

Abstract
Acute leukemias of ambiguous lineage (ALAL), including mixed phenotype acute leukemia (MPAL) and related entities such as early T-cell precursor acute leukemia (ETP-ALL), remain diagnostic and clinical challenges dues to limited understanding of pathogenesis, reliance of immunophenotyping to classify disease, and the lack of a rational approach to guide selection of appropriate therapy. Recent studies utilizing genomic sequencing and complementary approaches have provided key insights that are changing the way in which such leukemias are classified, and potentially, treated. Several recurrent genomic alterations define leukemias that straddle immunophenotypic entities, such as ZNF384-rearranged childhood B-ALL and B/myeloid MPAL, and BCL11B-rearranged T/myeloid MPAL, ETP-ALL and AML. In contrast, some cases of MPAL represent canonical ALL/AML entities exhibiting lineage aberrancy. For many cases of ALAL, experimental approaches indicate lineage aberrancy arises from acquisition of a founding genetic alterations into a hematopoietic stem and progenitor cell. Determination of optimal therapeutic approach requires genomic characterization of uniformly treated ALAL patients in prospective studies, but several approaches, including kinase inhibitors and BH3 mimetics may be efficacious in subsets of ALAL.
Keywords: ALAL, lineage ambiguous, 14q32, BCL11B, enhancer hijacking, mixed phenotype acute leukemia
Acute leukemias of ambiguous lineage (ALAL) encompass multiple poorly understood subtypes of acute leukemia that pose challenges in diagnosis and clinical management. Most ALAL cases are diagnosed as mixed phenotype acute leukemia (MPAL), with blasts exhibiting both a myeloid and T- or B-lymphoid immunophenotype or, more rarely, B/T or B/T/myeloid immunophenotypes. The remaining cases of ALAL lack lineage-defining markers and are diagnosed as acute undifferentiated leukemia (AUL) (Table 1). Together, MPAL and AUL account for 3–5% of newly diagnosed adult and pediatric acute leukemia cases [1–3]. Outcomes are generally poor [4,5], and due to their relative rarity, advances in our understanding of the genomic and cellular basis of ALAL have lagged behind more prevalent subtypes of acute leukemia. Moreover, diagnosis is based on few markers aimed to distinguish myeloid (e.g. MPO) and lymphoid (e.g. CD3, CD19) lineages [6]; however, recent data suggest that such immunophenotypic-based classifications often fail to capture biological similarities between different clinical entities of acute leukemia. For example, MPO is often used to distinguish T/myeloid MPAL from early T cell precursor acute lymphoblastic leukemia (ETP-ALL), despite the fact that both entities express myeloid and stem cell markers [7–9], frequently harbor similar mutations (e.g. in WT1, RUNX1, and FLT3) [8,10,11], and exhibit similar gene expression profiles [12]. Molecular similarities between leukemias with myeloid and T-lymphoid phenotypes have led to the proposal of entirely new diagnostic entities to capture these “interface” cases [13,14]. However, whether this is warranted requires specific investigation of the genomic and cellular basis of diverse ALAL and related (e.g. ETP-ALL, acute myeloid leukemia (AML)) cases.
Table 1. Flow cytometry-based diagnostic criteria for major acute leukemia subtypes.
Cell surface and intracellular markers used in the diagnosis of acute leukemia are shown, based on the WHO classification system[2]. The immunophenotype of cases with BCL11B SVs is shown at the bottom (“BCL11B deregulated AL”). Note that MPO, which is used to distinguish ETP-ALL from T/myeloid MPAL, is variable in this new subgroup.
| Diagnostic entity | Immunophenotypic criteria |
|---|---|
| AUL | Lack of CD3, CD19, MPO, positive for CD117/CD34, positive for 1–2 of CD13/CD33, frequently CD7 and/or TdT positive |
| AML | MPO positive and/or positive for multiple markers of myeloid/monocytic differentiation (CD13/CD33/CD11b/CD11c/CD14/CD15/CD64/CD65), negative for cCD3, CD19, often CD117 and/or CD34 positive, variably CD7 positive |
| T/myeloid MPAL | Myeloid lineage: MPO or at least 2 markers associated with monocytic differentiation (CD11c, CD14, CD64). |
| ETP-ALL | cytoplasmic CD3 positive, but CD1a and CD8 negative, CD5 on <75% of blasts, variable for CD2, usually CD117 and/or CD34+, CD13/CD33/CD11b positive, negative for MPO |
| Typical T-ALL | Surface CD3 positive, CD8 positive, CD5 >75% of blasts, variable for CD34, infrequent for CD13/33/11b, negative for MPO |
| B/myeloid MPAL | Myeloid lineage: MPO or at least 2 markers associated with monocytic differentiation. |
| Typical precursor B-ALL | Positive for CD19/CD10/CD79a, negative for CD117, frequently CD34+, negative for CD3, myeloid markers |
| BCL11B deregulated AL | CD117 and/or CD34+, usually cCD3+, always CD2+, always CD5−, always CD8−, variable for MPO, positive for other myeloid markers |
Several recent studies have performed genomic analyses of ALAL/MPAL. In a study of 8 pediatric and 18 adult patients, Xiao and colleagues identified recurrent and largely mutually exclusive alterations in PHF6, DNMT3A, and WT1 in MPAL with a T-lineage immunophenotype [15]. Notably, NOTCH1 mutation was only identified in a single case. In contrast, a study focused exclusively on 31 cases of adult MPAL identified NOTCH1 mutations in 29% of T/myeloid MPAL cases [16]. This study also found recurrent alterations of DNMT3A, as well as IDH2, NRAS, and KRAS. However, in contrast to Xiao et al, no WT1 or PHF6 mutations were identified. Both studies identified BCR-ABL1 and KMT2A (MLL) rearrangements in B/myeloid cases. Discrepancies between these studies, for example regarding the prevalence of NOTCH or WT1 mutations, could be due to small numbers of cases profiled or differences in the genetic origins of pediatric and adult ALAL.
We recently performed whole exome and genome sequencing in a cohort of 115 pediatric MPAL/ALAL cases [12] which confirmed that WT1 mutations are among the most recurrent alteration in pediatric T/myeloid MPAL (20/49 cases, 40.8%). We also identified activating FLT3 alterations in 42.9% of cases (including 70% of WT1-mutated cases) as well as mutations in RUNX1 and PHF6. In B/myeloid MPAL, fusion genes involving the ZNF384 transcription factor gene were identified in nearly 50% of cases. ZNF384 fusions are also common in B-ALL [17,18] which had an overlapping gene expression profile with ZNF384-rearranged B/myeloid MPAL, supporting the notion that genomic alteration, rather than immunophenotype, more accurately classifies certain subtypes of leukemia. These fusions were not identified in adult B/myeloid cases [16] confirming that subsets of pediatric and adult MPAL have distinct genetic origins. DNMT3A mutations, for example, were only observed in adults and suggest that mutations associated with age-related clonal hematopoiesis might contribute to adult-onset MPAL. Importantly, all mutations described in these studies also occur in AML and/or ALL, leaving open the question of whether genomic alterations exist that specifically drive a lineage ambiguous phenotype.
Towards a transcriptional and genomic roadmap of acute leukemia
Although these studies identified several recurrent genomic alterations associated with ALAL, an important limitation was the selection of cases using current immunophenotypic criteria for ALAL/MPAL, precluding the identification of leukemia subtypes that might transcend diagnostic boundaries, and often, limited genomic characterization and a focus on protein coding alterations and fusion genes. Motivated by these challenges, we sought to perform an unbiased analysis of all acute leukemia subtypes in order to delineate biological heterogeneity, irrespective of diagnostic classification or immunophenotype, with an emphasis on comprehensively characterizing the genomic and molecular features that define lineage-ambiguous leukemia in the context of other leukemia subtypes. Here we summarize our pan-leukemia study that identified multiple unrecognized subtypes of acute leukemia, with a key finding being identification of distinct subgroup of lineage ambiguous leukemia driven by structural variants deregulating BCL11B that spans current diagnostic categories [19]. We discuss the implications of these findings on the cellular origins of lineage ambiguous leukemias, clinical diagnosis, and treatment.
