

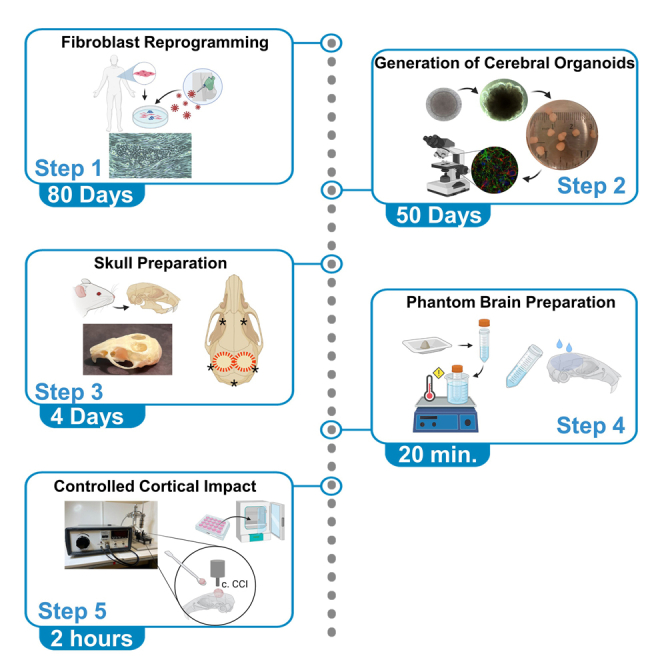
Summary
Modeling traumatic brain injury (TBI) has been a challenge. Rodent and cellular models have provided relevant contributions despite their limitations. Here, we present a protocol for a TBI model based on the controlled cortical impact (CCI) performed on human cerebral organoids (COs), self-assembled 3D cultures that recapitulate features of the human brain. Here, we generate COs from iPSCs obtained from reprogrammed fibroblasts.
For complete details on the use and execution of this protocol, please refer to Ramirez et al. (2021).
Subject areas: Neuroscience, Stem Cells, Organoids
Graphical abstract
Highlights
-
•
Steps for generating human cerebral organoids (COs)from iPSCs
-
•
Protocol to adapt controlled cortical impact system for use with human COs
-
•
Enables the study of traumatic brain injury in human COs
Modeling traumatic brain injury (TBI) has been a challenge. Rodent and cellular models have provided relevant contributions despite their limitations. Here, we present a protocol for a TBI model based on the controlled cortical impact (CCI) performed on human cerebral organoids (COs), self-assembled 3D cultures that recapitulate features of the human brain. Here, we generate COs from iPSCs obtained from reprogrammed fibroblasts.
Before you begin
This protocol describes a procedure to generate iPSC from fibroblast to further develop COs and perform TBI using the controlled cortical impact procedure. The execution of this protocol requires experience in advanced cell culture techniques, a sterile workspace, and specialized equipment. Further, it requires knowledge of mouse skull anatomy, experience on stereotaxic frames and the vernier scale, experience with controlled cortical impact procedures, and histology.
Choosing starting points
Cerebral organoids can be grown from iPSCs (Velasco et al., 2019). iPSCs can be derived from fibroblast (Malik and Rao, 2013). Fibroblast and human iPSC are commercially available. This protocol allows you to start from fibroblast or readily derived iPSCs.
Choosing the cerebral organoid culture strategy
TBI pathology is complex and involves multiple cell types present in the brain. Several in vitro models of TBI have recently been developed based on human iPSC-derived 2D cell culture models. However, using these models, it is difficult to comprehensively elucidate the changes at the level of network connections and the interactions of diverse nerve cell types, which is a key aspect of TBI pathology.
Cerebral organoids, derived from human iPSC, generate diverse cell types present in the human brain. Furthermore, COs can develop different brain-like regions, where diverse cell types maintain their brain-like positional organization and interactions to some extent. To generate COs, we followed an optimized method developed by StemCell Technologies. The reagents necessary to generate COs using this method are available to the scientific community in a kit format. Several groups, including the Lancaster group (Pellegrini et al., 2020; Giandomenico et al., 2019) and others (such as Zhao et al., 2020) have recently used this protocol to reliably generate COs.
Working with a high risk of culture contamination
Cerebral organoid cultures are maintained throughout all the processes in absence of antimicrobial reagents. Sterile conditions in your workspace are thus essential. To accomplish this protocol, you will require an autoclave, a hydrogen peroxide steamer for gas sterilization, and a biosafety cabinet.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MAP2 (1:400) | BD Pharmingen | Cat#556320 |
| Anti-Cleaved Caspase-3 (1:500) | Abcam | Cat#ab2302 |
| NSE (1:1000) | ProteinTech | Cat#10149-1-AP |
| Anti-Tubulin β 3 (TUBB3)/ TUJ1 (1:400) | BioLegend | Cat#801201 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 (1:500) | Invitrogen | Cat#A32744 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 (1:500) | Invitrogen | Cat#A32790 |
| Anti-SOX2 (1:200) | Abcam | Cat#ab97959 |
| Anti-FOXG1 (1:400) | Abcam | Cat#ab18259 |
| Anti-GFAP (1:500) | Abcam | Cat#ab7260 |
| OCT4 (1:200) | Stemgent | Cat#09-0023 |
| Anti-SSEA4 (1:200) | Abcam | Cat#ab16287 |
| Anti-TBR1 (1:200) | EMD Millipore | Cat#AB10554 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM High glucose medium | Sigma-Aldrich | Cat#D5796 |
| DMEM / F12 (1:1) (1X) | Gibco | Cat#11330-032 |
| 10% Fetal Bovine Serum | Gibco | Cat#10082147 |
| Glutamax | Gibco | Cat#25030081 |
| MEM-NEAA | Gibco | Cat#11140-050 |
| 0.05% Trypsin-EDTA | Gibco | Cat#25300054 |
| 1× PBS | HyClone | Cat#SH30256LS |
| Accutase | Sigma-Aldrich | Cat#SCR005 |
| Matrigel hESC-Qualified Matrix, ∗LDEV-Free | Corning | Cat#354277 |
| ReproTeSR medium | STEMCELL Technologies | Cat#05920 |
| mTeSR1 medium | STEMCELL Technologies | Cat#85850 |
| ReLeSR reagent | STEMCELL Technologies | Cat#05872 |
| HyClone FBS | Cytiva | Cat#SH300070.02 |
| Dimethyl sulphoxide | Sigma-Aldrich | Cat#D2650 |
| 10% Neutral Buffered Formalin v/v | Fisher-brand | Cat#032059 |
| Triton X-100 | Sigma-Aldrich | Cat#X100-500ML |
| FluorSave mounting medium | Merck Millipore | Cat#345789 |
| Gelatin from Porcine Skin | Sigma-Aldrich | Cat#G1890-500G |
| Agarose LE | Thomas Scientific | Cat#C748D75 |
| Xylene | Fisher scientific | Cat#X3S-4 |
| 100X Sodium Citrate Buffer | Abcam | Cat#ab64236 |
| 30% hydrogen peroxide solution | Sigma-Aldrich | Cat# CAS 7722-84-1 |
| ROCK Inhibitor Y-27632 | STEMCELL Technologies | Cat#72302 |
| TrypLE Express Enzyme (1X), no phenol red | Gibco | Cat#12604013 |
| Bovine Serum Albumin, heat shock fraction, suitable for RIA, pH 5.2, ≥96% | Sigma-Aldrich | Cat#A7888-50G |
| Critical commercial assays | ||
| CytoTune-iPS 2.0 Sendai virus (SeV) Reprogramming Kit | Thermo Fisher Scientific | Cat#A16517 |
| StemAb Alkaline Phosphatase Staining Kit II | Stemget | Cat#00-0055 |
| STEMdiff Cerebral Organoid Kit | STEMCELL Technologies | Cat#08570 |
| Experimental models: Cell lines | ||
| MRC-G Fibroblast culture | ATCC | Cat#ATCC® CCL171 |
| Experimental models: Organisms/strains | ||
| Mouse model | Skull only, strain C56Bl6; Male ∼3 Months old. | Cat#JAX-000664 |
| Other | ||
| Cell culture incubator-grade orbital shaker | Thermo Scientific | Cat#88881101 |
| Corning 96-Well, Cell Culture-Treated, Flat-Bottom Microplate | Fisher Scientific | Cat#07-200-627 |
| Corning 96-Well Clear Ultra Low Attachment Microplate | Fisher Scientific | Cat#07-201-680 |
| Corning 24-Well Clear Ultra Low Attachment Microplate | Fisher Scientific | Cat#07-200-602 |
| Corning 6-Well Clear Ultra Low Attachment Microplate | Fisher Scientific | Cat#07-200-601 |
| Nalgene Mr. Frosty isopropanol freezing container | Sigma-Aldrich | Cat#C1562 |
| Neubauer cell counting chamber | Hausser Scientific | Cat#5971R30 |
| Embedding surface | STEMCELL technologies | Cat#08579 |
| 0.11 μm PES membrane filter unit | N/A | N/A |
Materials and equipment
CCI Protocol: The Leica impactor one CCI device allows to calibrate 3 relevant parameters: speed, dwell time, and depth. Variations on these protocols may be necessary to adapt your model. (Osier and Dixon, 2017). We suggest a speed of 4 m/s, dwell time of 200 ms, and 1 mm depth as previously performed by our group (Edwards et al., 2020).
