

Abstract
Background
Apoptosis plays a pivotal role in cardiac rupture after myocardial infarction (MI), and p53 is a key molecule in apoptosis during cardiac rupture. Hif‐1α (hypoxia‐inducible factor‐1α), upregulated under hypoxia, is a known p53 inducer. However, the role of Hif‐1α in the regulatory mechanisms underlying p53 upregulation, apoptosis, and cardiac rupture after MI is unclear.
Methods and Results
We induced MI in mice by ligating the left anterior descending artery. Hif‐1α and p53 expressions were upregulated in the border zone at day 5 after MI, accompanied by apoptosis. In rat neonatal cardiomyocytes, treatment with cobalt chloride (500 μmol/L), which mimics severe hypoxia by inhibiting PHD (prolyl hydroxylase domain‐containing protein), increased Hif‐1α and p53, accompanied by myocyte death with caspase‐3 cleavage. Silencing Hif‐1α or p53 inhibited caspase‐3 cleavage, and completely prevented myocyte death under PHD inhibition. In cardiac‐specific Hif‐1α hetero‐knockout mice, expression of p53 and cleavage of caspase‐3 and poly (ADP‐ribose) polymerase were reduced, and apoptosis was suppressed on day 5. Furthermore, the cleavage of caspase‐8 and IL‐1β (interleukin‐1β) was also suppressed in hetero knockout mice, accompanied by reduced macrophage infiltration and matrix metalloproteinase/tissue inhibitor of metalloproteinase activation. Although there was no intergroup difference in infarct size, the cardiac rupture and survival rates were significantly improved in the hetero knockout mice until day 10 after MI.
Conclusions
Hif‐1α plays a pivotal role in apoptosis, inflammation, and cardiac rupture after MI, in which p53 is a critical mediator, and may be a prospective therapeutic target for preventing cardiac rupture.
Keywords: apoptosis, cardiac rupture, Hif‐1α, myocardial infarction, p53
Subject Categories: Basic Science Research, Cell Biology/Structural Biology, Myocardial Biology, Myocardial Infarction
Nonstandard Abbreviations and Acronyms
- caHetKO
cardiac‐specific Hif‐1α hetero‐knockout
- Hif‐1α
hypoxia‐inducible factor‐1α
- IL‐1β
interleukin‐1β
- MMP
matrix metalloproteinase
- PARP
poly (ADP‐ribose) polymerase
- PHD
prolyl hydroxylase domain‐containing protein
- TIMP
tissue inhibitor of metalloproteinase
Clinical Perspective
What Is New?
Hif‐1α (hypoxia‐inducible factor‐1α) protects against ischemic injuries such as myocardial infarction (MI) and ischemia reperfusion injury; however, excessive and prolonged Hif‐1α expression causes apoptosis through p53 induction and triggers cardiac rupture following MI.
Loss of cardiomyocytes due to apoptosis in the MI border zone leads to heart‐tissue vulnerability, resulting in cardiac rupture.
Hif‐1α also activates caspase‐8, an inflammatory caspase, thereby exacerbating inflammation accompanied by IL‐1β (interleukin‐1β) processing and enhanced extracellular matrix reconstruction through the matrix metalloproteinase/tissue inhibitor of metalloproteinase system. Apoptosis and inflammation, triggered by Hif‐1α and p53, are major pathophysiologies of post‐MI cardiac rupture.
What Are the Clinical Implications?
Mechanical complications, including cardiac rupture following MI, have become less common because primary percutaneous coronary intervention markedly reduces their onset following acute MI.
The current COVID‐19 pandemic has delayed primary percutaneous coronary intervention in acute MI, and thus mechanical complications have reportedly increased worldwide.
Excessive and sustained ischemia can trigger cardiac rupture through the Hif‐1α–p53 axis, which causes apoptosis and inflammation via caspase‐3 and caspase‐8, respectively. Our findings support clinical evidence indicating that late successful reperfusion therapy and use of β‐blockers following MI reduce mechanical complications during acute MI, underscoring their significance in delayed clinical cases.
Cardiac rupture is a fatal complication after myocardial infarction (MI), and is associated with high hospital mortality, although early revascularization via percutaneous coronary intervention (PCI) reduces its occurrence. 1 Several potential molecular mechanisms, including inflammation and the matrix metalloproteinase (MMP)/tissue inhibitor of metalloproteinase (TIMP) system, have been demonstrated to be the key pathophysiologic processes of cardiac rupture. 2 , 3 , 4 , 5 , 6 Apoptosis is also a key pathological feature in cardiac rupture after MI. 2 , 7 , 8 We previously reported that targeted deletion of p53 suppressed apoptosis and prevented cardiac rupture after MI, suggesting that p53 and p53‐dependent apoptosis play a key role in the pathogenesis of cardiac rupture. 7 However, the molecular mechanism underlying the regulation of p53 and apoptosis during MI is not fully understood.
p53 is a proapoptotic transcriptional factor regulated by several stressors, such as DNA damage, nutrient depletion, reactive oxygen species, and heat shock. 9 Particularly, Hif‐1α (hypoxia‐inducible factor‐1α), which is upregulated under hypoxic conditions, also induces p53. 10 Under well‐oxygenated conditions, PHDs (prolyl hydroxylase domain‐containing proteins) hydroxylate Hif‐1α, after which von Hippel‐Lindau binds to hydroxylated Hif‐1α and recruits ubiquitin ligase, resulting in ubiquitination and proteasome degradation of Hif‐1α. 11 In contrast, under hypoxic conditions, PHDs cannot hydroxylate Hif‐1α, interrupting the ubiquitination and degradation of Hif‐1α. Thus, Hif‐1α accumulates intracellularly and induces hypoxic responses as a transcriptional and nontranscriptional factor. 10 , 11 , 12 , 13 Because the myocardium during MI is exposed to sustained severe ischemia, we hypothesized that excessive Hif‐1α under PHD inhibition due to sustained ischemia upregulates p53 in the MI border zone, and thereby induces apoptosis and cardiac rupture during MI.
In the present study, we analyzed MI mice and cultured cardiomyocytes under PHD inhibition to investigate the role of Hif‐1α in p53 upregulation and apoptosis, and the significance of p53 in Hif‐1α–induced apoptosis. We also analyzed the myocardium in the MI border zone and the prognosis after MI in cardiomyocyte‐specific Hif‐1α hetero‐knockout (caHetKO) mice to examine the role of Hif‐1α in cardiac rupture.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Murine MI Model
All procedures involving animals and animal care protocols were approved by the Committee on Ethics of Animal Experiments of the Kyushu University Faculty of Medical and Pharmaceutical Sciences (A27‐327, A29‐150, and A19‐028), and were performed in accordance with the Guideline for Animal Experiments of Kyushu University and Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised in 2011). To retain homogeneity in the MI model, male C57BL/6J mice (Kyudo, Saga, Japan) were used in this study. The animals were housed in a temperature‐ and humidity‐controlled room, fed a commercial diet (CRF‐1; Oriental Yeast, Tokyo, Japan), and given free access to water. Hif‐1α‐floxed mice and Myh6‐Cre mice (stock no. 007561 and 011038, respectively; Jackson Laboratory, Bar Harbor, ME) 14 , 15 were crossed to generate cardiomyocyte‐specific Hif‐1α caHetKO mice. Mice that did not possess sequences of Hif‐1α‐flox and Myh6‐Cre were used as control; 10‐ to 14‐week‐old mice were anesthetized with a mixture of medetomidine (0.3 mg/kg), midazolam (4 mg/kg), and butorphanol tartrate (5 mg/kg) (Wako Chemicals, Osaka, Japan) via intraperitoneal administration according to institutional recommendations. Under mechanical ventilation, the intercostal space was opened, and MI was induced by ligating the left coronary artery as described previously. 16 Sham mice underwent operation without arterial ligation. Mice that showed myocardial pallor during ligation and survived 24 hours after ligation were enrolled in the survival protocol (n=40 and n=45 in control and caHetKO groups, respectively). Mice with no histological evidence of transmural infarction based on Masson's trichrome staining (the base, mid, and apex regions) were excluded (n=2 and n=1 in control and caHetKO groups, respectively). Sample size was determined based on previous studies on cardiac rupture after MI. 8 , 16 Cardiac rupture was diagnosed by intrathoracic hematoma and/or fissure of the heart during autopsy. Rupture rates and survival were considered as primary and secondary end points, respectively. These end points were assessed objectively without blinding because their definitions were clear and rigorous.
