

Abstract
Background
Obesity is associated with long‐term health consequences including cardiovascular disease. Separating the independent effects of childhood and adulthood obesity on cardiovascular disease risk is challenging as children with obesity typically remain overweight throughout the lifecourse.
Methods and Results
This study used 2‐sample univariable and multivariable Mendelian randomization to estimate the effect of childhood body size both independently and after accounting for adult body size on 12 endpoints across the cardiovascular disease disease spectrum. Univariable analyses identified strong evidence of a total effect between genetically predicted childhood body size and increased risk of atherosclerosis, atrial fibrillation, coronary artery disease, heart failure, hypertension, myocardial infarction, peripheral artery disease, and varicose veins. However, evidence of a direct effect was weak after accounting for adult body size using multivariable Mendelian randomization, suggesting that childhood body size indirectly increases risk of these 8 disease outcomes via the pathway involving adult body size.
Conclusions
These findings suggest that the effect of genetically predicted childhood body size on the cardiovascular disease outcomes analyzed in this study are a result of larger body size persisting into adulthood. Further research is necessary to ascertain the critical timepoints where, if ever, the detrimental impact of obesity initiated in early life begins to become immutable.
Keywords: cardiovascular disease, genetic epidemiology, lifecourse, Mendelian randomization, obesity
Subject Categories: Cardiovascular Disease, Genetics, Obesity, Risk Factors
Nonstandard Abbreviations and Acronyms
- FDR
false discovery rate
- MR
Mendelian randomization
Clinical Perspective
What Is New?
This Mendelian randomization investigation provides univariable evidence that genetically predicted childhood obesity increases risk of 8 types of cardiovascular disease.
Evidence of total effects substantially attenuated upon accounting for body size in adulthood.
What Are the Clinical Implications?
These findings suggest that childhood obesity influences cardiovascular disease risk as a result of the long‐term effect of adiposity persisting into adulthood.
Risks associated with childhood adiposity may therefore potentially be reversed by early resolution of obesity in prepubertal children.
Further research is required to elucidate whether there are critical timepoints in the lifecourse where the effects of childhood obesity may be reversed through lifestyle modifications.
Approximately 2.6 million people worldwide die as a result of obesity related diseases each year, including cardiovascular diseases (CVDs), such as heart disease and stroke. 1 The World Health Organization posits that obesity results from and might influence social disparities. 2 Therefore, decreasing the frequency of obesity and thus reducing associated long‐term adverse health consequences is key to reducing health inequity, with the goal of achieving health attainment as well as poverty reduction globally.
The extent and prevalence of obesity have been growing steadily in both pediatric and adult populations over the past 4 decades. 3 In a 2017 meta‐analysis, pooled estimates from 21 cohort and case‐control studies suggest that childhood obesity may be a risk factor for selected adult CVD risk factors; however, causal effects independent of adult adiposity could not be established. 4 This is in part due to the inherent problems with observational investigations, in which standard statistical techniques used to adjust for confounding do not fully negate bias, which may lead to spurious findings. 5 Furthermore, whether prepubertal childhood obesity has a lasting effect on different types of cardiovascular disease or whether those who become nonobese by adulthood have a similar risk to individuals who were never obese 6 , 7 is yet to be determined.
