

Abstract
Background
Stress has emerged as an important risk factor for heart disease in women. Stress levels have been shown to correlate with delayed recovery and increased mortality after a myocardial infarction. Therefore, we sought to investigate if the observed sex‐specific effects of stress in myocardial infarction may be partly attributed to genomic interactions between the female sex hormones, estrogen (E2), and the primary stress hormones glucocorticoids.
Methods and Results
Genomewide studies show that glucocorticoids inhibit estrogen‐mediated regulation of genes with established roles in cardiomyocyte homeostasis. These include 5‐HT2BR (cardiac serotonin receptor 2B), the expression of which is critical to prevent cardiomyocyte death in the adult heart. Using siRNA, gene expression, and chromatin immunoprecipitation assays, we found that 5‐HT2BR is a primary target of the glucocorticoid receptor and the estrogen receptor α at the level of transcription. The glucocorticoid receptor blocks the recruitment of estrogen receptor α to the promoter of the 5‐HT2BR gene, which may contribute to the adverse effects of stress in the heart of premenopausal women. Using immunoblotting, TUNEL (terminal deoxynucleotidal transferase–mediated biotin–deoxyuridine triphosphate nick‐end labeling), and flow cytometry, we demonstrate that estrogen decreases cardiomyocyte death by a mechanism relying on 5‐HT2BR expression. In vitro and in vivo experiments show that glucocorticoids inhibit estrogen cardioprotection in response to hypoxia/reoxygenation injury and exacerbate the size of the infarct areas in myocardial infarction.
Conclusions
These results established a novel mechanism underlying the deleterious effects of stress on female cardiac health in the setting of ischemia/reperfusion.
Keywords: cardiomyocytes, estrogen, gene regulation, glucocorticoids, serotonin receptor 2b
Subject Categories: Cardiomyopathy, Heart Failure
Nonstandard Abbreviations and Acronyms
- 5‐HT2B
5‐HTR2 subtype B
- 5‐HT2BR
cardiac serotonin receptor 2B
- ER
estrogen receptor
- EREs
estrogen responsive elements
- ERα
estrogen receptor α
- ERβ
estrogen receptor β
- GR
glucocorticoid receptor
- GREs
glucocorticoid responsive elements
- I/R
ischemia/reperfusion
- NTC
nontarget control
- SLC25A4
solute carrier family 25
Clinical Perspective
What Is New?
Women are more reactive to the effects of mental stress, yet the mechanisms underlying the harmful effects of stress are unknown.
Increased mental stress has been shown to correlate with delayed recovery and increased mortality after a myocardial infarction in women.
In our study, we proposed for the first time a novel mechanism whereby stress negatively influences cardiac outcomes in women; we found that the genomic interactions between estrogen and glucocorticoids mediate the adverse effects of stress on the female heart.
What Are the Clinical Implications?
This study highlights that stress has profound effects on the female heart by inhibiting estrogen cardioprotection.
Our findings suggest that decreasing stress signaling in the female heart may provide a therapeutic approach to prevent/improve cardiac outcomes after a heart attack.
Cardiovascular disease is a leading cause of death for both men and women; however, cardiovascular disease incidence and mortality have increased significantly in young women (<50 years old) compared with men of the same age. 1 Although cardiovascular disease risk factors (eg, smoking, hypertension, diabetes mellitus, central adiposity, diet, physical activity, alcohol consumption, lipids, and psychosocial factors) are considered to be the same for both men and women, the effects of these risk factors may differ between the sexes. 2 Clinical data show that women are more susceptible to psychological factors, such as depression, anxiety, and trauma. 3 , 4 , 5 , 6 , 7 , 8 , 9 The stress associated with these psychological factors has become a good predictor for cardiovascular risk in young women. 10 , 11 , 12 , 13 Recent studies by Vaccarino et al showed that mental stress has a significant effect on cardiovascular risk and outcomes in young women with early‐onset ischemic heart disease. 1 However, the molecular mechanisms underlying the deleterious effects of mental stress on cardiac health are unknown.
In the present study, we investigated the effects of glucocorticoids on estrogen transcriptional responses in cardiomyocytes. We found that the combination of synthetic glucocorticoid dexamethasone and estradiol (the most abundant estrogen in premenopausal women) leads to significant changes in estrogen‐regulated gene expression. Our data show that estrogen regulation of several serotonin receptor (5‐HTR) subtypes is inhibited by cotreatment with glucocorticoids. Among these receptors, we identified 5‐HT2BR (cardiac serotonin receptor 2B), which has been shown to be a critical survival factor in cardiomyocytes. 14 5‐HTR2B gene expression is directly regulated by estrogen via binding of the ERα (estrogen receptor α) to a newly identified estrogen responsive element (ERE) located in the promoter of the 5‐HTR2B gene. This regulation is blocked by the glucocorticoid receptor (GR) binding to the same genomic element and blocking gene transcription in the presence of both dexamethasone and estrogen. Moreover, our results show that hormone cotreatment has profound effects on cardiomyocyte viability in response to hypoxia/reoxygenation injury and that some of the cardioprotective effects of estrogen are in part mediated via the regulation of the 5‐HTR2B gene in cardiomyocytes. Moreover, in vivo data showed that chronic elevation in systemic corticosterone levels blocks the natural protection of estrogen to ischemia/reperfusion (I/R) injury. These effects correlate with the levels of 5‐HTR2B expression in the heart. Together, these findings suggest that downregulation of the estrogen‐dependent 5‐HTR2B gene expression by the coactivation of GR may be 1 of the underlying mechanisms that exacerbate cardiomyocyte death and progression to heart failure in women after a heart attack.
Methods
Data Availability Disclosure Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.
Reagents and Antibodies
Dexamethasone (1,4‐pregnadien‐9α‐fluoro‐16α‐methyl‐11β, 17, 21‐triol‐3, 20‐dione; ≥98% by thin‐layer chromatography) and estrogen (17‐β‐estradiol; ≥98% thin‐layer chromatography) were purchased from Steraloids (Newport, RI). Cycloheximide was purchased from Millipore Sigma (St. Louis, MO). The anti‐5‐HTR2B (5‐hydroxytryptamine receptor 2B) antibody, anti‐ERα, and anti‐ERβ antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). The anti‐GR antibody was purchased from Cell Signaling (Danvers, MA). The anti‐G protein‐coupled receptor 30 (GPR30) and anti‐GAPDH antibodies were purchased from Abcam (Boston, MA).
The estrogen receptor (ER) antagonists ICI (182 780; fulvestrant) and PHTPP (2‐phenyl‐3‐(4‐hydroxyphenyl)‐5,7‐bis (trifluoromethyl)‐pyrazolo[1,5‐a] pyrimidine,4‐[2‐phenyl‐5,7 bis(trifluoromethyl)pyrazolo[1,5‐a]‐pyrimidin‐3‐yl]phenol) were purchased from Millipore Sigma, and G‐15 ([3aS,4R,9bR]‐4‐[6‐bromo‐1,3‐benzodioxol‐5‐yl]‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta[c]quinoline) was purchased from Cayman Chemical (Ann Arbor, MI).
Animals
For in vivo experiments, 2‐month‐old intact and gonadectomized C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). At 1 week after surgery, sham and gonadectomized mice were shipped to our facility, and upon arrival, all animals were maintained in accordance with the Louisiana State University Health Sciences Center Shreveport directives for the care and use of laboratory animals. Mice were treated as previously described. 15 , 16 Briefly, dexamethasone was dissolved in saline with sonication, and estrogen was prepared by first dissolving in 100% ethanol and then diluting in saline. Mice were treated with saline, 1 mg/kg of dexamethasone, 10 μg/kg of estrogen, or dexamethasone (1 mg/kg)+estrogen (10 μg/kg) via intraperitoneal injection. At 6 hours after the treatment, mice were euthanized by cervical dislocation and hearts were harvested for RNA and protein extraction.
For the corticosterone studies, 2‐month‐old intact female mice were housed in groups of 4 to synchronize their estrous cycle. Mice were placed under corticosterone treatment (2.5 μg/mL in water) or regular drinking water for 3 months as previously described. 17 Serum corticosterone were measured using the commercially available Corticosterone ELISA kit (Arbor Assays, Ann Arbor, MI) as previously described. 17 Vaginal smear cytology was used to determine the estrous cycle phases as previously described. 18 Before the I/R procedure, smears were done, and mice were classified according to their phase on the estrous cycle. Mice were subjected to I/R at the estrus (low basal estradiol levels) and proestrus (high estradiol levels) phases.
I/R injury was performed following the protocol described by Xu et al. 19 Briefly, each mouse individually was placed in an induction chamber and anesthetized using 5% isoflurane and oxygen with a flow rate of 0.4 L/minute until loss of righting reflex and then maintain the animal with 2% isoflurane in 100% oxygen with a flow of 0.4 L/minute using a nosecone tube connected to the anesthesia apparatus until the tracheal tube was inserted. Following intubation, the animal was placed on a surgical platform, and the chest was shaved and prepared with betadine and alcohol before the chest incisions were made. The left anterior descending artery was located on the surface of the heart through a dissection microscope and ligated by passing a 6‐0 silk suture underneath the left anterior descending artery and making a loose double knot with the suture, leaving a 2‐mm to 3‐mm diameter loop through which a 2‐mm to 3‐mm long piece of polyethylene tubing size 10 (PE‐10) tubing was placed. The loop was tightened around the artery and tubing. The occlusion of the left anterior descending artery was confirmed by the appearance of a paler color in the anterior wall of the left ventricle. After 60 minutes of the ischemia period, the knot was untied, and the PE‐10 tubing was removed. After confirming reperfusion by the appearance of a pink‐red color after 15 to 20 seconds. The chest cavity was then closed by sewing shut the incision in the third intercostal space with 4‐0 silk suture. When suturing was completed, the isoflurane flow was ceased, the mouse was removed from the ventilator, and the tube was carefully removed. The mouse was observed for 5 minutes and then returned to the cage for recovery. Animals were continuously monitored for any pain of distress during the 48 hours of reperfusion. These studies were approved by the Louisiana State University Health Sciences Center's Animal Care and Use Committee.
Heart Histology
Mice were euthanized by overdosing with isoflurane, and whole hearts were sliced using an acrylic mouse heart slicer matrix. Coronal sections (~1.0 mm) obtained after cutting were used for different experimental protocols. The second and third slices from the apex were used for percentage of tetrazolium chloride staining and other histological protocols, respectively, which was kept consistent throughout for all the animals. The slices for tetrazolium chloride staining were then incubated in 1% tetrazolium chloride at a temperature of 37 ℃ for 10 minutes, followed by scanning under the same settings by the placement of the slices between 2 glass slides. The red color indicated live tissue, whereas white/pale areas represented the infarct area. Infarct areas were quantified using ImageJ color threshold mode to differentiate infarcted from viable tissue (https://imagej.nih.gov/ij/docs/menus/analyze.html). The heart slices obtained for the histological protocols were transferred to histological cassettes (Leica Biosystems 3802765) and fixed in 10% formalin overnight. Formalin‐fixed, paraffin‐embedded tissue blocks were sectioned at 5 µm, deparaffinized in xylene, hydrated in a graded ethanol series, rinsed with distilled water, and washed with PBS. Sections were then stained for hematoxylin‐eosin and Masson trichrome staining.
