

Abstract
Orthohantavirus, a zoonotic virus responsible for causing human cardio-pulmonary disease, is proven to be a fatal disease. Due to the paucity of regimens to cure the disease and efficient management to eradicate this deadly virus, there is a constant need to expand in-silico approaches belonging to immunology domain to formulate best feasible peptide-based vaccine against it. In lieu of that, we have predicted and validated an epitope of nine-residue-long sequence “MIGLLSSRI”. The predicted epitope has shown best interactions with HLA alleles of MHC Class II proteins, namely HLA DRB1_0101, DRB1_0401, DRB1_0405, DRB1_0701, DRB1_0901, DRB1_1302, and DRB1_1501. The structure of the epitope was modeled by deploying PEPFOLD 3.5 and verified by Ramachandran plot analysis. Molecular docking and simulation studies reveal that this epitope has satisfactory binding scores, ACE value and global energies for docked complexes along with selectable range of RMSD and RMSF values. Also, the predicted epitope “MIGLLSSRI” exhibits population coverage of more than 62% in world population and maximum of 70% in the United States of America. In this intensive study, we have used many tools like AllergenFP, NETMHCII 3.2, VaxiJen, ToxinPred, PEPFOLD 3.5, DINC, IEDB-Population coverage, MHCPred and JCat server. Most of these tools are based on modern innovative statistical algorithms like HMM, ANN, ML, etc. that help in better predictions of putative candidates for vaccine crafting. This innovative methodology is facile, cost-effective and time-efficient, which could facilitate designing of a vaccine against this virus.
Keywords: Peptide, Epitope, Docking, Simulation, Surface glycoprotein, Vaccine
Introduction
Hantavirus belongs to the family of Bunyaviridae with key features including enveloped single-stranded negative sense RNA; they are mostly associated with zoonosis (Guo et al. 2013). Hantavirus pulmonary syndrome and hemorrhagic fever renal syndrome are two common ailments associated with interference of human systems with Hantavirus. The causative agents of two human illnesses are rodent-borne hantaviruses (genus: Orthohantavirus): hemorrhagic fever with kidney disorders (HFRS) in Europe and hantavirus pulmonary syndrome (HPS) in North and South America. HPS mortality rates in South America can range from 35 to 50% (Maes et al. 2009; Ferro et al. 2020). An MHC class II molecule binds to epitopes that are produced after proteolytic processes on antigens coming from pathogens within the antigen-presenting cells (APCs), including B cells, macrophages and dendritic cells. Aftermath, MHC-II holding pathogenic epitopes binds to CD4+ T cells to trigger an adaptive immune response against similar kind of pathogens. Hantavax, a vaccine derived from rodent brain formulations with adjuvant like aluminum hydroxide, was used in Korea, but its production was halted and discouraged by many other countries due to use of rodent brain (Schmaljohn 2009). The proteasome is largely responsible for the processing of cytosolic proteins. TAP transports the short peptides into the ER, where they are assembled with MHC-I molecules, CD4+ T cell epitopes are moreover less variant in comparison to B cells and CD8+ T cell epitopes, also availability of data supports the multiple MHC Class II HLA variants makes this choice preferable (Blum et al. 2013; Bibi et al. 2021; Sanchez-Trincado et al. 2017). Exogenous proteins can also be supplied into this pathway via retro-translocation from phagosomes in some antigen-presenting cells, notably dendritic cells, a process known as cross-presentation. The retro-translocation channels may be recruited from the ER, where they are utilized to degrade misfolded membrane—spanning or cytoplasmic proteins via ER-associated degradation. MHC-II molecules, on the other hand, are largely responsible for presenting exogenous proteins. Antigens are absorbed through phagocytosis, macro-pinocytosis, and endocytosis, and then transported to a mature or late endosomal compartment, in which they are handled and placed into MHC-II molecules. Ribavirin (1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide), another potential drug, was also not approved by Food and Drug Administration (FDA) because of poor experimental results (Mahmud-Al-Rafat et al. 2015). So, there is a great challenge and scope for developing T cell epitopes that can possibly interact directly with MHC molecules and elicit an immunogenic response against Hantavirus. This study is primarily focused in developing epitope-based vaccine against Hantavirus by deploying modern in-silico immunoinformatic approaches. Adaptive immunity plays a crucial role in annihilating viral agents and it is more established with epitope-based peptide vaccines (Zhang 2018). The spread of Hantaviruses has also been reported to occur from contaminated aerosols inhalation (Jiang et al. 2014).