Echocardiography
Echocardiographic parameters were measured in 2‐dimensional targeted M‐mode images obtained from the short‐axis view at the papillary muscle level using a Vevo2100 ultrasonography system (Visual Sonics, Toronto, Canada) under light anesthesia (1%–2% isoflurane). 17
Histopathology
After in vivo studies, such as survival evaluation and echocardiography, were conducted, pentobarbital overdoses were administered for euthanasia, and the mouse hearts were excised. Hearts were stored in 10% formalin for at least 4 days for histological analysis, cut at 3 levels (base, mid, apex), and embedded in paraffin. The 3‐mm sectioned samples were stained with Masson's trichrome reagent as described previously. 16 Infarct lengths were measured along the endocardial and epicardial surfaces in each of the Masson's trichrome‐stained cardiac sections, and values from all specimens were summed. Infarct sizes (in percentages) were calculated as the total infarct circumference divided by the total cardiac circumference.
Terminal Deoxynucleotidyl Transferase‐Mediated dUTP Nick End‐Labeling Staining
Left ventricular tissue sections were stained by terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling (TUNEL) (Takara, Shiga, Japan), as described previously. 18 Border lines were drawn according to inflammatory cell invasion. Border zones were defined as points 100 μm from the border line on both sides. TUNEL‐positive nuclei were counted in the border zone, and the border zone was measured using ImageJ software (National Institutes of Health, Bethesda, MD). The data were normalized in counts per square millimeter.
Cultured Neonatal Rat Ventricular Cardiomyocytes
Primary cultures of isolated neonatal rat ventricular cardiomyocytes were prepared from the ventricles of neonatal Sprague‐Dawley rats as described previously, with some modifications. 19 , 20 Neonatal rats were euthanized by isoflurane overdose, and the hearts were rapidly excised. After digesting the myocardial tissue with trypsin (Wako) and collagenase type 2 (Worthington Biochemical, Freehold, NJ), the cells were suspended in DMEM (Sigma‐Aldrich, St. Louis, MO; D6046) containing 10% FBS (Hyclone, Logan, UT; SH30396, lot no. ABB213138) and penicillin/streptomycin (Nacalai Tesque, Kyoto, Japan; 26253‐84). Cells were plated twice in 100‐mm culture dishes (Cellstar; Greiner Bio‐One, Kremsmünster, Austria) for 70 minutes each to reduce the number of nonmyocytes. Nonadherent cells were plated in culture dishes (Primaria; Corning, Corning, NY) at a density of 2.5×105 cells/mL for each experiment. Isolated cardiomyocytes were maintained at 37℃ in humidified air with 5% CO2 for 24 to 36 hours after plating on the culture dishes. Images of cultured cardiomyocytes were obtained using an Olympus IX71 microscope (Olympus, Tokyo, Japan).
Transfection of Cultured Cardiomyocytes With Small Interfering RNA
Cultured cardiomyocytes were transfected with small interfering RNA (siRNA) targeting each gene using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA) as described previously. 20 siRNAs targeting Hif‐1α (RS0035615 and RS0035615; Takara) and p53 (s128541; Thermo Fisher Scientific) were used in this study. Briefly, after changing the penicillin/streptomycin‐free DMEM containing 10% FBS, Opti‐MEM (Gibco, Grand Island, NY; 31985‐070) containing each siRNA and Lipofectamine RNA iMAX was added to each well to achieve a final concentration of 1 nmol/L siRNA. The medium was changed to one containing 10% FBS and penicillin/streptomycin at 24 hours, and then CoCl2 was added to inhibit PHDs and mimic severe hypoxia in vitro starting at 48 hours after siRNA transfection.
Quantitative Reverse Transcription Polymerase Chain Reaction
Total RNA extraction, reverse transcription, and quantitative reverse transcription‐ polymerase chain reaction were performed as described previously, with some modifications. 21 , 22 Briefly, total RNA was extracted using an RNeasy Mini Kit (Qiagen, Hilden, Germany), and was converted to cDNA using a ReverTra Ace quantitative reverse transcription‐polymerase chain reaction kit (Toyobo, Osaka, Japan). The reactions were performed in an Applied Biosystems QuantStudio3 with THUNDERBIRD SYBR qPCR Mix (Toyobo) in accordance with the manufacturer's instructions (denaturation, 95℃ for 15 seconds; annealing and elongation, 60℃ for 45 seconds). The forward and reverse primer sequences are presented in Table S1.
Western Blotting
Western blotting was performed as previously described. 20 , 21 Briefly, frozen myocardial tissue samples were homogenized in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific) containing a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), 1 mmol/L NaF, and 0.1 mmol/L Na3VO4. Equal amounts of protein (20 μg per lane) were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis, and then electrophoretically transferred onto a nitrocellulose membrane using a Trans‐Blot apparatus (Bio‐Rad). After blocking for 1 hour with skim milk in Tris‐buffered saline containing 1% Tween 20, the membrane was incubated with primary antibody at 4 °C overnight, followed by incubation with the appropriate secondary antibody at room temperature for 1 hour. Primary antibodies against the following proteins were used: Hif‐1α (36169; Cell Signaling Technology [CST], Danvers, MA), p53 (32532; CST), cleaved caspase‐3 (9579; CST), cleaved poly (ADP‐ribose) polymerase (PARP) (9548; CST), cleaved caspase‐1 (67314; CST), caspase‐8 (9429; CST), cleaved caspase‐8 (4790; CST), CD107b (Mac‐3; 550292; BD Biosciences, Franklin Lakes, NJ), MMP‐2 (87809; CST), MMP‐9 (ab228402; Abcam, Cambridge, UK), TIMP‐1 (ab179580; Abcam), and TIMP‐2 (5738; CST). An antibody against GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase; sc‐32233; Santa Cruz Biotechnology, Dallas, TX) was used as a control. Anti‐rabbit, anti‐mouse, and anti‐rat immunoglobulin G antibodies (7074, 7076, and 7077, respectively; CST) were used as secondary antibodies. We acquired images with a Western Lighting ECL Pro (Perkin Elmer, Waltham, MA) or Chemi‐Lumi One Ultra (11644‐40; Nacalai Tesque, Kyoto, Japan) using Fusion (Vilber Lourmat, Marne‐la‐Vallée, France).
Cell Viability Assay
Cell viability assays were performed using CellTiter‐Blue (G808A; Promega, Madison, WI) in accordance with the manufacturer's instructions. After 48 hours of treatment with CoCl2 (500 μmol/L), the medium (100 μL/well) was changed to FBS‐free DMEM, and 20 μL of CellTiter‐Blue was added to each well of a 96‐well plate. After 1 hour incubation at 37℃, the fluorescence intensity (excitation 560 nm, emission 590 nm) was measured with a Varioskan LUX (VLB000D0; Thermo Fisher Scientific). Data are shown as a percentage of the control.
Statistical Analysis
All data are shown as the mean±standard error of the mean. To compare groups, we used the Student t test, Pearson χ2 test, or 1‐way ANOVA, followed by the Tukey or Dunnett post hoc test. Survival analysis was performed using the Kaplan‐Meier method, and differences in survival between groups were evaluated with the log‐rank test. Results with P<0.05 were considered statistically significant. The JMP15 Pro software (SAS Institute, Cary, NC) was used for statistical analysis.
Results
Hif‐1α, p53, and Apoptosis Were Induced in the Border Zone of MI
The myocardium in the MI border zone, which is the most frequent site at which cardiac rupture occurs after MI induction, was analyzed biochemically on day 5, as previously described. 16 , 23 Both Hif‐1α and p53 were significantly increased in the border zone of MI at day 5 after MI (Figure 1A and 1B). Cleaved PARP and caspase‐3, representing apoptosis, were also induced, and TUNEL‐positive cells were significantly increased in the border zone of MI (Figure 1C and 1D).
Figure 1. Characterization of the myocardium in the myocardial infarction (MI) border zone at day 5 after MI.