To perform this study, we compiled transcriptome sequencing (RNA-seq) data from 2,573 cases spanning the major leukemia subtypes: major subgroups of B-ALL [20] and T-ALL [11], ETP-ALL, AML, MPAL, and AUL samples. We identified biologically-defined subgroups within each leukemia subtype through gene expression clustering analyses, using both unsupervised hierarchical clustering and t-distributed stochastic neighbor embedding (tSNE) approaches. As reported previously [20], B-ALL cases segregated into over 20 subgroups, each with defining genomic alterations (Figure 1A). Of note, the majority of immunophenotypically-defined B/myeloid MPAL cases clustered with B-ALL subgroups with matched genomic alterations, indicating that B/myeloid MPAL as a single distinct diagnostic entity may be misguided. The majority of T-ALL and AML cases also clustered according to genomic alteration, albeit less distinctly as compared to B-ALL cases, with many subtypes reported previously [11,21]. However, this analysis identified two novel gene expression clusters, highlighting the power of large, diverse cohorts to resolve biological heterogeneity (Figure 1B). The first cluster included 1.7% of T-ALL cases and was defined by LMO2 rearrangement to non-T cell receptor loci, most frequently STAG2. The second cluster had an unknown genomic driver and encompassed 30–40% of T/myeloid MPAL and ETP-ALL cases, in addition to 3.8% of AML and 50% of AUL cases. Identification of this new subgroup, which spanned at least 4 different diagnostic entities, has broader implications for diagnosis and classification of leukemias with stem, myeloid and T-lymphoblastic features.
Figure 1. Gene expression profiling of acute leukemia reclassifies lineage ambiguous cases.
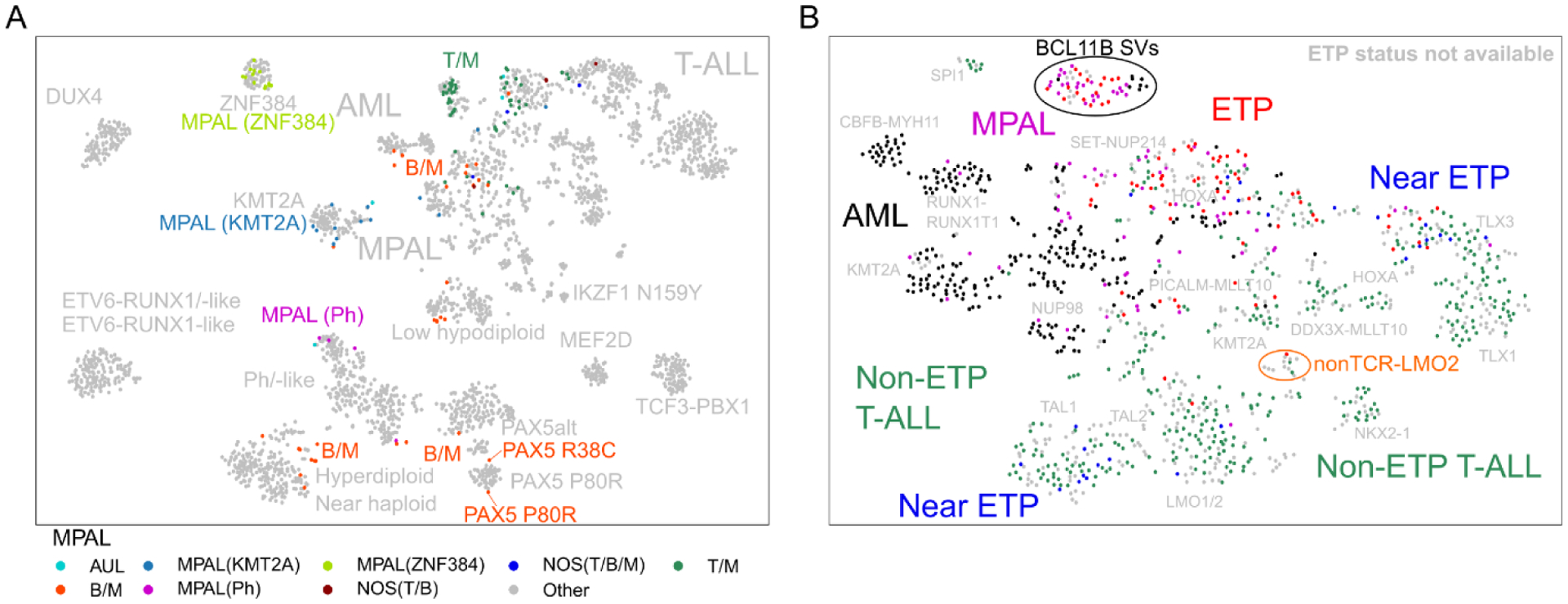
(A) tSNE analysis of 2,573 acute leukemia samples. All cases diagnosed as MPAL are shown according to MPAL subtype and/or known genomic alteration. The majority of B/myeloid MPAL cases cluster with B-ALL subtypes with shared genomic alteration. (B) tSNE analysis of all non-B-ALL cases. Two previously undescribed subgroups were identified, including cases with BCL11B SVs (circled in black) and cases with non-TCR LMO2 rearrangements (circled in orange). Figure reproduced from ref. [19].
BCL11B deregulation defines a new subtype of ALAL
Initially, the only notable recurrent alterations identified in this subgroup were WT1 mutations (16/61, 26.2%) and activating FLT3 alterations (50/61, 82%) as previously reported in T/myeloid MPAL [12] and ETP-ALL [10,11]. As they were not exclusive to this subgroup, we suspected the presence of non-coding alterations that had eluded standard transcriptome and exome-based analysis. Using cis-X [22], a tool that uses matched transcriptome and whole-genome sequencing (WGS) data to identify genes with allele-specific expression (ASE), we found that all 53 cases in this subgroup with matched RNA-seq and WGS data exhibited ASE of the T cell transcription factor gene BCL11B, and harbored non-coding structural variants (SVs) with breakpoints near the BCL11B gene on chromosome 14q32.2. These SVs included translocations to chromosomes 2,3,6,7,8,12 and 21, as well as cases with a focal amplification in cis with BCL11B (Figure 2A). Collectively, these genomic data nominated BCL11B as the likely driver of this gene expression subgroup.
Figure 2. BCL11B SVs result in deregulated BCL11B expression through various enhancer-mediated mechanisms.