A lifecourse approach crucially investigates the contribution of early and later life factors together to identify risk and protective mechanisms across the lifespan. 8 The issues described relating to observational studies make separating the effects of obesity at different stages throughout the lifecourse challenging, which is one of the motivations behind using human genetics through the application of an approach known as Mendelian randomization (MR). 9 MR is often implemented as an instrumental variable analysis that exploits the random assortment of genetic variants at birth. 10 Thus, by using this approach, it is possible to estimate the causal effects between closely related exposures and disease outcomes of interest, while mitigating the influence of common observational epidemiological issues relating to confounding and reverse causation. 5
Multivariable MR is an extension of this approach that uses multiple genetic variants associated with multiple exposures to concurrently estimate the causal effect of each risk factor on an outcome. It has previously been applied to separate the independent effects of childhood and adult body size on disease risk, 9 using genetic instruments for self‐reported perceived body size at age 10 and body mass index (BMI) in adulthood (average age 56.5 years). 11 , 12 In addition, these childhood effects have been replicated using measured BMI data from the Young Finns Study 13 and the HUNT Study (Nord‐Trøndelag Health Study). 14 Childhood body size has been shown to have an effect on coronary artery disease using univariable MR (odds ratio [OR], 1.49; 95% CI, 1.33–1.68). However, after accounting for adult body size using multivariable MR, this effect attenuated (OR, 1.02; 95% CI, 0.86–1.22), suggesting that the childhood effects are because of sustained effects of adiposity over time. 9 This approach has not yet been applied to comprehensively evaluate end points throughout the CVD spectrum, however.
This investigation sets out to examine whether genetically predicted childhood body size has a direct effect on a comprehensive list of CVD outcomes, independent of adult body size, that have been linked previously to obesity. Initially, univariable MR was applied to estimate the “total effect” of early life body size on 12 CVD outcomes (ie, without accounting for adult body size). Multivariable MR was then used to estimate the “direct” (Figure 1A) and “indirect” (Figure 1B) effect of early life body size on the CVD outcomes. Estimating the independent effects of childhood and adult body size on a spectrum of CVD outcomes is of public health importance to better understand whether risks associated with childhood adiposity are lasting or can be minimized, suspended, or even reversed by resolution of obesity in early life. 15
Figure 1. Directed acyclic graphs indicating 2 scenarios to explain the causal effect between childhood body size and disease outcomes in later life.
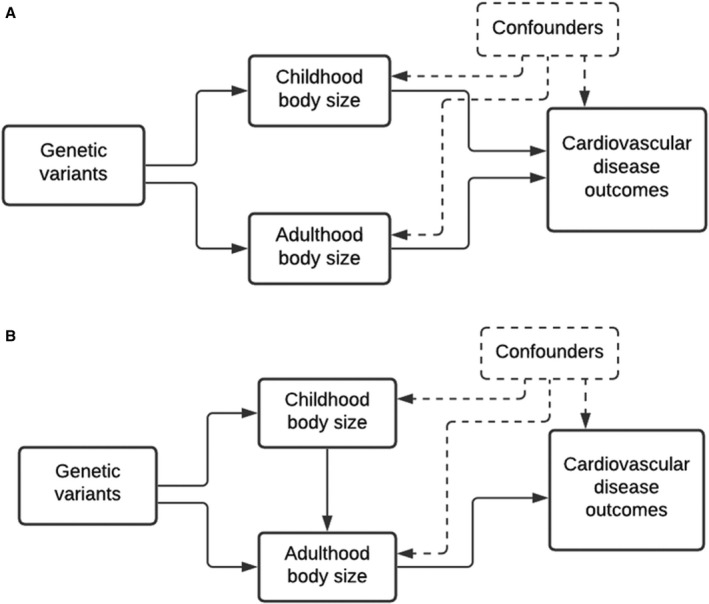
Childhood body size has a direct effect on cardiovascular disease (CVD) risk independent of body size in adulthood (A), and childhood body size has an indirect effect on CVD risk, through body size in adulthood (B).
Methods
The data that support the findings of this study are all available from within the supplementary materials or publicly accessible from the resources cited. Data were obtained from the UKBB (UK Biobank) study, which obtained ethics approval from the Research Ethics Committee (REC; approval number: 11/NW/0382) and informed consent from all participants enrolled in UKBB.’
