

Introduction
Mutations of RAS genes drive cancer more frequently than any other oncogene. The recent explosion of cancer genomic analysis has revealed the paramount role of the mitogen activated protein kinase (MAPK) pathway in RAS-mediated oncogenesis. RAS proteins integrate signals from a wide array of receptors and initiate MAPK signaling, making them an exceedingly attractive target for cancer drug discovery. RAS proteins are fundamentally binary molecular switches in which the off/on state is determined by the binding of GDP or GTP, respectively. As such, the intrinsic and regulated nucleotide binding and hydrolytic properties of the RAS GTPase were historically believed to account for the entirety of the regulation of RAS signaling. However, it is increasingly clear that RAS proteins are also regulated by a vast array of posttranslational modifications (PTMs) (Fig. 1). The first PTMs recognized were those of the C-terminus of RAS that convert a globular, hydrophilic protein into one that is lipidated and has affinity for cellular membranes. Subsequently a large assortment of PTMs of the GTP-binding (G) domain (aa 1–165) have been reported. The current challenge is to understand what are the functional consequences of these modifications and which are physiologically relevant. Because direct inhibitors of RAS have been difficult to develop and because PTMs are catalyzed by enzymes that may offer targets for drug discovery, the study of RAS PTMs has been a high priority for RAS biologists.
Figure 1. Posttranslational modifications of RAS proteins.
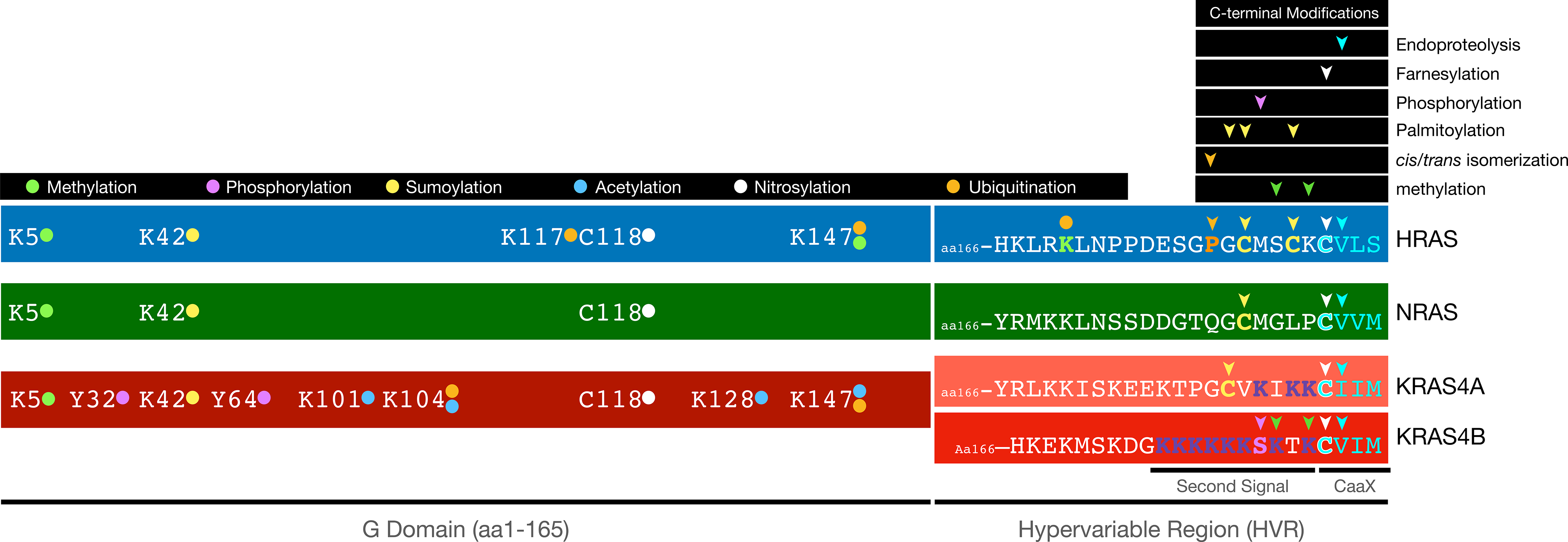
All reported and validated modifications of the four mammalian RAS proteins are indicated. Prenylation and palmitoylation of the C-terminal portion of the hypervariable region (HVR) create affinity for phospholipid bilayers whereas phosphorylation and lysine methylation of KRAS4B modulate that affinity. Methylation, phosphorylation, sumoylation, nitrosylation and mono-, di- and polyubiquitination of the G domains regulate trafficking, GDP/GTP exchange and degradation.
CaaX Processing
The three mammalian RAS genes give rise to four proteins because the KRAS locus harbors alternative 4th exons [1]. Whereas RAS proteins are 97% identical in their G domains (aa1–165), the 3’ half of the fourth exons encode a hypervariable region (HVR) that mediates subcellular trafficking and membrane association. This is accomplished by posttranslational modification (PTM) of this region that converts nascent RAS from a globular, hydrophilic protein to a peripheral membrane protein with a hydrophobic C-terminus that affords affinity for phospholipid bilayers [2].
The last four amino acids of each HVR consist of a CaaX sequence (cysteine, aliphatic x 2, variable) found in >200 peripheral membrane proteins [3]. RAS and other CaaX proteins are synthesized in the cytosol on free polysomes. Upon termination of translation, CaaX sequences are recognized by a prenyltransferase that attaches a C15 farnesyl or C20 geranylgeranyl polyisoprene lipid to the CaaX cysteine. These enzymes utilize as substrates farnesyl or geranylgeranyl pyrophosphates that are intermediates or derivatives, respectively, in the cholesterol biosynthetic pathway [4]. Whereas protein acylation is accomplished on cysteines via a labile thioester linkage, prenyl lipids are attached via a stable thioether linkage that is irreversible and persists throughout the life of the protein. Prenyltransferases are cytosolic, heterodimeric enzymes composed of an α and β subunit. Four α subunits combine with three β subunits to generate four holoenzymes, farnesyltransferase (FTase) and geranylgeranyltransferase (GGTase) types 1–3 [5]. GGTase 2 modifies RAB proteins that lack CaaX sequences and adds two geranylgeranyl lipids to C-terminal CXC or CC sequences. The other three enzymes modify CaaX proteins. The X position of the CaaX sequence determines which prenyltransferase modifies the protein. RAS proteins end with CaaM or CaaS and are substrates for FTase [6]. Many RAS-related small GTPases, including most RHO family proteins, end in a CaaL sequence, which directs modification by GGTase I [7].
Once CaaX processing of RAS was elucidated more than three decades ago, FTase became a logical target for anti-RAS drug discovery and numerous FTIs were developed [8]. Although these drugs were highly potent, orally bioavailable, hit their target in vivo, and inhibited HRAS-driven tumors, they failed in the clinic as anti-cancer drugs. This unfortunate result was shown to be a consequence of the fact that the vast majority of RAS mutant human cancer is driven by KRAS. Although KRAS is normally a substrate only for FTase, in the presence of an FTI it can be modified by GGTase 1 such that its membrane association and signaling function are preserved [9,10].
Prenylation is the first of three sequential modifications of the CaaX sequence. It is followed immediately by endoproteolytic removal of the aaX amino acids by RAS converting enzyme 1 (RCE1) and methylesterification of the newly C-terminal prenylcysteine by isoprenylcysteine carboxylmethyltransferase (ICMT). These latter modifications position the lipid moiety at the extreme C-terminus of the protein and neutralize the negative charge on the C-terminal carboxylate adding hydrophobicity and allowing the C-terminus to intercalate into phospholipid bilayers. Interestingly, before proteolysis and carboxyl methylation, prenylated CaaX sequences appear to be targeted specifically to the membranes of the endoplasmic reticulum (ER). The biophysical properties of the ER membrane that supports this affinity are unknown and some have argued that it is simply the vast surface area of the ER relative to that of the plasma membrane (PM) that accounts for the observed distribution [11]. However, at all expression levels, green fluorescent protein (GFP) extended with CaaX sequences lacking other membrane targeting motifs decorate the ER but are not observed on endosomes, mitochondria or the PM [12]. The mechanism of targeting to the ER notwithstanding, it is on this compartment that both RCE1 [13] and ICMT [14], both polytopic membrane proteins, are restricted such that we can be certain that fully processed CaaX proteins must visit the cytoplasmic face of the ER before trafficking to the PM. Like FTase, RCE1 and ICMT have been considered targets for anti-RAS drug discovery but no inhibitors have reached clinical testing.
CaaX processing (prenylation, proteolysis and carboxyl methylation) occurs immediately after translation and is highly efficient. Several observations support this view. First, top down proteomic analysis of endogenous KRAS4B in tumor cells revealed only farnesylated species, although the degree of carboxyl methylation varied with or without an activating mutation [15]. Second, unprocessed RAS proteins (e.g. those that include a C>S substitution in the CaaX sequence or those in cells treated with high concentrations of FTIs) have a slower electrophoretic mobility allowing quantification of processed and unprocessed species. Whereas markedly overexpressed RAS proteins migrate with both mobilities, endogenous RAS migrates only as fully processed protein [16–18]. Thus, CaaX processing appears to be a constitutive feature of the proteome. Nevertheless, some evidence suggests that CaaX processing can be modulated. Carol Williams’ group has shown that splice variants of small GTP-binding protein GDP-dissociation stimulator (SmgGDS), a guanine nucleotide exchange factor active against a wide array of small GTPases, can regulate RAS prenylation [19,20]. Noncoding RNAs have also been reported to modulate access of KRAS to FTase [21]. Interestingly, Drosophila Ras1 is inefficiently prenylated as a consequence of a lysine in the a1 position of its CaaX sequence, but the unprocessed form can nevertheless support eye development [22]. Although FTase activity may be constitutive, recently it was shown that its expression is regulated by scaffold association factor B (SAFB), a nuclear DNA and RNA binding protein required for the transcription of FNTA, the α subunit of FTase [23]. Silencing SAFB sensitized KRAS-driven tumor cells to growth inhibition by FTIs [23].