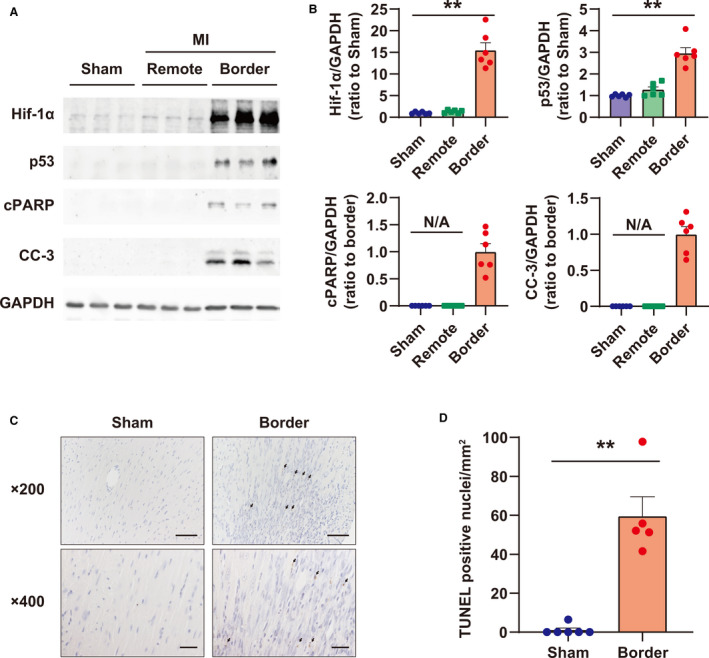
A, Western blots of Hif‐1α (hypoxia‐inducible factor‐1α), p53, cPARP (cleaved poly [ADP‐ribose] polymerase), and CC‐3 (cleaved caspase‐3) in the MI border zone. GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) was used as a loading control. B, Quantification of Western blots shown in (A); n=6 in each group. **P<0.01, analyzed by Dunnett test vs sham. Data are shown as the ratio to sham group average or border group average. C, Representative images of terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling (TUNEL) staining in the MI border zone on day 5. The arrow indicates TUNEL‐positive nuclei. Bar = 50 μm in 200× images, and 20 μm in 400× images. D, Quantification of TUNEL‐positive cells in the MI border zone per square millimeter. Sham: n=6; MI border: n=5. **P<0.01 vs Sham, analyzed by Student t test. N/A indicates not applicable.
Excessive Hif‐1α Induced p53 Upregulation and Apoptosis in Isolated Cardiomyocytes
To clarify the role of Hif‐1α in p53 upregulation and apoptosis in cardiomyocytes, the expression of Hif‐1α, p53, and cleaved caspase‐3 at 24 hours, as well as cell survival at 48 hours, after CoCl2 treatment were examined using siRNA targeting Hif‐1α in cultured cardiomyocytes. Inhibition of PHDs by CoCl2, mimicking severe hypoxia during ischemia via PHD inhibition, increased Hif‐1α in a CoCl2 concentration‐dependent manner, whereas p53 was steadily increased at 500 μmol/L in our preliminary experiments (Figure S1A and S1B). Therefore, we used 500 μmol/L of CoCl2 in this study. CoCl2 treatment did not affect the expression of Tp53 at the transcriptional level (Figure 2A), whereas it excessively increased Hif‐1α and p53 at the protein level, and induced caspase‐3 cleavage in cardiomyocytes (Figure 2B and 2C), indicating that excessive Hif‐1α increased p53 protein by stabilizing p53, as reported previously by An et al. 10 In contrast, silencing of Hif‐1α significantly inhibited the increase in p53 induced by CoCl2 treatment and fully inhibited the cleavage of caspase‐3 (Figure 2B and 2C). Although prolonged inhibition of PHDs (48 hours) led to myocyte death in vitro in cultured cardiomyocytes, silencing of Hif‐1α completely prevented myocyte death induced by CoCl2 treatment (Figure 2D and 2E).
Figure 2. Role of Hif‐1α (hypoxia‐inducible factor 1‐α) in upregulating p53 expression and apoptosis induced by PHD (prolyl hydroxylase domain) inhibition using CoCl2 in cultured rat cardiomyocytes.
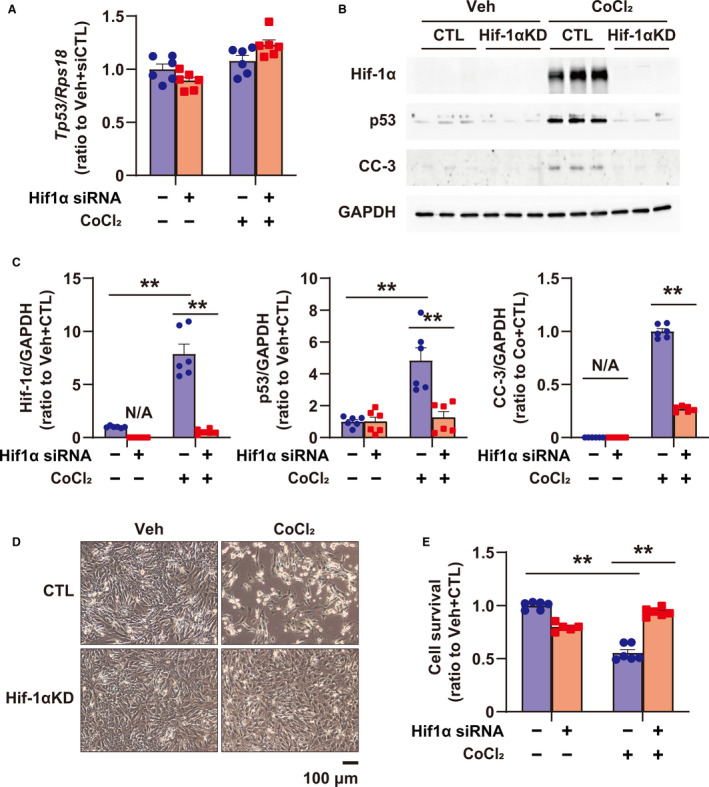
A, Tp53 expression in cultured cardiomyocytes treated with vehicle (Veh) and CoCl2 (500 μmol/L) for 24 hours, or transfected with small interfering RNA (siRNA) for control (CTL) (scramble siRNA, CTL) and Hif‐1α, was quantified by quantitative reverse transcription‐polymerase chain reaction; n=6 in each group. B, Western blots of Hif‐1α, p53, and CC‐3 (cleaved caspase‐3) in cardiomyocytes treated with Veh and CoCl2 (500 μmol/L) for 24 hours, or transfected with siRNA for CTL (scramble siRNA, CTL) and Hif‐1α. GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) was used as a loading CTL. C, Quantification of Western blots shown in (B); n=6 in each group. Data are shown as a ratio to the average of Veh+CTL. D, Representative images of cardiomyocytes treated with Veh and CoCl2 (500 μmol/L), or transfected with siRNA for CTL (scramble siRNA, CTL) and Hif‐1α. E, Cellular viability using cultured cardiomyocytes treated with Veh and CoCl2 (500 μmol/L), or transfected with siRNA for CTL (scramble siRNA, CTL) and Hif‐1α by CellTiterBlue Assay; n=5‒6 in each group. **P<0.01, analyzed by 1‐way ANOVA, followed by Tukey post hoc test. KD indicates knockdown; and N/A, not applicable.
p53 Was a Major Mediator of Hif‐1α‐Induced Apoptosis
To investigate the role of p53 in Hif‐1α–induced apoptosis, the expression of Hif‐1α, p53, and cleaved caspase‐3 at 24 hours and cell survival at 48 hours after CoCl2 treatment were examined after treatment with siRNA targeting p53 in cultured cardiomyocytes (Figure 3A). Silencing of p53 inhibited the cleavage of caspase‐3, even in the presence of excess Hif‐1α (Figure 3B and 3C), and fully prevented Hif‐1α–induced cell death in cardiomyocytes (Figure 3D and 3E). These results suggest that p53 is a major mediator of Hif‐1α–induced apoptosis in cardiomyocytes.
Figure 3. Role of p53 in apoptosis induced by PHD (prolyl hydroxylase domain) inhibition using CoCl2 in cultured rat cardiomyocytes.