In present day times, immunotherapy is an amazing and proficient procedure to control harmful ailments. The prediction of peptide-based epitope vaccine is always a good approach for the treatment of tumors or viral diseases. There are numerous multi-epitope vaccines targeting various diseases including diseases caused by HIV, dengue (Krishnan et al. 2021; Sun et al. 2017), hepatitis B and C, Ebola, Chikungunya, Avian flu-A (H7N9), Zikavirus (Sharma et al. 2020), classical pig fever, Nipah virus (Kaushik 2020; Liang et al. 2018; Qu et al. 2018), and Norovirus. A perfect multi-epitope vaccine candidate can be used to incorporate a progressive immunological change and it can also evoke either a cellular or a humoral resistant reaction against the infection. A well-designed epitope-based peptide vaccine ought to turn into incredible prophylactic agents against the viral infections. In this investigation, a progression of immunoinformatic approaches has been applied against the proteome of Hantavirus for crafting an epitope-based vaccine. We have vigorously screened the repertoire of epitope by computational methods and validated by molecular simulation of the selected epitope. In addition, the codon usage and in-silico cloning, and the molecular simulations of the best antibody receptor docked complex were successively considered in our study. In present study, we crafted epitope-based vaccine strategy against this harmful pathogen by deploying best modern in-silico tools based on artificial intelligence and neural networks. The aim of study was to predict epitope-based vaccine peptide that holds therapeutic properties against Hantavirus disease. It is a fast and effective way for developing vaccine over hit-and-trial approaches of wet-lab analysis. It provides ease of doing for wet-lab validations and testing for novel vaccination programs.
Materials and methods
Protein database analysis
Primarily, the IEDB (Kim et al. 2012) and ViPR databases were explored to determine essential proteins related to Orthohantavirus. Coding sequence for surface glycoprotein (Accession no. KF537002.1) of Orthohantavirus, and NCBI-genbank accession number (AIA08876.1) for selected protein (SG). Molecular weight of SG Protein was found as 126,191 Da, and Theoretical pI as 7.73 using ExPASY ProtParam tool. The coding DNA sequence and the amino acid sequences were retrieved from NCBI-GenBank database. After that, RSCU values and codon usage analysis of selected coding sequence was done using DnaSP6 software (Rozas et al. 2017) to analyze more validation in selection. For the coding sequence under consideration, ENC-effective number of codons (its value should be in the range of 20–61; 20 indicates high biasness, while 61 shows no biasness) (Wright 1990) was computed.
Allergenicity, antigenicity and epitope screening
AllergenFP server was deployed to analyze allergenicity of selected protein. NETMHC-II Pan 3.2 server (Jensen et al. 2018) was used to screen out single best interacting epitope to maximum number of HLA-DRB alleles, it is based on artificial neural network (ANN) algorithms. Predicted epitope was subjected to VaxiJen server (Doytchinova and Flower 2007) for deriving best antigenicity criterion (> 1.1).
Structural analysis of epitope and HLA allele
After intensive analysis of epitope sequence, its structure was constructed using PEPFOLD 3.5 server (Lamiable et al. 2016). The obtained modeled structure was validated and observed for biochemical and toxicity analysis by deploying MolProbity (Chen et al. 2010), Ramachandran plot analysis and ToxinPred (Gupta et al. 2013), QM and SVM scores for toxicity analysis.
Molecular docking and simulation analysis
RCSB-PDB database (Berman et al. 2000) was deployed to get MHC Class II HLA alleles’ structure that exhibits strong interaction to screened and analyzed epitopes. Then molecular docking analysis was performed by DINC server (Antunes et al. 2017) and PatchDock—Firedock server (Schneidman-Duhovny et al. 2005) to obtain atomic contact energies and binding scores for docked complexes. Molecular simulation studies were performed using Gromacs software (Van Der Spoel et al. 2005) used for coarse-grained Brownian dynamics in GROMOS 9643a1 force field environment. GROMOS 9643a1 force field was utilized to evaluate thermodynamic properties throughout the MD simulation procedure. To eliminate very large energy configuration (steric conflicts or bonded components far off from the equilibrium), energy reduction was used. Some big protein structures can result in poor connections and unphysical fluctuations in total energy in later simulations; therefore, a quick reduction can eliminate these issues. After that, the subsystems were subjected through a 100-picosecond NVT ensemble MD_Simulation at 300 K temperature. Consequently, at 300 K and 1 bar pressure, NPT equilibrium was reached. In the binding procedure, a 10 ns MD_Simulation was performed on the selected conformational geometry of the HLA receptor to epitope vaccine complex. Trajectory analysis for 10 ns was used to evaluate RMSD values and RMSF analysis of docked complex (Joshi et al. 2020b).