Aside from insertion into phospholipid bilayers, CaaX processing also establishes affinity for prenyl-binding proteins that serve as cytosolic shuttling factors with the ability to shield the C-terminus of prenylated proteins from the aqueous environment [24]. The best studied of these is RhoGDI that sequesters RHO, RAC, CDC42 and other RHO family proteins in the cytosol where they are poised for redistribution to cellular membranes as part and parcel of their activation cycle [25]. RabGDI serves a similar function for RAB family small GTPases [26]. The δ subunit of phosphodiesterase type 6 (PDE6δ) serves this function for RAS and many other RAS-related GTPases [27,28]. In photoreceptors PDE6δ binds PDE6α and PDE6β, both prenylated CaaX proteins, and allows rapid transport to the outer segment where the enzyme is integral to visual signal transduction [29]. Interestingly, although there is little homology between RhoGDI and PDE6δ at the amino acid sequence level, there is marked structural homology whereby the prenyl group is inserted into a hydrophobic cleft bounded on both sides by β sheets suggesting convergent evolution [30,31]. Somewhat surprisingly, PDE6δ can accommodate both a C15 farnesyl or C20 geranylgeranyl polyisoprene lipid in its binding pocket [32]. RhoGDI is postulated to be regulated by RHO GTPase releasing factors (GRFs) that promote discharge of the GTPase from its carrier protein to allow insertion into membranes [25]. Whereas no GRF has been reported for RhoGDI, GTP-bound ARL2/3, a small GTPase lacking a CaaX sequence, serves as the GRF for PDE6δ [31] suggesting that the localization of ARL proteins marks a subcellular compartment as an acceptor membrane for RAS. PDE6δ is not the only protein that functions as a prenyl binding protein for RAS. Others include prenylated RAB acceptor protein 1 [33], SmgGDS [34], galectin-1 [35], and VPS35 [18].
Palmitoylation
CaaX processing is necessary but not sufficient for delivery of RAS proteins to the PM. Also required is the so called “second signal” immediately upstream of the CaaX sequence in the HVR. The second signal imparts both higher affinity for the phospholipid bilayer as well as the trafficking information required to transport RAS from the cytosolic face of the ER to the PM (Fig. 2). There are two types of second signal. One requires no further PTM and consists of a stretch of basic amino acids that allows for an electrostatic interaction with the negatively charged phospholipid headgroups of the inner leaflet of the PM. The second is additional PTM with one or two palmitoyl acyl chains that modify cysteines. Whereas a farnesyl only modification imparts relatively low affinity to membranes, each of these second signals adds the affinity required for trafficking and stable association with membranes [36].
Figure 2. Postprenylation modification and trafficking of RAS.
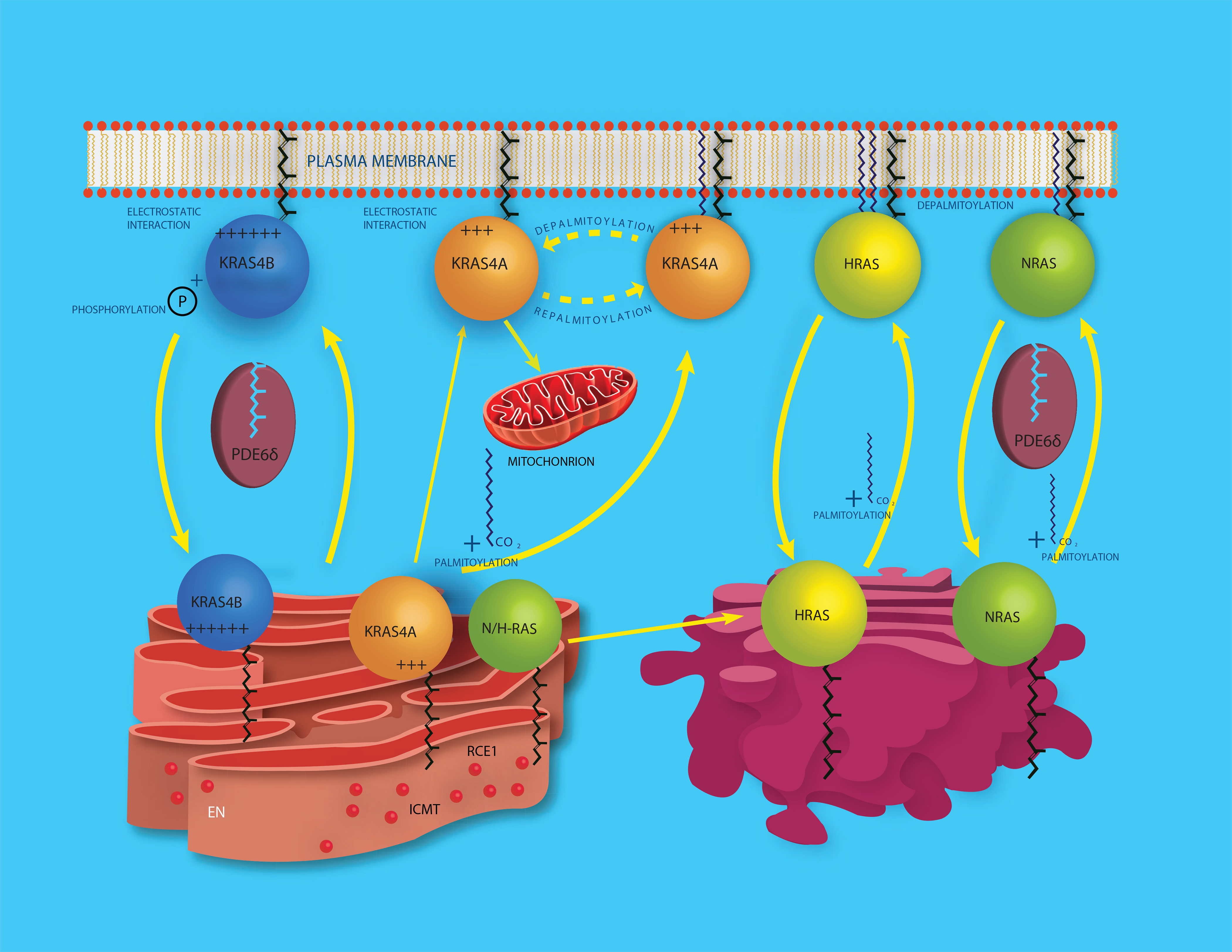
All RAS proteins are prenylated in the cytosol and then delivered to the cytosolic face of the endoplasmic reticulum (ER) where CaaX processing is completed through the actions of RAS converting enzyme 1 (RCE1) and isoprenylcysteine carboxylmethyltransferase (ICMT). CaaX processed NRAS and HRAS then traffic to the cytosolic face of the Golgi apparatus where they are palmitoylated and thereby gain enough affinity for membranes to engage in vesicular traffic to the plasma membrane (PM). NRAS also traffics through the cytosol in complex with chaperones such as PDE6δ and VPS35. Depalmitoylation occurs at the PM allowing NRAS and HRAS to cycle back to the Golgi for another round of palmitoylation. KRAS4A is palmitoylated, although the location for this modification has not been determined. KRAS4A is also depalmitoylated at the PM and moves to endomembranes that include the outer mitochondrial membrane. KRAS4B has a strong polybasic region that substitutes for palmitoylation, the prenylated protein traffics in complex with chaperones like PDE6δ and loses affinity for the PM upon phosphorylation of S181 in its HVR.
S-acylation of cysteines via a thioester linkage is an exceedingly common PTM and may affect up to 10% of the proteome [37]. Although acyl chains of various lengths have been documented, by far the most common is C16 palmitate. Importantly, no consensus sequence for cysteine S-acylation has been defined. Early studies of acylated proteins employed metabolic labeling with [3H]palmitate but more recently, acyl exchange chemistry and click-chemistry labeling have afforded both quantification of acylation and affinity capture of acylated proteins [38–41]. Whereas NRAS and KRAS4A have one HVR cysteine modified by palmitate, HRAS has two. HRAS was among the first proteins characterized with this modification [42]. The enzymes that modify proteins with palmitate are known as palmitoyl acyltransferases (PATs). These enzymes all possess a DHHC sequence in their catalytic sites. Twenty-three PATs are encoded in the human genome with a variety of subcellular localizations [43]. PATs are typically promiscuous in that they modify a large spectrum of proteins. DHHC9 in complex with GCP16 was identified as a Golgi-localized PAT that modifies NRAS and HRAS [44]. However, silencing of DHHC9 diminished but did not eliminate NRAS palmitoylation demonstrating that RAS proteins can be modified by more than one PAT [45]. The PAT responsible for the PTM of KRAS4A has not been identified.