Data Resources
Genetic Variants for Childhood and Adult Body Size
Genetic instruments for childhood and adult body size were identified previously, where 295 and 557 independent genetic variants were identified, respectively (using P<5×10‐8 and r 2<0.001) from a large‐scale genome‐wide association study (GWAS) of 453 169 participants in the UKBB study. 11 , 12 Adulthood measured BMI (mean age=56.5 years) was obtained at baseline using height (measured in cm) and weight (to the nearest 0.1 kg). In addition, participants were asked the question “When you were 10 years old, compared to average would you describe yourself as thinner, about average, or plumper?” For comparability purposes, BMI in adults was transformed into a categorical variable comprising 3 groups indicating body sizes indicative of being “thinner” (21.1–25 kg/m2), “about average” (25–31.7 kg/m2), and “plumper” (31.7–59.9 kg/m2). Throughout this study, these measures are described as childhood and adult body size, respectively. The estimates derived in our analysis thus reflect a change in the odds of each change in weight category within childhood and adult groups (eg, from “thinner” to “about average” or from “about average” to “plumper”).
GWAS were conducted on individuals who had both measures available adjusting for age, sex, and the genotyping chip used to measure genetic data in the UKBB. This analysis applied linear regression and thus assumes that the effect of a single nucleotide polymorphism from the lowest to the middle category of the body size variables is the same as the effect from the middle to the highest. The GWAS used for childhood body size was also adjusted for month of birth. Conditional F‐statistics generated for childhood (F=13.6) and adult (F=16.0) body size instruments suggested that weak instrument bias was unlikely to influence findings from these analyses. 9 Validation and simulation analyses for measures have been performed and comparisons of genome‐wide effect estimates between early life and adult body size in the UKBB made 9 (Table S1). Scores have been validated in the Young Finns Study. 13 This study indicated that the genetic score for childhood body size was a stronger predictor of childhood BMI compared with the adult body size score (area under the curve coefficients, 0.74; 95% CI, 0.65–0.82 versus 0.62, 0.53–0.72; P=0.02) and the adult genetic score was a stronger predictor of adult BMI than the childhood body size score (area under the curve coefficients, 0.62; 95% CI, 0.58–0.65 versus 0.57, 0.54–0.60; P=0.02). In addition, findings from the HUNT Study validated the childhood and adult gene scores for BMI with repeated BMI measurements of a Norwegian population aged 12 to 70 over 6 decades, confirming that both polygenetic risk scores were valid instruments. This study showed that the predictive performance of the childhood score was better in those aged 12 to 15.9 years compared with 24 to 29.9 years (polygenic risk scores 6.7% versus 2.4%) and that of the adult score was better in those aged 24 to 29.9 years compared to 12 to 15.9 years (3.9% versus 3.6%). 14 Thus, the predictive ability of these BMI scores have been validated in 2 further population groups in addition to validation analyses undertaken in the original study that conducted the GWAS. 13 , 14
Genetic Effects on Cardiovascular Disease Outcomes
The CVD end points analyzed in this study comprised those available from GWAS that BMI has been shown to influence previously. 16 , 17 , 18 Genetic estimates on coronary artery disease and myocardial infarction were obtained from the Coronary Artery Disease Genome‐wide Replication and Meta‐analysis plus the Coronary Artery Disease Genetics (CARDIOGRAMplusC4D) consortium. 19 Effect estimates on stroke were obtained from a GWAS undertaken by the MEGASTROKE consortium. 20 These GWAS were additionally selected as they did not include the UKBB study, to avoid overlapping samples with our instrument identification data set. Genetic estimates for the remaining CVD outcomes (all N>1000 cases) had also been associated with BMI in the literature and were obtained from the FinnGen (FinnGen‐tutkimushanke vie suomalaiset löytöretkelle genomitietoon) study (freeze 4), which brings together the nationwide network of Finnish biobanks and digital health care data. International Classification of Diseases, Tenth Revision (ICD‐10) codes are available for these outcomes 21 (Table S2).
Statistical Analysis
The “total effects” of genetically predicted childhood body size on each of the 12 CVD outcomes were estimated in a 2‐sample setting using the “TwoSampleMR” R package. 22 We initially applied the inverse variance weighted method, which provides an overall weighted estimate of causal effects of an exposure on an outcome by combining estimates using each variant in a fixed effect meta‐analysis model. 23 Weighted median and MR‐Egger methods were additionally used in this study to assess the robustness of univariable results to horizontal pleiotropy, the phenomenon whereby genetic variants influence multiple traits or disease outcomes via independent biological pathways. 24 , 25
To mitigate false positive rates due to multiple testing, false discovery rate (FDR) corrections were used to adjust the P values computed for the univariable MR estimates using the inverse variance weighted method. For comparative purposes, we also undertook univariable MR analyses of adult body size for all 12 outcomes.