Cell Culture and Hormone Treatment
The cardiomyocyte cell line HL‐1 was purchased from Millipore Sigma and cultured in claycomb medium supplemented with 10% fetal bovine serum, 100 U/L penicillin/streptomycin, 0.1 mmol/L norepinephrine, and 2 mmol/L L‐glutamine. 20 All treatments of cells were performed in serum‐free complete media unless otherwise specified. Cells were starved of serum overnight and washed 3 times with PBS before all the hormone assays. HL‐1 cardiomyocytes were treated with vehicle (PBS), 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone (100 nmol/L)+estrogen (10 nmol/L) as previously described 15 and incubated with each hormone combination for the time points indicated in each figure legend. For all experiments, cells were pretreated with estrogen for 1 hour to mimic the in vivo situation. The doses selected for this study are based on publications by Whirledge et al 15 and our laboratory experience. 17 Estrogen and dexamethasone concentrations at 10 and 100 nmol/L, respectively, were found to lead to significant biological effects in cell lines that mimic physiological changes in mice. 15 , 16
RNA Extraction and Quantitative Real Time‐Polymerase Chain Reaction
Total RNA was isolated from tissues and cells using the RNeasy Mini Kit and RNase‐Free DNase Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions with the deoxyribonuclease treatment performed on the column. RNA purity and yield were assessed by evaluating the A260/A280 ratio and concentration using the NanoDrop One Spectrophotometer (ThermoFisher Scientific, Waltham, MA). The One‐Step RT‐PCR Universal Master Mix reagent (ThermoFisher Scientific) was used to quantify mRNA levels of the selected target genes. Quantitative real‐time polymerase chain reaction (PCR) was performed with the CFX96 Real‐Time System C1000 Touch Thermal Cycler (Bio‐Rad, Hercules, CA) using predesigned primer–probe sets (ThermoFisher Scientific) for 5‐HTR2B (Mm00434123_m1), SLC25A4 (solute carrier family 25), also known as ANT‐1 (adenine nucleotide translocase 1; Mm01207393_m1), and the reference gene PPIB (peptidylprolyl isomerase B; Mm00478295_m1) in a 10‐μL reaction volume. The thermocycling parameters for each reaction were 48 ℃ for 30 minutes and 95 ℃ for 10 minutes followed by 40 cycles of 95 ℃ for 15 seconds and 60 ℃ for 60 seconds. Values measured for each primer/probe set were normalized to PPIB.
Western Blotting Analysis
Heart tissue and cells were homogenized and lysed in Tris glycine SDS sample buffer (Invitrogen, Waltham, MA) supplemented with 2.5% β‐mercaptoethanol as previously described. 21 Membranes with equivalent amounts of protein were incubated with primary antibodies and developed using the ChemiDoc Imaging System (Bio‐Rad).
RNA Interference and Cycloheximide Experiments
Nontarget control (NTC), GR, ERα and ERβ, and GPR30 siRNAs were purchased from SMARTpool siRNA (ThermoScientific). HL‐1 cells were transfected with 50 nmol/L of each siRNA using Dharmafect1 transfection reagent (ThermoScientific). At 48 hours after transfection, cells were replated and treated with vehicle (PBS), 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone (100 nmol/L)+estrogen (10 nmol/L). RNA was isolated from these cells 6 hours following hormone treatments. 5‐HTR2B mRNA levels were measured by quantitative real‐time PCR and compared with NTC.
For the cycloheximide experiments, HL‐1 cells were treated with 10 μm cycloheximide for 3 hours before hormone treatment. Cells were then administered with 100 nm dexamethasone, 10 nm estrogen, or dexamethasone (100 nmol/L)+estrogen (10 nmol/L) for 6 hours, and 5‐HTR2B mRNA levels were measured by quantitative real‐time PCR and compared with vehicle.
Promoter Analysis of the 5‐HTR2B Gene
The JASPAR Database (http://jaspar.genereg.net) and Ensembl (https://useast.ensembl.org/index.html) were used to identify predicted GR and ERα binding sites within 5000 bp upstream of the transcriptional start site and 700 bp downstream of the transcriptional start site. Sequences were scanned at a profile score threshold of 80% and 85%.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation assays were performed in HL‐1 cells following a previously described protocol. 15 , 21 , 22 Briefly, HL‐1 cells were plated on 150‐mm dishes in 30 mL of medium supplemented with 10% dextran‐coated, charcoal‐stripped fetal calf serum and grown to 90% confluence. Cells were starved of serum overnight and washed 3 times with PBS before all hormone assays. Cells were treated with vehicle or hormone treatment for 3 hours. Cells were then scraped, resuspended in cell lysis buffer (50 mM HEPES‐KOH at pH 8, 1 mM EDTA, 140 mM NaCl, 10% glycerol, 0.5% NP‐40, 0.25% Triton X‐100, protease inhibitor cocktail), and subjected to nutation for 30 minutes at 4 ℃. The crude nuclei were collected by centrifugation (600g for 5 minutes at 4 ℃), resuspended in shearing buffer (10 mM Tris‐HCl at pH 8, 1 mM EDTA, 140 mM NaCl, 1% SDS, 0.1% sodium deoxycholate, 1% Triton X‐100, protease inhibitor cocktail), and sonicated using a Branson Sonifier 150 at setting 4. Sheared chromatin was precleared with rabbit immunoglobulin G and protein A agarose/salmon sperm DNA (Millipore, Billerica, MA) and then immunoprecipitated overnight with 10 μL of anti‐GR (Cell Signaling Technology), 10 μl of anti‐ERα antibody (Santa Cruz Biotechnology), and 2 μL of immunoglobulin G (Millipore). After elution of protein:DNA complexes and DNA purification, PCR analysis was performed on immunoprecipitated and input DNA. PCR analysis of the glucocorticoid response elements (GREs) and estrogen response elements (EREs) of 5‐HTR2B used the primers shown in the Results section. Percentage input was calculated using the following equations: adjusted input 100%=(Ct input−6.64) and 100*2(Adjusted input−Ct [input]).
Microarray Studies
HL‐1 cells were treated with vehicle (PBS), 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone (100 nmol/L)+estrogen (10 nmol/L) for 6 hours, and total RNA from each treatment was isolated. Gene expression analysis was conducted as previously described 21 using Agilent Whole Mouse Genome 4×44 multiplex format oligo arrays (014868; Agilent Technologies, Santa Clara, CA) following the Agilent 1‐color microarray‐based gene expression analysis protocol. Starting with 500 ng of total RNA, Cy3‐labeled cRNA was produced per the manufacturer’s protocol. For each sample, 1.65 µg of Cy3‐labeled cRNAs was fragmented and hybridized for 17 hours in a rotating hybridization oven. Slides were washed and then scanned with an Agilent Scanner. Data were obtained using the Agilent Feature Extraction software (version 12) using the 1‐color defaults for all parameters. The Agilent Feature Extraction Software performed error modeling, adjusting for additive and multiplicative noise. To identify differentially expressed probes, an ANOVA was used to determine if there was a statistical difference between the means of groups. Using OmicSoft Array Studio (version 9.0) software (Qiagen) we compared treatment group vehicle versus hormone treatments (dexamethasone, estrogen, and dexamethasone+estrogen) using ANOVA analyses with a P value cutoff of P<0.05. The microarray data are available in the Gene Expression Omnibus repository at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE143461).
A heat map was generated using Heatmapper Online Software (http://www2.heatmapper.ca). The lists of probe sets generated were visually sorted by using a Venn diagram generator and further analyzed with Pathway Analysis version 6.5 (Ingenuity Systems, Redwood City, CA).
Hypoxia/Reoxygenation Injury Model and Cell Viability
To induce an ischemic injury, vehicle‐treated and hormone‐treated HL‐1 cells were incubated for 17 hours in an anaerobic chamber (Coy Hypoxic Chamber, Coy, Grass Lake, MI) with 1% oxygen levels (ischemia phase) in hypoxic media (1 mM KH2PO4, 10 mM NaHCO3, 1.2 mM MgCl2, 25 mM HEPES, 74 mM NaCl, 16 mM KCl, 1.2 mM CaCl2, 20 mM sodium lactate at pH 6.2). Cells were pretreated for 6 hours with vehicle or hormones before subjecting them to hypoxia. The media was replaced, and hormones at the same concentrations were added to the hypoxia buffer. The hypoxia challenge was done in the presence and absence of hormones. Following hypoxia, cells were reoxygenated (reperfusion phase) for 2 hours in normoxic medium at 95% oxygen/37 ℃. The time control group consisted of cells without the hypoxic stimulus kept in complete medium (claycomb medium, 2 mmol/L L‐glutamine, 100 U/L antibiotics, and 0.1 mM norepinephrine) in a normoxic incubator at 37 ℃. Cell viability was measured by assessing plasma membrane integrity by flow cytometry analysis of 10 μg/mL of propidium iodide (Invitrogen) exclusion. A total of 10 000 cells were analyzed using a BD LSRII flow cytometer (San Jose, CA) equipped with FACSDiVa software. Cells were excited with a 561‐nm laser, and propidium iodide fluorescence was detected at 585 nm. A gate was drawn on a propidium iodide histogram for the control sample to determine the percent of viable and dead experimental cells. The effect of hypoxia‐induced and reoxygenation‐induced cell apoptosis was examined using the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer’s instructions. After incubation with anti–digoxigenin conjugate, cells were mounted using VECTASHIELD antifade mounting medium with 4´,6‐diamidino‐2‐phenylindole. Images were obtained on a Leica TCS SP5 Spectral Confocal Microscope equipped with a ×40 (oil) objective. TUNEL (terminal deoxynucleotidal transferase–mediated biotin–deoxyuridine triphosphate nick‐end labeling)–positive cells were quantified in ImageJ using the TUNEL Cell Counter method developed by Maidana et al. 23
5‐HTR2B Knockdown by siRNA
NTC and 5‐HTR2B siRNAs were purchased from SMARTpool siRNA (ThermoScientific). HL‐1 cells were transfected with 50 nm of each siRNA using Dharmafect1 transfection reagent (ThermoScientific) as described previously (RNA interference and cycloheximide experiments). At 48 hours after transfection, cells were replated and treated with hormones or vehicle control and subjected to hypoxia/reoxygenation as described previously. Cell viability was measured using the LIVE/DEAD Cell Vitality Assay Kit (ThermoFisher Scientific). This assay provides a 2‐color fluorescence assay that distinguishes live cells from injured and dead cells. The assay uses C12‐resazurin to red fluorescent (live and uninjured cells) and SYTOX green dye (dead cells) to distinguish between metabolic intact cells (red), injured cells (reduced red and green fluorescence), and dead cells (green fluorescence). Between 5000 and 10 000 cells were analyzed using a BD LSRII flow cytometer (San Jose, CA) equipped with FACSDiVa software. Cells were excited with a 488‐nm laser. A gate was drawn on a histogram for the control sample to determine the percentage of live and dead cells.
Statistical Analysis
Data are represented as mean±SEM. Statistical analysis was performed with the GraphPad Prism software version 7 (GraphPad Software, San Diego, CA). Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments. Differences were considered to be statistically significant when P<0.05. For all studies, we used n=3 to 5 independent experiments unless otherwise specified in the figure legend.
