

Abstract
Background
Human mutations in the X‐linked lysosome‐associated membrane protein‐2 (LAMP2) gene can cause a multisystem Danon disease or a primary cardiomyopathy characterized by massive hypertrophy, conduction system abnormalities, and malignant ventricular arrhythmias. We introduced an in‐frame LAMP2 gene exon 6 deletion mutation (denoted L2Δ6) causing human cardiomyopathy, into mouse LAMP2 gene, to elucidate its consequences on cardiomyocyte biology. This mutation results in in‐frame deletion of 41 amino acids, compatible with presence of some defective LAMP2 protein.
Methods and Results
Left ventricular tissues from L2Δ6 and wild‐type mice had equivalent amounts of LAMP2 RNA, but a significantly lower level of LAMP2 protein. By 20 weeks of age male mutant mice developed left ventricular hypertrophy which was followed by left ventricular dilatation and reduced systolic function. Cardiac electrophysiology and isolated cardiomyocyte studies demonstrated ventricular arrhythmia, conduction disturbances, abnormal calcium transients and increased sensitivity to catecholamines. Myocardial fibrosis was strikingly increased in 40‐week‐old L2Δ6 mice, recapitulating findings of human LAMP2 cardiomyopathy. Immunofluorescence and transmission electron microscopy identified mislocalization of lysosomes and accumulation of autophagosomes between sarcomeres, causing profound morphological changes disrupting the cellular ultrastructure. Transcription profile and protein expression analyses of L2Δ6 hearts showed significantly increased expression of genes encoding activators and protein components of autophagy, hypertrophy, and apoptosis.
Conclusions
We suggest that impaired autophagy results in cardiac hypertrophy and profound transcriptional reactions that impacted metabolism, calcium homeostasis, and cell survival. These responses define the molecular pathways that underlie the pathology and aberrant electrophysiology in cardiomyopathy of Danon disease.
Keywords: autophagy, calcium transients, cardiomyopathy, Danon disease, mouse model
Subject Categories: Animal Models of Human Disease, Arrhythmias, Genetically Altered and Transgenic Models, Cardiomyopathy, Calcium Cycling/Excitation-Contraction Coupling
Nonstandard Abbreviations and Acronyms
- CsA
cyclosporine A
- L2Δ6
LAMP2 gene with in‐frame deletion of exon 6
- LAMP2
lysosome‐associated membrane protein‐2
- LC3
microtubule‐associated protein light chain 3
- WT
wild‐type
Clinical Perspective
What Is New?
We developed a mouse model of Danon disease containing a mutation found in a human family; mice develop cardiomyopathy and conduction system disease comparable with cardiac manifestations in the affected humans.
Unlike previous statements associating Danons disease with absence of LAMP2 (lysosome‐associated membrane protein‐2 ), ie null allele, we demonstrate the presence of abnormal protein in mutant mice. In addition to accumulating autophagosomes, mice suffer from abnormal lysosomal localization and dysfunction.
Cardiomyocytes from mutant mice demonstrate prolongation of contraction and relaxation, abnormal calcium transients and increased sensitivity to catecholamines.
What Are the Clinical Implications?
Different effects of LAMP2 mutations on protein expression may explain the phenotypic diversity of Danon disease.
Presence of abnormal LAMP2 protein may give rise to a predominant cardiac phenotype.
Physiological and pharmacological factors affecting autophagic flux may modify the clinical course of cardiomyopathy in Danon disease.
Human mutations in the lysosome‐associated membrane protein‐2 (LAMP2) gene encoded on chromosome X produce a range of severe clinical manifestations. LAMP2 mutations that encode null alleles cause multisystem Danon disease with neurologic, hepatic, skeletal and cardiac muscle abnormalities. 1 In some patients/families, LAMP2 mutations primarily cause cardiomyopathy, which accounts for ≈1%–3% of unexplained cardiac hypertrophy in adolescents and young men. 2 , 3 , 4 LAMP2 cardiomyopathy exhibits ventricular hypertrophy, conduction system defects and ventricular arrhythmias, 1 , 5 , 6 resulting in heart failure and early death in young males and in women of diverse ages. We assumed that some of these mutations lead to production of certain amounts of abnormal LAMP2 protein, thereby accounting for the phenotypic differences. 2
LAMP2 is a 410 amino acid, highly glycated protein that together with LAMP1 constitutes about 50% of lysosomal membrane proteins. While its precise functions remain obscure, LAMP2 is thought to participate in lysosomal biogenesis, particularly the sequestration of enzymes, and in mediating lysosomal fusion with autophagosomes. This vesicular structure contains aged and defective cellular material targeted for autophagy, ie, degradation and recycling. 7 , 8 , 9 How defects in these cellular processes result in the malignant cardiomyopathy of Danon disease is not completely understood.
A previously reported LAMP2‐null mouse 10 , 11 , 12 , 13 recapitulates the multisystem manifestations of Danon disease, demonstrating prominent accumulation of autophagic vacuoles in visceral organs and striated muscles, resulting in early lethality. 12 , 13 To study how hypomorphic LAMP2 mutations cause cardiomyopathy, we targeted the endogenous murine LAMP2 gene by introducing an in‐frame deletion of exon 6 (designated L2Δ6). This mutation was previously identified by us in a human family with cardiomyopathy. 2 We hereby report the phenotype and molecular analyses of L2Δ6 mice, defining novel hypertrophic pathways that are clearly distinct from molecular signals responsible for cardiac hypertrophy by sarcomere protein gene mutations or storage diseases.
Methods
Mouse Model
Mouse studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Harvard Medical School. The data that support the findings of this study are available from the first author upon reasonable request.
The mouse LAMP2 gene was identified in murine CITB CJ7‐129 Sv BAC library (Invitrogen, Al). Using ET cloning technology, 14 a 10450bp fragment encoding exons 4–6 and flanking intron sequence (Figure 1A) was subcloned into a modified Bluescript vector. A KpnI‐SacII fragment from PK11 vector containing the neomycin resistance gene with flanking Frt sites 15 and a LoxP site was introduced into the XmaI site in intron 6. Another LoxP site was introduced by standard mutagenesis into intron 5, located 200 bp 5’of exon 6. The targeting construct (Figure 1A) was subcloned into the thymidine kinase vector between the XhoI and Not I sites.
Figure 1. Construction of in‐frame LAMP2 gene exon 6 deletion (L2Δ6) mice.
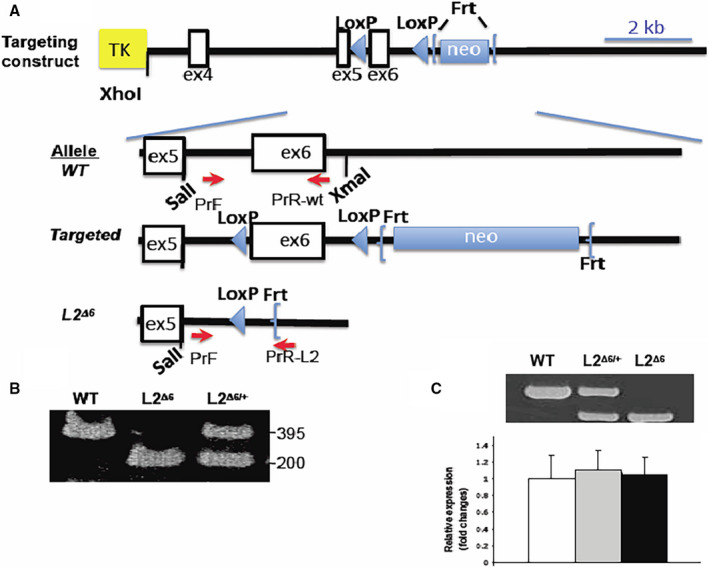
A, Illustration of the L2Δ6 targeting construct shows the neomycin resistance gene surrounded by 2 flippase recognition target (Frt) sites with exon (ex) 6 flanked by 2 locus of X‐over P1 (LoxP) sites, ligated to the thymidine kinase vector, between XhoI and NotI restriction sites. The charts below show on a larger scale the wild‐type (WT) allele, the engineered allele and the final L2Δ6 allele after applying Frt‐recombinase and Cre‐recombinase. Primers used for genotyping (see Methods) are denoted by red arrows. TK, thymidine kinase; neo, neomycin resistance gene B, Genotypes of WT, heterozygous female L2Δ6/+ and male L2Δ6 mice were determined by size characterization of polymerase chain reaction‐amplification of exon 6 using a common primer F together with R(WT) or R(L2Δ6) in 1 reaction. Amplicons of a WT (395 bp), L2Δ6 hemizygous male (≈200 bp) and of heterozygous female are shown. C, Lysosome‐associated membrane protein‐2 gene (LAMP2) reverse transcriptase‐polymerase chain reaction shows L2Δ6 RNA shorter by 123 bases compared with the WT transcript. Both WT and L2Δ6 transcripts are present in female (heterozygous) heart. Sequence analyses of polymerase chain reaction products confirmed the absence of exon 6 inL2Δ6 transcripts (data not shown). Real‐time quantitative polymerase chain reaction showed comparable levels of LAMP2 transcripts in the hearts from WT (white bar), heterozygous L2Δ6/+ (grey bar) and female L2Δ6/Δ6 (black bar) mice, mean±SEM. L2Δ6 indicates in‐frame LAMP2 gene exon 6 deletion; and WT, wild‐type.