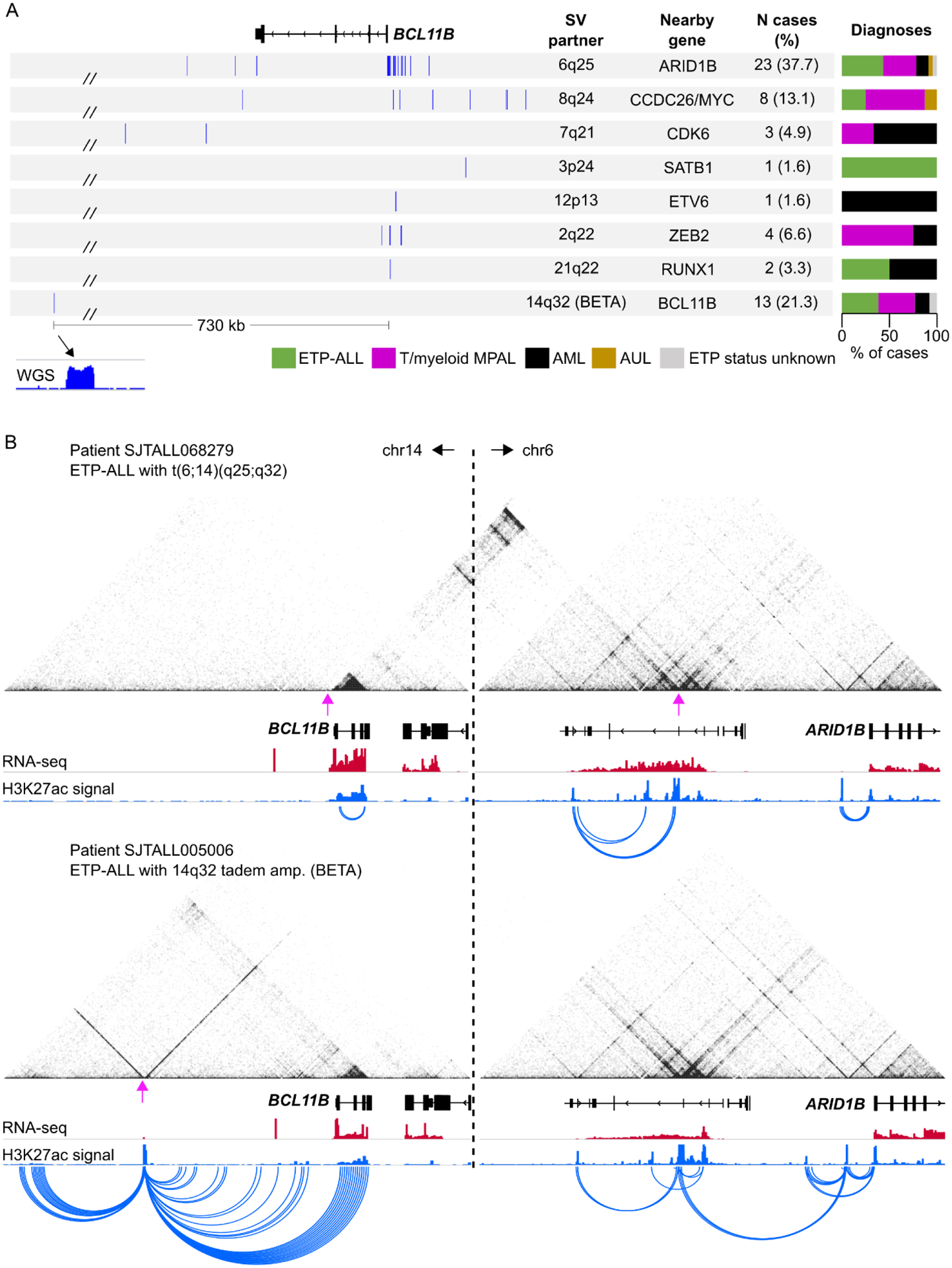
(A) Summary of patient-specific breakpoints organized by SV partner locus. Only breakpoints within the viewing region are shown and only for cases with an identifiable SV (6 cases excluded that lacked WGS). (B) H3K27ac HiChIP data for two patient samples showing enhancer hijacking by the ARID1B enhancer at the 6q25 locus (top) and de novo enhancer formation (BETA) resulting in BCL11B activation (bottom). The same genomic windows are shown for each sample to demonstrate that chromatin interactions between chromosome 14 and chromosome 6 are only observed in the case with the t(6;14) translocation (top). Magenta arrows show the breakpoint positions.
BCL11B as regulator of T lineage differentiation and oncogene
BCL11B plays important roles in both normal and malignant hematopoiesis. BCL11B encodes a C2H2 zinc finger transcription factor first expressed in CD34+CD7-CD1a- thymic progenitor cells (corresponding to the double negative 2, or DN2, stage of murine thymocyte development) [23,24]. BCL11B expression continues to increase during subsequent maturation stages [23]. Acting as both a transcriptional activator and repressor, BCL11B controls the expression of hundreds of genes in a developmental context-specific manner [25], including upregulation of T-lineage genes (e.g. Rag1/2, Tcf7, Gata3, Cd8a/b, Lck, Cd3d) and downregulation of stem and progenitor cell genes (e.g. CD34, Kit, Spi1) during T cell commitment [26,27], and genes related to positive selection, apoptosis, and alternate lineage fates in mature T cell populations [28,29]. Progenitor T cells lacking BCL11B are not able to progress past the DN2 stage and fail to recombine T cell receptor genes; moreover, these cells retain myeloid and natural kill cell differentiation potential which is otherwise blocked in BCL11B-expressing T cells [29,30]. BCL11B is therefore a critical “gate-keeper” of the T cell lineage.
BCL11B was first shown to have a tumor suppressor role in 2003 when nearly 50% of gamma ray-induced T cell lymphomas in mice were found to harbor Bcl11b mutations [31]. Shortly thereafter, BCL11B mutations were identified in human T-ALL and are now known to occur with a frequency ranging from ~9–15% [11,32–34]. BCL11B mutations typically result in amino acid substitutions within the zinc finger domains which are predicted to interfere with, or abrogate, DNA binding and thus disrupt BCL11B function. Loss of BCL11B through deletion is also reported [33]. Notably, most BCL11B-mutated T-ALLs retain a wild-type BCL11B allele, indicating that complete loss is either not tolerated or is otherwise not tumorigenic; rather, functional haploinsufficiency—either through deletion or mutation of one allele—confers BCL11B with tumor suppressor properties. Interestingly, and consistent with this notion, low BCL11B expression was associated with induction failure [33] and poor overall survival [34]. Despite clear evidence as a tumor suppressor, BCL11B knock-out in several T-ALL cell lines resulted in apoptosis and cell death [35], suggesting that BCL11B may also possess oncogenic functions in some contexts.
The nature of BCL11B alterations in ALAL
Unlike the BCL11B alterations described for T-ALL (primarily amino acid substitutions and deletions), the 14q32 structural variants identified in lineage ambiguous leukemias leave the BCL11B coding region intact, suggesting that these alterations may influence BCL11B expression. The observation of allele-specific, generally high expression of BCL11B suggested the SVs deregulated expression of one BCL11B allele, potentially in a hematopoietic stem and progenitor cell (HSPC) where BCL11B is normally repressed, resulting in lineage aberrancy. Enhancer hijacking is one such mechanism whereby SVs (e.g. translocations, deletions, or inversions) result in the “re-wiring” of enhancer-promoter interactions [36,37]. Because enhancers typically only regulate genes within one megabase, these rearrangements frequently lead to the aberrant activation or upregulation of newly acquired target genes. Using chromatin-based assays in patient samples, we confirmed that the BCL11B SVs did indeed result in enhancer hijacking, and we could use these data to glean further insights into the cellular origins of BCL11B-deregulated leukemia, as discussed below (Figure 2B).
Active enhancers are readily identified by high levels of the post-translational histone modification acetylation of histone H3 at lysine 27 (H3K27ac) [38–40]. Using publicly available H3K27ac chromatin immunoprecipitation followed by sequencing (ChIP-seq) data from human CD34+ HSPCs [41], we found that the SV breakpoints of all BCL11B-group patient samples occurred within several hundred kilobases (kb) of highly active CD34+ HSPC enhancers—also known as super-enhancers [42,43]. To interrogate the resulting enhancer-promoter re-wiring, we performed H3K27ac Hi-ChIP [44] in 5 patient samples to simultaneously survey H3K27ac and long-range chromatin interactions which identified chromatin interactions between the BCL11B gene and rearranged super-enhancers in all cases, even when the genomic distance was >300 kb. Additionally, all 7 CD34+ HSPC super-enhancers involved in BCL11B SVs showed activity in each patient sample, supporting the epigenetic similarities between BCL11B-deregulated ALAL and HSPCs.