Microscope set up: the fluorophores used in this study are suitable for several fluorescence microscopes loaded with laser options at 405 nm, 458 nm, 488 nm, or 594 nm wavelengths. Scanning capabilities and confocal microscopy are highly recommended.
MEF culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM high glucose medium (Sigma D5796) | N/A | 440 mL |
| FBS (Gibco™ Cat.10082147) | 10% | 50 mL |
| Glutamax (Gibco 25030081) | 1X | 5 mL |
| MEM-NEAA (Gibco 11140-050) | 1X | 5 mL |
| Total | n/a | 500 mL |
Store at 4°C max 2 weeks.
Cryopreservation medium for iPSCs 2X
| Reagent | Final concentration | Amount |
|---|---|---|
| HyClone FBS (Cytiva SH300070.0 | 60% | 6 mL |
| mTeSR1 medium | 20% | 2 mL |
| Dimethyl sulphoxide (Sigma-Aldrich D2650) | 20% | 2 mL |
| Total | n/a | 10 mL |
Filter the medium using a 0.22 μm filter (Millex® SLGP033RS), Store at 4°C
EB formation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| STEMdiff™ Cerebral Organoid Basal Medium 1 | 80% | 40 mL |
| STEMdiff™ Cerebral Organoid Supplement A | 20% | 10 mL |
| Total | n/a | 50 mL |
Included in STEMdiff Cerebral Organoid Kit, StemCell Technologies 08570, Store at 4°C max 2 weeks.
EB Induction medium
| Reagent | Final concentration | Amount |
|---|---|---|
| STEMdiff™ Cerebral Organoid Basal Medium | 99% | 49.5 mL |
| STEMdiff™ Cerebral Organoid Supplement B | 1% | 0.5 mL |
| Total | n/a | 50 mL |
Included in STEMdiff Cerebral Organoid Kit, StemCell Technologies 08570, Store at 4°C max 2 weeks.
EB seeding medium
| Reagent | Final concentration | Amount |
|---|---|---|
| EB formation medium | X | 15 mL |
| ROCK inhibitor (10 mM stock) | 10 μM final | 15 μL |
| Total | n/a | 15 mL |
Keep at 4°C and use fresh.
Resuspension medium
| Reagent | Final concentration | Amount |
|---|---|---|
| mTeSR™ medium | X | 9 mL |
| ROCK inhibitor (10 mM) | 10 μM∗ | 10 μL |
| Total | n/a | 9 mL |
∗ Consider 1 mL of TrypLE™ existing on 6 well plates from the previous step. Keep at 4°C and use fresh
EB Expansion medium
| Reagent | Final concentration | Amount |
|---|---|---|
| STEMdiff™ Cerebral Organoid Basal Medium 2 | 97% | 24.25 mL |
| STEMdiff™ Cerebral Organoid Supplement C | 1% | 0.25 mL |
| STEMdiff™ Cerebral Organoid Supplement D | 2% | 0.5 mL |
| Total | n/a | 25 mL |
Included in STEMdiff Cerebral Organoid Kit, StemCell Technologies 08570, Store at 4°C max 2 weeks.
EB Maturation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| STEMdiff™ Cerebral Organoid Basal Medium 2 | 98% | 98 mL |
| STEMdiff™ Cerebral Organoid Supplement E | 2% | 2 mL |
| Total | n/a | 100 mL |
Included in STEMdiff Cerebral Organoid Kit, StemCell Technologies 08570, Store at 4°C max 2 weeks.
Sodium citrate buffer 1X
| Reagent | Final concentration | Amount |
|---|---|---|
| 100X Sodium Citrate Buffer | 1 X | 1 mL |
| ddH2O | 99 X | 100 mL |
| Total | n/a | 100 mL |
Store at 20°C to 25°C, adjust pH to 6.
Ethanol Solutions
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Ethanol | 70, 80, 90 and 95% | 350 mL; 400 mL; 450 mL; 475 mL |
| ddH2O | 30, 20, 10, and 5% | 150 mL; 100 mL; 50 mL; 25 mL |
| Total | n/a | 500 mL |
Anhydrous Histological, Fisher scientific, A405P-4. Store at 20°C to 25°C.
Phantom brain
| Reagent | Final concentration | Amount |
|---|---|---|
| 1× PBS | X | 30 mL |
| Agarose LE | 0.3% | 2.4 mg |
| Gelatin from porcine skin | 0.8% | 0.9 mg |
| Total | n/a | 30 mL |
Do not store, single use.
Step-by-step method details
Generation of iPSCs
Timing: 80 days
Throughout this section, you will find step-by-step instructions to successfully generate iPSC cells derived from fibroblast, including details of the necessary techniques and cell culture strategies (Figure 1). Further, you will also be able to generate cerebral organoids from the previously derived iPSCs. The major aim of this section is to consistently generate cerebral organoids.
-
1.MRC-5 fibroblast cell line cultureNote: Fibroblast cells (Figure 1A) are cultured onto 0.1% gelatin-coated in MEF culture medium (DMEM high glucose medium (Sigma D5796), 10% FBS (Gibco™ Cat.10082147), Glutamax (Gibco 25030081) and MEM-NEAA (Gibco 11140-050)) at 37°C in 5% CO2. Passages should be performed every 4–5 days using 0.05% Trypsin-EDTA.
-
a.Fibroblast passage
-
i.Coat 100 mm plates with 5 mL of 0.1% gelatin (Sigma™ G1890-500G), and incubate them for at least 2 h at 37°C.
-
ii.Wash cells with 5 mL of 1X PBS. Remove the PBS and add 1 mL of 0.05% Trypsin-EDTA, incubate for 5 min at 37°C.
-
iii.During the incubation, add 9 mL of MEF medium in a 15 mL plastic tube once the incubation is completed.
-
iv.Using a 5 mL serological pipette take 3 mL from the previous tube and resuspend the cells.
-
v.Transfer the cells back to the 15 mL tube and mix with the remaining 6 mL of medium.
-
vi.Centrifuge the 15 mL tube at 200 g for 5 min at 20°C–25°C and discard the supernatant.
-
vii.Resuspend the pellet in 1 mL of MEF medium. Tap the bottom of the tube to detach the cells and add 9 mL to reach a total volume of 10 mL.
-
viii.Remove the gelatin solution from the 100 mm plate previously prepared.
-
ix.Add 9 mL of MEF medium and 1 mL of the resuspended cells.
-
x.Shake the plate softly drawing an “8” shape at the incubator to homogeneously distribute the cells.
