

Abstract
Background
Adhesion of vascular endothelial cells to the underlying basement membrane potently modulates endothelial cells to cells' inflammatory activation. The normal basement membrane proteins laminin and collagen IV attenuate inflammatory signaling in part through integrin α2β1. In contrast, fibronectin, the provisional matrix protein found in injured, remodeling or inflamed vessels, sensitizes endothelial cells to inflammatory stimuli through integrins α5β1and and αvβ3. A chimeric integrin in which the cytoplasmic domain of α5 is replaced with that of α2 pairs with β1 and binds fibronectin but signals like α2β1.
Methods and Results
Here, we examined mice in which integrin α5 is replaced with the α5/2 chimera, using the transverse aortic constriction and partial carotid ligation models of vessel remodeling. Following transverse aortic constriction and partial carotid ligation surgery, wild‐type mice showed increased fibronectin deposition and expression of inflammatory markers, which were strongly attenuated in a5/2 mice. α5/2 mice also showed reduced artery wall hypertrophy in the transverse aortic constriction model and diminished inward remodeling in the partial carotid ligation model. Acute atherosclerosis after partial carotid ligation in hyperlipidemic ApoE−/− mice on a high fat diet was dramatically decreased in α5/2 mice.
Conclusions
Fibronectin and integrin α5 signaling is a key element of pathological vascular remodeling in acute models of both hypertension and disturbed flow. These results underscore the key role for integrin α5 signaling in pathological vascular remodeling associated with hypertension and atherosclerosis and support its potential as a therapeutic target.
Keywords: artery wall remodeling, atherosclerosis, extracellular matrix, hypertension, inflammation, integrin signaling, transverse aortic constriction
Subject Categories: Atherosclerosis, Vascular Disease, Hemodynamics, Inflammation, Hypertension
Nonstandard Abbreviations and Acronyms
- ECM
extracellular matrix
- PCL
partial carotid ligation
- TAC
transverse aortic constriction
Clinical Perspective
What Is New?
Extracellular matrix remodeling with increased deposition of fibronectin amplifies inflammatory signaling and worsens outcomes in several models of vascular remodeling through the integrin α5 cytoplasmic domain.
The finding that hypertensive remodeling depends so strongly on fibronectin‐integrin α5 signaling is entirely novel and not predictable from past studies on responses to fluid shear stress.
What Are the Clinical Implications?
These results thus identify integrin α5 and downstream partners as potential therapeutic targets.
Arteries remodel in response to physiological stresses such as changes in blood pressure or fluid shear stress, and to pathological stresses such as low and disturbed shear stress, hyperlipidemia, diabetes mellitus, or oxidative stress among others. 1 , 2 Physiological remodeling is characterized by homeostasis, in which key variables return to their original levels or set point after a perturbation. 1 , 3 , 4 For example, increases in blood pressure induce hypertrophic thickening of the vessel wall to restore tensile wall stress to close to original values. Increased or decreased blood flow through a vessel with concomitant changes in fluid shear stress results in outward or inward remodeling of vessel diameters to restore shear stress to close to original values. Inflammation is an essential component of all remodeling. 5 Cells of the vascular wall that register changes in homeostatic variables (such as wall stress, the tension per unit area from blood pressure, or fluid shear stress, the frictional force exerted by blood flow) activate inflammatory pathways, resulting in recruitment of leukocytes, most prominently monocytes. These cells contribute to these processes by extracellular matrix (ECM) production and degradation, and cytokine/chemokine secretion. Restoration of wall or fluid shear stress to homeostatic levels leads to suppression of inflammatory signaling and restoration of the normal state.
Pathological remodeling, exemplified by atherosclerotic lesions, resembles physiological remodeling in many respects but differs in that the initiating stimuli are never terminated and the inflamed state never resolves. 4 , 6 But ECM remodeling is a central element of both physiological and pathological processes. 6 , 7 Vascular remodeling is universally accompanied by expression and assembly of provisional ECM proteins such as fibronectin, thrombospondin, or fibrin, whereas vessel stabilization and quiescence are associated with loss of these components and assembly of basement membranes where collagen IV and laminin are the main protein components. 8 , 9 Cells interact with provisional ECM proteins through RGD (Arg‐Gly‐Asp) binding integrins, of which integrins α5β1 and αvβ3 are the most prominent, whereas RGD (Arg‐Gly‐Asp) independent integrins α2β1, α6β1, and α6β4 are the main receptors for collagens and laminins. 10
Importantly, ECM remodeling also modulates inflammatory activation. 9 In endothelial cells, laminin/collagen IV basement membranes limit inflammatory responses through signaling by integrin α2β1, while fibronectin enhances responses to inflammatory stimuli, including disturbed fluid shear stress, interleukin‐1β, and oxidized LDL through integrin α5β1 and αvβ3. 11 , 12 , 13 , 14 , 15 Though less studied, collagen/laminin basement membranes also promote smooth muscle cell quiescence and differentiation. 16 , 17 The effects of these different ECM proteins were traced to the integrin α subunit cytoplasmic domains: a chimeric integrin in which the α5 cytoplasmic domain was replaced with that of α2 binds fibronectin and supports cell adhesion and cytoskeletal organization normally but signals as if the cells were on laminin or collagen. 17 A mouse in which this mutation was inserted into the α5 locus developed normally and is healthy and fertile but showed reduced endothelial inflammatory marker expression in regions of disturbed flow, developed smaller and less inflamed atherosclerotic lesions in hyperlipidemia, and showed improved recovery from hindlimb ischemia. 17 , 18
In the current study, we analyzed integrin α5/2 mice in 3 new models of vascular remodeling. We report that these mice show dramatic decreases in inflammation and remodeling, both physiological and pathological.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Partial Carotid Ligation Model
Ten‐ to twelve‐week‐old C57BL6, α5/2 integrin knock‐in mice and ApoE−/− mice were used for all animal studies. ApoE−/− mice and α5/2 integrin knock‐in mice in ApoE−/− are homozygous. All the mice experiments were performed according to the approved protocol of the IACUC (Institutional Animal Care and Use Committee) at Yale University. Mice were anesthetized with ketamine and xylazine and surgery was performed. Briefly, 3 out of 4 branches of the left common carotid artery (left external carotid, internal carotid, and occipital artery) were ligated with suture, while superior thyroid artery was left intact. Mice were euthanized after isoflurane inhalation, perfused, and vascular tree isolated along with carotid arteries at the indicated time points.