The targeting construct was electroporated into mouse embryonic stem cells. Targeted embryonic stem cell clones were identified by Southern blot analyses: DNA was digested with SacII and probed with a fragment corresponding to 665 bases 5' of the targeting construct.
The L2Δ6 mice were made in 129SVEv embryonic stem cells. Germline LAMP2‐Neo mice were sequentially bred with mice expressing Frt‐recombinase (Jackson Laboratory 005703, B6. Cg‐Tg(ACTFLPe)9205Dym/J) to remove the neo cassette in the germline. Then they were bred with mice expressing Cre‐recombinase (Jackson Laboratory 003328, 129S/Sv‐Tg(Prm‐cre)58Og/J) to generate the germline deletion L2Δ6 mice. The recombinase transgenes were removed during multiple subsequent backcrosses into the 129S6/SvEvTac background and there was no residual Cre activity.
All experiments were performed on male mice with germline L2Δ6 mice or their wild‐type (WT) littermates. Heterozygous females had no manifest phenotype and were thus used for breeding. Mice were genotyped by polymerase chain reaction (PCR) using the following primers (denoted by red arrows Figure 1A): F‐CCTCATTTGGACACCAGAGGGAGTG; R(WT)‐ GACAGCTGCCGGTGAAGTTGGTTG or R(L2Δ6)‐TCACCGCGGTGGCGGCCG. To demarcate the conductive system, L2Δ6 mice were bred to MinK‐lacZ reporter mice. 16
Quantitative Real‐Time Reverse Transcriptase‐PCR
Two micrograms of ventricular RNA was isolated from 20‐ to 30‐week‐old L2Δ6, L2Δ6/+ heterozygous female and WT male mice using the TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed with random hexamer primers and MultiScribe reverse transcriptase (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Foster City, CA). Oligonucleotide primers F(LAMP2): 5’‐AGGCGCAAAGCTCATCCTG‐3’; R(LAMP2): 5’‐CCCATTCTGCATAAAGGCAAGTA‐3’; F (Hprt): 5’‐GTTAAGCAGTACAGCCCCAAA‐3’; R(Hprt): 5’‐AGGG‐CATATCCAACAACAAACTT‐3’ were designed using Primer3 (http://frodo.wi.mit.edu/) or retrieved from Primer Bank (http://pga.mgh.harvard.edu/primerbank/). Fifteen microliter quantitative‐PCR reactions were performed by 7500 Fast Real‐Time PCR System (Applied Biosystems, Foster City, CA) containing Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), 7.5 pmol of each primer, and 2 µL of cDNA template. Levels of mRNA expression were normalized using hypoxanthine guanine phosphoribosyl transferase‐1 (Hprt1) as control and calculated according to the ∆CT method. 17 The dissociation curve analysis was performed after each complete quantitative PCR to confirm an expected DNA product.
Protein Analysis
Lysosome protein extracts were prepared using sucrose density centrifugation based on the method of Harms et al. 18 Following tissue lysis in NP40‐containing buffer, ventricular cytoplasmic and sarcoplasmic reticulum fractions were separated by differential centrifugation as described. 19 Protein concentrations were determined by the Bradford method (Bio‐Rad) or BCA assay (Pierce). Western blots of cell fractions were performed using standard procedures with the following antibodies: rabbit monoclonal anti‐LAMP1(1:500, Sigma), rabbit monoclonal anti‐LAMP2 (1:500, Sigma), goat monoclonal anti‐Cathepsin B (1:1000, R&D systems), goat monoclonal anti‐Cathepsin D (1:1000, R&D systems), rabbit polyclonal anti‐microtubule‐associated protein light chain 3 (LC3)‐I and LC3‐II (1:1000, Cell Signaling Technology), rabbit polyclonal anti‐calsequestrin (1:2500, Affinity BioReagents), mouse monoclonal anti‐SERCA2 ATPase (1:5000, Affinity BioReagents), rabbit polyclonal anti‐calstabin (FKBP12.6) (1:1000; Affinity BioReagents), mouse monoclonal anti‐PLN (1:1000; Millipore), rabbit polyclonal anti–RyR2 DeP Ser2809 (un‐phosphorylated at serine residue 2809; 1:1000; Badrilla), rabbit polyclonal anti‐RYR2 P Ser2809 (phosphorylated at serine residue 2809; 1:1000; Badrilla). Horseradish peroxidase‐conjugated secondary antibodies were used for chemiluminescence detection. Western blot signals were quantified using National Institutes of Health ImageJ software (http://rsb.info.nih.gov/). LC3 western blots were performed at baseline and after 48 hours of starvation (only drinking water was given). High‐sensitivity cardiac troponin T levels were measured in serum with Elecsys STAT kit (Roche Diagnostics).
Echocardiography
Images were acquired using the Vevo 770 High‐Resolution In Vivo Micro‐Imaging System and RMV 707B scanhead (VisualSonics Inc.) at heart rates of 500 to 550 bpm as described. 20 , 21 Images were obtained as 2‐dimensional (left parasternal long and short axes) and M‐mode (left parasternal short axis). Left ventricular (LV) end‐diastolic diameter, LV end‐systolic diameter, and wall thickness were determined by averaging measurements of 3 consecutive cardiac cycles. LV fractional shortening (%) was calculated as (LV end‐diastolic diameter – LV end‐systolic diameter)/LV end‐diastolic diameter ×100. Two experienced observers who were blinded to genotypes conducted the measurements.
Electrophysiology
Multi‐lead ECG recordings, electrophysiological studies, and continuous telemetry were performed as previously described, 22 , 23 by 2 experienced electrophysiologists who were blinded to mouse genotypes.
Histopathology and Electron Microscopy
Mouse ventricular histology was performed after phosphate buffered saline (PBS) washes, fixation in 10% formaldehyde, and paraffin embedding. Five‐micrometer sections were stained with hematoxylin and eosin, Masson trichrome and special stains. Human ventricular tissue from a male patient with LAMP2 mutation Y109Ter was fixed in formalin, and 6 µm sections were stained (as s reference) with Masson trichrome.
Conduction system histology was performed in 25‐week‐old L2Δ6 and WT hearts carrying the MinK‐LacZ allele as described. 16 β‐Galactosidase activity in these hearts was detected by 5‐bromo‐4‐chloro‐3‐indolyl‐beta‐d‐galactopyranoside (X‐Gal– dark blue) staining. Quantification of X‐Gal positive cells was obtained using National Institutes of Health ImageJ software (http://rsb.info.nih.gov/). For transmission electron microscopy, ventricular myocardium from 25‐week‐old L2Δ6 and WT mice (3 mice per group) were fixed with glutaraldehyde using standard protocols.