Unlike prenylation, S-palmitoylation is readily reversible and the lability of this PTM is integral to its biological function allowing for the establishment of palmitoylation/deplamitoylation cycles that direct protein trafficking. Although some depalmitoylation may occur non-enzymatically, a variety of esterases can accelerate this process. Acylprotein thioesterase 1 and 2 (APT1/2) were the first such esterases shown to have RAS depalmitoylating activity [46]. More recently the ABHD17 family of serine hydrolases have been shown to be physiologically relevant RAS thioesterases [47,48]. In addition to S-acylation, KRAS4A has been reported to be N-acylated on lysines in the HVR [49]. Although N-acylation is not readily reversible, SIRT2 can remove the acyl modification and alter the signaling potential of oncogenic KRAS4A [49].
Upon palmitoylation RAS proteins acquire a 100-fold greater affinity for membranes than that of the prenylated only protein [50,51]. This increased affinity creates a kinetic trap that enriches NRAS and HRAS at the cytoplasmic face of Golgi membranes from where these RAS proteins can engage vesicular transport and gain access to other membranes, including the PM [52]. Hancock and colleagues have shown that another consequence of RAS palmitoylation is to promote partition into PM microdomains such that RAS forms nanoclusters that promote signaling [53–55]. Bastiaens and colleagues have developed models wherein the continuous cycle of palmitoylation at the Golgi and depalmitoylation at the PM counteracts the entropy that would otherwise distribute RAS evenly among membranes and insures a sufficient concentration of RAS proteins at the PM to allow for signaling [56–58]. Since the dwell time on the PM of palmitoylated RAS proteins is critical for signaling, the regulation of depalmitoylation may play an important role, but is poorly understood. Ahearn found that prolyl isomerization in the HVR of HRAS catalyzed by FKBP12 promotes depalmitoylation and therefore operates as an internal timer for PM association [59]. Compartmentalized signaling of RAS proteins from distinct subcellular localizations is a well-established paradigm in RAS biology [60–63] and the palmitoylation/depalmitoylation cycle is among the most important regulators of this system. The best characterized functional consequence of the palmitoylation/depalmitoylation cycles of RAS proteins is the regulation of hexokinase 1 on the outer mitochondrial membrane (OMM) by KRAS4A [64]. Whereas palmitoylated KRAS4A is expressed primarily at the PM, depalmitoylated KRAS4A traffics to endomembranes, including the OMM, where it encounters hexokinase 1 and reverses its allosteric regulation by glucose-6-phosphate [64]. Other differential biological outcomes controlled by palmitoylation-driven RAS localization include positive and negative selection of thymocytes [65]. Recently Shannon et al. demonstrated in vivo that palmitoylation is required for Nras-driven myeloid transformation adding encentive to target this pathway in cancer [66].
Phosphorylation
Reversible phosphorylation of signaling molecules is an exceedingly common mode of regulation. HRAS, NRAS and KRAS4B have been shown to be phosphoproteins although the functional consequences of this PTM remain poorly defined. Ora Rosen and colleagues reported in 1987 that KRAS4B is a substrate for protein kinase C (PKC) [67]. Around the same time Arimura et al. reported that HRAS is a substrate for protein kinase A (PKA), although the stoichiometry of phosphate incorporation was too low to be physiologically meaningful [68]. More recently Kim et al. found that phosphorylation of HRAS by glycogen synthase kinase 3 β (GSK3β) on threonine residues 144 and 148 led to polyubiquitination by β-transducin repeat-containing protein (β-TrCP) and degradation [69]. Yin et al. recently reported that NRAS is phosphorylated on S89 by STK19. This PTM activates NRAS signaling and inhibitors of STK19 are effective in a mouse model of NRAS-driven melanoma [70].
Phosphorylation of KRAS4B in its HVR is the best characterized modification of this type. Bivona et al. found that PKC phosphorylated KRAS4B on serine 181, which weakened the electrostatic interaction of the polybasic HVR with the PM (Fig. 2) and allowed the GTPase to translocate to endomembranes in a process designated the farnesyl-electrostatic switch [71] that is analogous to the myristoyl electrostatic switch of the MARKS protein [72]. Importantly, phosphorylation of oncogenic KRAS4B on serine 181 was associated with decreased cell survival as a consequence of interference with the ability of Bcl-XL to modulate calcium flux between ER and mitochondria [73]. Wang et al. showed that phosphorylation of KRAS4B on serine 181 inhibits tumor initiation by blocking interaction with calmodulin and thereby abrogating suppression of non-canonical Wnt signaling [74]. Thus, phosphorylation of KRAS4B appears to be a negative regulatory event. One contrary result was reported by Alvarez-Moya who found that activated KRAS4B that could not be phosphorylated was impaired as an oncogene [75]. However, we have found that KRAS4B-12V,181A is as potent in vivo as KRAS4B-12V,181S in producing tumors (unpublished results) strongly arguing against a requirement for phosphorylation. Phosphorylation of KRAS4B on serine 181 also affects its ability to partition into membrane microdomains [76,77]. In addition to PKC, phosphorylation of serine 181 of KRAS4B was shown to catalyzed by cyclic GMP-dependent protein kinases 2 (PKG2) [78]. Recently, Iijima has made the remarkable discovery that KRAS4B is also phosphorylated on serine 181 by PKA downstream of insulin signaling and that phospho-KRAS4B combines with RHOA to regulate the mTORC2 complex (unpublished results).
In addition to serine/threonine phosphorylation, RAS proteins have also been reported to be phosphorylated on tyrosine 32 and 64 [79]. The non-receptor tyrosine kinase SRC has long been known to cooperate with oncogenic RAS in driving cell growth and tumorigenesis. In 2014, Michael Ohh and colleagues reported that SRC directly phosphorylates GTP-bound RAS proteins on tyrosine 32 and thereby reciprocally regulates binding to RAF1 and RasGAP to inhibit RAS signaling [80]. These authors went on to show that the ubiquitous tyrosine phosphatase SHP2 dephosphorylates RAS at position 32 and thereby potentiates signaling [81]. Most recently, this group found that tyrosine 64 was also phosphorylated by SRC such that the kinase modifies residues in both the switch I and II regions and that SRC and SHP2 cooperate to take RAS proteins in and out of a “dark state.” [82]. The availability of clinically useful SHP2 inhibitors makes this a very exciting model. However, because numerous investigators have interrogated the RAS/MAPK pathway for decades using anti-phosphotyrosine antibodies without observing phospho-RAS, these observations await confirmation.
RAS Cysteine Oxidation
The cellular redox state plays an important role in numerous signaling pathways that regulate cellular growth. When this balance is disrupted, aberrant proliferation can occur [83]. Indeed, protein modification by reactive oxygen species constitutes another class of PTMs that affect RAS. Redox homeostasis requires a balance of cellular oxidation and reduction. The reactive intermediates involved in these processes include reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive thiols. While cellular redox reactions are diverse and can generate many products, in this review, we will focus on regulation of RAS proteins by reversible thiol oxidation. The earliest studies on RAS modulation by ROS and RNS date back to 1995. Studies both in vitro and in T cells [84,85] support enhanced RAS activation by reactive intermediates. In vitro analyses indicated that incubation of ROS and RNS with recombinant RAS stimulated exchange of GDP for GTP, suggesting a mechanism for activation. The reaction site was a later shown to be a solvent accessible cysteine (Cys118) [84,86] within the NKCD nucleotide binding motif. Intriguingly, RAS activation by ROS and RNS occurred via a cysteine thiol radical intermediate rather than stable cysteine oxidation or nitrosation [86–90]. Reactive species that promote RAS Cys118 thiol radical intermediate led to oxidation of the guanine nucleotide and destabilization of nucleotide binding, which in turn stimulated guanine nucleotide exchange and RAS activation. These studies also led to identification of a variant of RAS, RAS C118S that retains RAS structure yet is redox insensitive [91]. The RAS C118S redox insensitive variant proved useful for a variety of cell-based studies aimed at discriminating direct or indirect effects of ROS/RNS on RAS activity. While RNS and ROS have been shown in many studies to regulate RAS activation and downstream signaling [92,93], a land mark study by the Counter lab, linked endothelial nitric oxide synthase (eNOS) to NO-mediated RAS activation and tumorigenesis in mice [94]. Oncogenic RAS activates the phosphoinositide 3-kinases (PI3K) pathway, which results in AKT-mediated eNOS phosphorylation. Lim et al. identified a feedback mechanism whereby eNOS-generated nitric oxide (NO) activates wild type RAS and stimulates downstream signaling. Consistent with their findings, introducing the RAS C118S mutation in HRAS and NRAS circumvented PI3K pathway activation and reduced tumor growth. In addition to RAS C118, three cysteines within the C-terminal hypervariable region RAS can undergo modification by RNS and ROS both in vitro and in mouse fibroblasts [93]. While these C-terminal cysteines may be protected from modification due to lipid PTM, redox conditions in which cysteine oxidation occurs at these sites could prevent lipid modification that facilitates membrane association and RAS activity. Hence, modification of these residues may downregulate RAS function by preventing proper membrane localization, in contrast to the upregulation of RAS by RNS/ROS modification of C118.
Whereas RAS is regulated by redox-driven PTMs, the reciprocal is also true in that RAS signaling modulates oxidant and anti-oxidant production and consequently redox balance. Oncogenic RAS can drive redox mediated tumorigenesis [95].
Lysine modifications (ubiquitylation, SUMOlyation, acetylation, methylation)
Ubiquitylation
RAS proteins are substrates for ubiquitination. Distinct ubiquitin linkages (mono-, di- and polyubiquitination) have been identified at multiple lysine residues in RAS [96]. The type and site of ubiquitin modification can alter RAS function by at least three different mechanisms; RAS subcellular localization, protein–protein interactions, and degradation (Fig. 3).
Figure 3. Three modes of RAS regulation by ubiquitination and SUMOylation.