Transverse Aortic Constriction
Ten‐ to twelve‐week‐old mice anesthetized with ketamine and xylazine and hair was removed. A Para median incision was introduced with 1‐mm sternum and each rib was cut individually, to expose heart and thymus, which was retracted to expose the aorta and carotid arteries. A curved 22‐gauge needle with 9‐0 sutures passed under aorta between carotid arteries and 27‐gauze needle, space was placed on the arch before securing the ligature. The space was then removed, thus leaving a desired stenosis. The chest was closed with 3 to 5 simple interrupted sutures (5.0) followed by skin closure. Buprenorphine was given subcutaneously at 8‐hour intervals following surgery. Mice were euthanized after isoflurane inhalation, perfused, and vascular tree isolated along with carotid arteries at indicated times.
Immunofluorescence and Histochemistry
Carotid arteries were embedded in optimal cutting temperature, frozen on dry ice, and stored at −80 °C. Carotid arteries were sectioned on cryostat to generate 10‐μm sections. Cryosections were fixed in acetone for 10 minutes at −20 °C, blocked in IHC Tek antibody diluent for 1 hour at room temperature, and incubated with indicated antibodies in IHC Tek antibody diluent buffer. Antibodies were phospho‐NF‐ĸβ p65 (Ser536) [phospho‐nuclear factor ĸβ]‐P65 (1; 100, Cat # 3033 Cell signaling); fibronectin (1:400; Cat # F3648, Sigma); VCAM1[Vascular cell adhesion molecule‐1] (1:200, Cat # 553330, BD Bioscience); ICAM1 [Intercellular adhesion molecule‐1] (1:200, Cat # 116101, Biolegend); CD68 (1:200, Cat # AB125212, Abcam); CD45 (1:200, Cat # 550539, BD Biosciences). Sections were washed 3 times with PBS and incubated with Alexa Fluor 598‐conjugated donkey anti rabbit or rat secondary antibody for 1 hour at room temperature. Slides were washed with PBS and mounted in VECTASHIELD with DAPI. Images were acquired using Nikon 4 laser confocal microscope.
Statistical Analysis
National Institutes of Health ImageJ program was used for quantification. Lumen diameter, vessel area, and staining area were calculated from carotid artery sections. For measuring the lumen and exact vessel area, paraformaldehyde fixed carotid artery sections were stained with PECAM (platelet endothelial cell adhesion molecule) antibody and mounted with VECTASHIELD DAPI. Using ImageJ program, first we measured the lumen area and total vessel area for each section. By subtracting the lumen area from total vessel area, we determined the exact vessel area. For the quantification of specific antibody‐stained area, images were converted to grayscale in ImageJ and segmented the endothelial stained area using the thresholding. Finally, we measured the thresholded area. For CD45, CD68, and phospho‐NF‐ĸβ p65 we counted the number of positively stained cells in the endothelium. For the measurement of signal intensity, images converted to grayscale in ImageJ and stained area thresholded. Signal intensity measured in the thresholded area. Statistical analysis was performed using SigmaPlot 14.5. When variances were unequal, we used non‐parametric Kruskal–Wallis test with multiple pairwise comparisons (Student‒Newman‒Keule method). For data with equal variances, we used parametric 1‐way ANOVA with multiple pairwise comparisons (Student–Newman‒Keule method). For data with unequal sample size, we used Dunn multiple pairwise comparisons followed by Kruskal–Wallis test. P<0.05 was considered statistically significant. All data are expressed as mean±SEM. We computed the effect size for pairwise comparisons using Cohen d calculator (the difference between 2 means divided by an SD for the data). Effect size score was assigned as follows, >0.2 small, >0.5 medium, and >0.8 large effect size. All graphs were created using GraphPad Prism software (San Diego, CA).