To study the effect of cyclosporine A (CsA) on left ventricular hypertrophy, 32‐week‐old mice (n=3/group), were injected subcutaneously 15 μg/g of bodyweight twice per day, for 2 weeks. Echocardiographic measurements were done before and after treatment, as described.
Isolated Cardiomyocyte Studies
Adult ventricular myocytes were isolated via a Langendorff hanging heart preparation and enzymatic digestion as previously described. 24 , 25 For immunofluorescence studies, the isolated myocytes were fixed in 1% paraformaldehyde and spun onto slides using Cytospin3 (Shandon). After PBS washes, myocytes were permeabilized with 0.2% Triton X‐100 and blocked with 5% fetal bovine serum in PBS. Slides were incubated overnight with goat monoclonal antibodies against lysosomal enzymes anti‐Cathepsin B (1:50, R&D systems) or anti‐Cathepsin D (1:50, R&D systems) in 1% fetal bovine serum. Slides were washed and incubated with Alexa fluor 488 or 594 florescence labeled rabbit anti goat antibodies (1:100, Invitrogen), counterstained with Alexa Fluor 488 or 647 florescence labeled phalloidin (Invitrogen) and DAPI. Images were acquired using a Leica TCS SP2 confocal microscope.
For calcium transient analysis, the isolated ventricular myocytes were loaded with 1.2 mmol/L Ca2+ and 1 µmol/L fura‐2/AM Ca2+ indicator (Invitrogen) as previously described. 23 , 26 Only rod‐shaped cells with striations and resting sarcomere length >1.6 µm were studied. Myocytes were quiescent in the absence of electrical stimulation or were paced with1 Hz using platinum wires. Sarcomere contraction and fura‐2 fluorescence ratios were simultaneously recorded from discrete positions in myocytes using a dual‐excitation fluorescence imaging/contractility recording system (IonWizard SarcLen detection and PMT Acquisition fluorescence system; IonOptix Corp.). Sarcomere length and Ca2+ transient recordings were performed in Tyrode buffer, and where indicated, with 5.5 mmol/L Epinephrine using IonOptix transient analysis software (version 5.0; IonWizard).
RNA Sequence
RNAseq libraries were constructed as described, 27 except for libraries not normalized with the Duplex‐specific enzyme or fragmented. Reads were aligned on the mouse genome using TOPHAT and BOWTIE and expression profiles were constructed as previously described. 27
Statistical Analysis
Data are expressed as mean±SEM. Significance was assessed using ANOVA, unpaired Student’s t tests for continuous variables and Fisher exact test for categorized variables, as appropriate. Generalized Estimating Equation was used when numerous myocytes or sections were analyzed from the same mouse. P values <0.05 were considered significant. A change in gene expression was defined by at least ≥1.5 or <0.67 ratio in the read count (for increase or decrease respectively, at a P value of <0.001).
Results
The in‐frame exon 6 deletion in the endogenous LAMP2 gene (Figure 1A and 1B) resulted in neonatal death in ≈25% of hemizygous male L2Δ6 mice. Surviving mutant males, while initially smaller than WT littermates, achieved comparable size and weight by 12 weeks of age (≈30 g), and exhibited normal voluntary activity and survival (Figure S1).
LV tissues from L2Δ6 and WT mice had equivalent amounts of LAMP2 RNA, although the L2Δ6 transcript was smaller due to the deletion of 123 bp encoding exon 6 (Figure 1C). Western blots of LV extracts (Figure 2A) showed significantly lower levels of LAMP2 protein in L2Δ6 compared with WT mice (5.6‐fold±2.6; P=0.002). SDS‐PAGE gels indicated that mutant and WT LAMP2 protein had comparable sizes (≈110kd), implying that both molecules underwent post‐translational glycation.
Figure 2. In‐frame LAMP2 gene exon 6 deletion (L2Δ6) decreases LAMP2 levels and causes cardiomyopathy.
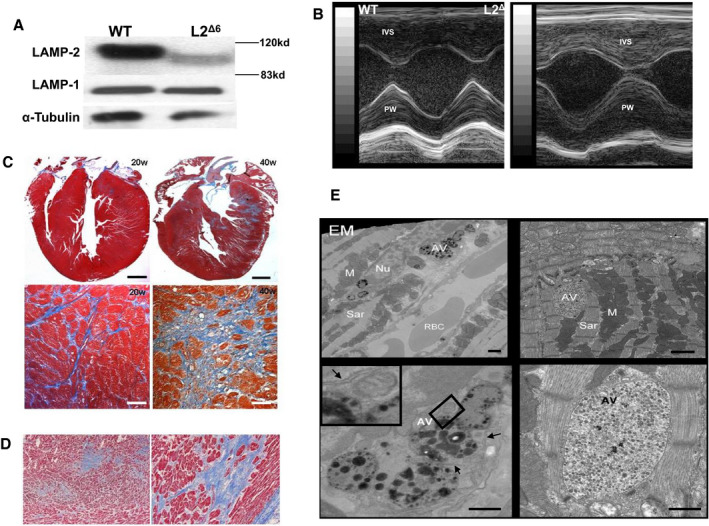
A, Western blots revealed reduced LAMP2 but normal LAMP1 levels in lysosomal extracts from L2Δ6 hearts compared with wild‐type (WT) hearts (n=4 per genotype). The smaller L2Δ6 protein reflected deletion of 41 amino acids encoded by exon 6. B, Echocardiograms show prominent hypertrophy of the intra‐ventricular septum and posterior wall in 25‐week L2Δ6 mice, see also Table 1. C, Progressive fibrosis in L2Δ6 heart. At 20‐weeks, fibrosis (stained blue) is interstitial, but by 40weeks hearts show both interstitial and areas of scarring. Focal fibrosis surrounds residual L2Δ6 myocytes with vacuoles. Upper panels, scale=500 mm lower panels, scale=50 mm. D, Heart section from a 14‐year‐old human patient with LAMP2 cardiomyopathy (Y109Ter mutation) who died suddenly (Maron et al 20095) demonstrating marked interstitial and replacement fibrosis (Masson trichrome). E, Electron microscopy (EM) of heart from 25‐week‐old L2Δ6 mice show large autophagic vacuoles that are interspersed among sarcomeres, containing partially degraded particles. Upper panels, scale=1 mm, lower panels, scale=500 nm. Autophagic vacuoles typically have double membrane (marked with black arrows) as shown more in details in the inset of the left lower panel. AV indicates autophagic vacuoles; IVS, intraventricular septum; L2Δ6, in‐frame LAMP2 gene exon 6 deletion; PW, posterior wall; RBC, red blood cells; and WT, wild‐type.
Development of Cardiomyopathy in L2Δ6 Mice
Significant cardiac hypertrophy developed after 20 weeks of age in male L2Δ6 mice (Figure 2B) with preserved or even increased systolic function (Table 1). These mice had a pronounced elevation of serum cardiac troponin T (484±246, range 150‐920 ng/L, compared with non‐detectable levels (<13) in WT, P=0.001, n=7/group) indicating an ongoing myocardial injury. The fractional shortening decreased in aged (≈40 weeks) compared with younger L2Δ6 mice while it did not change with aging in WT animals (Table 1). Heart to body weight at 40 weeks increased in L2Δ6 compared with WT mice (7.44 and 6.03, respectively; n=6, P<0.05) and myocardial fibrosis was strikingly increased (Figure 2C), recapitulating findings of human LAMP2 cardiomyopathy (Figure 2D). Cell death in L2Δ6 was indicated by troponin elevation and degenerative changes in some cardiomyocytes (Figure S2), and was presumably followed by myocardial fibrosis.
Table 1.