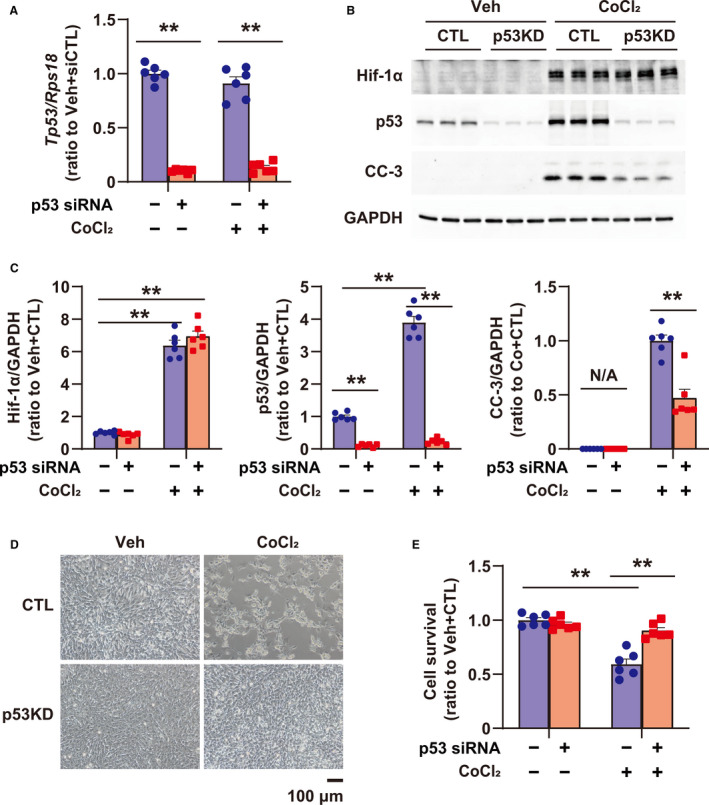
A, Tp53 expression in cultured cardiomyocytes, treated with vehicle (Veh) and CoCl2 (500 μmol/L) for 24 hours, or transfected with small interfering RNA (siRNA) for control (CTL) (scramble siRNA, CTL) and p53, was quantified by quantitative reverse transcription‐polymerase chain reaction; n=6 in each group. B, Expression of Hif‐1α, p53, and CC‐3 (cleaved caspase 3) in cardiomyocytes treated with Veh and CoCl2 (500 μmol/L) for 24 hours, or transfected with siRNA for CTL (scramble siRNA, CTL) and p53. GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) was used as a loading CTL. C, Quantification of Western blots shown in (B); n=6 in each group. Data are shown as a ratio to the average of Veh+CTL. D, Images of cardiomyocytes treated with Veh and CoCl2 (500 μmol/L), or transfected with siRNA for CTL (scramble siRNA, CTL) and p53. E, Cellular viability using cultured cardiomyocytes treated with Veh and CoCl2 (500 μmol/L), or transfected with siRNA for CTL (scramble siRNA, CTL) and p53 by CellTiterBlue Assay; n=6 in each group. **P<0.01, analyzed by 1‐way ANOVA, followed by Tukey post hoc test. KD indicates knockdown; and N/A, not applicable.
Cardiomyocyte‐Specific Hetero Deletion of Hif‐1α Reduced p53 and Apoptosis in the Border Zone of MI
To investigate the role of Hif‐1α as an inducer of p53 and apoptosis in the border zone after MI, cardiac‐specific Hif‐1α (+/−)/Myh6Cre (+/−) mice (caHetKO) were prepared by crossing Hif‐1α‐floxed mice and Myh6‐Cre mice. Echocardiography showed no differences in the left ventricular ejection fraction and left ventricular end‐diastolic diameter between control mice and caHetKO at day 5 after MI, whereas left ventricular ejection fraction was reduced and left ventricular end‐diastolic diameter was increased in MI of both wild type (WT) and caHetKO mice (Figure S2A). In addition, heart and lung weights were comparable between the 2 groups in MI (Figure S2B). In contrast, Hif‐1α was decreased, and p53, cleaved PARP, and caspase‐3 were significantly reduced in the MI border zone in caHetKO mice on day 5 (Figure 4A and 4B). Furthermore, TUNEL‐positive cells after MI were significantly suppressed in caHetKO mice (Figure 4C and 4D).
Figure 4. Hif‐1α (hypoxia‐inducible factor 1‐α), p53, and apoptosis in the myocardial infarction (MI) border zone of cardiomyocyte‐specific Hif‐1α hetero‐knockout (caHetKO) mice on day 5.
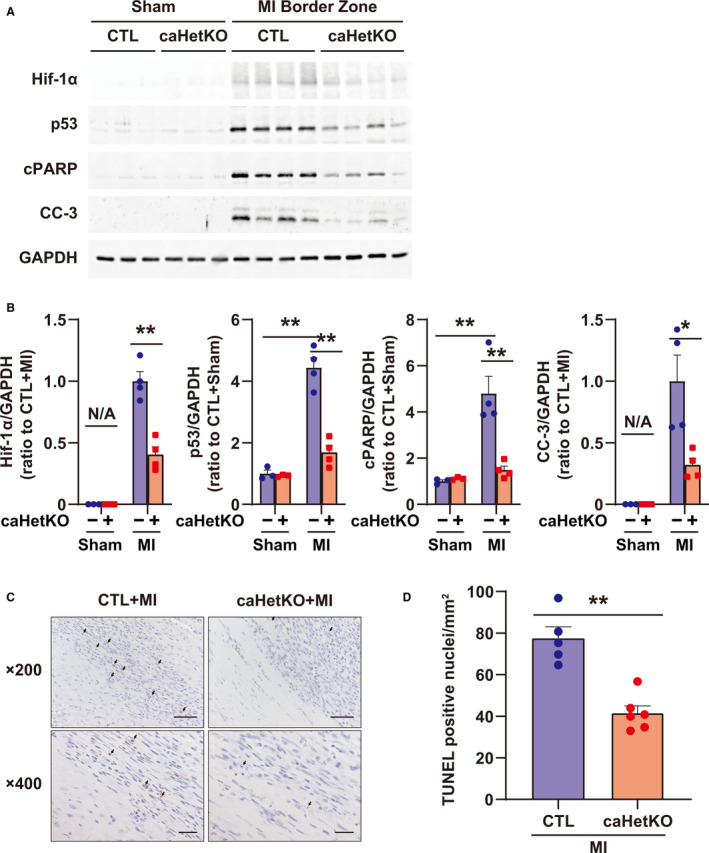
A, Expression of Hif‐1α, p53, cPARP (cleaved poly [ADP‐ribose] polymerase), and CC‐3 (cleaved caspase 3) in the MI border zone at day 5 after MI. GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) was used as a loading control (CTL). B, Quantification of Western blots shown in (A); n=4 in each group. Data are shown as a ratio to CTL+sham or CTL+MI mice. C, Representative images of terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling (TUNEL)‐positive cells in CTL and caHetKO mice at day 5 after MI. D, Quantification of TUNEL‐positive cell count per square millimeter in CTL and caHetKO mice; n=5–6 in each group. The arrow indicates TUNEL‐positive nuclei. Bar = 50 μm in 200× images and 20 μm in 400× images. Data are shown as a ratio to the average of CTL+MI group. *P<0.05, **P<0.01 vs CTL, analyzed by 1‐way ANOVA, followed by Tukey post hoc test or Student t test (for Hif‐1α and CC‐3 in B and D). N/A indicates not applicable.
Cleavage of Caspase‐8 Was Suppressed in caHetKO Mice
MMP‐2 and MMP‐9, secreted from inflammatory cells such as Mac‐3–positive macrophages, regulate extracellular matrix reconstruction and are predominantly responsible for the induction of cardiac rupture after MI. 3 , 4 , 5 Caspase‐8 is an inflammatory caspase that can drive apoptosis as well as inflammation through IL‐1β processing, 24 and p53 regulates caspase‐8 activation. 25 Cleavage of caspase‐8 and IL‐1β was increased in the MI border zone, whereas it was suppressed in caHetKO mice (Figure 5A and 5B). Consistent with these findings, Mac‐3 representing macrophage invasion in the MI border zone was reduced, and the upregulation of MMP‐2 and MMP‐9, as well as TIMP‐1 and TIMP‐2, was suppressed in the border zone (Figure 5A and 5B). In cultured cardiomyocytes, CoCl2 treatment also enhanced the cleavage of caspase‐8, which was inhibited by silencing Hif‐1α and p53 (Figure S3). In contrast, CoCl2 treatment did not upregulate the gene expression of either MMPs or TIMPs. Although Hif‐1α or p53 downregulated expression of the Mmp9 or Timp4 genes, respectively, silencing Hif‐1α and p53 did not directly affect expression of other genes (Figure S4A and S4B). Collectively, these results indicate that inflammation, which is regulated by Hif‐1α and p53‐dependent caspase‐8 activation and IL‐1β processing, contributes to exacerbating extracellular matrix reconstruction through MMP/TIMP activation.
Figure 5. Role of Hif‐1α (hypoxia‐inducible factor 1‐α) in CC‐8 (cleaved caspase‐8), inflammation, and matrix metalloproteinase (MMP)/tissue inhibitor of metalloproteinase (TIMP) in the myocardial infarction (MI) border zone on day 5.