Multivariable MR using the inverse variance weighted method was conducted to fit childhood and adult body size as simultaneous risk factors on the remaining CVD outcomes, which were robust to FDR corrections in the previous analysis. This enabled the estimation of the “direct” effect of childhood body size on the CVD outcomes after accounting for adult body size.
Forest plots were generated using the R package “ggplot2.” 26 All analyses were undertaken using R (version 3.5.1).
Results
The total effects through univariable analyses indicated strong evidence of an effect between genetically predicted childhood body size and atherosclerosis (OR, 1.76; 95% CI, 1.37–2.27; P=1.25×10‐5), atrial fibrillation and flutter (OR, 1.69; 95% CI, 1.39–2.06; P=2.00×10‐7), coronary artery disease (OR, 1.53; 95% CI, 1.36–1.72; P=1.61×10‐12), heart failure (OR, 1.72; 95% CI, 1.45–2.05; P=5.5×10‐10), hypertension (OR, 1.77; 95% CI, 1.53–2.04; P=6.30×10‐15), myocardial infarction (OR, 1.51; 95% CI, 1.36–1.74; P=8.85×10‐11), peripheral artery disease (OR, 1.89; 95% CI, 1.48–2.42; P=4.69×10‐7), and varicose veins (OR, 1.57; 95% CI, 1.29–1.91; P=6.7×10‐6). These 8 outcomes were robust to FDR <0.05 corrections. Additionally, there was little evidence of an effect between genetically predicted childhood body size and pulmonary heart disease (OR, 1.28; 95% CI, 0.97–1.69; P=0.09), angina pectoris (OR, 1.15; 95% CI, 0.95–1.37; P=0.15), pulmonary embolism (OR, 1.27; 95% CI, 0.95–1.72; P=0.11), and stroke (OR, 1.57; 95% CI, 0.93–1.16; P=0.50) (Table S3) based on FDR <0.05. For comparison, univariable estimates for adult body size on these 12 CVD end points can be found in Table S4.
Consistent patterns of associations were observed using the weighted median method employed for robustness (Table S5). The MR‐Egger method did not suggest that horizontal pleiotropy was responsible for the estimates derived, with the possible exception of the estimate between childhood body size and atrial fibrillation and flutter (Table S6).
Upon accounting for adult body size using multivariable MR, estimates for all 8 end points that survived FDR corrections in the univariable analysis substantially attenuated (Figure 2A). These estimates provided little evidence of a direct effect between genetically predicted childhood body size and atherosclerosis (OR, 1.13; 95% CI, 0.81–1.59; P=0.468), atrial fibrillation and flutter (OR, 0.91; 95% CI, 0.71–1.17; P=0.462), coronary heart disease (OR, 1.05; 95% CI, 0.89–1.25; P=0.551), heart failure (OR, 0.87; 95% CI, 0.69–1.09; P=0.224), hypertension (OR, 0.96; 95% CI, 0.90–1.15; P=0.650), myocardial infarction (OR, 0.98; 95% CI, 0.82–1.18; P=0.848), peripheral artery disease (OR, 1.25; 95% CI, 0.90–1.73; P=0.118), and varicose veins (OR, 1.17; 95% CI, 0.92–1.50; P=0.211). In contrast, multivariable analyses provided strong evidence that adult body size directly increases risk of the 8 CVD disease outcomes analyzed (Figure 2B). Lastly, although there was little evidence of an effect between childhood body size and the 4 outcomes not included in multivariable analyses, adult body size provided strong evidence of a total effect on angina pectoris (OR, 1.67; 95% CI, 1.36–2.05; P=1.28×10‐6), pulmonary embolism (OR, 1.61; 95% CI, 1.15–2.25; P=0.005), pulmonary heart disease (OR, 1.77; 95% CI, 1.30–2.42; P=2.96×10‐4), and stroke (OR, 1.56; 95% CI, 1.36–1.79; P=1.36×10‐10) (Table S7).
