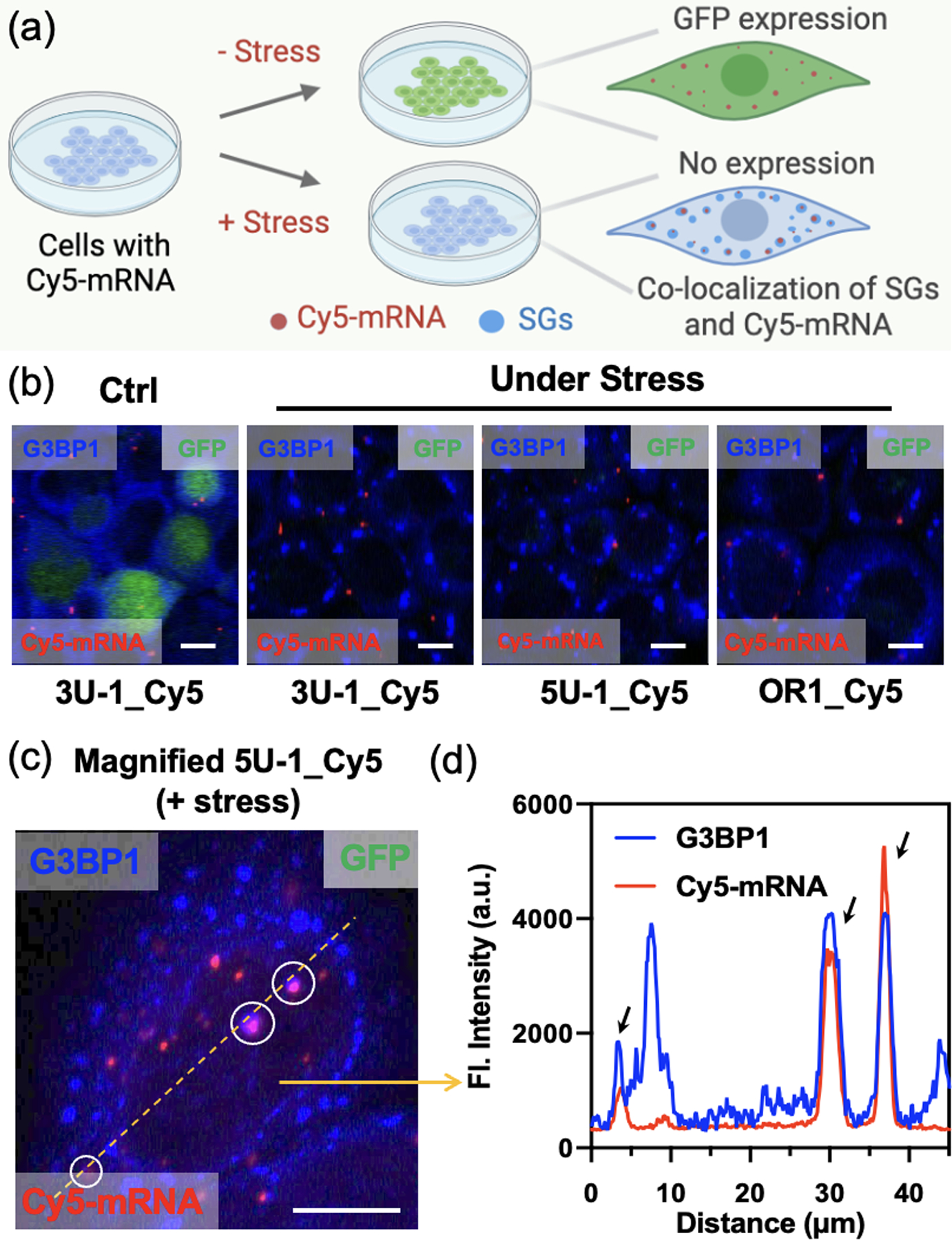
(a) Schematics of stress granule (SG) RNA association studied by monitoring translation and mRNA visualization in the presence/absence of stress. (b) Immunofluorescence images for evaluating translation level of Cy5-labeled mRNAs in the presence/absence of SGs. Red: Cy5-labeled mRNA; Blue: G3BP1-labeled SGs. Scale bar: 12.5 μm. (c) Magnified immunofluorescence image for tracking Cy5-labeled mRNA localization in the presence of SGs. SGs were formed by treating with 200 μM sodium arsenite. Red: Cy5-labeled mRNA; Blue: G3BP1-labeled SGs. Scale bar: 13 μm. (d) Plot profile of the fluorescence intensity along the yellow line in (c), showing three peaks of co-localization of Cy5-mRNA and G3BP1-SGs.