Population coverage and in-silico cloning assessment
Finally, IEDB population coverage analysis (> 50%) based on restriction database (Bui et al. 2006) and MHCPred 2.0 server (Guan et al. 2003) logIC50 (1–10) quantitative prediction analysis was deployed to get more validation of selected putative T cell epitope for vaccine crafting. JCat (Java Codon Adaptation Tool) (Grote et al. 2005) server was deployed to obtain optimized codon sequence for epitope in E. coli k12 strain to get CAI near to best score 1.0 and GC-content codon percentage in 30–70% range to ensured high expression rate of vaccine candidate in E. coli k12 strain.
Results
Protein selection and codon usage analysis
Surface glycoprotein of Orthohantavirus was found to be significant for our research study as it is present on membrane of virus and it is involved in membrane fusion while targeting host cells for gaining entry. It was selected by analyzing ViPR and NCBI-GenBank database. Coding sequence for surface glycoprotein (Accession no. KF537002.1) of Orthohantavirus, and NCBI-genbank accession number (AIA08876.1) for selected protein (SG). Molecular weight of SG Protein was found as 126,191 Da, and Theoretical pI as 7.73 using ExPASY ProtParam tool. Allergenicity based on Tanimoto-similarity index for surface glycoprotein (SG) (Levanov et al. 2019) with accession number AIA08876.1 was found to be 0.82 (i.e., non-allergen), so suitable for further analysis. Coding sequence for this protein was retrieved from NCBI database to determine codon usage and RSCU value analysis. The multiple parameters of codon usage that were used to analyze natural selection and mutational pressure on genomic considerations were calculated for coding sequence of SG protein, i.e., ENC 51.142, CBI 0.264, G + C2 0.443, G + C3s 0.432 and S-Chi2 0.228. We were mainly focused on RSCU values and codon usage of coding sequence for surface glycoprotein of Orthohantavirus. Figure 1 clearly reveals the codons and amino acids that are more prevalent in this RSCU plot for all 64 codons.
Fig. 1.

Relative synonymous codon usage (RSCU) for coding sequence of surface glycoprotein of Orthohantavirus
Epitope selection and its structural prediction
NETMHC-II Pan 3.2 server (Jensen et al. 2018) was used to screen out single best interacting Epitope to maximum number of HLA-DRB alleles, it is based on artificial neural network (ANN) algorithms. Predicted epitope subjected to VaxiJen server (Doytchinova and Flower 2007) for deriving best antigenicity criterion (> 1.1). NETMHC-II PAN 3.2 server and VaxiJen results in Table 1 depict the 1-log50k value, affinity, and antigenicity criterion of selection for epitope and HLA alleles. Here, we found MIGLLSSRI as a putative epitope that exhibits interactions with all selected HLA-DRB alleles of MHC-II. As the NETMHC server for screening epitopes is based on ANN, makes it efficient in prediction of epitopes with considered HLA alleles. Figure 2 is a graphical representation of 1-log50k (affinity) values for better understanding of epitope selection.
Table 1.
Selected epitope from surface glycoprotein interacting with MHC-II HLA alleles
| Protein name | MHC-II allele | Epitope | 1-Log50k (aff) | Affinity (nM) | VaxiJen score (antigenicity) | Consideration |
|---|---|---|---|---|---|---|
| Surface-glycoprotein (AIA08876.1) | HLA-DRB1*01:01 | MIGLLSSRI | 0.539 | 147.39 | 1.2848 | Antigen |
| HLA-DRB1*04:01 | MIGLLSSRI | 0.254 | 3192.94 | 1.2848 | Antigen | |
| HLA-DRB1*04:05 | MIGLLSSRI | 0.259 | 3018.98 | 1.2848 | Antigen | |
| HLA-DRB1*07:01 | MIGLLSSRI | 0.390 | 734.34 | 1.2848 | Antigen | |
| WLLIPTVTL | 0.415 | 563.87 | 0.5306 | Non-antigen | ||
| HLA-DRB1*09:01 | MIGLLSSRI | 0.372 | 893.83 | 1.2848 | Antigen | |
| HLA-DRB1*13:02 | MIGLLSSRI | 0.389 | 746.43 | 1.2848 | Antigen | |
| HLA-DRB1*15:01 | MIGLLSSRI | 0.318 | 1607.18 | 1.2848 | Antigen |
Fig. 2.
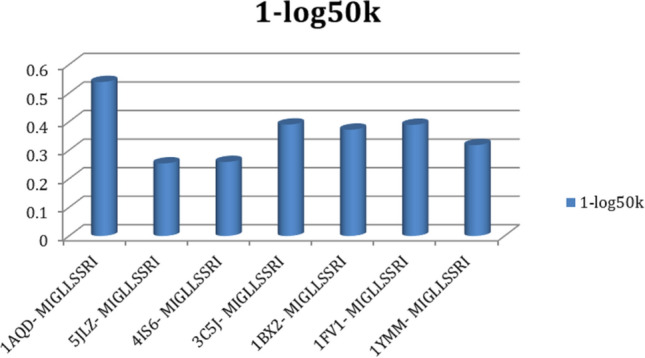
1-log50k values plotted against epitope MIGLLSSRI and HLA alleles
The structure for epitope MIGLLSSRI is obtained from the fast structure prediction De Novo method by deploying PEP-FOLD3.5, it was found to be helical in shape. Figure 3 shows its predicted structure viewed in PyMOL and probability plot obtained from PEP-FOLD server.