Cardiac Size and Function of L2Δ6 Mice
| L2Δ6 | WT | ANOVA | |||||
|---|---|---|---|---|---|---|---|
| Mutant | Age | Interaction | |||||
| Age (w) |
Adult 25.1±2.3 |
Aged 39.7±6.5 |
Adult 28.5±1.7 |
Aged 37.5±6.5 |
|||
| n | 6 | 6 | 5 | 8 | |||
| IVSd (mm) | 0.79±0.09 | 0.92±0.10 | 0.60±0.05 | 0.71±0.06 | 0.03 | 0.17 | 0.92 |
| LVPWd (mm) | 0.97±0.05 | 0.99±0.10 | 0.70±0.05 | 0.82±0.05 | 0.006 | 0.35 | 0.5 |
| LVEDd (mm) | 3.93±0.18 | 4.37±0.60 | 3.70±0.28 | 3.85±0.22 | 0.3 | 0.46 | 0.72 |
| FS (%) | 39.3±4.9* | 26.7±6.1* | 31.2±3.7 | 32.5±4.5 | 0.8 | 0.31 | 0.2 |
Mice were studied at 2 age categories. All measurements were performed at heart rate of 500 to 550 bpm. L2Δ6 mice tended to have slower heart rate, but that was not statistically significant. There was no apparent difference in the negative chronotropic effect of the anesthesia between mutant and wild‐type mice.
Data are expressed as mean±SEM. FS indicates fractional shortening; IVSd, interventricular septum diameter; L2Δ6, in‐frame LAMP2 gene exon 6 deletion; LVEDd, left ventricular end‐diastolic diameter; LVPWd, left ventricular posterior wall diameter; and WT, wild‐type.
ANOVA indicates a significant increase in the wall thickness in mutant mice without effect on left ventricular dimension or shortening fraction. There was no significant effect of age or interaction of age with L2Δ6 mutation.
In a post‐hoc analysis, aging was associated with a decrease in the shortening fraction of L2Δ6 mice (*P<0.01 comparing aged to young L2Δ6 mice) but no such change was seen in wild‐type mice.
Hearts of L2Δ6 mice contained ≈x2 glycogen compared with WT. This accumulation is insufficient to be considered “storage” 28 and no PAS‐positive inclusions were identified in histological sections from L2Δ6 mice. Transmission electron microscopy of LV specimens from WT and L2Δ6 showed that mutant myocytes had abundant bilayer membrane vesicles containing inclusions composed of cytoplasmic remnants, partially degraded organelles, and nonspecific amorphous materials. These features characterize autophagosomes, a cytoplasmic structure that deliver sequestered cargo to lysosomes for final degradation and recycling (Figure 2E). These large structures disrupted the normal sarcomere organization in L2Δ6 mice (Figure 2E, right lower panel).
Heart rhythm telemetry demonstrated no difference in average sinus heart rate between WT and L2Δ6 mice (data not shown). Cardiac electrophysiology showed PR prolongation and intermittent atrio‐ventricular block in L2Δ6 compared with WT mice (Figure 3A and 3B). Atrio‐ventricular block occurred in 60% of L2Δ6 and 8% of WT mice (P=0.02). L2Δ6 mice did not exhibit ventricular pre‐excitation or spontaneous arrhythmias but pacing induced ventricular arrhythmias in 75% of L2Δ6 mice (but in (<10% WT mice; P=0.01). We then assessed whether fibrosis contributed to electrophysiological abnormalities by examining the histopathology of compound L2Δ6/ MinK‐LacZ mice that selective express β‐galactosidase in their cardiac conduction system myocytes. 16 Compound mutant mice showed marked fibrotic encasement and vacuolation of the atrio‐ventricular node, less conduction system myocytes and disorganization of the proximal bundle of His (Figure 3C and 3D).
Figure 3. Conduction system physiology and histology of in‐frame LAMP2 gene exon 6 deletion (L2Δ6) hearts.
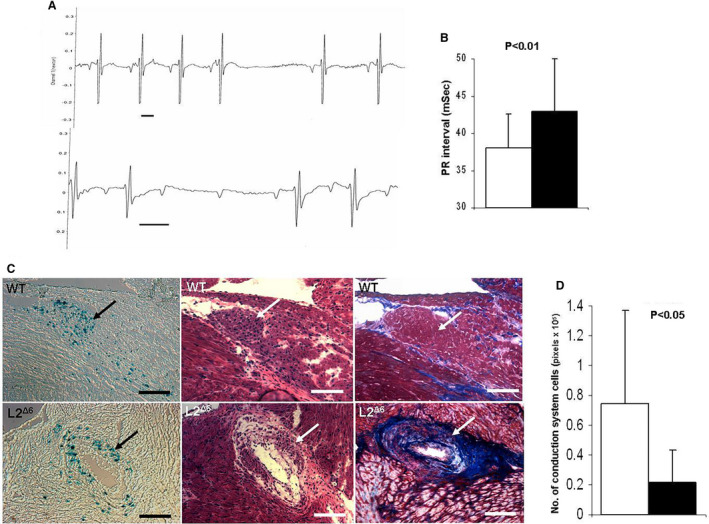
A, Representative traces recorded by telemetry in 15‐week‐old L2Δ6 mice: upper panel showed transient sinus node arrest and middle panel shoes transient atrioventricular block, Scale bars represent 50mSec. B, Average PR intervals (the time from the onset of the P wave to the start of the QRS complex on the electrocardiogram) of 15 weeks old WT (white bar) and L2Δ6 (black bar) mice, n=10/genotype, mean±SEM. C, Histology of the atrioventricular node and surrounding tissues in 15‐week‐old MnK/WT mouse (upper panels) and MnK/L2Δ6 mouse (lower panels). The hearts were stained with X‐Gal to identify β‐galactosidase activity in the conduction system cells (Blue dots in left panels), and then counterstained with hematoxylin–eosin (middle panels) and with Masson Trichrome (right panels). The atrioventricular node (arrow) in the MnK/ L2Δ6 hearts was more disorganized than MnK/WT hearts. Fibrosis and large vacuoles were detected in the atrioventricular region of MnK/L2Δ6 but not in the MnK/WT hearts. Scale bars represent 200μM. D, Quantitation of positive X‐Gal staining which represents conduction system cells. MnK/L2Δ6 hearts had less conduction cells (black bar) compared with MnK/WT (white bar). Analysis was done in consecutive section containing the atrioventricular node from 4 mice aged 15 to 20 weeks of each genotype, 3 sections per heart. Generalized Estimating Equation was used for statistical comparison. L2Δ6 indicates in‐frame LAMP2 gene exon 6 deletion; and WT, wild‐type.
Functional Assessment of Autophagy in L2Δ6 Myocytes
We used western blot analysis to assess the levels of microtubule‐associated protein light chain 3 (LC3‐II), which demarcates autophagosome, 29 the pre‐lysosomal fusion step of autophagy. Mice were studied in a fed state to assess basal levels of autophagy and after food deprivation to stimulate autophagy. Compared with WT, LC3‐II levels were substantially greater in hearts from fed L2Δ6 mice and comparable to levels found after 24 hours of food‐deprived WT mice (Figure 4A and 4B). Food deprivation farther increased LC3‐II levels in L2Δ6 mice, implying that L2Δ6 mice retained a capacity for further increases in the basal stages of the autophagic pathway.
Figure 4. Disrupted autophagy in in‐frame LAMP2 gene exon 6 deletion (L2Δ6) hearts.