Results
Transverse Aortic Constriction
To assess the role of integrin α5 signaling in pressure‐induced artery remodeling, we subjected mice to transverse aortic constriction (TAC) or to sham surgery as a control. This was accomplished by placing a clamp between the innominate artery and the left carotid, a well‐established experimental method to increase pressure in the right carotid artery 19 , 20 , 21 (Figure S1). Left and right carotid arteries were collected 1‐week postsurgery. We observed significant increases in vessel area and lumen diameter of the right carotid artery compared with sham, or with the contralateral left carotid. These changes were accompanied by strongly elevated fibronectin deposition in the endothelial layers of right carotids but not in contralateral left carotids and sham carotids (Figure 1A). Comparison of fibronectin staining area between sham and wild‐type (WT) TAC right carotid showed a large effect size, whereas the comparison between WT TAC left and sham carotid showed only a small effect size (Table S1).
Figure 1. Transverse aortic constriction‐induced artery remodeling and fibronectin deposition in α5/2 mice.
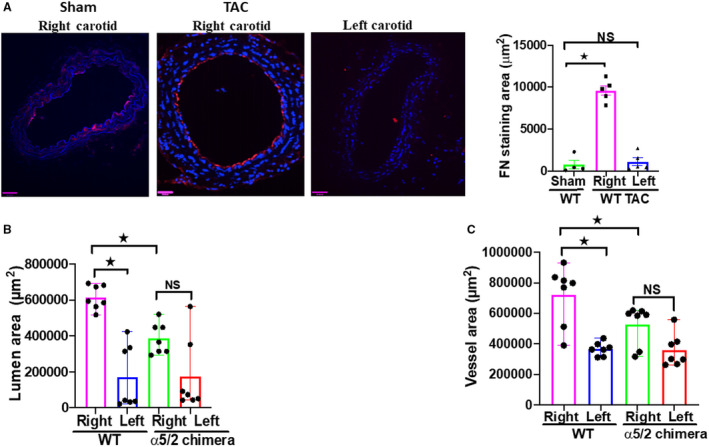
A, Transverse aortic constriction induced deposition of the fibronectin in right carotid arteries compared with sham controls and the left carotids. Nuclei were counterstained with DAPI (blue). Wild‐type (WT) sham mice, n=5; WT transverse aortic constriction surgery mice, n=5; NS, not significant. B and C, Vessel area and lumen diameter in carotid arteries from WT vs α5/2 knock‐in mice. WT mice, n=7; α5/2 mice, n=7; Statistical analysis: Kruskal–Wallis 1‐way analysis with Student–Newman–Keuls multiple pairwise comparison. TAC indicates transverse aortic constriction; and WT, wild‐type. Mean±SEM; *P<0.05, compared with WT mice). Scale bars=35 μm.
TAC in α5/2 Mice
To investigate the role of fibronectin‐integrin α5 signaling in TAC‐induced structural remodeling of the carotid arteries, we compared these results to integrin α5/2 knock‐in mice. 14 Right carotids from α5/2 knock‐in mice enlarged less than WT mice (Figure 1B and 1C). Comparison of the WT left and right carotids produced large effect size for lumen area and medium effect size for vessel area. Comparison of WT and α5/2 mice right carotid produced large effect size for lumen area and medium effect size for vessel area. Comparison of right and left carotids in α5/2 mice produced medium effect sizes for both lumen area and vessel area (Table S2). The accumulation of fibronectin seen in WT mice was largely suppressed in α5/2 carotids (Figure 2A). Staining for the leukocyte adhesion receptors and inflammatory markers VCAM1 and ICAM1 showed strong elevation in the endothelium of the right carotid in WT mice but only minor changes in α5/2 mice (Figure 2B and 2C). Staining for CD68 as a marker for monocyte/macrophages showed strong inflammatory cell recruitment to the right carotid from WT mice, but only small changes in α5/2 mice (Figure 2D). Finally, we stained these tissues for phospho‐NF‐ĸβ p65 (nuclear factor kappa β) to assess activation of this critical inflammatory transcription factor. TAC induced strong phospho‐NF‐κB p65 activation in the right carotid from WT mice but no significant changes in α5/2 mice (Figure 2E). Left carotids from both genotypes showed only minor changes in any of these markers. Quantifying signal intensities instead of area for these markers also showed attenuation in α5/2 mice (Figure S2). Comparison of the staining area between left and right carotids of WT mice produced large effect sizes for fibronectin, ICAM, CD68, and phospho‐NF‐ĸβ p65; and medium effect size for VCAM. Comparison of staining area between right carotid of WT and α5/2 mice produced large effect size for VCAM, ICAM, CD68, and phospho‐NF‐ĸβ p65; and medium effect size for fibronectin. Comparison of the staining area between right and left carotid of α5/2 mice produced small effect size for VCAM, ICAM, CD68 and phospho‐NF‐ĸβ p65; and medium effect size for fibronectin (Table S3). Together, these data show that inflammation and structural remodeling in this model of acute hypertension are drastically reduced in α5/2 chimera mice, consistent with a role of integrin α5.
Figure 2. Transverse aortic constriction‐induced fibronectin deposition and endothelial inflammation.