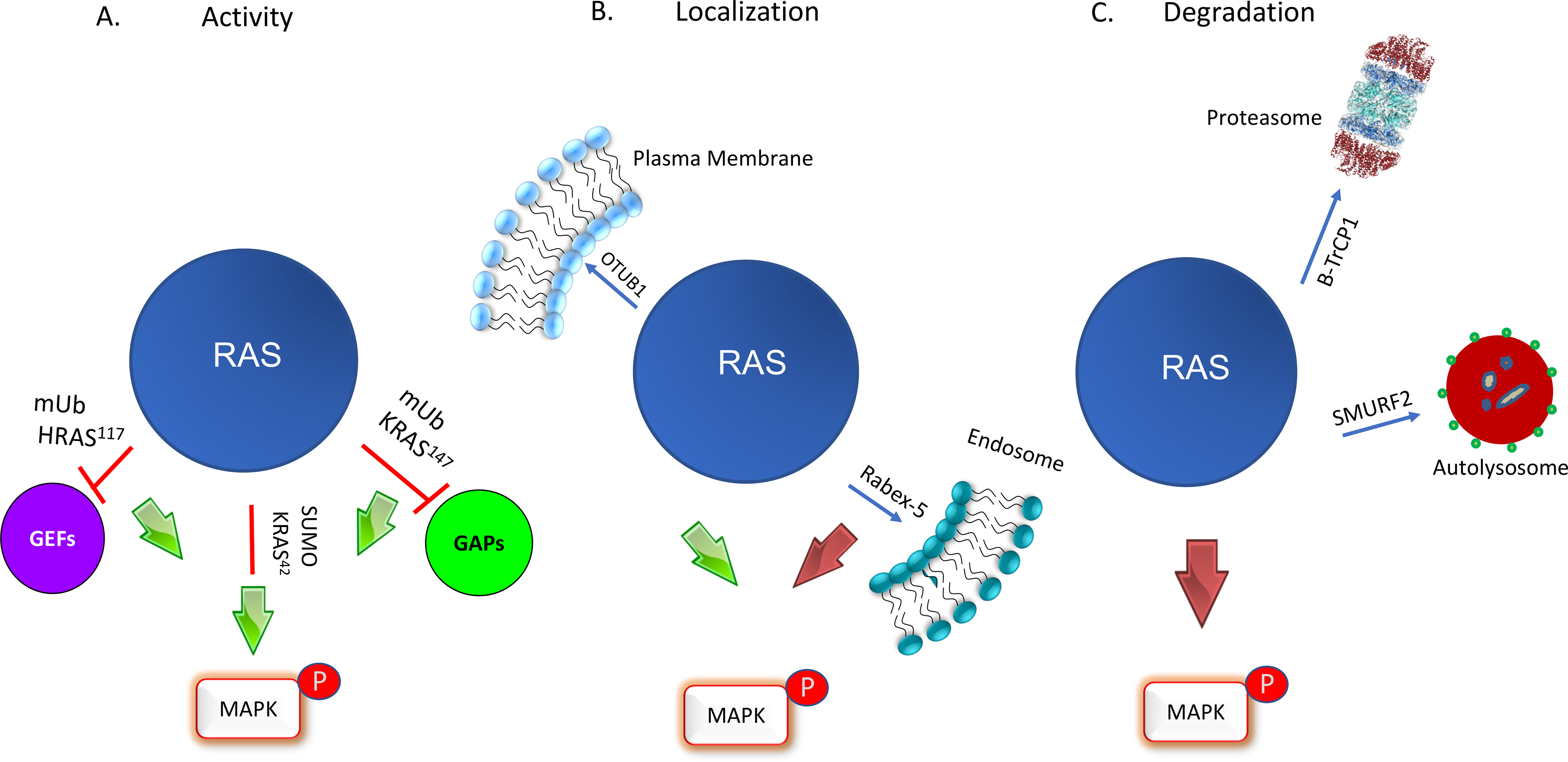
A. RAS activity is regulated by ubiquitination. Primary sites of monoubiquitination occur at residues 147 in KRAS, whereas in HRAS it is at 117. Monoubiquitination of KRAS at 147 upregulates RAS activity through a GAP defect leading to enhanced MAPK activation. In contrast, monoubiquitination of HRAS at 117 induces fast exchange and activates RAS in a GEF-independent manner. All RAS isoforms undergo SUMOylation at Lys 42 which upregulates downstream signaling by an unknown mechanism. B. RAS localization is regulated by ubiquitination. Rabex-5 promotes mono- and diubiquitination of HRAS and NRAS resulting in endosome localization and reduced MAPK signaling. The deubiquitinase, OTUB1, removes ubiquitin from RAS and promotes plasma membrane localization and MAPK signaling. C. Ubiquitination by β-TrCP1 and SMURF2 promote RAS degradation through proteasome and autolysosomes resulting in reduced MAPK signaling.
The earliest studies examining the effects of RAS ubiquitination identified the RAB5 exchange factor, Rabex-5, as a key regulator of RAS function. Studies performed in both mammalian and Drosophila cells showed that Rabex-5, which contains both ubiquitin binding and E3 ligase domains, catalyzes mono- and diubiquitination of NRAS and HRAS but not KRAS [97,98]. Ubiquitination of either HRAS or KRAS protein promoted endosomal membrane localization and reduced MAPK signaling. It is unclear whether the ability of Rabex-5 to selectively ubiquitinate HRAS and NRAS but not KRAS in CHO-K cells is due to specific colocalization of Rabex-5 complexes with these RAS isoforms or through another mechanism. These findings indicate that Rabex-5 downregulates RAS function through directing ubiquitin-mediated relocalization of RAS to endosomes. However, the sites of ubiquitination that drive endosomal localization have yet to be established. It is also unclear whether mono- versus diubiquitination have distinct effects of HRAS and NRAS signaling and whether pathways other than MAPK are affected. On the flip-side, the deubiquitinase OTUB1 functions a negative regulator of RAS mono- and diubiquitination by sequestering RAS on the plasma membrane. OTUB1 is commonly overexpressed in non-small cell lung carcinoma and can promote RAS activation and tumorigenesis in wild- type RAS cells [99].
While these studies revealed a role of ubiquitination in localization and suppression of HRAS and NRAS function, KRAS monoubiquitination was found to alter RAS interactions with regulatory proteins and effectors and up-regulate its activity. In one study comparing KRAS to HRAS, sites of ubiquitination in RAS were identified by mass spectrometry following isolation of ubiquitinated KRAS and HRAS from HEK293T cells [100]. Lysine 147 was identified as the most frequent site of KRAS ubiquitination whereas lysine 117 was the most populated site in HRAS. Compared with unmodified KRAS, the ubiquitinated subpopulation was predominantly in the activated GTP-bound state and showed increased association with the downstream effectors RAF, PI3K, and RalGEF. Mutation of lysine 147 to prevent KRAS ubiquitination impaired tumor growth in a mouse xenograft model system. In vitro biochemical and pulldown studies suggested that KRAS monoubiquitination at 147 impairs GAP-mediated down-regulation and enhances nucleotide independent RAF binding [101]. In contrast, ubiquitin modification at lysine 117 in HRAS promotes RAS activation due to faster nucleotide cycling, rather than by disrupting GAP interactions [102]. Intriguingly, the covalently bound ubiquitin does not appear to make specific and stable intramolecular contacts with RAS, but rather transient interactions that modulate RAS structure and dynamics and consequently recognition by regulatory factors and downstream effectors [103,104]. This appears to be a novel mechanism of RAS activation. Taken together, these findings indicate that ubiquitination can activate RAS-mediated signaling and tumorigenesis.
The above studies support a role of mono- and diubiquitination in the regulation of RAS localization and protein–protein interactions. However, a more classically recognized mechanism by which ubiquitin mediates protein function is through degradation. Proteasomal degradation of HRAS has been reported to occur in response to activation by the Wnt/α-catenin signaling pathway [69,105,106] by recruitment of the E3 ligase β-TrCP after HRAS phosphorylation by glycogen synthase kinase 3β. Conversely, inhibition of this pathway enhances RAS expression levels and RAS-induced colorectal tumorigenesis. In an effort to develop anticancer drugs targeting RAS, small molecules have been identified that enhance formation of the α-catenin destruction complex to induce degradation of α-catenin and/or RAS [105,107–109]. In addition, evidence for KRAS isoform specific degradation has been observed [110]. The Smad ubiquitination regulatory factor 2 (SMURF2) can promote monoubiquitination of E2 ubiquitin-conjugating enzyme 5 (UBCH5) to form an active E3/E2 complex. This complex polyubiquitinates and degrades, in an activation-dependent manner, the β-TrCP1 E3 ligase that modulates KRAS protein stability [110]. Here, degradation appears dependent on lysosomal proteolysis, as a lysosome inhibitor protected KRAS from SMURF2, whereas proteasomal inhibition was ineffective, consistent with previous observations that KRAS can undergo lysosomal degradation [111]. Thus, both proteasome- and lysosome-mediated degradation mechanisms modulate KRAS levels and do so in a stimulus-dependent manner.