Results
Dexamethasone Treatment Inhibits Estradiol (Estrogen)‐Induced Changes in Global Gene Expression in HL‐1 Cardiomyocytes
To determine the effects of dexamethasone on the global transcriptional response to estrogen in cardiomyocytes, we performed microarray analysis of HL‐1 cells treated for 6 hours with vehicle, 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone (100 nmol/L)+estrogen (10 nmol/L). Heat maps of averaged sample replicates within treatment groups showed that both dexamethasone and estrogen led to specific changes in gene expression in HL‐1 cells: 489 genes were differentially regulated by estrogen and 574 genes changed in response to dexamethasone compared with vehicle‐treated cells (Figure 1A). Cotreatment with dexamethasone+estrogen led to alterations in the expression of a similar number of genes (632 genes) in HL‐1 cells compared with that in the dexamethasone or estrogen treatments alone. However, the combination of dexamethasone+estrogen resulted in significant alterations in the patterns of gene expression associated with each individual hormone (Figure 1A). Venn diagram analysis was used to identify changes in each hormone‐specific gene expression signature and those genes that are unique and common among the 3 hormone treatments (Figure 1B). Of the 489 and 574 genes that were significantly dysregulated in response to estrogen and dexamethasone, respectively, 132 genes were coregulated by dexamethasone and estrogen, 172 were common to dexamethasone and dexamethasone+estrogen, and 168 were common to estrogen and dexamethasone+estrogen (Figure 1B). Only 68 genes were commonly regulated by dexamethasone, estrogen, and dexamethasone+estrogen (Figure 1B). Of the 489 estrogen genes, 257 genes were unique to estrogen (Figure 1B), suggesting that the combination of dexamethasone and estrogen abolished approximately 50% of the genes regulated by estrogen in HL‐1 cardiomyocytes. To identify the pathways in which dexamethasone, estrogen, or a combination of dexamethasone+estrogen gene regulation was involved, Ingenuity Pathway Analysis was performed (Figure 1C). Each hormone treatment led to changes in genes involved in different pathways (Figure 1C). Serotonin receptor signaling and G‐protein coupled receptor signaling are listed as the top pathways for estrogen‐regulated genes in HL‐1 cells, whereas fibroblast growth factor signaling and G‐protein G12/G13 signaling are the top pathways for dexamethasone‐regulated genes in these cells (Figure 1C). Interestingly, dexamethasone+estrogen cotreatment led to gene changes associated with distinct pathways compared with those altered by estrogen and dexamethasone alone (Figure 1C). Serotonin (5‐hydroxytryptamine) via its receptors (5‐HTRs) is involved in a myriad of physiological processes. Among those effects, 5‐hydroxytryptamine elicits mitogenic and secretory responses in the cardiovascular system, affecting endothelial cells, smooth muscle cells, fibroblasts, and cardiomyocytes. 24 Figure 1D illustrates how the expression of 4 of the 6 known classes of 5‐HTRs are regulated by estrogen, dexamethasone, and dexamethasone+estrogen. Estrogen treatment upregulates the expression of 5‐HTR2B (5‐HTR2 subtype B), 5‐HTR2C (5‐HTR2 subtype C), and 5‐HTR5A. Estrogen downregulates the expression of 5‐hydroxytryptamine‐Receptor 6 (5‐HTR6). Dexamethasone treatment only has effects on the gene expression of 5‐HTR6 (Figure 1D). Dexamethasone+estrogen abolished estrogen effects on 5‐HTR2B, 5‐HTR2C, and 5‐HTR5A gene expression (Figure 1D). These data suggest that dexamethasone combination with estrogen leads to significant alterations in estrogen‐targeted pathways in HL‐1 cardiomyocytes; in particular, dexamethasone blocks estrogen gene regulation of 5‐HTR2B, which has been shown to play a role in regulating differentiation and proliferation in the developing heart as well as in preserving heart structure and function in the adult heart. 25 , 26 , 27
Figure 1. Glucocorticoids alter estrogen transcriptional effects in HL‐1 cardiomyocytes.
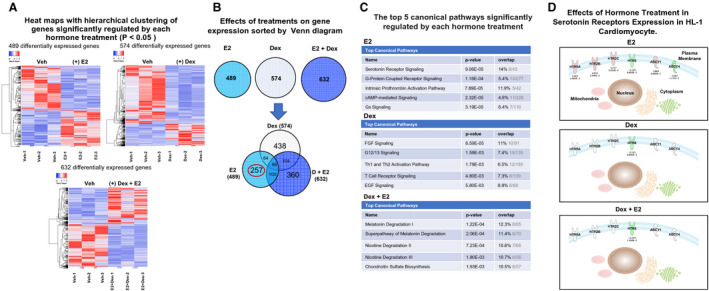
HL‐1 cells were treated with vehicle (Veh), 10 nmol/L estradiol (estrogen [E2]), 100 nmol/L dexamethasone (Dex), or 100 nmol/L dexamethasone+10 nmol/L estrogen (Dex+E2). mRNA was isolated and analyzed using a whole mouse genome 4×44 multiplex format oligo array (Agilent) for gene expression. A, Heat maps were generated with the online software Heatmapper (http://www2.heatmapper.ca) to visualize the differences in the gene expression of cells treated with each hormone and their combination. Blue indicates downregulation, and red indicates upregulation. B, The estrogen‐regulated, dexamethasone‐regulated, and estrogen+dexamethasone–regulated genes within each group were sorted by Venn diagrams that were generated using the Ingenuity Pathway Analysis software. Each hormone treatment regulated a similar number of genes; however, each treatment led to the regulation of a significant number of unique genes, and dexamethasone+estrogen significantly altered the transcriptional response to estrogen and dexamethasone alone. The hormone combination treatment inhibited the effects of dexamethasone in the regulation of 438 genes and of estrogen in the regulation of 257 genes (circled in red). In addition, dexamethasone+estrogen had unique effects on the expression of 360 genes. C, Results from microarray analysis of each hormone treatment were loaded into Ingenuity Pathway Analysis software. Estrogen treatment significantly regulated an important number of genes associated with serotonin receptor (5‐HTR) signaling (ranked top 1). Dexamethasone treatment led to the regulation of genes involved in different cellular and molecular biological pathways compared with estrogen. Dexamethasone+estrogen cotreatment alter the pathways that are regulated by a single estrogen treatment in cardiomyocytes. D, Estrogen altered the expression of 4 subtypes of 5‐HTRs in HL‐1 cardiomyocytes: 5‐HTR2B (5‐HTR2 subtype B), 5‐HTR2C (5‐HTR2 subtype C), and 5‐HTR5 (5‐HTR2 subtype A) and 5‐HTR6. The figure displays the effects of each hormone treatment on the expression of each gene. Estrogen also affects the expression of ADCY1 and ADCY4. Red indicates upregulation, and green indicates downregulation. ADCY1 indicates adenylate cyclase 1; ADCY4, adenylate cyclase 4; cAMP, cyclic adenosine monophosphate; EGF, endothelial growth factor; FGF, fibroblast growth factor; Th1, T helper 1; and Th2, T helper 2.
Dexamethasone Treatment Inhibits Estradiol (Estrogen) Upregulation of 5‐HTRs in HL‐1 Cardiomyocytes
We validated estrogen regulation of 5‐HTR2B and assessed whether dexamethasone+estrogen inhibits estrogen upregulation of 5‐HTR2B in HL‐1 cardiomyocytes using independent samples at different time points (Figure 2A). HL‐1 cardiomyocytes were treated with vehicle, 100 nmol/L dexamethasone, 10 nmol/L estrogen, and 100 nmol/L dexamethasone+ 10 nmol/L estrogen (same concentrations as those used in the microarray), and RNA was then isolated at different time points (0–24 hours). Increases in 5‐HTR2B gene expression were observed in HL‐1 cells in response to estrogen at 3, 6, and 12 hours (Figure 2A). In agreement with the microarray data, estrogen was not able to upregulate 5‐HTR2B gene expression in cells treated concomitantly with dexamethasone and estrogen at any time point (Figure 2A). Dexamethasone treatment has statistically significant effects on 5‐HTR2B expression at the 3‐hour and 6‐hour time points, but no significant effects on 5‐HTR2B gene expression in response to dexamethasone were found at the later time points (Figure 2A). To determine whether these alterations in gene expression translated to changes at the protein level, we evaluated dexamethasone, estrogen, and dexamethasone+Estradiol effects on 5‐HTR2B protein levels by Western blotting at 0, 6, and 24 hours (Figure 2A). Although estrogen leads to significant increases in 5‐HTR2B mRNA at early time points (3 and 6 hours), no substantial changes in protein levels of 5‐HTR2B were detected by Western blot at those time points. However, estrogen led to statistically significant increases in 5‐HTR2B protein levels at 24 hours (Figure 2A). Cotreatment with dexamethasone abolished estrogen upregulation of 5‐HTR2B protein levels at the same time point (Figure 2A). No significant increases in 5‐HTR2B protein levels were observed in response to dexamethasone treatment alone (Figure 2A). However, based on the protein data at 24 hours, estrogen can significantly increase 5‐HTR2B protein levels, and dexamethasone inhibits this effect. Overall, the mRNA and protein data support the gene expression studies in HL‐1 cells and show that estrogen treatment triggers significant increases in 5‐HTR2B mRNA and protein levels, whereas coadministration of dexamethasone inhibits these effects.
Figure 2. Estradiol (estrogen) regulates 5‐HTR2B (5‐HTR2 subtype B) gene expression and protein levels in HL‐1 and adult primary mouse cardiomyocytes.
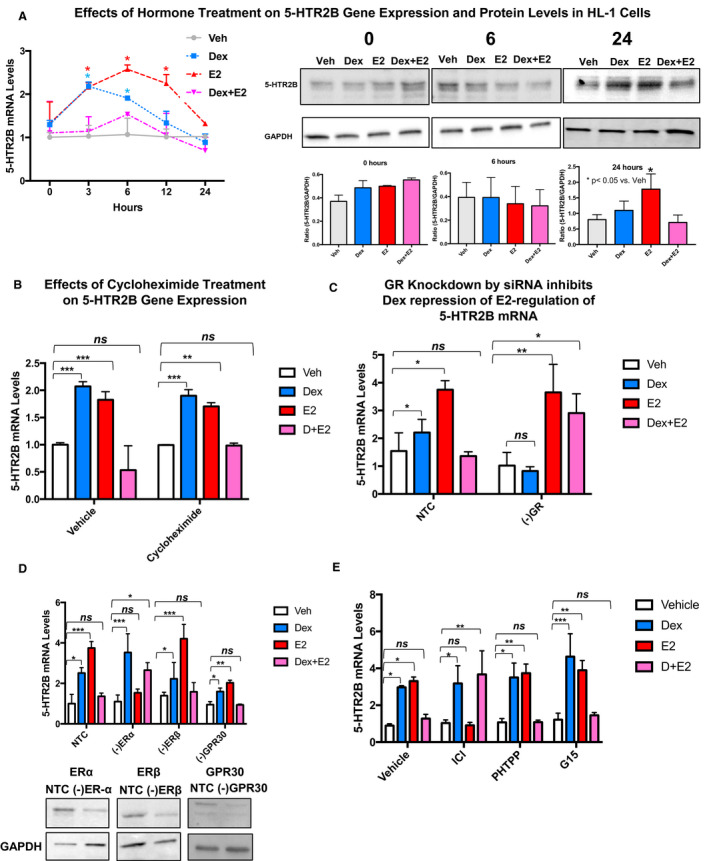
A, HL‐1 cells were treated with vehicle (Veh; gray line), 100 nmol/L dexamethasone (Dex; blue), estradiol (estrogen [E2]; red), and dexamethasone+estrogen (Dex+E2; pink) for the indicated times (0–24 hours), and 5‐HTR2B levels were analyzed by quantitative real‐time polymerase chain reaction and Western blotting. Estrogen steadily increased the mRNA expression of 5‐HTR2B in the time course experiment from 3 to 12 hours. Dexamethasone led to 5‐HTR2B increased expression at the 3‐hour time point. Dexamethasone+estrogen prevented estrogen‐induced upregulation of 5‐HTR2B expression at all time points. No significant changes in protein levels were observed at baseline (0 hours) or 6 hours following hormone treatment. The 5‐HTR2B protein levels were significantly increased in response to estrogen at 24 hours. Cotreatment of HL‐1 cells with dexamethasone+estrogen abolished estrogen effects on 5‐HTR2B at the protein level. Levels of 5‐HTR2B were normalized to the loading control GAPDH. B, HL‐1 cells were pretreated for 3 hours with vehicle (control) or 10 μg/mL of cycloheximide. Then cells were exposed to 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone+estrogen for 6 hours. Cycloheximide treatment did not have significant effects on 5‐HTR2B regulation by estrogen, suggesting that 5‐HTR2B is a direct target of estrogen. C, HL‐1 cells were transfected with NTC siRNA or glucocorticoid receptor (GR) siRNA (‐GR). At 48 hours after transfection, cells were treated with vehicle, 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone+estrogen for 3 hours. Downregulation of GR levels significantly blocked dexamethasone effects on 5‐HTR2B mRNA levels, showing that GR is required for dexamethasone effects. D, HL‐1 cells were also transfected with ERα (estrogen receptor α) siRNA (‐ERα), ERβ (‐ERβ) siRNA, and GPR30 (G protein‐coupled estrogen receptor 1) siRNA (‐GPR30) as described previously. Knocking down ERα abolished estrogen regulation of 5‐HTR2B, demonstrating that 5‐HTR2B regulation by estrogen is dependent on ERα expression. E, HL‐1 cells were treated with the following 3 estrogen receptor antagonists: ICI (182,780; fulvestrant; ERα antagonist), PHTPP (4‐[2‐Phenyl‐5,7‐bis(trifluoromethyl) pyrazolo[1,5‐a]pyrimidin‐3‐yl]phenol; ERβ antagonist), and G‐15 ([3aS,4R,9bR]‐4‐[6‐bromo‐1,3‐benzodioxol‐5‐yl]‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta[c]quinoline; G‐protein‐coupled estrogen receptor [GPER/GPR30] antagonist). After 1 hour of antagonist treatment, cells were subjected to hormone treatments. ICI significantly inhibited the estrogen upregulation of 5‐HTR2B. For the cycloheximide, siRNA, and antagonists experiments, 5‐HTR2B levels were measured by quantitative real‐time polymerase chain reaction. mRNA was normalized to cyclophillin B for all mRNA experiments. We used the same doses of hormones for all treatments. The figure includes representative Western blots confirming the changes in 5‐HTR2B protein levels and the knockdown of each individual receptor. Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments unless otherwise specified. Data represent the mean±SE (n=3–5 independent samples per group). *P<0.05; **P<0.01; ***P<0.001, ns=not significant.