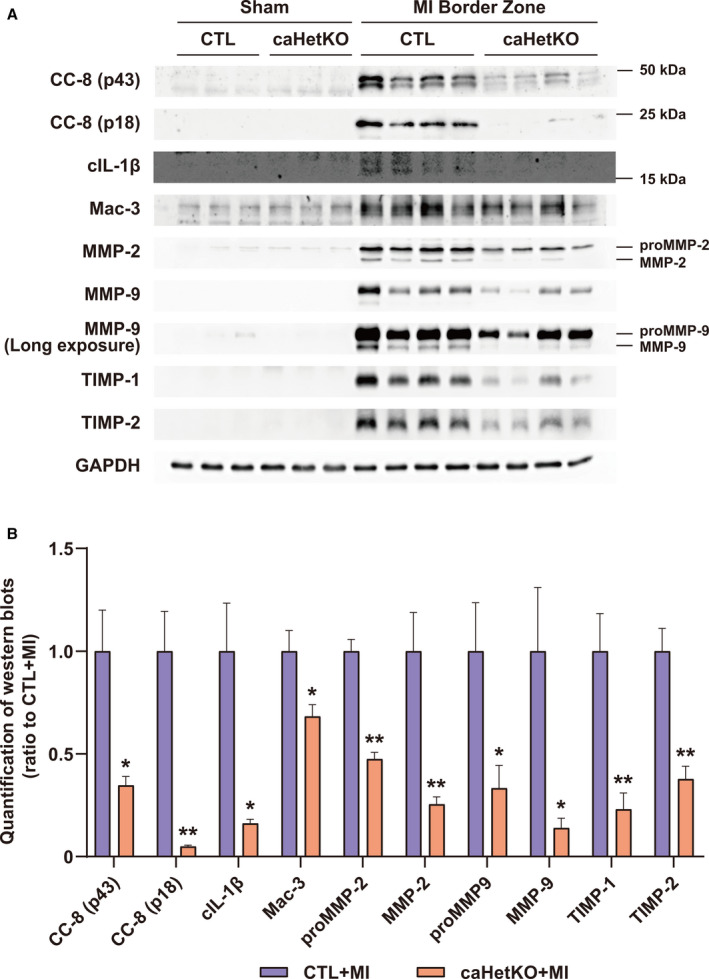
A, Expression of CC‐8, cleaved interleukin‐1β (cIL‐1β), Mac‐3, MMP‐2, MMP‐9, TIMP‐1, and TIMP‐2 in the MI border zone at day 5 after MI. GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) was used as a loading control (CTL). B, Quantification of Western blots shown in (A); n=4 in each group. Data are shown as a ratio to CTL+MI. *P<0.05, **P<0.01 vs CTL+MI, analyzed by Student t test. caHetKO indicates cardiac‐specific Hif‐1α hetero‐knockout; and N/A indicates not applicable.
Cardiomyocyte‐Specific Hetero Deletion of Hif‐1α Prevented Cardiac Rupture and Improved the Prognosis After MI
To investigate the effect of Hif‐1α deletion on cardiac rupture, we observed MI mice until day 10 and analyzed the surviving mice. Echocardiographic images of the left ventricular end‐diastolic diameter and left ventricular ejection fraction, and the weights of organs such as the heart and lung, did not differ between the groups of surviving mice at day 10 after MI (Figure S5A and S5B). In addition, infarct size was comparable between control and caHetKO mice (Figure 6A). Nevertheless, cardiac rupture was significantly prevented, and survival was improved in caHetKO mice (survival rate, 47% in control and 75% in caHetKO mice; Figure 6B, Table). These results suggest that excessive Hif‐1α is a major inducer of p53, inflammation, and apoptosis in the MI border zone during sustained cardiac ischemia, which can be suppressed by reduction in Hif‐1α expression in the MI border zone, allowing the prevention of cardiac rupture. Collectively, excessive Hif‐1α is a major pathological feature of cardiac rupture after MI, as summarized in Figure 7.
Figure 6. Infarct size and survival after myocardial infarction (MI) in cardiomyocyte‐specific Hif‐1α hetero‐knockout mice (caHetKO mice) until day 10.
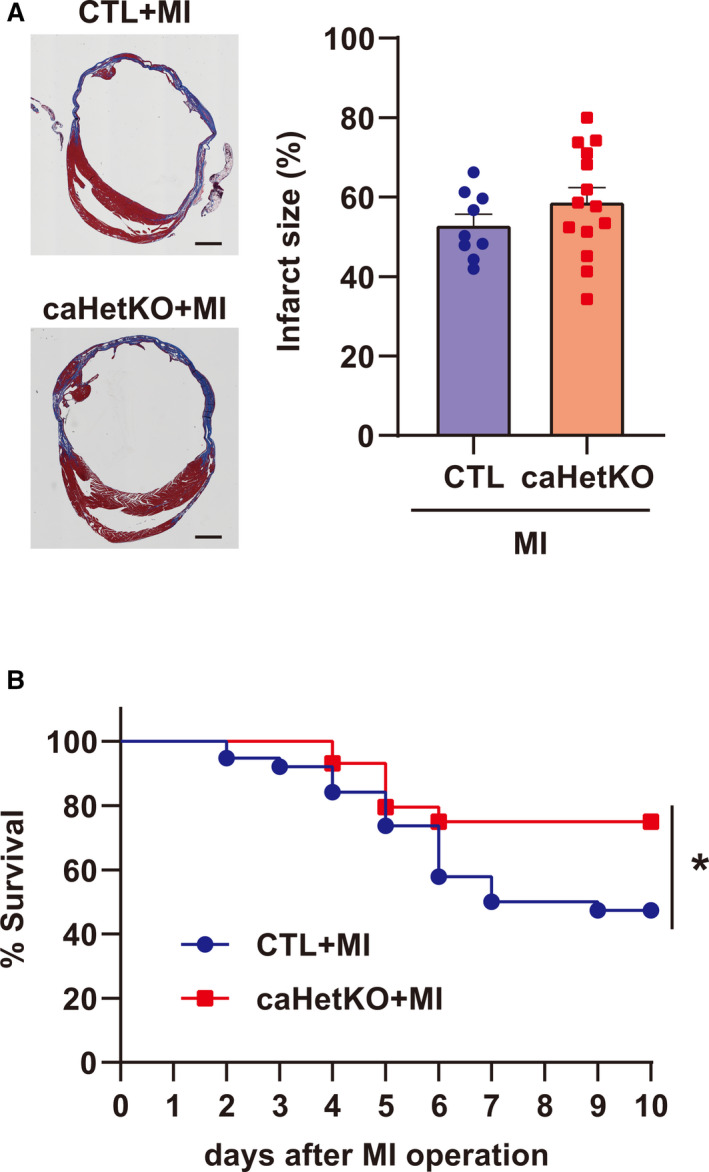
A, Representative images demonstrating infarct size, and quantification of infarct size in control (CTL) and caHetKO mice after MI. CTL mice: n=9; caHetKO mice: n=14. Bar = 1 mm. B, Survival of CTL and caHetKO mice after MI. CTL mice: n=38; caHetKO mice: n=44. Survival analysis was performed using the Kaplan‐Meier method. Between‐group differences in survival were evaluated by the log‐rank test. *P<0.05 vs CTL+MI mice. Hif‐1α indicates hypoxia‐inducible factor 1‐α.
Table 1.
Rupture and Nonrupture Death Rates in Control and Cardiomyocyte‐Specific Hif‐1α Hetero‐Knockout Mice
| Variables | CTL‐MI | caHetKO‐MI | P value |
|---|---|---|---|
| Summary of survival | |||
| No. | 38 | 44 | |
| Rupture rate, % | 45 (17/38) | 23 (10/44) | 0.03 |
| Rupture site, border/infarct/undetermined | 13/1/3 | 10/0/0 | 0.25 |
| Nonrupture death rate, % | 8 (3/38) | 2 (1/43) | 0.23 |
The rupture site was determined by locating the hole or fissure during autopsy. The rupture sites in 3 mice could not be determined due to tearing after adhesion of the heart to the chest wall during autopsy. Rupture and nonrupture death rates were analyzed by Pearson χ2 test. caHetKO‐MI indicates cardiomyocyte‐specific Hif‐1α hetero‐knockout–myocardial infarction; CTL‐MI, control‐myocardial infarction; and Hif‐1α, hypoxia‐inducible factor‐1α.
Figure 7. Schematic showing the mechanistic role of the Hif‐1α‐p53 axis in cardiac rupture after myocardial infarction (MI).