Fig. 3.

Structural prediction-related aspects of Epitope obtained from surface glycoprotein of Hantavirus by deploying Pepfold-3.5 A MIGLLSSRI epitope structure view in PyMOL. B Probability plot obtained during structural prediction
Structural analysis and toxicity prediction
Toxicity analysis and Ramachandran plot analysis provide essential information about peptide toxicity and secondary structure. ToxinPred server designing peptide module allows us to generate all possible scores and help in predicting whether the considered peptide is toxic or not, it is based on QM (− 33.50) and SVM (− 0.56) scores (Table 2). Molprobity was deployed to generate Ramachandran plot for selected epitope, and it was found to have good acceptance (more than 85% in allowed region) of structure (Fig. 4). Also, half-life prediction was estimated to 30 h in mammalian reticulocytes by deploying ExPASy tool (ProtParam) (Gasteiger et al. 2005).
Table 2.
Biochemical prediction and toxicity analysis (on the basis of QM and SVM score) for selected Epitope using ProtParam and ToxinPred tools
| Peptide | QM-SCORE | SVM SCORE | Prediction | GRAVY-Score | Half-Life (Mammalian reticulocytes) | Theoretical pI | Charge | Molecular weight |
|---|---|---|---|---|---|---|---|---|
| MIGLLSSRI | − 33.50 | − 0.56 | Non-toxin | 1.33 | 30 h | 9.50 | 1.00 | 989.38 |
Fig. 4.

Ramachandran plot analysis for screened Epitope MIGLLSSRI using MolProbity web server
Structures of HLA-DRB alleles were retrieved from RCSB-PDB database enlisted in Table 3. The selected HLA alleles are DRB*01:01, DRB*04:01, DRB*04:05, DRB*07:01, DRB*09:01, DRB*13:01, DRB*15:01.
Table 3.
Structure PDB ID for HLA-Allele DRB1 (MHC Class II)
| MHC-II allele | RCSB-PDB code | Structure |
|---|---|---|
| HLA-DRB1*01:01 | 1AQD | Crystal structure |
| HLA-DRB1*04:01 | 5JLZ | Crystal structure |
| HLA-DRB1*04:05 | 4IS6 | Crystal structure |
| HLA-DRB1*07:01 | 3C5J | Model |
| HLA-DRB1*09:01 | 1BX2 | Crystal structure |
| HLA-DRB1*13:02 | 1FV1 | Crystal structure |
| HLA-DRB1*15:01 | 1YMM | Crystal structure |
Molecular docking analysis
Molecular docking studies assist in examining the best binding scores, atomic contact energies and global energies for docked complexes (Table 4). For this, DINC server was used as it is based on Autodock software and also Patch-Dock along with Fire-Dock screening to generate more effective aspects of interaction. A comparison of energy values against docking complexes is graphically represented in Fig. 5.
Table 4.
Docking results obtained from DINC server and PatchDock-Firedock analysis
| Docking-complex | Binding score (kcal/mol) | ACE value | Global energy |
|---|---|---|---|
| 1AQD-MIGLLSSRI | − 5.90 | − 8.79 | − 49.07 |
| 5JLZ-MIGLLSSRI | − 6.50 | − 3.46 | − 15.16 |
| 4IS6-MIGLLSSRI | − 6.20 | − 22.89 | − 55.35 |
| 3C5J-MIGLLSSRI | − 6.40 | − 7.06 | − 33.44 |
| 1BX2-MIGLLSSRI | − 7.00 | − 10.89 | − 53.94 |
| 1FV1-MIGLLSSRI | − 7.40 | − 10.87 | − 52.59 |
| 1YMM-MIGLLSSRI | 6.60 | − 6.46 | − 46.19 |
Fig. 5.