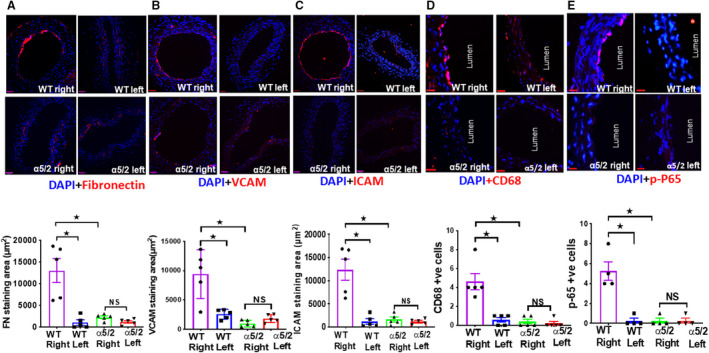
Wild‐type and α5/2 knock‐in mice were subjected to transverse aortic constriction surgery. One‐week postsurgery, carotid artery sections were stained for: A, Fibronectin; (B) VCAM‐1 (Vascular cell adhesion molecule‐1); (C) ICAM‐1 (Intercellular adhesion moleucle‐1); (D) CD68; (E) phospho‐NF‐ĸβ p65 (nuclear factor kappa β ) p‐65. Nuclei are counterstained with DAPI (blue). Wild‐type mice, n=5; α5/2 knock‐in mice, n=5; For phospho‐NF‐ĸβ p65 staining, wild‐type mice, n=4; α5/2 knock‐in mice, n=4; endothelial cell staining for these markers was then quantified. Statistical analysis: Kruskal–Wallis 1‐way analysis with Student‐Newman–Keuls multiple pairwise comparison. WT indicates wild‐type. *P<0.05, compared with wild‐type mice. Scale bars=35 μm.
Partial Carotid Ligation
Partial carotid ligation (PCL) in mice decreases blood flow magnitude and introduces disturbances into the flow patterns in the common carotid, resulting in inflammatory activation of the endothelium and reduced lumen volume. 22 , 23 We first subjected WT mice to PCL surgery and examined carotid arteries at 1‐week postsurgery (Figure S3). PCL resulted in reduced lumen diameter of the left carotid compared with the right carotid (Figure 3A). Comparison of left and right carotids of WT mice produced medium effect size for lumen area, whereas comparison of left and right carotids of α5/2 mice produced small effect size for lumen area (Table S4). We also observed a dramatic increase in fibronectin staining in the endothelial layer of the left but not right carotid arteries (Figure 3B). These observations prompted us to test the involvement of integrin α5 in these events by performing PCL in α5/2 mice. In contrast to WT mice, integrin α5/2 mice showed no change in the lumen area (Figure 3A). Further, ICAM1 and VCAM1 were strongly induced in the left carotid in WT mice but changed little in α5/2 mice (Figure 4A and 4B). Consistent with this finding, inflammatory cell recruitment marked by CD45 and CD68 was also attenuated in the left carotid of α5/2 mice (Figure 4C and 4D) as was activation of phospho‐NF‐ĸβ p65 (Figure 4E). The contralateral right carotids of WT and α5/2 knock‐in mice remained negative. Quantifying signal intensities for theses markers also showed attenuation in α5/2 mice (Figure S4). Comparison of the staining area between left and right carotids of WT mice produced large effect size for VCAM1, ICAM1, and CD45; and medium effect size for fibronectin, CD68, and p‐NFĸβ. Comparison of staining area between left carotid of WT and α5/2 mice produced large effect size for VCAM, ICAM, and CD45; and medium effect size for fibronectin, CD68, and p‐NFĸβ. Comparison of the staining area between WT left and α5/2 mice right carotids produced large effect sizes for VCAM1, ICAM1, and CD45; and medium effect sizes for fibronectin, CD68, and phospho‐NF‐ĸβ p65 (Table S5). PCL‐induced artery inflammation and structural remodeling thus depend strongly on fibronectin signaling through integrin α5.
Figure 3. Partial carotid ligation‐induced artery remodeling.
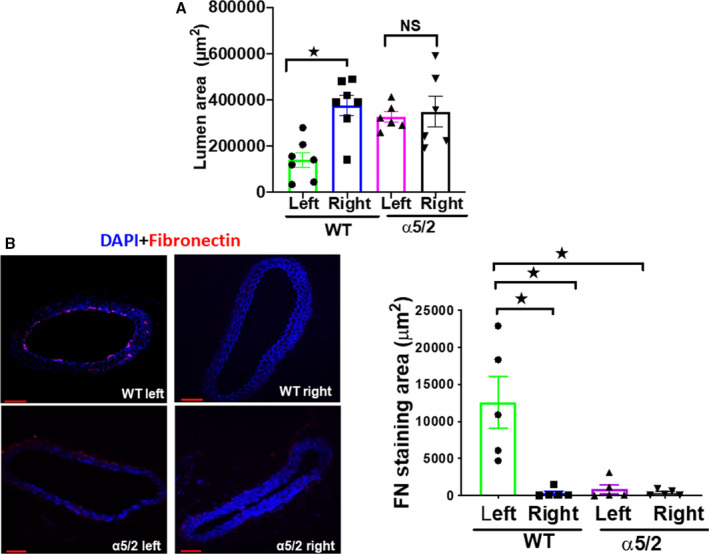
A, Wild‐type (WT) and α5/2 knock‐in mice were subjected to partial carotid ligation surgery. One‐week post‐surgery, carotid artery lumen diameters were measured. Left carotid arteries from WT mice have reduced lumen diameter compared with right carotid arteries form WT mice. In contrast to WT mice, integrin α5/2 mice showed no change in the lumen area. WT mice, n=7; α5/2 knock‐in mice, n=7. B, Fibronectin staining in left carotids in WT and α5/2 mice. Nuclei were counterstained with DAPI (blue). WT mice, n=5; α5/2 knock‐in mice, n=5; Statistical analysis: Kruskal–Wallis 1‐way analysis with multiple pairwise comparisons. (Dunn test for lumen area, A and Student–Newman Keuls for fibronectin staining area B). WT indicates wild‐type. Mean±SEM; *P<0.05, compared with WT mice. Scale bars=35 μm.