-
xi.Incubate at 37°C until the next passage.
 Pause point: Culture until 90% confluence before continuing, this may take 4–5 days.
Pause point: Culture until 90% confluence before continuing, this may take 4–5 days.
-
i.
-
a.
-
2.Fibroblast reprogramming strategy with SeV Cocktail. Troubleshooting 1
-
a.Prepare the cells for the CytoTune-iPS 2.0 Sendai virus (SeV) Reprogramming Kit (Thermo Fisher, A16517).
 CRITICAL: Before starting, coat a 6 well plate with 2 mL of 0.01% gelatin in MEF medium and allow it to polymerize for at least 2 h at 37°C.CRITICAL: Fibroblast culture is required to reach 90% of confluence before starting the reprogramming protocol.
CRITICAL: Before starting, coat a 6 well plate with 2 mL of 0.01% gelatin in MEF medium and allow it to polymerize for at least 2 h at 37°C.CRITICAL: Fibroblast culture is required to reach 90% of confluence before starting the reprogramming protocol.-
i.Wash the cells with 5 mL of 1X PBS, remove the PBS and add 1 mL of Accutase® and incubate for 4 min at 37°C.Note: Use of Accutase® instead of Trypsin is recommended at this stage by the reprogramming kit provider. TrypLE Express Enzyme can be used as well, although the investigator needs to optimize the incubation time.
-
ii.Prepare a 15 mL tube with 9 mL of MEF medium.
-
iii.Take 3 mL of MEF medium from the 15 mL tube to resuspend the cells. Use a 5 mL serological pipette.
-
iv.Transfer the cells back to the 15 mL tube and centrifuge at 200 g for 5 min at 20°C–25°C.
-
v.Discard the supernatant and resuspend the pellet in 1 mL of MEF medium.
-
vi.Add 9 mL more to reach a total of 10 mL.
-
vii.Count the cells and calculate the proper volume to take 1, 1.5, and 2 × 105 cells/well. Prepare 2 wells for each quantity of cells.Note: Different numbers of cells will give you three options to choose the best plate that is 30%–60% confluent the next day.
-
viii.Transfer each number of cells to an individual well in the previously coated 6-well plate.
-
ix.Resuspend the cells in a final volume of 2 mL per well.
-
x.Distribute the cells homogeneously by shaking the plate softly in an “8” shape at the incubator.CRITICAL: It is necessary to generate a homogeneous distribution of the cells.
-
xi.Culture the cells for 18 h at 37°C.CRITICAL: The cells should be at 30%–60% confluency.
-
i.
-
b.Cells’ transduction with CytoTune-iPS 2.0 Sendai virus (SeV) Reprogramming Kit (Thermo Fisher, A16517).Note: This kit is composed of three reprogramming viral vectors that express the four Yamanaka factors (Oct, Sox2, Klf4, and c-Myc). Prepare the cocktail in MEF medium following manufacturer instructions.Note: To compare the efficiency of Sendai virus-based reprogramming with other integration-free reprogramming methods (please see Schlaeger et al., 2015).
-
i.Add the proper amount of the cocktail to 2 mL of MEF medium to reach a ratio of hKOS, hc-cMyc, and hKlf4 of 5:5:3 MOI in respect to fibroblast cells number in each well.
-
ii.Incubate at 37°C for 24 h.Note: To estimate the cell number in each well detach the cells in one well of each concentration plated the day before, resuspend the cells in MEF medium and count them. Use the other well seeded at the same concentration of cells to perform the transduction.Note: The titer of the vectors is lot dependent. To make a proper calculation visit thermofisher.com/cytotune and search for the certificate of analysis by product lot number to have the correct titer for each vector.Note: This formula is provided by the manufacturer to do the calculations. MOI or multiplicity of infection, and CIU or cell infectious units:
Volume of virus (uL): MOI (CIU/cell) × number of cells Titer of virus (CIU/mL) × 10−3 (mL/μL) Calculation example: 7.5 uL of viral stock is required to infect 1.5 × 105 cells with MOI of 5 if the viral titer is 108 CIU/mL.CRITICAL: From this step until the iPSC colony passage, pre-treat all the used material with 10% bleach before discarding it.Note: Soon after transfection, iPSC colonies could be visible in a mixed population with fibroblast cells (Figure 1B). It is important to pipette softly during the passages, to not detach fibroblasts from the wells. This will allow keeping iPSC cultures free of fibroblasts. -
iii.Replace with 2 mL of fresh MEF medium daily for 5 days.
-
iv.On day 4, coat two 100 mm plates with hESC qualified Matrigel® according to manufacturer instructions and incubate 18 h at 37°C.CRITICAL: Matrigel® should be utilized in cold conditions, it will turn into a gel once over 10°C. Pre-cold 200 μL tips at −20°C for at least 2 h before using them. Defrost an aliquot of Matrigel® in ice for 1–2 h or at 4°C for 18 h; keep it on ice during the whole procedure. Use cold DMEM-F12 medium to dilute Matrigel®. Dilution factor (DF) is lot dependent. Find DF in the quality certificate. Half the amount of DF is enough to prepare 25 mL of the coating solution. Twelve mL of this solution is sufficient for one 100 mm plate. This Matrigel® dilution preparation will be required later on step 23.
-
v.On day 5, remove the medium and perform a passage using steps 8–10.
-
vi.Discard the supernatant and resuspend the cells in 1 mL of medium.
-
vii.Count the cells and take 150,000 and 200,000 cells and seed them in each Matrigel-coated plate.
-
viii.Culture seeded plates at 37°C for 18 h in a final volume of 2 mL MEF medium.
-
ix.On day 6, remove MEF medium and add 2 mL per well of ReproTeSR medium (StemCell technologies 05920) and perform daily medium changes until day 13.
- x.
-
i.
-
c.Isolation and culture of iPSC colonies
-
i.Prepare 12 well plates coated with Matrigel previously diluted in DMEM/F12 using 500 μL per well and incubate for 18 h at 37°C before reprogrammed cells transfer.Note: To perform the iPSC colonies transference, a microscope inside the biosafety cabinet is required.
-
ii.Using a 27G needle make squares over the selected colony to break it into pieces (Figure 1E).
-
iii.To transfer the cells, use a 200 μL tip and set the micropipette to 50 μL.
-
iv.Transfer each colony to a different well.
-
v.Incubate at 37°C for 18 h.CRITICAL: Select isolated colonies to ensure they are clones from one cell.Note: Keep culturing the cells in Matrigel-coated plates using mTeSR1 medium and perform passages at 60%–80% confluence.
-
i.
-
d.iPSC PassageNote: To perform iPSC passage, use ReLeSR reagent (StemCell technologies 05872).
-
i.First, remove mTeSR1 medium and wash the cells with 1 mL 1× PBS.
-
ii.Add 200 μL of ReLeSR to each well and incubate the plate for 1 min at 20°C to 25°C
-
iii.Remove ReLeSR and incubate for 2 min at 37°C.Note: Once the cells are placed into the new well, resuspend the cells softly 3 times to break the cells into smaller colonies (50–100 μm).CRITICAL: To avoid the transfer of fibroblast during the first set of passage, incubation with ReLeSR is recommended to be performed at 20°C–25°C, since iPSC will be released faster than fibroblast at this temperature. Also, in the following passages, you can see differentiated cells, and apply the same strategy to exclude them. Examples of differentiated cells in Figure 1G.
-
iv.Detach the cells using 500 μL of mTeSR1 medium.
-
v.Resuspend softly, no more than 2 times.
-
vi.In parallel, add 1 mL of mTeSR1 medium into a Matrigel-coated well plate after removing the coating medium and transfer the appropriate number of cells.CRITICAL: The number of iPSCs transferred into the new well will depend on the cellular confluence at the source well. With 65%–80% or higher confluence, 1:10 of the 500 μL (50 μL) can be transferred to the new well loaded with 1 mL of fresh mTeSR1. In a lower confluence (50%–65%) 1:5 (100 μL) is recommended.Note: Once all the fibroblasts have been eliminated, you can start culturing them in a bigger amount (e.g., 6 well plates). Keep using mTeSR1 medium.Note: Perform at least 10 passages before iPSC characterization and frozen stocks.