Enhancer hijacking in acute leukemia
Enhancer hijacking as a mechanism of oncogenic gene expression has been described previously in acute leukemia and lymphoma, including aberrant activation or upregulation of MN1 [45], EVI1/MECOM [46–48], and FLT3 [49] in AML, and TAL1/2, TLX3/5, LYL1, and LMO1/2 in T-ALL [50]. Additionally, most translocations in leukemia involving the immunoglobulin genes (i.e. T cell receptor (TCR) and immunoglobulin heavy/light chain (IGH/IGK/IGL) genes) result in overexpression of oncogenes through the activity of tissue-specific enhancers in these loci. For example, Burkitt lymphoma often results from t(8;14) translocations which juxtapose enhancer elements within the 3’ IGH locus near the MYC gene to drive MYC overexpression [51,52], and subsets of Philadelphia chromosome-like (“Ph-like”) B-ALL harbor IGH rearrangements to CRLF2, resulting in CRLF2 overexpression and JAK/STAT pathway activation [53,54]. As most previously described enhancer hijacking events are driven by one or two recurrently targeted enhancers, the diverse array of at least 7 distinct enhancer hijacking events described for BCL11B was striking, and might reflect a particularly susceptible window in early hematopoietic development for the acquisition of translocations involving these highly transcriptionally active loci.
BETA: a novel mechanism of oncogenic enhancer formation in cancer
The translocations described above occur in ~80% of the BCL11B-deregulated group. The remaining 20% of cases lacked an identifiable translocation involving 14q32; instead, these cases harbored a high-copy amplification event of a relatively short (~2.5 kb) non-coding region located 730 kb downstream of BCL11B. We were not able to identify any cell type in which this region corresponded to a highly active enhancer, although weak H3K27ac of this amplified region was present in normal HSPCs and T cell progenitors. Using H3K27ac HiChIP, we discovered that this region contained extremely high levels of H3K27ac in patient samples with the amplification, which showed evidence of looping to the BCL11B gene. Using long-read PacBio sequencing, we confirmed that the 2.5 kb element was amplified in tandem, ruling out the possibility that multiple copies were scattered throughout the genome. We term this amplification “BCL11B Enhancer Tandem Amplification”, or BETA. Other reported cases of enhancer amplifications in acute leukemia have been described and primarily occur within the ~2 megabase MYC locus, resulting in MYC overexpression. These include duplication of the NOTCH1-driven enhancer (NMe) in T-ALL [55] and amplifications of a more distal enhancer cluster, originally called “E1–E5” [56] and later as the B cell Enhancer Cluster, or BENC [57], in AML [58,59]. To our knowledge, BETA represents the first description of oncogenic enhancer formation by amplification of a genomic region not previously demarcated with high levels of H3K27ac.
14q32 rearrangements in mixed-lineage leukemia
Although we formally identified the BCL11B-deregulated leukemia subgroup through unbiased genomic approaches, several prior reports described chromosomal rearrangements consistent with BCL11B-deregulating SVs: three case reports published between 1990 and 2003 described a total of 13 pediatric T/myeloid mixed-lineage leukemia cases with 14q32 translocations identified from karyotyping, most notably t(6;14)(q25;q32) [60–62]. BCL11B was subsequently identified as the likely target of these rearrangements through fluorescent in situ hybridization (FISH) analysis of bacterial artificial chromosome (BAC) probes [63], followed by description of 5 additional cases, all with t(6;14) and a T/myeloid leukemia phenotype, including the first reports in adult patients [64–66]. Abbas and colleagues were the first to suggest that BCL11B might be the target of aberrant gene regulatory activity following 14q32 rearrangements, and the first to report the striking co-incidence with FLT3 mutations [67]. The first translocation event not involving chromosome 6 was reported in 2018, with identification of two patients harboring rearrangement to 8q24 (i.e. distal to MYC) [68]. More recently, elevated BCL11B protein expression was identified in around 50% of T/myeloid MPAL cases examined in one study, although 14q32 rearrangements were only identified in a small subset [69]. The authors hinted at the possibility of other cryptic SVs that might upregulate BCL11B but which eluded karyotype and FISH analyses. Indeed, coincident with our study, Di Giacomo and colleagues reported on a further two 14q32 rearrangement events involving 2q22.2 (i.e. ZEB2), and 7q21.2 (i.e. CDK6), in addition to previously reported 8q24.2 and 6q25 [70]. The authors also identified that 14q32 rearrangements defined a unique gene expression group comprised of 20 T/myeloid MPAL, ETP-ALL and AML samples. Our study identified a further 3 previously undescribed translocation events, including 3p24.3 (near SATB1), 12p13.2 (within ETV6), and 21q22.12 (within RUNX1). Moreover, we identified the novel amplification event, BETA, which we now know accounts for 20% of BCL11B-deregulated cases. Because of the small size (35–50 kb after amplification), this event would likely escape detection by standard FISH analyses. Importantly, through mapping 3D chromatin contacts in patient samples we were able to uncover the mechanism by which 14q32 rearrangements deregulate BCL11B. Knowing that aberrant enhancer activity drives oncogenic BCL11B expression paves the way to investigate therapeutics that directly target this mechanism, for example by globally repressing enhancer function through bromodomain inhibitors [42], or by targeting the factors that mediate hijacked enhancer activity, which remain undefined.
Is a new entity of acute leukemia warranted?
Molecular similarities between acute leukemias with myeloid and T-lymphoid features have led to the proposal of new clinical entities termed “acute myeloid/T lymphoblastic leukemia” (AMTL) [13] or “interface acute leukemia” (IAL) [14] which would encompass all ETP-ALL and T/myeloid MPAL samples, as well as the subset of AML cases with T cell phenotypes (e.g. T cell receptor rearrangements), and the subset of T-ALL with myeloid phenotypes, irrespective of genomic driver. We argue that this broad classification is inappropriate, as it continues to give primacy to immunophenotype, rather than (genomic) leukemogenic drivers to guide classification, without providing useful insight into disease biology that may meaningfully impact diagnosis or therapy. As shown in multiple recent genomic studies of AML and ALL, designation of new subtypes should be determined by improved knowledge of the genomic drivers of disease, which are commonly distilled into distinct leukemic gene expression profiles. As we have shown, BCL11B-deregulating SVs effectively segregate 30–40% of T/myeloid MPAL and ETP-ALL into one biological group. Thus, classifying all ETP-ALL and T/myeloid MPAL into a single entity would mis-represent the impact of BCL11B SVs on driving distinct leukemia biology, particularly as other ETP-ALL and T/myeloid MPAL cases are driven by very different oncogenic events. Our analysis of outcome data suggested that ETP-ALL patients with BCL11B-deregulating SVs have better outcomes compared to those lacking these alterations, although these results require confirmation as the number of uniformly treated cases with outcome data was small. Future work will determine whether existing or novel therapeutic interventions will be effective against BCL11B-deregulated leukemia; however, based on the collective observations that BCL11B SVs are a recurrent, subgroup-defining genomic alteration that have not been identified in any cancer sample outside of this subgroup, we support the definition of 14q32 SVs targeting BCL11B as a new entity of acute leukemia. This recommendation is further supported by confirmation of the gene expression profile, characteristic immunophenotype and recurrent BCL11B alterations in an independent cohort of T, T/myeloid MPAL and ETP-ALL cases treated on Eastern Cooperative Oncology Group protocols.
Implications for the cell of origin of acute leukemias of ambiguous lineage
Multiple hypotheses have been advanced for the basis of lineage ambiguity/plasticity of lineage ambiguous leukemias. These include: 1) that oncogenic transformation occurs in a cell that retains myeloid and lymphoid differentiation potential (i.e. an HSPC) [71]; or 2) the cellular transformation, whether in a stem or committed progenitor, leads to oncogenic deregulation of gene expression programs that manifest, in part, as aberrant multi-lineage marker expression [72]; or 3) that transformation occurs in a committed T-lineage cell followed by de-/cross differentiation to acquire stem and myeloid markers [13]; or 4) that leukemia samples with multiple immunophenotypic populations are comprised of at least two genetically distinct and independently derived leukemias in a single sample; that is, that immunophenotypic variegation is driven by mutational evolution.