Based on our microarray and time course data for our next experiments we mainly focused on early time points because changes in 5‐HTR2B mRNA seems to be an early and direct effect of estrogen treatment in these cells. Focusing on later time points will add other cofounding factors that may be influencing the regulation of 5‐HTR2B by estrogen.
Dexamethasone Inhibition of Estrogen Upregulation of 5‐HTR2B in HL‐1 Cardiomyocytes Is Mediated at the Transcriptional Level and Depends on GR and ERα
To elucidate the mechanism(s) underlying estrogen regulation of 5‐HTR2B and whether the inhibitory effects of dexamethasone are direct or indirect, HL‐1 cells were treated with the protein synthesis inhibitor cycloheximide for 1 hour before hormone treatment (3 hours). The rationale for using a short treatment with cycloheximide and the different hormone combinations is that HL‐1 cells are very sensitive to cycloheximide effects and significant increase in cell death has been observed in this cell line if incubated for longer periods of time. 21 This assay measured whether dexamethasone diminishes estrogen upregulation of 5‐HTR2B mRNA levels by de novo protein synthesis of a “third factor” that disrupts the estrogen effects on 5‐HTR2B gene expression in cardiomyocytes. Cycloheximide did not alter repression by dexamethasone of estrogen‐induced 5‐HTR2B expression (Figure 2B). In addition, our data suggest that 5‐HTR2B is a direct target of estrogen because cycloheximide did not have significant effects on the increase in 5‐HTR2B mRNA levels in response to estrogen (Figure 2B). To determine if GR is required for dexamethasone inhibition of estrogen induction of 5‐HTR2B, HL‐1 cells were transfected with NTC or siRNA against GR and treated for 6 hours with vehicle, 100 nm dexamethasone, 10 nm estrogen, or dexamethasone+estrogen. Total RNA isolated from each condition was examined for 5‐HTR2B expression by quantitative real‐time PCR. Our results show that GR is required for dexamethasone inhibition of estrogen induction of 5‐HTR2B mRNA (Figure 2C). Using the same approach, we next investigated which estrogen is necessary for estrogen regulation of 5‐HTR2B gene expression in HL‐1 cells.
HL‐1 cells were transfected with NTC or siRNA against ERα, ERβ, or GPR30 and treated for 6 hours with vehicle, 100 nm dexamethasone, 10 nm estrogen, or dexamethasone+estrogen. Transfection with siRNA against each ER led to a significant reduction in ERα, ERβ, and GPR30 protein levels (representative immunoblot, Figure 2D). ERα knockdown by siRNA significantly abolished the upregulation of 5‐HTR2B (Figure 2D), whereas ERβ or GPR30 knockdown by siRNA did not significantly change the effects of estrogen treatment on the expression of 5‐HTR2B (Figure 2D). To further confirm these results, cells were treated with 3 estrogen antagonists: fulvestrant (ICI; ERα antagonist), PHTPP, and G‐15 (GPER/GPR30 [G‐protein‐coupled ER] antagonist). HL‐1 cells were pretreated for 1 hour with each antagonist as previously described 15 and then incubated for 6 hours with vehicle, 100 nm dexamethasone, 10 nm estrogen, or dexamethasone+estrogen. Our results showed that treatment with PHTPP or G‐15 did not affect estrogen effects on 5‐HTR2B gene expression. In contrast, ICI significantly inhibited estrogen upregulation of 5‐HTR2B (Figure 2E).
These results suggest that estrogen signaling via ERα is responsible for the increases in 5‐HTR2B expression in cardiomyocytes.
GR Inhibits ERα Regulation of 5‐HTR2B by Competing for the Same DNA Binding Site Located on the Gene Promoter of 5‐HTR2B
One of the mechanisms by which ERα and GR modulate gene transcription is through direct binding to DNA sequences known as EREs 28 , 29 and GREs, 30 respectively. Analysis of the 5‐HTR2B promoter and gene sequence revealed several putative EREs and GREs (Figure 3A). One of these putative EREs (ERE1, −740 kb downstream of the transcription start site) was also identified as a potential GRE (GRE1; Figure 3A). The location of the identified GREs and EREs are displayed in Figure 3A.
Figure 3. The estrogen receptor α (ERα) and the glucocorticoid receptor (GR) are recruited to a functional estrogen responsive element (ERE)/glucocorticoid responsive element (GRE) located in the promoter of the 5‐HTR2B (5‐HTR2 subtype B) gene.
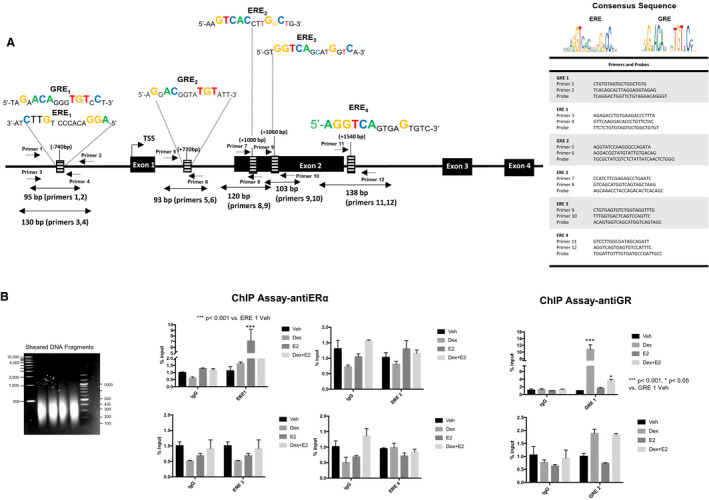
A, Schematic representation of the mouse 5‐HTR2B gene. Sequences of the consensus EREs and GREs identified within the predicted promoter, targeting areas of the designed primers, and the gene sequence of 5‐HTR2B. The table displaying the sequences of the primers and probes used for the assay. B, HL‐1 cells were treated with vehicle (Veh; black bars), 100 nmol/L dexamethasone (Dex; gray bars), 10 nmol/L estrogen (E2; dark gray bars), or dexamethasone+estrogen (Dex+E2; light gray bars) for 2 hours, and chromatin immunoprecipitation (ChIP) assays were performed with equivalent amounts of rabbit immunoglobulin G or rabbit anti‐GR or ERα antibody. Coimmunoprecipitated DNA was analyzed by real‐time polymerase chain reaction using primers and probes to the identified EREs and GREs located in the 5‐HTR2B promoter and gene sequence. Sheared DNA fragment sizes were between 200 and 500 bp (left panel). Results are plotted as a function of input DNA. Percentage input was calculated using the following equations: adjusted input 100%=(Ct input−6.64) and 100*2(Adjusted input−Ct (input)). Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments unless otherwise specified. Data represent mean±SE for 4 independent experiments per treatment group. *P<0.05 and ***P<0.001 vs vehicle in each group.
Chromatin immunoprecipitation assays were performed on genomic DNA from HL‐1 cells treated with vehicle, 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone+estrogen for 1 hour to test whether both ERα and GR were recruited to this common ERE/GRE binding site. Primers and probes used for the assays are listed in Figure 3A. DNA‐protein complexes were sheared into ~200 to 300 bp DNA fragments by sonication (Figure 3A). Figure 3B shows that both GR and ERα are individually recruited to the identified ERE1/GRE1 in the 5‐HTR2B promoter in a hormone‐dependent manner, and no significant recruitment to the additional identified EREs or GREs was found (Figure 3B). In the presence of the hormone‐combined treatment, no significant recruitment of ERα to ERE1/GRE1 was found (Figure 3B). Similarly, although GR was significantly associated with the chromatin at the ERE1/GRE1 in response to dexamethasone, this recruitment was decreased in the presence of dexamethasone and estrogen (Figure 3B). However, GR recruitment was still statistically significant compared with its vehicle control (Figure 3B). Recruitment of GR to the promoter of 5‐HTR2B by dexamethasone suggests that both hormones can regulate the expression of this gene. These results support the hypothesis that activated ERα and GR compete for the occupancy of the same binding site on the 5‐HTR2B promoter, which decreases the ERα upregulation of 5‐HTR2B mRNA levels in HL‐1 cardiomyocytes.
Dexamethasone Inhibits 5‐HTR2B Induction by Estrogen in Male and Female Hearts at the mRNA Level
To assess whether estrogen treatment induces 5‐HTR2B expression and dexamethasone inhibits this effect in the heart, male and female adrenalectomized and gonadectomized C57Bl/6 mice were given a single intraperitoneal injection of PBS (vehicle), 1 mg/kg of dexamethasone, 10 μg/kg of estrogen, or 1 mg/kg of dexamethasone+10 μg/kg, and hearts were harvested 6 hours later for RNA and protein analyses. Our results show that a single injection of estrogen statistically significantly increases 5‐HTR2B mRNA by ~1.5‐fold to 1.7‐fold in both male and female hearts compared with the vehicle‐treated counterparts (Figure 4A and 4B). Increases in 5‐HTR2B mRNA were strongly inhibited by dexamethasone cotreatment (Figure 4A and 4B). Although the changes in 5‐HTR2B mRNA in vivo mimic the responses in vitro, inconclusive effects were observed at the protein level (Figure 4A and 4B). However, dexamethasone+estrogen treatment seems to reduce 5‐HTR2B protein level (Figure 4A and 4B). Further studies are needed to clarify the effects of dexamethasone and estrogen antagonism in vivo. These findings indicate that 5‐HTR2B gene expression levels are controlled by estrogen in vivo and that dexamethasone blocks this regulation by estrogen in both men and women.