Figure 2. Univariable for childhood body size (A) and multivariable for childhood and adult body size (B) Mendelian randomization onto CVD outcomes.
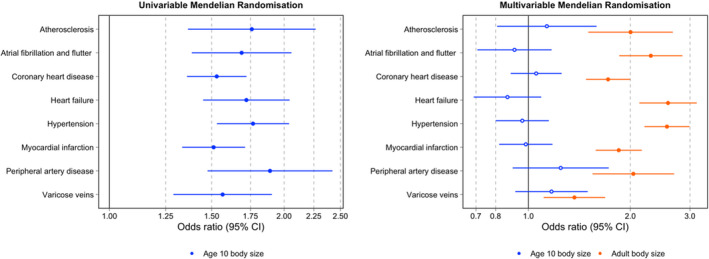
CVD indicates cardiovascular disease.
Discussion
In the present MR investigation, we applied a lifecourse approach to investigate the mechanisms underlying the causal effect of genetically predicted childhood body size on the risk of a spectrum of later life CVD outcomes linked to obesity. This was to determine whether childhood body size has an independent effect on CVD risk or whether its influence can be explained by accounting for body size in adulthood. We report results based on direct or indirect effects as described previously in the multivariable Mendelian randomization literature. 27 A consistent relationship between higher childhood body size and an increase in the odds of CVD was identified in univariable analyses. When accounting for adult body size, however, estimates attenuated to include the null (and in some cases reversed direction of effect), indicating weak evidence of a direct effect between childhood body size and CVD outcomes in multivariable analyses. There was strong evidence indicating the influence of adult body size on CVD outcomes in both univariable and multivariable analyses.
Results from this study are in line with previous clinical and epidemiological observational research, indicating that childhood adiposity is a risk factor for several disease outcomes, including type 2 diabetes mellitus, hypertension, dyslipidemia and carotid‐artery atherosclerosis, only if individuals remain overweight into puberty or later ages. 6 , 7 These findings therefore highlight the importance of prevention efforts to reduce adiposity in prepubescent children to help mitigate adverse cardiovascular consequences in later life. 28 Ensuring socially disadvantaged groups benefit from relevant public health interventions aimed at reducing obesity is especially key in both policy and practice, because obesity levels in high‐income countries disproportionately affect ethnic minority, low‐income, and other socially marginalized populations. 29 For example, in the context of the United States, the prevalence of a BMI above the 85th percentile for age and sex rose to 35% in Hispanic and Black children compared with 20% in their counterparts of European ancestry. 28
In addition, findings from this report highlight the advantages of considering the long‐term effects of childhood and early adult risk factors on later disease to elucidate processes operating across the lifecourse, that influence the development of disease risk. 8 Previous longitudinal research has investigated adverse outcomes in overweight individuals at multiple different stages throughout early life. For example, a retrospective cohort study comprising individuals born between 1930 and 1989 in Copenhagen, Denmark, assessed the adverse effects of weight gain in childhood (7 years of age), adolescence (13 years of age) and early adulthood on type 2 diabetes mellitus risk. 7 Whereas those who were overweight between 7 to 13 years who had subsequently maintained normal weight in early adulthood had a risk of type 2 diabetes mellitus similar to those with normal weights at all ages, those who were overweight between 13 and early adulthood had a risk of type 2 diabetes mellitus higher than those who had never been overweight. In addition, a prospective cohort study completed on the Israeli Defence Force Medical Corps revealed that elevated BMI in both adolescence (mean 17.44±0.46 years) and adulthood (mean 30.59±5.30 years) were independently associated with angiography‐proven coronary heart disease. 30 Furthermore, in this study we found that the effect sizes of childhood body size estimates on CVD were consistently smaller than adulthood body size in univariable analyses. Although the magnitude of total effect estimates are relevant from a lifecourse perspective, only through multivariable analyses can evidence of direct and indirect effects be inferred. Future investigations using genetics would benefit from developing genetic scores for multiple age groups to assess the causal effects of increased body size at specific ages on adverse outcomes related to obesity.