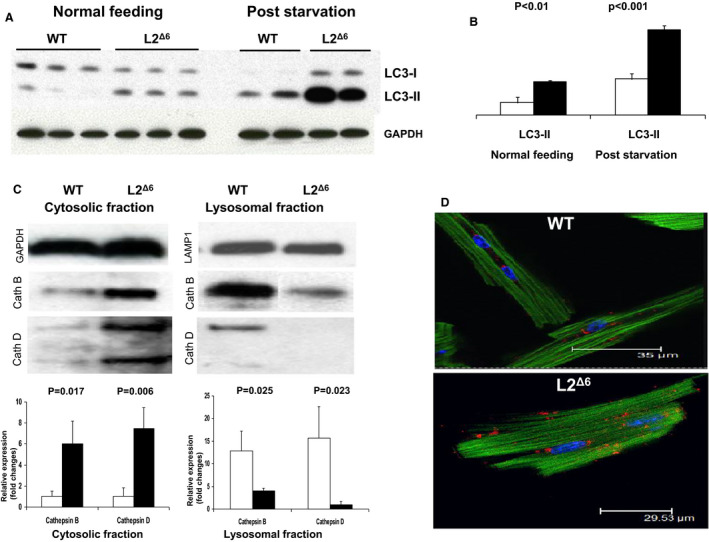
A, Western blot analysis of the microtubule‐associated protein light chain 3 (LC3) extracted from 20‐weeks old wild‐type (WT) and L2Δ6 mice hearts. Upper band represent the LC3‐I, which is cytosolic, and the lower LC3‐II is present on isolation membranes and autophagosomes. B, Densitometry (3 mice of each genotype) shows that L2Δ6 hearts (black bars) express higher levels of LC3‐II compared with WT (white bars). Markedly increased LC3‐II levels after starvation indicate farther accumulation of autophagosomes. C, Western blot of lysosomal enzymes cathepsin B and D in fractionated subcellular extracts. Densitometry (3 mice of each genotype) shows increased levels of cathepsin B and D in the cytosolic fraction of L2Δ6 hearts (black bars) compared with WT (white bars) and reciprocally decreased levels in the lysosomal fractions. D, Representative confocal micrographs of WT and L2Δ6 myocytes stained with immunofluorescent anti‐Cathepsin B antibody (red), DAPI (blue) and Phalloidin (green). Note a peri‐nuclear localization of lysosomes detected by the anti‐cathepsin antibody in WT myocytes. Cathepsin immunofluorescence is dispersed throughout the cytoplasm in L2Δ6 myocytes. Numerical data are expressed as mean±SEM. L2Δ6 indicates in‐frame LAMP2 gene exon 6 deletion; LC3, microtubule‐associated protein light chain 3; and WT, wild‐type.
To assess lysosome function in L2Δ6 mice, we capitalized on transcription data that indicated increased expression of 6 cathepsins (A‐D, L and Z). Using Western blot analyses of LV extracts we examined protein levels of cathepsins B and D. Both were increased in cytoplasmic fractions and diminished in lysosomal fractions of LV extracts from L2Δ6 hearts while the reverse was found in LV extracts from WT hearts (Figure 4C). Immunofluorescent labeling of cathepsin B and cathepsin D identified a perinuclear location in 83% of WT myocytes (18 myocytes from 3 mice) but in only 25% of L2Δ6 myocytes (16 myocytes from 3 mice, P<0.01). Cathepsin immunofluorescence in L2Δ6 myocytes was more diffuse within the cytosol (Figure 4D), suggesting mis‐localization of lysosomal enzymes.
In summary, transcriptional and functional analyses indicated significantly activated molecular signals that promote autophagosome and lysosome production and autophagy in L2Δ6 hearts. Despite these signals, there was an accumulation of autophagasome and mislocalization of lysosome enzymes in L2Δ6 hearts, defining a block in autophagy at the step of formation of autolysosomes.
Transcriptional Analysis in L2Δ6 Hearts
We then performed RNAseq 27 of LV tissues from 26‐ to 30‐week‐old L2Δ6 and WT mice, to identify transcriptional responses to the L2Δ6 mutation (illustrated for LAMP2 expression in Figure S3). The full results of the transcriptome analysis are provided at. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166731 (GEO accession number GSE166731 ). Among the LV transcripts that were significantly altered in L2Δ6 mice (4.0%, increased; 2.2% decreased), there was a significant upregulation of transcripts annotated in autophagy and apoptotic pathways (Table S1). Thus, among 793 mouse homologues of yeast autophagy genes that are expressed in the LV, 62 were increased (7.8%, P<0.001) in L2Δ6 compared with WT mice and among 1252 apoptotic markers, 85(6.8%) were increased (P<0.001). Similarly, among 399 human autophagy network proteins 30 that are expressed in the LV, 86 were significantly increased in L2Δ6 in comparison with WT hearts (P<0.0001).
The main genes in the autophagy and apoptotic pathways altered in L2Δ6 mice compared with WT are shown in Table S2.
Abnormal Relaxation and Altered Calcium Homeostasis in L2Δ6 Myocytes
To assess whether impaired autophagy altered an intrinsic contractile function and calcium homeostasis, we studied isolated myocytes from 20‐week‐old L2Δ6 and WT mice (Figure 5 and Table 2). Despite accumulations of autophagosomes among sarcomeres in mutant cardiomyocytes (Figure 2A), the extent of shortening or relaxation was not different compared with WT (Figure 5A). However, the durations of contraction and relaxation were markedly prolonged in L2Δ6 myocytes (Table 2).
Figure 5. Ca2+ abnormalities in in‐frame LAMP2 gene exon 6 deletion cardiomyocytes.
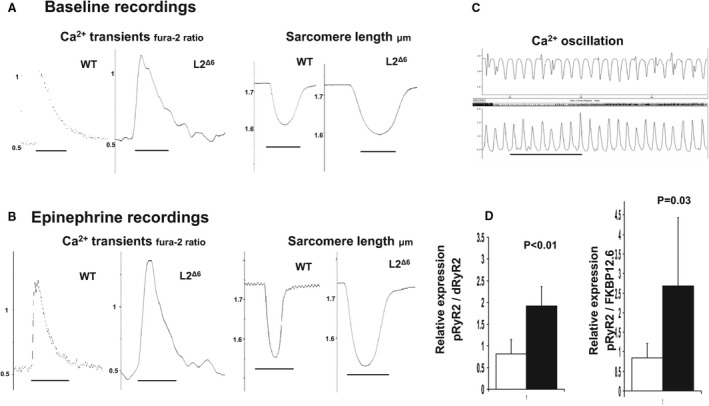
A, B, Representative calcium (Ca2+) transients (left panels) and sarcomere length (right panels) of wild‐type and in‐frame Lysosome‐associated membrane protein‐2 gene (LAMP2) gene exon 6 deletion myocytes in Ca2+ Tyrode solution without (a) or with (b) 5.5 mmol/L epinephrine. Traces represent the average of 15 contraction‐relaxation cycles at 1 Hz from 1 representative cell of each genotype. C, Epinephrine infusion produced spontaneous Ca2+ oscillations in‐frame LAMP2 gene exon 6 deletion myocytes (sarcomere length, upper trace; Ca2+ transient, lower trace). Scale=0.2 sec. D, Quantitative Western blot estimate of phosphorylated ryanodine receptor 2 (pRyR2) in SR extracts from in‐frame LAMP2 gene exon 6 deletion (black bar) and wild‐type (white bar) hearts. The relative amounts of pRyR2, de‐phosphorylated ryanodine receptor 2 (dRyR2), and FK506 binding protein 12.6 (FKBP12.6, calstabin‐2) were determined by densitometry of antibody signals (n=3 mice/group, mean±SEM). See Table 2 for comparisons. L2Δ6 indicates in‐frame LAMP2 gene exon 6 deletion; SR, sarcoplasmic reticulum, and WT, wild‐type.
Table 2.
Cardiomyocytes Properties in L2Δ6 Mice
| WT | L2Δ6 | P Value | |
|---|---|---|---|
| Isolated myocytes* | |||
| Resting myocyte length (µm) | 1.75±0.06 | 1.76±0.05 | NS |
| % Sarcomere shortening | 6.6±2.1 | 7.4±2.8 | NS |
| Time to 50% contraction (mSec) | 25±7 | 65±15 | <0.001 |
| Time to 50% relaxation (mSec) | 148±40 | 310±80 | <0.001 |
| Ca2+ transient amplitude (Fura2 ratio) | 0.45±0.15 | 0.71±0.24 | <0.001 |
| Time to 50% transient decay (mSec) | 118±13 | 201±69 | <0.001 |
| Ca2+ transient decay–Tau (mSec) | 124±22 | 178±52 | <0.001 |
| Catecholamine stimulated myocytes † | |||
| Resting myocyte length (µm) | 1.72±0.06 | 1.73±0.06 | NS |
| % Sarcomere shortening | 12.9±3.3 | 13.5±3.7 | NS |
| Time to 50% contraction (mSec) | 20±4 | 32±13 | <0.05 |
| Time to 50% relaxation (mSec) | 107±18 | 150±50 | <0.05 |
| Ca2+ transient amplitude (Fura2 ratio) | 0.56±0.17 | 0.92±0.3 | <0.01 |
| Time to 50% transient decay (mSec) | 92±16 | 105±25 | NS |
| Ca2+ transient decay–Tau (mSec) | 68±9 | 69±17 | NS |
Data are expressed as mean±SEM. L2Δ6 indicates in‐frame LAMP2 gene exon 6 deletion; LAMP2, lysosome‐associated membrane protein‐2; NS, non‐significant; and WT, wild‐type.