Recently, the leucine zipper-like transcription regulator 1 protein (LZTR1), which associates with the cullin 3 E3 ubiquitin ligase, has been postulated to regulate RAS. Although ubiquitination by LZTR1 was initially proposed to modulate RAS function by a nondegradative mechanism [*112,*113], a more recent study suggested that LZTR1-dependent proteolysis of RAS was prevented by treatment with a proteasome inhibitor, suggesting that LZTR1 facilitates polyubiquitination and degradation of RAS via the ubiquitin–proteasome pathway [*114] to modulate RAS protein levels. LZTR1 appears to downregulate RAS signaling, as LZTR1 haploinsufficiency in mice results in Noonan syndrome, a developmental disorder that arises from enhanced RAS and MAPK signaling. Moreover, loss of LZTR1 in Schwann cells promotes differentiation and proliferation [*113]. However, these findings have been challenged by Castel et al. who found that LZTR1 co-immunoprecipitates with RIT1 and MRAS but not HRAS, NRAS or KRAS [115]. Additional studies are needed to define the substrate specificity of LZTR1 and to determine whether LZTR1 directly regulates RAS function or affects RAS signaling indirectly.
RAS lysine SUMOylation
In addition to ubiquitination, RAS proteins are targets of SUMOylation [116,117]. While this small ubiquitin-like modifier (SUMO) is similar in size to ubiquitin, SUMOylation does not appear to initiate degradation. Rather, one well-known role of SUMOylation lies in regulation of cell growth, migration and tumorigenesis [118]. SUMOylation of RAS proteins occurs at a conserved lysine, Lys42, which is proximal to the effector region (residues 30–40). RAS modification by SUMO3 appears to be associated with activation as inhibition of SUMOylation or mutation of Lys42 results in downregulation of RAS signaling, reduced cell migration and invasion in multiple cell lines and tumor development in vivo. However, the mechanism of activation is unknown.
RAS lysine acetylation
Acetylation occurs through transfer of an acetyl CoA acetyl group by a cognate lysine acetyltransferase. Lysine acetylation modifies the lysine side chain, reduces the positive charge and can alter protein function and recognition. The best-studied example of this is the lysine modifications of histones that regulate chromatin. Acetylation of WT and G12V KRAS has been detected in the core GTPase domain at multiple lysine residues (K101, K104, K128 and K147) [119]. Two of these identified acetylation sites in RAS (K104 and K147) have been studied. Yang et al. generated a K104Q mutant to mimic acetylation and found that this substitution impaired SOS-mediated exchange [120]. This variant in the context of an oncogenic RAS G12V mutation showed reduced proliferation and clonogenic survival of NIH3T3 cells in a focus-formation assay relative to unmodified KRAS G12V. These findings indicated that KRAS K104 acetylation negatively regulates RAS function. The investigators [121] also found that knockdown of HDAC6 and SIRT2 reduced viability in NIH3T3 cells expressing K-RASG12V, but not in cells expressing KRASG12V/K104A, suggesting that these deacetylases modulate RAS acetylation. Using molecular dynamic (MD) simulations, they predicted that mutations at K104 disrupt the structure of helix 2 in switch 2 and consequently inhibited recognition by GEFs. However, subsequent NMR studies using the same K104Q mutant found only partial disruption of helix 2 [122], which explained both the GEF and GAP defects that were detected in biochemical assays. In contrast to previous findings, the KRAS G12V/K104Q variant did not significantly alter steady-state GTP levels, cellular growth or proliferation in NIH 3T3 cells, leading to the conclusion that the GEF and GAP defects were compensatory in nature, and the K104Q mutant likely did not impact overall Ras activity [122]. Moreover, K104-acetylated KRAS4B WT and G12V mutation did not alter SOS-catalyzed nucleotide exchange in vitro [119,122], indicating that glutamine substitution at K104 in RAS does not mimic acetylation. While both CBP and p300 lysine-acetyltransferases were able to acetylate both wildtype and G12V KRAS4B in vitro [119], Sirt2 and HDAC6 did not stimulate KRAS 104 deacetylation. Taken together, these somewhat conflicting studies indicate that the RAS K104Q variant does not mimic acetylation, and RAS acetylation may not alter RAS downstream function as originally proposed. RAS K147 acetylation has also been observed in KRAS4B, however, this modification does not alter intrinsic and the SOS-catalyzed nucleotide exchange, and the cellular consequences of acetylation at this site are unknown [119]. Given that intrinsic RAS function is not significantly altered upon acetylation K104 and K147, acetylation may serve as a docking site to promote new interactions. Additional studies are clearly needed to better understand the role of RAS acetylation in RAS-mediated signaling.
Lysine Methylation.
While lysine methylation has been primarily studied in the context of histone regulation, it is a critical PTM that can modulate the function of non-histone proteins [123]. Research in our lab and others have identified novel methylation sites (Lys5, Lys147) within the core RAS G-domain, but the functional role of these modifications is unclear. In contrast to acetylation, methylation does not alter the side chain charge and it is challenging to deduce its impact on protein structure by conventional or unnatural amino acid substitutions. Methylation of conserved lysine residues (Lys5, Lys16 and Lys117) in the core GTPase domain of RAS superfamily GTPases have been identified through an algorithm developed by the Sasaki lab called GoMADScan [124], suggesting that methylation at these sites may play a conserved role in GTPase function. To follow up on these observations, endogenous RAS was immunoprecipitated from HEK293T cells, and mass spectrometric analysis conducted. RAS residues, Lys5 and Lys147 were found to be di- and monomethylated. Lysine 147 methylation was unique to HRAS and the GoMADScan algorithm did not detect methylation at the equivalent position to Lys147 in other small GTPases [124]. Given that substitutions at Lys147 to alanine, cysteine, or leucine do not significantly alter RAS activity [102], it is tempting to speculate that Lys147 methylation may function to create a docking site or block other PTMs (acetylation, ubiquitination), rather than alter RAS structure and intrinsic function. Mutations at Lys5 have been identified in cancers and RASopathies [125–131], however, it is unclear how these mutations upregulate RAS function. To assess the impact of Lys5 dimethylation on RAS structure and dynamics, molecular dynamics (MD) simulations were conducted, which predicted that dimethylation of Lys-5 does not significantly alter RAS conformation. This suggests that Lys-5 methylation may alter existing protein interactions, create a docking site to foster new interactions or other PTMs at this position to modulate RAS function.
Conclusion
RAS is a highly regulated signaling node that plays a key role in controlling cellular growth. Dysregulation of accessory proteins (e.g. GEFS and GAPs) and point mutations in RAS can alter RAS activity and cause aberrant signaling and pathologies, resulting in RASopathies and cancer. PTMs represent yet another layer of RAS regulation. In comparison to GEFs and GAPs, modulation of RAS function by PTMs is poorly understood. Nevertheless, it is clear that a diverse array of often reversible PTMs can regulate RAS function by controlling membrane association and subcellular trafficking, that active state of RAS, its protein-protein interactions, and its expression levels. To better understand the role of various PTMs in RAS function, it will be important to identify the sites of modification in a site-specific manner and the enzymes responsible for both the PTMs and their reversal. While advances in mass spectrometry have identified a subset of PTMs and the sites of modification, PTMs can be transient and present at low levels thereby evading MS detection. Conversely, MS is sensitive enough to report PTMs that lack physiological significance. Indeed, one of the great challenges in the study of RAS PTMs has been to establish the stoichiometry of the modifications that must be relatively high to affect processes such as protein-protein interaction is a way to have significant impact. Another challenge has been how to study the effects of PTMs when they cannot be genetically encoded. PTMs are often probed by mutating the site of modification to block or mimic a modification. However, it has become increasingly apparent that blocking mutations may alter the structure of the protein or adjacent modification sites and that mimetic substitutions may not truly mimic the PTM of interest. While synthetic biology can unambiguously replicate some PTMs in vitro, the ability to express or modify proteins in cells remains limited. Extension of genetic, biochemical, and synthetic biological approaches are needed to better understand RAS PTM.
RAS has proven difficult to target for drug discovery due to the lack of druggable pockets and exceedingly high affinity of guanine nucleotides. Nevertheless, despite over 30 years of being dubbed an ‘undruggable’ target, progress has recently been made on this front, as covalent inhibitors of an oncogenic RAS cysteine containing mutant are showing efficacy in clinical trials. However, because the KRAS G12C mutant is only one of over 100 oncogenic RAS mutations found in various RAS-driven cancers, additional therapeutic intervention approaches are needed. Given that PTMs can regulate RAS activity by a variety of mechanisms, enzymes that control RAS PTMs may be viable targets. Indeed, early efforts to antagonize oncogenic RAS signaling relied on inhibition of farnesyltransferases that target RAS to the membrane, an approach that has recently been revived for HRAS-driven head and neck cancer [132].