Conclusion
Our experiments have developed and documented a general method that enables site-selective modification of long RNAs such as mRNAs at virtually any determined position based on stoichiometric 2’-OH acylation that is directed by complementary DNAs. This method, TRAIL, is convenient, rapid and low-cost, simply by tiling inexpensive and commercially synthesized DNA strands to the mRNA without any further engineering. The new results establish that tiling a pool of complementary DNAs can effectively protect mRNA from acylation at undesired positions, regardless of potentially interfering secondary structures in the mRNA. We further find that a possible antisense effect of the DNAs, which might come from remaining incompletely digested DNA fragments, can be eliminated with optimized DNase treatment and column purification. Our experiments also show that the mRNA can function properly after exposure to the tiling, digestion and purification steps. The method is efficient, giving an acylation yield of ~60–70%, and is not highly labor intensive, requiring a simple purification of DNase treatment and clean-up column. Thus we expect that the technology can be widely useful to many chemistry and biology researchers.
Using this approach, our studies reveal for the first time that localized acylation by a nicotinyl group at different sites of mRNA can have dramatic effects on translation, demonstrating the high flexibility of this method for both mRNA control and labeling. Recent studies have shown that 2’-O-methyl groups can lead to significant stalling of translation depending on their positions in the codon[32], and the current nicotinyl ester group is significantly larger. Interestingly, a 1990 report of acetylation of RNA reported unhindered translation[33], which is unexpected in light of our results but might also be explained by the larger nicotinate size. In any case, we find that acylation with NAI-N3 at specific coding regions generates a reversible adduct on mRNA that cages translation, and translation can be restored by the addition of phosphines in vitro or in cell culture. We observe that the localized adducts provide considerably better mRNA recovery compared with randomized (non-TRAIL) acylation. Additionally, adducts introduced into selected coding region sites induce specific ribosome stalling, which offers a simple approach for manipulating stalling for studies of translation and RNA quality control.
We further find that an azide group on the acylating agent is useful for click-based conjugation, which enables adding affinity labels and fluorescent labels at desired noninterfering sites in an mRNA with retention of translation capacity. We have shown that TRAIL can selectively introduce a biotin label to pull down mRNA-binding proteins and can precisely install a fluorescent label to simultaneously visualize mRNA and its translation product in cultured cells. We conclude that this simple approach could be employed in a wide variety of studies of mRNA interactions, functions and dynamics, and we expect that it could be broadly applied to many other mRNAs and to long noncoding RNAs as well.
Supplementary Material
Acknowledgements
We thank the U.S. National Institutes of Health (GM127295 and GM130704) for support.
References
- [1]_.(a) Buccitelli C, Selbach M, Nat. Rev. Genet 2020, 21, 630–644; [DOI] [PubMed] [Google Scholar]; (b) Shi Y, Nat. Rev. Mol. Cell Biol 2017, 18, 655–670; [DOI] [PubMed] [Google Scholar]; (c) Bracken CP, Scott HS, Goodall GJ, Nat. Rev. Genet 2016,17, 719–732; [DOI] [PubMed] [Google Scholar]; (d) Goodall GJ, Wickramasinghe VO, Nat. Rev. Cancer 2021, 21, 22–36. [DOI] [PubMed] [Google Scholar]
- [2].(a) Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ, Nat. Rev. Neurol 2012, 8, 518–530; [DOI] [PubMed] [Google Scholar]; (b) Lee TI, Young RA, Cell. 2013, 152, 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dammes N, Peer D, Trends Pharmacol. Sci 2020, 41, 755–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Pardi N, Hogan MJ, Porter FW, Weissman D, Nat. Rev. Drug Discov 2018, 17, 261–279; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sahin U, Karikó K, Türeci Ö, Nat. Rev. Drug Discov 2014, 13, 759–780. [DOI] [PubMed] [Google Scholar]
- [5].Chen X, Zhang D, Su N, Bao B, Xie X, Zuo F, Yang L, Wang H, Jiang L, Lin Q, Fang M, Li N, Hua X, Chen Z, Bao C, Xu J, Du W, Zhang L, Zhao Y, Zhu L, Loscalzo J, Yang Y, Nat. Biotechnol 2019, 37, 1287–1293. [DOI] [PubMed] [Google Scholar]
- [6].Urbanek MO, Galka-Marciniak P, Olejniczak M, Krzyzosiak WJ, RNA Biol. 2014, 11, 1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li F, Dong J, Hu X, Gong W, Li J, Shen J, Tian H, Wang J, Angew. Chem. Int. Ed 2015, 54, 4597–4602; Angew. Chem. 2015, 127, 4680–4685. [DOI] [PubMed] [Google Scholar]
- [8].Busby KN, Fulzele A, Zhang D, Bennett EJ, Devaraj NK, ACS Chem. Biol 2020, 15, 2247–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang Y, Kleiner RE, J. Am. Chem. Soc 2019, 141, 3347–3351. [DOI] [PubMed] [Google Scholar]