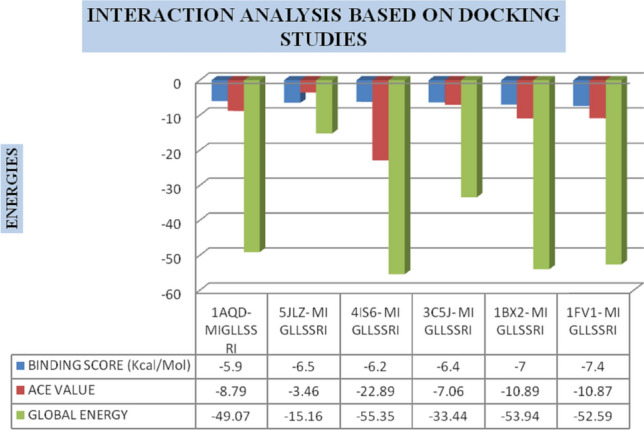
Interaction analysis based on docking studies: graphical representation of Binding score, ACE value, and global energy for docked complexes
The best docked interaction complex 1FV1-MIGLLSSRI is represented in Fig. 6, as it shows a binding score of − 7.4 kcal/mol, ACE value − 10.87, and global energy − 52.59. This epitope MIGLLSSRI exhibits better interactions with other HLA alleles too. This makes the epitope the most suitable candidate for crafting peptide-based vaccine.
Fig. 6.

Best docking result for 1FV1-MIGLLSSRI obtained by DINC web server. A Ligand in binding pocket (Far view). B Ligand in binding pocket (zoom view). C Docked epitope or ligand on binding pocket visualized in PyMOL
Molecular dynamics and simulation
Molecular simulation investigations assisted to reveal the interaction of MIGLLSSRI epitope on the basis of RMSD value per residue. RMSD values (0–1 nm) and RMSF values (1–1.5 nm) for docked complexes are represented by results of Gromacs tool in Fig. 7. Trajectory analysis for docked complex dynamic simulation was conducted for 10 ns. The RMSD and RMSF plots clearly represent less fluctuation under the simulation time span that makes the interaction more stable between HLA allele and selected epitope MIGLLSSRI.
Fig. 7.
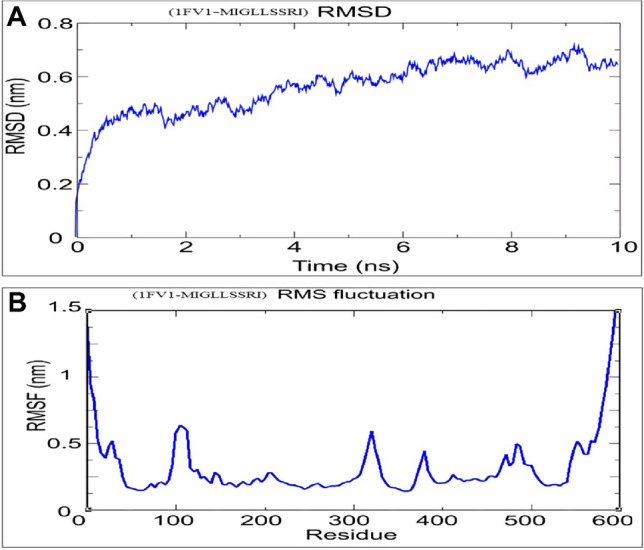
Simulation results for docked complex 1FV1-MIGLLSSRI. A RMSD value for docked complex at 10 ns. B RMSF plot for all amino acid side chains
Population coverage analysis
IEDB Population conservancy analysis outcomes were 62.96%, 70.16%, 66.48%, 59.61% population coverage for World, USA, Europe and India, respectively. These are satisfactory results for considering MIGLLSSRI epitope for crafting vaccine (Fig. 8). MHCPred server has three HLA-DRB1 alleles (01*01, 04*01, 07*01) and results on LogIC50 value (Table 5) were also found satisfactory and provide more confidence in epitope selection.
Fig. 8.
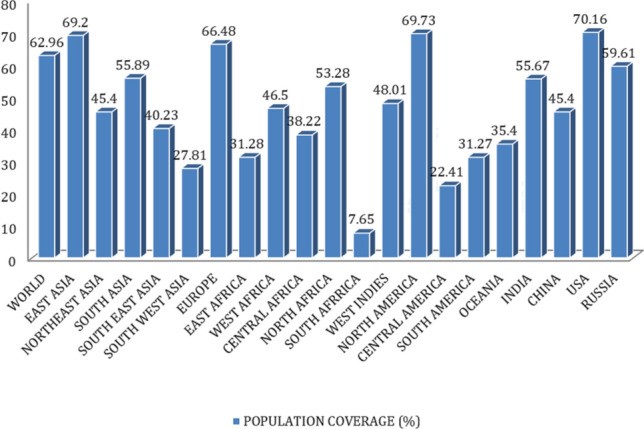
IEDB-population conservancy analysis of the selected epitope “MIGLLSSRI”
Table 5.