Enzymes in the native RAS ubiquitination pathways represent another class of targets. In addition, engaging E3 ligases that do not normally target RAS to create neomorphic RAS degraders is another approach that co-opts PTMs. These include both proteolysis targeting chimeric small molecules (PROTACS) [**133,134] and genetically encoded degraders [*135–137]. Palmitoyl acyltransferases, palmitoyl thioesterases, carboxyl methyltranferases, aaX endoproteases, kinases, phosphatases, acetyltransferases, deacetylases and methyltransferases may represent a rich tapestry of additional targets.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR: K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A 2015, 112:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright LP, Philips MR: Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res 2006, 47:883–891. [DOI] [PubMed] [Google Scholar]
- 3.Palsuledesai CC, Distefano MD: Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem Biol 2015, 10:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buhaescu I, Izzedine H: Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 2007, 40:575–584. [DOI] [PubMed] [Google Scholar]
- 5.Kuchay S, Wang H, Marzio A, Jain K, Homer H, Fehrenbacher N, Philips MR, Zheng N, Pagano M: GGTase3 is a newly identified geranylgeranyltransferase targeting a ubiquitin ligase. Nat Struct Mol Biol 2019, 26:628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hougland JL, Lamphear CL, Scott SA, Gibbs RA, Fierke CA: Context-dependent substrate recognition by protein farnesyltransferase. Biochemistry 2009, 48:1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid TS, Terry KL, Casey PJ, Beese LS: Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol 2004, 343:417–433. [DOI] [PubMed] [Google Scholar]
- 8.Cox AD, Der CJ, Philips MR: Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res 2015, 21:1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK: K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem 1997, 272:14459–14464. [DOI] [PubMed] [Google Scholar]
- 10.Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM: Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. Journal of biological chemistry 1997, 272:14093–14097. [DOI] [PubMed] [Google Scholar]
- 11.Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L, Bastiaens PI: KRas Localizes to the Plasma Membrane by Spatial Cycles of Solubilization, Trapping and Vesicular Transport. Cell 2014, 157:459–471. [DOI] [PubMed] [Google Scholar]
- 12.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR: Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 1999, 98:69–80. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S: Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proceedings of the National Academy of Sciences (USA) 1998, 95:11175–11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, Michaelis S, Philips MR: Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem 1998, 273:15030–15034. [DOI] [PubMed] [Google Scholar]
- 15.Ntai I, Fornelli L, DeHart CJ, Hutton JE, Doubleday PF, LeDuc RD, van Nispen AJ, Fellers RT, Whiteley G, Boja ES, et al. : Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc Natl Acad Sci U S A 2018, 115:4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohl NE, Mosser SD, deSolms SJ, Giuliani EA, Pompliano DL, Graham SL, Smith RL, Scolnick EM, Oliff A, Gibbs JB: Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science 1993, 260:1934–1937. [DOI] [PubMed] [Google Scholar]
- 17.Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM: Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem 1995, 270:26802–26806. [DOI] [PubMed] [Google Scholar]
- 18.Zhou M, Wiener H, Su W, Zhou Y, Liot C, Ahearn I, Hancock JF, Philips MR: VPS35 binds farnesylated N-Ras in the cytosol to regulate N-Ras trafficking. J Cell Biol 2016, 214:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuld NJ, Vervacke JS, Lorimer EL, Simon NC, Hauser AD, Barbieri JT, Distefano MD, Williams CL: The chaperone protein SmgGDS interacts with small GTPases entering the prenylation pathway by recognizing the last amino acid in the CAAX motif. J Biol Chem 2014, 289:6862–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Torres D, Fierke CA: The chaperone SmgGDS-607 has a dual role, both activating and inhibiting farnesylation of small GTPases. J Biol Chem 2019, 294:11793–11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siprashvili Z, Webster DE, Johnston D, Shenoy RM, Ungewickell AJ, Bhaduri A, Flockhart R, Zarnegar BJ, Che Y, Meschi F, et al. : The noncoding RNAs SNORD50A and SNORD50B bind K-Ras and are recurrently deleted in human cancer. Nat Genet 2016, 48:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung PJ, Rodrigues AB, Kleinberger A, Quatela SE, Bach EA, Philips MR: Cytosolic Ras supports eye development in Drosophila. Molecular and cellular biology 2010, 30:5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou M, Kuruvilla L, Shi X, Viviano S, Ahearn IM, Amendola CR, Su W, Badri S, Mahaffey J, Fehrenbacher N, et al. : Scaffold association factor B (SAFB) is required for expression of prenyltransferases and RAS membrane association. Proc Natl Acad Sci U S A 2020, 117:31914–31922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Philips MR: Ras hitchhikes on PDE6delta. Nat Cell Biol 2012, 14:128–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Mata R, Boulter E, Burridge K: The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 2011, 12:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfeffer S, Aivazian D: Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol 2004, 5:886–896. [DOI] [PubMed] [Google Scholar]
- 27.Nancy V, Callebaut I, El Marjou A, de Gunzburg J: The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J Biol Chem 2002, 277:15076–15084. [DOI] [PubMed] [Google Scholar]
- 28.Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M, Venkitaraman AR, et al. : The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol 2012, 14:148–158. [DOI] [PubMed] [Google Scholar]
- 29.Karan S, Zhang H, Li S, Frederick JM, Baehr W: A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vision Res 2008, 48:442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffman GR, Nassar N, Cerione RA: Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell 2000, 100:345–356. [DOI] [PubMed] [Google Scholar]
- 31.Ismail SA, Chen Y-X, Rusinova A, Chandra A, Blerbaum M, Gremer L, Trlola G, Waldmann H, Bastiaens PIH, Wittinghofer A: Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nature chemical biology 2011, 7:942–949. [DOI] [PubMed] [Google Scholar]
- 32.Dharmaiah S, Bindu L, Tran TH, Gillette WK, Frank PH, Ghirlando R, Nissley DV, Esposito D, McCormick F, Stephen AG, et al. : Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc Natl Acad Sci U S A 2016, 113:E6766–E6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Figueroa C, Taylor J, Vojtek AB: Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J Biol Chem 2001, 276:28219–28225. [DOI] [PubMed] [Google Scholar]
- 34.Berg TJ, Gastonguay AJ, Lorimer EL, Kuhnmuench JR, Li R, Fields AP, Williams CL: Splice variants of SmgGDS control small GTPase prenylation and membrane localization. J Biol Chem 2010, 285:35255–35266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rotblat B, Niv H, Andre S, Kaltner H, Gabius HJ, Kloog Y: Galectin-1(L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res 2004, 64:3112–3118. [DOI] [PubMed] [Google Scholar]
- 36.Leventis R, Silvius JR: Lipid-binding characteristics of the polybasic carboxy-terminal sequence of K-ras4B. Biochemistry 1998, 37:7640–7648. [DOI] [PubMed] [Google Scholar]
- 37.Sanders SS, Martin DD, Butland SL, Lavallee-Adam M, Calzolari D, Kay C, Yates JR 3rd, Hayden MR: Curation of the Mammalian Palmitoylome Indicates a Pivotal Role for Palmitoylation in Diseases and Disorders of the Nervous System and Cancers. PLoS Comput Biol 2015, 11:e1004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drisdel RC, Green WN: Labeling and quantifying sites of protein palmitoylation. Biotechniques 2004, 36:276–285. [DOI] [PubMed] [Google Scholar]
- 39.Drisdel RC, Alexander JK, Sayeed A, Green WN: Assays of protein palmitoylation. Methods 2006, 40:127–134. [DOI] [PubMed] [Google Scholar]
- 40.Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC: Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc 2009, 131:4967–4975. [DOI] [PubMed] [Google Scholar]
- 41.Yang YY, Ascano JM, Hang HC: Bioorthogonal chemical reporters for monitoring protein acetylation. J Am Chem Soc 2010, 132:3640–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hancock JF, Paterson H, Marshall CJ: A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63:133–139. [DOI] [PubMed] [Google Scholar]
- 43.Ko PJ, Dixon SJ: Protein palmitoylation and cancer. EMBO Rep 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME: DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem 2005, 280:31141–31148. [DOI] [PubMed] [Google Scholar]
- 45.Liu P, Jiao B, Zhang R, Zhao H, Zhang C, Wu M, Li D, Zhao X, Qiu Q, Li J, et al. : Palmitoylacyltransferase Zdhhc9 inactivation mitigates leukemogenic potential of oncogenic Nras. Leukemia 2016, 30:1225–1228. [DOI] [PubMed] [Google Scholar]
- 46.Kong E, Peng S, Chandra G, Sarkar C, Zhang Z, Bagh MB, Mukherjee AB: Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-ras product and growth-associated protein-43. J Biol Chem 2013, 288:9112–9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin DT, Conibear E: ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Remsberg JR, Suciu RM, Zambetti NA, Hanigan TW, Firestone AJ, Inguva A, Long A, Ngo N, Lum KM, Henry CL, et al. : ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat Chem Biol 2021, 10.1038/s41589-021-00785-8. * This study describes a specific inhibitor of the ABHD17 family of RAS depalmitoylating enzymes and shows effects on NRAS signaling and growth of NRAS-mutant AML cells.
- 49.Jing H, Zhang X, Wisner SA, Chen X, Spiegelman NA, Linder ME, Lin H: SIRT2 and lysine fatty acylation regulate the transforming activity of K-Ras4a. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shahinian S, Silvius JR: Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry 1995, 34:3813–3822. [DOI] [PubMed] [Google Scholar]
- 51.Schroeder H, Leventis R, Rex S, Schelhaas M, Nagele E, Waldmann H, Silvius JR: S-Acylation and plasma membrane targeting of the farnesylated carboxyl-terminal peptide of N-ras in mammalian fibroblasts. Biochemistry 1997, 36:13102–13109. [DOI] [PubMed] [Google Scholar]
- 52.Goodwin JS, Drake KR, Rogers C, Wright L, Lippincott-Schwartz J, Philips MR, Kenworthy AK: Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol 2005, 170:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abankwa D, Gorfe AA, Hancock JF: Ras nanoclusters: molecular structure and assembly. Semin Cell Dev Biol 2007, 18:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy S, Plowman S, Rotblat B, Prior IA, Muncke C, Grainger S, Parton RG, Henis YI, Kloog Y, Hancock JF: Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol Cell Biol 2005, 25:6722–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plowman SJ, Muncke C, Parton RG, Hancock JF: H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci U S A 2005, 102:15500–15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, Bastiaens PI: An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science 2005, 307:1746–1752. [DOI] [PubMed] [Google Scholar]
- 57.Rocks O, Gerauer M, Vartak N, Koch S, Huang ZP, Pechlivanis M, Kuhlmann J, Brunsveld L, Chandra A, Ellinger B, et al. : The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 2010, 141:458–471. [DOI] [PubMed] [Google Scholar]
- 58.Schmick M, Kraemer A, Bastiaens PI: Ras moves to stay in place. Trends Cell Biol 2015, 25:190–197. [DOI] [PubMed] [Google Scholar]
- 59.Ahearn IM, Tsai FD, Court H, Zhou M, Jennings BC, Ahmed M, Fehrenbacher N, Linder ME, Philips MR: FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol Cell 2011, 41:173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, Cox AD, Philips MR: Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 2002, 4:343–350. [DOI] [PubMed] [Google Scholar]
- 61.Onken B, Wiener H, Philips M, Chang EC: Compartmentalized signaling of Ras in fission yeast. Proc Natl Acad Sci U S A 2006, 103:9045–9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mor A, Philips MR: Compartmentalized Ras/MAPK Signaling. Annu Rev Immunol 2006, 24:771–800. [DOI] [PubMed] [Google Scholar]
- 63.Aran V, Prior IA: Compartmentalized Ras signaling differentially contributes to phenotypic outputs. Cell Signal 2013, 25:1748–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen WC, Zhou M, Court H, Shi J, Mendoza SL, Morten MJ, et al. : KRAS4A directly regulates hexokinase 1. Nature 2019, 576:482–486. ** This study shows that a palmitoylation/depalmitoylation cycle allows KRAS4A to associate with the outer mitochondrial membrane where it interacts with and activates hexokinase 1, a clear example of compartmentalized signaling of a RAS isoform regulated by a PTM.