Figure 4. Partial carotid ligation‐induced endothelial inflammation.
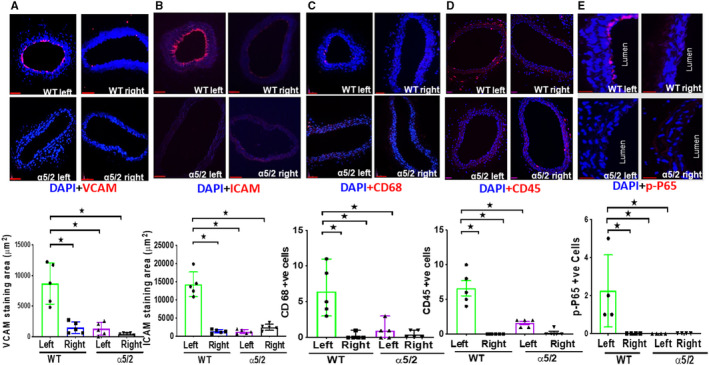
Wild‐type and α5/2 knock‐in mice were subjected partial carotid ligation surgery. One‐week post‐surgery, carotid artery sections were stained with antibodies to: (A) VCAM1 (Vascular cell adhesion molecule‐1); (B) ICAM1 (Intercellular adhesion molecule‐1); (C) CD68; (D) CD45; (E) phospho‐NF‐ĸβ p65. Nuclei were counterstained with DAPI (blue). Wild‐type mice, n=5; α5/2 knock‐in mice, n=5. For phospho‐NF‐ĸβ p65 staining, wild‐type mice, n=4; α5/2 knock‐in mice, n=4. Statistical analysis: Kruskal–Wallis 1‐way analysis with Student–Newman–Keuls multiple pairwise comparison. WT indicates wild‐type. Mean±SEM; *P<0.05, compared with wild‐type mice. Scale bars=35 μm.
Acute Atherosclerosis After PCL in Hypercholesterolemic ApoE−/− Mice
PCL in hypercholesterolemic mice is used to model acute disturbed shear‐induced atherosclerosis. 24 , 25 Lesions develop within a few weeks because of the low and strongly oscillatory shear stress induced by surgery. To test the role of integrin α5 in this model, we crossed α5/2 knock‐in mice with hypercholesterolemic ApoE−/− mice (Jackson laboratories, B6.129p2‐Apoetm1Unc/j; Cat#002052) to generated WT; ApoE−/− and α5/2; ApoE−/− mice. α5/2; ApoE−/− mice are homozygous for the α5/2 integrin. Mice at 8 to 10 weeks were subject to PCL and fed a Western diet (RD Western diet #D12079B, Open Source Diet) for 3 weeks. Hematoxylin and eosin staining demonstrated significant atherosclerosis in the LCA (Left carotid artery) of WT; ApoE−/− mice whereas plaque in α5/2; ApoE−/− mice was barely detected (Figure 5A). Staining lipids with Oil Red O confirmed the reduction in plaque in α5/2 mice (Figure 5B). Monocytes/macrophage (CD68+ cells) infiltration in the LCA were also markedly reduced in α5/2; ApoE−/− mice (Figure 5C). As before, the contralateral side was unaffected and showed negligible plaque formation on this time scale. Plasma lipid profiles of total cholesterol, LDL cholesterol, and triglycerides were not statistically different between WT and α5/2 mice (Figure 6). Comparison of the staining area between left carotid of WT; ApoE−/−and α5/2; ApoE−/−mice produced medium effect sizes for Oil red, CD68, and plaque area. Comparison of staining area between left and right carotid of WT; ApoE−/−mice produced large effect sizes for Oil red, CD68 and plaque area. Comparison of the staining area between WT; ApoE−/− left and α5/2; ApoE−/− right produced large effect size for Oil red, CD68, and plaque area (Table S6). Overall, these results revealed that the α5/2 knock‐in strongly attenuated flow‐dependent acute atherosclerosis.
Figure 5. Partial carotid ligation in hypercholesterolemic mice.