MHCPred 2.0 results for MHC-II HLA alleles
| Epitope-HLA allele | LogIC50 value (M) |
|---|---|
| MIGLLSSRI-HLA-DRB1*01:01 | 8.180 |
| MIGLLSSRI-HLA-DRB1*04:01 | 5.608 |
| MIGLLSSRI-HLA-DRB1*07:01 | 6.889 |
In-silico cloning
JCat server results show GC content of about 48.148% and CAI value 1.0 with improved or optimized codon sequence in E. coli K12, i.e., “ATGATCGGTCTGCTGTCTTCTCGTATC” for epitope MIGLLSSRI. This clearly indicates a high expression of vaccine candidate in E. coli K12 strain (Ali et al. 2017). So, this epitope can be easily used for vaccine formulations against Hantavirus after wet-lab validation.
Discussion
Orthohantavirus group of virus is a major etiological threat as associated with Hantavirus cardio-pulmonary syndrome in North American (Richardson et al. 2013), Korean and Chinese populations. Immunologically HLA haplotypes of hosts are major determinants to develop B-cell and T-cell immune response (Hooper et al. 2013). The current study primarily focuses on screening peptide from glycoprotein present in Orthohantavirus, which can exhibit interaction with MHC Class II HLA alleles, and elicit adaptive immune response in the human host domain. For this purpose, NETMHC-II pan 3.2 server, molecular docking, molecular simulations, and population coverage were applied. The surface or envelope glycoproteins create heterodimers, which then form tetrameric spikes, which are the lattice's building components. Viral entry is achieved by receptor-mediated endocytosis and endosomal membrane fusion, and glycoproteins are the exclusive targets of neutralizing antibodies (Serris et al. 2020), so we targeted SG protein to develop immunogenic therapeutic peptide. This study reveals the putative epitope for crafting vaccine against this harmful pathogen. It is an easy and robust methodology to apply and beneficial as it saves time consumption and money (Kaushik 2019; Sharma et al. 2020) along with restricting epidemic situations. In 2017, the repeat of worldwide cases of HFRS brings this old illness under research domain which truly compromises the human well-being. HFRS (Hemorrhagic fever with renal syndrome) in China was yet a regular central ailment with generally high dismalness and casualty; what is more, its appropriation and plague patterns had likewise changed. Observation measures, along with counteraction and control methodologies, ought to be improved and fortified to diminish HFRS disease in China. In this way, the best arrangement is to induce antibody production to forestall the spread of Hantavirus infection. Protein subunit vaccines are safe and simple to deliver, with adjuvant practices aiming obstruction between the segments of multivalent motifs of surface glycol protein of Hantavirus (Sun et al. 2017; Liang et al. 2018; Qu et al. 2018). To determine the above issues, we propose to build an all-inclusive hereditary designed novel subunit protein antibody against Hantavirus by consolidating bioinformatics strategies, i.e., viral surface protein structure science information. The most elevated antigenic proteins were exposed to the NetMHC server and an aggregate of 3769 epitopes with a joined score (1-log50k ≥ 0.6) were anticipated. The anticipated epitopes were considered to explore their antigenicity, immunogenicity, conservancy, harmfulness, and their separate MHC HLA alleles. From that point, the anticipated epitopes were subjected to docking-simulation parameters to screen whether the epitopes effectively fulfill all the rules. Afterward, these epitopes were tried for biochemical parameters testing and population conservancy analysis to determine the susceptible nature. Lastly, the best epitope (i.e., MIGLLSSRI) from considered surface glycoprotein was chosen as an immunization applicant that has attributes superior to other epitopes of that protein. In recent studies, it was found that SG protein was found to be main factor for immunogenic peptide development by deploying proteomic analysis study (Abdulla et al. 2021). Similar methodology was applied for successful prediction of SARS-CoV-2 epitopes in recent studies (Akhtar et al. 2020; Joshi et al. 2020a). Many other recent studies successfully demonstrated that epitope-based vaccine designing was successful against Dengue virus (Krishnan et al. 2020, 2021), Candida fungus (Akhtar et al. 2021a), Human cytomegalovirus (Akhtar et al. 2021b), Canine circovirus (Jain et al. 2021) and bacterium Tropheryma whipplei (Joshi and Kaushik 2021). This study will open new prospects in developing vaccine agents against Hantavirus as it is economically very beneficial and is a viable alternative to cumbersome, long hit-and-trial wet-lab experiments.
Conclusion
Immunoinformatic methodologies are very useful in the modern era, and can be easily implemented, but extensive studies in revealing further new epitopes and wet-lab screening is a must for any immunotherapy. Our predicted epitope “MIGLLSSRI” passes all in-silico parameters and makes a way directing modern vaccine designers to go for specificity rather than getting involved in long hit-and-trial experiments. The molecular dynamics and simulation studies along with population coverage and expressive behavior of this epitope mark it as a possible choice in crafting a vaccine against Hantavirus.
Acknowledgements
The authors acknowledge computational services and sound supervision provided by the Department of Bioengineering and Biosciences, Lovely Professional University, Jalandhar, Punjab.