-
i.
-
e.Freezing iPSC stock
-
i.Prepare cryopreservation medium for iPSCs
-
ii.If you are starting from a well in a 6-well plate, remove the medium and add 1 mL of PBS to wash the cells.
-
iii.Remove PBS and add 500 μL of ReLeSR solution. Incubate for 1 min at 20°C–25°C and 2 min at 37°C.
-
iv.Remove ReLeSR and add 2 mL of mTeSR1 medium.
-
v.Resuspend the cells 3 to 5 times to detach them from the well.
-
vi.Transfer the cells to a 15 mL tube and add 5 mL of fresh mTeSR medium.
-
vii.Centrifuge the cells at 200 g for 5 min at 20°C–25°C.
-
viii.Discard the supernatant and resuspend carefully the cells in 1 mL of mTeSR1 medium.Note: If the confluence of the cells is less than 70% add 500 μL instead of 1 mL of mTeSR and cryopreservation medium and transfer the mixture to only one cryovial as explained in the next steps.
-
ix.Add 1 mL or 500 μL of cryopreservation medium and resuspend 3 times or until the medium looks completely homogeneous.
-
x.Transfer 1 mL or 500 μL of the mixture to 1 cryovial previously labeled with the name of the cell line, passage number, and date.
-
xi.Freeze the cells using a Nalgene® Mr. Frosty isopropanol freezing container (Sigma Aldrich C1562) that allows the temperature to decrease by 1°C/min.
-
xii.Keep the cells at −80°C for 18 h.
-
xiii.Store the cells in a liquid nitrogen tank.Note: In parallel to the generation of frozen stocks, keep culturing the cells to perform iPSCs characterization.
-
i.
-
f.Fixation of iPSC in the culture plate for characterization.
-
i.Before passage, coat 18 mm glass coverslips in a 12 well plate and incubate for 18 h at 37°C.
-
ii.Perform a passage (as listed in steps 26–27) using the plate with Matrigel-coated coverslips, and culture for 1 or 2 days, or until individual and compact colonies can be identified.
-
iii.Remove the mTeSR medium and wash quickly with 1 mL of 1× PBS. Remove PBS and add 1 mL of 4% PFA in PBS previously warmed at 20°C–25°C, and incubate for 15 min at 37°C.
-
iv.Remove the fixative solution and wash with 1 mL PBS. Incubate for 5 min at 20°C–25°C and replace with fresh 1 mL PBS.Pause point: At this point, cells can be stored at 4°C until performing the immunofluorescence assay.
-
i.
-
g.Immunofluorescence assay on iPSC cellsNote: The immunofluorescence assay will be performed in the culture plate.
-
i.Wash the cells once, with 1 mL PBS for 5 min at 20°C–25°C.
-
ii.For blocking/permeabilization incubate each well with 500 μL of 3% bovine serum albumin (Sigma-Aldrich A7888) in PBS 0.05 Triton™ X-100 (Sigma-Aldrich T8787) for 1 h at 20°C–25°C.
-
iii.Remove the blocking/permeabilization solution.
-
iv.Incubate with 400 μL of the different antibodies diluted in blocking/permeabilization solution for 18 h at 4°C.
-
v.Wash each well with 500 μL of PBS for 5 min at 20°C–25°C.
-
vi.Repeat the previous step 2 times.
-
vii.Incubate with 400 μL of fluorescent secondary antibodies, diluted in PBS 0.05% Triton X-100. Incubate for 1 h at 20°C–25°C.
-
viii.Wash each well with 500 μL of PBS for 5 min at 20°C–25°C.
-
ix.Incubate with 500 μL of DAPI solution (300 μM, in PBS).
-
x.Wash each well with 500 μL of PBS for 5 min at 20°C–25°C.CRITICAL: Prepare slides correctly labeled with the name of the cell line, colony number, passage, and antibodies used.
-
xi.Add 1 drop of FluorSave mounting medium (Millipore 345789) over each slide.
-
xii.Carefully, remove each coverslip from the well using the bent tip of a needle to release the coverslip and a tweezer to take it out of the well.
-
xiii.Flip it over the slide and put it on top of the mounting medium drop.
-
xiv.Press softly with a paper towel to eliminate the excess of medium and let the mounting medium dry for 1–2 h at 20°C–25°C, or for 18 h at 4°C.
-
xv.Seal the coverslips with transparent nail polish and keep the slides at 4°C. Protect from light until imaging. Do not store the slides in a vertical position until the nail polish is completely dry.Note: Examples of the results are shown in Figures 1H and 1I.
-
i.
-
h.Alkaline phosphatase (AP) staining
-
i.Before starting the alkaline phosphatase (AP) staining, culture cells onto Matrigel-coated coverslips as previously explained in step f.Note: AP is an enzyme highly expressed in iPSC. Expression of AP is related to undifferentiated state and self-renewal potential. In this protocol, we have performed the AP assay using the StemAb Alkaline Phosphatase Staining Kit II 00-0055, following the manufacturer's instructions.
-
ii.Wash the cells in the well with 1 mL PBS and fix for 2–5 min using the fixative solution provided by the kit. Do not exceed this time, over fixation can diminish AP activity.
-
iii.Incubate the cells 5–15 min at 20°C–25°C with the AP substrate away from light.
-
iv.Check the purple-red color formation and stop the reaction aspirating the solution and perform two washes with 1 mL PBS each.Note: Do not incubate the cells for extended periods to avoid non-specific staining. Cells that do not have AP expression will remain colorless.
-
v.Take the coverslips out of the plate and mount them in a slide using a hydrophilic mounting medium (not provided in the kit).
-
vi.Evaluate the results using brightfield microscopy.
-
vii.Examples of the results are shown in Figure 1H.Note: After the initial screening based on pluripotency markers, the chromosomal integrity, and pluripotency of the cells should be tested in detail using karyotyping trilineage differentiation potency, and expression of pluripotency gene array. These techniques are outside the scope of this manuscript. For further reading on the detailed characterization of iPSCs please see Baghbaderani et al. (2016).Pause point: iPSCs stocks from characterized colonies are frozen, ready to proceed with the generation of cerebral organoids.Alternative starting point. iPSCs are commercially available, if this is your case, proceed with the generation of cerebral organoids.
-
i.
-
a.
Figure 1.

Fibroblast reprogramming, iPSC colony picking, and iPSCs characterization for pluripotency
(A) Fibroblast culture.
(B) Morphological changes in reprogrammed fibroblast organized as small clusters of rounded cells, surrounded by fibroblast in culture.
(C) Early stage iPSC colony.
(D) Large iPSCs colony at day 21–28 after reprogramming procedure ready to be picked (Day 36 on the protocol).
(E) Scratching pattern for colony picking and the remaining cells after iPSC colony collection.
(F) iPSC colony after passages.
(G) A closed up of the edges of the iPSC colony. Arrows indicate differentiated cells around the iPSC colony.
(H and I) Markers of pluripotency in newly generated iPSCs; (H) Alkaline Phosphatase and, (I) SSEA4 and OCT4.
Generation of cerebral organoids
-
3.Embryoid body (EB) formation. Troubleshooting 2Note: Start with an iPSC culture with no more than 70%–80% confluence. If required, perform a starting passage as described in step 26.CRITICAL: Check pluripotency (compact colonies with well-defined edges, high nuclei to cytoplasm ratio) by morphology. Carefully remove differentiated cells by scraping them with a pipette tip, trying to leave iPSCs colonies intact.Note: Before starting cerebral organoid generation, prepare the EB formation medium, EB seeding medium and Resuspension medium.
-
a.Prepare the iPSC for the generation of EB
-
i.Aspirate iPSCs culture medium and wash the cells with PBS (1 mL for a 6 well plate).