Strengths and Limitations
Although the association between obesity and CVD risk has been investigated longitudinally in observational research, this is a unique study in that it estimates effects on various endpoints throughout the CVD disease spectrum, using genetically predicted exposures at separate timepoints in the lifecourse. Doing so means that our effect estimates should be more robust to reverse causation and confounding compared with previous observational estimates. In addition, this investigation leverages large sample sizes available through the UKBB study (n=453 169) for measures of early and later life body size and the CARDIOGRAMplusC4D consortium (n=185 000), MEGASTROKE consortium (n=521 612), and FinnGen study (n=269 077) for measures of CVD outcomes, increasing study power. This study also uses a 2‐sample approach, where the risk factor and outcome are taken from nonoverlapping data sets, minimizing the potential for bias from overfitting. 31 Furthermore, weighted median and MR‐Egger methods were used to assess the robustness of univariable results to horizontal pleiotropy.
This study, however, also has important limitations. First, participants’ self‐reporting of their body size in childhood may have resulted in differential social desirability bias, with respect to retrospective weight recall at age 10. Furthermore, the age of participants in adulthood when reporting this information could potentially influence the childhood body size measurement, because individuals recalled their body weight at different ages in midlife, resulting in different lengths of time since they were children. To account for this, GWAS were conducted on individuals who had both measures available adjusting for age, as well as sex and the genotyping chip. Though, as described earlier using measured BMI data from the Young Finns Study 13 and the HUNT Study, 14 the genetic score for early life body size has been shown to be a better predictor of childhood BMI and the score for adult body size a better predictor of adult BMI. Second, although effect estimates in this study were positive, there was weak evidence of a genetically predicted effect between childhood body size and 4 of the 12 risk factors in univariable analyses. This may be because of a lack of power based on the number of cases for these endpoints in the FinnGen dataset. However, this could also potentially indicate that having a larger body size does not begin to exert its effect on angina pectoris, pulmonary embolism, pulmonary heart disease, and stroke as early in the lifecourse as other outcomes assessed. This requires further investigation. Third, this study uses genetically determined body size, which may not translate directly into weight loss or gain from lifestyle reforms. 9 Fourth, selection bias using the UKBB is an important limitation, because participants in the UKBB are more likely to be older, female, and live in less socioeconomically deprived areas than participants in nationally representative data sources. 32 Underrepresentation of younger, male, nonbinary, or any other gender identity and the lowest socioeconomic position group is therefore problematic, with the potential to cause issues for instrumental variable analyses. 33 Furthermore, because allele frequencies and disease or exposure rates vary between different subgroups of the population, introducing the potential for confounding, this study performs analyses in homogeneous populations of European ancestry. 34 This therefore provides evidence of the causal effects identified only within this single ancestry group and may thus not be generalizable to other ancestry populations. Future research should therefore investigate estimates derived in this study across a broader range of different ancestries. Lastly, genetic correlation has been shown to exist in the UKBB between birth location and several health outcomes relevant to our study, after adjusting for population structure, including hypertension and BMI. 35 Future work to assess the extent to which environmental factors confounded with location may influence findings from UKBB analyses would be worthwhile.
Conclusions
These findings provide evidence that the total effect of childhood body size on CVD outcomes is a result of larger body size persisting into adulthood, suggesting that childhood obesity is unlikely to have a direct causal effect on the CVD outcomes assessed in this work. Importantly, elevated risk of these types of CVD associated with childhood adiposity may therefore putatively be mitigated or potentially reversed by early resolution of obesity in early life.