Figure 4. 5‐HTR2B (5‐HTR2 subtype B) mRNA and protein levels are regulated by estradiol in the adult mouse heart.
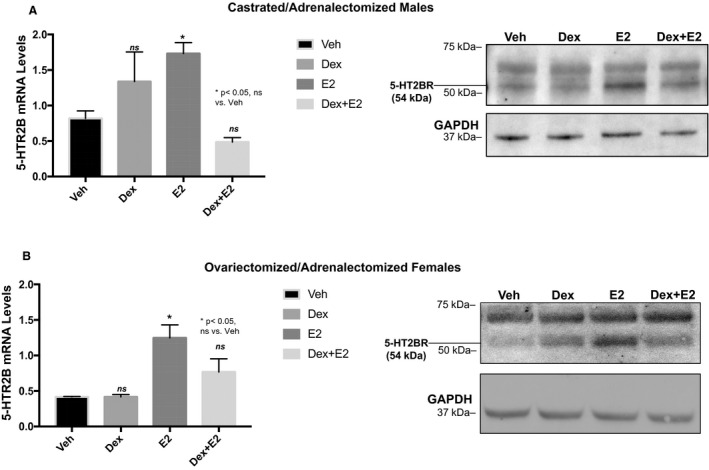
Gonadectomized and adrenalectomized C57BL/6 male and female mice were injected with vehicle (Veh), 1 mg/kg of dexamethasone (Dex), or 10 μg/kg estradiol (estrogen [E2]). RNA and protein were isolated at 6 hours after the treatment. mRNA and representative Western blots of 5‐HTR2B in each treatment group for male (A) and female (B) hearts. mRNA was normalized to cyclophillin B, and protein was normalized to GAPDH. Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments unless otherwise specified. Data represent the mean±SE (n=3–5 mice per group). *P<0.05; ns=not significant.
ERα Regulation of 5‐HTR2B Is Critical to Estrogen‐Associated Cardiomyocyte Protection in Hypoxia/Reoxygenation–Induced Cell Death
To establish the physiological relevance of ERα regulation of 5‐HTR2B in cardiomyocytes, we examined the effects of blocking estrogen induction of 5‐HTR2B in cardiomyocytes by coadministered dexamethasone in an in vitro model of I/R. HL‐1 cells were subjected to 17 hours of hypoxia followed by 2 hours of reoxygenation in the presence and absence of hormones as described in the Methods section. Following these treatments, cell death was measured by membrane integrity by flow cytometry (propidium iodide exclusion). Changes in membrane integrity are characteristic of cell death. In response to hypoxia/reoxygenation, we observed an ~25% increase in cell death in vehicle‐treated cells (Figure 5A). Similar increases in cell death were observed in dexamethasone‐treated (~22%) and dexamethasone+estrogen (~23%)–treated cells (Figure 5A). Estrogen treatment protected HL‐1 cells from hypoxia/reoxygenation–induced cell death (Figure 5A). To further support these findings, a TUNEL assay was performed. As shown in the representative images show in Figure 5B, dexamethasone+estrogen treatment exacerbated HL‐1 DNA fragmentation (characteristic of late phase of apoptosis) in response to hypoxia/reoxygenation. Quantification of TUNEL‐positive cells showed that although estrogen administration significantly decreased the number of TUNEL‐positive cells, dexamethasone+estrogen led to a statistically significant increase in positive cells compared with the vehicle‐treated cells (Figure 5B, lower panel). In association with these results of cell viability, we found that 5‐HTR2B mRNA levels were increased by estrogen, whereas dexamethasone+estrogen significantly diminished 5‐HTR2B gene expression in cells exposed to hypoxia/reoxygenation (Figure 5C). In agreement with the increase in cell death of HL‐1 cells exposed to hypoxia/reoxygenation and treated dexamethasone+estrogen, we found that the mRNA levels of SLC25A4, also known as ANT‐1, were significantly elevated by dexamethasone+estrogen (Figure 5C). SLC25A4 is a mitochondrial carrier with a role as a gated pore that translocates adenosine diphosphate from the cytoplasm into the mitochondrial matrix and adenosine triphosphate from the mitochondrial matrix into the cytoplasm. Mutations in SLC25A4 have been associated with cardiomyopathies and increased apoptosis. 31
Figure 5. Dexamethasone blocks estradiol protection of cardiomyocytes against cell death.
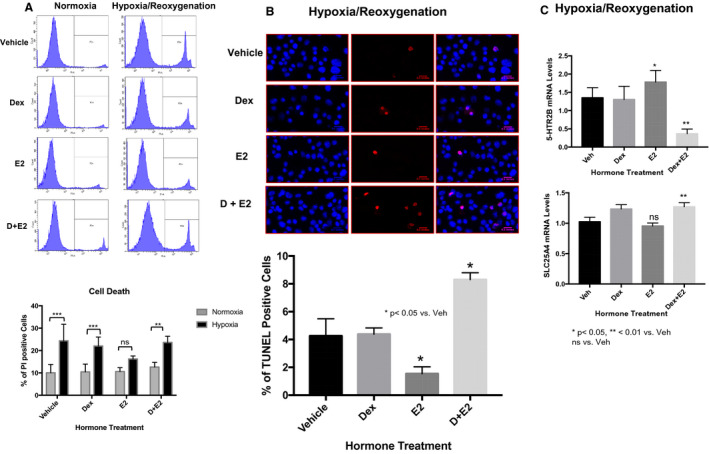
HL‐1 cells were pretreated for 6 hours with vehicle (Veh), 100 nmol/L dexamethasone (Dex), 10 nmol/L estradiol (estrogen [E2]), or dexamethasone+estrogen (Dex+E2). Following these treatments, the cells were subjected to 17 hours of hypoxia followed by 1 hour of reoxygenation in the presence and absence of hormones. A, Cell viability was then analyzed by flow cytometry using propidium iodide staining. The plot represents the percentage of dead cells in each treatment group. B, Apoptotic cardiomyocytes stained with TUNEL (terminal deoxynucleotidal transferase–mediated biotin–deoxyuridine triphosphate nick‐end labeling; red) and 4´,6‐diamidino‐2‐phenylindole (blue nuclear staining). The percentage of TUNEL‐positive cells was calculated by quantifying the TUNEL‐positive cells in ImageJ using the TUNEL Cell Counter method as described in the Methods section. C, mRNA levels of 5‐HTR2B (5‐HTR2 subtype B) and SLC25A4 (adenine nucleotide translocase 1) in each treatment group after hypoxia/reoxygenation. mRNA was normalized to cyclophillin B. Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments unless otherwise specified. Data are represented as the mean±SE from 3 independent experiments. *P<0.05; **P<0.01; ns=not significant vs vehicle in each group.
To test if the effects of estrogen and dexamethasone+estrogen on cell viability were mediated by 5‐HTR2B, siRNA was used to knockdown 5‐HTR2B expression in HL‐1 cardiomyocytes (Figure 6A). HL‐1 cells were transfected with NTC or siRNA against 5‐HTR2B as described in the Methods section. Following 48 hours after transfection, cells were subjected to hypoxia (17 hours) followed by reoxygenation (2 hours) in the presence and absence of hormones. Cell viability was assessed by flow cytometry using a LIVE/DEAD Cell Viability Assay as described in the experimental procedures section. Live and dead cells were quantified in the NTC and siRNA groups treated with vehicle, 100 nmol/L dexamethasone, 10 nmol/L estrogen, or dexamethasone+estrogen and exposed to hypoxia/reoxygenation. Our data quantification showed that both dexamethasone and estrogen protected the NTC cells from injury and cell death, whereas dexamethasone+estrogen treatment decreased the number of live/uninjured cells in response to hypoxia/reoxygenation (Figure 6A). Although dexamethasone treatment was able to significantly protect the cells from death and injury in the siRNA 5‐HTR2B group, the protection of estrogen was significantly decreased in absence of normal levels of 5‐HTR2B expression (Figure 6A). In contrast to the NTC cells, dexamethasone+estrogen treatment did not decrease cell viability in the siRNA 5‐HTR2B group (Figure 6A). In terms of changes in gene expression, we found that 5‐HTR2B mRNA levels were increased by estrogen, whereas dexamethasone+estrogen significantly diminished 5‐HTR2B expression in the NTC cells (Figure 6B). No changes in 5‐HTR2B were observed in the siRNA 5‐HTR2B cells (Figure 6B). In agreement with our previous data regarding hormones in hypoxia/reoxygenation (Figure 5C), we found that the mRNA levels of SLC25A4 were significantly elevated by dexamethasone+estrogen (Figure 6B). No changes were found in cells expressing lower 5‐HTR2B levels. Together, these data indicate that 1 potential mechanism by which dexamethasone antagonizes estrogen protection HL‐1 cardiomyocytes from hypoxia/reoxygenation–induced cell death is through the regulation of 5‐HTR2B.
Figure 6. Estradiol regulation of 5‐HTR2B (5‐HTR2 subtype B) protects cardiomyocytes against cell death.
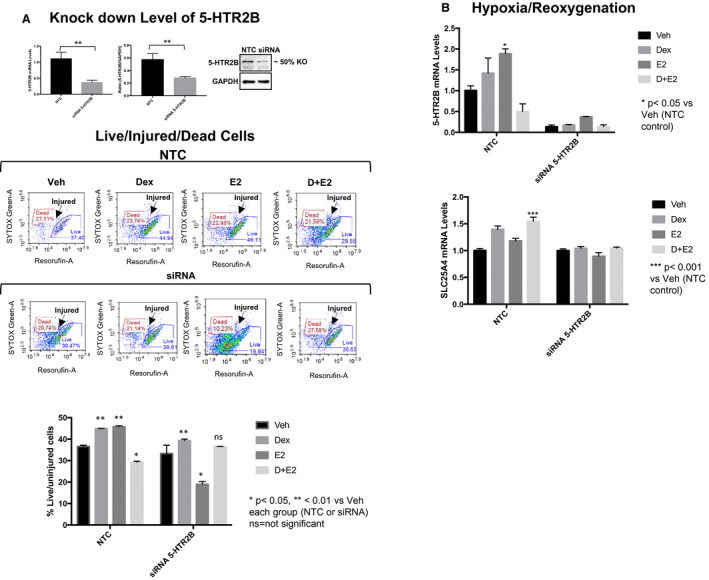
5‐HTR2B siRNA was used to knockdown 5‐HTR2B expression in HL‐1 cardiomyocytes. HL‐1 cells were transfected with nontarget (NTC) siRNA or 5‐HTR2B siRNA. At 48 hours after transfection, cells were treated with vehicle, 100 nmol/L dexamethasone (DEX), 10 nmol/L estrogen (E2), or dexamethasone+estrogen (D+E2) for 6 hours and then subjected to hypoxia/reoxygenation. A, 5‐HTR2B levels were knocked down to 50% at the mRNA and protein levels. Live and dead cells were quantified using the LIVE/DEAD Cell viability assay as described in the Methods section. Estrogen failed to protect HL‐1 cells from dead in response hypoxia/reoxygenation in 5‐HTR2B knockdown cells. B, 5‐HTR2B and SLC25A4 mRNA levels in NTC and 5‐HTR2B siRNA cells. mRNA was normalized to cyclophillin B. Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments unless otherwise specified. Data are represented as the mean±SE from 3 independent experiments. *P<0.05; **P<0.01; ***P<0.001; ns=not significant vs vehicle in each group.