- 65.Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, Werlen G, Hollander GA, Gascoigne NR, Palmer E: Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature 2006, 444:724–729. [DOI] [PubMed] [Google Scholar]
- 66. Zambetti NA, Firestone AJ, Remsberg JR, Huang BJ, Wong JC, Long AM, Predovic M, Suciu RM, Inguva A, Kogan SC, et al. : Genetic disruption of N-RasG12D palmitoylation perturbs hematopoiesis and prevents myeloid transformation in mice. Blood 2020, 135:1772–1782. ** In this study that authors knocked in a conditional, activated but palmitoylation-deficient Nras allele into a mouse model of myeloid leukemia and found that the lack of palmitoylation abrogated myeloid transformation. This compelling in vivo result establishes palmitoylation as a promising therapeutic target for NRAS-driven cancers.
- 67.Ballester R, Furth ME, Rosen OM: Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem 1987, 262:2688–2695. [PubMed] [Google Scholar]
- 68.Arimura S, Nakata H, Tomiyama K, Watanabe Y: Phosphorylation of H-ras proteins by protein kinase A. Cell Signal 1997, 9:37–40. [DOI] [PubMed] [Google Scholar]
- 69.Kim SE, Yoon JY, Jeong WJ, Jeon SH, Park Y, Yoon JB, Park YN, Kim H, Choi KY: H-Ras is degraded by Wnt/beta-catenin signaling via beta-TrCP-mediated polyubiquitylation. J Cell Sci 2009, 122:842–848. [DOI] [PubMed] [Google Scholar]
- 70. Yin C, Zhu B, Zhang T, Liu T, Chen S, Liu Y, Li X, Miao X, Li S, Mi X, et al. : Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell 2019, 176:1113–1127 e1116. ** These authors identify STK19 as a kinase that phosphorylates NRAS on serine 89 and thereby enhances downstream signaling. A D89N mutation in STK19, often found in melanoma, enhances binding to NRAS and an STK19 inibitor blocked NRAS-driven melanocyte transformation establishing a novel therpeutic target.
- 71.Bivona TG, Quatela SE, Bodemann BO, Ahearn IO, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, et al. : PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell 2006, 21:481–493. [DOI] [PubMed] [Google Scholar]
- 72.McLaughlin S, Aderem A: The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci 1995, 20:272–276. [DOI] [PubMed] [Google Scholar]
- 73.Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, Foskett JK, Philips MR: Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci U S A 2013, 110:20593–20598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang MT, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, McCormick F: K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163:1237–1251. [DOI] [PubMed] [Google Scholar]
- 75.Alvarez-Moya B, Lopez-Alcala C, Drosten M, Bachs O, Agell N: K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29:5911–5922. [DOI] [PubMed] [Google Scholar]
- 76.Barcelo C, Paco N, Beckett AJ, Alvarez-Moya B, Garrido E, Gelabert M, Tebar F, Jaumot M, Prior I, Agell N: Oncogenic K-ras segregates at spatially distinct plasma membrane signaling platforms according to its phosphorylation status. J Cell Sci 2013, 126:4553–4559. [DOI] [PubMed] [Google Scholar]
- 77.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, Nussinov R, Gaponenko V: Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J Biol Chem 2015, 290:9465–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cho KJ, Casteel DE, Prakash P, Tan L, van der Hoeven D, Salim AA, Kim C, Capon RJ, Lacey E, Cunha SR, et al. : AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol Cell Biol 2016, 36:3086–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buday L, Vas V: Novel regulation of Ras proteins by direct tyrosine phosphorylation and dephosphorylation. Cancer Metastasis Rev 2020, 39:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bunda S, Heir P, Srikumar T, Cook JD, Burrell K, Kano Y, Lee JE, Zadeh G, Raught B, Ohh M: Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc Natl Acad Sci U S A 2014, 111:E3785–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, Raught B, Zhang ZY, Zadeh G, Ohh M: Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 2015, 6:8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kano Y, Gebregiworgis T, Marshall CB, Radulovich N, Poon BPK, St-Germain J, Cook JD, Valencia-Sama I, Grant BMM, Herrera SG, et al. : Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat Commun 2019, 10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chio IIC, Tuveson DA: ROS in Cancer: The Burning Question. Trends Mol Med 2017, 23:411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A: Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem 1995, 270:7017–7020. [DOI] [PubMed] [Google Scholar]
- 85.Lander HM, Ogiste JS, Teng KK, Novogrodsky A: p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem 1995, 270:21195–21198. [DOI] [PubMed] [Google Scholar]
- 86.Williams JG, Pappu K, Campbell SL: Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A 2003, 100:6376–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heo J, Campbell SL: Mechanism of p21Ras S-nitrosylation and kinetics of nitric oxide-mediated guanine nucleotide exchange. Biochemistry 2004, 43:2314–2322. [DOI] [PubMed] [Google Scholar]
- 88.Heo J, Campbell SL: Superoxide anion radical modulates the activity of Ras and Ras-related GTPases by a radical-based mechanism similar to that of nitric oxide. J Biol Chem 2005, 280:12438–12445. [DOI] [PubMed] [Google Scholar]
- 89.Heo J, Prutzman KC, Mocanu V, Campbell SL: Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol 2005, 346:1423–1440. [DOI] [PubMed] [Google Scholar]
- 90.Heo J, Campbell SL: Ras regulation by reactive oxygen and nitrogen species. Biochemistry 2006, 45:2200–2210. [DOI] [PubMed] [Google Scholar]
- 91.Mott HR, Carpenter JW, Campbell SL: Structural and functional analysis of a mutant Ras protein that is insensitive to nitric oxide activation. Biochemistry 1997, 36:3640–3644. [DOI] [PubMed] [Google Scholar]
- 92.Mitchell L, Hobbs GA, Aghajanian A, Campbell SL: Redox regulation of Ras and Rho GTPases: mechanism and function. Antioxid Redox Signal 2013, 18:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Messina S, De Simone G, Ascenzi P: Cysteine-based regulation of redox-sensitive Ras small GTPases. Redox Biol 2019, 26:101282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lim KH, Ancrile BB, Kashatus DF, Counter CM: Tumour maintenance is mediated by eNOS. Nature 2008, 452:646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lim JKM, Leprivier G: The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis 2019, 10:955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dohlman HG, Campbell SL: Regulation of large and small G proteins by ubiquitination. J Biol Chem 2019, 294:18613–18623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yan H, Jahanshahi M, Horvath EA, Liu HY, Pfleger CM: Rabex-5 ubiquitin ligase activity restricts Ras signaling to establish pathway homeostasis in Drosophila. Curr Biol 2010, 20:1378–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu L, Lubkov V, Taylor LJ, Bar-Sagi D: Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr Biol 2010, 20:1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baietti MF, Simicek M, Abbasi Asbagh L, Radaelli E, Lievens S, Crowther J, Steklov M, Aushev VN, Martinez Garcia D, Tavernier J, et al. : OTUB1 triggers lung cancer development by inhibiting RAS monoubiquitination. EMBO Mol Med 2016, 8:288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Pandolfi PP, Cantley LC: Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal 2011, 4:ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baker R, Lewis SM, Sasaki AT, Wilkerson EM, Locasale JW, Cantley LC, Kuhlman B, Dohlman HG, Campbell SL: Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat Struct Mol Biol 2013, 20:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baker R, Wilkerson EM, Sumita K, Isom DG, Sasaki AT, Dohlman HG, Campbell SL: Differences in the regulation of K-Ras and H-Ras isoforms by monoubiquitination. J Biol Chem 2013, 288:36856–36862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hobbs GA, Gunawardena HP, Baker R, Campbell SL: Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Small GTPases 2013, 4:186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yin G, Zhang J, Nair V, Truong V, Chaia A, Petela J, Harrison J, Gorfe AA, Campbell SL: KRAS Ubiquitination at Lysine 104 Retains Exchange Factor Regulation by Dynamically Modulating the Conformation of the Interface. iScience 2020, 23:101448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cho YH, Cha PH, Kaduwal S, Park JC, Lee SK, Yoon JS, Shin W, Kim H, Ro EJ, Koo KH, et al. : KY1022, a small molecule destabilizing Ras via targeting the Wnt/beta-catenin pathway, inhibits development of metastatic colorectal cancer. Oncotarget 2016, 7:81727–81740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeong WJ, Ro EJ, Choi KY: Interaction between Wnt/beta-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of beta-catenin and RAS by targeting the Wnt/beta-catenin pathway. NPJ Precis Oncol 2018, 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cha PH, Cho YH, Lee SK, Lee J, Jeong WJ, Moon BS, Yun JH, Yang JS, Choi S, Yoon J, et al. : Small-molecule binding of the axin RGS domain promotes beta-catenin and Ras degradation. Nat Chem Biol 2016, 12:593–600. [DOI] [PubMed] [Google Scholar]
- 108.Lee SK, Cho YH, Cha PH, Yoon JS, Ro EJ, Jeong WJ, Park J, Kim H, Il Kim T, Min DS, et al. : A small molecule approach to degrade RAS with EGFR repression is a potential therapy for KRAS mutation-driven colorectal cancer resistance to cetuximab. Exp Mol Med 2018, 50:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park J, Cho YH, Shin WJ, Lee SK, Lee J, Kim T, Cha PH, Yang JS, Cho J, Min DS, et al. : A Ras destabilizer KYA1797K overcomes the resistance of EGFR tyrosine kinase inhibitor in KRAS-mutated non-small cell lung cancer. Sci Rep 2019, 9:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shukla S, Allam US, Ahsan A, Chen G, Krishnamurthy PM, Marsh K, Rumschlag M, Shankar S, Whitehead C, Schipper M, et al. : KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated beta-TrCP1 degradation. Neoplasia 2014, 16:115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lu A, Tebar F, Alvarez-Moya B, Lopez-Alcala C, Calvo M, Enrich C, Agell N, Nakamura T, Matsuda M, Bachs O: A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J Cell Biol 2009, 184:863–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bigenzahn JW, Collu GM, Kartnig F, Pieraks M, Vladimer GI, Heinz LX, Sedlyarov V, Schischlik F, Fauster A, Rebsamen M, et al. : LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362:1171–1177. * These four papers highlight a putative role for the LZTR1 in downregulating RAS or RIT GTPase function. LZTR1 is thought to act as a tumor suppressor. Association of RAS or RIT with the LZTR1/ CUL3 ubiquitin ligase (Cullin-Based Ubiquitin Ligase 3) complex promotes downregulation of GTPase signaling.