-
ii.Add 1 mL of TrypLE™ (per 6 well plate) and incubate for approximately 3–5 min at 37°C.
-
iii.Gently dissociate the cells (3–5 times) in TrypLE using a 1 mL pipette and transfer the cell suspension to a 50 mL conical tube containing resuspension medium (9 mL).
-
iv.Centrifuge the cells at 300 g for 5 min.Note: At this step, the ROCK inhibitor in the resuspension medium will reach a final concentration of 10 μM.CRITICAL: Avoid over-pipetting the cells as it can induce damage.
-
v.Aspirate the supernatant and resuspend the cells in 1 mL of EB seeding medium.CRITICAL: Pipette up and down several times until single-cell suspension is obtained.
-
vi.Count the cells and calculate the appropriate volume of EB Seeding Medium needed to obtain 90,000 cells/mL.
-
vii.Plate 100 μL of single-cell suspension (Figure 2A) into each well of an ultra-low attachment 96-well round-bottom plate using a multichannel pipette.
-
viii.Place the plate in a humidified incubator at 37°C and 5% CO2.CRITICAL: Mix well before seeding. Resuspend cells every so often to avoid cell settling and to ensure an equal number of cells are seeded. Do not disturb the plate for 24 h.Note: Observe EBs under the microscope after 24 h, they should be round in shape with defined clear borders. Some dead cells can be seen surrounding the EBs (Figure 2B).
-
ix.On day 2, gently agitate the medium by pipetting up and down once, to detach dead cells around EBs.
-
x.Add 100 μL of EB Formation medium to the existing medium to each well using a multichannel pipette.
-
xi.On day 4, repeat the procedure as in day 2.
-
i.
-
b.EB induction
-
i.Prepare the EB Induction medium
-
ii.Add 500 μL of EB Induction medium to each well of an ultra-low attachment 24-well.
-
iii.Carefully transfer one EB to each well of an ultra-low attachment 24-well using a 200 μL wide-bore pipette tip.CRITICAL: To avoid transferring any formation medium, collect the EBs by pipetting up and let them fall by gravity until they settle at the opening of the tip. Gently, insert the tip into the well-containing Induction medium and without ejecting, let the EBs fall.CRITICAL: Avoid using tips with a small diameter as they can damage the EBs.
-
iv.Place the plate in a humidified incubator at 37°C and 5% CO2.Note: Observe EB under the microscope after 24 h, they should be round with defined and translucent edges (Figure 2B).
-
i.
-
c.EB embeddingCRITICAL: 1 day before starting the EB embedding procedure, thaw Matrigel® in ice at 4°C for 18 h or until completely melted (15 μL/EB, total of 1.44 mL for 96 COs). To prevent unwanted polymerization, keep Matrigel® on ice.
-
i.Prepare the EB Expansion medium and embedding surfaceNote: Prepare the embedding surface by cutting a piece of Parafilm® (approximately 4x4 cm), place it on a 96-well plate and apply digital pressure to create 16 small indentations (Figure 2C) To sterilize, immerse each Parafilm® piece in 70% ethanol, transfer to a 60 mm sterile cell culture plate and expose to UV radiation for 18 h. Alternatively, purchase an embedding surface from StemCell technologies (08579).
-
ii.Using a 200 μL wide-bore pipette tip, carefully transfer one EB to each indentation in the Parafilm®.
-
iii.Gently remove the excess medium, without disturbing EBs
-
iv.Add 15 μL of Matrigel® onto each EB.
-
v.Verify that EBs are in the center of the Matrigel®. If not in the center, gently move Matrigel® around EBs using a tip to move them.
-
vi.Incubate for 30 min at 37°C with 5% CO2.CRITICAL: Embed one plate at a time to prevent EBs from drying.
-
vii.Add 3 mL/well of Expansion medium to an ultra-low attachment 6-well plate.
-
viii.Using sterile forceps hold Parafilm® surface above one well and carefully wash off Matrigel® containing EBs using a 1 mL pipette with Expansion medium.
-
ix.Distribute EBs evenly among wells by incubating up to 12–16 EBs per well.
-
x.Place the plate in a humidified incubator at 37°C and 5% CO2.
-
i.
-
d.Organoid MaturationNote: The extension of the maturation stage can rank from 30 days to over 300 days, where they can achieve several millimeters in size (Figure 2F). For practical purposes, we will complete the maturation stage at 30 div. However, for the TBI model 80 div to 220 div are recommended.Optional: Maturation of organoids can be done in presence of antibiotics.
-
i.Prepare the EB Maturation medium
-
ii.Carefully, aspirate the medium using a sterile serological pipette from wells containing organoids. Do not disturb Matrigel® embedded organoids.Note: Medium change can also be performed with a glass vacuum pipette; however, we recommend this with bigger organoids as they can be accidentally aspirated.
-
iii.Replace the medium with 3 mL/well of maturation medium and place the plate with organoids on an orbital shaker (see below for rpm calculation) in a humidified incubator at 37°C and 5% CO2.Note: Calculate the rpm using the equation below.
Where:Rpm = shaker speed (revolutions per minute)RCF = relative centrifugal force (g)Throw = shaking diameter (mm), as specified by the manufacturerFor 6-well plates use the following data:Culture ware Volume of medium (mL) Recommended Shaker speed (rpm) Relative Centrifugal Force (RCF) † (g) 6-well plate
60 mm dish3.0 65 0.11808 -
iv.Change the maturation medium every 3–4 days.Note: Cerebral organoids will continue to grow and reach a size of approximately 3–4 mm in diameter after 30 days in Maturation Medium.
-
i.
-
e.Organoid Fixation
-
i.Transfer the number of organoids required, using a sterile metal surgical spoon to a 5 mL conical tube.
-
ii.Carefully, remove the remaining medium and wash once with 1 mL of PBS.CRITICAL: Make sure organoids are not damaged during the transfer. Alternatively, cut the tip of a 1000 μL pipette tip with sterile scissors to obtain a bigger tip opening.
-
iii.Aspirate PBS and gently resuspend in 1 mL of 10% neutral buffered formalin v/v (Fisher-brand, 032059).
-
iv.Remove formalin and add 4 mL of fresh formalin.
-
v.Incubate horizontally (to prevent organoid attachment) at 20°C–25°C for 24 h.Note: After fixation, organoids can be stored at 4oC in PBS, if required.
-
vi.After fixation, remove formalin, wash organoids carefully with 1 mL of 70% ethanol (Fisher scientific, A405P-4) and incubate for 24 h with 4 mL of 70% ethanol.
-
vii.Remove 70% ethanol and add incubate for 1.5 h with 4 mL of 80% ethanol, followed by 90% ethanol and 95% ethanol.
-
viii.Aspirate ethanol and incubate with 4 mL 95% ethanol for 18 h.
-
ix.Remove 95% ethanol and incubate two times with 4 mL of 100% ethanol for 1.5 h, discard the ethanol and perform two incubations with xylene (Fisher Scientific X3S-4) for 2 h.
-
x.Aspirate xylene and add 4 mL of fresh xylene, incubate for 18 h.
-
i.
-
f.Paraffin embedding
-
i.Aspirate xylene and add enough paraffin (Fisher Scientific T56-5) to cover organoids.
-
ii.Incubate at 70°C for 2 h.
-
iii.Repeat incubation with paraffin another 2 times before including organoids in molds (Fisher brand, 22-363-553).
-
iv.Let dry the molds for 18 h at 20°C–25°C before microtome sectioning.
-
v.Perform 10 μm sections.Pause point: Paraffin blocks can be stored until use.Optional: Alternatively, cerebral organoids can be fixed and cryopreserved for cryosection.
-
i.
-
g.Immunofluorescence. Troubleshooting 3
-
i.Place slides in histopathological staining racks and incubate for 20 min at 60°C.CRITICAL: Incubation at 60°C prevents sample detachment compared with higher temperatures.