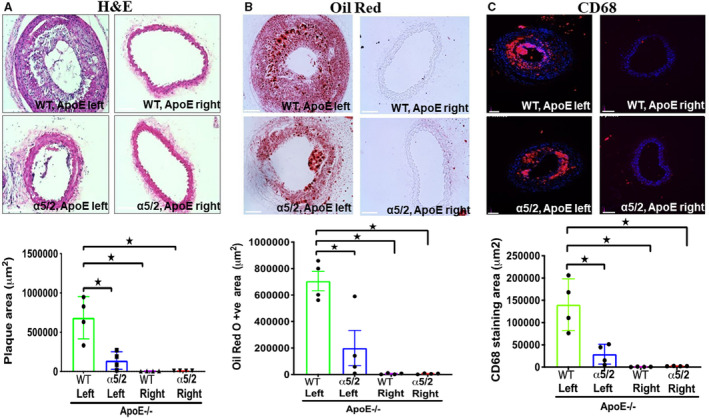
Eight‐ to ten‐week‐old wild‐type and integrin α5/2 mice on the ApoE−/− background were subjected to partial carotid ligation surgery. After the surgery, mice were fed a Western diet for 3 weeks and carotid arteries examined. A, Hematoxylin and eosin staining. B, Oil Red O staining. C, CD68 staining. Wild‐type; ApoE−/− mice, n=4; α5/2; ApoE−/− mice, n=4; Statistical analysis: Kruskal–Wallis 1‐way analysis with Student–Newman–Keuls multiple pairwise comparison. H&E indicates hematoxylin and eosin; and WT, wild‐type. Mean±SEM; *P<0.05, compared with wild‐type mice. Scale bars=35 μm.
Figure 6. Plasma lipids.
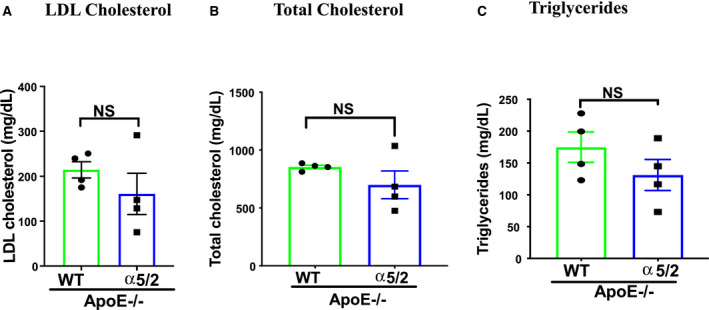
Plasma lipid profiles in wild‐type; ApoE−/− and α5/2; ApoE−/− mice: (A) Plasma low‐density lipoprotein cholesterol; (B) total cholesterol; (C) triglycerides. Statistical analysis: 2‐tailed Student t test. No significant differences were observed. LDL indicates low‐density lipoprotein; WT, wild‐type.
Discussion
In this study, we report that the integrin α5/2 mutation in mice, which abolishes the inflammatory effects of fibronectin, strongly affects vascular remodeling. In the TAC model of acute hypertension in the right carotid artery, α5/2 mice show markedly less fibronectin deposition, less inflammatory activation of the endothelium, and less artery wall thickening. It should be noted that C57Bl/6 mice show excessive adventitial thickening in hypertension, perhaps related to the susceptibility of this strain to inflammatory stimuli. 26 , 27 These responses may thus be regarded as partially pathological in that remodeling overshoots the homeostatic goal. This effect was abolished by the α5/2 mutation, consistent with its ability to limit inflammation.
PCL is principally a model of flow‐induced remodeling attributable to induction of low and oscillatory flow in the affected common carotid artery. Altered flow leads to inflammatory activation of the endothelium and inward remodeling to reduce lumen diameter, sometimes to near closure, a feature more in keeping with pathological than physiological processes, likely because of the oscillatory flow component. Consistent with high fibronectin deposition in the endothelial layer of WT mice, inflammatory activation and inward remodeling were strongly blunted in α5/2 mice. To expand on these findings, we then examined effects of PCL in hypercholesterolemic mice, a model of acute flow‐induced atherosclerotic plaque formation. This model typically looks at short times after initiation of disturbed flow, thus, represents an acute, mechanistic model rather than a model of chronic disease. 24 Plaque size, lipid accumulation, and inflammatory cell recruitment were all greatly decreased. These results strongly support a major role for inflammatory fibronectin signaling in disturbed flow‐induced vessel pathology.
We observed strong inhibition of inflammatory activation and remodeling in α5/2 mice despite the fact that endothelial cells express other fibronectin‐binding integrins such as αvβ3. It seems likely that this is because the chimeric integrin strongly disrupts fibronectin matrix assembly (Figures 3A and 4B), leaving little fibronectin for other integrins to bind. While α5β1 plays a primary role in fibronectin‐matrix assembly, αv integrins can partially substitute, such that total loss is only seen after deletion of both α5 and αv. 28 We suspect that the dramatic loss of fibronectin matrix with the α5/2 chimera reflects active inhibition instead of mere loss of function; this speculation is consistent with a previous study in endothelial cells that showed inhibition of αvβ3 activation by signaling through integrin α2β1. These results raise the question, are the improved outcomes because of altered integrin signaling, less fibronectin deposition, or both? We suggest that our results are best understood in the context of the inflammatory and remodeling pathways that control fibronectin gene expression and matrix assembly. Fibronectin can be induced through NF‐ĸβ, β‐catenin, and Smad2/3 transcription factors, 29 , 30 , 31 , 32 creating a positive feedback loop in which the elevated fibronectin promotes further inflammation, which amplifies these pathways. 32 Thus, reducing the inflammatory effects of fibronectin by mutation of the integrin α5 cytoplasmic domain disrupts this regulatory loop and reduces fibronectin expression.