Author contributions
All authors (AJ, NMR, VK, AKU, and JS) equally contributed in conducting experiments and writing manuscript. Validation and supervision of manuscript was done by VK, AKU and JS.
Declarations
Conflict of interest
The authors hereby declare that they have no conflict of interest.
Ethical standards and approval
The authors did not perform any experiments on human or animals.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Amit Joshi, Email: amit34655@gmail.com.
Nillohit Mitra Ray, Email: nillohit.22151@lpu.co.in.
Joginder Singh, Email: joginder.15005@lpu.co.in.
Atul Kumar Upadhyay, Email: atul.upadhyay@thapar.edu.
Vikas Kaushik, Email: vikas.14664@lpu.co.in.
References
- Abdulla F, Nain Z, Hossain MM, Syed SB, Khan MSA, Adhikari UK. A comprehensive screening of the whole proteome of hantavirus and designing a multi-epitope subunit vaccine for cross-protection against hantavirus: structural vaccinology and immunoinformatics study. Microb Pathog. 2021;150:104705. doi: 10.1016/j.micpath.2020.104705. [DOI] [PubMed] [Google Scholar]
- Akhtar N, Joshi A, Singh B, Kaushik V. Immuno-Informatics quest against COVID-19/SARS-COV-2: determining putative T-cell epitopes for vaccine prediction. Infect Disord Drug Targets. 2020 doi: 10.2174/1871526520666200921154149. [DOI] [PubMed] [Google Scholar]
- Akhtar N, Joshi A, Kaushik V, Kumar M, Mannan MAU. In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb Pathog. 2021;155:104879. doi: 10.1016/j.micpath.2021.104879. [DOI] [PubMed] [Google Scholar]
- Akhtar N, Joshi A, Singh J, Kaushik V. Design of a novel and potent multivalent epitope based human cytomegalovirus peptide vaccine: an immunoinformatics approach. J Mol Liq. 2021 doi: 10.1016/j.molliq.2021.116586. [DOI] [Google Scholar]
- Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK. Exploring dengue genome to construct a multi-epitope-based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep. 2017;7(1):1–13. doi: 10.1038/s41598-017-09199-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes DA, Moll M, Devaurs D, Jackson KR, Lizée G, Kavraki LE. DINC 2.0: a new protein–peptide docking webserver using an incremental approach. Cancer Res. 2017;77(21):e55–e57. doi: 10.1158/0008-5472.CAN-17-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Altunkaya A. RCSB Protein Data Bank: structural biology views for basic and applied research. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibi S, Ullah I, Zhu B, Adnan M, Liaqat R, Kong WB, Niu S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci Rep. 2021;11(1):1–16. doi: 10.1038/s41598-020-80899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7(1):153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8(1):4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro I, Bellomo CM, López W, Coelho R, Alonso D, Bruno A, Martinez VP. Hantavirus pulmonary syndrome outbreaks associated with climate variability in Northwestern Argentina, 1997–2017. PLoS Negl Trop Dis. 2020;14(11):e0008786. doi: 10.1371/journal.pntd.0008786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E. et al. (2005) Protein Identification and Analysis Tools on the ExPASy Server. In: Walker J.M. (eds) The Proteomics Protocols Handbook. Springer Protocols Handbooks. Humana Press pp 571–607
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33(suppl_2):W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan P, Doytchinova IA, Zygouri C, Flower DR. MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003;31(13):3621–3624. doi: 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo WP, Lin XD, Wang W, Tian JH, Cong ML, Zhang HL, Xu J. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013;9(2):e1003159. doi: 10.1371/journal.ppat.1003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JW, Josleyn M, Ballantyne J, Brocato R. A novel Sin Nombre virus DNA vaccine and its inclusion in a candidate pan-hantavirus vaccine against hantavirus pulmonary syndrome (HPS) and hemorrhagic fever with renal syndrome (HFRS) Vaccine. 2013;31(40):4314–4321. doi: 10.1016/j.vaccine.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Joshi A, Akhtar N, Krishnan S, Kaushik V. An immunoinformatics study: designing multivalent T-cell epitope vaccine against canine circovirus. J Genet Eng Biotechnol. 2021 doi: 10.1186/s43141-021-00220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Nielsen M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154(3):394–406. doi: 10.1111/imm.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Wang PZ, Yu HT, Zhang Y, Zhao K, Du H, Bai XF. Development of a SYBR Green I based one-step real-time PCR assay for the detection of Hantaan virus. J Virol Methods. 2014;196:145–151. doi: 10.1016/j.jviromet.