- 113. Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, Zhang M, Jang H, Renzi F, Cai Y, et al. : Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362:1177–1182. * These four papers highlight a putative role for the LZTR1 in downregulating RAS or RIT GTPase function. LZTR1 is thought to act as a tumor suppressor. Association of RAS or RIT with the LZTR1/ CUL3 ubiquitin ligase (Cullin-Based Ubiquitin Ligase 3) complex promotes downregulation of GTPase signaling.
- 114. Abe T, Umeki I, Kanno SI, Inoue SI, Niihori T, Aoki Y: LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ 2020, 27:1023–1035. * These four papers highlight a putative role for the LZTR1 in downregulating RAS or RIT GTPase function. LZTR1 is thought to act as a tumor suppressor. Association of RAS or RIT with the LZTR1/ CUL3 ubiquitin ligase (Cullin-Based Ubiquitin Ligase 3) complex promotes downregulation of GTPase signaling.
- 115. Castel P, Cheng A, Cuevas-Navarro A, Everman DB, Papageorge AG, Simanshu DK, Tankka A, Galeas J, Urisman A, McCormick F: RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science 2019, 363:1226–1230. * These four papers highlight a putative role for the LZTR1 in downregulating RAS or RIT GTPase function. LZTR1 is thought to act as a tumor suppressor. Association of RAS or RIT with the LZTR1/ CUL3 ubiquitin ligase (Cullin-Based Ubiquitin Ligase 3) complex promotes downregulation of GTPase signaling.
- 116.Choi BH, Philips MR, Chen Y, Lu L, Dai W: K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J Biol Chem 2018, 293:17574–17581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Choi BH, Chen C, Philips M, Dai W: RAS GTPases are modified by SUMOylation. Oncotarget 2018, 9:4440–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xie M, Yu J, Ge S, Huang J, Fan X: SUMOylation homeostasis in tumorigenesis. Cancer Lett 2020, 469:301–309. [DOI] [PubMed] [Google Scholar]
- 119.Knyphausen P, Lang F, Baldus L, Extra A, Lammers M: Insights into K-Ras 4B regulation by post-translational lysine acetylation. Biol Chem 2016, 397:1071–1085. [DOI] [PubMed] [Google Scholar]
- 120.Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, Philips MR, Shan Y, Shaw DE, Bar-Sagi D, et al. : Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci U S A 2012, 109:10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang MH, Laurent G, Bause AS, Spang R, German N, Haigis MC, Haigis KM: HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol Cancer Res 2013, 11:1072–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yin G, Kistler S, George SD, Kuhlmann N, Garvey L, Huynh M, Bagni RK, Lammers M, Der CJ, Campbell SL: A KRAS GTPase K104Q Mutant Retains Downstream Signaling by Offsetting Defects in Regulation. J Biol Chem 2017, 292:4446–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cornett EM, Ferry L, Defossez PA, Rothbart SB: Lysine Methylation Regulators Moonlighting outside the Epigenome. Mol Cell 2019, 75:1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yoshino H, Yin G, Kawaguchi R, Popov KI, Temple B, Sasaki M, Kofuji S, Wolfe K, Kofuji K, Okumura K, et al. : Identification of lysine methylation in the core GTPase domain by GoMADScan. PLoS One 2019, 14:e0219436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee SH, Lee JW, Soung YH, Kim HS, Park WS, Kim SY, Lee JH, Park JY, Cho YG, Kim CJ, et al. : BRAF and KRAS mutations in stomach cancer. Oncogene 2003, 22:6942–6945. [DOI] [PubMed] [Google Scholar]
- 126.Zenker M, Lehmann K, Schulz AL, Barth H, Hansmann D, Koenig R, Korinthenberg R, Kreiss-Nachtsheim M, Meinecke P, Morlot S, et al. : Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet 2007, 44:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bertola DR, Pereira AC, Brasil AS, Albano LMJ, Kim CA, Krieger JE: Further evidence of genetic heterogeneity in Costello syndrome: involvement of the KRAS gene. J Hum Genet 2007, 52:521–526. [DOI] [PubMed] [Google Scholar]
- 128.Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, Aoki Y, Matsubara Y, Arveiler B, Lacombe D, et al. : Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet 2007, 44:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lo FS, Lin JL, Kuo MT, Chiu PC, Shu SG, Chao MC, Lee YJ, Lin SP: Noonan syndrome caused by germline KRAS mutation in Taiwan: report of two patients and a review of the literature. Eur J Pediatr 2009, 168:919–923. [DOI] [PubMed] [Google Scholar]
- 130.Bertola DR, Pereira AC, Brasil AC, Suzuki L, Leite C, Falzoni R, Tannuri U, Poplawski AB, Janowski KM, Kim CA, et al. : Multiple, diffuse schwannomas in a RASopathy phenotype patient with germline KRAS mutation: a causal relationship? Clin Genet 2012, 81:595–597. [DOI] [PubMed] [Google Scholar]
- 131.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, Pineda M, Gindin Y, Jiang Y, Reinhold WC, et al. : The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res 2013, 73:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ho AL, Brana I, Haddad R, Bauman J, Bible K, Oosting S, Wong DJ, Ahn MJ, Boni V, Even C, et al. : Tipifarnib in Head and Neck Squamous Cell Carcinoma With HRAS Mutations. J Clin Oncol 2021, 10.1200/JCO.20.02903:JCO2002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM: Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent Sci 2020, 6:1367–1375. ** In these studies, the authors report the first PROTACs capable of degrading an oncogenic KRAS mutant that contains a reactive cysteine (KRASG12C). These bifunctional PROTAC utilize warheads that covalently targets KRAS G12C and engage the E3 ligases VHL or CRBN, respectively.
- 134. Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, Nabet B, Gero TW, Feru F, Li L, et al. : Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem Biol 2020, 27:19–31 e16. ** In these studies, the authors report the first PROTACs capable of degrading an oncogenic KRAS mutant that contains a reactive cysteine (KRASG12C). These bifunctional PROTAC utilize warheads that covalently targets KRAS G12C and engage the E3 ligases VHL or CRBN, respectively.
- 135. Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C, Hou X, Zhang Y, Neel BG, O’Bryan JP, et al. : Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat Commun 2021, 12:2656. * These studies also target KRAS for degradation by recruiting an E3 ligase, but unlike the reports of PROTACs, these utilize genetically encoded constructs that will require further development to generate practical therapeutics.
- 136. Simpson LM, Macartney TJ, Nardin A, Fulcher LJ, Roth S, Testa A, Maniaci C, Ciulli A, Ganley IG, Sapkota GP: Inducible Degradation of Target Proteins through a Tractable Affinity-Directed Protein Missile System. Cell Chem Biol 2020, 27:1164–1180 e1165. * These studies also target KRAS for degradation by recruiting an E3 ligase, but unlike the reports of PROTACs, these utilize genetically encoded constructs that will require further development to generate practical therapeutics.
- 137. Bery N, Miller A, Rabbitts T: A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat Commun 2020, 11:3233. * These studies also target KRAS for degradation by recruiting an E3 ligase, but unlike the reports of PROTACs, these utilize genetically encoded constructs that will require further development to generate practical therapeutics.