-
ii.To remove paraffin from the samples, immerse slides in xylene for 8 min, remove and immerse again in fresh xylene for an additional 8 min.
-
iii.Continue to rehydrate samples by immersing slides in decreasing ethanol concentrations as follows: 100% ethanol for 3 min, remove and incubate with fresh 100% ethanol for an additional 3 min, followed by 95%, 90%, 80%, and 70% ethanol for 3 min each, respectively.
-
iv.Wash three times with 100 mL of PBS for 5 min each time with gentle agitation using the rinse container.
-
v.During the last washing step, heat 1X sodium citrate buffer (ab64236, Abcam) in the microwave until a temperature of 80°C is reached.Note: Use plastic staining jars for this procedure, as glass containers can crack.CRITICAL: Change microwave parameters to low power for the following step.
-
vi.Immerse the slides in a sodium citrate buffer and microwave for 10 min at low power.
-
vii.Remove from the microwave, let stand for another 20 min to cool to 20°C–25°C.
-
viii.Rinse slides once with warm tap water.
-
ix.Wash slides three times with PBS for 5 min each time with gentle agitation.
-
x.Remove slides from staining jars and carefully remove excess PBS without disturbing samples.
-
xi.Create a hydrophobic barrier by drawing around the sample using a PAP pen.
-
xii.Carefully, add enough blocking solution (3% BSA, 0.5% Triton X-100) to cover samples (approximately 200 μL) and incubate for 1–2 h at 20°C–25°C in a wet chamber.
-
xiii.Prepare primary antibodies in blocking solution and incubate at 20°C–25°C for 18 h in a wet chamber. Dilute primary antibodies according to the manufacturer’s instructions.Note: Examples and suggested dilutions for primary antibody: SOX2 (1:200, ab97959; Abcam), Tubulin β3 (1:400, 801201; BioLegend), FoxG1 (1:400, ab34735; Abcam), GFAP (1:500, ab7260; Abcam), MAP2 (1:400, 556320; BD Pharmingen) Cleaved Caspase 3 (1:500, ab2302; Abcam) and NSE (1:1000, 10149-1-AP; Proteintech).
-
xiv.Wash slides three times with PBS for 5 min each time with gentle agitation.
-
xv.Prepare the secondary antibodies in 0.2% Triton X-100 and incubate for 2 h at 20°C–25°C in a wet chamber
-
xvi.Incubate for 1 h at 20°C–25°C with the respective secondary antibodies. Anti-Mouse Alexa-594 (1:500, Invitrogen™ A32744) or Anti Rabbit Alexa-488 (1:500 Invitrogen™ A32790) antibodies. Repeat the washing step
-
xvii.Prepare DAPI (300 μM, in PBS) and immerse slides for 10 min at 20°C–25°C with gentle agitation. Wash with PBS for 5 min. Mount using FlourSave™ (Sigma 34788920ML) and visualize using a fluorescent microscope.Note:Figure 3 shows examples of staining of early (40 DIV) and late (200 DIV) organoids with different markers including SOX2, Tuj1 (Figure 3A), Ki67 (Figure 3B), Foxg1 (Figure 3C), TBR1 (Figure 3D), GFAP and MAP2 (Figure 3E). For a description of the listed markers, see previous publications (Lancaster et al., 2013; Gonzalez et al., 2018).
-
i.
-
a.
Figure 2.

Generation of cerebral organoids
(A) Single-cell suspension.
(B) EBs at day 1 (Day 85 of the protocol), revealing small and normal amounts of dead cells around the EB.
(C) Embedding surface.
(D) Picture of an organoid at day 8, 24 h after Matrigel® embedding showing a budding morphology (arrow), an indication of neuroepithelial formation (Day 90 of the protocol).
(E) An organoid on day 13 (Day 95 of the protocol), with greatly expanded neuroepithelia (arrow).
(F) Cerebral organoids at 40 DIV after EBs embedding (Day 120 of the protocol).
Figure 3.

Characterization of cerebral organoids
Paraffin sections of cerebral organoids at 40 DIV (early) and 220 DIV (late) were analyzed.
(A) 40 DIV organoids showing SOX2 (ventricular zone) and TUJ1 (inmature neurones) markers.
(B) 40 DIV organoids showing Ki67 and TUJ1 markers.
(C) 40 DIV organoids showing forebrain FOXG1 marker.
(D) 40 DIV organoids developing cortical preplate marked by TBR1 delineated by a dotted white line.
(E) 220 DIV organoids showing markers for astrocytes (GFAP) and neurons (MAP2).
Controlled cortical impact
Through this section, you will find step-by-step instructions to generate a mouse skull and a phantom brain, to successfully perform the controlled cortical impact on cerebral organoids under sterile conditions.
-
4.Skull Preparation.
-
a.Skull cleaning
-
i.Euthanize a C57Bl6 male mouse following your approved guidelines.
-
ii.With surgical tools (forceps, scissors) remove as much tissue as possible (Figure 4).
-
iii.Prepare a 50 mL plastic tube with 30% hydrogen peroxide (H2O2) solution and incubate for 18 h at 20°C–25°C (Sigma-Aldrich CAS 7722-84-1).Note: This procedure will allow you to remove soft tissue and any possible biological fluid or bloodstains from the bone surface.Note: H2O2 is classified as oxidizing, corrosive, and irritant. Safety measurements must be taken. Specific instructions are also required for disposal. Before use, consult the biosafety data sheet (https://www.sigmaaldrich.com/US/en/sds/sigma/h1009).
-
iv.Remove the sample from the 50 mL plastic tube and wash it 3 times with PBS for 5 min each.
-
v.Place the sample on a clean surface and complete the removal of soft tissue. Repeat steps 102–103 if necessary, with 10% hydrogen peroxide.Note: Microdissection tools are recommended to avoid damage to the skull.
-
vi.Once the skull is free of soft tissue let it dry at 20°C–25°C for 2 h.
-
vii.Store in a 15 mL plastic tube.
-
i.
-
b.Skull craniotomy and Sealing.CRITICAL: It is relevant to have basic knowledge of the mouse skull anatomy. For further references, we highly recommend reading Carretero et al. (2017).
-
i.Using a pencil, draw a circle with 3–4 mm of diameter on each parietal bone.
-
ii.Using a 1 mm drill bit with a speed of 5000–10000 RPM, carefully proceed with the skull opening following the drawing.
-
iii.Remove all debris using compressed air spray and remaining soft tissue from the inside of the skull with surgical tools.
-
iv.Prepare the dental cement following manufacturer recommendations.
-
v.Seal the palatine process, Cranio-pharyngeal channel, tympanic bulla, and the foramen Magnus.
-
vi.Leave the external auditory meatuses uncovered to fit the ear bars from the stereotactic frame as shown in Figure 4C.
-
vii.Store the skull in a 15 mL plastic tube until use.
-
i.
-
c.Gas sterilization.Note: Cerebral organoids are cultured in absence of any antimicrobial compound. Thus, sterility is key to the successful growth of organoids, and to perform the CCI procedure. Cleared bones with the previously explained method will help to provide a sample free of bacteria, although it is highly recommended to perform a sterilization method to secure the sterility of our sample. High-temperature strategies are not recommended to sterilize bones, thus gas-based methods are suitable for this purpose. Here we have successfully used hydrogen peroxide steamed gas, to perform the sterilization of skull and stereotactic equipment simultaneously.
-
i.Disassemble the stereotaxic frame and place all the parts inside a plastic box without the lid. Put the lid on a side with the inner surface facing up.
-
ii.Uncap the tube where the skull previously sealed and drilled was stored.
-
iii.Locate the tube with the skull in the plastic box with the stereotaxic frame.
-
iv.Apply the steamed gas sterilization procedure.