Sarcomere lengths and shortening of myocytes (n=21 and n=29 cells from 4 wild‐type (WT) and 5 in‐frame LAMP2 exon 6 deletion (L2Δ6)mice, respectively). Ca2+ transients were measured in myocytes (n=30 cells and n=18) from 5 WT and 3 L2Δ6 mice, respectively.
Catecholamine‐stimulated sarcomere lengths and shortening were measured in myocytes (n=11 and n=10) from 4 WT and 3 L2Δ6 mice, respectively. Catecholamine‐stimulated Ca2+ transients were measured in myocytes (n=12 and n=12 cells from 4 WT and 3 L2Δ6 mice, respectively).
Ca2+ transient studies revealed prominent abnormalities in L2Δ6 myocytes. The mean amplitude of Ca2+ transients was strikingly higher in L2Δ6 (0.71±0.24) than in WT (0.45±0.15) myocytes (Table 2 and Figure 5A). Catecholamines further increased mean Ca2+ transient amplitudes to levels that were significantly higher than those found in catecholamine‐stimulated WT myocytes (Figure 5B). Catecholamines also produced spontaneous and frequent sarcoplasmic reticulum Ca2+ release events in ≈75% of L2Δ6 myocytes (Figure 5C); these events were triggered in only 8% of WT myocytes (P=0.009).
Ca2+ decay in L2Δ6 myocytes was slower than in WT myocytes (Table 2). However, because catecholamines did normalize Ca2+ decay times in mutant myocytes (Table 2), we deduced that the sarcoplasmic reticular pump, Ca2+ATPase (Serca2a), remained functional in L2Δ6 myocytes.
We also tested if abnormal Ca2+ homeostasis is associated with altered expression of Ca2+‐handling proteins. Transcripts encoding the cardiac ryanodine receptor (RyR2) and the RyR2‐stabilizing protein FKBP12.6 showed coordinate increase above their levels in WT mice (both with P<0.0001). In contrast, transcript levels of SERCA2 and PLN (phospholamban) were unchanged while the expression of the sarcoplasmic reticulum calcium binding proteins CASQ1/2 (calsequestrin), were decreased. As predicted from RNA data, protein levels of the cardiac ryanodine receptor were increased in L2Δ6 hearts as were the levels of phosphorylated cardiac ryanodine receptor (pRyR2, Figure 5D). On the other hand, the levels of SERCA2a, total PLN, phosphorylated PLN, and CASQ2 were comparable between L2Δ6 and WT hearts (Figure S4A), similar to LAMP2‐null hearts. 12 Collectively, a marked increase in ryanodine receptor phosphorylation (pRyR2) in L2Δ6 hearts, unopposed by FKBP12.6 (Figure 5D and Figure S4B), would promote increased Ca2+ leak from the RyR2 and might account for delayed after‐depolarizations and arrhythmias.
Hypertrophic Signaling
Cardiac hypertrophy in L2Δ6 mice was associated with alteration in gene expression related to myocardial stretch and various forms of hypertrophic remodeling. Genes such as nppb (x4.2), nppa (x3.8) and acta1 (x2.5), markedly and significantly increased (all at P<0.0001). Because increased frequency of spontaneous Ca2+ transients could also affect molecular signaling through either the calcineurin/NFAT (Nuclear factor of activated T‐cells) pathway 31 or by mitochondrial autophagy we hypothesized that calcineurin 32 might have an effect on L2Δ6 cardiomyopathy. Indeed, calcineurin RNA (ppp3ca) is increased 1.4‐fold (P=0.0008) in L2Δ6 left ventricles. We therefore treated adult L2Δ6 mice with CsA that blocks mitochondrial autophagy and modulates cardiac hypertrophy. After 2 weeks of CsA treatment, there was a reduction in LV wall thickness in L2Δ6 hearts compared with untreated mice (Figure S5). However, neither the fractional shortening, nor the fibrosis burden, were affected by CsA treatment.
Discussion
L2Δ6 mice expressing a defective LAMP2 allele missing the 41 amino acids of exon 6, constitute a useful model recapitulating the human cardiomyopathy associated with Danon disease. L2Δ6 hearts contain considerably less LAMP2 protein than WT hearts in the presence of normal mRNA levels (Figure 1C), suggesting that the mutant LAMP2Δ6 protein is unstable (Figure 2). Whether cardiomyopathy results from reduced amounts of protein or altered activity of the mutant protein remains uncertain. Nevertheless, the fact that in the presence of certain amounts of protein, L2Δ6 mice appear to have a better survival compared with the null‐allele LAMP2 mice, 10 suggests that the expressed mutant protein retains some of its function. Further, deficiency in mutant protein, rather than its toxic effect, is responsible for the phenotype. As opposed to a complete LAMP2 gene deficiency, 10 the hypomorphic LAMP2 allele in L2Δ6 mutant mice allow the animals to survive to adulthood and develop significant cardiomyopathy.
Characterization of the cardiac phenotype of L2Δ6 mice provides further insights into the mechanisms of LAMP2‐mediated cardiomyopathy. L2Δ6 mice demonstrate that lysosomal dysfunction appears to be a powerful stimulus for hypertrophic remodeling, heart failure, and cardiac arrhythmias. Of the multiple ensuing consequences, failed autophagy appears to be a fundamental cause of progressive cardiomyopathy. Studies demonstrating that activation of autophagy can ameliorate load‐induced hypertrophy 31 support our conclusion. This pathway differs from "hypertrophic" sarcomere protein mutations that enhance contractile parameters, 32 structural protein mutations (eg, desmin, alphaB‐crystallin, ZASP) that accumulate abnormal protein aggregates, 33 or defects in metabolic genes accrue glycogen, glycolipids, or their degradation products (eg, PRKAG2, 34 GAA 35 GLA). 36 Importantly, there is a growing appreciation of the role of a secondary dysfunction of autophagy in pathophysiology of lysosomal storage diseases. 37 Unlike in the original description of Danon disease defining it as “Pompe” with normal acid maltase or GSD IIb, we did not find evidence of significant glycogen storage. That could result from differences in glycogen metabolism and lower levels of glycogen in murine heart compared with humans. 28 Differences in glycogen accumulation could explain why humans but not mice with Danon disease often develop massive left ventricular hypertrophy.
The L2Δ6 mutation causes incomplete lysosome biogenesis. Lysosomes in L2Δ6 myocytes had mistargeting of cathepsins B and D and glycolytic enzymes. Consistent with these markers of incomplete maturation, L2Δ6 lysosomes were dispersed throughout the cytosol (Figure 4), whereas normal lysosomes clustered in perinuclear regions. Autophagosomes containing aggregated proteins and membrane components accumulated in L2Δ6 myocytes. The expression of 62 RNAs encoding autophagy‐associated proteins was increased. More importantly LC3‐II, a marker of autophagosomes, was significantly overexpressed in L2Δ6 hearts compared with WT (Figure 4). Processing of autophagosome requires docking and fusion with mature lysosomes, to yield phagolysosomes that move via microtubules to perinuclear regions. 38 Collectively, autophagy appears to be stimulated in L2Δ6 myocytes, but the later stages in the process are blocked, resulting in accumulated autophagosomes, absent phagolysosomes, and myocytes unable to clear cellular debris or effectively recycle proteins and organelles. Myocytes from fasted L2Δ6 mice expressed significantly more LC3‐II than myocytes from non‐fasted L2Δ6 mice. These results indicate that food deprivation, an autophagy‐stimulating stress, causes additional autophagosome accumulation and may be expected to aggravate cardiac disease in L2∆6 animals. To summarize, tissue injury and hypertrophy in L2∆6 mice probably triggered by accumulation of autophagosomes and multiple partially degraded cytosolic organelles. Failed autophagy apparently promotes myocyte apoptosis 39 (Table S1) or necrosis, which are followed by fibrosis and arrhythmogenicity.