Corticosterone Treatment Blocks Estrogen Cardioprotective Effects in I/R Injury In Vivo
To test whether the in vitro data reflect the physiological effects of an increase in stress hormones in vivo, female C57BL/6 mice were treated with regular drinking water or corticosterone (25 μg/mL) in water for 3 months as previously described. 17 Mice treated with corticosterone in water have significantly higher levels of serum corticosterone as compared with the mice maintained in drinking water (1394 ng/mL±321.3 versus 163.1 ± 44.84; Figure 7A). Mice in the 2 treatment groups were further classified according to their estrous cycle phase using vaginal smear cytology. 18 Mice in the estrus phase (basal estrogen levels, cornified squamous epithelial cells) and proestrus phase (higher estrogen levels, predominance of nucleated epithelial cells) were subjected to ischemia for 60 minutes and reperfusion for 48 hours. Following reperfusion, hearts were harvested and evaluated for the size of the infarct area, inflammation and fibrosis, and changes in gene expression. No significant or mild morphological changes were observed in response to I/R on mice drinking regular water regardless of the cycle phase (Figure 7Be,f). Similar findings were observed in corticosterone‐estrus mice. In contrast, corticosterone‐treated mice undergoing the proestrus phase of the cycle presented a statistically significant increase in the infarct area compared with their sham counterparts, estrus‐corticosterone, and water‐treated mice (Figure 7B). Hematoxylin‐eosin and Masson trichrome staining showed a more prominent infarct area, signs of edema and inflammation, and fibrosis in heart sections from proestrus‐corticosterone mice that were challenged in I/R compared with their sham controls and to mice taking regular drinking water (Figure 7C and 7D). The Table describes the main histological findings in the water and corticosterone groups in response to I/R injury in the proestrus group. In correlation with these data, we also found that 5‐HTR2B mRNA levels were significantly elevated in I/R in water‐treated mice independently of the phase of the cycle (Figure 7E and 7F). The expression of 5‐HTR2B was also significantly elevated in response to I/R in corticosterone‐treated mice in the estrus phase (Figure 7E and 7F). In contrast, no changes in 5‐HTR2B expression were found in corticosterone‐proestrus mice challenged in I/R. These results suggest that high levels of corticosterone in combination with higher than baseline physiological levels of estrogen leads to adverse effects in myocardial infarction. The expression of 5‐HTR2B significantly increases in I/R, and its levels correlate with the severity of the infarct. Also, increased in corticosterone levels significantly blocked 5‐HTR2B gene expression during the proestrus phase, suggesting that glucocorticoids in vivo can affect the transcriptional effects of estrogen in the heart.
Figure 7. Corticosterone administration exacerbates ischemia/reperfusion (IR) injury in female C57BL/6 mice in the proestrus phase of the estrus cycle.
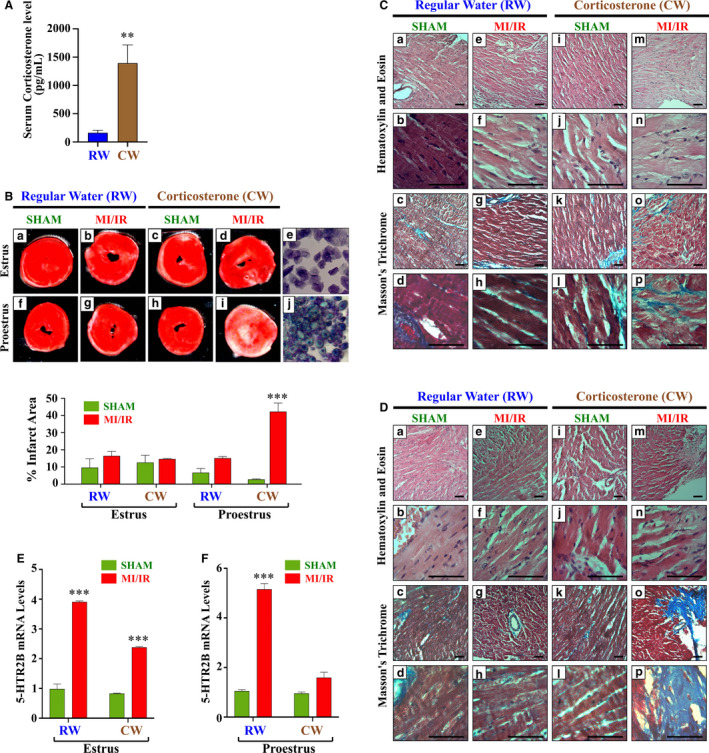
Female C57BL/6 mice were administered regular water (RW) or corticosterone water (CW; 25 μg/mL) for 3 months and subjected to 60 minutes of ischemia and 48 hours of reperfusion at 2 different phases of the estrus cycle, estrus and proestrus. A, Serum levels of corticosterone in mice drinking RW or CW. B, Representative photographs of triphenyltetrazolium chloride heart sections harvested from sham and myocardial infarction (MI)/IR groups at 48 hours after surgery. The infarct areas (percentage) were quantified using ImageJ color threshold mode. C and D, Representative micrographs of hematoxylin‐eosin Masson trichrome–stained hearts harvested from water and corticosterone sham and MI/IR groups in the estrus and proestrus phases (scale bar=50 µm). See the Table for a full description. E and F, 5‐HTR2B mRNA levels in each experimental group. mRNA was normalized to cyclophillin B. Ordinary 1‐way ANOVA with Dunnett multiple comparisons analysis was used to evaluate differences among group treatments unless otherwise specified. The graphs show the mean±SEM for 3 to 4 repeated experiments performed in duplicate. ***P<0.001 vs sham in RW or CW controls.
Table 1.
Main Histological Findings of Female Mice in the Proestrus Cycle (Figure 7) Exposed to Water and Corticosterone Treatments in Response to Ischemia‐Reperfusion Injury Compared With Sham Mice
| Figures | Gross appearance | Microscopy | Phenotype (proestrus) |
|---|---|---|---|
| TTC staining | |||
| Figure 7Bf | No evidence of infarction | No TTC‐negative areas observed | Sham‐RW |
| Figure 7Bg | Pallor of infarcted region | TTC‐negative (white) areas indicate MI area | MI/IR‐RW |
| Figure 7Bh | No evidence of infarction | No TTC‐negative areas observed | Sham‐CW |
| Figure 7Bi | Prominent infarcted region | Substantial TTC‐negative areas indicating dead tissue | MI/IR‐CW |
| Hematoxylin‐eosin staining | |||
| Figure 7Da,b | No evidence of infarction | Cardiac muscle fibers, nucleus located in the center, blood vessel enclosed in fine endothelium | Sham‐RW |
| Figure 7De,f | Pallor of infarcted region | Pockets of PMN inflammatory cells infiltration, loss of individual cardiomyocyte boundary | MI/IR‐RW |
| Figure 7Di,j | Pathological infiltration of inflammatory cells | Pockets of PMN inflammatory cells infiltration | Sham‐CW |
| Figure 7Dm,n | Prominent infarcted region | Indication of edema, fibrosis, and scar formation; wavy fibers with elongation and narrowing | MI/IR‐CW |
| Masson trichrome staining | |||
| Figure 7Dc,d | No evidence of infarction | Cardiac muscle fibers stained in red, collagenous tissue stained in blue enclosing the blood vessel | Sham‐RW |
| Figure 7Dg,h | Infarcted region evident | Focal nucleomegaly, acute inflammation | MI/IR‐RW |
| Figure 7Dk,l | No evidence of infarction | Faint loss of cardiac cell striations | Sham‐CW |
| Figure 7Do,p | Prominent evidence of infarct | Dense granulation tissue with early scar formation; blue represents region with replacement fibrosis | MI/IR‐CW |
All descriptions are mentioned for proestrus; however, similar characteristics were observed with less severity for the estrus‐staged hearts. CW indicates corticosterone water; IR, ischemia reperfusion; MI, myocardial infarction; PMN, polymorphonuclear cells; RW, regular water; and TTC, triphenyltetrazolium chloride.
Discussion
Exposure to stress is associated with an increased risk for cardiovascular events, including myocardial ischemia, heart failure, arrhythmia/conduction disorder, and stroke. 32 Women, particularly young women, have an elevated risk of cardiovascular complications associated with stress. 1 , 6 , 33 , 34 However, the molecular mechanisms underlying the increased cardiovascular risk in women are unknown.
In the present study, we show that stress may increase the risk for myocardial infarction and worsen the outcomes after myocardial infarction in the female heart by a mechanism involving genomic interaction between the GR and ER (ERα). Our data show that cardiomyocyte cotreatment with glucocorticoids altered estradiol (estrogen)‐transcriptional response in these cells (Figure 1) and led to significant changes in the cardiac pathways regulated by estrogen. We found that the top pathway regulated by estrogen in HL‐1 cardiomyocytes is the serotonin receptor signaling pathway (Figure 1) and that cotreatment with dexamethasone inhibited this regulation.
Serotonin (5‐hydroxytryptamine) via its receptors (5‐HTRs) is involved in a myriad of physiological processes. Among those effects, 5‐hydroxytryptamine elicits mitogenic and secretory responses in the cardiovascular system, affecting endothelial cells, smooth muscle cells, fibroblasts, and cardiomyocytes.1 5‐HTR2B has been implicated in cardiac differentiation and proliferation of cardiomyocytes during embryogenesis and development. 26 In the adult heart, mutations in the 5‐HT2B receptor led to cardiomyopathy with a loss of ventricular mass attributed to a reduction in number and size of cardiomyocytes. In addition, the ventricles of the 5‐HT2B receptor‐mutant mice exhibit structural abnormalities characterized by Z‐stripe enlargement and N‐cadherin downregulation, which leads to left ventricular dilatation and decreased systolic function in these mice. 25 Molecular studies using cardiomyocytes showed that the 5‐HT2B receptor protects cardiomyocytes against apoptosis triggered by serum deprivation by a mechanism involving the activation of the phosphatidylinositol‐3 kinase/protein kinase B and extracellular signal‐regulated kinase 1/2 signaling pathways, suggesting that 5‐HT2B receptor signaling in the heart is cardioprotective in response to stress. 14 However, overexpression of the 5‐HT2B receptor in the heart leads to cardiac hypertrophy resulting from abnormal mitochondrial proliferation and production of pro‐oxidant enzymes. 35 Therefore, based on these loss/gain of function studies, it is clear that the 5‐HT2B receptor plays a role in cardiac development and cardiomyopathy by a mechanism associated with pathways involved in cardiomyocyte proliferation, survival, and mitochondria function.
Our microarray data showed that estrogen treatment increases the expression of 5‐HT2BR by 2‐fold, and the addition of dexamethasone blocks this effect (Figure 1). This finding was further validated using independent samples in a time course experiment. Increases in 5‐HTR2B gene expression were observed in HL‐1 cells in response to both estrogen and dexamethasone at early time points (3 and 6 hours) and only estrogen at 12 hours (Figure 2A). In agreement with the microarray data, estrogen did not upregulate 5‐HTR2B gene expression in the presence of coadministered dexamethasone (Figure 2). Similar effects were found at the protein level (Figure 2A); however, we only observed a significant increase in protein levels in response to estrogen at 24 hours. In contrast, dexamethasone alone was not able to significantly increase 5‐HTR2B protein levels (Figure 2A). Cotreatment of estrogen and dexamethasone inhibit 5‐HTR2B increase in protein levels at 24 hours (Figure 2A). A potential explanation for these results is that synthesis and degradation rates of a protein and its encoding mRNA can be different. Also, it has been observed that changes in mRNA can be detected before changes at the protein level can be measured.It has also been reported that no changes in mRNA levels are detected despite clear increases in the corresponding protein. It is still challenging to find perfect correlations between transcripts and their corresponding proteins. 36 However, overall, our findings show that 5‐HTR2B is a target for both estrogen and dexamethasone and that a combination of these 2 hormones inhibit this 5‐HTR2B upregulation at the mRNA and protein levels.