Several observations suggest the first hypothesis is most likely in many cases. The most direct observation is that immunophenotype may shift during disease progression, e.g. from ETP-ALL to AML or T-ALL, or B/myeloid ALL to B-ALL with retention of the truncal oncogenic drivers, as exemplified by cases with ZNF384 rearrangements [17,73]. This suggests that lineage plasticity is inherent to the leukemia, but does not exclude mutational evolution as a driver of lineage shift/switch. However, experimentally, we and others have shown that the same somatically-acquired mutations were present in immunophenotypically-defined subpopulations of MPAL samples harboring multiple subpopulations, including hematopoietic stem cells (HSCs) [12,74]; moreover, these subpopulations individually recapitulated the immunophenotypic diversity of the bulk tumor sample upon xenografting [12]. These data are consistent with a model where transformation occurs in an HSC or early progenitor that retains multi-lineage differentiation capacity. In our current work, epigenetic and gene expression analyses provide further support to an HSPC as the cell of origin (Figure 3). Namely, all 7 CD34+ HSPC super-enhancers implicated in 14q32 translocations showed enhancer activity (H3K27ac ChIP-seq signal) in patient tumor samples, including the ARID1B super-enhancer that was only active in HSPCs and not T cell progenitors. Additionally, the T cell enhancer elements ThymoD (which is required for normal BCL11B expression [75,76]) and NMe are only activated in recently immigrated thymic progenitor cells, yet these elements were inactive in all patient tumor samples. Assuming the activity status of these enhancers faithfully reflects the cell of origin, these data would support that transformation occurred in a cell that had yet to enter the thymus. Single cell analyses supported these inferences, with patient tumors showing the strongest similarity in both gene expression and open chromatin profile with activated HSPCs, not T lineage cells. The universal lack of T cell receptor rearrangements, which were present in 56.7% and 37.2% of non-BCL11B group ETP-ALL and T/myeloid MPAL samples, respectively, further ruled out the possibility that a T lineage cell underwent de-differentiation. These data support a model where 14q32 SVs occurring in an HSPC result in the aberrant activation of BCL11B to drive a lineage-ambiguous stem cell leukemia (Figure 3).
Figure 3. Model of BCL11B-mediated lineage ambiguous leukemia guided by epigenetic analysis.
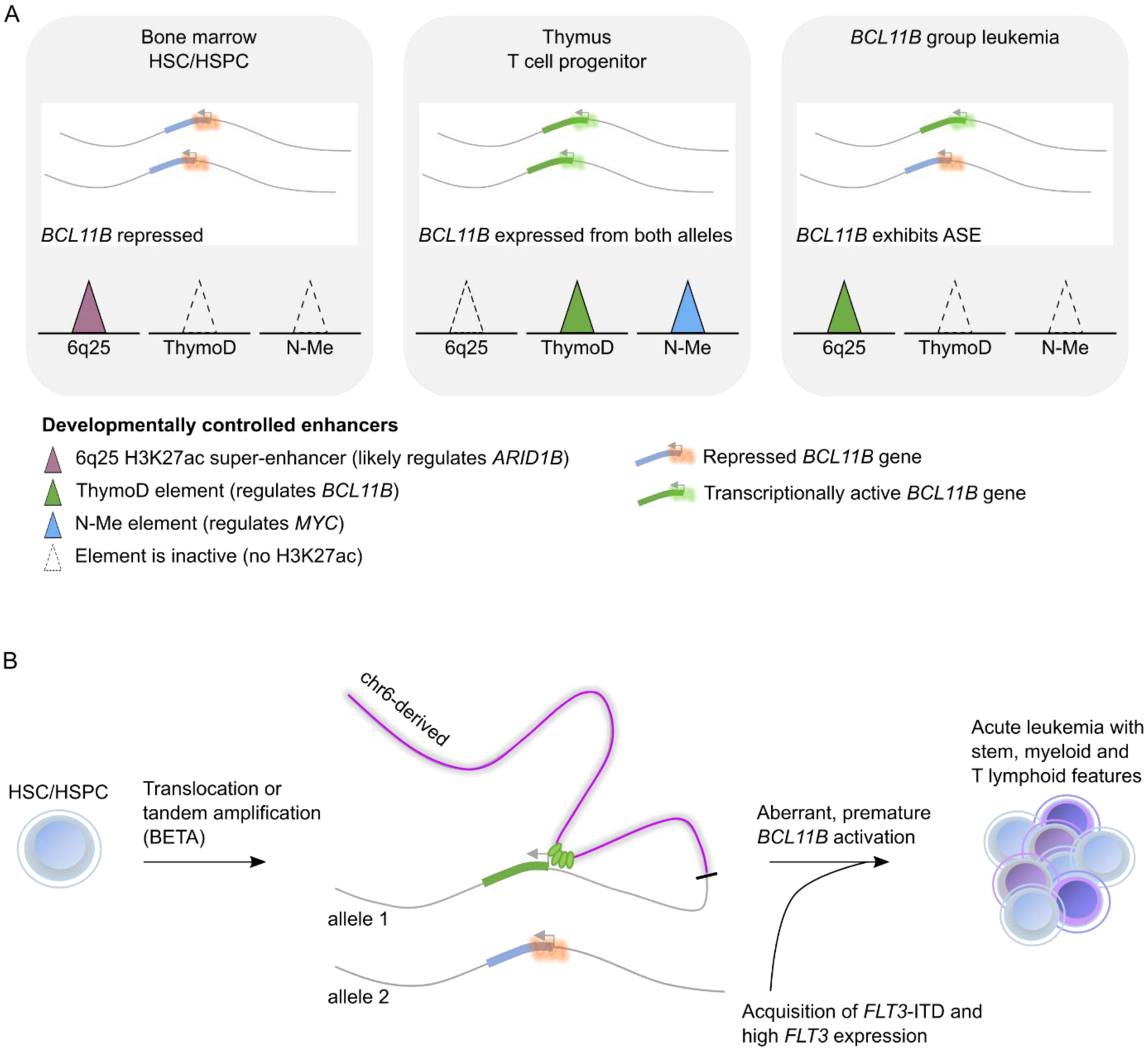
(A) representation of the two BCL11B alleles in normal and leukemic cells. In bone marrow HSCs/HSPCs (left panel), both alleles are actively repressed and the two T-lineage enhancers, ThymoD (which regulates BCL11B) and N-Me (which regulates MYC) are not active (dotted triangles), whereas the ARID1B enhancer with an unknown function is active (purple filled triangle). In normal T cell progenitors that have immigrated to the thymus (middle panel), BCL11B is expressed from both alleles, and the T lineage enhancers are active whereas the ARID1B enhancer no longer shows H3K27ac signal. In BCL11B-deregulated leukemia (right panel), BCL11B is only expressed from one allele, and the enhancer landscape reflects that of normal HSCs/HSPCs, suggesting BCL11B expression reflects an abnormal gene regulatory state. (B) Working model that depicts the mechanism of aberrant BCL11B expression. SVs result in enhancer hijacking (e.g. of the ARID1B enhancer depicted) or de novo enhancer formation (BETA). Together with constitutive FLT3 activation, either through mutation or upregulation, these genomic events drive development of a lineage ambiguous acute leukemia with stem, myeloid and T lineage features that result from a combination of the cell of origin (HSPC) and driving genomic lesion (BCL11B).
Implications for therapy
Does expression of both T and myeloid features in these leukemias reflect the differentiation capacity of HSPCs, or is this expression aberrant and rather reflects oncogenic transcription factor activity? Would distinguishing between these possibilities have implications for therapy, which relies primarily on the decision between AML- or ALL-directed regimens [5,77]? Using viral overexpression, we demonstrated that BCL11B is sufficient to upregulate T cell differentiation gene expression programs in CD34+ HSPCs which was accompanied by cytoplasmic CD3 detection in vitro, raising the possibility that the T lineage phenotype could be driven exclusively by BCL11B activity in an otherwise stem/myeloid leukemia.