- [10].(a) Custer TC, Walter NG, Protein Sci. 2017, 26, 1363–1379; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baladi T, Nilsson JR, Gallud A, Celauro E, Gasse C, Levi-Acobas F, Sarac I, Hollenstein MR, Dahlén A, Esbjörner EK, Wilhelmsson LM, J. Am. Chem. Soc 2021, 143, 5413–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Croce S, Serdjukow S, Carell T, Frischmuth T, ChemBioChem. 2020, 21, 1641–1646. [DOI] [PubMed] [Google Scholar]
- [12].Mamot A, Sikorski PJ, Warminski M, Kowalska J, Jemielity J, Angew. Chem. Int. Ed 2017, 56, 15628–15632; Angew. Chem. 2017, 129, 15834–15838. [DOI] [PubMed] [Google Scholar]
- [13].Anhäuser L, Hüwel S, Zobel T, Rentmeister A, Nucleic Acids Res. 2019, 47: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].(a) Maghami MG, Dey S, Lenz A-K, Höbartner C, Angew. Chem. Int. Ed 2020, 59, 9335–9339; Angew. Chem. 2020, 132, 9421–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maghami MG, Scheitl CPM, Höbartner C, J. Am. Chem. Soc 2019, 141, 19546–19549. [DOI] [PubMed] [Google Scholar]
- [15].Ovcharenko A, Weissenboeck FP, Rentmeister A, Angew. Chem. Int. Ed 2021, 60, 4098–4103; Angew. Chem. 2021, 133, 4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kurz M, Gu K, Lohse PA, Nucleic Acids Res. 2000, 28: e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Graveley BR, Mol. Cell 2016, 63, 186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].(a) Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM, J. Am. Chem. Soc 2005, 127, 4223–4231; [DOI] [PubMed] [Google Scholar]; (b) Spitale RC, Flynn RA, Torre EA, Kool ET, Chang HY, WIREs RNA. 2014, 5, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].(a) Velema WA, Kool ET, Nat. Rev. Chem 2020, 4, 22–37; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kadina A, Kietrys AM, Kool ET, Angew. Chem. Int. Ed 2018, 57, 3059–3063; Angew. Chem. 2018, 130, 3113–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Velema WA, Kietrys AM, Kool ET, J. Am. Chem. Soc 2018, 140, 3491–3495; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Park HS, Kietrys AM, Kool ET, Chem. Commun 2019, 55, 5135–5138; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Habibian M, McKinlay C, Blake TR, Kietrys AM, Waymouth RM, Wender PA, Kool ET, Chem. Sci 2020, 11, 1011–1016; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang S-R, Wu L-Y, Huang H-Y, Xiong W, Liu J, Wei L, Yin P, Tian T, Zhou X, Nat. Commun 2020, 11: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].(a) Ursuegui S, Chivot N, Moutin S, Burr A, Fossey C, Cailly T, Laayoun A, Fabis F, Laurent A, Chem. Commun 2014, 50, 5748–5751; [DOI] [PubMed] [Google Scholar]; (b) Fessler AB, Fowler AJ, Ogle CA, Bioconjugate Chem 2021, 32, 904–908. [DOI] [PubMed] [Google Scholar]
- [21].Xiao L, Habibian M, Kool ET, J. Am. Chem. Soc 2020, 142, 16357–16363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Protozanova E, Yakovchuk P, Frank-Kamenetskii MD, J. Mol. Biol 2004, 342, 775–785. [DOI] [PubMed] [Google Scholar]
- [23].Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung J-W, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY, Nature. 2015, 519, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].(a) Fauster K, Hartl M, Santner T, Aigner M, Kreutz C, Bister K, Ennifar E, Micura R, ACS Chem. Biol 2012, 7, 581–589; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Govan JM, Young DD, Lusic H, Liu Q, Lively MO, Deiters A, Nucleic Acids Res. 2013, 41, 10518–10528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ando H, Furuta T, Tsien RY, Okamoto H, Nat. Genet 2001, 28, 317–325. [DOI] [PubMed] [Google Scholar]
- [26].Zhang D, Zhou CY, Busby KN, Alexander SC, Devaraj NK, Angew. Chem. Int. Ed 2018, 57, 2822–2826; Angew. Chem. 2018, 57, 2822–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chandrasekaran V, Juszkiewicz S, Choi J, Puglisi JD, Brown A, Shao S, Ramakrishnan V, Hegde RS, Nat. Struct. Mol. Biol 2019, 26, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ramanathan M, Majzoub K, Rao DS, Neela PH, Zarnegar BJ, Mondal S, Roth JG, Gai H, Kovalski JR, Siprashvili Z, Palmer TD, Carette JE, Khavari PA, Nat. Methods 2018, 15, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mangus DA, Evans MC, Jacobson A, Genome Biol. 2003, 4: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].(a) Wheeler JR, Matheny T, Jain S, Abrisch R, Parker R, eLife. 2016, 5: e18413; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wolozin B, Ivanov P, Nat. Rev. Neurosci 2019, 20, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].(a) Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R, Mol. Cell 2017, 68, 808–820; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCormick C, Khaperskyy DA, Nat. Rev. Immunol 2017, 17, 647–660. [DOI] [PubMed] [Google Scholar]
- [32].Choi J, Indrisiunaite G, DeMirci H, Ieong K-W, Wang J, Petrov A, Prabhakar A, Rechavi G, Dominissini D, He C, Ehrenberg M, Puglisi JD, Nat. Struct. Mol. Biol 2018, 25, 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ovodov SY, Alakhov YB, FEBS Lett. 1990, 270, 111–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.