Our studies using siRNA and specific antagonists for GR and ERs showed that the effects of estrogen and dexamethasone on 5‐HTR2B levels are directly mediated by GR and ERα (Figure 2B through 2E). These data correlate with studies showing that glucocorticoids can block estrogen responses in vitro and in vivo by a mechanism depending on activation of GR. 37 Studies by Whirledge et al showed that glucocorticoids and estrogen act antagonistically to regulate gene expression by a mechanism involving the recruitment of both GR and ERα to hormone‐responsive elements in the DNA and to the transcription start site. 15 Molecular studies have also shown that there is cross‐talk between ER and GR at the promoter of several genes involved in inflammation. 38 These studies provided evidence that glucocorticoids and estrogen can have additive effects and repress the same target genes, but they also showed that GR and ER can each antagonize the actions of the other by a mechanism that involves the recruitment of both receptors to the same binding sites on the target genes and by interacting with different coactivators/corepressors. 38 This study also showed that ERα can displace GR coactivators, preventing GR effects on gene transcription. 38 In addition, recent data from Vahrenkamp et al showed that activated ERα and GR have opposing effects on uterine growth and that ER and GR have some genomic binding site overlap. Upon simultaneous induction, they compete for the same binding sites. 39 These results are in agreement with our chromatin immunoprecipitation assay data, which show that there is recruitment of GR and ERα to the GRE/ERE located at −749 bp from the transcription start site (TSS) of the 5‐HTR2B gene, suggesting molecular interplay between ERα and GR at this promoter. Recruitment of ER and GR to this site is diminished in the presence of both dexamethasone and estrogen, suggesting that the presence of both hormones prevents ER and GR promoter binding. These results suggest that activation of both receptors with their ligands interferes with the recruitment of the other as well as with transcriptional activity. These findings correlate with our time course experiments showing that dexamethasone alone can upregulate 5‐HTR2B expression. Together, these results suggest that the combination of hormones negatively influences 5‐HTR2B gene expression in cardiomyocytes.
To further study the biological relevance of the molecular interplay of estrogen and glucocorticoids, single and combined hormone treatments were administered to male and female adrenalectomized and gonadectomized C57BL/6 mice. Estrogen administration significantly increased 5‐HTR2B mRNA and protein levels in the hearts of both male and female mice (Figure 4), and this effect was abolished by dexamethasone cotreatment with estrogen. Dexamethasone administration alone did not significantly alter 5‐HTR2B levels in the heart (Figure 4). These data correlated with the in vitro findings and confirmed that the combination of glucocorticoids and estrogen significantly represses 5‐HTR2B expression in the heart. Based on these results, we next sought to elucidate the molecular signaling and functional effects of 5‐HTR2B repression in cardiomyocytes in a model of hypoxia/reoxygenation.
We found that exposure to hypoxia/reoxygenation led to significant increases in cell death in vehicle‐treated cells (Figure 5A). Estrogen treatment significantly improved cell viability, whereas dexamethasone+estrogen abolished the protective effects of estrogen. Similar results were obtained by TUNEL assays, where cells treated with estrogen were protected from cell death. In contrast, cotreatment with dexamethasone+estrogen increased the number of TUNEL‐positive cells (Figure 5B). In association with estrogen cardioprotective effects in response to hypoxia/reoxygenation injury, we found an increase in 5‐HTR2B mRNA levels in response to estrogen (Figure 5C). The increase in 5‐HTR2B expression was abolished by dexamethasone+estrogen, which also correlated with a more pronounced cell death (Figure 5A and 5B). Positive TUNEL staining is an indication of cell death, potentially apoptosis. 40 Apoptosis results from mitochondrial dysfunction characterized by changes in membrane permeability. 41 The SLC25A4 gene encodes ANT‐1, which forms a channel in the inner membrane of mitochondria. This channel allows adenosine diphosphate into mitochondria and adenosine triphosphate out of mitochondria to be used as energy for the cell and it is part of the mitochondrial permeability transition pore. 42 Overexpression of the SLC25A4 gene is associated with opening of the mitochondrial permeability pore, which induces the release of CYCS (cytochrome c, somatic) and DIABLO/Second mitochondria‐derived activator caspase (diablo Inhibitor of apoptosis‐binding mitochondrial) proteins into the cytoplasm. The release of these factors triggers caspase activation as well as the translocation to the nucleus of the AIFM1/AIF (apoptosis‐inducing factor mitochondrial‐associated 1) and ENDOG (endonuclease G), which directly induce DNA damage and fragmentation. 43 We found that dexamethasone+estrogen treatment significantly upregulates the expression of the SLC25A4 gene (Figure 5C), suggesting that the hormone combination alters mitochondrial membrane permeability, which in turn induces an increase in cardiomyocyte apoptosis in response to hypoxia/reoxygenation injury.
To investigate if the protective effects of estrogen on cell viability were directly mediated by 5‐HTR2B, siRNA was used to knockdown 5‐HTR2B expression in HL‐1 cardiomyocytes (Figure 6A). Our results showed that decreased levels in 5‐HTR2B in cardiomyocytes blocked the positive effects of estrogen in cell viability in response to hypoxia/reoxygenation (Figure 6A). On the other hand, dexamethasone treatment significantly protected HL‐1 cardiomyocytes from injury and cell death in 5‐HTR2B knockdown cells. Also, no negative effects were observed in the dexamethasone+estrogen group in siRNA‐treated cells (Figure 6A). These results suggest that 5‐HTR2B regulation by estrogen is in part 1 of the potential mechanisms by which estrogens exert cardioprotection and that concurrent elevation in glucocorticoids may block estrogen‐positive actions via inhibition of 5‐HTR2B expression. Also, our data show that dexamethasone can exert protective effects in HL‐1 cardiomyocytes when 5‐HTR2B levels are decreased, even in the presence of estrogen. The cardioprotective effects of dexamethasone are in agreement in published data that showing that glucocorticoids can protect cardiomyocytes from apoptosis in response to stressors. 44 In normal physiology, activation of 5‐HTR2B has been shown to protect cardiomyocytes from apoptosis by activating the phosphatidylinositol‐3 kinase/Akt and extracellular signal‐regulated kinase pathways. 14 Activation of extracellular signal‐regulated kinase inhibits Bcl‐2‐associated X protein expression, whereas Akt activation represses the expression of ANT‐1, 14 therefore preventing changes in mitochondrial membrane permeability and cytochrome c release. Future studies are needed to elucidate if estrogen regulation of 5‐HTR2B exert cardioprotective mechanisms in hypoxia/reoxygenation injury through this downstream signaling pathway.
To further validate the in vitro studies and created a situation more analogous to the in vivo situation in humans exposed to higher glucocorticoid levels, we performed in vivo experiments in which corticosterone was administered to cycling female mice for 3 months. Mice were then classified in 2 groups, estrus and proestrus. Administration of corticosterone in water led to a significant elevation in circulating corticosterone. The progression in the estrus cycle was not influence by corticosterone. Previous studies have shown that female mice are more tolerant to I/R injury than their male counterparts. 45 , 46 , 47 , 48 , 49 , 50 This resistance has been associated with the cardioprotective effects of estrogen. 50 In agreement with these studies, we found that female mice in the regular water group presented smaller infarct areas and few histological changes in response to I/R injury, independent if they were in the estrus or proestrus phase of their cycle (Figure 7B). In contrast, corticosterone treatment had significantly deleterious effects on the female myocardium in response to I/R, characterized by bigger infarct areas and larger areas of edema, fibrosis, and scar formation (Figure 7D through 7d, Table). These negative effects were more pronounced in the proestrus phase of the cycle (Figure 7D). In correlation, with our histological findings in proestrus, corticosterone administration inhibited 5‐HTR2B mRNA expression. The results obtained in these experiments suggest that corticosterone blocks estrogen cardioprotection, and this effect may be in part mediated by 5‐HTR2B.
In conclusion, our data show for the first time a potential mechanism underlying the deleterious effects of stress on the female heart. We show that the molecular interplay of glucocorticoids and estrogen mediated by the GR and ERα has significant effects on cardiomyocyte viability following I/R injury. The combination of dexamethasone+estrogen significantly downregulated the expression of 5‐HT2BR and its prosurvival signaling in cardiomyocytes and in the whole heart. These results provide molecular evidence that glucocorticoids significantly alter estrogen cardioprotection in the heart and highlight the possibility that modulation of 5‐HT2B receptor signaling in women after myocardial infarction may be beneficial in preventing cardiomyocyte death and progression to heart failure.
Sources of Funding
This research was supported by Louisiana State University Health Sciences Center‐Shreveport; National Heart, Lung, and Blood Institute (NHBLI) 5K01HL144882‐0 (D.C.‐T.); Center for Redox Biology and Cardiovascular Disease Grant 3P20GM121307‐03S1 (A.W.O., D.C.‐T., H.D., N.G.C., and S.A.); National Institutes of Health Grants HL098435, HL133497, and HL141155 (A.W.O.); and the Intramural Research Program of the National Institutes of Health (J.A.C.).
Disclosures
None.
Acknowledgments
We thank Louisiana State University Health Sciences Center Shreveport Microscopy and Flow Cytometry Core Facilities and the Redox Molecular Signaling Core of the Center for Redox Biology and Cardiovascular Disease for their assistance with image acquisition, flow cytometry assays, and hypoxia/reoxygenation experiments. We also acknowledge Dr Kevin Gerrish from the Molecular Genomics Core at the National Institute of Environmental Health Sciences.
For Sources of Funding and Disclosures, see page 19.