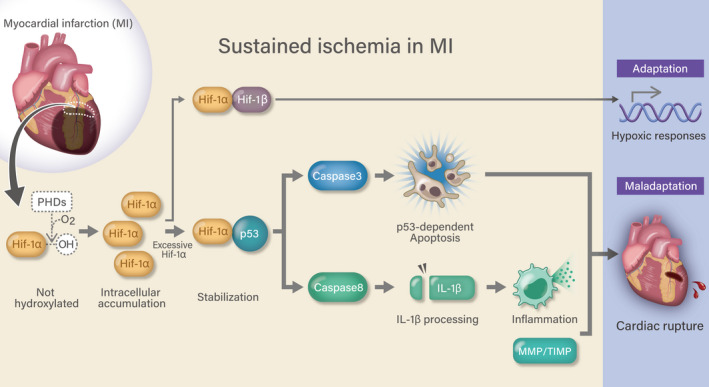
Hif‐1α indicates hypoxia‐inducible factor 1‐α; Hif‐1β, hypoxia‐inducible factor 1‐β; IL‐1β, interleukin‐1β; MMP/TIMP, matrix metalloproteinase/tissue inhibitor of metalloproteinase; OH, hydroxy group; and PHD, prolyl hydroxylase domain‐containing protein.
Discussion
Our primary findings were as follows: (1) Hif‐1α is a major inducer of p53 and apoptosis in cultured cardiomyocytes and the myocardium in the MI border zone in vivo. (2) p53 is a critical mediator of Hif‐1α–induced apoptosis. (3) A reduction in Hif‐1α leads to prevention of cardiac rupture after MI, accompanied by suppression of p53, inflammation, and apoptosis. We previously reported that deletion of p53 suppressed apoptosis in the MI border zone and prevented cardiac rupture, indicating that p53 plays a major role in apoptosis and cardiac rupture during MI. 7 However, the regulatory mechanisms of p53 and apoptosis during MI are not clear. We demonstrated that an excessive level of Hif‐1α was a major inducer of p53 expression, apoptosis, inflammation, and cardiac rupture during MI. This is the first report describing the role of Hif‐1α in cardiac rupture after MI.
Hif‐1α primarily regulates metabolism, cell proliferation, and cell growth in response to hypoxia as an adaptive mechanism. 26 Studies by our group and others demonstrated that Hif‐1α induces ischemic tolerance, and protects against ischemia/reperfusion injury and MI induced by permanent ligation of a coronary artery. 18 , 27 , 28 , 29 , 30 , 31 , 32 Notably, our findings indicate that excessive and prolonged activation of Hif‐1α leads to myocyte death through p53 upregulation and apoptosis in the MI border zone after MI. These phenomena may appear contradictory, but apoptosis might be a reasonable choice for myocytes exposed to severe and sustained ischemia. When myocytes are exposed to hypoxia, Hif‐1α initially lowers oxygen consumption through metabolic reprogramming, which is the switch from aerobic to anaerobic metabolism, to ensure myocyte survival even under hypoxia as an adaptation. 18 , 33 However, Hif‐1α may also drive some myocytes to cell death through p53 upregulation and apoptosis to rescue other myocytes by ultimately reducing oxygen consumption when myocytes are exposed to severe or sustained hypoxia. These aspects of Hif‐1α may enable regulation of cell survival and death, because apoptosis is a programmed cell death pathway for ensuring organ survival and saving the whole organism through self‐sacrifice. 34 However, silencing p53 failed to completely abolish the cleavage of caspase‐3 and caspase‐8 induced by CoCl2 treatment in cultured cardiomyocytes (Figure 3, Figure S3), indicating that the other mechanism excluding p53 partly mediated Hif‐1α–induced apoptosis. Thus, further investigation is needed to fully reveal the molecular mechanism of Hif‐1α–induced apoptosis in cardiomyocytes.
In this study, we also showed that the hetero deletion of Hif‐1α suppressed the inflammation and subsequent extracellular matrix reconstruction through the MMP/TIMP system in the MI border zone. Based on the evidence that the activation of the MMP/TIMP system was suppressed in caHetKO mice, we first hypothesized that Hif‐1α or p53 transcriptionally regulated the gene expression of MMP/TIMP. However, CoCl2 treatment with Hif‐1α and p53 silencing in cultured cardiomyocytes could not elucidate the mechanism by which the MMP/TIMP system was suppressed in caHetKO mice (Figure S4A and S4B). Although apoptosis is classically recognized as regulated cell death unaccompanied by inflammation, inflammatory caspases, such as caspase‐1 and caspase‐8, have been reported to induce inflammation through the processing and secretion of IL‐1β. 24 In particular, caspase‐1, which causes pyroptosis, is a representative caspase that can cleave IL‐1β; however, cleaved caspase‐1 was not increased in the MI border zone (Figure S6). Caspase‐8 is another caspase responsible for the cleavage of IL‐1β and is known to be regulated by p53. 25 Cleaved caspase‐8 was markedly increased in the MI border zone, and this increase was suppressed in caHetKO of Hif‐1α (Figure 5). Moreover, CoCl2‐induced cleavage of caspase‐8 was prevented by silencing Hif‐1α and p53 (Figure S3). These results suggested that the Hif‐1α–p53 axis also activated caspase‐8 and IL‐1β processing, and thereby induced inflammation with extracellular matrix reconstruction through the activation of the MMP/TIMP system, which critically contributed to the induction of cardiac rupture after MI. Furthermore, Hif‐1α might directly regulate the inflammation, independent of p53, because Hif‐1α is also well known to regulate inflammation. 35 Although we were not able to separate the roles of Hif‐1α and p53 in inflammation in the MI border zone, we deduced that excessive Hif‐1α in the MI border zone would also induce inflammation through caspase‐8 and IL‐1β directly or via p53 induction.
Nevertheless, there was no reduction in infarct size in caHetKO mice, although apoptosis was significantly suppressed in the MI border zone. This may be because Hif‐1α has a protective effect on infarct size after MI, as previously described, 29 and thus, deletion of Hif‐1α abolishes its protective effect. As mentioned above, Hif‐1α exerts opposing effects by protecting against or promoting cell death. The detrimental effect of Hif‐1α deletion may mask its protective effect against apoptosis in the MI border zone, resulting in comparable infarct sizes between the 2 groups.
In addition to cardiac rupture, Hif‐1α–induced apoptosis may play a critical role in the progression of cardiomyopathy in heart failure (HF). Hif‐1α is upregulated in the myocardium in the end stage of HF, as demonstrated in previous studies, 36 , 37 whereas prolonged activation of Hif‐1α, by the transgene of Hif‐1α or PHD inhibition in cardiomyocytes, induces susceptibility to mechanical stress‐related impairment, resulting in cardiac dysfunction, and excessively overexpressed Hif‐1α induces cardiomyopathy. 36 , 37 , 38 , 39 , 40 Taken together, these reports indicate that excessive or prolonged Hif‐1α expression contributes to the progression of cardiomyopathy in HF. Although the molecular mechanism by which excessive or prolonged Hif‐1α activation induces cardiac dysfunction requires further analysis, 40 p53‐dependent apoptosis triggered by Hif‐1α expression may contribute to aggravation of cardiomyopathy in the end stage of HF because p53 is the key molecule determining the failed phenotype of cardiomyocytes in HF. 41
Early primary PCI reduces cardiac rupture, and mechanical complications including cardiac rupture have clinically become rare. 1 , 42 Nevertheless, the COVID‐19 pandemic has delayed primary PCI for acute MI worldwide, 43 , 44 , 45 and thus, an increase in mechanical complications is currently creating concerns. 46 , 47 , 48 Our findings may provide the rationale of potential strategies for preventing mechanical complications in the delayed presentation of MI. Primary PCI maximally lessens myocardial damage within 90 minutes after the onset of MI. 49 , 50 , 51 However, delayed PCI may also be effective for preventing mechanical complications by releasing sustained ischemia in the MI border zone and potentially suppressing apoptosis and inflammation induced by excessive Hif‐1α. This strategy is supported by clinical evidence showing that successful late reperfusion by primary PCI is associated with a reduced risk of mechanical complications in patients with AMI. 52 Moreover, reducing oxygen consumption with β‐blockers may also be effective for the prevention of mechanical complications. Clinical studies showed that β‐blockers clinically prevent cardiac rupture after MI. 53 , 54 Recently, we demonstrated that heart rate reduction with ivabradine markedly reduced apoptosis in the MI border zone and prevented cardiac rupture, improving the survival after MI in mice. 8 Given that heart rate reduction with ivabradine can reduce oxygen consumption in the myocardium and prolong the perfusion time, 55 , 56 apoptosis suppressed by ivabradine may be achieved partly by attenuating excessive hypoxic responses. Because delayed PCI does not frequently result in sufficient perfusion in the myocardium due to coronary microvascular occlusion, 57 a pharmacological approach as well as reperfusion may be important for preventing mechanical complications.