Vascular remodeling in response to altered mechanical forces derived from blood flow and pressure is physiological when key variables such as tensional wall stress or fluid shear stress are restored to close to their initial levels. 2 , 3 Physiological remodeling requires inflammation and participation of immune cells but is carefully regulated and resolves when key variables return to their initial value or set point. By contrast, remodeling is pathological when initial set points are not restored because of either insufficiency or, more often, overcompensation. For example, artery walls in hypertension thicken too much and lose elasticity, which exacerbates hypertension and its consequences for the microvasculature. Importantly, excessive or uncontrolled inflammation is the principal cause of pathological remodeling, typified by formation of atherosclerotic lesions in regions of low/disturbed shear stress. 33 Adventitial fibrosis and stiffening in hypertension provides another example. 27
The current results identify fibronectin and integrin α5 signaling as key elements of pathological vascular remodeling in acute models of both hypertension and disturbed flow. We note that mice in these experiments were total knock‐ins, thus many cell types may contribute to the observed effects. Assessing effects in cell‐type specific conditional knock‐ins is an important direction for future work. While the ultimate goal of these studies is the identification of therapeutic targets, fibronectin, or integrin α5 themselves are essential for physiological processes and thus seem unlikely candidates for direct intervention. However, elements of downstream pathways have been discovered, including the interaction of phosphodiesterase 4D with the α5 cytoplasmic domain and the interaction of phosphodiesterase 4D with the B55 subunit of protein phosphatase 2A. 14 , 15 Blocking these downstream pathways may be more promising avenues for translation, as are further investigations into mechanisms of α5 inflammatory signaling in atherosclerosis and hypertension.
Sources of Funding
This work was supported by National Institutes of Health (NIH) (RO1135582‐05) to M.A.S.
Disclosures
None.
Supporting information
Table S1–S6
Figure S1–S4
Acknowledgments
We thank Tristan P. Driscoll, FAMU—FSU (Florida A&M University ‐ Florida State University) College of Engineering, for advice on statistical analysis. Lipid analysis was done by the Yale Mouse Phenotypic Center. M.B. designed the study, performed all experiments, and analyzed data and wrote the article. M.B. performed most of the studies at Yale and completed the analysis at University of Texas Health Sciences, San Antonio. J.Z. performed the mouse surgeries. M.A.S. initiated the project, co‐wrote the article, and provided financial support.
For Sources of Funding and Disclosures, see page 9.
Preprint posted on BioRxiv January 08, 2021. doi: 10.1101/2021.01.06.425577.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.021160
References
- 1. Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209–H1224. doi: 10.1152/ajpheart.01047.2006 [DOI] [PubMed] [Google Scholar]
- 2. Humphrey JD. Mechanisms of arterial remodeling in hypertension: coupled roles of wall shear and intramural stress. Hypertension. 2008;52:195–200. doi: 10.1161/HYPERTENSIONAHA.107.103440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Humphrey JD. Mechanisms of vascular remodeling in hypertension. Am J Hypertens. 2020;34:432–441. doi: 10.1093/ajh/hpaa195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. 2016;126:821–828. doi: 10.1172/JCI83083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201 [DOI] [PubMed] [Google Scholar]
- 6. Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878 [DOI] [PubMed] [Google Scholar]
- 7. Astrof S, Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis. 2009;12:165–175. doi: 10.1007/s10456-009-9136-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finney AC, Stokes KY, Pattillo CB, Orr AW. Integrin signaling in atherosclerosis. Cell Mol Life Sci. 2017;74:2263–2282. doi: 10.1007/s00018-017-2490-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yurdagul A Jr, Orr AW. Blood brothers: hemodynamics and cell‐matrix interactions in endothelial function. Antioxid Redox Signal. 2016;25:415–434. doi: 10.1089/ars.2015.6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- 11. Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF‐kappab activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Orr AW, Stockton R, Simmers MB, Sanders JM, Sarembock IJ, Blackman BR, Schwartz MA. Matrix‐specific p21‐activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol. 2007;176:719–727. doi: 10.1083/jcb.200609008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yurdagul A Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. Alpha5beta1 integrin signaling mediates oxidized low‐density lipoprotein‐induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yun S, Budatha M, Dahlman JE, Coon BG, Cameron RT, Langer R, Anderson DG, Baillie G, Schwartz MA. Interaction between integrin alpha5 and pde4d regulates endothelial inflammatory signalling. Nat Cell Biol. 2016;18:1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yun S, Hu R, Schwaemmle ME, Scherer AN, Zhuang Z, Koleske AJ, Pallas DC, Schwartz MA. Integrin alpha5beta1 regulates PP2A complex assembly through pde4d in atherosclerosis. J Clin Invest. 