REFERENCES
- 1. Vaccarino V, Sullivan S, Hammadah M, Wilmot K, Al Mheid I, Ramadan R, Elon L, Pimple PM, Garcia EV, Nye J, et al. Mental stress‐induced‐myocardial ischemia in young patients with recent myocardial infarction: sex differences and mechanisms. Circulation. 2018;137:794–805. DOI: 10.1161/CIRCULATIONAHA.117.030849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta LS, Beckie TM, DeVon HA, Grines CL, Krumholz HM, Johnson MN, Lindley KJ, Vaccarino V, Wang TY, Watson KE, et al.; American Heart Association Cardiovascular Disease in W, Special Populations Committee of the Council on Clinical Cardiology CoE, Prevention CoC, Stroke N, Council on Quality of C, Outcomes R . Acute myocardial infarction in women: a scientific statement from the American Heart Association. Circulation. 2016;133:916–947. DOI: 10.1161/CIR.0000000000000351. [DOI] [PubMed] [Google Scholar]
- 3. Shah AJ, Veledar E, Hong Y, Bremner JD, Vaccarino V. Depression and history of attempted suicide as risk factors for heart disease mortality in young individuals. Arch Gen Psychiatry. 2011;68:1135–1142. DOI: 10.1001/archgenpsychiatry.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nabel EG. Heart disease prevention in young women: sounding an alarm. Circulation. 2015;132:989–991. DOI: 10.1161/CIRCULATIONAHA.115.018352. [DOI] [PubMed] [Google Scholar]
- 5. Vaccarino V, Goldberg J, Magruder KM, Forsberg CW, Friedman MJ, Litz BT, Heagerty PJ, Huang GD, Gleason TC, Smith NL. Posttraumatic stress disorder and incidence of type‐2 diabetes: a prospective twin study. J Psychiatr Res. 2014;56:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaccarino V, Shah AJ, Rooks C, Ibeanu I, Nye JA, Pimple P, Salerno A, D’Marco L, Karohl C, Bremner JD, et al. Sex differences in mental stress‐induced myocardial ischemia in young survivors of an acute myocardial infarction. Psychosom Med. 2014;76:171–180. DOI: 10.1097/PSY.0000000000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei J, Pimple P, Shah AJ, Rooks C, Bremner JD, Nye JA, Ibeanu I, Murrah N, Shallenberger L, Raggi P, et al. Depressive symptoms are associated with mental stress‐induced myocardial ischemia after acute myocardial infarction. PLoS One. 2014;9:e102986. DOI: 10.1371/journal.pone.0102986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dreyer RP, Smolderen KG, Strait KM, Beltrame JF, Lichtman JH, Lorenze NP, D'Onofrio G, Bueno H, Krumholz HM, Spertus JA. Gender differences in pre‐event health status of young patients with acute myocardial infarction: a VIRGO study analysis. Eur Heart J Acute Cardiovasc Care. 2016;5:43–54. DOI: 10.1177/2048872615568967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smolderen KG, Strait KM, Dreyer RP, D'Onofrio G, Zhou S, Lichtman JH, Geda M, Bueno H, Beltrame J, Safdar B, et al. Depressive symptoms in younger women and men with acute myocardial infarction: insights from the VIRGO study. J Am Heart Assoc. 2015;4:e001424. DOI: 10.1161/JAHA.114.001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bremner JD, Campanella C, Khan Z, Shah M, Hammadah M, Wilmot K, Al Mheid I, Lima BB, Garcia EV, Nye J, et al. Brain correlates of mental stress‐induced myocardial ischemia. Psychosom Med. 2018;80:515–525. DOI: 10.1097/PSY.0000000000000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hammadah M, Sullivan S, Pearce B, Al Mheid I, Wilmot K, Ramadan R, Tahhan AS, O'Neal WT, Obideen M, Alkhoder A, et al. Inflammatory response to mental stress and mental stress induced myocardial ischemia. Brain Behav Immun. 2018;68:90–97. DOI: 10.1016/j.bbi.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mallik S, Spertus JA, Reid KJ, Krumholz HM, Rumsfeld JS, Weintraub WS, Agarwal P, Santra M, Bidyasar S, Lichtman JH, et al.; Investigators PR . Depressive symptoms after acute myocardial infarction: evidence for highest rates in younger women. Arch Intern Med. 2006;166:876–883. DOI: 10.1001/archinte.166.8.876. [DOI] [PubMed] [Google Scholar]
- 13. Wokhlu A, Pepine CJ. Mental stress and myocardial ischemia: young women at risk. J Am Heart Assoc. 2016;5:e004196. DOI: 10.1161/JAHA.116.004196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nebigil CG, Etienne N, Messaddeq N, Maroteaux L. Serotonin is a novel survival factor of cardiomyocytes: mitochondria as a target of 5‐HT2B receptor signaling. Faseb J. 2003;17:1373–1375. [DOI] [PubMed] [Google Scholar]
- 15. Whirledge S, Cidlowski JA. Estradiol antagonism of glucocorticoid‐induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology. 2013;154:499–510. DOI: 10.1210/en.2012-1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whirledge SD, Kisanga EP, Oakley RH, Cidlowski JA. Neonatal genistein exposure and glucocorticoid signaling in the adult mouse uterus. Environ Health Perspect. 2018;126:047002. DOI: 10.1289/EHP1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cruz‐Topete D, Myers PH, Foley JF, Willis MS, Cidlowski JA. Corticosteroids are essential for maintaining cardiovascular function in male mice. Endocrinology. 2016;157:2759–2771. DOI: 10.1210/en.2015-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caligioni CS, Franci CR. Oxytocin secretion induced by osmotic stimulation in rats during the estrous cycle and after ovariectomy and hormone replacement therapy. Life Sci. 2002;71:2821–2831. DOI: 10.1016/S0024-3205(02)02139-2. [DOI] [PubMed] [Google Scholar]
- 19. Xu ZB, Alloush J, Beck E, Weisleder N. A murine model of myocardial ischemia‐reperfusion injury through ligation of the left anterior descending artery. Jove‐J Vis Exp. 2014. DOI: 10.3791/51329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr. HL‐1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. DOI: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cruz‐Topete D, He B, Xu X, Cidlowski JA. Kruppel‐like factor 13 is a major mediator of glucocorticoid receptor signaling in cardiomyocytes and protects these cells from DNA damage and death. J Biol Chem. 2016;291:19374–19386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramamoorthy S, Cidlowski JA. Ligand‐induced repression of the glucocorticoid receptor gene is mediated by an NCoR1 repression complex formed by long‐range chromatin interactions with intragenic glucocorticoid response elements. Mol Cell Biol. 2013;33:1711–1722. DOI: 10.1128/MCB.01151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maidana DE, Tsoka P, Tian B, Dib B, Matsumoto H, Kataoka K, Lin HJ, Miller JW, Vavvas DG. A novel IMAGEJ macro for automated cell death quantitation in the retina. Invest Ophth Vis Sci. 2015;56:6701–6708. DOI: 10.1167/iovs.15-17599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hutcheson JD, Setola V, Roth BL, Merryman WD. Serotonin receptors and heart valve disease–it was meant 2B. Pharmacol Ther. 2011;132:146–157. DOI: 10.1016/j.pharmthera.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nebigil CG, Hickel P, Messaddeq N, Vonesch JL, Douchet MP, Monassier L, Gyorgy K, Matz R, Andriantsitohaina R, Manivet P, et al. Ablation of serotonin 5‐HT(2B) receptors in mice leads to abnormal cardiac structure and function. Circulation. 2001;103:2973–2979. [DOI] [PubMed] [Google Scholar]
- 26. Nebigil CG, Maroteaux L. A novel role for serotonin in heart. Trends Cardiovasc Med. 2001;11:329–335. DOI: 10.1016/S1050-1738(01)00135-9. [DOI] [PubMed] [Google Scholar]
- 27. Nebigil CG, Etienne N, Schaerlinger B, Hickel P, Launay JM, Maroteaux L. Developmentally regulated serotonin 5‐ht2b receptors. Int J Dev Neurosci. 2001;19:365–372. [DOI] [PubMed] [Google Scholar]
- 28. Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. DOI: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends Endocrinol Metab. 2004;15:73–78. DOI: 10.1016/j.tem.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 30. Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA. 2004;101:15603–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gutierrez‐Aguilar M, Baines CP. Physiological and pathological roles of mitochondrial slc25 carriers. Biochem J. 2013;454:371–386. DOI: 10.1042/BJ20121753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song H, Fang F, Arnberg FK, Mataix‐Cols D, Fernandez de la Cruz L, Almqvist C, Fall K, Lichtenstein P, Thorgeirsson G, Valdimarsdottir UA. Stress related disorders and risk of cardiovascular disease: population based, sibling controlled cohort study. BMJ. 2019;365:l1255. DOI: 10.1136/bmj.l1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Felix AS, Lehman A, Nolan TS, Sealy‐Jefferson S, Breathett K, Hood DB, Addison D, Anderson CM, Cené CW, Warren BJ, et al. Stress, resilience, and cardiovascular disease risk among black women. Circ Cardiovasc Qual Outcomes. 2019;12:e005284. DOI: 10.1161/CIRCOUTCOMES.118.005284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garcia M, Mulvagh SL, Merz CN, Buring JE, Manson JE. Cardiovascular disease in women: clinical perspectives. Circ Res. 2016;118:1273–1293. DOI: 10.1161/CIRCRESAHA.116.307547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nebigil CG, Jaffre F, Messaddeq N, Hickel P, Monassier L, Launay JM, Maroteaux L. Overexpression of the serotonin 5‐ht2b receptor in heart leads to abnormal mitochondrial function and cardiac hypertrophy. Circulation. 2003;107:3223–3229. [DOI] [PubMed] [Google Scholar]
- 36. Edfors F, Danielsson F, Hallstrom BM, Kall L, Lundberg E, Ponten F, Forsstrom B, Uhlen M. Gene‐specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016;12:883. 10.15252/msb.20167144. PMID: 27951527; PMCID: PMC5081484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, Gao J, Song WC, DeFranco DB, Cheng SY, et al. Glucocorticoids antagonize estrogens by glucocorticoid receptor‐mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68:7386–7393. DOI: 10.1158/0008-5472.CAN-08-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cvoro A, Yuan C, Paruthiyil S, Miller OH, Yamamoto KR, Leitman DC. Cross talk between glucocorticoid and estrogen receptors occurs at a subset of proinflammatory genes. J Immunol. 2011;186:4354–4360. DOI: 10.4049/jimmunol.1002205. [DOI] [PubMed] [Google Scholar]
- 39. Vahrenkamp JM, Yang CH, Rodriguez AC, Almomen A, Berrett KC, Trujillo AN, Guillen KP, Welm BE, Jarboe EA, Janat‐Amsbury MM, et al. Clinical and genomic crosstalk between glucocorticoid receptor and estrogen receptor alpha in endometrial cancer. Cell Rep. 2018;22:2995–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu H, Che X, Xu X, Zheng M, Zhao Y, He W, Yu J, Xiong J, Li W. Insulin protects apoptotic cardiomyocytes from hypoxia/reoxygenation injury through the sphingosine kinase/sphingosine 1‐phosphate axis. PLoS One. 2013;8:e80644. DOI: 10.1371/journal.pone.0080644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. DOI: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Echaniz‐Laguna A, Chassagne M, Ceresuela J, Rouvet I, Padet S, Acquaviva C, Nataf S, Vinzio S, Bozon D, Mousson de Camaret B. Complete loss of expression of the ant1 gene causing cardiomyopathy and myopathy. J Med Genet. 2012;49:146–150. [DOI] [PubMed] [Google Scholar]
- 43. Bloemberg D, Quadrilatero J. Autophagy, apoptosis, and mitochondria: molecular integration and physiological relevance in skeletal muscle. Am J Physiol Cell Physiol. 2019;317:C111–C130. DOI: 10.1152/ajpcell.00261.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ren RQ, Oakley RH, Cruz‐Topete D, Cidlowski JA. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology. 2012;153:5346–5360. DOI: 10.1210/en.2012-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Booth EA, Flint RR, Lucas KL, Knittel AK, Lucchesi BR. Estrogen protects the heart from ischemia‐reperfusion injury via cox‐2‐derived PGI(2). J Cardiovasc Pharm. 2008;52:228–235. DOI: 10.1097/FJC.0b013e3181824d59. [DOI] [PubMed] [Google Scholar]
- 46. Booth EA, Lucchesi BR. Estrogen‐mediated protection in myocardial ischemia‐reperfusion injury. Cardiovasc Toxicol. 2008;8:101–113. DOI: 10.1007/s12012-008-9022-2. [DOI] [PubMed] [Google Scholar]
- 47. Ostadal B, Netuka I, Maly J, Besik J, Ostadalova I. Gender differences in cardiac ischemic injury and protection‐experimental aspects. Exp Biol Med. 2009;234:1011–1019. DOI: 10.3181/0812-MR-362. [DOI] [PubMed] [Google Scholar]
- 48. Deschamps AM, Murphy E, Sun JH. Estrogen receptor activation and cardioprotection in ischemia reperfusion injury. Trends Cardiovas Med. 2010;20:73–78. DOI: 10.1016/j.tcm.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murphy E, Amanakis G, Fillmore N, Parks RJ, Sun JH. Sex differences inmetabolic cardiomyopathy. Cardiovasc Res. 2017;113:370–377. DOI: 10.1093/cvr/cvx008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ross JL, Howlett SE. Age and ovariectomy abolish beneficial effects of female sex on rat ventricular myocytes exposed to simulated ischemia and reperfusion. PLoS One. 2012;7:e38425. DOI: 10.1371/journal.pone.0038425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.