If BCL11B-group leukemias are immature myeloid leukemias with aberrant T cell marker expression, might one expect that AML-directed therapy would be more effective compared to ALL-directed therapy? Multiple retrospective analyses support that ALL-directed regimens result in superior outcomes compared to AML regimens in MPAL patients[4,77,78]. Thus, even if the T lineage phenotypes characteristic of BCL11B group leukemias are truly aberrant (i.e. do not reflect the cell of origin), these phenotypes are not necessarily clinically irrelevant and might confer the requisite lymphoid characteristics that make ALL-directed therapy more effective in treating T/myeloid MPAL.
Despite the improved outcomes of patients treated with ALL- versus AML-directed therapy, overall survival of MPAL patients in general is poor and targeted therapies are greatly needed. Activating mutations in the FMS-like tyrosine kinase 3 gene, FLT3, were identified in 80.1% of BCL11B group samples. This frequency greatly exceeds that reported for AML, where FLT3 mutations are the most common alteration (15–30%) [79–83], as well as T/myeloid MPAL (11–37%)[12,16,84] and ETP-ALL (26–35%) [10,11,85]. This striking co-occurrence is notable, as it suggests these leukemias are dependent on FLT3 signaling and raises the possibility that FLT3 inhibitors might be therapeutically relevant in BCL11B-deregulated leukemia. The FLT3-specific tyrosine kinase inhibitor gilteritinib achieved Food and Drug Administration (FDA) approval for relapsed/refractory AML in 2018, and other non-selective FLT3/tyrosine kinase inhibitors have shown efficacy in multiple AML clinical trials [86–89]. No clinical trial has yet evaluated the efficacy of FLT3 inhibitors in T/myeloid MPAL or ETP-ALL, and case reports are scarce, although one report details the successful treatment of two adult T/myeloid MPAL patients with non-selective FLT3 inhibitors (midostaurin and sorafenib) [90].
Another promising therapeutic option includes the small molecule BH3 mimetics that block antiapoptotic activity in multiple leukemia cell types [91–93] (reviewed in ref. [94]). The finding that ETP-ALL is selectively dependent on the BCL-2 family of antiapoptotic proteins nominated the BCL-2-specific BH3 mimetic venetoclax for clinical use [95]. The first successful treatment of two patients with refractory ETP-ALL treated with venetoclax were reported in 2018 [96] and a recent study reported favorable outcomes in a phase I dose escalation study of venetoclax in combination with low-dose navitoclax, which targets both BCL-2 and BCL-XL, in relapsed/refractory T-ALL [97]. Notably, this study included 5 patients with ETP-ALL that had failed prior therapy, highlighting the potential of BCL-2 inhibitors in leukemias with an ETP immunophenotype. Similar to FLT3 inhibitors, venetoclax has yet to be tested in patients diagnosed with T/myeloid MPAL. Given the unknown genetic basis of most of these cases, genotype-response correlation of BH3 mimetics and FLT3 inhibitors is needed.
Conclusions
Recent transcriptomic and mutational profiling studies emphasize the limitations of using immunophenotype alone to classify individual leukemias with multi-lineage markers. The increasingly widespread use of clinical RNA and genome sequencing suggests that the diagnostic approach to classifying leukemia should be revisited, and specifically, these new entities of lineage ambiguous/overlap leukemias be incorporated. Indeed, recognition of the clinical importance of identification of such entities provides increasing support for adoption of such genomic diagnostic approaches. While identification of this entity required analysis of a very large data set [19,98], one can now readily identify the entity on a case-by-case basis. Moreover, the BCL11B entity may be suspected from the characteristic immunophenotype (Table 1).
Moving forward, it will be important to define clinical cases with 14q32 SVs to enable future evaluation of therapeutic approaches and outcomes. However, even with therapeutic strategies in practice today, outcomes of patients with ALAL remain poor, especially for adults, underscoring the need to develop faithful preclinical models that recapitulate the mixed-lineage phenotypes associated with these diseases. In conclusion, by focusing on genomic and transcriptional alterations, we have identified a clinically relevant demarcation in lineage-ambiguous leukemias that provides clarity on cellular origins and possible novel therapeutic approaches.
Funding
The authors are supported by the St. Jude Children’s Research Hospital Chromatin Collaborative, the Henry Schueler 41&9 Foundation (to C.G.M.), a St. Baldrick’s Foundation Robert H. Arceci Innovation award (to C.G.M.) and NCI NRSA F32 CA254140 (to L.E.M)
Declaration of competing interests
Lindsey Montefiori declares no conflict of interest. Charles Mullighan has received research funding from Abbvie, Pfizer and Loxo Oncology, and consulting fees from Illumina.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Owaidah TM, Al Beihany A, Iqbal MA, Elkum N, Roberts GT. Cytogenetics, molecular and ultrastructural characteristics of biphenotypic acute leukemia identified by the EGIL scoring system. Leukemia 2006;20:620–6. 10.1038/sj.leu.2404128. [DOI] [PubMed] [Google Scholar]
- [2].Weinberg OK, Arber DA. Mixed-phenotype acute leukemia: historical overview and a new definition. Leukemia 2010;24:1844–51. 10.1038/leu.2010.202. [DOI] [PubMed] [Google Scholar]
- [3].Yan L, Ping N, Zhu M, Sun A, Xue Y, Ruan C, et al. Clinical, immunophenotypic, cytogenetic, and molecular genetic features in 117 adult patients with mixed-phenotype acute leukemia defined by WHO-2008 classification. Haematologica 2012;97:1708–12. 10.3324/haematol.2012.064485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rubnitz JE, Onciu M, Pounds S, Shurtleff S, Cao X, Raimondi SC, et al. Acute mixed lineage leukemia in children: The experience of St Jude Children’s Research Hospital. Blood 2009;113:5083–9. 10.1182/blood-2008-10-187351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Matutes E, Pickl WF, Veer MVNT, Morilla R, Swansbury J, Strobl H, et al. Mixed-phenotype acute leukemia: Clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 2011;117:3163–71. 10.1182/blood-2010-10-314682. [DOI] [PubMed] [Google Scholar]
- [6].Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405. 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- [7].Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009;10:147–56. 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481:157–63. 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Noronha EP, Marques LVC, Andrade FG, Sardou-Cezar I, Santos-Bueno FV Dos, Da Paz Zampier C, et al. T-lymphoid/myeloid mixed phenotype acute leukemia and early T-cell precursor lymphoblastic leukemia similarities with NOTCH1 mutation as a good prognostic factor. Cancer Manag Res 2019;11:3933–43. 10.2147/CMAR.S196574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Neumann M, Coskun E, Fransecky L, Mochmann LH, Bartram I, Farhadi Sartangi N, et al. FLT3 Mutations in Early T-Cell Precursor ALL Characterize a Stem Cell Like Leukemia and Imply the Clinical Use of Tyrosine Kinase Inhibitors. PLoS One 2013;8:e53190. 10.1371/journal.pone.0053190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 2017;49:1211–8. 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018;562:373–9. 10.1038/s41586-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gutierrez A, Kentsis A. Acute myeloid/T-lymphoblastic leukaemia (AMTL): a distinct category of acute leukaemias with common pathogenesis in need of improved therapy. Br J Haematol 2018;180:919–24. 10.1111/bjh.15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bond J, Krzywon A, Ludovic Lhermitte, Roumier C, Roggy Anne, Belhocine M, et al. A transcriptomic continuum of differentiation arrest identifies myeloid interface acute leukemias with poor prognosis. Leukemia 2021;35:724–36. 10.1038/s41375-020-0965-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao W, Bharadwaj M, Levine M, Farnhoud N, Pastore F, Getta BM, et al. PHF6 and DNMT3A mutations are enriched in distinct subgroups of mixed phenotype acute leukemia with T-lineage differentiation. Blood Adv 2018;2:3526–39. 10.1182/bloodadvances.2018023531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Takahashi K, Wang F, Morita K, Yan Y, Hu P, Zhao P, et al. Integrative genomic analysis of adult mixed phenotype acute leukemia delineates lineage associated molecular subtypes. Nat Commun 2018;9:2670. 10.1038/s41467-018-04924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhong CH, Prima V, Liang X, Frye C, McGavran L, Meltesen L, et al. E2A-ZNF384 and NOL1-E2A fusion created by a cryptic t(12;19)(p13.3; p13.3) in acute leukemia. Leukemia 2008;22:723–9. 10.1038/sj.leu.2405084. [DOI] [PubMed] [Google Scholar]