-
v.Once completed, seal the plastic box with the lid and sterilize the outer surface.CRITICAL: This procedure needs to be performed by trained personnel. Protection equipment is required. Hydrogen peroxide gas is a chemical hazard. The following equipment and sterilization protocol is recommended: SteraMist, (TOMI Environmental Solutions) using 7.8% hydrogen peroxide solution (“BIT Solution” – EPA registration 90150-2) sprayed through a cold plasma arc to form an aerosol of ionized hydrogen peroxide (iHP). The iHP is sprayed from 18-24” away from the surface. 5–10 s of contact time per 1 square foot of surface is required to be disinfected.
-
vi.Transfer the equipment and skull into a biosafety cabinet.CRITICAL: To perform this properly, place the container in front of a biosafety cabinet and transfer the frame pieces and the skull. UV light treatment can be performed for 18 h.
-
vii.Proceed with the assembly of the stereotactic frame.
-
viii.Open sterile PBS inside the biosafety cabinet and wash the skull for 10 min. Let it dry for 15 min inside the biosafety cabinet.
-
i.
-
a.
-
5.Phantom brain preparation.
-
a.Gelatin and agarose mixture preparation.CRITICAL: Prepare the mixture in a biosafety cabinet.
-
i.Add the agarose LE (Thomas Scientific™C748D75) and gelatin from porcine skin (Sigma™ G1890-500G) together into a 50 mL tube with sterile PBS to reach a final concentration of 0.3% agarose and 0,8% gelatin in a total volume of 30 mL.
-
ii.Boil the mixture on a hot plate until complete melting.Note: As an alternative, a microwave can be used to melt the mixtures. Extra precautions are required to avoid contamination.CRITICAL: Leave the tube lid partially open during the boiling procedure.
-
i.
-
b.Skull filling with the phantom brain. Troubleshooting 4
-
i.Transfer the phantom brain mixture in the liquid phase into the skull, using approximately 1 mL.
-
ii.Leave it to solidify for 15–20 min.Note: Use a 5 mL sterile syringe loaded with a needle no smaller than 20G. Secure the skull filled with the phantom brain in the stereotactic frame, using the ear and tooth bars. Figure 5B.Note: At this stage, the stereotactic frame and skull preparation is ready to receive the cerebral organoids for the CCI procedure. Before transferring the COs, clear your workspace by removal of all unnecessary equipment.
-
i.
-
a.
Figure 4.

Mouse skull preparation for the phantom brain
(A–D) (A) picture of the skull after removal of all soft tissue. The palatine process, Cranio-pharyngeal channel (B∗), tympanic bulla (C), and the foramen Magnus (D). The external auditory meatuses (C-#) (also known as acoustic pore) must be left uncovered to fit the ear bars from the stereotactic frame.
Figure 5.

Controlled cortical impact setup
(A) CCI equipment used in our experiments was Leica™ impact one device for electromagnetic control of the impact attached to a Stoelting™ stereotactic frame.
(B) Skull positioning using the ear bars and teeth bar.
(C) Positioning of the cerebral organoids using sterile spoons into the skull filled with phantom brain mixture.
(D) The ear bar takes direct contact with the phantom brain, allowing the equipment to determine the contact point between the cerebral organoid and impactor tip (zero Z-axis). Impactor and skull ready for impact. The red star identifies the contact sensor, which is clamped to the ear bar.
-
6.Controlled cortical impact.CRITICAL: CCI equipment calibration must be previously performed based on the researcher's aim.CRITICAL: Autoclave the impactor tip together with the stainless spoon previously mentioned, these tools will make direct contact with the organoids.
-
a.Positioning the cerebral organoids into the skull. Troubleshooting 5
-
i.Transfer the COs from the incubator to the biosafety cabinet. Proceed carefully.
-
ii.In the biosafety cabinet, carefully remove and replace the old media with fresh media.
-
iii.Using a sterilized stainless spoon, pick up a single organoid and place it on top of the phantom brain through the skull window to perform the CCI as shown in Figure 5C.
-
i.
-
b.Calibration of CCI equipment and impact procedure. Troubleshooting 6
-
i.Proceed with the impact and transfer the organoids back to a petri dish with 3 mL of fresh maturation media until the CCI procedure is completed.Note: The impact parameters must be carried as desired by the researcher, considering the impact speed, dwelling time, and depth. i.e., to induce a moderate to severe TBI, use the following parameters on the CCI equipment: (4 m/s), dwell time (200 ms), and depth (1mm).Note: Calibrate the impact depth using the vernier scale on the stereotactic frame in the Z-axis. Perform this depth adjustment after setting the zero with the contact sensor in the CCI equipment (Figure 5D).
-
i.
-
c.Cerebral organoid recovery and analysis.
-
i.After the CCI procedure, remove the media and replace it with 3 mL of fresh maturation media.
-
ii.Transfer the organoids back to the incubator.
-
iii.Keep the organoids in the incubator for the desired recovery time.Note: The extent of recovery time before analysis depends on the experimental design. However, 7 days were sufficient to evaluate the events associated with cellular response to primary injury after CCI. Long-term recovery times (100–180 days post-impact) can be also performed. In our experience, organoids impacted at early developmental stages (40–50 days in vitro) released cells to the media, however, we were able to culture them for up to 180–200 days after CCI.
-
i.
-
a.
Expected outcomes
In our experience, up to 80%–85% of the COs generated in this protocol can contain over 70% neuroepithelial tissue (Figure 2E). The efficiency can be further increased by prescreening organoids containing more than 70% or higher content of neuroepithelial structure at day 13 (Figure 2E). The investigator can decide the time to apply the CCI procedure based on the hypothesis tested. For example, we recommend performing the CCI in COs not earlier than 150 days in vitro (DIV) if the investigator wants to study the consequences of CCI in mature postmitotic neurons and astrocytes. At this stage, cerebral organoids should have developed postmitotic neurons and begun generating astrocytes. This will allow the study of changes on GFAP expression, all along with cellular morphology to address astrogliosis; reduction in MAP2 as an indicator of neuronal loss; upregulation of the neuron-specific enolase (NSE) as makers of metabolic changes related to neuronal damage; cleaved caspase 3 to evaluate cellular death.
Limitations
The protocol uses an agarose-based polymer to reproduce the stiffness and density of the brain. Although our approach requires an open skull, further development to apply the impact as a closed skull injury is desirable. However, for agarose-gelatin-based models, it is technically difficult since the liquid phase at high temperature could affect COs biology.
COs can produce a rich diversity of cells and brain-like structures. However, this model, as it is, currently lacks cell types and structural components necessary to model the complete spectrum of TBI pathology. In addition, lack of nutrient delivery tends to produce a necrotic core in the COs. Nevertheless, this is a rapidly developing area. Brain organoids can be enriched with microglia (Abud et al., 2017; Brownjohn et al., 2018) which is crucial for TBI pathology. Functional vasculature can be developed in brain organoids (Cakir et al., 2019; Shi et al., 2020), which can eventually help us model the cerebrovascular pathology associated with TBI.
Troubleshooting
Problem 1
Inappropriate coating due to premature polymerization of Matrigel®
Potential solution
Before starting a passage, check the Matrigel-coated plate under a microscope. Matrigel should look like small punctuated polymers homogeneously distributed in the plate. If they do not look homogeneously distributed, or bigger and darker particles are present, this could impair the iPSC culture. To avoid this problem, keep Matrigel always in ice and take it out of ice only to be added to the medium to prepare the plates. Use pre-cooled 200 μL tips to pipette Matrigel® and be aware to have DMEM/F-12 medium at 4°C to dilute it. Serological pipettes that will be used to pipette the diluted Matrigel also can be pre-cooled at 4°C for 15 min before being used. Prepared plates should be kept immediately at 4°C to be stored for up to 2 weeks or incubated at 37°C to be used.
Problem 2
Many dead cells around the EBs.