Sources of Funding
This work was in part supported by the MRC Integrative Epidemiology Unit which receives funding from the UK Medical Research Council and the University of Bristol (MC_UU_00011/1). Davey Smith conducts research at the National Institute for Health Research Biomedical Research Centre at the University Hospitals Bristol National Health Service (NHS) Foundation Trust and the University of Bristol. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health. Power is supported by grant MR/N0137941/1 for the GW4 Biomed Doctoral Training Programme, awarded to the Universities of Bath, Bristol, Cardiff, and Exeter from the Medical Research Council (MRC)/UKRI. Richardson was a UK Research and Innovation Innovation Research Fellow while contributing to this study (MR/S003886/1). Frayling has received funding from the Medical Research Council, MR/T002239/1 and the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 875534. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and European Federation of Pharmaceutical Industries and Associations and T1D Exchange, JDRF, and Obesity Action Coalition. Tyrell is supported by an Academy of Medical Sciences (AMS) Springboard award, which is supported by the AMS, the Wellcome Trust, GCRF, the Government Department of Business, Energy and Industrial Strategy, the British Heart Foundation and Diabetes UK (SBF004\1079).
Disclosures
T.G.R. has begun a part‐time post with Novo Nordisk since this article was submitted. The remaining authors have no disclosures to report.
Supporting information
Table S1–S7
Acknowledgments
We thank the UK Biobank study and all participants who contributed to it, as well as the authors of all the GWAS who made their summary statistics available for the benefit of this work. Estimates on childhood and adult body size were derived previously using data from the UK Biobank (app #15825).
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.021503
For Sources of Funding and Disclosures, see page 7.
Contributor Information
Grace M. Power, Email: grace.power@bristol.ac.uk.
Tom G. Richardson, Email: tom.g.richardson@bristol.ac.uk.
References
- 1. Chan RSM, Woo J. Prevention of overweight and obesity: how effective is the current public health approach. Int J Environ Res Public Health. 2010;7:765–783. DOI: 10.3390/ijerph7030765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sacks G, Swinburn B, Xuereb G. Population‐based approaches to childhood obesity prevention. World Health Organization (WHO). 2012.
- 3. Branca F, Nikogosian H, Lobstein T. The challenge of obesity in the WHO European Region and the strategies for response: summary. World Health Organization (WHO). 2007.
- 4. Umer A, Kelley GA, Cottrell LE, Giacobbi P Jr, Innes KE, Lilly CL. Childhood obesity and adult cardiovascular disease risk factors: a systematic review with meta‐analysis. BMC Public Health. 2017;17:683. DOI: 10.1186/s12889-017-4691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol. 2003;32:1–22. DOI: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 6. Juonala M, Magnussen CG, Berenson GS, Venn A, Burns TL, Sabin MA, Srinivasan SR, Daniels SR, Davis PH, Chen W, et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med. 2011;365:1876–1885. DOI: 10.1056/NEJMoa1010112. [DOI] [PubMed] [Google Scholar]
- 7. Bjerregaard LG, Jensen BW, Ängquist L, Osler M, Sørensen TIA, Baker JL. Change in overweight from childhood to early adulthood and risk of type 2 diabetes. N Engl J Med. 2018;378:1302–1312. DOI: 10.1056/NEJMoa1713231. [DOI] [PubMed] [Google Scholar]
- 8. Kuh D, Ben‐Shlomo Y, Lynch J, Hallqvist J, Power C. Life course epidemiology. J Epidemiol Community Health. 2003;57:778. DOI: 10.1136/jech.57.10.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richardson TG, Sanderson E, Elsworth B, Tilling K, Davey Smith G. Use of genetic variation to separate the effects of early and later life adiposity on disease risk: mendelian randomisation study. BMJ. 2020;369:m1203. DOI: 10.1136/bmj.m1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–R98. DOI: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. DOI: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. DOI: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richardson TG, Mykkanen J, Pahkala K, Ala‐Korpela M, Bell JA, Taylor K, Viikari J, Lehtimäki T, Raitakari O, Davey Smith G. Evaluating the direct effects of childhood adiposity on adult systemic metabolism: a multivariable mendelian randomization analysis. medRxiv. 