In conclusion, excessive Hif‐1α is a major inducer of p53 upregulation, inflammation, apoptosis, and cardiac rupture after MI. Thus, the Hif‐1α‐p53 axis may be a critical therapeutic target for cardiac rupture.
Sources of Funding
This work was supported by the Japan Society for the Promotion of Science KAKENHI grants (M.I.: JP16H07049: JP18K15892: JP21K16090: JP20K08426), and a research grant from the Japan Foundation for Applied Enzymology (Vascular Biology of Innovation), the MSD Life Science Foundation, the Public Interest Incorporated Foundation, and Novartis Pharma Grants for Basic Research 2020 (M.I.).
Disclosures
Dr Tsutsui received grants from Daiichi Sankyo, Mitsubishi‐Tanabe Pharma, Nippon Boehringer, IQVIA Services Japan, Omron Healthcare, and MEDINET; personal fees from AstraZeneca, Ono Pharmaceutical, Otsuka Pharmaceutical, Daiichi Sankyo, Mitsubishi‐Tanabe Pharma, Teijin Pharma, Nippon Boehringer Ingelheim, Novartis Pharma, Bayer Yakuhin, Pfizer Japan, Bristol‐Myers Squibb, Kowa, and Nippon Rinsho. The remaining authors have no disclosures to report.
Supporting information
Table S1
Figures S1–S6
Acknowledgments
The authors thank A. Hanada for her excellent experimental techniques. The authors also appreciate T. Tohyama for consultation on the statistical analysis.
Author contributions: M.I., T.T., H.D.M., S.I., K.O., A.I., M.S., K.A., S.F., and K.I. performed the experiments. M.I., T.I., S.M., and H.T. interpreted the data obtained from the experiments. M.I. and T.I. designed experimental protocols, wrote the article, and prepared the figures. M.I. and T.I. conceived the project. H.T. approved and supervised the project.
For Sources of Funding and Disclosures, see page 13.
Contributor Information
Masataka Ikeda, Email: ikeda-m@cardiol.med.kyushu-u.ac.jp.
Tomomi Ide, Email: tomomi_i@cardiol.med.kyushu-u.ac.jp.
References
- 1. Figueras J, Alcalde O, Barrabes JA, Serra V, Alguersuari J, Cortadellas J, Lidon RM. Changes in hospital mortality rates in 425 patients with acute ST‐elevation myocardial infarction and cardiac rupture over a 30‐year period. Circulation. 2008;118:2783–2789. DOI: 10.1161/CIRCULATIONAHA.108.776690. [DOI] [PubMed] [Google Scholar]
- 2. Sane DC, Mozingo WS, Becker RC. Cardiac rupture after myocardial infarction: new insights from murine models. Cardiol Rev. 2009;17:293–299. DOI: 10.1097/CRD.0b013e3181bf4ab4. [DOI] [PubMed] [Google Scholar]
- 3. Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens J, Shipley M, Angellilo A, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–11342. DOI: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 4. Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, et al. Targeted deletion of matrix metalloproteinase‐9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. DOI: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayashidani S, Tsutsui H, Ikeuchi M, Shiomi T, Matsusaka H, Kubota T, Imanaka‐Yoshida K, Itoh T, Takeshita A. Targeted deletion of MMP‐2 attenuates early left ventricular rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;285:H1229–H1235. DOI: 10.1152/ajpheart.00207.2003. [DOI] [PubMed] [Google Scholar]
- 6. Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, Liu PP. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110:3221–3228. DOI: 10.1161/01.CIR.0000147233.10318.23. [DOI] [PubMed] [Google Scholar]
- 7. Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of p53 prevents cardiac rupture after myocardial infarction in mice. Cardiovasc Res. 2006;70:457–465. DOI: 10.1016/j.cardiores.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 8. Ikeda M, Ide T, Furusawa S, Ishimaru K, Tadokoro T, Miyamoto HD, Ikeda S, Okabe K, Ishikita A, Abe KO, et al. Heart rate reduction with ivabradine prevents cardiac rupture after myocardial infarction in mice. Cardiovasc Drugs Ther. 2021. Jan 7 [epub ahead of print]. DOI: 10.1007/s10557-020-07123-5. [DOI] [PubMed] [Google Scholar]
- 9. Meek DW. Regulation of the p53 response and its relationship to cancer. Biochem J. 2015;469:325–346. DOI: 10.1042/BJ20150517. [DOI] [PubMed] [Google Scholar]
- 10. An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild‐type p53 by hypoxia‐inducible factor 1alpha. Nature. 1998;392:405–408. [DOI] [PubMed] [Google Scholar]
- 11. Semenza GL. Hypoxia‐inducible factors in physiology and medicine. Cell. 2012;148:399–408. DOI: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. DOI: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 13. Villa JC, Chiu D, Brandes AH, Escorcia FE, Villa CH, Maguire WF, Hu CJ, de Stanchina E, Simon MC, Sisodia SS, et al. Nontranscriptional role of Hif‐1alpha in activation of gamma‐secretase and notch signaling in breast cancer. Cell Rep. 2014;8:1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia‐inducible factor‐1α is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- 15. Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac restricted, site‐specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. DOI: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inoue T, Ikeda M, Ide T, Fujino T, Matsuo Y, Arai S, Saku K, Sunagawa K. Twinkle overexpression prevents cardiac rupture after myocardial infarction by alleviating impaired mitochondrial biogenesis. Am J Physiol Heart Circ Physiol. 2016;311:H509–H519. DOI: 10.1152/ajpheart.00044.2016. [DOI] [PubMed] [Google Scholar]
- 17. Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, Tyynismaa H, Yamasaki T, Yamada K‐I, Kang D, et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One. 2015;10:e0119687. DOI: 10.1371/journal.pone.0119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deguchi H, Ikeda M, Ide T, Tadokoro T, Ikeda S, Okabe K, Ishikita A, Saku K, Matushima S, Tsutsui H. Roxadustat markedly reduces myocardial ischemia reperfusion injury in mice. Circ J. 2020;84:1028–1033. DOI: 10.1253/circj.CJ-19-1039. [DOI] [PubMed] [Google Scholar]
- 19. Fujino T, Ide T, Yoshida M, Onitsuka K, Tanaka A, Hata Y, Nishida M, Takehara T, Kanemaru T, Kitajima N, et al. Recombinant mitochondrial transcription factor A protein inhibits nuclear factor of activated T cells signaling and attenuates pathological hypertrophy of cardiac myocytes. Mitochondrion. 2012;12:449–458. DOI: 10.1016/j.mito.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 20. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada K‐I, et al. Mitochondria‐dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5:e132747. DOI: 10.1172/jci.insight.132747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ikeda M, Ide T, Fujino T, Matsuo Y, Arai S, Saku K, Kakino T, Oga Y, Nishizaki A, Sunagawa K. The Akt‐mTOR axis is a pivotal regulator of eccentric hypertrophy during volume overload. Sci Rep. 2015;5:15881. DOI: 10.1038/srep15881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arai S, Ikeda M, Ide T, Matsuo Y, Fujino T, Hirano K, Sunagawa K, Tsutsui H. Functional loss of DHRS7C induces intracellular Ca2+ overload and myotube enlargement in C2C12 cells via calpain activation. Am J Physiol Cell Physiol. 2017;312:C29–C39. DOI: 10.1152/ajpcell.00090.2016. [DOI] [PubMed] [Google Scholar]
- 23. Gao XM, Xu Q, Kiriazis H, Dart AM, Du XJ. Mouse model of post‐infarct ventricular rupture: time course, strain‐ and gender‐dependency, tensile strength, and histopathology. Cardiovasc Res. 2005;65:469–477. DOI: 10.1016/j.cardiores.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 24. Opdenbosch NV, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50:1352–1364. DOI: 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding HF, Lin YL, McGill G, Juo P, Zhu H, Blenis J, Yuan J, Fisher DE. Essential role for caspase‐8 in transcription‐independent apoptosis triggered by p53. J Biol Chem. 2000;49:38905–38911. DOI: 10.1074/jbc.M004714200. [DOI] [PubMed] [Google Scholar]