- [18].Hirabayashi S, Ohki K, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017;102:118–29. 10.3324/HAEMATOL.2016.151035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Montefiori LE, Bendig S, Gu Z, Chen X, Polonen P, Ma X, et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage ambiguous stem cell leukemia. Cancer Discov 2021:candisc.0145.2021. 10.1158/2159-8290.CD-21-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 2019:1. 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Valk PJM, Verhaak RGW, Beijen MA, Erpelinck CAJ, van Doorn-Khosrovani SB van W, Boer JM, et al. Prognostically Useful Gene-Expression Profiles in Acute Myeloid Leukemia. N Engl J Med 2004;350:1617–28. 10.1056/nejmoa040465. [DOI] [PubMed] [Google Scholar]
- [22].Liu Y, Li C, Shen S, Chen X, Szlachta K, Edmonson MN, et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat Genet 2020;52:811–8. 10.1038/s41588-020-0659-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ha VL, Luong A, Li F, Casero D, Malvar J, Kim YM, et al. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia 2017;31:2503–14. 10.1038/leu.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kueh HY, Yui MA, Ng KKH, Pease SS, Zhang JA, Damle SS, et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol 2016;17:956–65. 10.1038/ni.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sidwell T, Rothenberg EV. Epigenetic Dynamics in the Function of T-Lineage Regulatory Factor Bcl11b. Front Immunol 2021;12. 10.3389/fimmu.2021.669498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hosokawa H, Romero-Wolf M, Yui MA, Ungerbäck J, Quiloan MLG, Matsumoto M, et al. Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat Immunol 2018;19:1427–40. 10.1038/s41590-018-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hosokawa H, Romero-Wolf M, Yang Q, Motomura Y, Levanon D, Groner Y, et al. Cell type–specific actions of Bcl11b in early T-lineage and group 2 innate lymphoid cells. J Exp Med 2020;217. 10.1084/jem.20190972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Albu DI, Feng D, Bhattacharya D, Jenkins NA, Copeland NG, Liu P, et al. BCL11B is required for positive selection and survival of double-positive thymocytes. J Exp Med 2007;204:3003. 10.1084/JEM.20070863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, et al. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science (80-) 2010;329:85–9. 10.1126/science.1188063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science (80-) 2010;329:89–93. 10.1126/science.1188989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wakabayashi Y, Inoue J, Takahashi Y, Matsuki A, Kosugi-Okano H, Shinbo T, et al. Homozygous deletions and point mutations of the Rit1/Bcl11b gene in γ-ray induced mouse thymic lymphomas. Biochem Biophys Res Commun 2003;301:598–603. 10.1016/S0006-291X(02)03069-3. [DOI] [PubMed] [Google Scholar]
- [32].De Keersmaecker K, Real PJ, Gatta G Della, Palomero T, Sulis ML, Tosello V, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med 2010;16:1321–7. 10.1038/nm.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 2011;118:4169–73. 10.1182/blood-2010-11-318873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bartram I, Gökbuget N, Schlee C, Heesch S, Fransecky L, Schwartz S, et al. Low expression of T-cell transcription factor BCL11b predicts inferior survival in adult standard risk T-cell acute lymphoblastic leukemia patients. J Hematol Oncol 2014;7:1–10. 10.1186/s13045-014-0051-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Grabarczyk P, Przybylski GK, Depke M, Völker U, Bahr J, Assmus K, et al. Inhibition of BCL11B expression leads to apoptosis of malignant but not normal mature T cells. Oncogene 2007;26:3797–810. 10.1038/sj.onc.1210152. [DOI] [PubMed] [Google Scholar]
- [36].Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nat 2014 5117510 2014;511:428–34. 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015;161:1012–25. 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009;459:108–12. 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL, et al. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat Biotechnol 2015;33:345–6. 10.1038/nbt.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].ENCODE Project Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang X, Jeong M, Huang X, Wang XQ, Wang X, Zhou W, et al. Large DNA Methylation Nadirs Anchor Chromatin Loops Maintaining Hematopoietic Stem Cell Identity. Mol Cell 2020;78:506–521.e6. 10.1016/j.molcel.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes 2013. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed]
- [44].Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 2016. 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Riedel SS, Lu C, Xie HM, Nestler K, Vermunt MW, Lenard A, et al. Intrinsically disordered Meningioma-1 stabilizes the BAF complex to cause AML. Mol Cell 2021;81:2332–2348.e9. 10.1016/j.molcel.2021.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 2014;25:415–27. 10.1016/j.ccr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gröschel S, Sanders MA, Hoogenboezem R, De Wit E, Bouwman BAM, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in Leukemia. Cell 2014;157:369–81. 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- [48].Schwartz JR, Ma J, Kamens J, Westover T, Walsh MP, Brady SW, et al. The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat Commun 2021;12. 10.1038/s41467-021-21255-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yang M, Safavi S, Woodward EL, Duployez N, Olsson-Arvidsson L, Ungerbäck J, et al. 13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking. Blood 2020;136:946–56. 10.1182/blood.2019004684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016;16:494–507. 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- [51].Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985;318:533–8. 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- [52].Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic Mechanisms in Burkitt Lymphoma. Cold Spring Harb Perspect Med 2014;4. 10.1101/CSHPERSPECT.A014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood 2009;114:2688–98. 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- [54].Harvey RC, Mullighan CG, Chen I-M, Wharton W, Mikhail FM, Carroll AJ, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010;115:5312. 10.1182/BLOOD-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014;20:1130–7. 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe J-S, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 2013;27:2648–62. 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bahr C, von Paleske L, Uslu VV., Remeseiro S, Takayama N, Ng SW, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018;553:515–20. 10.1038/nature25193. [DOI] [PubMed] [Google Scholar]
- [58].Radtke I, Mullighan CG, Ishii M, Su X, Cheng J, Ma J, et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci U S A 2009;106:12944–9. 10.1073/pnas.0903142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kühn MWM, Radtke I, Bullinger L, Goorha S, Cheng J, Edelmann J, et al. High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 2012;119:e67–75. 10.1182/blood-2011-09-380444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hayashi Y, Pui C, Behm F, Fuchs A, Raimondi S, Kitchingman G, et al. 14q32 translocations are associated with mixed-lineage expression in childhood acute leukemia. Blood 1990;76:150–6. 10.1182/blood.v76.1.150.150. [DOI] [PubMed] [Google Scholar]