2019;129:4863–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Orr AW, Lee MY, Lemmon JA, Yurdagul A Jr, Gomez MF, Bortz PD, Wamhoff BR. Molecular mechanisms of collagen isotype‐specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2009;29:225–231. doi: 10.1161/ATVBAHA.108.178749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2010;47:168–180. doi: 10.1159/000250095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Budatha M, Zhang J, Zhuang ZW, Yun S, Dahlman JE, Anderson DG, Schwartz MA. Inhibiting integrin alpha5 cytoplasmic domain signaling reduces atherosclerosis and promotes arteriogenesis. J Am Heart Assoc. 2018;7:e007501. doi: 10.1161/JAHA.117.007501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X, Feng T, Zeng X‐X, Liang H, Situ BO, Zhang Q, Zhou F, Chen Y, Wang T, Cai DU, et al. Identification of transcriptional variation in aortic remodeling using a murine transverse aortic constriction (TAC) model. Front Cardiovasc Med. 2020;7:581362. doi: 10.3389/fcvm.2020.581362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richards DA, Aronovitz MJ, Calamaras TD, Tam K, Martin GL, Liu P, Bowditch HK, Zhang P, Huggins GS, Blanton RM. Distinct phenotypes induced by three degrees of transverse aortic constriction in mice. Sci Rep. 2019;9:5844. doi: 10.1038/s41598-019-42209-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eberth JF, Gresham VC, Reddy AK, Popovic N, Wilson E, Humphrey JD. Importance of pulsatility in hypertensive carotid artery growth and remodeling. J Hypertens. 2009;27:2010–2021. doi: 10.1097/HJH.0b013e32832e8dc8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korshunov VA, Solomatina MA, Plekhanova OS, Parfyonova YV, Tkachuk VA, Berk BC. Plasminogen activator expression correlates with genetic differences in vascular remodeling. J Vasc Res. 2004;41:481–490. doi: 10.1159/000081804 [DOI] [PubMed] [Google Scholar]
- 23. Yuan S, Yurdagul A Jr, Peretik JM, Alfaidi M, Al Yafeai Z, Pardue S, Kevil CG, Orr AW. Cystathionine gamma‐lyase modulates flow‐dependent vascular remodeling. Arterioscler Thromb Vasc Biol. 2018;38:2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535–H1543. doi: 10.1152/ajpheart.00510.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar S, Kang DW, Rezvan A, Jo H. Accelerated atherosclerosis development in C57BL6 mice by overexpressing AAV‐mediated PCSK9 and partial carotid ligation. Lab Invest. 2017;97:935–945. doi: 10.1038/labinvest.2017.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taherzadeh Z, VanBavel E, de Vos J, Matlung HL, van Montfrans G, Brewster LM, Seghers L, Quax PH, Bakker EN. Strain‐dependent susceptibility for hypertension in mice resides in the natural killer gene complex. Am J Physiol Heart Circ Physiol. 2010;298:H1273–H1282. doi: 10.1152/ajpheart.00508.2009 [DOI] [PubMed] [Google Scholar]
- 27. Bloss CS, Wineinger NE, Peters M. A prospective randomized trial examining health care utilization in individuals using multiple smartphone‐enabled biosensors. bioRxiv. Preprint posted online October 28, 2015. doi: 10.1101/029983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Flier A, Badu‐Nkansah K, Whittaker CA, Crowley D, Bronson RT, Lacy‐Hulbert A, Hynes RO. Endothelial alpha5 and alphav integrins cooperate in remodeling of the vasculature during development. Development. 2010;137:2439–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang J, Zeng Z, Wu T, Yang Z, Liu B, Lan T. Emodin attenuates high glucose‐induced TGF‐beta1 and fibronectin expression in mesangial cells through inhibition of NF‐Kappab pathway. Exp Cell Res. 2013;319:3182–3189. [DOI] [PubMed] [Google Scholar]
- 30. Jagadeeshan S, Krishnamoorthy YR, Singhal M, Subramanian A, Mavuluri J, Lakshmi A, Roshini A, Baskar G, Ravi M, Joseph LD, et al. Transcriptional regulation of fibronectin by p21‐activated kinase‐1 modulates pancreatic tumorigenesis. Oncogene. 2015;34:455–464. doi: 10.1038/onc.2013.576 [DOI] [PubMed] [Google Scholar]
- 31. Ventura E, Weller M, Macnair W, Eschbach K, Beisel C, Cordazzo C, Claassen M, Zardi L, Burghardt I. TGF‐beta induces oncofetal fibronectin that, in turn, modulates TGF‐beta superfamily signaling in endothelial cells. J Cell Sci. 2018;131.1–12. doi: 10.1242/jcs.209619 [DOI] [PubMed] [Google Scholar]
- 32. Feaver RE, Gelfand BD, Wang C, Schwartz MA, Blackman BR. Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation. Circ Res. 2010;106:1703–1711. doi: 10.1161/CIRCRESAHA.109.216283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1–S6
Figure S1–S4