- 26. Majmundar AJ, Wong WJ, Simon MC. Hypoxia‐inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. DOI: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bishop T, Ratcliffe PJ. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ Res. 2015;117:65–79. DOI: 10.1161/CIRCRESAHA.117.305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cai Z, Zhong H, Bosch‐Marce M, Fox‐Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioning‐induced cardioprotection in mice with partial deficiency of HIF‐1 alpha. Cardiovasc Res. 2008;77:463–470. DOI: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- 29. Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, Thistlethwaite PA. Hypoxia‐inducible factor 1‐alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. DOI: 10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 30. Adluri RS, Thirunavukkarasu M, Dunna NR, Zhan L, Oriowo B, Takeda K, Sanchez JA, Otani H, Maulik G, Fong GH, et al. Disruption of hypoxia‐inducible transcription factor‐prolyl hydroxylase domain‐1 (PHD‐1‐/‐) attenuates ex vivo myocardial ischemia/reperfusion injury through hypoxia‐inducible factor‐1alpha transcription factor and its target genes in mice. Antioxid Redox Signal. 2011;15:1789–1797. DOI: 10.1089/ars.2010.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holscher M, Silter M, Krull S, von Ahlen M, Hesse A, Schwartz P, Wielockx B, Breier G, Katschinski DM, Zieseniss A. Cardiomyocyte‐specific prolyl‐4‐hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J Biol Chem. 2011;286:11185–11194. DOI: 10.1074/jbc.M110.186809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie L, Pi X, Wang Z, He J, Willis MS, Patterson C. Depletion of PHD3 protects heart from ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis. J Mol Cell Cardiol. 2015;80:156–165. DOI: 10.1016/j.yjmcc.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF‐1‐dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci. 2019;20:238. DOI: 10.3390/ijms20020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin SJ. Apoptosis: suicide, execution or murder? Trends Cell Biol. 1993;3:141–144. DOI: 10.1016/0962-8924(93)90128-N. [DOI] [PubMed] [Google Scholar]
- 35. Palazon A, Goldrath A, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41:518–528. DOI: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, Signoretti S, Liao R, Kaelin WG Jr. Loss of hypoxia‐inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation. 2010;122:1004–1016. DOI: 10.1161/CIRCULATIONAHA.109.922427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holscher M, Schafer K, Krull S, Farhat K, Hesse A, Silter M, Lin Y, Pichler BJ, Thistlethwaite P, El‐Armouche A, et al. Unfavourable consequences of chronic cardiac HIF‐1alpha stabilization. Cardiovasc Res. 2012;94:77–86. [DOI] [PubMed] [Google Scholar]
- 38. Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–3244. DOI: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bekeredjian R, Walton CB, MacCannell KA, Ecker J, Kruse F, Outten JT, Sutcliffe D, Gerard RD, Bruick RK, Shohet RV. Conditional HIF‐1alpha expression produces a reversible cardiomyopathy. PLoS One. 2010;5:e11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heusch G. HIF‐1alpha and paradoxical phenomena in cardioprotection. Cardiovasc Res. 2012;96:214–215; 216–219. DOI: 10.1093/cvr/cvs145. [DOI] [PubMed] [Google Scholar]
- 41. Nomura S, Satoh M, Fujita T, Higo T, Sumida T, Ko T, Yamaguchi T, Tobita T, Naito AT, Ito M, et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat Commun. 2018;9:4435. DOI: 10.1038/s41467-018-06639-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elbadawi A, Elgendy IY, Mahmoud K, Barakat AF, Mentias A, Mohamed AH, Ogunbayo GO, Megaly M, Saad M, Omer MA, et al. Temporal trends and outcomes of mechanical complications in patients with acute myocardial infarction. JACC Cardiovasc Interv. 2019;12:1825–1836. DOI: 10.1016/j.jcin.2019.04.039. [DOI] [PubMed] [Google Scholar]
- 43. Garcia S, Albaghdadi MS, Meraj PM, Schmidt C, Garberich R, Jaffer FA, Dixon S, Rade JJ, Tannenbaum M, Chambers J, et al. Reduction in ST‐segment elevation cardiac catheterization laboratory activations in the United States during COVID‐19 pandemic. J Am Coll Cardiol. 2020;75:2871–2872. DOI: 10.1016/j.jacc.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mafham MM, Spata E, Goldacre R, Gair D, Curnow P, Bray M, Hollings S, Roebuck C, Gale CP, Mamas MA, et al. COVID‐19 pandemic and admission rates for and management of acute coronary syndromes in England. Lancet. 2020;396:381–389. DOI: 10.1016/S0140-6736(20)31356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Rosa S, Spaccarotella C, Basso C, Calabro MP, Curcio A, Filardi PP, Mancone M, Mercuro G, Muscoli S, Nodari S, et al. Reduction of hospitalizations for myocardial infarction in Italy in the COVID‐19 era. Eur Heart J. 2020;41:2083–2088. DOI: 10.1093/eurheartj/ehaa409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ahmed T, Nautiyal A, Kapadia S, Nissen SE. Delayed presentation of STEMI complicated by ventricular septal rupture in the era of COVID‐19 pandemic. JACC Case Rep. 2020;2:1599–1602. DOI: 10.1016/j.jaccas.2020.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alsidawi S, Campbell A, Tamene A, Garcia S. Ventricular septal rupture complicating delayed acute myocardial infarction presentation during the COVID‐19 pandemic. JACC Case Rep. 2020;2:1595–1598. DOI: 10.1016/j.jaccas.2020.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Atreya AR, Kawamoto K, Yelavarthy P, Arain MA, Cohen DG, Wanamaker BL, El Ela AA, Romano MA, Grossman PM. Acute myocardial infarction and papillary muscle rupture in the COVID‐19 era. JACC Case Rep. 2020;2:1637–1641. DOI: 10.1016/j.jaccas.2020.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McNamara RL, Wang Y, Herrin J, Curtis JP, Bradley EH, Magid DJ, Peterson ED, Blaney M, Frederick PD, Krumholz HM; NRMI Investigators . Effect of door‐to‐balloon time on mortality in patients with ST‐segment elevation myocardial infarction. J Am Coll Cardiol. 2006;47:2180–2186. DOI: 10.1016/j.jacc.2005.12.072. [DOI] [PubMed] [Google Scholar]
- 50. Brodie BR, Gersh BJ, Stuckey T, Witzenbichler B, Guagliumi G, Peruga JZ, Dudek D, Grines CL, Cox D, Parise H, et al. When is door‐to‐balloon time critical? Analysis from the HORIZONS‐AMI (Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction) and CADILLAC (Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications) trials. J Am Coll Cardiol. 2010;56:407–413. DOI: 10.1016/j.jacc.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 51. Lambert L, Brown K, Segal E, Brophy J, Rodes‐Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST‐elevation myocardial infarction. JAMA. 2010;303:2148–2155. DOI: 10.1001/jama.2010.712. [DOI] [PubMed] [Google Scholar]
- 52. Nakatani D, Sato H, Kinjo K, Mizuno H, Hishida E, Hirayama A, Mishima M, Ito H, Matsumura Y, Hori M; Osaka Acute Coronary Insufficiency Study Group . Effect of successful late reperfusion by primary coronary angioplasty on mechanical complications of acute myocardial infarction. Am J Cardiol. 2003;92:785. DOI: 10.1016/S0002-9149(03)00883-X. [DOI] [PubMed] [Google Scholar]
- 53. Mechanisms for the early mortality reduction produced by beta‐blockade started early in acute myocardial infarction: ISIS‐1. ISIS‐1 (First International Study of Infarct Survival) Collaborative Group. Lancet. 1988;1:921–923. [PubMed] [Google Scholar]
- 54. Gong W, Feng S, Wang X, Fan J, Li A, Nie SP. Beta‐blockers reduced the risk of cardiac rupture in patients with acute myocardial infarction: a meta‐analysis of randomized control trials. Int J Cardiol. 2017;232:171–175. DOI: 10.1016/j.ijcard.2017.01.035. [DOI] [PubMed] [Google Scholar]
- 55. Sunagawa G, Saku K, Arimura T, Nishikawa T, Mannoji H, Kamada K, Abe K, Kishi T, Tsutsui H, Sunagawa K. Mechano‐chronotropic unloading during the acute phase of myocardial infarction markedly reduces infarct size via the suppression of myocardial oxygen consumption. J Cardiovasc Transl Res. 2019;12:124–134. DOI: 10.1007/s12265-018-9809-x. [DOI] [PubMed] [Google Scholar]
- 56. Dillinger JG, Maher V, Vitale C, Henry P, Logeart D, Manzo Silberman S, Allee G, Levy BI. Impact of ivabradine on central aortic blood pressure and myocardial perfusion in patients with stable coronary artery disease. Hypertension. 2015;66:1138–1144. DOI: 10.1161/HYPERTENSIONAHA.115.06091. [DOI] [PubMed] [Google Scholar]
- 57. Niccoli G, Scalone G, Lerman A, Crea F. Coronary microvascular obstruction in acute myocardial infarction. Eur Heart J. 2016;37:1024–1033. DOI: 10.1093/eurheartj/ehv484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figures S1–S6