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Joshi A, Kaushik V. In-silico proteomic exploratory quest: crafting T-cell epitope vaccine against Whipple’s disease. Int J Pept Res Ther. 2021;27:169–179. doi: 10.1007/s10989-020-10077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Joshi BC, Mannan MAU, Kaushik V. Epitope based vaccine prediction for SARS-COV-2 by deploying immuno-informatics approach. Inform Med Unlocked. 2020 doi: 10.1016/j.imu.2020.100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Krishnan GS, Kaushik V. Molecular docking and simulation investigation: effect of beta-sesquiphellandrene with ionic integration on SARS-CoV2 and SFTS viruses. J Genet Eng Biotechnol. 2020;18(1):1–8. doi: 10.1186/s43141-020-00095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik V. In silico identification of epitope-based peptide vaccine for Nipah virus. Int J Peptide Res Ther. 2020;26(2):1147–1153. doi: 10.1007/s10989-019-09917-0. [DOI] [Google Scholar]
- Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Nielsen M. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40(W1):W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S, Joshi A, Kaushik V. T cell epitope designing for dengue peptide vaccine using docking and molecular simulation studies. Mol Simul. 2020;46(10):787–795. doi: 10.1080/08927022.2020.1772970. [DOI] [Google Scholar]
- Krishnan S, Joshi A, Akhtar N, Kaushik V. Immunoinformatics designed T cell multi epitope dengue peptide vaccine derived from non structural proteome. Microb Pathog. 2021;150:104728. doi: 10.1016/j.micpath.2020.104728. [DOI] [PubMed] [Google Scholar]
- Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44(W1):W449–W454. doi: 10.1093/nar/gkw329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanov L, Iheozor-Ejiofor RP, Lundkvist Å, Vapalahti O, Plyusnin A. Defining of MAbs-neutralizing sites on the surface glycoproteins Gn and Gc of a hantavirus using vesicular stomatitis virus pseudotypes and site-directed mutagenesis. J Gen Virol. 2019;100(2):145–155. doi: 10.1099/jgv.0.001202. [DOI] [PubMed] [Google Scholar]
- Liang H, Yang R, Liu Z, Li M, Liu H, Jin X. Recombinant Zika virus envelope protein elicited protective immunity against Zika virus in immunocompetent mice. PLoS One. 2018;13(3):e0194860. doi: 10.1371/journal.pone.0194860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes P, Clement J, Van Ranst M. Recent approaches in hantavirus vaccine development. Expert Rev Vaccines. 2009;8(1):67–76. doi: 10.1586/14760584.8.1.67. [DOI] [PubMed] [Google Scholar]
- Mahmud-Al-Rafat A, Sobhani ME, Taylor-Robinson A (2015) Effects of anthropogenic events and viral persistence on rodent reservoirs of hantavirus infection understanding host-pathogen interactions facilitates novel approaches to intervention strategies. American Journal of Infectious Diseases and Microbiology 3(2): 77-86
- Qu P, Zhang W, Li D, Zhang C, Liu Q, Zhang X, Zhong J. Insect cell-produced recombinant protein subunit vaccines protect against Zika virus infection. Antivir Res. 2018;154:97–103. doi: 10.1016/j.antiviral.2018.04.010. [DOI] [PubMed] [Google Scholar]
- Richardson KS, Kuenzi A, Douglass RJ, Hart J, Carver S. Human exposure to particulate matter potentially contaminated with Sin Nombre virus. EcoHealth. 2013;10(2):159–165. doi: 10.1007/s10393-013-0830-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- Sanchez-Trincado, Jose L.; Gomez-Perosanz, Marta; Reche, Pedro A. (2017). Fundamentals and Methods for T- and B-Cell Epitope Prediction. Journal of Immunology Research, 2017, 1–14. [DOI] [PMC free article] [PubMed]
- Schmaljohn C. Vaccines for hantaviruses. Vaccine. 2009;27:D61–D64. doi: 10.1016/j.vaccine.2009.07.096. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(suppl_2):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serris A, Stass R, Bignon EA, Muena NA, Manuguerra JC, Jangra RK, Guardado-Calvo P. The hantavirus surface glycoprotein lattice and its fusion control mechanism. Cell. 2020;183(2):442–456. doi: 10.1016/j.cell.2020.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Kaur R, Upadhyay AK, Kaushik V. In-silico prediction of peptide based vaccine against Zika virus. Int J Pept Res Ther. 2020;26(1):85–91. doi: 10.1007/s10989-019-09818-2. [DOI] [Google Scholar]
- Sun J, Li M, Wang Y, Hao P, Jin X. Elaboration of tetravalent antibody responses against dengue viruses using a subunit vaccine comprised of a single consensus dengue envelope sequence. Vaccine. 2017;35(46):6308–6320. doi: 10.1016/j.vaccine.2017.09.063. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Zhang L. Multi-epitope vaccines: a promising strategy against tumors and viral infections. Cell Mol Immunol. 2018;15(2):182–184. doi: 10.1038/cmi.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]