Additional mechanisms likely contribute to development of LAMP2 cardiomyopathy. Activation of hypertrophy associated genes indicate myocardial stretch in hypertrophic signaling. Recent studies confirm the critical role of defective mitophagy, oxidative stress, and energetic deficiency in the pathogenesis of LAMP2 cardiomyopathy. 40 , 41 , 42 CsA attenuates cardiac hypertrophy caused by pressure overload but aggravates it when caused by sarcomere protein gene mutations. 32 , 43 Carreira et al reported that changes in mitochondrial membrane permeability under stress conditions can trigger autophagy, a process called mitochondrial permeability transition. 44 Because CsA is a potent inhibitor of the Ca2+‐dependent pore in heart mitochondria, 45 it could block mitochondrial permeability transition and autophagy in cardiomyocytes. Treatment of adult L2∆6 mice with CsA, a calcineurin inhibitor, results in attenuation of cardiac hypertrophy without affecting systolic function or decreasing fibrosis (Figure S5). These data do support the involvement of calcineurin/NFAT (Nuclear factor of activated T‐cells) pathway 43 in cardiac remodeling in Danon disease. It is possible that the effect of CsA could be more pronounced if given in younger mice before development of cardiomyopathy and fibrosis.
Another prominent feature of LAMP2 cardiomyopathy is conduction block, which was associated in our model with infiltration and pathological alterations in the conduction system (Figure 3). The death of L2Δ6 myocytes could arise from toxic accumulations autophagosomes and/or energy deficits. Additionally, the absence of recycling of subcellular components would necessitate that L2Δ6 myocytes to expend more energy on de novo protein synthesis. While the long‐term survival of most L2Δ6 mice indicated that cardiomyocyte death and energy deficits were tolerable to a certain extent, these parameters likely contribute to the uniform progression of human LAMP2 cardiomyopathy to heart failure.
Both humans with LAMP2 cardiomyopathy and L2Δ6 mice exhibit ventricular arrhythmias. Although fibrosis is thought to be one cause of such arrhythmias, L2Δ6 mice exhibit ventricular arrhythmias early in life even before developing fibrosis (Figure 2), suggesting that other mechanisms are likely involved in arrhythmia induction. An unexpected property of L2Δ6 myocytes helps to explain this conundrum: L2Δ6 myocytes had markedly increased Ca2+ transient amplitudes, prolonged contraction and relaxation times, despite having normal sarcomere shortening (Table 2, Figure 5). Augmented Ca2+ transients mediated by the RyR2 channel activation may evolve in L2Δ6 myocytes to allow these cells to maintain their contractility despite ultrastructural damage. Unfortunately, these compensatory changes in Ca2+ management cause the myocytes to become electrically unstable. Ca2+ signals are likely to account for increased ventricular arrhythmogenicity in L2Δ6 mice. Because mutant mice developed arrhythmias before histopathologic abnormalities and isolated L2Δ6 myocytes had spontaneous Ca2+ oscillations upon catecholamine stimulation (Figure 5C), arrhythmogenicity appears to be a fundamental property of L2Δ6 myocytes, and not a secondary response to hypertrophy and fibrosis.
Notably, Ca2+ transients in L2Δ6 myocytes were substantially bigger than those triggered by sarcomere gene mutations 46 possibly explaining the more severe arrhythmic phenotypes seen in patients with LAMP2 mutations compared with arrhythmias observed in patients with hypertrophic cardiomyopathy. Hyperphosphorylation of RyR2 channels, observed in L2Δ6 mice, is a key feature, implicating Ca2+ leak from the sarcoplasmic reticulum, spontaneous calcium release, and delayed after‐depolarizations to explain arrhythmia in this model. 23 , 47 , 48
In Summary
Autophagy, a lysosomal‐associated degradation of the cells’ own constituents, plays an important role in the renewal and survival of cardiomyocytes. L2Δ6 hearts develop progressive cardiomyopathy because of abnormal lysosomal biogenesis, inability of mutant lysosomes to dock with autophagosomes, and failed autophagy. Accumulation of autophagy intermediates and of undigested contents, causes myocyte injury, triggers hypertrophy, followed by cell death, myocardial fibrosis, and decreasing systolic function. Calcium dysregulation, abnormal calcium release, and increased sensitivity to catecholamine stimulation contribute to lethal arrhythmia in LAMP2 cardiomyopathy.
Limitations and Perspectives
By precisely introducing a “human” mutation into mouse genome, we generated a model of cardiomyopathy in Danon disease. These mice recapitulate the course of cardiomyopathy in human patients, as well as many of the histological and arrhythmic features. Important issues to be resolved are the function of the mutant protein LAMP2b, of the isoform LAMP2a involved in chaperon‐mediated autophagy, as well as LAMP1 which apparently fails to compensate for LAMP2 deficiency. Studying the effect of pharmacological agents on autophagic flux, lysosomal function and calcium handling may clarify the link between LAMP2 defect and disease evolvement. Most importantly, this model should be used to study potential therapies to modify the course of this lethal condition.
Sources of Funding
This work was supported by grants from Howard Hughes Medical Institute (C.E.S.), National Institutes of Health (J.G.S., C.E.S.), from the Machiah Foundation/Jewish Community Foundation Program (R.A.) and from the Chief Scientist Office, Israeli Ministry of Health (R.A.).
Disclosures
M.A. and D.Y. received a research support from Genzyme Corporation for a project entitled “Phenotype Modification and Developing Therapy in Cardiac Danon Disease.” The remaining authors have no disclosures to report.
Supporting information
Tables S1–S2
Figures S1–S5
For Sources of Funding and Disclosures, see page 12.