- [61].Batanian JR, Dunphy CH, Gale G, Havlioglu N. Is t(6;14) a non-random translocation in childhood acute mixed lineage leukemia? Cancer Genet Cytogenet 1996;90:29–32. 10.1016/0165-4608(96)00030-1. [DOI] [PubMed] [Google Scholar]
- [62].Wu SQ, Kuo J, Chen XR, Chen SA, Quinn JJ. Translocation (6;14) in childhood acute mixed lineage leukemia [3]. Cancer Genet Cytogenet 2003;141:178–9. 10.1016/S0165-4608(02)00790-2. [DOI] [PubMed] [Google Scholar]
- [63].Bezrookove V, Van Zelderen-Bhola SL, Brink A, Szuhai K, Raap AK, Barge R, et al. A novel t(6;14)(q25~q27;q32) in acute myelocytic leukemia involves the BCL11B gene. Cancer Genet Cytogenet 2004;149:72–6. 10.1016/S0165-4608(03)00302-9. [DOI] [PubMed] [Google Scholar]
- [64].Georgy M, Yonescu R, Griffin CA, Batista DAS. Acute mixed lineage leukemia and a t(6;14)(q25;q32) in two adults. Cancer Genet Cytogenet 2008;185:28–31. 10.1016/j.cancergencyto.2008.04.010. [DOI] [PubMed] [Google Scholar]
- [65].Zhao XF, Gojo I, York T, Ning Y, Baer MR. Diagnosis of biphenotypic acute leukemia: A paradigmatic approach. Int J Clin Exp Pathol 2010;3:75–86. [PMC free article] [PubMed] [Google Scholar]
- [66].Kobayashi S, Taki T, Nagoshi H, Chinen Y, Yokokawa Y, Kanegane H, et al. Identification of novel fusion genes with 28S ribosomal DNA in hematologic malignancies. Int J Oncol 2014;44:1193–8. 10.3892/ijo.2014.2291. [DOI] [PubMed] [Google Scholar]
- [67].Abbas S, Sanders MA, Zeilemaker A, Geertsma-Kleinekoort WMC, Koenders JE, Kavelaars FG, et al. Integrated genome-wide genotyping and gene expression profiling reveals BCL11B as a putative oncogene in acute myeloid leukemia with 14q32 aberrations. Haematologica 2014;99:848–57. 10.3324/haematol.2013.095604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pallavajjala A, Kim D, Li T, Ghiaur G, Jones RJ, Burns KH, et al. Genomic characterization of chromosome translocations in patients with T/myeloid mixed-phenotype acute leukemia. Leuk Lymphoma 2018;59:1231–8. 10.1080/10428194.2017.1372577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang W, Beird H, Kroll CJ, Hu S, Bueso-Ramos CE, Fang H, et al. T(6;14)(q25;q32) involves BCL11B and is highly associated with mixed-phenotype acute leukemia, T/myeloid. Leukemia 2020;34:2509–12. 10.1038/s41375-020-0761-9. [DOI] [PubMed] [Google Scholar]
- [70].Di Giacomo D, La Starza R, Gorello P, Pellanera F, Kalender Atak Z, De Keersmaecker K, et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T and myeloid immature acute leukemia. Blood 2021. 10.1182/blood.2020010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Greaves MF, Chan LC, Furley AJ, Watt SM, Molgaard HV. Lineage promiscuity in hemopoietic differentiation and leukemia. Blood 1986;67:1–11. [PubMed] [Google Scholar]
- [72].Smith L, McCulloch E. Lineage infidelity following exposure of T lymphoblasts (MOLT-3 cells) to 5-azacytidine. Blood 1984;63:1324–30. 10.1182/blood.V63.6.1324.1324. [DOI] [PubMed] [Google Scholar]
- [73].Oberley MJ, Gaynon PS, Bhojwani D, Pulsipher MA, Gardner RA, Hiemenz MC, et al. Myeloid lineage switch following chimeric antigen receptor T-cell therapy in a patient with TCF3-ZNF384 fusion-positive B-lymphoblastic leukemia. Pediatr Blood Cancer 2018;65. 10.1002/pbc.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kotrova M, Musilova A, Stuchly J, Fiser K, Starkova J, Mejstrikova E, et al. Distinct bilineal leukemia immunophenotypes are not genetically determined. Blood 2016;128:2263–6. 10.1182/blood-2016-07-725861. [DOI] [PubMed] [Google Scholar]
- [75].Li L, Zhang JA, Dose M, Kueh HY, Mosadeghi R, Gounari F, et al. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood 2013;122:902–11. 10.1182/blood-2012-08-447839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, et al. Non-Coding Transcription Instructs Cohesin-Dependent Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 2017;171:103. 10.1016/J.CELL.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Maruffi M, Sposto R, Oberley MJ, Kysh L, Orgel E. Therapy for children and adults with mixed phenotype acute leukemia: A systematic review and meta-analysis. Leukemia 2018;32:1515–28. 10.1038/s41375-018-0058-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mejstrikova E, Volejnikova J, Fronkova E, Zdrahalova K, Kalina T, Sterba J, et al. Prognosis of children with mixed phenotype acute leukemia treated on the basis of consistent immunophenotypic criteria. Haematologica 2010;95:928–35. 10.3324/haematol.2009.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Network TCGAR. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med 2013;368:2059–74. 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 2014;28:1586–95. 10.1038/leu.2014.55. [DOI] [PubMed] [Google Scholar]
- [81].Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21. 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Tarlock K, Alonzo TA, Gerbing RB, Raimondi SC, Hirsch BA, Sung L, et al. Gemtuzumab ozogamicin reduces relapse risk in FLT3/ITD acute myeloid leukemia: A report from the children’s oncology group. Clin Cancer Res 2016;22:1951–7. 10.1158/1078-0432.CCR-15-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 2018;24:103–12. 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Eckstein OS, Wang L, Punia JN, Kornblau SM, Andreeff M, Wheeler DA, et al. Mixed-phenotype acute leukemia (MPAL) exhibits frequent mutations in DNMT3A and activated signaling genes. Exp Hematol 2016;44:740–4. 10.1016/j.exphem.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Neumann M, Heesch S, Schlee C, Schwartz S, Gökbuget N, Hoelzer D, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 2013;121:4749–52. 10.1182/blood-2012-11-465138. [DOI] [PubMed] [Google Scholar]
- [86].Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005;105:54–60. 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- [87].Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol 2015;16:1691–9. 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- [88].Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol 2017;18:1061–75. 10.1016/S1470-2045(17)30416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017;377:454–64. 10.1056/nejmoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Andrews C, Lam W, Sibai H. The successful use of FLT3 inhibitors in FLT3-positive mixed phenotype acute leukemia. Leuk Lymphoma 2020;61:3275–7. 10.1080/10428194.2020.1802451. [DOI] [PubMed] [Google Scholar]
- [91].Tsao T, Shi Y, Kornblau S, Lu H, Konoplev S, Antony A, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol 2012;91:1861–70. 10.1007/s00277-012-1537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008;68:3421–8. 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- [93].Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 2012;30:488–96. 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Leverson JD, Sampath D, Souers AJ, Rosenberg SH, Fairbrother WJ, Amiot M, et al. Found in translation: How preclinical research is guiding the clinical development of the BCL2-selective inhibitor venetoclax. Cancer Discov 2017;7:1376–93. 10.1158/2159-8290.CD-17-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, et al. Maturation Stage of T-cell Acute Lymphoblastic Leukemia Determines BCL-2 versus BCL-XL Dependence and Sensitivity to ABT-199. Cancer Discov 2014;4:1074–87. 10.1158/2159-8290.CD-14-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Numan Y, Alfayez M, Maiti A, Alvarado Y, Jabbour EJ, Ferrajoli A, et al. First Report of Clinical Response to Venetoclax in Early T-Cell Precursor Acute Lymphoblastic Leukemia. JCO Precis Oncol 2018:1–6. 10.1200/po.18.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Pullarkat VA, Lacayo NJ, Jabbour E, Rubnitz JE, Bajel A, Laetsch TW, et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov 2021;11:1440–53. 10.1158/2159-8290.CD-20-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Di Giacomo D, La Starza R, Gorello P, Pellanera F, Kalender Atak Z, De Keersmaecker K, et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T and myeloid immature acute leukemia. Blood 2021. 10.1182/blood.2020010510. [DOI] [PMC free article] [PubMed] [Google Scholar]