Potential solution
It is normal to find some dead cells around the EBs. However, too many dead cells can be a problem. This may happen due to (1) inadequate ROCK inhibition; (2) harsh handling of the cells while generating the single-cell suspension. iPSCs tend to become apoptotic while cultured as single cells. It is important to have the apoptosis inhibitor ROCK in the media as early as the single-cell suspension is generated. Please make sure that the ROCK inhibitor stock is not freeze-thawed multiple times. We recommend the use of freshly aliquoted ROCK inhibitors each time. Also, optimize minimum and gentle trituration to generate the single-cell suspension.
Problem 3
The COs generated contain mostly non-neuroepithelial tissues.
Potential solution
The generation of good quality COs depends on the quality of iPSC and the single-cell suspension. Even different clones of the same iPSC lines may produce COs with different efficiency. This is particularly the case for a protocol, such as this one, which is based on the spontaneous generation of neuroepithelium in the absence of any exogenously added patterning direction. For a new laboratory establishing this protocol, we recommend starting with a line, previously published to generate COs before moving into other lines of interest. The researcher must confirm that the iPSC quality is optimum before starting the procedure. The morphological quality of the iPSC colony can be evaluated by the compactness of the iPSC colony, defined ages, and high nucleolus to cytoplasm ratio. This evaluation requires experience in iPSC culture. It is critical to remove differentiated cells from iPSC culture before CO generation. The quality of the single-cell suspension is also crucial for quality CO generation. The researcher must ensure that single-cell suspension does not have a cluster of cells (Figure 2A). In addition, please make sure that single cells maintain spherical morphology with a regular border (Figure 2A). An irregular border may indicate apoptotic blebbing. Clusters and irregular morphology can be identified while counting cells using a hemocytometer. Use single-cell suspension with spherical cells and with no clusters. If these do not resolve the issue, please recheck the pluripotency and chromosomal integrity of the iPSC colony.
Problem 4
Leakage of non-solidified phantom brain mixture from the skull.
Potential solution
To avoid this situation, precaution measures are key. Before sterilization procedures, perform a brief test filling the skull with PBS. However, if the sterilization procedure was conducted and the skull is being prepared to receive the cerebral organoids and leaks of the liquid stage of the phantom brain mix are observed, do not complete the total volume. Instead, load only a few drops and keep the skull steady, wait for 3–5 min, and continue adding the phantom brain mix. Superficial tension and solidification of the small amount used will help to overcome the problem.
Problem 5
Cerebral organoids trapped inside the skull after impact.
Potential solution
If an organoid is trapped after impact, try to recover it by adding sterile PBS to the skull window. Excessive and direct manipulation of the sample is not desired. After adding PBS, remove the skull from the stereotaxic frame and try to transfer the sample by flipping the skull upside down. To avoid this situation, double-check the volume of phantom brain mix was adequate, and no pockets were left between the skull bone and the solidified mixture.
Problem 6
Rupture of brain organoids after impact.
Potential solution
The benefit of the CCI technique is the possibility of calibration of impact parameters. Double-check the deepness of impact on the CCI setup and recalibrate accordingly. Consider including a trial to evaluate the best parameters for your organoids, also consider including organoid transfer procedures to the skull and back to the culture plates. This will help you to train the fine motor skills required in this procedure.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Claudio Soto PhD., (Claudio.soto@uth.tmc.edu).
Materials availability
Materials and resources will be made fully available to not for-profit research institutions upon signature of a standard Materials Transfer Agreement.
Acknowledgments
We would like to acknowledge Dr. Jean Kim, the director of Human Stem Cell core at the Baylor college of medicine, for her valuable consultation during the generation of the iPSC line. We are also grateful to Dr. Brett M. Haltiwanger PhD., Safety manager at UTHealth, who provided access to the SteraMist equipment and service of gas sterilization to all our CCI equipment and Skull.
This work was partially funded by a Zenith Award from the Alzheimer’s Association to CS.
Author contributions
A.M., experimental design, protocol reviewer, cerebral organoid expert. S.E.S., cell culture, fibroblast reprogramming, iPSC culture, cerebral organoids generation. C.G., cerebral organoid culture, immunofluorescence studies, cerebral organoid protocol reviewer. .N.B.-V., cerebral organoid maintenance and CCI procedure. A.B.-C., cerebral organoid protocol reviewer. S.R., Experimental design, research coordinator, phantom brain developer, skull preparation, CCI procedure, imaging, data analysis and interpretation, manuscript preparation. C.S., experimental design and project supervisor, manuscript reviewer.
Declaration of interests
The authors declare no competing interests.
Data and code availability
Not applicable.
References
- Abud E.M., Ramirez R.N., Martinez E.S., Healy L.M., Nguyen C., Newman S.A., Yeromin A.V., Scarfone V.M., Marsh S.E., Fimbres C., et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron. 2017;94:278–293.e9. doi: 10.1016/j.neuron.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghbaderani B.A., Syama A., Sivapatham R., Pei Y., Mukherjee O., Fellner T., Zeng X., Rao M.S. Detailed characterization of human induced pluripotent Stem cells manufactured for therapeutic applications. Stem Cell Rev. Rep. 2016;12:394–420. doi: 10.1007/s12015-016-9662-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownjohn P.W., Smith J., Solanki R., Lohmann E., Houlden H., Hardy J., Dietmann S., Livesey F.J. Functional studies of missense TREM2 mutations in human stem cell-derived microglia. Stem Cell Rep. 2018;10:1294–1307. doi: 10.1016/j.stemcr.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakir B., Xiang Y., Tanaka Y., Kural M.H., Parent M., Kang Y.J., Chapeton K., Patterson B., Yuan Y., He C.S., et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods. 2019;16:1169–1175. doi: 10.1038/s41592-019-0586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero A., Ruberte J., Navarro M., Nacher V., Mendes-Jorge L. In: Morphological Mouse Phenotyping: Anatomy, Histology and Imaging. 1a ed. Ruberte J., Carretero A., Navarro M., editors. 2017. Osteology; pp. 7–53. [Google Scholar]
- Edwards G., Zhao J., Dash P.K., Soto C., Moreno-Gonzalez I. Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma. 2020;37:80–92. doi: 10.1089/neu.2018.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giandomenico S.L., Mierau S.B., Gibbons G.M., Wenger L., Masullo L., Sit T., Sutcliffe M., Boulanger J., Tripodi M., Derivery E., et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019;22:669–679. doi: 10.1038/s41593-019-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C., Armijo E., Bravo-Alegria J., Becerra-Calixto A., Mays C.E., Soto C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry. 2018;23:2363–2374. doi: 10.1038/s41380-018-0229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster M.A., Renner M., Martin C.A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N., Rao M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013;997:23–33. doi: 10.1007/978-1-62703-348-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osier N., Dixon C.E. Mini review of controlled cortical impact: a well-suited device for concussion research. Brain Sci. 2017;1462:172–192. doi: 10.3390/brainsci7070088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L., Bonfio C., Chadwick J., Begum F., Skehel M., Lancaster M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science. 2020;369:eaaz5626. doi: 10.1126/science.aaz5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S., Mukherjee A., Sepulveda S.E., Becerra-Calixto A., Bravo-Vasquez N., Gherardelli C., Chavez M., Soto C. Modeling traumatic brain injury in human cerebral organoids. Cells. 2021;10:2683. doi: 10.3390/cells10102683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger T.M., Daheron L., Brickler T.R., Entwisle S., Chan K., Cianci A., DeVine A., Ettenger A., Fitzgerald K., Godfrey M., et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Sun L., Wang M., Liu J., Zhong S., Li R., Li P., Guo L., Fang A., Chen R., et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. Plos Biol. 2020;18:e3000705. doi: 10.1371/journal.pbio.3000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco S., Kedaigle A.J., Simmons S.K., Nash A., Rocha M., Quadrato G., Paulsen B., Nguyen L., Adiconis X., Regev A., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570:523–527. doi: 10.1038/s41586-019-1289-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Fu Y., Yamazaki Y., Ren Y., Davis M.D., Liu C.C., Lu W., Wang X., Chen K., Cherukuri Y., et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer's disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020;11:5540. doi: 10.1038/s41467-020-19264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.