2020;2020.08.25.20181412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brandkvist M, Bjørngaard JH, Ødegård RA, Åsvold BO, Davey Smith G, Brumpton B, Hveem K, Richardson TG, Åberge VG. Separating the genetics of childhood and adult obesity: a validation study of genetic scores for body mass index in adolescence and adulthood in the HUNT Study. Hum Mol Genet. 2021;29:3966–3973. DOI: 10.1093/hmg/ddaa256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malhotra S, Sivasubramanian R, Singhal V. Adult obesity and its complications: a pediatric disease? Curr Opin Endocrinol Diabetes Obes. 2021;28:46–54. DOI: 10.1097/MED.0000000000000592. [DOI] [PubMed] [Google Scholar]
- 16. Zhu J, Su X, Li G, Chen J, Tang B, Yang Y. The incidence of acute myocardial infarction in relation to overweight and obesity: a meta‐analysis. Arch Med Sci. 2014;10:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Csige I, Ujvárosy D, Szabó Z, Lőrincz I, Paragh G, Harangi M, Somodi S. The impact of obesity on the cardiovascular system. J Diabetes Res. 2018;2018:3407306. DOI: 10.1155/2018/3407306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ohlsson C, Bygdell M, Sondén A, Jern C, Rosengren A, Kindblom JM. BMI increase through puberty and adolescence is associated with risk of adult stroke. Neurology. 2017;89:363–369. DOI: 10.1212/WNL.0000000000004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. DOI: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten‐Jacobs L, Giese A‐K, van der Laan SW, Gretarsdottir S, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. DOI: 10.1038/s41588-018-0058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. FinnGen . The FinnGen project: Institute for Molecular Medicine Finland (FIMM) at the University of Helsinki. 2020. [updated 2020 9 November; cited 2021 3 January]. Available from: http://r4.finngen.fi.
- 22. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR‐Base platform supports systematic causal inference across the human phenome. Elife. 2018;7. DOI: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665. DOI: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. DOI: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. DOI: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ginestet C. ggplot2: elegant graphics for data analysis. J R Stat Soc Series A. 2011;174:245–246. DOI: 10.1111/j.1467-985X.2010.00676_9.x. [DOI] [Google Scholar]
- 27. Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single‐sample and two‐sample summary data settings. Int J Epidemiol. 2018;48:713–727. DOI: 10.1093/ije/dyy262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goran MI, Ball GD, Cruz ML. Obesity and risk of type 2 diabetes and cardiovascular disease in children and adolescents. J Clin Endocrinol Metab. 2003;88:1417–1427. DOI: 10.1210/jc.2002-021442. [DOI] [PubMed] [Google Scholar]
- 29. Kumanyika SK. A framework for increasing equity impact in obesity prevention. Am J Public Health. 2019;109:1350–1357. DOI: 10.2105/AJPH.2019.305221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tirosh A, Shai I, Afek A, Dubnov‐Raz G, Ayalon N, Gordon B, Derazne E, Tzur D, Shamis A, Vinker S, et al. Adolescent BMI trajectory and risk of diabetes versus coronary disease. N Engl J Med. 2011;364:1315–1325. DOI: 10.1056/NEJMoa1006992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two‐sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608. DOI: 10.1002/gepi.21998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, Collins R, Allen NE. Comparison of sociodemographic and health‐related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–1034. DOI: 10.1093/aje/kwx246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hughes RA, Davies NM, Davey Smith G, Tilling K. Selection bias when estimating average treatment effects using one‐sample instrumental variable analysis. Epidemiology. 2019;30:350–357. DOI: 10.1097/EDE.0000000000000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sekula P, Del Greco MF, Pattaro C, Köttgen A. Mendelian randomization as an approach to assess causality using observational data. J Am Soc Nephrol. 2016;27:3253. DOI: 10.1681/ASN.2016010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook JP, Mahajan A, Morris AP. Fine‐scale population structure in the UK Biobank: implications for genome‐wide association studies. Hum Mol Genet. 2020;29:2803–2811. DOI: 10.1093/hmg/ddaa157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1–S7