References
- 1. Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP‐2 deficiency causes X‐linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406:906–910. doi: 10.1038/35022604 [DOI] [PubMed] [Google Scholar]
- 2. Arad M, Maron BJ, Gorham JM, Johnson WH, Saul JP, Perez‐Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–372. doi: 10.1056/NEJMoa033349 [DOI] [PubMed] [Google Scholar]
- 3. Musumeci O, Rodolico C, Nishino I, Di Guardo G, Migliorato A, Aguennouz M, Mazzeo A, Messina C, Vita G, Toscano A. Asymptomatic hyperCKemia in a case of Danon disease due to a missense mutation in Lamp‐2 gene. Neuromuscul Disord. 2005;15:409–411. doi: 10.1016/j.nmd.2005.02.008 [DOI] [PubMed] [Google Scholar]
- 4. Yang Z, McMahon CJ, Smith LR, Bersola J, Adesina AM, Breinholt JP, Kearney DL, Dreyer WJ, Denfield SW, Price JF, et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation. 2005;112:1612–1617. doi: 10.1161/CIRCULATIONAHA.105.546481 [DOI] [PubMed] [Google Scholar]
- 5. Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, Wright GB, Almquist AK, Baffa JM, Saul JP, Ho CY, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301:1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brambatti M, Caspi O, Maolo A, Koshi E, Greenberg B, Taylor MRG, Adler ED. Danon disease: gender differences in presentation and outcomes. Int J Cardiol. 2019;286:92–98. doi: 10.1016/j.ijcard.2019.01.020 [DOI] [PubMed] [Google Scholar]
- 7. Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55–64. doi: 10.1172/JCI73943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marques ARA, Saftig P. Lysosomal storage disorders – challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci. 2019;132:jcs221739. doi: 10.1242/jcs.221739. PMID: 30651381. [DOI] [PubMed] [Google Scholar]
- 10. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann‐Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP‐2‐deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595 [DOI] [PubMed] [Google Scholar]
- 11. Saftig P, Tanaka Y, Lüllmann‐Rauch R, von Figura K. Disease model: LAMP‐2 enlightens Danon disease. Trends Mol Med. 2001;7:37–39. doi: 10.1016/S1471-4914(00)01868-2 [DOI] [PubMed] [Google Scholar]
- 12. Stypmann J, Janssen PML, Prestle J, Engelen MA, Kögler H, Lüllmann‐Rauch R, Eckardt L, von Figura K, Landgrebe J, Mleczko A, et al. LAMP‐2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic Res Cardiol. 2006;101:281–291. doi: 10.1007/s00395-006-0591-6 [DOI] [PubMed] [Google Scholar]
- 13. Manso AM, Hashem SI, Nelson BC, Gault E, Soto‐Hermida A, Villarruel E, Brambatti M, Bogomolovas J, Bushway PJ, Chen C, et al. Systemic AAV9.LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease. Sci Transl Med. 2020;12:eaax1744. doi: 10.1126/scitranslmed.aax1744. PMID: 32188720. [DOI] [PubMed] [Google Scholar]
- 14. Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering‐based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre‐ and Flp‐mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136 [DOI] [PubMed] [Google Scholar]
- 16. Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD, Magnuson MA, Roden DM. Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circ Res. 1999;84:146–152. [DOI] [PubMed] [Google Scholar]
- 17. Karlen Y, McNair A, Perseguers S, Mazza C, Mermod N. Statistical significance of quantitative PCR. BMC Bioinformatics. 2007;8:131. doi: 10.1186/1471-2105-8-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harms E, Kartenbeck J, Darai G, Schneider J. Purification and characterization of human lysosomes from EB‐virus transformed lymphoblasts. Exp Cell Res. 1981;131:251–266. doi: 10.1016/0014-4827(81)90230-5 [DOI] [PubMed] [Google Scholar]
- 19. Mas‐Oliva J, Williams A, Nayler WG. Separation of isolated cardiac sarcolemma into inside‐out and right‐side‐out vesicles by affinity chromatography. Biochem Soc Trans. 1979;7:950–952. doi: 10.1042/bst0070950 [DOI] [PubMed] [Google Scholar]
- 20. McConnell BK, Fatkin D, Semsarian C, Jones KA, Georgakopoulos D, Maguire CT, Healey MJ, Mudd JO, Moskowitz IPG, Conner DA, et al. Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ Res. 2001;88:383–389. doi: 10.1161/01.RES.88.4.383 [DOI] [PubMed] [Google Scholar]
- 21. Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, et al. The L‐type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI200214677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolf CM, Arad M, Ahmad F, Sanbe A, Bernstein SA, Toka O, Konno T, Morley G, Robbins J, Seidman JG, et al. Reversibility of PRKAG2 glycogen‐storage cardiomyopathy and electrophysiological manifestations. Circulation. 2008;117:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner DA, Berul CI, Eldar M, Seidman CE, Seidman JG. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2007;117:1814–1823. doi: 10.1172/JCI31080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim CC, Apstein CS, Colucci WS, Liao R. Impaired cell shortening and relengthening with increased pacing frequency are intrinsic to the senescent mouse cardiomyocyte. J Mol Cell Cardiol. 2000;32:2075–2082. doi: 10.1006/jmcc.2000.1239 [DOI] [PubMed] [Google Scholar]
- 25. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires Tgf‐β. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagata K, Liao R, Eberli FR, Satoh N, Chevalier B, Apstein CS, Suter TM. Early changes in excitation‐contraction coupling: transition from compensated hypertrophy to failure in Dahl salt‐sensitive rat myocytes. Cardiovasc Res. 1998;37:467–477. doi: 10.1016/S0008-6363(97)00278-2 [DOI] [PubMed] [Google Scholar]
- 27. Christodoulou DC, Wakimoto H, Onoue K, Eminaga S, Gorham JM, DePalma SR, Herman DS, Teekakirikul P, Conner DA, McKean DM, et al. 5’RNA‐Seq identifies Fhl1 as a genetic modifier in cardiomyopathy. J Clin Invest. 2014;124:1364–1370. doi: 10.1172/JCI70108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, Yan J, Wakimoto H, Morita H, Arad M, et al. Aberrant activation of AMP‐activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest. 2007;117:1432–1439. doi: 10.1172/JCI30658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 30. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rothermel BA, Hill JA. Autophagy in load‐induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seidman JG, Seidman C. The genetic basis for cardiomyopathy. Cell. 2001;104:557–567. doi: 10.1016/S0092-8674(01)00242-2 [DOI] [PubMed] [Google Scholar]
- 33. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arad M, Benson DW, Perez‐Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–362. doi: 10.1172/JCI0214571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bijvoet AG, van de Kamp EH, Kroos MA, Ding JH, Yang BZ, Visser P, Bakker CE, Verbeet MP, Oostra BA, Reuser AJ, et al. Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum Mol Genet. 1998;7:53–62. doi: 10.1093/hmg/7.1.53 [DOI] [PubMed] [Google Scholar]
- 36. Matsuzawa F, Aikawa S, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in alpha‐galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005;117:317–328. [DOI] [PubMed] [Google Scholar]
- 37. Lim J‐A, Meena NK, Raben N. Pros and cons of different ways to address dysfunctional autophagy in Pompe disease. Ann Transl Med. 2019;7:279. doi: 10.21037/atm.2019.03.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mackeh R, Perdiz D, Lorin S, Codogno P, Poüs C. Autophagy and microtubules – new story, old players. J Cell Sci. 2013;126:1071–1080. doi: 10.1242/jcs.115626 [DOI] [PubMed] [Google Scholar]
- 39. Xing R, Liu D, Cheng X, Tian X, Yan C, Han Y. MiR‐207 inhibits autophagy and promotes apoptosis of cardiomyocytes by directly targeting LAMP2 in type 2 diabetic cardiomyopathy. Biochem Biophys Res Commun. 2019;520:27–34. doi: 10.1016/j.bbrc.2019.09.092 [DOI] [PubMed] [Google Scholar]
- 40. Hashem SI, Perry CN, Bauer M, Han S, Clegg SD, Ouyang K, Deacon DC, Spinharney M, Panopoulos AD, Izpisua Belmonte JC, et al. Brief report: oxidative stress mediates cardiomyocyte apoptosis in a human model of danon disease and heart failure. Stem Cells. 2015;33:2343–2350. doi: 10.1002/stem.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hashem SI, Murphy AN, Divakaruni AS, Klos ML, Nelson BC, Gault EC, Rowland TJ, Perry CN, Gu Y, Dalton ND, et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J Mol Cell Cardiol. 2017;108:86–94. doi: 10.1016/j.yjmcc.2017.05.007 [DOI] [PubMed] [Google Scholar]
- 42. Chi C, Leonard A, Knight WE, Beussman KM, Zhao Y, Cao Y, Londono P, Aune E, Trembley MA, Small EM, et al. LAMP‐2B regulates human cardiomyocyte function by mediating autophagosome‐lysosome fusion. Proc Natl Acad Sci USA. 2019;116:556–565. doi: 10.1073/pnas.1808618116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: role of calcineurin/NFAT as Ca2+ signal integrators. Proc Natl Acad Sci USA. 2008;105:2859–2864. doi: 10.1073/pnas.0712316105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carreira RS, Lee Y, Ghochani M, Gustafsson ÅB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6:462–472. doi: 10.4161/auto.6.4.11553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+‐dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 46. Kim SJ, Iizuka K, Kelly RA, Geng YJ, Bishop SP, Yang G, Kudej A, McConnell BK, Seidman CE, Seidman JG, et al. An alpha‐cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am J Physiol. 1999;276:H1780–H1787. [DOI] [PubMed] [Google Scholar]
- 47. Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BEC, Horton KD, Weissman NJ, Holinstat I, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lehnart SE, Terrenoire C, Reiken S, Wehrens XHT, Song L‐S, Tillman EJ, Mancarella S, Coromilas J, Lederer WJ, Kass RS, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci USA. 2006;103:7906–7910. doi: 10.1073/pnas.0602133103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S5